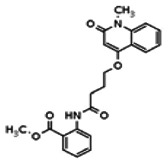
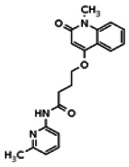
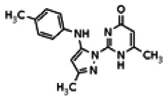
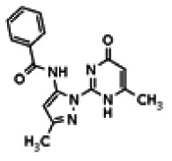
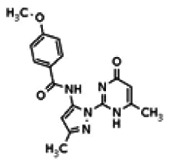
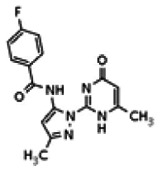
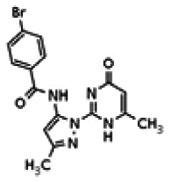
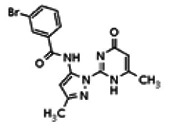
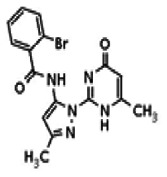
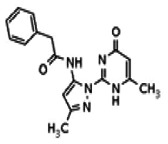
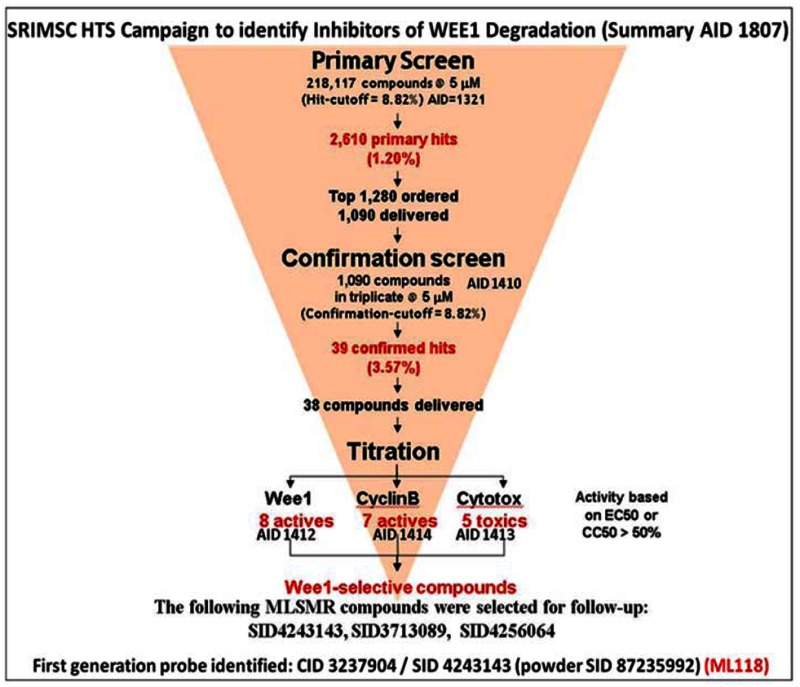
The tyrosine kinase Wee1 is part of a key cellular sensing mechanism that signals completion of DNA replication, ensuring proper timing of entry into mitosis. Wee1 acts as an inhibitor of mitotic entry by phosphorylating cyclin-dependent kinase CDK1. Wee1 activity is mainly regulated at the protein level through its phosphorylation and subsequent degradation by the ubiquitin proteasome pathway. To facilitate identification of small molecules preventing Wee1 degradation, a homogeneous cell-based assay was developed using HeLa cells transiently transfected with a Wee1-luciferase fusion protein. To ensure ultra-high-throughput screening (uHTS) compatibility, the assay was scaled to a 1536-well plate format and cells were transfected in bulk and cryopreserved. This miniaturized homogeneous assay demonstrated robust performance, with a calculated Z′ factor of 0.65 +/− 0.05. The assay was screened against a publicly available library of approximately 218,000 compounds to identify Wee1 stabilizers. Nonselective, cytotoxic, and promiscuous compounds were rapidly triaged through the use of a similarly formatted counterscreen that measured stabilization of an N-cyclin B-luciferase fusion protein, as well as execution of viability assessment in the parental HeLa cell line. This screening campaign led to the discovery of 4 unrelated cell-permeable small molecules that showed selective Wee1-luciferase stabilization with micromolar potency. One of these compounds, SID-4243143 (ML118), was shown to inhibit cell cycle progression, underscoring the importance of Wee1 degradation to the cell cycle. This probe was found to be inactive in a whole-cell assay against its anti-target, cyclin B. In contrast, the current state-of-the-art probe, MG132, inhibits degradation of both Wee1 and cyclin B in the same assays. More importantly, flow-cytometry assays confirm that the probe is able to induce an increase in the G2/M population after cell treatment, without increasing the sub-G1 population, suggesting the probe is not toxic to cells. These results suggest that this uHTS approach is suitable for identifying selective chemical probes that prevent Wee1 degradation and generally applicable to discovering inhibitors of the ubiquitin proteasome pathway.
Assigned Assay Grant #: 1R21NS056991-01
Screening Center Name & PI: Scripps Research Institute Molecular Screening Center (SRIMSC), H. Rosen
Chemistry Center Name & PI: SRIMSC, H. Rosen
Assay Submitter & Institution: Nagi Ayad, The Scripps Research Institute (TSRI)
PubChem Summary Bioassay Identifier (AID): 1807
Recommendations for scientific use of the probe (ML118)
Limitations in state of the art. Although selective inhibitors of Wee1 kinase activity have been reported, none have been shown to inhibit Wee1 turnover. For example, inhibition of the Wee1 kinase activity by compounds that abrogate the G2/M checkpoint improve the cytotoxic effects of DNA damaging agents on p53-negative cells [1–4]. However, these compounds are dual Src/Wee1 kinase inhibitors (i.e. they are not inhibitors of Wee1 degradation). In other efforts, the assay provider screened LOPAC and other libraries that contain Src inhibitors and identified no hits. Thus, Src activity is unrelated to inhibition of Wee1 degradation.
