

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology [Internet]. 3rd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015-2017. doi: 10.1101/glycobiology.3e.047


Essentials of Glycobiology [Internet]. 3rd edition.
Show details
Altered glycosylation is a universal feature of cancer cells, and certain glycans are well-known markers of tumor progression. This chapter discusses glycan biosynthetic pathways that are frequently altered in cancer cells; correlations between altered glycosylation, diagnosis, and clinical prognosis; the genetic bases of some of these changes; and studies indicating their pathological importance.
HISTORICAL BACKGROUND
The earliest evidence of altered glycosylation as a hallmark of cancer was that some plant lectins show enhanced binding and agglutination of tumor cells. Next, it was found that transformation of cultured cells is frequently accompanied by a general increase in the size of radiolabeled glycopeptides of cell-surface glycoproteins. With the advent of monoclonal antibody technology, many “tumor-specific” antibodies directed against glycan epitopes were found, especially those borne on glycolipids. In many cases, these epitopes represented “onco-fetal antigens”—that is, glycan epitopes expressed on tumor cells and embryonic tissues. Like normal cells during embryogenesis, tumor cells undergo rapid growth, adhere to other cells and cell matrices, and invade tissues. Correlations between certain types of altered glycosylation and the prognosis of tumor-bearing animals or patients increased interest in glycan changes. In vitro cellular assays and in vivo animal studies have now supported the conclusion that glycan changes are critical to several aspects of tumor cell behavior. This chapter outlines the most common tumor-associated glycan changes in relation to biosynthetic mechanisms and their relevance to cancer.
GLYCOSYLATION CHANGES IN CANCER ARE NONRANDOM
Glycan changes in malignant cells take a variety of forms: loss of expression or excessive expression of certain glycans, increased expression of incomplete or truncated glycans, and, less commonly, the appearance of novel glycans. However, this is not simply the random consequence of disordered biosynthesis in tumor cells. It is striking that a very limited subset of changes are frequently correlated with malignant transformation and tumor progression. Given that cancer is a “microevolutionary” process in which only the fittest cells survive, it is likely that these specific glycan changes are selected for during tumor progression.
ALTERED BRANCHING AND FUCOSYLATION OF N-GLYCANS
Classic reports of increased size of tumor cell glycopeptides have now been partly explained by an increase in β1-6 branching of N-glycans (Figure 47.1), which results from enhanced expression of N-acetylglucosaminyltransferase V (GlcNAc-TV, GnT-V, MGAT5) (Chapter 9). Increased transcription of the MGAT5 gene is induced by various oncogenic transcription factors, as well as viral- and chemical-induced carcinogenesis. Cells with increased MGAT5 expression show an increased frequency of metastasis in mice, and spontaneous revertants lacking MGAT5 lose the metastatic phenotype. Some human tumors show increased staining with the plant lectin L-phytohemagglutinin (L-PHA) (Chapter 48), which preferentially recognizes branched N-glycans induced by MGAT5. Overexpression of Mgat5 cDNA in cultured cells causes a transformed phenotype (i.e., colony formation in soft agar, increased cell spreading, enhanced invasiveness through membranes, and ability to form tumors in immunocompromised mice). Most convincingly, Mgat5-deficient mice show a striking reduction in the growth and metastasis of mammary tumors induced by a viral oncogene. Likewise, metabolic inhibition of N-glycan synthesis by the plant alkaloid swainsonine (which blocks α-mannosidase II [MAN2A1], thereby preventing complete processing of N-glycans and preventing addition of the β1-6 GlcNAc branch; Chapter 55) inhibits tumor formation. Increased expression of MGAT4 to form a β1-4 branched tetra-antennary N-glycan also enhances tumor progression. Possible mechanisms by which increased N-glycan branching and extension enhances progression include lattice formation via galectin binding to poly-N-acetyllactosamines (LacNAc) leading to prolonged growth-factor signaling, and the generation of sialyl Lewis x (SLex) recognized by selectins because of increased outer-chain fucosylation (Figure 47.1). Increased expression of FUT8 that transfers the core Fuc to N-glycans (Figure 47.1) is also observed in solid tumors and promotes tumor progression in lung cancer cells. In contrast, enhanced expression of N-acetylglucosaminyltransferase III (GlcNAc-TIII, GnT-III, MGAT3) and the bisecting GlcNAc (Figure 47.1), observed in rat hepatomas and mouse mammary tumors, suppresses tumor progression and metastasis. Mice lacking MGAT3 show increased mammary tumors and lung metastases, and high MGAT3 expression correlates with better relapse-free survival in human breast cancer. However, liver tumors are reduced in mice lacking MGAT3. Thus, the effects of MGAT3 on cancer progression depend on tissue context.
