Neurodegeneration with Brain Iron Accumulation Disorders Overview
Allison Gregory, MS, CGC and Susan Hayflick, MD.
Author Information and AffiliationsInitial Posting: February 28, 2013; Last Update: October 21, 2019.
Estimated reading time: 19 minutes
Summary
The purpose of this overview is to increase the awareness of clinicians regarding neurodegeneration with brain iron accumulation (NBIA) disorders and their genetic causes and management.
The following are the goals of this overview.
Goal 2.
Review the genetic causes of neurodegeneration with brain iron accumulation.
Goal 3.
Provide an evaluation strategy to identify the genetic cause of neurodegeneration with brain iron accumulation in a proband (when possible).
Goal 4.
Inform genetic counseling of family members of an individual with neurodegeneration with brain iron accumulation.
Goal 5.
Review high-level management of neurodegeneration with brain iron accumulation.
1. Clinical Characteristics of Neurodegeneration with Brain Iron Accumulation
Neurodegeneration with brain iron accumulation (NBIA) is a group of inherited neurologic disorders characterized by abnormal accumulation of iron in the basal ganglia (most often in the globus pallidus and/or substantia nigra) (see Table 2).
Generalized cerebral atrophy and cerebellar atrophy are frequently observed.
The hallmark clinical manifestations of NBIA are progressive dystonia and dysarthria, spasticity, parkinsonism, neuropsychiatric abnormalities, and optic atrophy or retinal degeneration. Although cognitive decline occurs in some genetic types, more often cognition is relatively spared.
Onset ranges from infancy to adulthood. Progression can be rapid or slow with long periods of stability.
Diagnosis of NBIA
The quality of the neuroimaging, including magnet strength and spacing of image slices, can limit the ability to accurately identify abnormal brain iron. Iron-sensitive sequences, such as SWI, GRE, and T2*, should be used as a first-line diagnostic investigation to identify the characteristic changes in NBIA. By the time clear neurologic features are present, the brain MRI almost always shows characteristic changes, although iron may be visible only later in the disease course.
Neuropathologic findings include axonal spheroids in the CNS and, in some types, in peripheral nerves.
Differential Diagnosis of NBIA
The differential diagnosis of NBIA is usually based on a brain MRI that raises the suspicion of abnormal iron accumulation (Table 1).
Table 1.
Conditions with Brain MRI Findings or Clinical Findings that Resemble NBIA
View in own window
| Findings | Condition(s) | Gene(s) 1 |
|---|
| T2-weighted hyperintensity in globus pallidus reminiscent of early PKAN | Fucosidosis (OMIM 230000) |
FUC1A
|
|
Glutaric aciduria I
|
GCDH
|
| Leigh encephalopathy / pyruvate dehydrogenase deficiency | Several nuclear (see Nuclear Gene-Encoded Leigh Syndrome Overview; Primary Pyruvate Dehydrogenase Complex Deficiency Overview) & mitochondrial (see Mitochondrial DNA-Associated Leigh Syndrome and NARP) |
| Cerebellar & cerebral atrophy, thin corpus callosum; optic atrophy | Spastic paraplegia 30; hereditary sensory neuropathy IIC |
KIF1A
|
| T2-weighted hypointensity in globus pallidus; childhood-onset dystonia; prominent cervical, cranial, & laryngeal dystonia | KMT2B-related dystonia (DYT28, DYT-KMT2B) |
KMT2B
|
| Iron accumulation in basal ganglia (case report); early-onset dystonia-parkinsonism | Early-onset dystonia-parkinsonism |
PSEN1
|
| Susceptibility-weighted signal dropout in globus pallidus & substantia nigra; adult-onset gait change & ↑ tone | SCPx deficiency (OMIM 613724) |
SCP2
|
| T2-weighted hypointensity of globus pallidus & striatum that is hyperintense on T1-weighted imaging; childhood-onset dystonia & spasticity | SLC39A14 deficiency (SLC39A14-related early-onset dystonia-parkinsonism) |
SLC39A14
|
| T2-weighted hypointensity of globus pallidus, mild cerebellar atrophy, childhood onset; childhood onset of ataxia, dystonia, & gaze palsy | Neurodegeneration w/ataxia, dystonia, & gaze palsy, childhood onset (NADGP) (OMIM 617145) |
SQSTM1
|
| Cerebellar atrophy, thinning of corpus callosum, dysarthria, gait change | UBTF-related neuroregression (OMIM 617672) |
UBTF
|
| Acanthocytes, oral dystonia |
Chorea-acanthocytosis
|
VPS13A
|
- 1.
