

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Bast RC Jr, Kufe DW, Pollock RE, et al., editors. Holland-Frei Cancer Medicine. 5th edition. Hamilton (ON): BC Decker; 2000.


The heterogeneous group of acute leukemic disorders of myeloid hematopoietic cells has been called a variety of names including acute myelogenous leukemia, acute myelocytic leukemia, acute myeloid leukemia, acute myeloblastic leukemia, acute granulocytic leukemia, and acute nonlymphocytic leukemia. The myeloid character of the malignant bone blasts can be determined by detection of characteristic morphologic and immunologic findings. A National Cancer Institute-sponsored workshop has suggested that the term acute myeloid leukemia (AML) is preferred, and this name will be used in this chapter.1
AML is the most common variant of acute leukemia occurring in adults, comprising approximately 80 to 85% of cases of acute leukemia diagnosed in individuals greater than 20 years of age. Striking advances in transfusion and infectious disease supportive care2,3 as well as serial evaluations of different chemotherapeutic approaches have eradicated the therapeutic nihilism that characterized many editorials and reports as late as the mid-1960s. Currently, more than 80% of young adults and 60% of all patients can achieve complete remission (CR). Varying with patient age and other factors, from 15 to 50% of these complete responders can be expected to achieve long-term survival with the likelihood that most of these individuals are cured of their disease (Fig. 124.1).
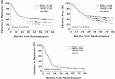
Figure 124.1
Data demonstrating the effect of different ara-C doses and schedules in patients of different ages with AML in remission. Top left, CR duration in patients < 60 years old; Top right, survival of CR patients < 60 years old; bottom, CR duration (more...)
During the past 10 years, there has been dramatic growth in the ability to apply a variety of laboratory techniques to the study of patients with AML. Immunophenotyping and cytogenetic analyses are now routinely performed on specimens from most patients with AML. Due to identification of genes at cytogenetic breakpoints in balanced chromosomal translocations, the pathophysiology of certain AML subtypes has been identified. Prognostic groupings and disease heterogeneity can be defined using these approaches, which provide the eventual potential for individualizing therapy to a greater extent than has been possible heretofore.
Because repetitive sampling of blood and bone marrow for in vitro correlative studies can more easily be accomplished, the acute leukemias have frequently served as the initial experimental model from which findings were extended to other types of cancers. Unfortunately, however, in contrast to many of the solid tumors, it has been difficult to clone leukemic cells reliably so as to permit in vitro experimentation and serial evaluations in the same patient. Similarly, there are relatively few stable leukemia cell lines, despite continued attempts by many laboratories to establish such lines from leukemia patients.4 Indeed, the question arises whether conclusions derived from the study of these unusual cell lines can be readily extrapolated to human leukemias.
AML affects adults of all ages but is especially common in older adults. This point is emphasized by the observation that the median age of patients with de novo AML entered on recent cooperative group studies is approximately 55 years, with some studies having a median age of 60 years.5 It can present either as a de novo leukemia without an apparent antecedent illness or as an evolution from obvious marrow disorders such as myelodysplasia, aplastic anemia, and Fanconi’s anemia6 or after the administration of therapy for other types of cancers or nonmalignant disorders. An acute leukemic phase uniformly terminates chronic myelogenous leukemia (CML) and occasionally in the other myeloproliferative disorder subtypes. The blast crisis of CML may be morphologically indistinguishable from AML, although it can generally be diagnosed by history or cytogenetic analysis. The natural history of AML arising de novo in young adults is believed to be a disease of committed stem cells that is vastly different than the same disease arising from a primitive stem cell, typified by secondary AML.7
The proper care of patients with AML is a multidisciplinary effort, benefiting from a team approach. Expertise in transfusion medicine, infectious disease, placement and care of indwelling catheters, nutrition, antineoplastic drug pharmacology, and the availability of sophisticated diagnostic laboratory facilities and psychosocial counseling for both patients and their families are required. These disciplines are described elsewhere in this text, but their critical importance in the care of the leukemia patient cannot be overestimated.