The new probe ML118 acts as an inhibitor of cell cycle progression, but, unlike MG132, is not a proteasome inhibitor since it did not affect turnover of another proteasome substrate, N-cyclin B-luciferase. One other small molecule has been reported in the literature to stabilize Wee1 in HeLa cells: the dihydropteridinone BI 2536 [5]. The mechanism by which BI 2536 leads to Wee1 stabilization is unknown. BI 2536 is an inhibitor of Plk-1, which phosphorylates many proteins associated with cell cycle progression and mitotic entry. A sample of BI 2536 was obtained from commercial sources [Selleck Chemicals, catalog number S1109] and was determined by the assay provider to be inactive in the Wee1 degradation inhibition assay. Thus, dissecting the role of BI 2536 on Wee1 biology relative to its multiple other cellular roles is an exceedingly challenging process. A second commercially available Plk-1 inhibitor, [5-(5,6-dimethoxybenzimidazol-1-yl)-3-(4-methanesulfonyl-benzyloxy)-thiophene-2-carboxamide, purchased from EMD Chemicals, catalog number 528282-5MG] was determined by the assay provider to be inactive in the Wee1 degradation inhibition assay. Moreover, probe compound SID-4243143/CID-3237904/powder SID-87235992 does not inhibit Plk-1, so its mechanism of action for Wee1 stabilization is distinct from that of BI 2536.
Probe Applications. The probe can be used to elucidate the specific role of Wee1 in cell cycle-related tumorigenesis. The importance of this probe is underscored by recent reports suggesting that Wee1 is shuttled from the nucleus to the cytoplasm, and thus there may be different pools of Wee1 that are located in different intracellular compartments and turned over via distinct mechanisms [6]. In this regard, the probe can also be used to elucidate Wee1 turnover in different intracellular compartments. This is not possible with siRNA mediated knockdown of intracellular components that generally inhibit turnover of all proteins since such treatments are often toxic and nonspecific.
Expected end-users of the probe in the research community. The probe can be used by academic researchers studying cell cycle biology, cancer biology, cellular kinases, and development. Thus it is conceivable that scientists in diverse fields will be able to apply this chemical probe to elucidate the role of Wee1 in these cellular pathways.
Relevant biology of the probe. Wee1 is a kinase that phosphorylates the Cdk1/cyclin B complex and delays entry of cells into mitosis (M phase). The phosphatase Cdc25 counteracts Wee1 by dephosphorylating Cdk1/cyclin B. Wee1 is also a kinase substrate. As G2 progresses phosphorylated Wee1 accumulates and is recognized by E3 ubiquitin ligases and is degraded via the proteosome. Cdc25 is then able to remove the inhibitory phosphorylation on Cdk1 and mitotic entry proceeds. Thus, the goal of this probe development project was to identify small molecules that prevent mitotic entry, via a novel whole-cell HTS assay that measures Wee1-K328M-luciferase degradation.
1. Introduction
Cell cycle progression and entry into mitosis are regulated by a highly conserved cellular process known as checkpoint signaling. Wee1 is a highly conserved tyrosine kinase that inhibits mitotic entry by inactivating the mitosis-specific kinase Cdk1/cyclin B complex during the S and G2 phases through Cdk1 phosphorylation at tyrosine 15. [7] By contrast, the phosphatase Cdc25 abrogates the Wee1-mediated effect by removing Cdk1 phosphorylation.[8, 9] Therefore, there is a competition between Wee1 and Cdc25 in controlling Cdk1/cyclin B complex activity, which ultimately determines mitotic entry or division arrest.[10] Upon the onset of mitosis, Wee1 is inactivated both by protein phosphorylation on specific residues and subsequent degradation via the ubiquitin proteasome pathway.[11, 12] This mechanism tips the balance in favor of Cdc25, triggering a positive feedback loop driven by activated Cdc25 and Cdk1/cyclin B, thus conferring unidirectionality to mitosis.[13] Maintaining the right amount of Wee1 is essential for cell growth and proliferation, and hence Wee1 is likely to participate in tumor progression. Lung cancer biopsies have low levels of Wee1 protein.[14] By contrast, increasing Wee1 levels by reducing its degradation in a prostate cancer model was beneficial as it limited cell growth.[15] Moreover, an anticancer compound that increases the steady-state levels of Wee1 by inhibiting Plk-1 dependent Wee1 turnover entered phase I clinical trials.[5] In addition, many cancer cells are lacking Wee1-dependent checkpoint pathways needed to ensure proper correction of DNA defects prior to mitosis, causing the cells to divide with incompletely replicated DNA.[16] Tight regulation of Wee1 activity in these cells may prevent the genomic instability caused by premature mitosis entry. Taken together, these studies suggest Wee1 is a promising target in cancer and the regulation of its degradation to be point of choice for chemotherapeutic intervention. In addition to providing potential novel drug leads, small-molecule inhibitors of Wee1 degradation could yield valuable probes to decipher pathways controlling Wee1 turnover and cell cycle transit. However, no effort to identify such small molecule probes has been reported thus far.
In this probe report, we describe a novel homogeneous 1536-well plate assay to monitor Wee1 degradation using cryopreserved transiently transected cells. We also demonstrate the excellent performance of this assay in the context of an ultra-high-throughput screening (uHTS) campaign that led to the identification of potential selective cell-permeable Wee1-Luc stabilizers, and a novel probe, ML118 [17].
Table
PubChem BioAssay Table.
Table
Table of Assay Rationale and Description.
2. Materials and Methods
2.1. Assays
(Click on the hyperlinks to obtain itemized protocols directly from PubChem; also see Summary AID-1807 and reference [17])
WEE1 Degradation Inhibition Assays (PubChem AIDs 1321, 1410, 1412, and 2088)
The purpose of these assays is to identify compounds that act as inhibitors of Wee1 degradation. These assays employ HeLa cells transfected with a kinase negative mutant of Wee1 (Wee1K328M) fused to a luciferase reporter gene. As designed, compounds that increase Wee1K328M-luciferase stability and/or prevent its degradation will lead to increased well luminescence compared to untreated wells. Specifically, compounds that increase luminescence are considered Wee1 degradation inhibitors. Compounds were tested in singlicate (AID-1321) or triplicate (AID-1410) at a final nominal concentration of 5 μM, and in a 10-point, 1:3 serial dilutions starting at a nominal test concentration of 50 micromolar (AIDs 1412 and 2088).