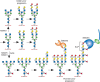
FIGURE 47.1.
N-Glycans increase in size on transformation, in part because of increased MGAT4 and MGAT5 activity, which catalyze GlcNAc branching of N-glycans. This may lead to increased numbers of LacNAc units, which can also be sialylated and fucosylated. The resulting (more...)
INCREASED MUCINS AND TRUNCATED O-GLYCANS
Overexpression of mucins in carcinomas was first observed in classic studies of episialin (now known as MUC1) on mouse tumor cells. Mucins are large glycoproteins that carry many O-GalNAc glycans on Ser or Thr in tandem repeat regions (Chapter 10). In normal polarized epithelium, mucins are expressed exclusively in the apical membrane, facing the lumen of a hollow organ, and soluble mucins are secreted exclusively into the lumen. Loss of adhesion junctions and topology in malignant epithelial cells destroys polarization, allowing soluble mucins to enter the extracellular space and the blood. The rod-like structure of mucins and their negative charge are thought to repel intercellular interactions and sterically inhibit other adhesion molecules such as cadherins and integrins from carrying out their functions. Thus, in some instances, mucins may act as “antiadhesins” that can also promote displacement of a cell from the primary tumor during the initiation of metastasis. Tumor mucins bearing selectin ligands facilitate several aspects of cancer progression (see below). Evidence suggests that they might also physically block interactions between blood-borne carcinoma cells and host cytolytic cells such as natural killer cells. In addition, mucins may mask presentation of antigenic peptides by major histocompatibility complex (MHC) molecules.
A hallmark abnormal feature of carcinoma mucins is their incomplete glycosylation, leading to the expression of Tn and T antigens (Figure 47.2). Secreted mucins expressing T, Tn, or sialyl-Tn (STn) often appear in the bloodstream of patients with cancer. Because these glycans occur infrequently in normal tissues, they provoke immune responses in the patient, which may be of use both prognostically and therapeutically. Clinical trials to deliberately provoke or enhance such immune responses are underway. Importantly, the best immune responses are elicited by the truncated glycans presented in arrays, exactly as on the mucins. The appearance of excessive Tn and STn on tumor cells has been shown in some instances to be correlated with silencing of the COSMC gene (C1GALT1C1). The β1-3 galactosyltransferase (C1GALT1) that synthesizes Galβ1-3GalNAcα1-Ser/Thr requires the chaperone C1GALT1C1 for activity. The gene for C1GALT1C1 is on the X chromosome, so a single mutation may be sufficient to eliminate expression. In other instances, STn accumulation results from overexpression of the sialyltransferase ST6GALNAC1 (Figure 47.2). The expression of Tn and STn can enhance tumorigenic and invasive properties.

FIGURE 47.2.
Loss of normal topology and polarization of epithelial cells in cancer results in secretion of mucins with truncated O-GalNAc glycans such as STn and Tn into the bloodstream. Tumor cells invading tissues and the bloodstream also present mucins on their (more...)
INCREASED AND ALTERED SIALIC ACID EXPRESSION
Early observations of wheat germ agglutinin binding to animal tumor cells are likely explained by an overall increase in cell-surface sialic acid (Sia), which binds factor H to limit complement activation, engages inhibitory Siglecs (Chapter 35) to suppress innate immune responses, and reduces attachment of tumor cells to the matrix, facilitating metastasis. Increases in α2-6 Sia on outer LacNAc units by up-regulation of ST6GAL1 enhances tumor progression by multiple mechanisms, including suppression of galectin-3-induced apoptosis, up-regulation of integrin activity, focal adhesion activity and cell motility, suppression of Fas death receptor functions, and generation of resistance to certain chemotherapeutic agents. However, α2-6 Sia may also inhibit epidermal growth factor (EGF) binding to the EGF receptor and thereby inhibit tumor progression. There can also be changes in Sia modifications in tumor cells. Sia 9-O-acetylation can be increased (e.g., 9-O-acetylated GD3 [Figure 47.3] in melanoma cells) or decreased (e.g., on the O-glycans of colon carcinomas). Some tumor cells express small amounts of de-N-acetyl (deNAc) gangliosides (Figure 47.3). O-Acetylation of gangliosides appears to protect tumor cells from apoptosis, and de-N-acetylgangliosides may stimulate tyrosine phosphorylation of the EGF receptor.