Pathogenesis and Etiology
The pathophysiology of AML can be explained by acquired genetic changes in bone marrow stem cells that cause a complete or partial block in normal hematopoietic stem cell maturation. The genetic changes may involve mutations that lead to activation of growth-promoting proto-oncogenes, inactivation of tumor suppressor genes, or alterations in transcription factors.8 Mutations in codons 12, 13, or 61 of the N-ras gene encoding a 21 kd guanosine nucleotide binding protein involved in signal transduction have been noted in up to 30% of those with AML.9 Such abnormalities may be more common in the monocytic subtypes of AML.10 Overexpression of the fit-3 growth factor receptor has been reported,11 suggesting the possible role of autocrine stimulation. Mutations of the tumor suppressor gene Rb12 and p5313 have been observed and appear to confer an inferior prognosis.
The greatest insight regarding AML pathogenesis has been provided by their identification of genes at cytogenetic breakpoints involved in balanced translocation. The fusion proteins generated by the translocation generally results in disruption of transcription factors believed to be critical in myeloid differentiation.14 Many of theses chromosomal abnormalities [e.g., t(8;21), t (15;17), inv 16] are associated with specific AML subtypes and carry prognostic importance. Each will be discussed in turn later in this chapter. However, the genetic changes that account for poor prognosis when loss of all or parts of chromosomes occur remain to be elucidated.
Although the acquired genetic lesions that lead to leukemia are being rapidly defined, DNA damage from a known cause account for a small fraction of patients with AML. Nonetheless, leukemias clearly occur with increased frequency after military15 or therapeutic16 radiation exposure, after certain types of chemotherapy, and with heavy and continuous occupational exposures to benzene.17,18 It is now recognized that there are two types of chemotherapy-related leukemias: (a) the classic alkylating agent-induced type19 in which the leukemia is preceded by a myelodysplastic prodrome and is characterized by clonal abnormalities of chromosome 5 and/or 7 and (b) an epipodophyillotoxin-associated type with shorter (2-yr vs 5-yr) incubation period that is associated with myelomonocytic or monocytic differentiation and abnormalities at the 11q23 region.20
There have been a number of large, survey-type epidemiologic studies that have attempted to link a variety of environmental exposures to leukemia incidence.21 Many of these studies contain relatively small sample sizes and identify modest, if any, increases in risk ratio. In addition, older and retrospective studies may be somewhat suspect in that the pathologic diagnoses have usually not been reviewed and classified using contemporary criteria, and therefore the distinctions between myeloid, lymphoid, and perhaps even acute versus chronic leukemia may not be accurate. Furthermore, as in many epidemiologic studies, it is difficult to quantify the degree of exposure to various environmental insults. There may be a small increased risk associated with cigarette smoking,22,23 whereas exposure to electromagnetic radiation24 seems an unlikely cause of AML. However, occupational exposures, particularly to benzene17,18 or petrochemicals25,26 have been implicated in the development of AML. Alkaylating agents used in Hodgkin’s disease, multiple myeloma, ovarian cancer, and colon cancer have been associated with –5/–7 chromosome type, whereas tenoposide use in childhood acuate lymphoblastic leukemia (ALL) and high-dose anthracycline/cyclophosphamide combinations have caused 11q23-associated disease. With the few exceptions noted above, however, there is no clear relationship between environmental exposures and the occurrence of acute leukemia.
Nor are the acute leukemias a familial or inherited disorder. Except for the increased incidence of acute leukemia occurring during the first 6 months after diagnosis in the identical twin of an affected child,27,28 the occurrence of multiple cases of acute leukemia in a given family is extremely rare. Indeed, it is quite important to reassure family members in this regard because this is often an unasked question when leukemia is first diagnosed in a relative. A few families have been described in which leukemia incidence is increased.29–32 In some of these families, multiple different types of leukemias and other cancers have been found. In other, potentially more informative families, the morphologic or clinical characteristics of the leukemia have been similar, suggesting a common, heritable genetic mutation. A large family has recently been described in which seven cases of erythroleukemia or myelodysplasia have been noted in three successive generations.31 Most of these cases antedated current molecular biologic techniques, and the available cytogenetic data did not suggest a common chromosomal abnormality. There was no association of leukemia with specific HLA types in this family, a finding similar to that noted in large surveys of HLA typing among patients with leukemia.33,34
Morphologic Classification and Clinical and Laboratory Correlates
The diagnosis of AML depends on the examination by experienced observers of well-prepared specimens of peripheral blood and bone marrow. Both bone marrow aspirates and biopsies should be evaluated. Although the biopsy is usually not helpful in identifying individual cells, it provides the best assessment of cellularity, can occasionally identify aggregates of leukemic cells not seen on aspirate, and is necessary to evaluate marrow fibrosis. In 1976, a group of morphologists from France, the United States, and Great Britain (FAB) suggested a classification system designed to quantify and standardize definitions of the sometimes clinically and biologically distinct subtypes of AML and ALL.35 This FAB classification has been serially modified in an effort to improve concordance among different observers35,36 and incorporate new findings from more recent immunologic and cytogenetic studies.1,37–39 It is widely used in all studies of leukemia treatment but may be modified once again as the critical influences of cytogenetics on prognosis become clear.