Cyclin B Degradation Inhibition Counterscreen (AIDs 1414 and 2088)
The purpose of these assays is to determine whether compounds identified as active in a previous set of experiments entitled, “Primary cell-based high throughput screening assay for inhibitors of Wee1 degradation” (PubChem AID-1321), and that confirmed activity in a set of experiments entitled, “Confirmation cell-based high throughput screening assay for inhibitors of Wee1 degradation” (PubChem AID-1410), were non-selective inhibitors of protein degradation, as measured by inhibition of cyclin B degradation. These assays employ HeLa cells transfected with a plasmid that encodes a cyclin B-luciferase fusion protein to monitor cyclin B levels. The cyclin B-luciferase complex is rapidly turned over in these cells. As designed, compounds that inhibit cyclin B degradation will increase cyclin B-luciferase stability, leading to increased well luminescence. Compounds were tested in triplicate using a 10-point, 1:3 dilution series, starting at a nominal concentration of 50 μM (AIDs 1414 and 2088).
Cytotoxicity Counterscreen (AIDs 1413)
The purpose of this assay is to determine the cytotoxicity of compounds identified as active in a previous set of experiments entitled, “Primary cell-based high throughput screening assay for inhibitors of Wee1 degradation” (PubChem AID-1321), and that confirmed activity in a set of experiments entitled, “Confirmation cell-based high throughput screening assay for inhibitors of Wee1 degradation” (PubChem AID-1410). The assay employs the CellTiter-Glo luminescent reagent, which contains luciferase to catalyze the oxidation of beetle luciferin to oxyluciferin and light in the presence of cellular ATP. As designed, cytotoxic compounds will reduce viable cell numbers and ATP levels, resulting in decreased well luminescence. Compounds were assayed in a 10-point 1:3 dilution series starting at a nominal concentration of 50 μM.
G2/M Arrest Assays (AIDs 2088)
In order to determine whether the identified probe candidate could inhibit cell cycle progression, cell cycle analyses were made by performing FACS analysis. This assay was performed by the assay provider. Compound-treated HeLa cells were resuspended in 70% ethanol, incubated at −20C overnight, and washed with 10ml cold PBS. The supernatant was removed and the cell pellet was resuspended in 38 mM sodium citrate containing 69 mM of propidium iodide and 19 mg/mL of RNase A. FACS analysis was performed on a BD Bioscience LSR II system and analyzed using Flowjo 8.7.3 software. The compound was tested in triplicate using a 3-point, 1:10 dilution series, starting at a nominal concentration of 5 micromolar.
2.2. Probe Chemical Characterization
Synthetic route. Synthesis schemes for probe (Scaffold 1) and representative compounds from Scaffolds 2 and 3:

Figure
Scaffold 1 (PROBE). SID-87235992 Synthesis of SID-87235992 (liquid SID-4243143)

Figure
Scaffold 2. SID-87235990 Synthesis of SID-87235990 (liquid SID-3713089)

Figure
Scaffold 3. SID-87235991 Synthesis of SID-87235991 (liquid SID-4256064)
Probe chemical structure including stereochemistry. Separation of diastereomers (if necessary)
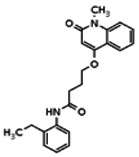
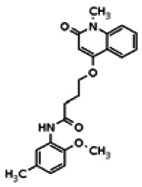
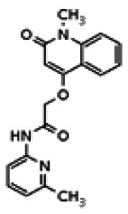
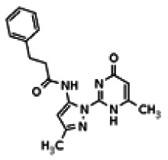
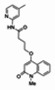
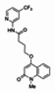
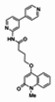
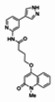
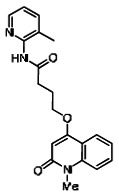
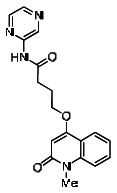
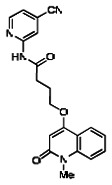
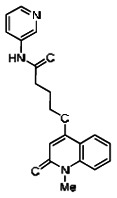
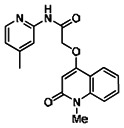
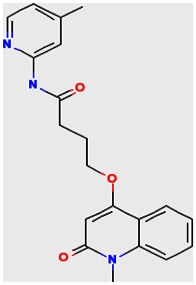
The structure of the probe ML118, is shown below. ML118 is an achiral molecule, and has no stereochemistry or stereoisomers.
Structure verification with 1H NMR and LCMS results
Probe ML118 was obtained as an oil with 99% purity (HPLC analysis): 1H NMR (DMSO-d6, 400MHz) δ 10.6 (br, s, 1H), 8.08-8.06 (m, 1H, 7.81-7.72 (m, 2H), 7.56-7.52 (m, 1H, 7.42-7.34 (m, 1H), 7.06-7.02 (m, 1H), 6.94-6.92 (m, 1H), 5.96 (s, 1H), 4.12 (t, 2H, J = 6), 3.48 (s, 3H), 2.59 (t, 2H, J = 7.2), 2.27 (s, 3H), 2.12-2.06 (m, 2H). 13C NMR (DMSO-d6, 100MHz) δ 172.6, 162.3, 160.9, 152.4, 150.3, 144.0, 139.4, 131.3, 122.7, 121.2, 120.6, 115.5, 114.5, 114.3, 96.6, 67.9, 33.0, 28.5, 23.8, 21.2. IR (neat) 2945, 1720, 1636, 1579, 1505, 1368 cm−1. LCMS 352.01 (M+H+). HRMS (ESI) 374.1484 m/z (calc M+Na+ C20H21N3O3Na 374.1475).
Solubility. The solubility of the probes was measured in phosphate buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate dibasic, 2 mM potassium phosphate monobasic and a pH of 7.4) at room temperature (23°C). The solubility of probe ML118 was found to be 58 μM.
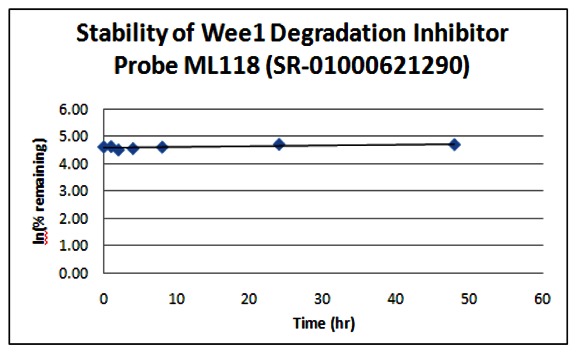
Stability. The stability of the probe was measured at room temperature (23ºC) in PBS (no antioxidants or other protectants; DMSO concentration below 0.1%). The stability, represented by the half-life, was found to be > 48 hours. Below is a graph showing loss of compound with time over a 48 hour period with a minimum of 6 time points. The table indicates the percent of compound remaining at the end of the 48 hours.