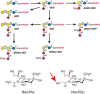
FIGURE 47.3.
Gangliosides expressed in human neuroectodermal tumors. Heavy arrows indicate up-regulated reactions; dashed arrow indicates a potential reaction. O-acetylation of Sia can occur at the 7- or 9-position. An O-acetyl group at position 7 will migrate to (more...)
Many years ago, patients with cancer were reported to express “Hanganutziu–Deicher” antibodies, which agglutinated animal red blood cells and were later shown to be directed against gangliosides carrying the nonhuman sialic acid N-glycolylneuraminic acid (Neu5Gc), which was also found in human tumors. This unusual phenomenon has now been explained by the metabolic incorporation of diet-derived Neu5Gc into human glycans (Chapter 15). This Sia differs from the usual human Sia, Neu5Ac, by addition of a single oxygen atom (Figure 47.3). Humans show variable levels of circulating antibodies against Neu5Gc-containing epitopes. The resulting weak immune response can promote tumor growth by enhancing chronic inflammation and angiogenesis. Given this selective advantage, it is not surprising that tumor cells are better at accumulating Neu5Gc in the face of an increased immune response. Because the primary dietary source of Neu5Gc is red meat (muscle from mammals), this may help explain consistent reports of increased cancer risk for those who consume red meat.
INCREASED SELECTIN LIGAND EXPRESSION
SLex and sialyl-Lewis a (SLea) glycan epitopes (Chapter 14) were first identified as tumor antigens on glycosphingolipids. Expression of these antigens by epithelial carcinomas correlates with metastatic potential in mice and tumor progression, metastatic spread, and poor prognosis in humans. SLex and SLea epitopes on ligand glycoproteins are key recognition determinants of the selectins (Chapter 34). Indeed, selectin ligands are also expressed on carcinoma cells, and mucin-like tumor antigens carrying SLex and SLea are found in the blood of carcinoma patients (Figure 47.4). Transgenic overexpression of E-selectin in mouse liver redirects syngeneic carcinoma cells that normally colonize the lung to liver, and metastasis is attenuated in mice lacking P-selectin or L-selectin. Selectin interactions also help explain the classic observation that cancer cells entering the bloodstream form thromboemboli with platelets and leukocytes, which facilitate arrest in the vasculature, assist extravasation through the endothelium, and help in evasion of the immune system. Indeed, the aberrant entry of tumor mucins with selectin ligands into the circulation can help explain some forms of “Trousseau's syndrome,” in which thrombotic phenomena precede the eventual diagnosis of an occult tumor. Of practical relevance is the finding that the glycosaminoglycan heparin (Chapter 17) is a potent inhibitor of P- and L-selectin interactions, and this is consistent with reports of benefits of heparin therapy in cancer and cancer-associated thrombosis. Another therapeutic strategy involves small-molecule inhibitors of selectin ligand formation (Chapter 34).
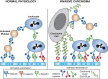
FIGURE 47.4.
In normal physiology platelets, leukocytes and endothelial cells interact via selectins and selectin ligands. In invasive carcinoma, interactions that promote invasion occur between tumor cells expressing selectin ligands and selectins on endothelial (more...)
ALTERED EXPRESSION OF BLOOD GROUPS
Loss of AB blood group expression in the context of carcinoma (accompanied by exposure of underlying H and Ley epitopes) (Chapter 14) is associated with poor prognosis in several studies. The Sda (or Cad) antigen, a blood group glycan abundantly expressed in the colon, is lost in colon carcinoma. Sulfation of the C-3 position of terminal Gal residues is also reduced in cancers. Sialyl-6-sulfo Lewis x and disialyl Lewis a, expressed on colonic epithelial cells, are reduced in colon cancer cells. These changes may reflect the enhanced production of SLex and SLea in cancers. DNA methylation and histone deacetylation, epigenetic mechanisms for suppression of gene transcription commonly observed in cancers, are proposed to underlie these glycan alterations. There are also rare instances in which a tumor may present a “forbidden” blood group structure (i.e., expression of a B blood group antigen in an A-positive patient) (Chapter 14). Regardless of the underlying mechanism, tumor regression has been noted in a few such cases, presumably mediated by the naturally occurring endogenous antibodies directed against the foreign structure. The appearance of P antigen in cancers of rare P blood type individuals is another example.