Genes are listed alphabetically.
2. Genetic Causes of Neurodegeneration with Brain Iron Accumulation
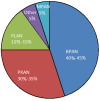
The ten genes known to be associated with types of NBIA are ATP13A2, C19orf12, COASY, CP, DCAF17, FA2H, FTL, PANK2, PLA2G6, and WDR45 (Table 2).
Table 2.
Neurodegeneration with Brain Iron Accumulation: Genetic Types
View in own window
AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance; XL = X-linked
- 1.
Genes are listed alphabetically.
- 2.
- 3.
Autosomal dominant inheritance and de novo dominant pathogenic sequence variants have been reported in some families [Gregory et al 2019].
- 4.
Based on prevalence in the International Registry for NBIA and Related Disorders from the Hayflick laboratory
- 5.
- 6.
- 7.
The distinctive clinical and neuroimaging findings of the ten genetically defined NBIA types are summarized in Tables 3 and 4, respectively.
Table 3.
Neurodegeneration with Brain Iron Accumulation: Clinical Characteristics by Genetic Type
View in own window
| NBIA Genetic Type | Onset | Typical Presentation | Other Key Clinical Manifestations |
|---|
|
PKAN
| Classic
PKAN | Early (w/rapid progression) | Gait abnormalities at age ~3 yrs | Progressive dystonia, dysarthria, rigidity, spasticity, hyperreflexia, & extensor toe signs Retinal degeneration is common & may be detected by ERG several yrs before onset of visual symptoms. Neuropsychiatric symptoms (more frequent in later-onset form)
|
Atypical
PKAN | >10 yrs (w/slower progression) | Dysarthria |
PLAN
(Parkinson disease 14; PARK14) | Infantile
PLAN
(INAD) | 6 mos - 3 yrs | Developmental regression, initial hypotonia, progressive psychomotor delay, & progressive spastic tetraparesis | Progressive cognitive decline Strabismus, nystagmus, & optic atrophy Rapid disease progression
|
Juvenile
PLAN 1 | Typically early childhood | Gait instability, ataxia, or speech delay & autistic features |
|
Adult
PLAN | Early adulthood | Gait disturbance or neuropsychiatric changes |
|
| MPAN 2 | Childhood to early adulthood | Children: gait abnormalities, limb spasticity, optic atrophy
Adults: gait abnormalities, acute neuropsychiatric changes | Progressive cognitive decline in most individuals (unlike most NBIA) Neuropsychiatric changes Spasticity (more prominent than dystonia), motor neuronopathy w/early upper-motor neuron findings followed by signs of lower-motor neuron dysfunction Optic atrophy Slowly progressive course w/survival well into adulthood
|
|
BPAN
| Childhood | DD w/slow motor & cognitive gains & little to no expressive language | Seizures of various types more prominent in childhood & may resolve in later adolescence Abnormal behaviors similar to ASD & Rett syndrome Motor dysfunction incl broad-based or ataxic gait, hypotonia, mild spasticity During late adolescence or adulthood, relatively sudden onset of progressive dystonia-parkinsonism & dementia
|
|
FAHN
| Childhood | Most common presentation is subtle change in gait that may lead to increasingly frequent falls | Slowly progressive ataxia, dysarthria, dystonia, & tetraparesis Optic atrophy leading to progressive loss of visual acuity Seizures in later disease course Progressive intellectual decline in most affected individuals
|
Kufor-Rakeb syndrome 3
(Parkinson disease 9; PARK9) | Juvenile | Gait abnormalities, neuropsychiatric changes |
|
| Neuroferritinopathy 5 | Adult | May be similar to Huntington disease w/chorea or dystonia & cognitive changes |
|
|
Aceruloplasminemia
| Adult (25-60 yrs) | Clinical triad of retinal degeneration, diabetes mellitus, & neurologic disease | Facial & neck dystonia, dysarthria, tremors, chorea, ataxia, & blepharospasm ↓ serum concentrations of copper & iron & ↑ serum concentrations of ferritin can distinguish aceruloplasminemia from other forms of NBIA.
