Clinical Description
To date, more than 100 individuals have been identified with a pathogenic variant in FTL. The following description of the phenotypic features associated with this condition is based on these reports [Curtis et al 2001, Wills et al 2002, Chinnery et al 2003, Maciel et al 2005, Mancuso et al 2005, Mir et al 2005, Chinnery et al 2007, Ohta et al 2008, Devos et al 2009, Kubota et al 2009, Batey et al 2010, Ondo et al 2010, Cassidy et al 2011, Shah et al 2012, Fatima et al 2013, Storti et al 2013, Moutton et al 2014, Nishida et al 2014, Maccarinelli et al 2015, Brugger et al 2016, Ni et al 2016, Yoon et al 2019].
Table 2.
Neuroferritinopathy: Frequency of Select Features
View in own window
| Category | Feature | % of Persons w/Feature 1 | Comment |
|---|
Presenting
phenotype
| Chorea | 50% |
|
| Dystonia | 43% |
| Parkinsonism | 8% |
| Asymmetry of mvmt disorder | 63% |
|
Motor issues
| Bradykinesia | 35% | Motor issues typically progressive |
| Dystonia | 83% |
| Chorea | 70% |
| Normal strength in nondystonic limbs | 100% |
| ↑ tendon reflexes | 18% |
|
Oral motor issues
| Dysarthria | 78% | |
| Dysphonia | 48% | |
| Orolingual dyskinesia | 65% | |
| Dysphagia | 40% | |
|
Eye issues
| Abnormal EOM | 8% | |
|
Cognitive deficits
| Impaired verbal learning & executive function | Frequency
not known | Typically subtle but progress over time |
|
Behavioral issues
| Disinhibition, emotional lability, aggression | |
|
Brain MRI features
| Excess brain iron accumulation
on T2-weighted MRI | 100% | |
|
Serum ferritin levels
| Low serum ferritin concentrations (<20 µg/L) in most males & postmenopausal females |
| Typically w/in normal limits in premenopausal females |
EOM = extraocular muscle (function)
- 1.
Movement disorder. The two presenting phenotypes are typically chorea or dystonia affecting one or two limbs, although one individual presented with late-onset parkinsonism [Curtis et al 2001, Burn & Chinnery 2006, Chinnery et al 2007] and two families with cerebellar features [Vidal et al 2004, Devos et al 2009] (see Table 2). The age of onset of movement abnormalities is during adulthood (mean: 40 years, range: second to seventh decade) [Chinnery et al 2007].
Oral motor manifestations
Cognitive deficits
Cognitive issues are usually subtle but progressively worsen through the course of the disease [
Crompton et al 2005]. These issues most commonly include impaired verbal fluency and verbal learning and impaired executive function [
Keogh et al 2013].
Behavioral issues. Emotional lability and aggression have been reported [Keogh et al 2013].
Prognosis. To date, there is limited information regarding prognosis in individuals with neuroferritinopathy. However, demise has been reported in individuals in mid-to-late adult life.
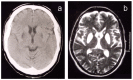
Neuroimaging. From disease onset, all affected individuals have evidence of excess brain iron accumulation on T2-weighted MRI. The iron deposition may be missed on other MR sequences in early stages of the disease. Later stages are associated with high signal on T2-weighted MRI in the caudate, globus pallidus, putamen, substantia nigra, and red nuclei, followed by cystic degeneration in the caudate and putamen. Neuroferritinopathy has a characteristic appearance, distinguishing it from other disorders associated with brain iron accumulation [McNeill et al 2008] and associated with progressive iron accumulation on MRI [McNeill et al 2012], including the "eye of the tiger" sign [McNeill et al 2012] and other radiologic features [Batla et la 2015].
Histopathologic examination. Of three individuals with the pathogenic FTL variant c.460dupA, histopathologic examination confirmed evidence of abnormal iron accumulation throughout the brain and particularly in the basal ganglia [Hautot et al 2007]. Affected regions contain iron and ferritin-positive spherical inclusions, often colocalizing with microglia, oligodendrocytes, and neurons. Axonal swellings (neuroaxonal spheroids) that were immunoreactive to ubiquitin, tau, and neurofilaments were also present. Mancuso et al [2005] report similar neuropathologic findings in an individual with FTL pathogenic variant c.442dupC.
Serum ferritin. Serum ferritin concentrations were low (<20 µg/L) in most males and postmenopausal females but within normal limits for premenopausal females [Chinnery et al 2007].