ALTERED EXPRESSION OF GLYCOSPHINGOLIPIDS
Many of the “tumor-specific” monoclonal antibodies raised against cancer cells recognize the glycan portion of glycosphingolipids. Some glycolipids are highly enriched in specific cancers (e.g., Gb3/CD77 in Burkitt's lymphoma and GM3, GD2, and GD3 in melanomas) (Figure 47.3). Several types of tumors (particularly melanoma and neuroblastoma) are characterized by the synthesis of very high levels of gangliosides (Chapter 11). Some of these (e.g., GD2) are not normally found at high levels in extraneural tissues and are therefore targets for both passive immunotherapy (monoclonal antibody infusion) and active immunotherapy (immunization with purified glycolipids). In some cases, gangliosides are also major carriers of modified Sias (Figure 47.3). Studies in cell culture suggest that some gangliosides promote cell growth. In contrast, strong immunosuppressive effects observed in vivo correlate with large quantities of gangliosides “shed” by some tumors.
LOSS OF GPI-ANCHOR EXPRESSION
A complete loss of glycosylphosphatidylinositol (GPI)-anchored proteins is seen in some cases of malignant and premalignant states involving the hematopoietic system. This results from acquired mutations in hematopoietic stem cells in the PIGA gene (required for an early step in GPI-anchor biosynthesis (Chapter 12). The PIGA gene is on the X chromosome (Xp22.1). GPI-anchored proteins are lost in the progeny of a single hematopoietic stem cell-mutant clone. This clone often gradually replaces healthy cells, giving rise to a syndrome called paroxysmal nocturnal hemoglobinuria or PNH (Chapter 12). Although the mutation itself does not confer a malignant phenotype, the presence of such an affected clone is sometimes a harbinger of leukemia.
CHANGES IN HYALURONAN
Many classes of malignant tumors express high levels of hyaluronan, a very large negatively charged polysaccharide composed of the repeating disaccharide unit [GlcAβ1-3GlcNAcβ1-4]n (Chapter 16). In carcinomas, hyaluronan is usually enriched in tumor-associated stroma (i.e., connective tissue, immune cells, and blood vessels). This stroma is more or less prominent depending on tumor type; for example, it is usually prominent in breast cancer. However, hyaluronan is also localized around the tumor cell surface. In normal tissues, hyaluronan serves at least three functions, which may also contribute to tumor progression. First, it increases levels of tissue hydration, which can facilitate movement of cells through tissues. Second, it is intrinsic to the assembly of extracellular matrices through specific interactions with other macromolecules, and thus it participates in tumor cell–matrix interactions that facilitate or inhibit tumor cell survival and invasion. Finally, hyaluronan interacts with several types of cell-surface receptors, especially CD44, which mediate or modify cell signaling pathways. These interactions, notably with alternatively spliced isoforms of CD44 that are elevated in most cancer cells, are often crucial to tumor malignancy and are a current target for novel therapies.
In normal adult tissues, hyaluronan appears to be relatively inert with respect to cell signaling and behavior. However, a very high molecular weight form of hyaluronan found in the naked mole rat, can inhibit spontaneous tumor formation. In addition, during embryonic development, tissue healing, and regeneration, and in various pathologies, hyaluronan-CD44 signaling becomes activated, possibly because of limited cleavage of hyaluronan by hyaluronidases. The consequences of this signaling are dramatic because they are essential to, or promote, proliferation, survival, epithelial–mesenchymal transition (EMT), and invasion, which are also key elements of the malignant phenotype (Figure 47.5). Hyaluronan–CD44 interaction at the tumor cell surface is required for the constitutive activation of some well-known oncogenes, especially receptor tyrosine kinases such as ErbB2, which is amplified or mutated in a large number of carcinomas. Accordingly, hyaluronan–CD44 interaction promotes downstream intracellular pathways that are also hallmarks of cancer, such as the phosphatidylinositol 3-kinase (PI3K)/AKT and mitogen-activated protein kinase pathways. Antagonists of hyaluronan–CD44 interaction cause inactivation of these pathways in malignant cells in culture and inhibit tumor growth and metastasis in animal models. Hyaluronan–CD44 interaction also stimulates multidrug resistance, and antagonists sensitize resistant cancer cells to chemotherapeutic drugs. Evidently, stimulation of receptor tyrosine kinase activities by hyaluronan leads to increased PI3K levels, which in turn stimulate new hyaluronan synthesis, thus setting up a positive feedback loop that amplifies antiapoptotic pathways. In addition, interactions of hyaluronan with its receptors are important for the activities of several types of multidrug transporters and metabolic transporters. These widespread effects may be due to stabilization of signaling platforms in the plasma membrane (e.g., lipid rafts) that are dependent on multivalent interactions of hyaluronan with CD44 or, in some cases, with another hyaluronan receptor (e.g., LYVE-1 or RHAMM/CD168) (Figure 47.5). Consequently, hyaluronan–receptor interactions are an important therapeutic target in cancer.