|
| Woodhouse-Sakati syndrome (hypogonadism, alopecia, diabetes mellitus, ID, & extrapyramidal syndrome) 6 | Childhood | Alopecia may be earliest symptom; ID & delayed puberty | Progressive extrapyramidal disorder, generalized & focal dystonia, dysarthria, & cognitive decline Endocrine abnormalities (hypogonadism, alopecia, & diabetes mellitus)
|
| CoPAN 7 | Childhood | Childhood-onset dystonia & spasticity w/cognitive impairment | Oromandibular dystonia, dysarthria, axonal neuropathy, parkinsonism, cognitive impairment, & obsessive-compulsive behavior Slow progression; non-ambulatory in 3rd decade
|
ASD = autism spectrum disorder; BPAN = beta-propeller protein-associated neurodegeneration; CoPAN = COASY protein-associated neurodegeneration; DD = developmental delay; ERG = electroretinography; FAHN = fatty acid hydroxylase-associated neurodegeneration; ID = intellectual disability; INAD = infantile neuroaxonal dystrophy; MPAN = mitochondrial membrane protein-associated neurodegeneration; NAD = neuroaxonal dystrophy; PKAN = pantothenate kinase-associated neurodegeneration; PLAN = PLA2G6-associated neurodegeneration
- 1.
Juvenile PLAN is less common than the infantile form (INAD).
- 2.
A common C19orf12 founder variant (NM_001031726.3: c.204_214del11; NP_001026896.2: p.Gly69ArgfsTer10) has been observed in persons of central European descent (mainly Polish).
- 3.
Proposed to be an NBIA based on findings described by Schneider et al [2010] in a family originally reported in 1994
- 4.
- 5.
A common FTL pathogenic variant in exon 4 has been found in approximately 80% of affected individuals.
- 6.
A founder pathogenic variant in DCAF17 accounts for the cases in the Saudi Arabian population.
- 7.
The CoPAN phenotype will continue to evolve as additional affected individuals are recognized.
Table 4.
Neurodegeneration with Brain Iron Accumulation: Neuroimaging Findings
View in own window
| NBIA Genetic Type | Brain MRI 1 |
|---|
| Pattern of iron distribution | Other key radiographic features |
|---|
|
PKAN
| GP, SN | Eye-of-the-tiger sign 2 in GP (virtually pathognomonic for PKAN 3) |
PLAN
(Parkinson disease 14; PARK14) | Variable GP & SN or no iron accumulation | Cerebellar atrophy & optic atrophy are hallmark features; other features incl cortical cerebellar hyperintensities, white matter abnormalities, thin vertically oriented corpus callosum, apparent hypertrophy of the clava. |
|
MPAN
| GP, SN | Cerebellar & cortical atrophy; on T2-weighted images hyperintense streaking of medial medullary lamina between globus pallidus interna & externa that could be mistaken for eye-of-the-tiger sign |
|
BPAN
| GP, SN
(SN iron > GP iron) | Early childhood hypomyelination & thin corpus callosum; later development of T2-weighted signal hypointensity in SN & GP & T1-weighted signal hyperintensity w/central band of hypointensity in SN. Cerebellar & cerebral atrophy may also be present at any age. |
|
FAHN
| GP, SN | Pontocerebellar atrophy, diffuse cerebral atrophy, thinning of corpus callosum, periventricular & subcortical white matter hyperintensity on T2-weighted images, optic atrophy |
Kufor-Rakeb syndrome
(Parkinson disease 9; PARK9) | Variable GP, putamen, caudate, or no iron accumulation 4 | Cerebral, cerebellar, & brain stem atrophy |
|
Neuroferritinopathy
| GP, putamen, caudate, dentate, SN & RN | Cystic changes & cavitation in caudate & putamen (unique to neuroferritinopathy); mild cortical & cerebellar atrophy |
|
Aceruloplasminemia
| GP, putamen, caudate, thalamus, RN, dentate 5 | Cerebellar atrophy |
| Woodhouse-Sakati syndrome (hypogonadism, alopecia, diabetes mellitus, ID, & extrapyramidal syndrome) | GP | White matter disease is common. |
| CoPAN | GP, SN | T2-weighted images: hyperintense caudate, putamina, medial & posterior thalami in early disease, 6 GP calcifications |
