A new version of this title is available
See the updated version of this chapter
This chapter provides a historical background to the field of glycobiology and an overview of this book. General terms found in the volume are considered, common monosaccharide units of glycoconjugates mentioned, and a uniform symbol nomenclature used for structural depictions presented. The major glycan classes discussed in the book are described, and an overview of the general pathways for their biosynthesis is provided. Topological issues relevant to biosynthesis and functions of glycoconjugates are also considered, and the growing role of these molecules in medicine, biotechnology, nanotechnology, bioenergy, and materials science is mentioned.
WHAT IS GLYCOBIOLOGY?
Defined in the broadest sense, glycobiology is the study of the structure, biosynthesis, biology, and evolution of saccharides (also called carbohydrates, sugar chains, or glycans) that are widely distributed in nature and of the proteins that recognize them. How does glycobiology fit into the modern concepts of molecular biology? The central paradigm driving research in molecular biology has been that biological information flows from DNA to RNA to protein. The power of this concept lies in its template-based precision, the ability to manipulate one class of molecules based on knowledge of another, and the patterns of sequence homology and relatedness that predict function and reveal evolutionary relationships. With ongoing sequencing of numerous genomes, spectacular gains in understanding the biology of nucleic acids and proteins have occurred. Thus, many scientists assume that studying just these molecules will explain the makeup of cells, tissues, organs, physiological systems, and intact organisms. In fact, making a cell requires many small molecule metabolites as well as two other major classes of macromolecules—lipids and carbohydrates—which serve as intermediates in generating energy and as signaling effectors, recognition markers, and structural components. Taken together with the fact that they encompass some of the major posttranslational modifications of proteins, lipids and carbohydrates help explain how the relatively small number of genes in the typical genome can generate the enormous biological complexities inherent in the development, growth, and functioning of diverse organisms.
The biological roles of carbohydrates are particularly prominent in the assembly of complex multicellular organs and organisms, which requires interactions between cells and the surrounding matrix. Without any known exception, all cells and numerous macromolecules in nature carry an array of covalently attached sugars (monosaccharides) or sugar chains (oligosaccharides), which are generically referred to in this book as “glycans.” Sometimes glycans can also be freestanding entities. Being on the outer surface of cellular and secreted macromolecules, many glycans are in a position to modulate or mediate a variety of events in cell–cell, cell–matrix, and cell–molecule interactions critical to the development and function of a complex multicellular organism. They can also mediate interactions between organisms (e.g., between host and a parasite, pathogen, or a symbiont). In addition, simple, rapidly turning over, protein-bound glycans are abundant within the nucleus and cytoplasm, where they can serve as regulatory switches. A more complete paradigm of molecular biology must therefore include glycans, often in covalent combination with other macromolecules (i.e., glycoconjugates, such as glycoproteins and glycolipids). In analogy to the current situation in Cosmology, glycans can be considered as the “dark matter” of the biological universe: a major and critical component that has yet to be fully incorporated into the “standard model” of biology.
The chemistry and metabolism of carbohydrates were prominent matters of interest in the first part of the 20th century. Although engendering much attention, they were primarily considered as a source of energy or as structural materials, apparently lacking other biological activities. Furthermore, during the molecular biology revolution of the 1970s, studies of glycans lagged far behind those of other major classes of molecules. This was in part because of their inherent structural complexity, the difficulty in determining their sequences, and the fact that their biosynthesis could not be directly predicted from a DNA template. The development of many new technologies for exploring the structures and functions of glycans has since opened a new frontier of molecular biology called “glycobiology”—a word first coined in the late 1980s to recognize the coming together of the traditional disciplines of carbohydrate chemistry and biochemistry with a modern understanding of the cell and molecular biology of glycans and, in particular, their conjugates with proteins and lipids. Glycobiology is now one of the more rapidly growing fields in the natural sciences, with broad relevance to many areas of basic research, biomedicine, and biotechnology. The field includes the chemistry of carbohydrates, the enzymology of glycan formation and degradation, the recognition of glycans by specific proteins, roles of glycans in complex biological systems, and their analysis or manipulation by various techniques. Research in glycobiology thus requires a foundation not only in the nomenclature, biosynthesis, structure, chemical synthesis, and functions of glycans but also in the general disciplines of molecular genetics, protein chemistry, cell biology, developmental biology, physiology, and medicine. This book provides an overview of the field, with relative emphasis on the glycans of animal systems. It is assumed that the reader has a basic background in advanced undergraduate-level chemistry, biochemistry, and cell biology. Some of the major investigators who influenced the early development of Glycobiology are shown in Figure 1.1, and more are listed in Online Appendix 1A. Many others have made major contributions as well, and a summary of the general principles gained from all this research is presented in Table 1.1.

FIGURE 1.1.
Nobel laureates in fields related to the history of glycobiology. Listed are the Laureates and their original Nobel citations: Hermann Emil Fischer (Chemistry, 1902), “in recognition of the extraordinary services he has rendered by his work on (more...)
TABLE 1.1.
