See "Essentials of Glycobiology, 4th Edition"
See the updated version of this chapter
This chapter focuses on the structure, biosynthesis, and general biology of proteoglycans. Topics include a description of the major families of proteoglycans, their characteristic polysaccharide chains (glycosaminoglycans), biosynthetic pathways, and general concepts about proteoglycan function. Proteoglycans, like other glycoconjugates, are enormously diverse and have many essential roles in biology.
HISTORICAL PERSPECTIVE
The study of proteoglycans dates back to the beginning of the 20th century with investigations of “chondromucoid” from cartilage and anticoagulant preparations from liver (heparin). From 1930 to 1960, great strides were made in analyzing the chemistry of the polysaccharides of these preparations (also known as “mucopolysaccharides”), yielding the structure of hyaluronan (see Chapter 15), dermatan sulfate (DS), keratan sulfate (KS), different isomeric forms of chondroitin sulfate (CS), heparin, and heparan sulfate (HS). Together, these polysaccharides came to be known as glycosaminoglycans (sometimes abbreviated as GAGs) to indicate the presence of amino sugars and other sugars in a polymeric form. Subsequent studies provided insights into the linkage of the chains to core proteins, and these structural studies paved the way for the biosynthetic studies that followed.
The 1970s marked a turning point in the field, when improved isolation and chromatographic procedures were developed to purify and analyze tissue proteoglycans and glycosaminoglycans. Density-gradient ultracentrifugation allowed separation of the large aggregating proteoglycans from cartilage, revealing a complex of proteoglycan, hyaluronan, and link protein (Figure 16.1). Also during this period, it was realized that the production of proteoglycans was a general property of animal cells and that proteoglycans and glycosaminoglycans were present on the cell surface, inside the cell, and in the extracellular matrix (ECM). This observation led to a rapid expansion of the field and the eventual appreciation of proteoglycan function in cell adhesion and signaling, as well as a host of other biological activities (see Chapter 35). Today, studies with somatic cell mutants (see Chapter 46) as well as experiments using gene knockout and silencing techniques in a variety of model organisms, including nematode worms (Caenorhabditis elegans), fruit flies (Drosophila melanogaster), African clawed frogs (Xenopus laevis), zebrafish (Danio rerio), and mice (Mus musculus), are aimed at extending our understanding of the role of proteoglycans in development and physiology (see Chapters 23–25). In turn, human diseases associated with aberrant biosynthesis or degradation of proteoglycans have been identified (see Chapters 38–44), and some are classified as congenital disorders of glycosylation. A variety of analytical techniques have been developed, including mass spectroscopic methods and glycan array applications, that provide new tools for understanding proteoglycan structure and function.
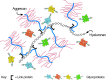
FIGURE 16.1
The large cartilage CS proteoglycan (aggrecan) forms an aggregate with hyaluronan and link protein (see Chapter 15). (Redrawn, with permission of Springer Science and Business Media, from Rodén L. 1980. In The biochemistry of glycoproteins and (more...)
PROTEOGLYCANS AND GLYCOSAMINOGLYCAN DIVERSITY
Proteoglycans consist of a core protein and one or more covalently attached glycosaminoglycan chains (Figure 16.2). Glycosaminoglycans are linear polysaccharides, whose disaccharide building blocks consist of an amino sugar (N-acetylglucosamine, glucosamine that is variously N-substituted, or N-acetylgalactosamine) and a uronic acid (glucuronic acid or iduronic acid) or galactose. Figure 16.3 depicts characteristic features of the major types of glycosaminoglycans found in vertebrates. Hyaluronan is shown for comparison: It does not occur covalently linked to proteoglycans, but instead interacts noncovalently with proteoglycans via hyaluronan-binding motifs (see Chapter 15). Generally, invertebrates produce the same types of glycosaminoglycans as vertebrates, except that hyaluronan is not present and the chondroitin chains tend to be nonsulfated. Most proteoglycans also contain O- and N-glycans typically found on glycoproteins (see Chapters 8 and 9). The glycosaminoglycan chains are much larger than these other types of glycans (e.g., a 20-kD glycosaminoglycan chain contains ~80 sugar residues, whereas a typical biantennary N-glycan contains 10–12 residues). Therefore, the properties of the glycosaminoglycans tend to dominate the chemical properties of proteoglycans (although N- or O-glycans on proteoglycans may have distinct biological properties as described for glycoproteins).
![FIGURE 16.2. Proteoglycans consist of a protein core (brown) and one or more covalently attached glycosaminoglycan chains ([blue] HS; [yellow] CS/DS).](/books/NBK1900/bin/ch16f2.gif)
FIGURE 16.2
Proteoglycans consist of a protein core (brown) and one or more covalently attached glycosaminoglycan chains ([blue] HS; [yellow] CS/DS). Membrane proteoglycans either span the plasma membrane (type I membrane proteins) or are linked by a GPI anchor. (more...)