BPAN = beta-propeller protein-associated neurodegeneration; CoPAN = COASY protein-associated neurodegeneration; FAHN = fatty acid hydroxylase-associated neurodegeneration; GP = globus pallidus; ID = intellectual disability; MPAN = mitochondrial membrane protein-associated neurodegeneration; PKAN = pantothenate kinase-associated neurodegeneration; PLAN = PLA2G6-associated neurodegeneration; RN = red nucleus; SN = substantia nigra
- 1.
For a more comprehensive overview of the radiographic findings in NBIA, see Kruer et al [2012].
- 2.
Eye-of-the-tiger sign = T2-weighted hypointense signal in the globus pallidus with a central region of hyperintensity
- 3.
- 4.
Similar to PLAN, it has been suggested that some individuals may not have iron accumulation, it may develop late in the disease course, or it may only be associated with more severe pathogenic variants [Chien et al 2011].
- 5.
Abnormal hypointensities in liver are common (liver iron content > basal ganglia iron content).
- 6.
In one individual early in disease, T2-weighted sequences showed hyperintense & swollen caudate nuclei & putamina & mild hyperintensity in the medial & posterior thalami, which may help distinguish CoPAN from other NBIA disorders if these features are observed in additional cases.
NBIA of unknown cause. A significant portion of individuals with clinical, radiographic, and sometimes neuropathologic evidence of NBIA do not have pathogenic variants identified in one of the ten known NBIA-related genes [Hogarth et al 2013] (see ). For the most part, these individuals do not stratify into clear phenotypic groups.
NBIA Types PKAN = pantothenate kinase-associated neurodegeneration
3. Evaluation Strategies to Identify the Genetic Cause of Neurodegeneration with Brain Iron Accumulation in a Proband
Establishing a specific genetic cause of NBIA:
Usually involves a medical history, physical examination, laboratory testing, family history, and genomic/genetic testing.
Medical history. See Table 3 for clinical characteristics and Table 4 for neuroimaging findings that could lead a clinician to suspect or consider a specific genetic type of NBIA (Table 2).
Physical examination. See Table 3 for clinical characteristics that could help the clinician identify key issues in the physical examination that would allow him/her to suspect and/or consider a specific genetic type of NBIA (Table 2).
Family history. A three-generation family history should be taken, with attention to relatives with manifestations of NBIA and documentation of relevant findings through direct examination or review of medical records, including results of molecular genetic testing.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing or multigene panel (Option 1) and comprehensive genomic testing (exome sequencing, exome array) (Option 2). Gene-targeted testing requires the clinician to hypothesize which gene(s) are likely involved, whereas genomic testing does not.
Option 1
Single-gene testing (sequence analysis of one gene, followed by gene-targeted deletion/duplication analysis) is useful when the neuroimaging and/or phenotype clearly point toward one NBIA-related gene. For example, an eye-of-the-tiger sign on brain MRI is nearly pathognomonic for PKAN caused by biallelic PANK2 pathogenic variants.
A multigene panel that includes some or all of the genes listed in Table 1 is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. Of note, given the rarity of some of the genes associated with NBIA, some panels may not include all the genes mentioned in this overview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive
genomic testing (which does not require the clinician to determine which gene[s] are likely involved) may be considered when clinical/neuroimaging findings have not suggested NBIA. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Note: For individuals in whom a genetic cause of NBIA cannot be established, participation in research studies may be considered as new technologies may facilitate identification of additional NBIA-related genes in the future (see Resources).