The probe was measured for its ability to form glutathione adducts. At concentrations of 100 μM reduced GSH, 10 μM of the probe does not appear to be a Michael acceptor [18, 19].
2.3. Probe Preparation
Detailed experimental procedures for the synthesis of Probe ML118 (SR-01000621290; CID-3237904; SID-4243143) follow.

tert-Butyl 4-bromobutanoate (2): Trifluoroacetic anhydride (6 mL) was added slowly to a −40 °C solution of 4-bromobutyric acid (1.7 g, 10.2 mmol) in dry THF (20 mL). The resulting solution was stirred at −40 °C for approximately 30 min. tert-Butanol (25 mL) was added and the solution allowed to warm up to ambient temperature and was stirred for an additional 16 h. The reaction mixture was poured slowly into a mixture of crushed ice and saturated sodium bicarbonate solution (50 mL). The product was extracted into EtOAc (2×100 mL) and the organic phase was washed with water (2×100 mL) and brine (2×100 mL). The organic extracts were dried over Na2SO4, then the solution was concentrated under reduced pressure to obtain an oil. The oil was dissolved in MTBE (100 mL) and filtered through a short pad of silica gel. The silica pad was washed with MTBE (100 mL). The combined filtrate was concentrated under reduced pressure to obtain 4.1 g (72%) of the knowni t-butyl ester 2 as a liquid: 1H NMR (CDCl3, 400MHz) δ 3.45 (t, 2H, J = 6.4 Hz), 2.40 (t, 2H, J = 7.2 Hz), 2.16-2.10 (m, 2H), 1.45 (s, 9H).

tert-Butyl 4-(1-methyl-2-oxo-1,2-dihydroquinolin-4-yloxy) butanoate (3). To a stirred solution of 4-hydroxy-1-methylquinolin-2(1H)-one (1.0 g, 5.71 mmol) in anhydrous DMF (10 mL) was added tert-butyl 4-bromobutanoate (1.25 g, 5.71 mmol) and cesium carbonate (2.23 g, 6.85 mmol). The reaction mixture was stirred at room temperature for 14 h. After completion of the reaction (monitored by LCMS), the solid was filtered off and DMF removed under vacuum. The residue was dissolved in ethyl acetate and washed successively with water and brine. The organic phase was dried over sodium sulfate and filtered. Removal of solvent by rotary evaporation gave crude 3 (1.59 g, 88% yield), which was directly used in the next step without further purification. LCMS 317.98 (M+H+).
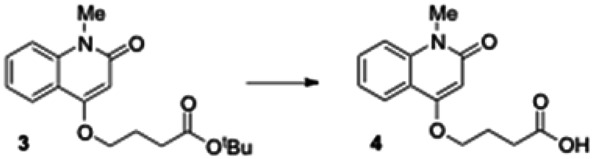
4-(1-Methyl-2-oxo-1,2-dihydroquinolin-4-yloxy) butanoic acid (4). Crude 3 from the previous experiment (500 mg, 1.58 mmol) was dissolved in anhydrous dichloromethane (5 mL). Triethylsilane (0.629 mL, 3.94 mmol) and trifluoroacetic acid (1.57 mL, 20.5 mmol) were then added to reaction mixture. The mixture was stirred for 5 h under nitrogen until the reaction was complete (monitored by LCMS). The solvent was removed under vacuum and the residue was dissolved in diethyl ether, causing the product to separate as a white solid which was filtered and dried under vacuum to give the carboxylic acid 4 (400 mg, quantitative yield): 1H NMR (CDCl3, 400MHz) δ 8.02 (dd, J = 8), 1H), 7.644-7.600 (m, 1H), 7.40-7.26 (m, 2H), 6.30 (s, 1H), 4.23 (t, 2H, J = 6.4), 3.72 (s, 3H), 2.67 (t, 2H, J = 6.8), 2.30-2.27 (m, 2H); LCMS 262.01 (M+H+).
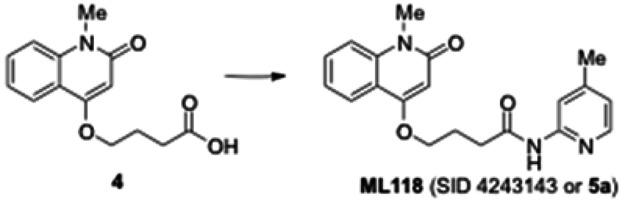
4-(1-Methyl-2-oxo-1,2-dihydroquinolin-4-yloxy)-N-(4-methylpyridin-2-yl)butanamide (ML118, SID-4243143, 5a). A mixture of carboxylic acid 4 (100 mg, 0.38 mmol), HATU (218mg, 0.57 mmol), and triethylamine (0.08 mL, 0.57 mmol) in anhydrous DMF (2 mL) was stirred under argon atmosphere for 15 min. 2-Amino-4-methylpyridine (49 mg, 0.46mmol) was then added. The reaction mixture stirred for 3 h. After completion of the reaction, DMF was removed by rotary evaporation, and the residue was dissolved in ethyl acetate. The organic phase was washed with saturated solution of NaHCO3 (50mL). Removal of solvent and purification of the residue by preparative HPLC afforded ML118 (69 mg, 51% yield) as an oil with 99% purity (HPLC analysis): 1H NMR (DMSO-d6, 400MHz) δ 10.6 (br, s, 1H), 8.08-8.06 (m, 1H, 7.81-7.72 (m, 2H), 7.56-7.52 (m, 1H, 7.42-7.34 (m, 1H), 7.06-7.02 (m, 1H), 6.94-6.92 (m, 1H), 5.96 (s, 1H), 4.12 (t, 2H, J = 6), 3.48 (s, 3H), 2.59 (t, 2H, J = 7.2), 2.27 (s, 3H), 2.12-2.06 (m, 2H). 13C NMR (DMSO-d6, 100MHz) δ 172.6, 162.3, 160.9, 152.4, 150.3, 144.0, 139.4, 131.3, 122.7, 121.2, 120.6, 115.5, 114.5, 114.3, 96.6, 67.9, 33.0, 28.5, 23.8, 21.2. IR (neat) 2945, 1720, 1636, 1579, 1505, 1368 cm−1. LCMS 352.01 (M+H+). HRMS (ESI) 374.1484 m/z (calc M+Na+ C20H21N3O3Na 374.1475).