FIGURE 16.3
Glycosaminoglycans consist of repeating disaccharide units composed of an N-acetylated or N-sulfated hexosamine and either a uronic acid (glucuronic acid or iduronic acid) or galactose. Hyaluronan lacks sulfate groups, but the rest of the glycosaminoglycans (more...)
Virtually all mammalian cells produce proteoglycans and secrete them into the ECM, insert them into the plasma membrane, or store them in secretory granules. The ECM determines the physical characteristics of tissues and many of the biological properties of the cells embedded in it. The major components of the ECM are fibrous proteins that provide tensile strength and elasticity (e.g., various collagens and elastins), adhesive glycoproteins (e.g., fibronectin, laminin, and tenascin), and proteoglycans that interact with other ECM components to provide a hydrated gel that resists compressive forces. Together, the various ECM components provide an extracellular environment that regulates cell proliferation and differentiation. Cells synthesize a diverse group of membrane proteoglycans. These typically have a type I orientation with a single membrane-spanning domain or a glycosylphosphatidylinositol (GPI) anchor (Figure 16.2). Additionally, some cells concentrate proteoglycans along with other secretory products in secretory granules. Secretory granule proteoglycans are thought to help sequester and regulate the availability of positively charged components, such as proteases and bioactive amines, through interaction with the negatively charged glycosaminoglycan chains.
The tremendous structural variation of proteoglycans is due to a number of factors. First, a large number of core proteins have been identified, and these can be substituted with one or two types of glycosaminoglycan chains. Some proteoglycans contain only one glycosaminoglycan chain (e.g., decorin), whereas others have more than 100 chains (e.g., aggrecan). Another source of variability lies in the stoichiometry of glycosaminoglycan chain substitution. For example, syndecan-1 has five attachment sites for glycosaminoglycans, but not all of the sites are used equally. Other proteoglycans can be “part time,” that is, they may exist with or without a glycosaminoglycan chain or with only a truncated oligosaccharide. A given proteoglycan present in different cell types often exhibits differences in the number of glycosaminoglycan chains, their lengths, and the arrangement of sulfated residues along the chains. Thus, a preparation of syndecan-1 represents a diverse population of molecules, each potentially representing a unique structural entity. These characteristics, typical of all proteoglycans, create diversity that may facilitate the formation of binding sites of variable density and affinity for different ligands.
The major classes of proteoglycans can be defined by distribution, homologies, and function. The following groupings provide an overview of the classes of proteoglycans currently known.
Interstitial Proteoglycans and the Aggrecan Family
A large number of proteoglycans are present in the ECM, and their distribution depends on the nature of the ECM. The interstitial proteoglycans represent a diverse class of molecules, differing in size and glycosaminoglycan composition. The small leucine-rich proteoglycans (SLRPs) contain leucine-rich repeats flanked by cysteines in their central domain. At least nine members of this family are known and they carry CS, DS, or KS chains (Table 16.1 and Table 16.2). These proteoglycans help to stabilize and organize collagen fibers, for example, in tendons. In the cornea, KS proteoglycans maintain the register of collagen fibers required for transparency. Decorin also can bind transforming growth factor β (TGF-β), serving as a sink to keep the growth factor sequestered in the matrix surrounding most cells.
TABLE 16.1
Examples of chondroitin sulfate proteoglycans
TABLE 16.2
Examples of keratan sulfate proteoglycans
The aggrecan family of proteoglycans consists of aggrecan, versican, brevican, and neurocan. In all four members, the protein moiety contains an amino-terminal domain capable of binding hyaluronan, a central region that contains covalently bound CS chains, and a carboxy-terminal domain containing a C-type lectin domain (see Chapter 31). Aggrecan is the best-studied member of this family, because it represents the major proteoglycan in cartilage. It contains as many as 100 CS chains and, in humans, it contains KS chains as well. Versican, which is produced predominantly by connective tissue cells, undergoes alternative splicing events that generate a family of proteins of differing complexity that may have a role in neural crest cell and axonal migration. Neurocan is expressed in the late embryonic central nervous system (CNS) and can inhibit neurite outgrowth. Brevican is expressed in the terminally differentiated CNS, particularly in perineuronal nets.
The interstitial proteoglycans and aggrecan family of proteoglycans appear to be unique to vertebrates. Other proteoglycans are expressed in C. elegans and D. melanogaster, suggesting that the core proteins have undergone enormous diversification during evolution.
Secretory Granule Proteoglycans
Serglycin is the major proteoglycan present in cytoplasmic secretory granules in endothelial, endocrine, and hematopoietic cells. Depending on the species, it has a variable number of glycosaminoglycan attachment sites that can carry CS or heparin chains. Heparin is a highly sulfated form of HS (discussed below) and is made exclusively on serglycin present in connective-tissue-type mast cells. Other granular proteoglycans may exist as well, such as chromogranin A, but the extent of glycosaminoglycan substitution appears to be substoichiometric, making them part-time proteoglycans.