4. Genetic Counseling of Family Members of an Individual with Neurodegeneration with Brain Iron Accumulation
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Modes of Inheritance
Seven of the ten genetically defined types of neurodegeneration with brain iron accumulation (NBIA) are inherited in an autosomal recessive manner. Exceptions:
Neuroferritinopathy, an autosomal dominant disorder caused by a pathogenic variant in FTL
Mitochondrial membrane protein-associated neurodegeneration (MPAN), an autosomal recessive or autosomal dominant disorder caused by mutation of C19orf12 (de novo and inherited pathogenic variants are known to be associated with autosomal dominant MPAN)
Beta-propeller protein-associated neurodegeneration (BPAN), an X-linked disorder caused by a de novo or inherited pathogenic variant in WDR45 with suspected male lethality (To date, most individuals with BPAN have been female.)
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
The parents of an affected individual are obligate heterozygotes (i.e., carriers of one NBIA-causing pathogenic variant).
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband
Individuals with autosomal recessive forms of NBIA rarely reproduce due to the severity of the condition.
Those with later-onset, atypical disease may have offspring; all offspring will be obligate heterozygotes.
Other family members. Each sib of the proband's parents is at 50% risk of being a carrier of an NBIA-causing pathogenic variant.
Carrier detection. Carrier testing for at-risk family members requires prior identification of the pathogenic variants in the family.
X-Linked Inheritance – Risk to Family Members
Parents of a proband
To date, the vast majority of reported individuals with BPAN have been simplex cases (i.e., a single occurrence in a family) and have had a de novo
WDR45 pathogenic variant.
Rarely, an individual with BPAN has the disorder as the result of an inherited pathogenic variant. In one family, an asymptomatic mother had the same
WDR45 pathogenic variant as her sons with significant X-chromosome inactivation skewed toward the normal allele in her leukocyte DNA [
Dufke et al 2014].
Recommended parental molecular genetic testing when a proband has an identified WDR45 pathogenic variant:
Female proband. Molecular genetic testing of both parents
Male proband. Molecular genetic testing of the mother only; the father of a male proband will neither have the disorder nor be hemizygous for the WDR45 pathogenic variant.
In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote. Note: If a woman has more than one affected child and no other affected relatives and if the
WDR45 pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism. Presumed maternal germline mosaicism has been reported in a family with two affected sibs [
Zarate et al 2016].
Sibs of a proband. To date, nearly all affected individuals have had a de novo
WDR45 pathogenic variant, suggesting a low risk to sibs.
Offspring of a proband
Other family members. Given that most probands with BPAN reported to date have the disorder as a result of a de novo
WDR45 pathogenic variant, the risk to other family members is presumed to be low.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
Neuroferritinopathy. Most individuals diagnosed with neuroferritinopathy have an affected parent. More rarely, a proband has the disorder as the result of a de novo pathogenic variant (the proportion of de novo cases in neuroferritinopathy is unknown).
Autosomal dominant MPAN. Rarely, individuals diagnosed with autosomal dominant MPAN have an affected parent. More often, a proband with autosomal dominant MPAN has the disorder as a result of a
de novo pathogenic variant in exon 3 of
C19orf12 that leads to a protein truncated after amino acid 79 [
Gregory et al 2019].
Recommendations for the evaluation of parents of a proband with an apparent de novo pathogenic variant in FTL include brain MRI, measurement of serum ferritin concentration, and molecular genetic testing for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
Offspring of a proband. Each child of an individual with neuroferritinopathy or autosomal dominant MPAN has a 50% chance of inheriting the pathogenic variant. There may be differences in the age of onset and rate of progression of the disorder between members of the same family.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected or has the NBIA-causing pathogenic variant, the parent's family members are at risk.