3. Results
3.1. Summary of Screening Results
Following primary HTS in singlicate to identify Wee1 degradation inhibitors (AID-1321), confirmation of hit activity in triplicate (AID-1410), titration assays to determine compound potency (AID-1412), cytotoxicity (AID-1413), and selectivity against cyclin B (AID-1414), compounds were identified as possible candidates for probe development. Powder assay data is available in late stage AID-2088.
3.2. Dose Response Curves for Probes
Probe ML118 (SR-01000621290; CID-3237904; SID-4243143).
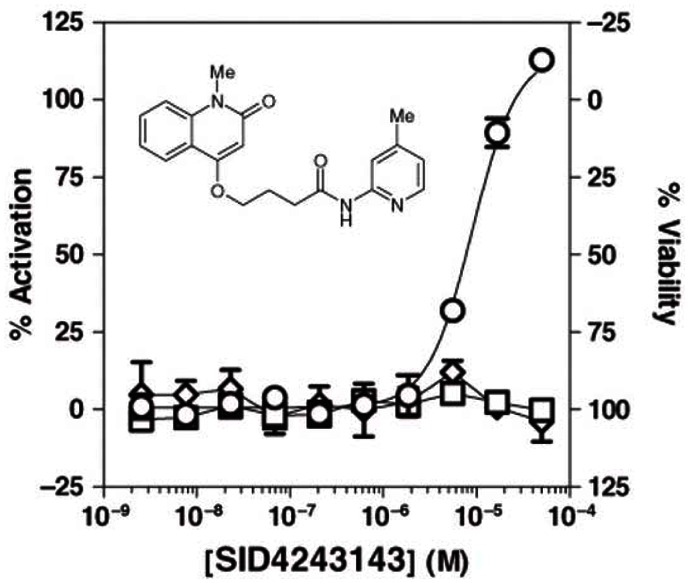
Dose-response curves for Wee1 Degradation inhibitor probe ML118 in three cell-based assays.
Ten-point, 1:3 serial dilution of probe compound ML118 (SID-4243143) were tested in triplicate in both the Wee1 (circles), N-Cyclin B (squares), and viability (diamonds) assays. Note the limited cytotoxicity and activity in the N-cyclin B assay at higher test concentrations. Data points represent mean ± SD (n = 3).
3.3. Scaffold/Moiety Chemical Liabilities
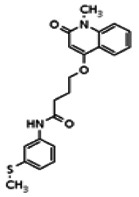
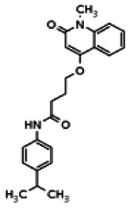
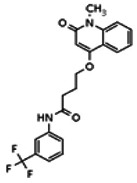
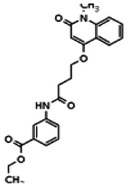
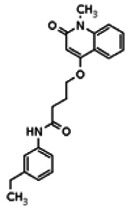
Describe SAR & chemistry strategy (including structure and data) that led to the probe. Scaffolds 1, 2 and 3 were the top hits from the HTS. The MLMSR was interrogated to identify structural analogs that were tested in the HTS, but found to be inactive, in order to begin generation of SAR information for all three series. Databases of commercial compounds were interrogated to identify additional analogs that were purchased for screening. Relatively few analogs of Scaffold 3 were identified. Efforts with scaffold 1 focused on substituent changes in the aminopyridine amide, the spacer chain length separating the aminopyridine from the 4-alkoxy quinolone ring. Very little structure diversity was encountered in the 4-alkoxy quinolone ring, so that unit was held constant. Because relatively few analogs with substituted aminopyridine units were available, we elected to examine substituted anilines (phenyl compounds) at this position.
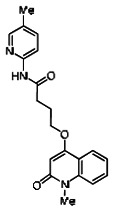
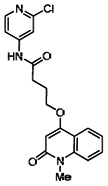
A reasonable number of analogs were identified from commercial sources in the pyrazolyl pyrimidine series, or were synthesized at TSRI, in order to probe the effect of substituents at different positions of the tricyclic ring system. Results of this SAR effort are summarized below.
Scaffold 1
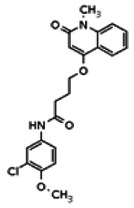
Analogs in the Scaffold 1 series were purchased in powder form and tested in dose response assays against both Wee1 and Cyclin B degradation. Results for these compounds are summarized in the SAR table below. Unfortunately, 100% of the analogs identified and obtained in this way were inactive in the Wee1 stabilization assay (IC50 > 25 μM in all cases). Thus, the 4-alkoxy quinolone, SID-4243143/CID-3237904 (powder SID-87235992), was the only compound in this series to display activity in the Wee1 stabilization assay and lack of activity in the Cyclin B assay. This compound was declared a probe (ML118) on the basis of its ability to induce G2/M arrest while not increasing sub G1. The activity of ML118/SID-4243143/CID-3237904 (powder SID-87235992) was confirmed by resynthesis, using the route summarized in Section 2.2 of this Probe Report. Five (inactive) analogs of SID-4243143/CID-3237904 (powder SID-87235992) were also synthesized for submission to the MLSMR, as we were unable to obtain sufficient quantities of these compounds from commercial sources.
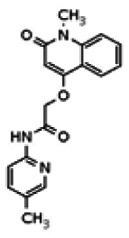
While the initial biological characterization of ML118/SID-4243143 was in progress, a preliminary chemical optimization effort was initiated. Preliminary results of a structure-activity relationship (SAR) study revealed an unusually stringent profile of compound ML118/SID-4243143. Data available from the SAR by purchase effort indicated that an aminopyridine carboxamide unit was required for activity. In addition, initial SAR efforts focused on moving the methyl to the 2-position of the pyridine ring resulted in complete loss of activity ((see SAR table, analog 6). Finally, amides based on 2-amino-6-methylpyridine and 2-amino-5-methylpyridine with only 1 methylene between the phenolic oxygen and carbonyl group were also found to be inactive (SAR table, analogs 15 and 16).