Basement Membrane Proteoglycans
The basement membrane is an organized layer of the ECM that lies flush against epithelial cells and consists largely of laminin, nidogen, collagens, and proteoglycans. Basement membranes contain at least four types of proteoglycans depending on tissue type: perlecan, agrin, and collagen type XVIII (Table 16.3), which carry HS chains (although perlecan has been shown to carry CS in cartilage), and leprecan, which carries CS chains (Table 16.1). Perlecan has a mass of 400 kD and consists of multiple domains that have numerous functions. It has a role in embryogenesis and tissue morphogenesis and a particularly important role in cartilage development. Agrin acts in neuromuscular junctions (where it aggregates acetylcholine receptors) and in renal tubules (where it has an important role in determining the filtration properties of the glomerulus).
TABLE 16.3
Examples of heparan sulfate proteoglycans
Membrane-bound Proteoglycans
The membrane proteoglycans are diverse (Tables 16.1–16.3). The syndecan family consists of four members, each with a short hydrophobic domain that spans the membrane, linking the larger extracellular domain containing the glycosaminoglycan attachment sites to a smaller intracellular cytoplasmic domain. Syndecan-1 and syndecan-3 carry CS chains on the membrane proximal regions and HS chains at the more distal sites further away from the membrane. In contrast, syndecan-2 and syndecan-4 carry only HS chains. These are expressed in a tissue-specific manner and facilitate cellular interactions with a wide range of extracellular ligands, such as growth factors and matrix molecules. Because of their membrane-spanning properties, the syndecans can transmit signals from the extracellular environment to the intracellular cytoskeleton via their cytoplasmic tails. For example, binding of a ligand to the HS chain can induce oligomerization of syndecans at the cell surface, which leads to recruitment of factors at their cytoplasmic tails, such as kinases (e.g., c-Src), PDZ-domain proteins, or cytoskeletal proteins. The recruitment of cytoplasmic proteins in turn triggers a signal that affects actin assembly. Proteolytic cleavage of the syndecans occurs by matrix metalloproteases, resulting in shedding of the ectodomains bearing the glycosaminoglycan chains. These ectodomains can have potent biological activity as well, for example, by binding the same ligands as cell-surface proteoglycans (see Chapter 35). C. elegans and D. melanogaster express only one syndecan.
Each member of the glypican family of cell-surface proteoglycans has a GPI anchor attached at the carboxyl terminus, which embeds these proteoglycans in the outer leaflet of the plasma membrane. Thus, the glypicans do not have a cytoplasmic tail like the syndecans. The amino-terminal portion of the protein has multiple cysteine residues and a globular shape that distinguishes the glypicans from the syndecan ectodomains, which tend to be extended structures (Figure 16.2). Glypicans carry only HS chains, which can bind a wide array of factors essential for development and morphogenesis. Six glypican family members exist in mammals, two in D. melanogaster and one in C. elegans. Glypican-3 (GPC3) is the best-studied member of the family in vertebrates. Humans lacking functional GPC3 exhibit Simpson-Golabi-Behmel syndrome, characterized as an overgrowth disorder. The overgrowth phenotype suggests that GPC3 normally functions to inhibit cell proliferation, but the mechanism by which this occurs is unknown.
In addition to these two gene families, a number of other membrane proteoglycans are expressed on the surface of many different cell types. The CS proteoglycan NG2 is a surface marker expressed on stem cell populations, cartilage chondroblasts, myoblasts, endothelial cells of the brain, and glial progenitors. CD44, a transmembrane cell-surface receptor present on leukocytes and other cells, has a role in processes as diverse as immune cell trafficking and function, axon guidance, and organ development. Only certain splice forms of CD44 carry a glycosaminoglycan chain, and, like the aggrecan family, it can bind hyaluronan (see Chapter 15). Phosphacan is expressed as three different splice variants in the CNS, and, depending on the isoform, it can carry KS or CS chains. One splice variant is present in the ECM, whereas two other forms represent short- and full-length versions of a protein-tyrosine-phosphatase type of transmembrane receptor.
Undoubtedly, new proteoglycans will be discovered as different tissues and model organisms are studied. Genomic analysis has shown that many of the same HS proteoglycans are present in vertebrates and invertebrates, but the CS-containing proteoglycans appear to vary. The application of proteomic technologies to C. elegans has revealed a new family of chondroitin-containing proteoglycans that do not have orthologs in vertebrates or other invertebrates. Thus, we can expect new species of proteoglycans to emerge in the future as genomic and proteomic methods are applied across phylogeny.
KERATAN SULFATE, A SULFATED POLY-N-ACETYLLACTOSAMINE
KS consists of a sulfated poly-N-acetyllactosamine chain. The poly-N-acetyllactosamine structure is identical to that found on conventional glycoproteins and mucins (see Chapters 8 and 9). There are two types of KS, distinguished by the nature of their linkage to protein. KS I, originally described in cornea, is found on an N-glycan core structure linked to protein through an asparagine residue. KS II (skeletal KS) is found on an O-glycan core 2 structure and is thus linked through N-acetylgalactosamine to serine or threonine. Examples of KS proteoglycans are shown in Figure 16.4 and Table 16.2.