The diagnosis of AML requires that myeloblasts constitute 30% (now 20% based on a recent World Health Organization classification system40) or more of bone marrow cells or circulating white blood cells, generally evaluated on Wright or Wright-Giemsa stained smears. Neoplastic promyelocytes, monoblasts, or promonocytes and megakaryoblasts are included in this percentage, and their presence defines the various FAB subtypes described below. An assortment of histochemical stains is routinely used to aid in subclassification and to distinguish AML from ALL. Monoclonal antibodies directed against antigen groups termed cluster designations (CD) considered to be restricted to cells committed to myeloid differentiation are also helpful in making this diagnostic distinction.40,41 Antibodies against CD11b, CD13, CD14, and CD33 are used most commonly. The antigens detected are found on normal hematopoietic elements and are not leukemia-specific. These antigens are not unique to different AML FAB subtypes and, generally, do not correlate with prognosis,41 with the probable exception of CD34. CD34 is detected on undifferentiated hematopoietic progenitors and can be found on the blasts of patients with either AML or ALL.42 There is evidence from at least four groups of investigators that patients with AML whose blasts strongly express CD34 have a poor outcome following treatment due to chemotherapy-resistant leukemia.43–46 This is particularly true in less morphologically differentiated leukemias in which other myeloid-associated antigens are less strongly expressed.
The FAB morphologic classification names the AML according to the normal marrow elements that they most closely resemble. This does not mean, however, that the leukemic event exclusively involves the cell lineage that is most prominently represented morphologically. Until recently, the involvement of other hematopoietic lineages could only be inferred by the presence of prominent morphologic abnormalities in these other cell lines. Thus, in patients with myelodysplasia or erythroleukemia, there is generally morphologic evidence of trilineage dysplasia with the inference that the initial cell that was malignantly transformed is a hematopoietic precursor with capability of multilineage maturation. Cytogenetic and in situ hybridization techniques using specific molecular probes capable of detecting abnormalities in morphologically identifiable cells,47,48 as well as the increased ability to grow colonies of different lineages in vitro to perform these tests, have enhanced the ability to pinpoint accurately the lineages involved in an individual patient’s leukemia. For example, Fialkow and colleagues,49 studying female patients with X-chromosome-linked polymorphisms of glucose-6-phosphate dehydrogenase, were able to demonstrate involvement of myeloid progenitors but not erythroid or megakaryocytic progenitors in some patients with myeloid leukemia. In contrast, using specific chromosomal probes, Keinanen and colleagues50 and Kibbelaar and co-workers51 demonstrated multiple lineage involvement in patients with erythroleukemia and myelodysplasia. Overall, there are relatively few studies of this type, and it is unknown how many lineages are affected in most patients with AML. In particular, patients with more differentiated types of AML such as acute prognulocytic leukemia (APL) or monocytic leukemia have not been systematically evaluated, although in a few patients with APL there is a suggestion that the mutation is restricted to the myeloid series. Such studies would be of considerable interest in further defining the fundamental biology of AML and may eventually suggest new approaches for therapy, particularly if new compounds capable of inducing differentiation in leukemia cells become available.