Predictive testing of at-risk asymptomatic adult family members requires prior identification of the NBIA-causing pathogenic variant in the family. See Neuroferritinopathy, Genetic Counseling for a review of issues related to predictive testing.
Prenatal Testing and Preimplantation Genetic Testing
Once the NBIA-causing pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
INADcure Foundation
Email: info@INADcure.org
NBIA Alliance
Email: Info@NBIAalliance.org
NBIAcure
Center of Excellence for NBIA Clinical Care and Research
International Registry for NBIA and Related Disorders
Oregon Health & Science University
Email: info@nbiacure.org
NBIA Disorders Association
Treat Iron-Related Childhood Onset Neurodegeneration (TIRCON)
Germany
Email: TIRCON@med.uni-muenchen.de
5. Management of Neurodegeneration with Brain Iron Accumulation
Treatment of Manifestations
Treatments for NBIA disorders are palliative and should be tailored to the specific NBIA disorder and to the affected individual. A consensus management guideline for PKAN has been published [Hogarth et al 2017]. The following treatments should be considered (some do not apply to all genetic types of NBIA):
Pharmacologic treatment of spasticity and seizures
Trial of oral or intrathecal baclofen for those with significant dystonia
Botulinum toxin for those with focal dystonia
L-DOPA treatment, which is beneficial in rare cases. Note: Some will have an initially dramatic response that usually diminishes over time; some will develop prominent dyskinesias early in the treatment.
Deep brain stimulation, used clinically for dystonia with increasing frequency, shows some evidence of benefit.
Psychiatric treatment for those with a later-onset, more protracted course accompanied by neuropsychiatric symptoms
As NBIA disorders progress, many affected individuals may experience episodes of extreme dystonia lasting for days or weeks. It is especially important during these episodes to evaluate for treatable causes of pain, which may include occult GI bleeding, urinary tract infections, and occult bone fractures. The combination of osteopenia in a non-ambulatory person with marked stress on the long bones from dystonia places many individuals with an NBIA disorder at high risk for fractures without apparent trauma.
Affected individuals benefit from services for the blind, educational programs, and assistive communication devices; adaptive aids (walkers, wheelchairs) may be required for gait abnormalities.
Swallowing evaluation and regular dietary assessments are indicated to assure adequate nutrition. Once the individual can no longer maintain an adequate diet orally, gastrostomy tube placement is indicated.
Gastric feeding tube and/or tracheostomy is appropriate as needed to prevent aspiration pneumonia.
Over-the-counter fiber supplements and/or stool softeners are indicated to treat constipation, which is likely caused by a combination of immobility, diet, and medications.
Surveillance
Evaluate for treatable causes of pain during episodes of extreme dystonia. The following should be performed on a regular basis:
Monitoring of height and weight using appropriate growth curves to screen children for worsening nutritional status
Ophthalmologic assessment
Assessment of ambulation and speech abilities
Chapter Notes
Revision History
21 October 2019 (bp) Comprehensive update posted live
24 April 2014 (aa) Revision: COASY protein-associated neurodegeneration added
28 February 2013 (me) Review posted live
23 April 2012 (ag) Original submission
References
Literature Cited
Al-Semari
A, Bohlega
S. Autosomal-recessive syndrome with alopecia, hypogonadism, progressive extra-pyramidal disorder, white matter disease, sensory neural deafness, diabetes mellitus, and low IGF1.
Am J Med Genet A.
2007;143A:149–60
[
PubMed: 17167799]
Behrens
MI, Brüggemann
N, Chana
P, Venegas
P, Kägi
M, Parrao
T, Orellana
P, Garrido
C, Rojas
CV, Hauke
J, Hahnen
E, González
R, Seleme
N, Fernández
V, Schmidt
A, Binkofski
F, Kömpf
D, Kubisch
C, Hagenah
J, Klein
C, Ramirez
A. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations.
Mov Disord.
2010;25:1929-37.