General principles of glycobiology
MONOSACCHARIDES ARE THE BASIC STRUCTURAL UNITS OF GLYCANS
Carbohydrates are defined as polyhydroxyaldehydes, polyhydroxyketones and their simple derivatives, or larger compounds that can be hydrolyzed into such units. A monosaccharide is a carbohydrate that usually cannot be hydrolyzed into a simpler form. It has a potential carbonyl group at the end of the carbon chain (an aldehyde group) or at an inner carbon (a ketone group). These two types of monosaccharides are therefore named aldoses and ketoses, respectively (for examples, see below, and for more details, see Chapter 2). Free monosaccharides can exist in open-chain or ring forms (Figure 1.2). Ring forms of the monosaccharides are the rule in oligosaccharides, which are linear or branched chains of monosaccharides attached to one another via glycosidic linkages (the term “polysaccharide” is typically used for large glycans composed of repeating oligosaccharide motifs; for examples, see Chapter 3). The ring form of a monosaccharide generates a chiral anomeric center at C-1 for aldo sugars or at C-2 for keto sugars (for details, see Chapter 2). A glycosidic linkage involves the attachment of a monosaccharide to another residue, typically via the hydroxyl group of this anomeric center, generating α-linkages or β-linkages that are defined based on the relationship of the glycosidic oxygen to the anomeric carbon and ring (Chapter 2). These two linkage types confer very different structural properties and biological functions on sequences that are otherwise identical in composition, as classically illustrated by the differences between starch and cellulose (both are homopolymers of glucose, the former largely α1-4-linked and the latter β1-4-linked throughout). A glycoconjugate is a compound in which one or more monosaccharide or oligosaccharide units (the glycone) are covalently linked to a noncarbohydrate moiety (the aglycone). An oligosaccharide that is not attached to an aglycone possesses the reducing power of the aldehyde or ketone in its terminal monosaccharide component, with the exception of oligosaccharides in which the sugars are linked together at their reducing ends, as in derivatives of sucrose or trehalose. The end of a glycan exposing the aldehyde or ketone group is therefore named the reducing terminus or reducing end, terms that tend to be used even when the sugar chain is attached to an aglycone and has thus lost its reducing power. Correspondingly, the opposite end of the chain tends to be called the nonreducing end (note the analogy to the amino and carboxyl ends of proteins, or the 5′ and 3′ ends of DNA and RNA).
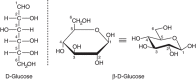
FIGURE 1.2.
Open-chain and ring forms of glucose. Changes in the orientation of hydroxyl groups around specific carbon atoms generate new molecules that have a distinct biology and biochemistry (e.g., galactose is the C-4 epimer of glucose). In the ring form, glucose (more...)
GLYCANS CAN CONSTITUTE A MAJOR PORTION OF THE MASS OF A GLYCOCONJUGATE
In naturally occurring glycoconjugates, the portion of the molecule comprising the glycans can vary greatly in contribution to its overall size. In many cases, the glycans comprise a substantial portion of the mass of glycoconjugates (for a typical example, see Figure 1.3). For this reason, the surfaces of all types of cells in nature (which are heavily decorated with different kinds of glycoconjugates) are effectively covered with a dense array of sugars, the so-called “glycocalyx.” This cell-surface structure was first observed many years ago by electron microscopists as a polysaccharide coat external to the cell surface membrane in bacteria, which could be stained with ruthenium red, and in animal cells, where the the anionic polysaccharides on the surface could be decorated with polycationic reagents such as cationized ferritin (for classic and recent examples, see Figure 1.4). The density of glycans in the glycocalyx can be remarkably high. For example, it has been calculated that the concentration of sialic acids in the glycocalyx of a typical human B lymphocyte may be >100 mm.
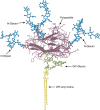
FIGURE 1.3.
Schematic representation of the Thy-1 glycoprotein including the three N-glycans (blue) and a glycosylphosphatidylinositol (GPI-glycan; green) lipid anchor whose acyl chains (yellow) would normally be embedded in the membrane bilayer. Note that the polypeptide (more...)
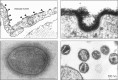
FIGURE 1.4.
(Upper left) Historical electron micrograph of endothelial cells from a blood capillary in the rat diaphragm muscle, showing the lumenal cell membrane of the cells (facing the blood) decorated with particles of cationized ferritin (arrowheads). These (more...)
MONOSACCHARIDES CAN BE LINKED TOGETHER IN MANY MORE WAYS THAN AMINO ACIDS OR NUCLEOTIDES
Nucleotides and proteins are linear polymers that can each contain only one basic type of linkage between monomers. In contrast, each monosaccharide can theoretically generate either an α- or a β-linkage to any one of several positions on another monosaccharide in a chain or to another type of molecule. Thus, while three different nucleotides or amino acids can only generate six trimers, three different hexoses could theoretically produce (depending on which of their forms are considered) anywhere from 1056 to 27,648 unique trisaccharides. This difference in complexity becomes even greater as the number of monosaccharide units found in natural glycan increases (now numbering in the hundreds). Fortunately for the student of glycobiology, naturally occurring biological macromolecules in a given species tend to contain relatively few of the possible monosaccharide units, and they are in a limited number of combinations. Of course, the great majority of glycans in most species have yet to be discovered and structurally defined. Thus, much of the possible diversity may yet exist in nature.
COMMON MONOSACCHARIDE UNITS OF GLYCOCONJUGATES
Although several hundred distinct monosaccharides are known in nature, only a minority of these are commonly found in well-studied glycans. Examples of common monosaccharides in vertebrate cells are listed below, along with their standard abbreviations (for details regarding their structures, see Chapter 2, and embedded links from the symbols in Online Appendix 1B).