FIGURE 47.5.
Glycosaminoglycans (GAGs) in cancer. (A) Hyaluronan interacts multivalently with CD44, which interacts with several types of signaling molecules in lipid rafts in the plasma membrane, thus promoting cell survival, proliferation, invasion, and epithelial–mesenchymal (more...)
CHANGES IN SULFATED GLYCOSAMINOGLYCANS
Proteoglycans comprise core proteins that are decorated with covalently attached, negatively charged, sulfated glycosaminoglycan (GAG) side chains—namely, chondroitin sulfate (CS), dermatan sulfate (DS), keratan sulfate (KS), and heparan sulfate (HS) (Chapter 17). Although the content and distribution of many proteoglycans are altered during tumorigenesis, only HS side chains of proteoglycans have been implicated in tumor pathogenesis in a convincing manner. HS chains vary with respect to number and location of sulfate groups. HS chains are covalently linked to the core protein of proteoglycans (e.g., syndecans, glypicans, and perlecan). Other proteoglycans such as decorin, biglycan, versican and lumican, carrying CS, DS, or KS chains are also present in most tissues, including many tumor types.
Major functions of HS proteoglycans relevant to tumor formation are to promote cell–cell and cell–matrix interactions important in tissue assembly and to bind a wide array of bioactive factors such as fibroblast growth factor-2 (FGF-2), vascular endothelial growth factor (VEGF), hepatocyte growth factor, WNT, and numerous other cytokines and chemokines. Thus, HS proteoglycans can serve to build either inhibitory barriers or permissive pathways for cell invasion and can either sequester factors away from their receptors or present them efficaciously to those receptors (Figure 47.5). Syndecan-1 well illustrates the multiple important roles of HS, especially in promoting metastasis and angiogenesis. Early work suggested that syndecan-1 helps maintain the normal differentiated state of epithelia and that low levels of tumor-associated syndecan-1 correlate with malignancy. Recent work highlights the importance of enzymatic processing by proteases and heparanase, an enzyme that cleaves the HS chains at specific sites. High levels of proteolytically shed HS-rich ectodomain of syndecan-1 are present in sera of patients with myeloma as well as some types of carcinoma and predicts poor prognosis. High levels of syndecan-1 ectodomain also accumulate in the tumor microenvironment, where they play an important role in activation of chemokine and growth factor signaling and consequently tumor cell behavior. Especially significant is the finding that ectodomains promote metastasis to bone tissue, which is characteristic of myeloma and certain carcinomas (e.g., of breast or prostate) in human patients. The mechanism of ectodomain shedding appears to involve members of the matrix metalloproteinase and ADAM protease families.
HS side chains of several HS proteoglycans are cleaved to fragments containing 10 to 20 sugar moieties by vertebrate heparanase and endo-β-glucuronidase. Heparanase is elevated in numerous types of cancer, and increased heparanase activity can lead to induction of angiogenesis and metastasis. HS cleavage promotes blood vessel remodeling required for angiogenesis and release of HS-bound angiogenic factors, growth factors, and chemokines. In addition, heparanase products are more bioactive compared with intact HS proteoglycan. The removal of sulfate moieties from HS by endosulfatases also amplifies growth factor activities and tumor growth. These studies have led to increased interest in HS and its modifying enzymes as therapeutic targets in cancers.
CHANGES IN CYTOPLASMIC AND NUCLEAR O-GlcNAc
O-GlcNAc levels regulated by the O-GlcNAc transferase OGT and the hexosaminidase OGA have been found to be elevated in many cancers. Increased O-GlcNAcylation leads to enhanced cancer cell metabolism due to alterations in the stability or activity of transcription factors and kinases. Specific examples of proteins modified by O-GlcNAc which are directly implicated in cancer progression include c-Myc, cyclin D1, NF-κB/p65, PFK1, and SNAIL.