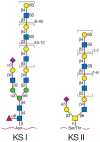
FIGURE 16.4
Keratan sulfates contain a sulfated poly-N-acetyllactosamine chain linked to either Asn or Ser/Thr residues. The actual order of the various sulfated and nonsulfated disaccharides occurs somewhat randomly along the chain. Not shown are sialic acids that (more...)
As mentioned above, KS proteoglycans maintain the even spacing of type I collagen fibrils in the cornea, allowing the passage of light without scattering. Defects in sulfation (macular corneal dystrophy) or chain formation (keratoconus) cause distortions in fibril organization and corneal opacity. In cartilage, the function of KS II is unclear. In humans and cows, the large CS proteoglycan found in cartilage (aggrecan) contains a segment of 4–23 hexapeptide repeats (E-E/L-P-F-P-S) where the KS chains are located, but aggrecan in rats and other rodents lacks this motif and does not contain KS.
The poly-N-acetyllactosamine of KS I can be quite long (~50 disaccharides, 20–25 kD) and contains a mixture of nonsulfated, monosulfated (Gal-GlcNAc6S), and disulfated (Gal6S-GlcNAc6S) disaccharide units. The biosynthesis of poly-N-acetyllactosamine and the underlying linkage structure is covered in Chapters 8 and 9. At least two classes of sulfotransferases, one or more GlcNAc 6-O-sulfotransferases, and one Gal 6-O-sulfotransferase catalyze the sulfation reactions. These enzymes, like other sulfotransferases, use activated sulfate (PAPS [3′-phosphoadenyl-5′-phosphosulfate]) as a high-energy donor (see Chapter 4). GlcNAc 6-O sulfation occurs only on the nonreducing terminal N-acetylglucosamine residue, whereas sulfation of galactose residues takes place on nonreducing terminal and internal galactose residues, with a preference for galactose units adjacent to a sulfated N-acetylglucosamine. Sulfation of a nonreducing terminal galactose residue blocks further elongation of the chain, providing a potential mechanism for controlling chain length. Only one galactose sulfotransferase has been identified, whereas multiple sulfotransferases catalyze the sulfation of N-acetylglucosamine residues. Mutations in the corneal GlcNAc 6-O-sulfotransferase result in macular corneal dystrophy. The relationship of enzymes involved in KS I and KS II sulfation is unclear. The chains can be fucosylated and sialylated as well (see Chapter 13).
Bacterial keratanases degrade KS at characteristic positions (Table 16.4). In animals, KS is degraded in lysosomes by the sequential action of exoglycosidases (β-galactosidase and β-hexosaminidase) after removal of the sulfate groups on the terminal residue by sulfatases (see Chapter 41).
TABLE 16.4
Keratanases
HEPARAN SULFATE AND CHONDROITIN SULFATE ARE LINKED BY XYLOSE TO SERINE
Two classes of glycosaminoglycan chains, CS/DS and HS/heparin, are linked to serine residues in core proteins by way of xylose (Figure 16.5). Xylosyltransferase initiates the process using UDP-xylose as donor. Two isoforms of the enzyme are known in vertebrates (XT-1 and XT-2), but only one isozyme exists in C. elegans and D. melanogaster. A glycine residue invariably lies to the carboxy-terminal side of the serine attachment site, but a perfect consensus sequence for xylosylation does not exist. At least two acidic amino acid residues are usually present, and they can be located on one or both sides of the serine, usually within a few residues. Several proteoglycans contain clustered glycosaminoglycan attachment sites, raising the possibility that xylosyltransferase could act in a processive manner. Xylosylation is an incomplete process in some proteoglycans, which may explain why proteoglycans with multiple potential attachment sites contain different numbers of chains in different cells. Variation in the degree of glycosaminoglycan substitution also might result from low levels of UDP-xylose, low activity of the xylosyltransferases, or competing reactions such as serine phosphorylation, acylation, or other forms of glycosylation.
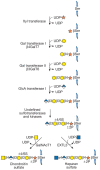
FIGURE 16.5
The biosynthesis of CS (left chain) and HS (right
chain) is initiated by the formation of a linkage region
tetrasaccharide. Addition of the first hexosamine commits the intermediate
to either CS (GalNAc) or HS (GlcNAc).
Symbol Key:

After xylose addition, a linkage tetrasaccharide assembles by the transfer of two galactose residues catalyzed by unique members of the β1–4 galactosyl-, β1–3 galactosyl-, and β1–3 glucuronosyltransferase families of enzymes (Figure 16.5). This intermediate can undergo phosphorylation at the C-2 position of xylose and sulfation of the galactose residues. In general, phosphorylation and sulfation occur substoichiometrically. The lack of chain specificity for phosphorylation would seem to exclude it as a signal for controlling composition. Phosphorylation may be transient, suggesting a role in processing or sorting. Galactose sulfation is found only in CS, but its role in chain initiation, polymerization, or turnover remains unclear.