[
PubMed: 20683840]
Brüggemann
N, Hagenah
J, Reetz
K, Schmidt
A, Kasten
M, Buchmann
I, Eckerle
S, Bähre
M, Münchau
A, Djarmati
A, van der Vegt
J, Siebner
H, Binkofski
F, Ramirez
A, Behrens
MI, Klein
C. Recessively inherited parkinsonism: effect of ATP13A2 mutations on the clinical and neuroimaging phenotype.
Arch Neurol.
2010;67:1357-63.
[
PubMed: 21060012]
Chien
HF, Bonifati
V, Barbosa
ER. ATP13A2-related neurodegeneration (PARK9) without evidence of brain iron accumulation.
Mov Disord.
2011;26:1364-5.
[
PubMed: 21469196]
Dick
KJ, Al-Mjeni
R, Baskir
W, Koul
R, Simpson
MA, Patton
MA, Raeburn
S, Crosby
AH. A novel locus for an autosomal recessive hereditary spastic paraplegia (SPG35) maps to 16q21-q23.
Neurology.
2008;71:248–52.
[
PubMed: 18463364]
Dick
KJ, Eckhardt
M, Paisán-Ruiz
C, Alshehhi
AA, Proukakis
C, Sibtain
NA, Maier
H, Sharifi
R, Patton
MA, Bashir
W, Koul
R, Raeburn
S, Gieselmann
V, Houlden
H, Crosby
AH. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35).
Hum Mutat.
2010;31:E1251–60.
[
PubMed: 20104589]
Dufke A, Grasshoff U, Dufke C, Kalscheuer V, Schroeder C, Beck-Wodl S, Tzschach A, Nagele T, Riess O, Bauer P, Krageloh-Mann I. NGS based whole X-exome analysis reveals a familial WDR45 missense mutation in 3 males with intellectual disability and brain iron accumulation. Abstract P08.53-S. Milan, Italy: European Society of Human Genetics Conference. 2014.
Dusi
S, Valletta
L, Haack
TB, Tsuchiya
Y, Venco
P, Pasqualato
S, Goffrini
P, Tigano
M, Demchenko
N, Wieland
T, Schwarzmayr
T, Strom
TM, Invernizzi
F, Garavaglia
B, Gregory
A, Sanford
L, Hamada
J, Bettencourt
C, Houlden
H, Chiapparini
L, Zorzi
G, Kurian
MA, Nardocci
N, Prokisch
H, Hayflick
S, Gout
I, Tiranti
V. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation.
Am J Hum Genet.
2014;94:11-22.
[
PMC free article: PMC3882905] [
PubMed: 24360804]
Edvardson
S, Hama
H, Shaag
A, Gomori
JM, Berger
I, Soffer
D, Korman
SH, Taustein
I, Saada
A, Elpeleg
O. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia.
Am J Hum Genet.
2008;83:643–8.
[
PMC free article: PMC2668027] [
PubMed: 19068277]
Eiberg
H, Hansen
L, Korbo
L, Nielsen
IM, Svenstrup
K, Bech
S, Pinborg
LH, Friberg
L, Hjermind
LE, Olsen
OR, Nielsen
JE. Novel mutation in ATP13A2 widens the spectrum of Kufor-Rakeb syndrome (PARK9).
Clin Genet.
2012;82:256-63.
[
PubMed: 21696388]
Evers
C, Seitz
A, Assmann
B, Opladen
T, Karch
S, Hinderhofer
K, Granzow
M, Paramasivam
N, Eils
R, Diessl
N, Bartram
CR, Moog
U. Diagnosis of CoPAN by whole exome sequencing: waking up a sleeping tiger’s eye.
Am J Med Genet.
2017;173:1878-86.
[
PubMed: 28489334]
Gregory
A, Lotia
M, Jeong
SY, Fox
R, Zhen
D, Sanford
L, Hamada
J, Jahic
A, Beetz
C, Freed
A, Kurian
MA, Cullup
T, van der Weijden
MCM, Nguyen
V, Setthavongsack
N, Garcia
D, Krajbich
V, Pham
T, Woltjer
R, George
BP, Minks
KQ, Paciorkowski
AR, Hogarth
P, Jankovic
J, Hayflick
SJ. Autosomal dominant mitochondrial membrane protein-associated neurodegeneration (MPAN).