MECHANISMS OF ALTERED GLYCAN EXPRESSION
The Cancer Genome Atlas (TCGA) has identified many alterations in the expression of glycosylation genes that correlate with cancers. The mechanistic basis is known in some cases. Thus, transcription of MGAT5 (Figure 47.1) is induced by v-src, H-ras, and v-fps and the transcription factor ets-1. Another example is enhanced expression of SLex on adult T-cell leukemia cells, which show extremely strong tissue infiltrative activity, likely mediated by selectins. The transcriptional activator protein Tax of HTLV-1, which causes leukemia, activates transcription of the FUT7 gene, which encodes the fucosyltransferase that generates SLex in leukocytes. Under the hypoxic conditions found in advanced tumors, hypoxia-resistant cancer cells survive by up-regulating the transcription factor, hypoxia-inducible factor (HIF). HIF induces transcription of several glycosylation genes, leading to altered glycans, including enhanced SLex and SLea expression in cancer cells. Tumor hypoxia may also affect the expression of Neu5Gc in human tumor cells by up-regulating transcription of the lysosomal sialin Sia transporter. Down-regulation of glycosylation genes by DNA methylation or histone deacetylation/trimethylation may silence glycosylation genes that suppress tumor progression like MGAT3 or ST6GAL1. Posttranscriptional regulation of glycosylation pathways through microRNAs is also involved in tumor progression. For instance, miR-30b/30d are known to promote the metastatic behavior of melanoma cells by targeting the GalNAc transferase GALNT7.
GLYCAN CHANGES IN CANCER STEM CELLS AND DURING EMT
Cancer stem cells, or tumor-initiating cells, are the small subpopulation of cancer cells that has the ability to initiate tumors. Several glycans that are specific markers for embryonic stem cells (stage-specific embryonic antigen-3 [SSEA-3], SSEA-3 with fucose [Globo H], and SSEA-4) are also expressed by cancer stem cells. SSEA-1, an embryonic stem cell marker in mice, is found in cancer stem cells in human gliomas. Thus the expression of these glycans appear to be associated with the “stemness” of cells. The hyaluronan receptor, CD44, is also a marker of cancer stem cells and is functionally important for their properties.
Cancer stem cells are often investigated in the context of EMT. Cells that have undergone EMT are highly similar to cancer stem cells. EMT is a critical event in tumor progression that prepares cancer cells for metastasis. It is governed by several well-defined transcription factors such as SNAIL and ZEB. Specific changes of glycan expression are known to occur on EMT of human cancer cells, including appearance of GD1 ganglioside, and decreased expression of Gg4 and GM2 glycolipids. Expression of known tumor-associated glycans, such as β1,6-GlcNAc branched N-glycans, SLex and SLea, is also enhanced in cancer cells undergoing EMT.
CLINICAL SIGNIFICANCE
Several glycan antigens are used for detecting and monitoring the growth status of tumors. CA19-9 or SLea is a cancer antigen and was discovered in serum of patients with colon or pancreatic cancers. It is used primarily to monitor pancreatic tumors. CA125 recognizes the mucin MUC16 and is used in monitoring ovarian cancer. Truncated O-GalNAc glycans Tn, STn, and T (Figure 47.2) attached to MUC16 from cancer cells enhance the specificity of diagnosis and monitoring of ovarian cancer. SLex-related glycans in lung and breast cancers can be used to monitor residual disease after surgery. In liver cancer, the core fucose on N-glycans can facilitate early diagnosis. In recent times, the failure of proteomics to deliver reliable cancer biomarkers has rekindled interest in glycomic profiling of serum and other body fluid glycoproteins. Classic findings of glycan-specific antibodies associated with cancer, such as the antibodies against the nonhuman Sia, Neu5Gc, are also being reinvestigated. Another potential advance lies in improving the specificity of known cancer biomarkers by defining glycoprotein glycoforms uniquely expressed by cancer cells (e.g., prostate-specific antigen).