The linkage tetrasaccharide lies at a bifurcation in the biosynthetic pathway. Two types of reactions occur: addition of β1–4GalNAc (initiation of CS) or addition of α1–4GlcNAc (initiation of HS) (Figure 16.5). Genetic evidence from studies of C. elegans suggests that N-acetylgalactosamine addition is mediated by the same enzyme that is involved in chain polymerization (SQV5), but biochemical evidence suggests that more than one enzyme may exist in vertebrates. In heparin/HS formation, the addition of the first N-acetylglucosamine residue is catalyzed by an enzyme called EXTL3, which differs from the transferases involved in heparan polymerization (called EXT1 and EXT2). These enzymes are important control points because they ultimately regulate the type of glycosaminoglycan chain that will assemble. Control of the addition of β4GalNAc or α4GlcNAc appears to be manifested at the level of enzyme recognition of the polypeptide substrate. In HS formation, EXTL3 recognizes the linear amino acid sequence immediately adjacent to the attachment site in the core protein. Attachment sites for HS formation usually contain a cluster of acidic residues within seven to nine residues of the serine. Several HS proteoglycans contain multiple contiguous Ser-Gly attachment sites. In the example of syndecan-2 shown below, the underlined sequence indicates the sites of glycosaminoglycan attachment and the boldface letters refer to the clustered acidic residues. Additionally, these sites often contain hydrophobic amino acids in close proximity (e.g., valine, tyrosine, and alanine in syndecan-2). Distant effects of polypeptide structure also can act as a negative factor, for example, by preventing the action of β1–4 N-acetylgalactosaminyltransferase.
-SSIEEASGVYPIDDDDYSSASGSGADEDIESPVLTTS-
CHONDROITIN SULFATE/DERMATAN SULFATE BIOSYNTHESIS
Vertebrate CS consists of repeating sulfate-substituted GalNAc-GlcA disaccharide units polymerized into long chains (see Figure 16.3). In contrast, invertebrates such as C. elegans and D. melanogaster make either nonsulfated or undersulfated chains, respectively. The assembly process for the backbone appears to be highly conserved, based on the presence of homologous genes for all of the reactions (see Chapters 23 and 24). As described above, the assembly process is initiated by the transfer of N-acetylgalactosamine to the linkage tetrasaccharide (Figure 16.5). In both vertebrates and invertebrates, the polymerization step is catalyzed by one or more bifunctional enzymes (chondroitin synthases) that have both β1–3 glucuronosyltransferase and β1–4 N-acetylgalactosaminyltransferase activities. Vertebrates also express homologs that can transfer individual sugars to the chain, but genetic data demonstrating the functionality of these isoforms have not yet been obtained. Chondroitin polymerization also requires the action of the chondroitin polymerizing factor, a protein that lacks independent activity but collaborates with the polymerases to enhance the formation of polymers.
Sulfation of chondroitin in vertebrates is a complex process, with multiple sulfotransferases involved in 4-O sulfation and 6-O sulfation of N-acetylgalactosamine residues (Figure 16.6). Additional enzymes exist for epimerization of glucuronic acid to iduronic acid in DS, sulfation at the C-2 position of the uronic acids, and other patterns of sulfation found in unusual species of chondroitin (Table 16.5). The location of sulfate groups is easily assessed using bacterial chondroitinases (ABC, B, and ACII) that cleave the chains into disaccharides. Many chains are hybrid structures containing more than one type of chondroitin disaccharide unit. For example, DS contains one or more iduronic acid–containing disaccharide units (CS B) as well as glucuronic acid–containing disaccharides (CS A and C). Animal cells also degrade CS in lysosomes using a series of exolytic activities (see Chapter 41).

FIGURE 16.6
Biosynthesis of CS/DS involves the polymerization of N-acetylgalactosamine and glucuronic acid units and a series of modification reactions including sulfation and epimerization of glucuronic acid to iduronic acid. Chain polymerization and modification (more...)
TABLE 16.5
Types of chondroitin sulfates
HEPARIN AND HEPARAN SULFATE BIOSYNTHESIS
Heparin and HS assemble as copolymers of GlcNAcα1–4GlcAβ1–4 (see Figure 16.3), which then undergoes extensive modification reactions. Heparin is produced exclusively by mast cells, whereas HS is made by virtually all types of cells. Heparin also differs from HS in the degree of modification of sugar residues, as described below. As the chains polymerize, they undergo a series of modification reactions catalyzed by at least four families of sulfotransferases and one epimerase (Figure 16.7). GlcNAc N-deacetylase/N-sulfotransferases act on a subset of N-acetylglucosamine residues to generate N-sulfated glucosamine units, some of which occur in clusters along the chain. Generally, the enzyme deacetylates N-acetylglucosamine and rapidly adds sulfate to the free amino group to form GlcNSO3. A small number of glucosamine residues with free amino groups are present, which may arise from incomplete N-sulfation. An epimerase, different from the one involved in DS synthesis, then acts on glucuronic acid residues immediately adjacent to and toward the reducing side of N-sulfated glucosamine units, followed by 2-O sulfation of some of the iduronic acid units generated. Some glucuronic units also undergo 2-O sulfation. The addition of 2-O-sulfate groups to glucuronic acid or iduronic acid blocks the epimerization reaction. Next, a 6-O-sulfotransferase adds sulfate groups to selected glucosamine residues. Finally, certain subsequences of sulfated sugar residues and uronic acid epimers provide targets for a 3-O-sulfotransferase.