- Pentoses: five-carbon neutral sugars—D-xylose (Xyl)
- Hexoses: six-carbon neutral sugars—for example, D-glucose (Glc)
- Hexosamines: hexoses with an amino group at the 2-position, which can be either free or, more commonly, N-acetylated—for example, N-acetyl-D-glucosamine (GlcNAc)
- 6-Deoxyhexoses: for example, L-fucose (Fuc)
- Uronic acids: hexoses with a carboxylate at the 6-position—for example, D-glucuronic acid (GlcA)
- Nonulosonic acids: family of nine-carbon acidic sugars, of which the most common in animals is the sialic acid N-acetylneuraminic acid (Neu5Ac, also sometimes called NeuAc or, historically, NANA) (see Chapter 15)
For simplicity, the symbols D- and L- are omitted from the full names of common monosaccharides from here on unless a less common variant occurs. This limited set of monosaccharides dominates the glycobiology of more recently evolved (so-called “higher”) animals, but several others have been found in “lower” animals (e.g., tyvelose; Chapters 25 and 26), bacteria and archea (e.g., keto-deoxyoctulosonic acid, rhamnose, L-arabinose, and muramic acid; Chapters 21 and 22), and plants (e.g., arabinose, apiose, and galacturonic acid; Chapter 24). A variety of modifications of glycans further enhance their diversity in nature and often serve to mediate specific biological functions. Thus, the hydroxyl groups of different monosaccharides can be subject to modifications such as phosphorylation, sulfation, methylation, O-acetylation, or fatty acylation. Although amino groups are commonly N-acetylated, they can be N-sulfated or remain unsubstituted. Carboxyl groups are occasionally subject to lactonization to nearby hydroxyl groups or even lactamization to nearby amino groups.
Details regarding the structural depiction of monosaccharides, linkages, and oligosaccharides are discussed in Chapter 2. Many figures in this volume use a symbolic depiction of sugar chains (see Online Appendix 1B and examples in Figure 1.5). This Symbol Nomenclature for Glycans (SNFG) is expanded from the Second Edition and has been vetted and adopted by many investigators. For reader convenience, a full table of symbols is reproduced on the inside cover of the book, is available for view at the NCBI Books website Online Appendix 1B, and can be downloaded in drawing and text formats from this website. Detailed notes regarding the logic used for the symbol nomenclature can also be found at the Online Appendix 1B, along with embedded links to online databases featuring details about each monosaccharide, as well as color settings information for artists who wish to use the system.
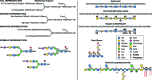
FIGURE 1.5.
Examples of symbols and conventions for drawing glycan structures. The monosaccharide symbol set from the Second Edition of Essentials of Glycobiology remains intact but has been extended to cover a wider range of monosaccharides found in nature. For (more...)
MAJOR CLASSES OF GLYCOCONJUGATES AND GLYCANS
The common classes of glycans are primarily defined according to the nature of the linkage to the aglycone (protein or lipid) (for common eukaryotic examples see Figures 1.6 and 1.7). A glycoprotein is a glycoconjugate in which a protein carries one or more glycans covalently attached to a polypeptide backbone, usually via N- or O-linkages. An N-glycan (N-linked oligosaccharide, N-[Asn]-linked oligosaccharide) is a sugar chain covalently linked to an asparagine residue of a polypeptide chain, commonly involving a GlcNAc residue in eukaryoates, and the consensus peptide sequence: Asn-X-Ser/Thr. Animal N-glycans also share a common pentasaccharide core region and can be generally divided into three main classes: oligomannose (or high-mannose) type, complex type, and hybrid type (Chapter 9). An O-glycan (O-linked oligosaccharide) is frequently linked to the polypeptide via N-acetylgalactosamine (GalNAc) to a hydroxyl group of a serine or threonine residue and can be extended into different structural core classes (Chapter 10). A mucin is a large glycoprotein that carries many O-glycans that are clustered (closely spaced). Several other types of O-glycans also exist (e.g., O-linked fucose, glucose, or mannose). A proteoglycan is a glycoconjugate that has one or more glycosaminoglycan (GAG) chains (see definition below) attached to a “core protein” through a typical core region that has at its reducing end a xylose residue linked to the hydroxyl group of a serine residue. The distinction between a proteoglycan and a glycoprotein is otherwise arbitrary, because some proteoglycan polypeptides can carry both GAG chains and different O- and N-glycans (Chapter 17). Many cytoplasmic and nuclear proteins have single GlcNAc residue on one or more of their serine or threonine residues (Chapter 19). Figure 1.7 provides a listing of known glycan–protein linkages in nature.
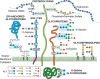
FIGURE 1.6.
Common classes of animal glycans. (Modified and updated from Varki A. 1997. FASEB J 11: 248–255; Fuster M, Esko JD. 2005. Nat Rev Can 7: 526–542, with permission from Macmillan; and Stanley P. 2011. Cold Spring Harb Perspect Biol 3: a005199.) (more...)
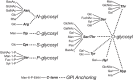
FIGURE 1.7.