Scaffold 2 (no probes resulted from this scaffold)
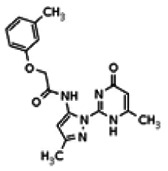
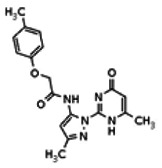
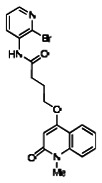
Of all the compounds in the pyrazolyl pyrimidone scaffold series that were examined, only the hit compound SID-3713089/CID-2408467 (powder SID-87235990) was confirmed to be active in the Wee1 degradation inhibition assay (see SAR table below). Its activity was confirmed by resynthesis. However, SID-3713089/CID-2408467 (powder SID-87235990) did not induce cell synchronization (G2/M arrest). Several compounds in this series that initially appeared to be active (e.g., compounds 2 and 4 in the pyrazolyl pyrimidine SAR table) did not confirm upon subsequent examination. Therefore, no probes were identified in the pyrazolyl pyrimidone series.
Scaffold 3 (no probes resulted from this scaffold)
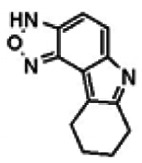
The only compound in the pyrrolo-benzoxadiazole N-oxide scaffold series that proved active in the Wee1 degradation inhibition assay was SID-4256064/CID-690911 (including related compounds in the screening deck). The activity of SID-4256064/CID-690911 (powder SID-87235991) was confirmed by resynthesis, but SID-4256064/CID-690911 failed to induce cell synchronization (G2/M arrest). Therefore, no probes were identified in the pyrrolo-benzoxadiazole N-oxide scaffold series.
3.4. SAR Tables
Table
WEE1 Degradation Inhibitor SAR Table (Scaffold 1: 4-alkoxy quinolone scaffold).
Table
Wee1 SAR Table (Scaffold 2: pyrazolyl pyrimidone; there are no probes in this scaffold).
Table
Wee1 SAR Table (Scaffold 3: pyrrolo-benzoxadiazole N-oxide; there are no probes in this scaffold).
Following the initial SAR effort, the SRIMSC synthesized an additional 20 analogs in a second round. The results are summarized in the tables below. All analogs are acylated 2-amino pyridine derivatives. The top 4 of these analogs (SR-03000001762, SR-03000001772, SR-03000001774, and SR-03000001775) appear to be more potent than the first generation Wee1 degradation inhibitor probe ML118. Three of these new analogs were submitted for solubility and stability testing; data are provided following the new SAR table. In addition, the assay provider repeated dose response assays on the top 3–4 inhibitors to confirm the potency of the individual compounds. Analysis of these results reveals that one of these new analogs (SR-03000001762-1) exhibited an EC50 of 346nM and is 10-fold more potent than the original probe ML118, and thus is an improvement over ML118. Moreover, data presented in Section 3.5 indicates that SR-03000001762 is more effective in arresting cells at the G2/M checkpoint.

3.5. Cellular Activity
Probe ML118 was tested in a variety of cell-based assays performed by the SRIMSC and assay provider. These assays were performed to determine the probe’s selectivity and mechanism of action. The results of these studies demonstrated that probe ML118 is a stabilizer of Wee1 that inhibits Wee1 degradation.
FACS analysis/G2/M Arrest Assays
In order to determine whether the identified inhibitors of Wee1 degradation inhibit cell cycle progression, the effect of probe compound ML118 (SID-4243143; powder SID-87235992/CID-44552613) on cell cycle was tested by performing FACS analysis. Compound-treated HeLa cells were resuspended in 70% ethanol, incubated at −20°C overnight, and washed with 10ml cold PBS. The supernatant was removed and the cell pellet was resuspended in 38 mM sodium citrate containing 69 mM of propidium iodide and 19 mg/mL of RNase A. FACS analysis was performed on a BD Bioscience LSR II system and analyzed using Flowjo 8.7.3 software.
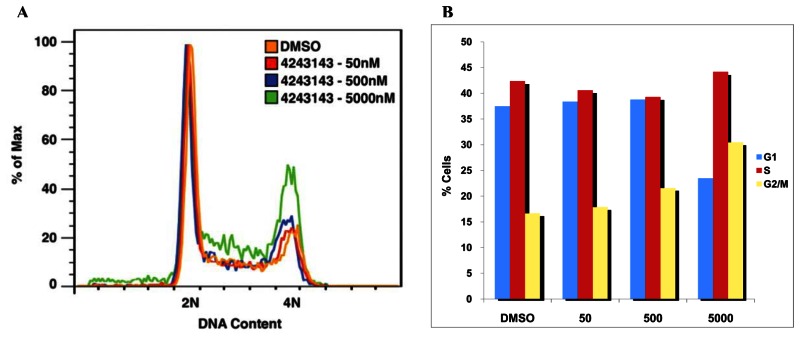
As shown in the figure below, probe compound ML118/SID-4243143/CID-3237904 (powder SID-87235992) induces G2/M arrest and does not increase sub G1. A) Cell sorting images of ML118/SID-4243143/CID-3237904 (powder SID-87235992) at 1:10 increasing concentrations of 50nM, 500nM, and 5000nM for 20 hours. B) Histogram showing the percentage of cells in each stage of the cell cycle. Whereas DMSO-treated cells contained 16.7% of cells in the G2/M phase, SID-4243143/CID-3237904 (powder SID-87235992)-treated cells contained 30.5%. Furthermore, no increase in the sub-G1 population after SID-4243143 treatment was observed, suggesting that compound ML118/SID-4243143/CID-3237904 (powder SID-87235992) is not toxic. While this compound acts as an inhibitor of cell cycle progression, it is not a proteasome inhibitor since it did not affect turnover of another proteasome substrate, N-cyclin B-luciferase.
FACS analysis/G2/M arrest assay data for the three most potent new analogs, SR-1762, DR-1772 and SR-1775, are summarized below. These experiments were performed using the test compounds at 1.11 μM. These data demonstrate that the new analogs, and especially SR-03000001772, are more potent than the original probe, ML118, in these experiments.