Representative examples of different FAB subtypes of AML are shown in the color photomicrographs in figures Fig. 124.2–11. The immunologic, cytogenetic, and (where they exist) typical correlates of these morphologic subtypes will be reviewed (Table 124.1).52
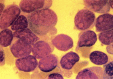
Figure 124.2
FAB M0. Marrow blasts from patients with this undifferentiated type of acute myelogenous leukemia can have variable amounts of agranular cytoplasms with Auer rods. Cells are peroxidase- and Sudan black-negative and can be confused with FAB M7 or FAB L2. (more...)
Table 124.1
Karotypic Abnormalities in AML.
Peripheral Blood
Most patients with AML present with anemia (median hemoglobin 8 g%), thrombocytopenia (median platelet count 40,000–50,000/μl), and leukocytosis (median white blood cell count 10,1000–20,000/μL). The red blood cell morphology is usually relatively normal. Large, sometimes hypogranular platelets can be seen, and functional defects can contribute to hemorrhagic manifestations. Most patients are neutropenic, and morphologic abnormalities (hypogranulation, nuclear hyperlobulation, Pelger-Huët anomaly) are often noted in the remaining neutrophils. Careful examination will detect blasts in most patients, although it can be difficult to distinguish among leukemia subtypes (or occasionally even to be confident of the diagnosis of acute leukemia) in patients with a low number of circulating blasts. In occasional patients, marked leukopenia at presentation (aleukemic leukemia) may obscure the diagnosis until a marrow examination is performed.
MO (Minimally Differentiated AML)
In most patients, it is relatively simple to distinguish between AML and ALL on morphologic grounds. In general, the blasts from patients with AML are larger, with more abundant cytoplasm and more prominent, often multiple nucleoli. The definitive diagnosis depends, however, on the presence of Auer rods, concentrations of myeloid-containing granules (present in 50% of those with AML), or demonstration of at least 3% granulated precursors in AML,35 usually visible on Wright stain and confirmed by staining with either peroxidase or Sudan black B. It should be noted that it can occasionally be difficult to distinguish between positivity in blasts versus staining in residual myeloid elements in patients with ALL; careful correlation with the Wright stain is mandatory.
As shown in Fig. 124.2, some patients have blasts that resemble myeloid blasts but are negative at the light microscopic level when examined with peroxidase, Sudan black, or other histochemical stains. The myeloid nature of these leukemias can be detected, however, by immunologic means or by election microscopy of peroxidase-stained preparations. Electron micrographs reveal ultrastructural peroxidase-positive granules, whereas immunologic phenotyping demonstrates reactivity with antibodies directed against myeloid antigens and nonreactivity with antibodies that characterize lymphoid differentiation.39 The cells are often reactive with antibodies directed against CD34. Terminal deoxynucleotidal transferase (TdT) is generally absent but can sometimes be detected in a minority of blasts.
Approximately 7% of patients with untreated AML reportedly have minimally differentiated AML (so-called MO AML),53,54 which is relatively treatment resistant. This undifferentiated leukemia can easily be confused with ALL, and the Cancer and Leukemia Group B (CALGB) has noted that occasional patients entered on ALL chemotherapy trials actually have minimally differentiated AML.55 This has undoubtedly occurred in other clinical trial settings and potentially influences interpretation of the clinical outcome. It is therefore critically important to obtain immunophenotyping of blasts from patients with morphologically undifferentiated leukemias.
Other than an apparent resistance to chemotherapy, this MO variant does not appear to be associated with other specific clinical findings. Although a number of patients have had complex karyotypic abnormalities, no distinctive cytogenetic pattern has been noted, with the possible exception of trisomy 13, which has been reported to occur in some patients with morphologically less differentiated leukemias.56
FAB M1 (Myeloid Leukemia without Maturation)
The blasts from patients with FAB M1 have round nuclei with moderate amounts of sometimes lightly granulated cytoplasm, which can contain Auer rods (Fig. 124.3). In contrast to FAB M2, there is little evidence of myeloid maturation, with <10% of cells beyond the level of the promyelocyte. There is no particular age, gender, clinical features, or characteristic cytogenetic abnormality associated with this morphologic variant. A retrospective analysis has suggested that the overall prognosis is poorer in patients in whom there is a lower fraction (50%) of blasts that are peroxidase-positive.57 Although this observation is compatible with the more refractory nature of MO AML, these results require confirmation.
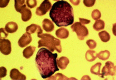
Figure 124.3
One of the blasts from this patient with FAB M1 AML contains a prominent Aver rod.