Glycan–protein linkages reported in nature. (Updated and redrawn, with permission of Oxford University Press, from Spiro RG. 2002. Glycobiology 12: 43R–56R.) Diagrammatic representation of six distinct types of sugar-peptide bonds that (more...)
A glycosylphosphatidylinositol anchor is a glycan bridge between phosphatidylinositol and a phosphoethanolamine that is in amide linkage to the carboxyl terminus of a protein. This structure typically constitutes the only anchor to the lipid bilayer membrane for such proteins (Chapter 12). A glycosphingolipid (often named a glycolipid) consists of a glycan usually attached via glucose or galactose to the terminal primary hydroxyl group of the lipid moiety ceramide, which is composed of a long chain base (sphingosine) and a fatty acid (Chapter 11). Glycolipids can be neutral or anionic. A ganglioside is an anionic glycolipid containing one or more residues of sialic acid. It should be noted that these represent only the most common classes of glycans reported in eukaryotic cells. There are several other less studied glycan types found on one or the other side of the cell membrane in animal cells (Chapters 13, 17, and 18) and many others in plants, algae, and prokaryotes.
Although different glycan classes have unique core regions by which they are distinguished, certain outer structural sequences are often shared among different classes of glycans. For example, animal N- and O-glycans and glycosphingolipids frequently carry the subterminal disaccharide Galβ1-4GlcNAcβ1-(N-acetyllactosamine or LacNAc) or, less commonly, GalNAcβ1-4GlcNAcβ1-(LacdiNAc) units. The LacNAc units can sometimes be repeated, giving extended poly-N-acetyllactosamines (sometimes incorrectly called “poly-lactosamines”) (Chapter 14). Less commonly, the LacdiNAc motif can also be repeated (termed polyLacdiNAc). Outer LacNAc units can be modified by fucosylation or by branching and are typically capped in vertebrates by sialic acids or, less commonly, by sulfate esters, Fuc, α-Gal, β-GalNAc, or β-GlcA units (Chapters 14 and 15). In contrast, eukaryotic glycosaminoglycans are linear copolymers of acidic disaccharide repeating units, each typically containing a hexosamine (GlcN or GalN) and a hexose (Gal) or hexuronic acid (GlcA or IdoA) (Chapter 17). The type of disaccharide unit defines the glycosaminoglycan as chondroitin or dermatan sulfate (GalNAcβ1-4GlcA/IdoA), heparin or heparan sulfate (GlcNAcα1-4GlcA/IdoA), or keratan sulfate (Galβ1-4GlcNAc). Keratan sulfate is actually a 6-O-sulfated form of poly-N-acetyllactosamine attached to an N- or O-glycan core, rather than to a typical Xyl-Ser-containing proteoglycan linkage region. Another type of glycosaminoglycan, hyaluronan (a polymer of GlcNAcβ1-4GlcA), appears to exist primarily as a free glycan unattached to any aglycone (Chapter 16). Some glycosaminoglycans have sulfate esters substituting either amino or hydroyxl groups (i.e., N- or O-sulfate groups). Another anionic polysaccharide that can be extended from LacNAc units is polysialic acid, a homopolymer of sialic acid that is selectively expressed only on a few proteins in vertebrates. Polysialic acids and hyaluronan are also found as the capsular polysaccharides of certain pathogenic bacteria (Chapter 15). For simplicity, this section has focused primarily on vertebrate glycans. Many other classes of glycans exist in other branches of the tree of life (Chapters 21–26).
GLYCAN STRUCTURES ARE NOT ENCODED DIRECTLY IN THE GENOME
Unlike protein sequences, which are primary gene products, glycan structures are not encoded directly in the genome and are secondary gene products. A few percent of known genes in the human genome are dedicated to producing the enzymes and transporters responsible for the biosynthesis and assembly of glycans (Chapter 8), typically as posttranslational modifications of proteins or by glycosylation of core lipids. The glycans themselves represent numerous combinatorial possibilities, generated by a variety of competing and sequentially acting glycosidases and glycosyltransferases (Chapter 6) and the subcompartmentalized “assembly-line” mechanisms of glycan biosynthesis in the Golgi apparatus of eukaryotes (Chapter 4). Thus, even with full knowledge of the expression levels of all relevant gene products, one cannot accurately predict the precise structures of glycans elaborated by a given cell type. Furthermore, small changes in environmental cues can cause dramatic changes in glycans produced by a given cell. It is this variable and dynamic nature of glycosylation that makes it a powerful way to generate and modulate biological diversity and complexity. Of course, it also makes glycans more difficult to study than nucleic acids and proteins.
SITE-SPECIFIC STRUCTURAL DIVERSITY IN PROTEIN GLYCOSYLATION
One of the most fascinating and yet frustrating aspects of protein glycosylation is the phenomenon of microheterogeneity. Thus, at any particular glycan attachment site on a given protein synthesized by a particular cell type, a range of variations might be found in the structures of the attached glycan (and in some instances, the glycan may be missing). Effectively, a given polypeptide encoded by a single gene can exist in numerous “glycoforms,” each constituting a distinct molecular species. For some glycoproteins the microheterogeneity at a particular site may be quite limited, whereas for other sites it may be extensive, even within the same glycoprotein species. Mechanistically, microheterogeneity might be generated by the rapidity with which multiple, sequential, partially competitive glycosylation and deglycosylation reactions take place in the endoplasmic reticulum (ER) and Golgi apparatus, through which a newly synthesized glycoprotein passes, along with the lack of a template for directing the synthesis and the accessibility of glycans at a site to the modifying enzymes (Chapter 4). An alternate possibility is that each individual cell or cell type is in fact exquisitely specific in the glycosylation it produces, but that intercellular variations result in the observed microheterogeneity of samples from natural multicellular sources. Whatever the origin of microheterogeneity, it explains the anomalous behavior of glycoproteins in analytical/separation techniques and makes complete structural analysis of a glycoprotein a difficult task. From a functional point of view, the biological significance of microheterogeneity remains unclear. It is possible that this is a type of diversity generator, intended for diversifying endogenous recognition functions and/or for evading microbes and parasites, each of which can bind with high specificity only to certain glycan structures (Chapters 37 and 42).