3.6. Profiling Assays
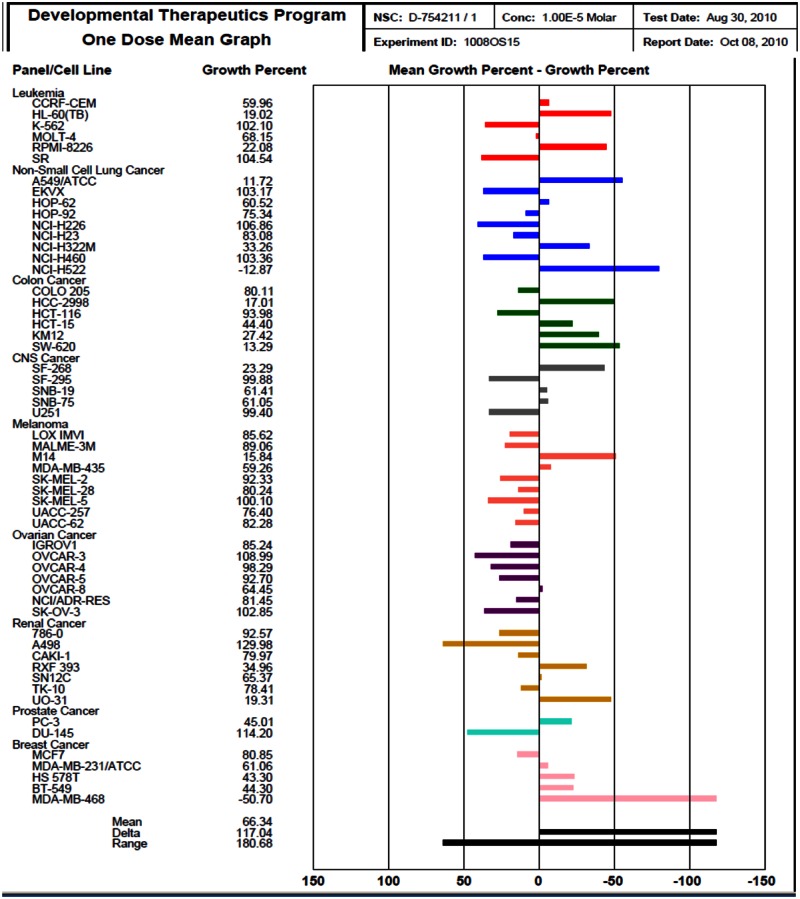
In addition to the above cellular assays, we have also obtained profiling results for probe ML118 at 10 μM using the NCI-60 DTP Human Tumor Cell Line Screen (Background and Methods can be found at http://dtp.nci.nih.gov/branches/btb/ivclsp.html). This compound advanced to dose response testing and has been assigned NIH code 754211. The profiling screen examined the effect of a single high concentration (10 μM) of the probe on cellular growth of 60 well-characterized cell lines during 48 hours of exposure http://dtp.nci.nih.gov/branches/btb/onedose_interp.html
The One-dose data is reported as a mean graph of the percent growth of treated cells. The number reported for the One-dose assay is growth relative to the no-drug control, and relative to the time zero number of cells. This allows detection of both growth inhibition (values between 0 and 100) and lethality (values less than 0). This is the same as for the 5-dose assay, described on http://dtp.nci.nih.gov/branches/btb/ivclsp.html. For example, a value of 100 means no growth inhibition. A value of 40 would mean 60% growth inhibition. A value of 0 means no net growth over the course of the experiment. A value of −40 would mean 40% lethality. A value of −100 means all cells are dead. Importantly, probe ML118 was able to inhibit the growth of the majority of cancer cell lines tested (mean growth value between 0 and 100). Thus, it is clear that this probe represents a useful tool for cell-based studies involving cell cycle regulation and cancer-related events.
Results of the NCI-60 profiling assays preformed with ML118 are on the next page.
Kinase profiling
Wee1 degradation has been shown to be regulated by complex pathways involving kinases and phosphatases.[20] In an attempt to identify potential new proteins involved in Wee1 phosphorylation, we profiled probe ML118/SID-4243143 against a panel of kinases involved in cell cycle progression, including Wee1 itself. The mitotic kinase profiling assay was performed by Reaction Biology Corporation (Malvern, PA) using a radiometric-based filtration binding assay as previously described.[21] Briefly, compound SID-4243143 was tested in duplicate at 10 μM on a panel of purified kinases in the presence of 32P-γ-ATP. Incorporation of 32P-γ-ATP into peptide substrates was measured and compared to DMSO controls. The kinase pan-inhibitor staurosporine was used as a positive control for these experiments. As shown below, the hit compound we identified significantly inhibited CDK9, either coupled to cyclin K or cyclin T1. As shown below, black bars indicate activities lower than the average of activities measured for all kinases minus 3 times their standard deviation. Error bars represent the standard deviation (n = 2).

IC50’s for the inhibition by ML118 of kinases reported in the literature to act on Wee1 are summarized below [5, 11]. The data for inhibition of CDK9/cyclin K and CDK9/cyclinT1 track well with the IC50 value for inhibition of Wee1 degradation by ML118. Because the pyridine carboxamide is essential for activity, a plausible hypothesis is that ML118 inhibits Wee1 degradation by inhibiting a kinase (or kinases) responsible for phosphorylation of Wee1 and thereby targeting Wee1 for degradation via the proteosome [5, 11].
4. Discussion
4.1. Comparison to existing art and how the new probe is an improvement
The new probe ML118 is a significant improvement over prior art compound MG132 because it is a selective inhibitor of Wee1 degradation, and unlike MG132, is not a generic proteosome inhibitor. Furthermore, given that it also inhibits CDK9/cyclin K and CDK9/cyclin T1 selectively relative to a panel of kinases tested, it is possible that stabilization of Wee1 mediated by ML118 is due to inhibition of CDK9/cyclin K and/or CDK9/cyclin T1. In this model, CDK9 would phosphorylate either Wee1 directly or a protein required for Wee1 degradation. Thus, ML118 would act upstream of the proteasome.
4.2. Mechanism of Action Studies
Our studies indicate that ML118 inhibits CDK9 activity in vitro. Since ML118 also inhibits Wee1 turnover, we hypothesize that it CDK9 activity may be necessary for Wee1 turnover. We will test this hypothesis using siRNAs targeting CDK9 and or Cyclin H or Cyclin K and measuring Wee1 degradation. We do not believe that any kinase inhibitor stabilizes Wee1 nonspecifically since we have screened several kinase inhibitors and have not observed any stabilization of Wee1. For instance, the Plk-1 inhibitor BI2536 does not stabilize Wee1-luciferase (our unpublished observations). This may indicate that Plk-1 mediated degradation of Wee1 occurs within mitosis and since we added BI2536 to asynchronous cells, which are mostly in interphase, we would not observe any effects of PLK-1 inhibition during interphase. By contrast, this may suggest that ML118 inhibits Wee1-luciferase degradation during interphase and not mitosis. Subsequent experiments where we synchronize cells in different phases of the cell cycle and treat with either ML118 or BI2536 will delineate the cell cycle phase where PLK-1 or CDK9 induces Wee1 degradation.