Specific therapies that target glycans are being developed. Most advanced is immunotherapy against certain tumor-associated gangliosides like GD2 in melanoma and against incompletely glycosylated mucins. With regard to the increased β1-6 branching of N-glycans, clinical trials of the N-glycan synthesis inhibitor swainsonine unfortunately revealed toxicity. The potent effects of heparin in attenuating tumor metastasis are most likely not due to its anticoagulant properties but rather to its ability to block P- and L-selectin binding to tumor and/or host ligands (Chapter 34). The recent move to supplant traditional unfractionated heparins with very low-molecular-weight heparins and heparinoids may be associated with a loss of selectin-inhibitory properties. A comparative clinical trial is needed. Several compounds have been shown to antagonize the protumorigenic activities of HS. For example, low-molecular-weight heparins may inhibit tumor progression not only by blocking selectins, but also by blocking heparanase activity or by interfering with constitutive HS activities. Other inhibitors of heparanase, such as suramin, laminarin sulfate, and the sulfated phosphomannopentaose PI-88, also inhibit angiogenesis and metastasis, but some of these compounds may also work by blocking selectins. Some of these compounds may act by competitively inhibiting HS binding and function. Low-molecular-weight oligosaccharides of hyaluronan could also be useful therapeutically, because they inhibit the pro-oncogenic influences of constitutive polymeric hyaluronan, especially drug resistance and signaling events induced by hyaluronan–CD44 interaction. Disaccharides that can enter the cell and act as decoys to divert glycosylation pathways are also showing promise (Chapter 55).
ACKNOWLEDGMENTS
The authors appreciate helpful comments and suggestions from Morten Thaysen-Andersen and Jeremy O'Sullivan.
FURTHER READING
- Feizi T. 1985. Demonstration by monoclonal antibodies that carbohydrate structures of glycoproteins and glycolipids are onco-developmental antigens. Nature 314: 53–57. [PubMed: 2579340]
- Fuster MM, Esko JD. 2005. The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat Rev Cancer 5: 526–542. [PubMed: 16069816]
- Borsig L, Stevenson JL, Varki A. 2007. Heparin in cancer: Role of selectin interactions. In Cancer-associated thrombosis (ed. AA Khorana, CW Francis., editors. ), pp. 95–111. Taylor-Francis, Boca Raton, FL.
- Kannagi R, Yin J, Miyazaki K, Izawa M. 2008. Current relevance of incomplete synthesis and neo-synthesis for cancer-associated alteration of carbohydrate determinants—Hakomori's concepts revisited. Biochim Biophys Acta 1780: 525–531. [PubMed: 17980710]
- Toole BP. 2009. Hyaluronan–CD44 interactions in cancer: Paradoxes and possibilities. Clin Cancer Res 15: 7462–7468. [PMC free article: PMC2796593] [PubMed: 20008845]
- Kannagi R, Sakuma K, Miyazaki K, Lim KT, Yusa A, Yin J, Izawa M. 2010. Altered expression of glycan genes in cancers induced by epigenetic silencing and tumor hypoxia: Clues in the ongoing search for new tumor markers. Cancer Sci 101: 586–593. [PubMed: 20085584]
- Boscher C, Dennis JW, Nabi IR. 2011. Glycosylation, galectins and cellular signaling. Curr Opin Cell Biol 23: 383–392. [PubMed: 21616652]
- Schultz MJ, Swindall AF, Bellis SL. 2012. Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev 31: 501–518. [PMC free article: PMC4079276] [PubMed: 22699311]
- Miwa HE, Koba WR, Fine EJ, Giricz O, Kenny PA, Stanley P. 2013. Bisected, complex N-glycans and galectins in mouse mammary tumor progression and human breast cancer. Glycobiology 23: 1477–1490. [PMC free article: PMC3816629] [PubMed: 24037315]
- Dobrenkov K, Cheung NK. 2014. GD2-targeted immunotherapy and radioimmunotherapy. Semin Oncol 41: 589–612. [PMC free article: PMC4254523] [PubMed: 25440605]
- Knelson EH, Nee JC, Blobe GC. 2014. Heparan sulfate signaling in cancer. Trends Biochem Sci 39: 277–288. [PMC free article: PMC4065786] [PubMed: 24755488]