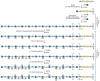
FIGURE 16.7
Heparan sulfate biosynthesis involves copolymerization of N-acetylglucosamine and glucuronic acid residues. A series of modification reactions including sulfation and epimerization of glucuronic acid to iduronic acid occurs; chain polymerization and modification (more...)
In contrast to chondroitin chains, which tend to have long tracts of fully modified disaccharides, the modification reactions in HS biosynthesis occur in clusters along the chain, with regions devoid of sulfate separating the modified tracts. This arrangement gives rise to segments referred to as N-acetylated (NA), N-sulfated (NS), and mixed domains (NA/NS) (Figure 16.8). In general, the reactions proceed in the order indicated, but they often fail to go to completion, resulting in tremendous chemical heterogeneity within the modified regions. The disaccharide composition of the chains can be readily assessed using bacterial heparin lyases (Table 16.6) or chemical degradation methods, which are more useful for differentiating glucuronic acid/iduronic acid, but direct sequencing of the chains has proven difficult because of their heterogeneity.

FIGURE 16.8
The heparan sulfate chain consists of different domains that vary in the extent of modification by sulfation and epimerization. (NS) N-sulfated domains; (NA/NS) domains of variable sulfation; (NA) N-acetylated domains. Modified domains make up binding (more...)
TABLE 16.6
Heparin lyases
The specific arrangement of sulfated residues and uronic acid epimers in heparin and HS gives rise to binding sequences for ligands (Figure 16.8). The two examples shown in Figure 16.8 demonstrate minimal sequences that can interact with fibroblast growth factors (FGFs) or antithrombin. More modified sequences can interact as well, and the binding of most FGFs is actually more sensitive to overall sulfation than to the specific position of the sulfate groups. Binding of glycosaminoglycans to proteins is described in greater detail in Chapters 15 and 35. A major question remains regarding how the enzymes and pathway of HS/heparin biosynthesis are regulated to achieve tissue-specific expression of ligand-binding sequences. During the last decade, all of the enzymes involved in HS synthesis have been purified and molecularly cloned from mammals. Several important features have emerged from these studies, which may shed light on how different binding sequences arise.
- Several of the enzymes appear to have dual catalytic activities. Thus, a single protein bearing two catalytic domains catalyzes N-deacetylation of N-acetylglucosamine residues and subsequent N-sulfation (NDSTs). The same is true of the copolymerase (EXT1/EXT2), which transfers N-acetylglucosamine and glucuronic acid from the corresponding UDP sugars to the growing polymer. In contrast, epimerase and 2-O-, 3-O-, and 6-O-sulfotransferase activities appear to be unique properties of independent enzymes.
- In several cases, multiple isozymes exist that can catalyze either a single or a pair of reactions. Thus, four N-deacetylase/N-sulfotransferases, three 6-O-sulfotransferases, and seven 3-O-sulfotransferases have been identified. Their tissue distribution varies and differences exist in substrate preference, which may cause differences in the pattern of sulfation. However, some overlap in expression and in substrate use occurs as well.
- The polymerization and polymer modification reactions probably colocalize in the same stacks of the Golgi complex. Thus, the enzymes may form a supramolecular complex that coordinates these reactions. The composition of these complexes may play a part in regulating the fine structure of the chains.
- In general, the composition of HS on a given proteoglycan varies more between cell types than that of HS on different core proteins expressed in the same cell. This observation suggests that each cell type may express a unique array of enzymes and potential regulatory factors. However, these general compositional differences may mask underlying differences in the arrangement of the various disaccharide units, which might confer differences in ligand binding.
- Recombinant enzymes are increasingly used to define substrate specificities. Chemoenzymatic methods are being used to generate saccharide products of predetermined structure, and glycan arrays are being developed to probe ligand-binding affinities and specificities.
THE DIFFERENCES BETWEEN HEPARIN AND HEPARAN SULFATE
Considerable confusion exists regarding the definition of heparin and HS; the major differences are summarized in Table 16.7. Heparin derived from porcine and bovine entrails is prepared commercially by selective precipitation and is sold by pharmaceutical companies as an anticoagulant due to its capacity to bind to antithrombin. Low-molecular-weight heparins (LMWH) are derived from commercial unfractionated heparin (UFH) by chemical and enzymatic cleavage, depending on the brand. The active sequence is a pentasaccharide shown in Figure 16.8, which is now sold as a purely synthetic anticoagulant (Arixtra). Selectively desulfated forms of heparin are also commercially available, some of which lack anticoagulant activity, but still retain other potentially useful properties (e.g., inhibition of inflammation and cell proliferation, and antimetastatic activity).