4.3. Planned Future Studies
The assay provider and SRIMSC are currently developing more potent novel compounds as Wee1 degradation inhibitors that represent an improvement over the current probe ML118. These efforts are discussed in the probe report for ML177.
We will test new analogs in both Wee1 stabilization assays and CDK9 kinase assays. In addition, we will perform kinase profiles of select compounds to determine the specificity of the compounds for CDK9 inhibition. If we find specific inhibition of CDK9 relative to many kinases, we will determine whether CDK9 indeed directly phosphorylates Wee1. We will then map phosphorylation sites on Wee1 mediated by CDK9 and determine whether mutation of these sites inhibits Wee1 degradation in cells.
5. References
- 1.
- Hashimoto O, Shinkawa M, Torimura T, Nakamura T, Selvendiran K, Sakamoto M, Koga H, Ueno T, Sata M. Cell cycle regulation by the Wee1 inhibitor PD0166285, pyrido [2,3-d] pyimidine, in the B16 mouse melanoma cell line BMC Cancer 20066292. [PMC free article: PMC1770931] [PubMed: 17177986]
- 2.
- Mizenina OA, Moasser MM. S-phase inhibition of cell cycle progression by a novel class of pyridopyrimidine tyrosine kinase inhibitors. Cell Cycle. 2004;3(6):796–803. [PubMed: 15136770]
- 3.
- Palmer BD, Smaill JB, Rewcastle GW, Dobrusin EM, Kraker A, Moore CW, Steinkampf RW, Denny WA. Structure-activity relationships for 2-anilino-6-phenylpyrido[2,3-d]pyrimidin-7(8H)-ones as inhibitors of the cellular checkpoint kinase Wee1. Bioorg Med Chem Lett. 2005;15(7):1931–5. [PubMed: 15780636]
- 4.
- Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, Sun Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001;61(22):8211–7. [PubMed: 11719452]
- 5.
- Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17(4):304–15. [PubMed: 17291761]
- 6.
- Li C, Andrake M, Dunbrack R, Enders GH. A bifunctional regulatory element in human somatic Wee1 mediates cyclin A/Cdk2 binding and Crm1-dependent nuclear export. Mol Cell Biol. 2010;30(1):116–30. [PMC free article: PMC2798281] [PubMed: 19858290]
- 7.
- Reed SI. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol. 2003;4(11):855–64. [PubMed: 14625536]
- 8.
- Gautier J, Solomon MJ, Booher RN, Bazan JF, Kirschner MW. cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell. 1991;67(1):197–211. [PubMed: 1913817]
- 9.
- Perry JA, Kornbluth S. Cdc25 and Wee1: analogous opposites? Cell Div 2007212. [PMC free article: PMC1868713] [PubMed: 17480229]
- 10.
- Kraft C. Mitotic entry: tipping the balance. Curr Biol. 2003;13(11):R445–6. [PubMed: 12781155]
- 11.
- Watanabe N, Arai H, Iwasaki J, Shiina M, Ogata K, Hunter T, Osada H. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc Natl Acad Sci U S A. 2005;102(33):11663–8. [PMC free article: PMC1187955] [PubMed: 16085715]
- 12.
- Watanabe N, Arai H, Nishihara Y, Taniguchi M, Hunter T, Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A. 2004;101(13):4419–24. [PMC free article: PMC384762] [PubMed: 15070733]
- 13.
- Pomerening JR, Kim SY, Ferrell JE Jr. Systems-level dissection of the cell-cycle oscillator: bypassing positive feedback produces damped oscillations. Cell. 2005;122(4):565–78. [PubMed: 16122424]
- 14.
- Yoshida T, Tanaka S, Mogi A, Shitara Y, Kuwano H. The clinical significance of Cyclin B1 and Wee1 expression in non-small-cell lung cancer. Ann Oncol. 2004;15(2):252–6. [PubMed: 14760118]
- 15.
- Kiviharju-af Hallstrom TMS, Jaamaa M, Monkkonen K, Peltonen LC, Andersson RH, Medema DM, Peehl, Laiho M. Human prostate epithelium lacks Wee1A-mediated DNA damage-induced checkpoint enforcement. Proc Natl Acad Sci U S A. 2007;104(17):7211–6. [PMC free article: PMC1855358] [PubMed: 17431037]
- 16.
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434(7035):907–13. [PubMed: 15829965]
- 17.
- Madoux F, Simanski S, Chase P, Mishra JK, Roush WR, Ayad NG, Hodder P. An Ultra-High Throughput Cell-Based Screen for Wee1 Degradation Inhibitors. J Biomol Screen. 2010 [PMC free article: PMC3082437] [PubMed: 20660794]
- 18.
- Li X, He Y, Ruiz CH, Koenig M, Cameron MD, Vojkovsky T. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37(6):1242–50. [PMC free article: PMC3202349] [PubMed: 19282395]
- 19.
- Li X, Kamenecka TM, Cameron MD. Bioactivation of the epidermal growth factor receptor inhibitor gefitinib: implications for pulmonary and hepatic toxicities. Chem Res Toxicol. 2009;22(10):1736–42. [PubMed: 19803472]
- 20.
- Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14(9):1878–91. [PMC free article: PMC398287] [PubMed: 7743995]
- 21.
- Ma H, Horiuchi KY, Wang Y, Kucharewicz SA, Diamond SL. Nanoliter homogenous ultra-high throughput screening microarray for lead discoveries and IC50 profiling. Assay Drug Dev Technol. 2005;3(2):177–87. [PubMed: 15871692]
Publication Details
Author Information and Affiliations
Authors
F Madoux,1 J Mishra,2 BA Mercer,1 N Ayad,3,* W Roush,2 P Hodder,1,4 and HR Rosen5.Affiliations
Publication History
Received: August 14, 2009; Last Update: March 25, 2011.
Copyright
Publisher
National Center for Biotechnology Information (US), Bethesda (MD)
NLM Citation
Madoux F, Mishra J, Mercer BA, et al. Small Molecule Inhibitors of Wee1 Degradation and Mitotic Entry. 2009 Aug 14 [Updated 2011 Mar 25]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.