- Ma Z, Vosseller K. 2014. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J Biol Chem 289: 34457–34465. [PMC free article: PMC4263853] [PubMed: 25336642]
- Samraj AN, Laubli H, Varki N, Varki A. 2014. Involvement of a non-human sialic acid in human cancer. Front Oncol 4: 33 [Corrigendum: Involvement of a non-human sialic acid in human cancer. Front Oncol 4: 83]. [PMC free article: PMC4001053] [PubMed: 24795860]
- Hakomori SI, Handa K. 2015. GM3 and cancer. Glycoconj J 32: 1–8. [PubMed: 25613425]
- Pinho SS, Reis CA. 2015. Glycosylation in cancer: Mechanisms and clinical implications. Nat Rev Cancer 15: 540–555. [PubMed: 26289314]
- Taniguchi N, Kizuka Y. 2015. Glycans and cancer: Role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv Cancer Res 126: 11–51. [PubMed: 25727145]
- Alisson-Silva F, Kawanishi K, Varki A. 2017. Human risk of diseases associated with red meat intake: Analysis of current theories and proposed role for metabolic incorporation of a non-human sialic acid. Mol Aspects Med 51: 16–30. [PMC free article: PMC5035214] [PubMed: 27421909]
- Pearce OM, Lübli H. 2017. Sialic acids in cancer biology and immunity. Glycobiology 26: 111–128. [PubMed: 26518624]
- Yu AL, Hung JT, Ho MY, Yu J. 2017. Alterations of glycosphingolipids in embryonic stem cell differentiation and development of glycan-targeting cancer immunotherapy. Stem Cells Dev 25: 1532–1548. [PubMed: 27507584]
- Afshar-Kharghan V. 2017. The role of the complement system in cancer. J Clin Invest 127: 780–489. [PMC free article: PMC5330758] [PubMed: 28248200]
- HISTORICAL BACKGROUND
- GLYCOSYLATION CHANGES IN CANCER ARE NONRANDOM
- ALTERED BRANCHING AND FUCOSYLATION OF N-GLYCANS
- INCREASED MUCINS AND TRUNCATED O-GLYCANS
- INCREASED AND ALTERED SIALIC ACID EXPRESSION
- INCREASED SELECTIN LIGAND EXPRESSION
- ALTERED EXPRESSION OF BLOOD GROUPS
- ALTERED EXPRESSION OF GLYCOSPHINGOLIPIDS
- LOSS OF GPI-ANCHOR EXPRESSION
- CHANGES IN HYALURONAN
- CHANGES IN SULFATED GLYCOSAMINOGLYCANS
- CHANGES IN CYTOPLASMIC AND NUCLEAR O-GlcNAc
- MECHANISMS OF ALTERED GLYCAN EXPRESSION
- GLYCAN CHANGES IN CANCER STEM CELLS AND DURING EMT
- CLINICAL SIGNIFICANCE
- ACKNOWLEDGMENTS
- FURTHER READING
- Review Glycosylation Changes in Cancer.[Essentials of Glycobiology. 2022]Review Glycosylation Changes in Cancer.Bellis SL, Reis CA, Varki A, Kannagi R, Stanley P. Essentials of Glycobiology. 2022
- Review Glycosylation Changes in Cancer.[Essentials of Glycobiology. 2009]Review Glycosylation Changes in Cancer.Varki A, Kannagi R, Toole BP. Essentials of Glycobiology. 2009
- Review Glycosylation markers in cancer.[Adv Clin Chem. 2019]Review Glycosylation markers in cancer.Silsirivanit A. Adv Clin Chem. 2019; 89:189-213. Epub 2019 Jan 17.
- Review Simple sugars to complex disease--mucin-type O-glycans in cancer.[Adv Cancer Res. 2015]Review Simple sugars to complex disease--mucin-type O-glycans in cancer.Kudelka MR, Ju T, Heimburg-Molinaro J, Cummings RD. Adv Cancer Res. 2015; 126:53-135. Epub 2015 Feb 7.
- Alterations in the glycome after HDAC inhibition impact oncogenic potential in epigenetically plastic SW13 cells.[BMC Cancer. 2019]Alterations in the glycome after HDAC inhibition impact oncogenic potential in epigenetically plastic SW13 cells.Montgomery MR, Hull EE. BMC Cancer. 2019 Jan 16; 19(1):79. Epub 2019 Jan 16.
- Glycosylation Changes in Cancer - Essentials of GlycobiologyGlycosylation Changes in Cancer - Essentials of Glycobiology
- Isopterygium ambiguum (0)Nucleotide
- Allium oreoscordum (2)Nucleotide
- transcription factor PRE6-like [Gossypium arboreum]transcription factor PRE6-like [Gossypium arboreum]gi|1050569959|ref|XP_017605733.1|Protein
- PREDICTED: zinc finger CCCH domain-containing protein 12-like [Camelina sativa]PREDICTED: zinc finger CCCH domain-containing protein 12-like [Camelina sativa]gi|727611034|ref|XP_010478659.1|Protein
Your browsing activity is empty.
Activity recording is turned off.
See more...