FAB M2 (Myeloid Leukemia with Maturation)
In contrast to FAB M1, there is obvious continued maturation in the myeloid series with the presence of promyelocytes, myelocytes, and often more mature myeloid elements. Granulation is generally more obvious, Auer rods are often prominent, and there is virtually never any difficulty in distinguishing this variant from ALL (Fig. 124.4). Approximately 20 to 25% of patients with FAB M2 AML have a characteristic translocation between chromosomes 8 and 21 [t(8; 21)(q22; 22)]; this translocation is seen almost exclusively in patients with FAB M2 and Auer rods. Such patients have a lower median age (approximately 30 years), a very high initial response rate to chemotherapy (>85% in most series), a lower relapse rate, and improved long-term disease-free survival compared with most other AML variants and with other patients with FAB M2.58–60 It has recently been suggested that the incidence of extramedullary granulocytic sarcomas, often in unusual sites, may be increased in patients with t(8;21).61 This extramedullary manifestation often presents as discrete tumor masses, often in paraspinous locations, and is distinct from the gingival and cutaneous involvement found in monocytic leukemia.
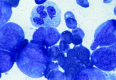
Figure 124.4
FAB M2. Leukemia is characterized evidence of continued myeloid differentiation with myelocytes and more mature myeloid elements present.
The t(8;21) has recently been cloned and involves what has been termed the AML 1 gene on chromosome 21 and the ETO gene on chromosome 8.62 The AML 1 gene has homology to the Drosophila runt gene and encodes the alpha chain of the heterodimeric transcriptional apparatus, core binding factor (CBF).63 Normal CBF function is critical for mammalian hematopoietic development.64 The AML-ETO fusion protein recruits nuclear corepressor molecules including histone deacetylase,65 which prevents transcription of genes required for myeloid differentiation. The AML-ETO fusion product can be detected by reverse transcriptase-polymerase chain reaction (RT-PCR) and used for both diagnostic testing and monitoring for minimal residual disease. Of note is that the t(8;21) has been detectable in the peripheral blood cells of some patients in long-term CR after treatment, suggesting that molecular detection by highly sensitive RT-PCR may not necessarily presage relapse.66
FAB M3 (Progranulocytic Leukemia)
APL is one of the most distinctive subtypes of AML with regard to morphologic, clinical, and cytogenetic features.67–70 In most patients, the morphologic diagnosis is straightforward, with the marrow being replaced by blasts that resemble abnormal, unusually heavily granulated progranulocytes. The nuclei are round, usually with obvious nucleoli, and the cytoplasm is filled with multiple, large, and often coalesced, azurophilic granules (Fig. 124.5). Auer rods are usually seen, and multiple Auer rods seen in sheaves (so-called faggot cells) are frequently noted. In a minority of patients, the blasts are hypogranular and granules sometimes can only be seen with electron microscopy. This M3 variant often has cells with bilobed or lobulated nuclei, which at first glance can sometimes be confused with monocytic variants of AML. In contrast to the typical leukopenic presentation, patients with the hypogranular variant tend to have a high white count. In both types of APL, staining with Sudan black or peroxidase is strongly positive.71 Class II HLA antigens (HLA DR), which are found on all hematopoietic precursors, are usually not detected or expressed on the surface of the malignant progranulocyte.72,73 The explanation and biologic implications of this finding are not known.
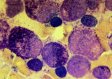
Figure 124.5
FAB M3. Progranulocytic leukemic cells usually have round nuclei with heavily granulated cytoplasm. Extracellular granules are often noted, and blasts with multiple Auer rods (not shown) are common. This leukemia has typical 15; 17 translocation and a (more...)