CELL BIOLOGY OF GLYCOSYLATION
Most well-characterized pathways for the biosynthesis of major classes of eukaryotic glycans are within ER and Golgi compartments (Chapter 4). Newly synthesized proteins originating in the ER are either cotranslationally or posttranslationally modified with glycans at various stages in their itinerary toward their final destinations. N-glycans are partially assembled on lipid donors on the cytoplasmic face of the ER and then flipped across the membrane where the oligosaccharide assembly is completed and transfer to the nascent protein occurs. This oligosaccharide is then trimmed and extended by the addition of one monosaccharide at a time as the protein passes through the ER and the Golgi. These glycosylation reactions use activated forms of monosaccharides (nucleotide sugars; Chapter 5) as donors for reactions that are catalyzed by glycosyltransferases (for details about their biochemistry, molecular genetics, and cell biology, see Chapters 4, 6, and 8). The nucleotide sugar donors are synthesized within the cytosolic or nuclear compartment from monosaccharide precursors of endogenous or exogenous origin and then actively transported across a membrane bilayer into the lumen of the ER and Golgi compartments (Chapter 5). Notably, the portion of the glycoconjugate that faces the inside of these compartments will ultimately face the outside of the cell or the inside of a secretory granule or lysosome and will be topologically unexposed to the cytosol. The biosynthetic enzymes (glycosyltransferases, sulfotransferases, etc.) responsible for catalyzing these reactions are well studied (Chapter 6), and their location has helped to define various functional compartments of the ER–Golgi pathway. A classical model envisioned that these enzymes are physically lined up along this pathway in the precise sequence in which they actually work. This model appears to be oversimplified, as there is considerable overlap in the distribution of these enzymes, and the actual distribution of a given enzyme depends on the cell type.
All topological considerations mentioned above are reversed with regard to nuclear and cytoplasmic glycosylation, because the active sites of the relevant glycosyltransferases face the cytosol, which is in direct communication with the interior of the nucleus. Until the mid-1980s, the accepted dogma was that glycoconjugates occurred exclusively on the outer surface of cells, on the internal (luminal) surface of intracellular organelles, and on secreted molecules. The cytosol and nucleus were assumed to be devoid of glycosylation capacity. However, it is now clear that certain distinct types of glycoconjugates are synthesized and reside within the cytosol and nucleus (Chapter 18). Indeed, one of them, named O-GlcNAc (Chapter 19), may well be numerically the most common type of glycoconjugate in many cell types. The fact that this major form of glycosylation was missed by so many investigators for so long serves to emphasize the relatively unexplored state of the field of glycobiology.
Like all components of living cells, glycans are constantly being turned over by degradation and the enzymes that catalyze this process cleave glycans either at the outer (nonreducing) terminus (exoglycosidases) or internally (endoglycosidases) (Chapters 4 and 44). Some terminal monosaccharide units such as sialic acids are sometimes removed and new units reattached during endosomal recycling, without degradation of the underlying chain. The final complete degradation of most eukaryotic glycans is generally performed by multiple glycosidases in the lysosome. Once degraded, their individual unit monosaccharides are then typically exported from the lysosome into the cytosol for reutilization (Figure 1.8). In contrast to the relatively slow turnover of glycans derived from the ER–Golgi pathway, the O-GlcNAc monosaccharide modifications of the nucleus and cytoplasm appear more dynamic (Chapter 19). In some instances, extracellular or intracellular free glycans can also serve as signaling molecules (Chapter 40).
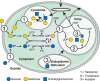
FIGURE 1.8.
Biosynthesis, use, and turnover of a common monosaccharide. This schematic shows the biosynthesis, fate, and turnover of galactose, a common monosaccharide constituent of animal glycans. Although small amounts of galactose can be taken up from the outside (more...)
TOOLS USED TO STUDY GLYCOSYLATION
Unlike oligonucleotides and proteins, glycans are not commonly found in a linear, unbranched fashion. Even when linear (e.g., GAGs), they often contain a variety of substituents, such as sulfate groups, which are not uniformly distributed. Thus, complete sequencing of glycans is usually impossible by a single method and requires iterative combinations of physical, chemical, and enzymatic approaches that together yield the details of structure (for a discussion of low- and high-resolution separation and analysis, including mass spectrometry and nuclear magnetic resonance [NMR], see Chapter 50). Less detailed information on structure may be sufficient to explore the biology of some glycans and can be obtained by simple techniques, such as the use of enzymes (endoglycosidases and exoglycosidases), lectins, and other glycan-binding proteins (Chapters 48 and 50), chemical modification or cleavage, metabolic radioactive labeling, antibodies, or cloned glycosyltransferases (Chapters 53 and 54). Glycosylation can also be perturbed in various ways, for example, by glycosylation inhibitors and primers (Chapters 55 and 56) and by genetic manipulation of glycosylation in intact cells and organisms (Chapter 49). Directed in vitro synthesis of glycans by chemical and enzymatic methods has also made great strides in recent years, providing many new tools for exploring glycobiology (Chapters 53, 54, and 57). The generation of complex glycan libraries by a variety of routes has further enhanced this interface of chemistry and biology (Chapters 53 and 54), including the generation of glycan microarrays.