TABLE 16.7
Major differences between heparin and heparan sulfate
Heparin is made solely as serglycin proteoglycan by connective-tissue-type mast cells, whereas HS is made by virtually all cells. HS can also contain anticoagulant activity, but typical preparations are much less active than heparin. During biosynthesis, heparin undergoes more extensive sulfation and uronic acid epimerization, such that more than 80% of the N-acetylglucosamine residues are N-deacetylated and N-sulfated and more than 70% of the uronic acid is converted to iduronic acid. Another way to distinguish heparin from HS is by susceptibility to bacterial (Flavobacterium) heparin lyases (Table 16.6).
PROTEOGLYCAN PROCESSING AND TURNOVER
As described above, cells secrete matrix proteoglycans directly into the extracellular environment (e.g., the basement membrane proteoglycans, SLRPs, serglycin, and members of the aggrecan family). However, others are shed from the cell surface through proteolytic cleavage of the core protein (e.g., the syndecans). Cells also internalize a large fraction of cell-surface proteoglycans by endocytosis (Figure 16.9). These internalized proteoglycans first encounter proteases that cleave the core protein and heparanase that cleaves the HS chains at a limited number of sites, depending on sequence. These smaller fragments eventually appear in the lysosome and undergo complete degradation by way of a series of exoglycosidases and sulfatases (see Chapter 41). The main purpose of intracellular heparanase may be to increase the number of target sites for exolytic degradative enzymes. CS and DS proteoglycans follow a similar endocytic route, but endoglycosidases that degrade the chains before the lysosome have not been described.
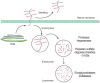
FIGURE 16.9
Heparan sulfate proteoglycans turn over by proteolytic shedding from the cell surface and endocytosis, as well as step-wise degradation inside lysosomes. (Brown) Core protein; (magenta) polysaccharide chains. (Adapted, with permission, from Yanagishita (more...)
Cells secrete heparanase as well (Figure 16.10). Extracellular heparanase can cleave HS chains at restricted sites, resulting in release of growth factors or chemokines immobilized on HS proteoglycans at cell surfaces or in the ECM. In particular, invading cells secrete heparanase, thought to help degrade the ECM. Thus, heparanase may act with matrix metalloproteases to remodel the ECM.
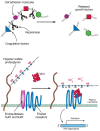
FIGURE 16.10
After secretion, cell-surface heparan sulfate proteoglycans can be processed further. Heparanase cleaves the HS chain, liberating oligosaccharides and bound ligands. The Sulfs remove sulfate groups from subsets of 6-O-sulfated N-sulfated glucosamine units (more...)
Recently, a family of plasma membrane endosulfatases has been described that can remove sulfate groups from internal 6-O-sulfated glucosamine residues (Sulfs; see Fig. 16.10). This postassembly processing of the chains at the cell surface results in altered response of cells to growth factors and morphogens. The discovery of this reaction and the action of extracellular heparanase suggests that other processing enzymes may exist that modify the structure and function of HS after secretion from the cell.
PROTEOGLYCANS HAVE DIVERSE FUNCTIONS
Several generalizations can be made regarding proteoglycan function.
- A common property of the interstitial proteoglycans containing CS and DS chains is their capacity to bind water and form hydrated matrices. Thus, these molecules fill the space between cells. In cartilage, the aggregates of proteoglycans and hyaluronan provide a stable matrix capable of absorbing high compressive loads by water desorption and resorption. Interstitial proteoglycans can interact with collagen, thus aiding in the structural organization of most tissues.
- Proteoglycans help to organize basement membranes, thus providing a scaffold for epithelial cell migration, proliferation, and differentiation. They can regulate the permeability properties of specialized basement membranes.
- Proteoglycans in secretory vesicles have a role in packaging granular contents, maintaining proteases in an active state, and regulating various biological activities after secretion, such as coagulation, host defense, and wound repair.
- Proteoglycans in the ECM can bind cytokines, chemokines, growth factors, and morphogens, protecting them against proteolysis. These interactions provide a depot of regulatory factors that can be liberated by selective degradation of the matrix. They also facilitate the formation of morphogen gradients essential for cell specification during development.
- Proteoglycans can act as receptors for proteases and protease inhibitors regulating their spatial distribution and activity.
- Membrane proteoglycans can act as coreceptors for various tyrosine-kinase-type growth factor receptors, lowering the threshold or changing the duration of signaling reactions.
- Membrane proteoglycans can act as endocytic receptors for clearance of bound ligands.
- Membrane proteoglycans cooperate with integrins and other cell adhesion receptors to facilitate cell attachment, cell–cell interactions, and cell motility. Proteoglycans in the ECM can regulate cell migration as well.
