Classic Pantothenate Kinase-Associated Neurodegeneration (PKAN)
The clinical features of classic PKAN are remarkably homogeneous. It presents in early childhood, usually before age six years (mean age: 3.4 years). The most common presenting symptom is impaired gait resulting from a combination of lower-extremity dystonia and spasticity, as well as restricted visual fields in those children with retinopathy. Some children have developmental delay, which is primarily motor but occasionally global. Early histories of ADHD or toe-walking are also common. Visual symptoms may bring children with PKAN to medical attention.
Neurologic signs and symptoms of classic PKAN are primarily extrapyramidal and include dystonia and dysarthria. Dystonia is always present and usually an early manifestation. Cranial dystonia and limb dystonia are frequent and may lead, respectively, to recurrent trauma to the tongue and to atraumatic long bone fracture from the combination of extreme bone stress and osteopenia. The resulting pain and distress can contribute to development of status dystonicus in a cycle that can be difficult to break.
Corticospinal tract involvement is common and includes spasticity, hyperreflexia, and extensor toe signs.
Seizures are rare.
Intellectual impairment is a feature of PKAN, particularly in children with young onset. A study of 16 children and adults with PKAN showed varied cognitive expression as measured by standardized evaluation tools, with skills ranging from high average to markedly below average. Age of onset had a strong inverse correlation with intellectual impairment (i.e., earlier onset was associated with greater impairment) [Freeman et al 2007]. However, children with classic disease retain cognitive abilities achieved and do not lose these skills in tandem with later motor function loss.
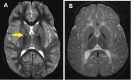
Retinal degeneration. Pigmentary retinal degeneration occurs in two thirds of affected individuals with classic PKAN. Retinopathy occurs early in the disease, although it is not often recognized until a full diagnostic evaluation including electroretinogram (ERG) and visual field testing is performed. The retinal degeneration follows a typical clinical course, with nyctalopia (night blindness) followed by progressive loss of peripheral visual fields and sometimes eventual blindness. Funduscopic changes initially include a flecked retina and later progress to bone spicule formation, conspicuous choroidal vasculature, and "bull's-eye" annular maculopathy. Individuals with a normal ophthalmologic examination at the time of diagnosis generally do not develop retinopathy later.
Abnormal eye movements, including vertical saccades and saccadic pursuits, are common. In one study, eight of ten individuals with PKAN had sectoral iris paralysis and partial loss of the pupillary ruff consistent with bilateral Adie's pupil [Egan et al 2005]. Optic atrophy is rarely seen in PKAN.
Prognosis. PKAN is a progressive disorder. Lost skills are usually not regained. The rate of progression correlates with age at onset: those with early symptoms decline more rapidly. As the disease advances, dystonia and spasticity compromise the child's ability to ambulate; most of those with early-onset disease are wheelchair bound by the mid-teens, and some much earlier. PKAN progresses at a non-uniform rate. Affected individuals experience episodes of rapid deterioration, often lasting one to two months, interspersed with longer periods of stability. Common causes of stress and catabolism do not appear to correlate with periods of decline, a phenomenon for which no cause has been found.
Premature death does occur. However, with improvements in medical care, a greater number of affected individuals are living into adulthood. Orofacial dystonia can result in the secondary effects of swallowing difficulty and poor nutrition. Premature death is more likely related to these secondary effects (e.g., nutrition-related immunodeficiency, aspiration pneumonia) than to the primary neurodegenerative process. In rare cases death occurs during status dystonicus.
Atypical PKAN
The clinical features of atypical PKAN are more varied than those of classic PKAN. Onset is in the first three decades (mean age: 13.6 years). Progression of the atypical form is slower, and presenting features are distinct, usually involving speech as either the sole presenting feature or part of the constellation of findings. The speech defects include palilalia (repetition of words or phrases), tachylalia/tachylogia (rapid speech of words and/or phrases), and dysarthria (poor articulation, slurring).
Psychiatric symptoms including personality changes with impulsivity and violent outbursts, depression, and emotional lability are common in atypical PKAN. Affected individuals may also exhibit motor and verbal tics, obsessive-compulsive behavior, and, rarely, psychotic symptoms [del Valle-López et al 2011].
As with classic PKAN, cognitive impairment may occur in individuals with atypical PKAN, but additional investigations are needed. Freeman et al [2007] found that later age of onset is correlated with less intellectual and adaptive behavior impairment.
Motor involvement is usually a later feature, although individuals with motor involvement often have been described as clumsy in childhood and adolescence. Spasticity, hyperreflexia, and other signs of corticospinal tract involvement are common and eventually limit ambulation. Conspicuously reminiscent of Parkinson disease, "freezing" during ambulation (especially when turning corners or encountering surface variations) is observed [Guimarães & Santos 1999].
An essential tremor-like syndrome has also been reported [Yamashita et al 2004].
Retinopathy is rare in atypical PKAN, and optic atrophy has not been associated with atypical PKAN.
HARP syndrome (hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration). Initially described as having a separate clinical entity, the two families described with HARP syndrome are now known to fall within the phenotypic spectrum of PKAN.