GLYCOMICS
Analogous to genomics and proteomics, glycomics represents the systematic methodological elucidation of the “glycome” (the totality of glycan structures) of a given cell type or organism (Chapters 51 and 52). In reality, the glycome is far more complex than the genome or proteome. In addition to the vastly greater structural diversity in glycans, one is faced with the complexities of glycosylation microheterogeneity (see above) and the dynamic changes that occur in the course of development, differentiation, metabolic changes, aging, malignancy, inflammation, or infection. Added diversity arises from intraspecies and interspecies variations in glycosylation. Thus, a given cell type in a given species can manifest a large number of possible glycome states. Glycomic analysis today generally consists of extracting entire cell types, organs, or organisms; releasing all the glycan chains from their linkages; and cataloging them via approaches such as mass spectrometry. In a variation called glycoproteomics, the glycans are analyzed while still attached to protease-generated fragments of glycoproteins. The results obtained represent a spectacular improvement over what was possible a few decades ago, but they are still analogous to cutting down all the trees in a forest and cataloging them, without attention to the layout of the forest and the landscape (Chapter 15 discusses this complex issue from the perspective of just one monosaccharide class, sialic acids; see Figure 15.3).
Glycomic analysis thus needs to be complemented by classical methods such as tissue-section staining or flow cytometry, using lectins or glycan-specific antibodies that aid in understanding the glycome by taking into account the heterogeneity of glycosylation at the level of the different cell types and subcellular domains in the tissue under study. This is even more important because of the common observation that removing cells from their normal milieu and placing them into tissue culture can result in major changes in the glycosylation machinery of the cell. However, such classical approaches suffer from poor quantitation and relative insensitivity to structural details. A combination of the two approaches is now potentially feasible via laser-capture microdissection of specific cell types directly from tissue sections, with the resulting samples being studied by mass spectrometry. New methods for in situ imaging and characterization of glycans in the intact “forest” are clearly needed.
Because most of the genes involved in glycan biosynthetic pathways have been cloned from multiple organisms, it is possible today to obtain an indirect genomic and transcriptomic view of the glycome in a specific cell type (Chapter 8). However, given the relatively poor correlation between mRNA and protein levels, and the complex assembly line and competitive nature of the cellular Golgi glycosylation pathways, even complete knowledge of the mRNA expression patterns of all relevant genes in a given cell cannot allow accurate prediction of the distribution and structures of glycans in that cell type. Thus, there is as yet no reliable indirect route toward elucidating the glycome, other than by actual analysis using an array of methods.
GLYCOSYLATION DEFECTS IN ORGANISMS AND CULTURED CELLS
Many mutant variants of cultured cell lines with altered glycan structures and specific glycan biosynthetic defects have been described, the most common of which are lectin-resistant (Chapter 49). Indeed, with few exceptions, mutants with specific defects at most steps of the major pathways of glycan biosynthesis have been found in cultured animal cells. The use of such cell lines has been of great value in elucidating the details of glycan biosynthetic pathways. Their existence implies that many types of glycans are not crucial to the optimal growth of single cells growing in the sheltered and relatively unchanging environment of the culture dish. Rather, most glycan structures must be more important in mediating cell–cell and cell–matrix interactions in intact multicellular organisms and/or interactions between organisms. In keeping with this supposition, genetic defects completely eliminating major glycan classes in intact animals all cause embryonic lethality (Chapter 45). As might be expected, naturally occurring viable animal mutants of this type tend to have disease phenotypes of intermediate severity and show complex phenotypes involving multiple systems. Less severe genetic alterations of outer chain components of glycans tend to give viable organisms with more specific phenotypes (Chapter 45). Overall, there is much to be learned by studying the consequences of natural or induced genetic defects in intact multicellular organisms, including humans (Chapter 45).