To a large extent, the biological functions of proteoglycans depend on the interaction of the glycosaminoglycan chains with different protein ligands. Table 16.8 lists examples of proteins known to interact with glycosaminoglycans (also see Chapter 35). Most of the proteins bind to HS or DS as opposed to CS, which may reflect the greater chemical diversity and capacity of these glycosaminoglycans to interact with proteins through the varied arrangements of sulfate groups and glucuronic acid/iduronic acid residues. Indeed, unusual CS species containing additional sulfate groups bind growth factors and matrix proteins more avidly. Proteins that bind to the sulfated GAG chains appear to have evolved by convergent evolution (i.e., they do not contain a specific fold present in all glycosaminoglycan binding proteins, in contrast to other groups of glycan binding proteins) (see Chapter 26).
TABLE 16.8
Examples of proteins that bind to sulfated glycosaminoglycans
These interactions have profound physiological effects. For example, injection of heparin into the bloodstream results in rapid anticoagulation because of binding and activation of antithrombin, release of lipoprotein lipase, transient blockade of P and L selectins, displacement of growth factors and chemokines, and undoubtedly a number of other activities. In some cases, the interaction depends on a very specific sequence of modified sugars in the glycosaminoglycan chain. The best-studied example is antithrombin-heparin, which depends on a specific pentasaccharide sequence (Figure 16.8). However, in other cases, the glycosaminoglycan sequences that interact with proteins, such as growth factors and their receptors, show preference for certain modifications or spatial arrangements of sulfated sugars rather than a specific sequence. As more refined methods become available for isolation of bioactive oligosaccharides and their sequence analysis, unusual arrangements of sulfated sequences may be discovered to confer selective ligand-binding and biological activity. This is an active area of research.
FURTHER READING
- Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. [PubMed: 10872465]
- Lindahl U. What else can “heparin” do? Haemostasis. 1999;29:38–47. [PubMed: 10629403]
- Yamaguchi Y. Lecticans: Organizers of the brain extracellular matrix. Cell Mol Life Sci. 2000;57:276–289. [PubMed: 10766023]
- Gallagher JT. Heparan sulfate: Growth control with a restricted sequence menu. J Clin Invest. 2001;108:357–361. [PMC free article: PMC209370] [PubMed: 11489926]
- Esko JD, Selleck SB. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. [PubMed: 12045103]
- Funderburgh JL. Keratan sulfate biosynthesis. IUBMB Life. 2002;54:187–194. [PMC free article: PMC2874674] [PubMed: 12512857]
- Song HH, Filmus J. The role of glypicans in mammalian development. Biochim. Biophys. Acta. 2002;1573:241–246. [PubMed: 12417406]
- Trowbridge JM, Gallo RL. Dermatan sulfate: New functions from an old glycosaminoglycan. Glycobiology. 2002;12:117R–125R. [PubMed: 12213784]
- Vlodavsky I, Goldshmidt O, Zcharia E, Atzmon R, Rangini-Guatta Z, Elkin M, Peretz T, Friedmann Y. Mammalian heparanase: Involvement in cancer metastasis, angiogenesis and normal development. Semin Cancer Biol. 2002;12:121–129. [PubMed: 12027584]
- Habuchi H, Habuchi O, Kimata K. Sulfation pattern in glycosaminoglycan: Does it have a code. Glycoconj J. 2004;21:47–52. [PubMed: 15467398]
- Kolset SO, Prydz K, Pejler G. Intracellular proteoglycans. Biochem J. 2004;379:217–227. [PMC free article: PMC1224092] [PubMed: 14759226]
- Iozzo RV. Basement membrane proteoglycans: From cellar to ceiling. Nat Rev Mol Cell Biol. 2005;6:646–656. [PubMed: 16064139]
- Volpi N, editor. Chondroitin sulfate: Structure, role and pharmacological activity. Adv Pharmacol. 2006;53:1–568.
- Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. [PubMed: 17460664]
- Gorsi B, Stringer SE. Tinkering with heparan sulfate sulfation to steer development. Trends Cell Biol. 2007;17:173–177. [PubMed: 17320398]
- Lamanna WC, Kalus I, Padva M, Baldwin RJ, Merry CL, Dierks T. The heparanome—The enigma of encoding and decoding heparan sulfate sulfation. J Biotechnol. 2007;129:290–307. [PubMed: 17337080]
- Vreys V, David G. Mammalian heparanase: What is the message. J Cell Mol. 2007;11:427–452. [PMC free article: PMC3922351] [PubMed: 17635638]
Publication Details
Author Information and Affiliations
Authors
Jeffrey D Esko, Koji Kimata, and Ulf Lindahl.Copyright
Publisher
Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY)
NLM Citation
Esko JD, Kimata K, Lindahl U. Proteoglycans and Sulfated Glycosaminoglycans. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology. 2nd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2009. Chapter 16.

![FIGURE 16.2. Proteoglycans consist of a protein core (brown) and one or more covalently attached glycosaminoglycan chains ([blue] HS; [yellow] CS/DS).](/books/NBK1900/bin/ch16f2.jpg)