Mol Genet Genomic Med.
2019;7:e00736.
[
PMC free article: PMC6625130] [
PubMed: 31087512]
Haack
TB, Hogarth
P, Kruer
MC, Gregory
A, Wieland
T, Schwarzmayr
T, Graf
E, Sanford
L, Meyer
E, Kara
E, Cuno
SM, Harik
SI, Dandu
VH, Nardocci
N, Zorzi
G, Dunaway
T, Tarnopolsky
M, Skinner
S, Frucht
S, Hanspal
E, Schrander-Stumpel
C, Héron
D, Mignot
C, Garavaglia
B, Bhatia
K, Hardy
J, Strom
TM, Boddaert
N, Houlden
HH, Kurian
MA, Meitinger
T, Prokisch
H, Hayflick
SJ. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA.
Am J Hum Genet.
2012;91:1144-9.
[
PMC free article: PMC3516593] [
PubMed: 23176820]
Hayflick
SJ, Westaway
SK, Levinson
B, Zhou
B, Johnson
MA, Ching
KH, Gitschier
J. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome.
N Engl J Med.
2003;348:33-40.
[
PubMed: 12510040]
Hogarth
P, Gregory
A, Kruer
MC, Sanford
L, Wagoner
W, Natowicz
MR, Egel
RT, Subramony
SH, Goldman
JG, Berry-Kravis
E, Foulds
NC, Hammans
SR, Desguerre
I, Rodriguez
D, Wilson
C, Diedrich
A, Green
S, Tran
H, Reese
L, Woltjer
RL, Hayflick
SJ. New NBIA subtype: Genetic, clinical, pathologic, and radiographic features of MPAN.
Neurology.
2013;80:268-75.
[
PMC free article: PMC3589182] [
PubMed: 23269600]
Hogarth
P, Kurian
MA, Gregory
A, Csanyi
B, Zagustin
T, Kmiec
T, Wood
P, Klucken
A, Scalise
N, Sofia
F, Klopstock
T, Zorzi, G, Nardocci
N, Hayflick
SJ. Consensus clinical management guildeline for pantothenate kinase-associated neurodegeneration (PKAN).
Mol Genet Metab.
2017;120(3):278-87.
[
PubMed: 28034613]
Kruer
MC, Boddaert
N, Schneider
SA, Houlden
H, Bhatia
KP, Gregory
A, Anderson
JC, Rooney
WD, Hogarth
P, Hayflick
SJ. Neuroimaging features of neurodegeneration with brain iron accumulation.
AJNR Am J Neuroradiol.
2012;33:407-14.
[
PMC free article: PMC7966445] [
PubMed: 21920862]
Kruer
MC, Paisán-Ruiz
C, Boddaert
N, Yoon
MY, Hama
H, Gregory
A, Malandrini
A, Woltjer
RL, Munnich
A, Gobin
S, Polster
BJ, Palmeri
S, Edvardson
S, Hardy
J, Houlden
H, Hayflick
SJ. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA).
Ann Neurol.
2010;68:611-8.
[
PMC free article: PMC6059612] [
PubMed: 20853438]
Rahbari
R, Wuster
A, Lindsay
SJ, Hardwick
RJ, Alexandrov
LB, Turki
SA, Dominiczak
A, Morris
A, Porteous
D, Smith
B, Stratton
MR, Hurles
ME, et al.
Timing, rates and spectra of human germline mutation.
Nat Genet.
2016; 48:126-33.
[
PMC free article: PMC4731925] [
PubMed: 26656846]
Schneider
SA, Paisán-Ruiz
C, Quinn
NP, Lees
AJ, Houlden
H, Hardy
J, Bhatia
KP. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation.
Mov Disord.
2010;25:979-84.
[
PubMed: 20310007]
Williams
DR, Hadeed
A, al-Din
AS, Wreikat
AL, Lees
AJ. Kufor Rakeb disease: autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia.
Mov Disord.
2005;20:1264-71.
[
PubMed: 15986421]