BIOLOGICAL ROLES OF GLYCANS ARE DIVERSE
A major theme of this volume is the exploration and elucidation of the biological roles of glycans. It is interesting to note that, in the short time since the first edition, we have gone from asking “what is it that glycans do anyway?” to having to explain a large number of complex and sometimes nonviable glycosylation-modified phenotypes in humans, mice, flies, and other organisms. Like any biological system, the optimal approach carefully considers the relationship of structure and biosynthesis to function (Chapter 7). As might be imagined from their ubiquitous and complex nature, the biological roles of glycans are remarkably varied. Indeed, asking what these roles are is akin to asking the same question about proteins. Thus, all of the proposed theories regarding glycan function turn out to be partly correct, and exceptions to each can also be found. Not surprisingly for such a diverse group of molecules, the biological roles of glycans also span the spectrum from those that are subtle to those that are crucial for the development, growth, function, or survival of an organism (Chapter 7). The diverse functions ascribed to glycans can be simply divided into two general categories: (i) structural and modulatory functions (involving the glycans themselves or their modulation of the molecules to which they are attached) and (ii) specific recognition of glycans by glycan-binding proteins. Of course, any given glycan can mediate one or both types of functions. The binding proteins in turn fall into two broad groups: lectins and sulfated GAG-binding proteins (Chapters 27, 28, and 38). Such molecules can be either intrinsic to the organism that synthesized the cognate glycans (e.g., see Chapters 31–36, 38, and 39) or extrinsic (see Chapters 37 and 42 for information concerning microbial proteins that bind to specific glycans on host cells). The atomic details of these glycan–protein interactions have been elucidated in many instances (Chapters 29 and 30). Although there are exceptions to this notion, the following general theme has emerged regarding lectins: monovalent binding tends to be of relatively low affinity, and such systems typically achieve their specificity and function by achieving high avidity, via interactions of multivalent arrays of glycans with cognate lectin-binding sites.
GLYCOSYLATION CHANGES IN DEVELOPMENT, DIFFERENTIATION, AND MALIGNANCY
Whenever a new tool (e.g., an antibody or lectin) specific for detecting a particular glycan is developed and used to probe its expression in intact organisms, it is common to find exquisitely specific temporal and spatial patterns of expression of that glycan in relation to cellular activation, embryonic development, organogenesis, and differentiation (see Chapter 41 for examples). Certain relatively specific changes in expression of glycans are also often found in the course of transformation and progression to malignancy (Chapter 47), as well as other pathological situations such as inflammation (Chapter 46). These spatially and temporally controlled patterns of glycan expression imply the involvement of glycans in many normal and pathological processes, the precise mechanisms of which are understood in only some cases.
EVOLUTIONARY CONSIDERATIONS IN GLYCOBIOLOGY
Remarkably little is still known about the evolution of glycosylation. There are clearly shared and unique features of glycosylation in different kingdoms and taxa. Among animals, there may be a trend toward increasing complexity of N- and O-glycans in more recently evolved (“higher”) taxa. Intraspecies and interspecies variations in glycosylation are also relatively common. It has been suggested that the more specific biological roles of glycans are often mediated by uncommon structures, unusual presentations of common structures, or further modifications of the commonly occurring saccharides themselves. Such unusual structures likely result from unique expression patterns of the relevant glycosyltransferases or other glycan-modifying enzymes. On the other hand, such uncommon glycans can be targets for specific recognition by infectious microorganisms and various toxins. Thus, at least some of the diversity in glycan expression in nature must be related to the evolutionary selection pressures generated by interspecies interactions (e.g., of host with pathogen or symbiont). In other words, the two different classes of glycan recognition mentioned above (mediated by intrinsic and extrinsic glycan-binding proteins) are in competition with each other with regard to a particular glycan target. The specialized glycans expressed by parasites and microbes that are of great interest from the biomedical point of view (Chapters 21, 22, 23, and 43) are themselves presumably subject to evolutionary selection pressures. These issues are further considered in Chapter 20, which also discusses the limited information concerning how various glycan biosynthetic pathways appear to have evolved and diverged in different life-forms.
GLYCANS IN MEDICINE AND BIOTECHNOLOGY
Numerous natural bioactive molecules are glycoconjugates, and the attached glycans can have dramatic effects on the biosynthesis, stability, action, and turnover of these molecules in intact organisms. For example, the sulfated glycosaminoglycan heparin and its derivatives are among the most commonly used drugs in the world. The aminoglycoside antibiotics all have carbohydrate components essential for activity. For this and many other reasons, glycobiology and carbohydrate chemistry have become increasingly important in modern biotechnology. Patenting a glycoprotein drug, obtaining FDA approval for its use, and monitoring its production all require knowledge of the structure of its glycans. Moreover, glycoproteins, which include monoclonal antibodies, enzymes, and hormones, are by now the major products of the biotechnology industry, with sales in the tens of billions of dollars annually, which continues to grow at an increasing rate. In addition, several human disease states involve changes in glycan biosynthesis that can be of diagnostic and/or therapeutic significance. The emerging importance of glycobiology in medicine and biotechnology is discussed in Chapters 56 and 57.
GLYCANS IN NANOTECHNOLOGY, BIOENERGY, AND MATERIALS SCIENCE
Although not traditionally considered part of “Glycobiology,” many natural and synthetic glycans are key components of Nanotechnology, Bioenergy, and Material Science. Glyconanomaterials (Chapter 58) have tunable chemical and physical properties and can be built on different scaffolds to probe cellular, tissue, and organismal interactions. Attached glycans can change nanomaterial properties, optimizing solubility and biocompatibility and lowering cytotoxicity. Glyconanomaterials have been used as imaging agents, spectroscopic tools, monitors of cellular systems, and vehicles for vaccination and drug delivery. Plant glycans are used for many purposes: energy sources, building materials, clothes, paper products, animal feed, and food and beverage additives (Chapter 59). Concerns about detrimental environmental effects and diminishing reserves of petroleum and its by-products have greatly renewed interest in using plant glycans for energy production, generation of polymers with improved or new functionalities, and as sources of high-value chemosynthetic precursors (Chapter 59).
ACKNOWLEDGMENTS
The authors acknowledge contributions to the previous version of this chapter from the late Nathan Sharon, as well as helpful comments and suggestions from all editors.
FURTHER READING
- Rademacher TW, Parekh RB, Dwek RA. 1988. Glycobiology. Annu Rev Biochem 57: 785–838. [PubMed: 3052290]
- Varki A. 1993. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 3: 97–130. [PMC free article: PMC7108619] [PubMed: 8490246]
- Drickamer K, Taylor ME. 1998. Evolving views of protein glycosylation. Trends Biochem Sci 23: 321–324. [PubMed: 9787635]
- Etzler ME. 1998. Oligosaccharide signaling of plant cells. J Cell Biochem Suppl 30–31: 123–128. [PubMed: 9893263]
- Gagneux P, Varki A. 1999. Evolutionary considerations in relating oligosaccharide diversity to biological function. Glycobiology 9: 747–755. [PubMed: 10406840]
- Roseman S. 2001. Reflections on glycobiology. J Biol Chem 276: 41527–41542. [PubMed: 11553646]
- Hakomori S-I. 2002. The glycosynapse. Proc Natl Acad Sci 99: 225–232. [PMC free article: PMC117543] [PubMed: 11773621]
- Spiro RG. 2002. Protein glycosylation: Nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12: 43R–56R. [PubMed: 12042244]
- Haltiwanger RS, Lowe JB. 2004. Role of glycosylation in development. Annu Rev Biochem 73: 491–537. [PubMed: 15189151]
- Sharon N, Lis H. 2004. History of lectins: From hemagglutinins to biological recognition molecules. Glycobiology 14: 53R–62R. [PubMed: 15229195]
- Ohtsubo K, Marth JD. 2006. Glycosylation in cellular mechanisms of health and disease. Cell 126: 855–867. [PubMed: 16959566]
- Patnaik SK, Stanley P. 2006. Lectin-resistant CHO glycosylation mutants. Methods Enzymol 416: 159–182. [PubMed: 17113866]
- Drickamer K, Taylor ME. 2006. Introduction to glycobiology, Vol. 2. Oxford University Press, Oxford.
- Kamerling J, Boons G-J, Lee Y, Suzuki A, Taniguchi N, Voragen AGJ. 2007. Comprehensive glycoscience, pp. 1–4. Elsevier Science, London.
- Freeze HH, Ng BG. 2011. Golgi glycosylation and human inherited diseases. Cold Spring Harb Perspect Biol 3: a005371. [PMC free article: PMC3181031] [PubMed: 21709180]
- Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. 2011. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu Rev Biochem 80: 825–858. [PMC free article: PMC3294376] [PubMed: 21391816]
- Sarrazin S, Lamanna WC, Esko JD. 2011. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 3: a004952. [PMC free article: PMC3119907] [PubMed: 21690215]
- Varki A. 2011. Evolutionary forces shaping the Golgi glycosylation machinery: Why cell surface glycans are universal to living cells. Cold Spring Harb Perspect Biol 3: a005462. [PMC free article: PMC3098673] [PubMed: 21525513]
- Aebi M. 2013. N-linked protein glycosylation in the ER. Biochim Biophys Acta 1833: 2430–2437. [PubMed: 23583305]
- Prasanphanich NS, Mickum ML, Heimburg-Molinaro J, Cummings RD. 2013. Glycoconjugates in host–helminth interactions. Front Immunol 4: 240. [PMC free article: PMC3755266] [PubMed: 24009607]
- Varki A. 2013. Omics: Account for the ‘dark matter’ of biology. Nature 497: 565. [PubMed: 23719451]
- Belardi B, Bertozzi CR. 2015. Chemical lectinology: Tools for probing the ligands and dynamics of mammalian lectins in vivo. Chem Biol 22: 983–993. [PMC free article: PMC4567249] [PubMed: 26256477]
- Endo T. 2015. Glycobiology of α-dystroglycan and muscular dystrophy. J Biochem 157: 1–12. [PubMed: 25381372]
- Misra S, Hascall VC, Markwald RR, Ghatak S. 2015. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front Immunol 6: 201. [PMC free article: PMC4422082] [PubMed: 25999946]
- Varki A, Cummings RD, Aebi M, Packer NH, Seeberger PH, Esko JD, Stanley P, Hart G, Darvill A, Kinoshita T, et al. 2015. Symbol nomenclature for graphical representations of glycans. Glycobiology 25: 1323–1324. [PMC free article: PMC4643639] [PubMed: 26543186]
- Aoki-Kinoshita K, Agravat S, Aoki NP, Arpinar S, Cummings RD, Fujita A, Fujita N, Hart GM, Haslam SM, Kawasaki T, et al. 2016. GlyTouCan 1.0—The international glycan structure repository. Nucleic Acids Res 44: D1237–D1242. [PMC free article: PMC4702779] [PubMed: 26476458]
Publication Details
Author Information and Affiliations
Authors
Ajit Varki and Stuart Kornfeld.Publication History
Published online: 2017.
Copyright
PDF files are not available for download.
Publisher
Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY)
NLM Citation
Varki A, Kornfeld S. Historical Background and Overview. 2017. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology [Internet]. 3rd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015-2017. Chapter 1. doi: 10.1101/glycobiology.3e.001