Patients with APL tend to be somewhat younger, with a median age of 30 to 40 years, although this variant is seen in patients of all ages. APL accounts for approximately 10% of AML and is somewhat more prevalent in Latinos74 and in obese people.75 APL is almost uniformly characterized by hypofibrinogenemia, variable depletion of other coagulation factors, elevated levels of fibrin degradation products, and accelerated consumption of endogenous and transfused platelets.76 The granular contents have been shown to contain potent procoagulants, and DIC is generally accelerated following lysis of blasts by chemotherapy, often with increased bleeding.77 In some patients, there is evidence that accelerated fibrinolysis may be the primary event triggering the coagulopathy.78 This type of AML is associated with the highest frequency of hemorrhagic morbidity and mortality, the latter usually related to intracranial hemorrhage. During chemotherapeutic treatment with accelerated cell lysis, these patients required aggressive support with platelet transfusions and often require transfusions 2 to 3 times per day during the first few days of treatment.79 The plasma accompanying the platelet transfusion is usually sufficient for coagulation factor replacement. Except for patients with very severe hypofibrinogenemia who may require supplementation with cryoprecipitate, most patients do not require supplemental support with clotting factors. Aggressive blood product support is probably as good as heparin, once frequently used to interrupt the clotting cascade during drug therapy.79–81 However, the heparin versus transfusion debate has been largely silenced because treatment with all-trans retinoic acid (ATRA) ameliorates DIC within 48 hours of its administration.82
Cytogenetic studies have demonstrated that almost all patients with APL have a characteristic translocation involving chromosomes 15 and 17 [t(15;17)(q22;q12)].68,83 This abnormality persists at relapse and may be associated with additional cytogenetic abnormalities, such as trisomy 8.84 The t(15;17) breakpoint has been cloned,85–87 thereby allowing detection of the fusion transcript as an assay for minimal residual disease as well as permitting the proper classification of the occasional patient with clinically and morphologically typical APL but with an apparently normal karyotype.88 The breakpoint on chromosome 17 is in the intron of the retinoic acid receptor alpha gene.85,86 A gene that has been termed PML, also with DNA-binding capability, is translocated from chromosome 15, resulting in the formation of a fusion protein that functions in a dominant fashion to block transcription of genes controlled by RAR-α, probably by recruiting nuclear co-repressor activity.89,90 It is likely that retinoic acid treatment relieves the co-repression activity, allowing transcription of genes involved in differentiation.90
Recently, a group of patients with a leukemia similar in morphology to APL but with a t(11;17)(q23;q21) have been described.91 Although there was a rearrangement of RAR-α, these patients failed to respond to ATRA. A novel zinc finger gene termed PZLF from chromosome 11 was translocated to the RAR-α, rather than the PML gene from chromosome 15, creating a fusion protein that does not allow the ATRA-mediated release of co-repressing transcriptional co-repression activity.92
Historically, the initial remission rate in patients with APL treated with chemotherapy was quite high. Initial drug resistance is very unusual, and most failures of therapy are related to hemorrhagic or infectious deaths. APL seems to be uniquely sensitive to anthracycline therapy, and CR rates in excess of 80% can be seen with anthracycline therapy alone.93 It is critical to recognize that in contrast to other types of AML, remission can be attained in APL with chemotherapy without producing bone marrow aplasia.93,94 Post-treatment bone marrows frequently remain cellular with abnormal progranulocytes, with follow-up marrows demonstrating disappearance of these cells and return of normal hematopoiesis; DIC does not reappear despite the apparent persistence of abnormal cells. Stable or improving blood counts and the return of erythroid and megakaryocytic precursors are other signs that further chemotherapy is not necessary. Undoubtedly, this unique feature of APL is in some way related to the equally unique sensitivity of APL to agents that have a differentiating and noncytotoxic mechanism of action (see “Differentiating Agent Therapy” below).
FAB M4 (Myelomonocytic Leukemia)
Myelomonocytic leukemia is characterized morphologically by a mixture of both myeloid and monocytic elements and represents about 15 to 20% of newly diagnosed patients with AML. According to the FAB criteria, greater than 20% of the leukemic cells must be monocytic in morphology to distinguish this variant from FAB M1 and particularly from FAB M2.38 The monocytic elements can be defined morphologically by the presence of partially differentiated monocytes with lightly granulated, grayish cytoplasm and folded nuclei, which are frequently seen in the peripheral blood (Fig. 124.6). Monocytic derivation can be confirmed by staining with nonspecific esterases such as alpha naphthyl acetate or alpha naphthyl butyrate.
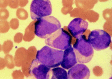
Figure 124.6
FAB M4. Myelomonocytic leukemia has blasts with both myeloid and monocytoid appearance.
There is no distinct clinical picture associated with this morphologic variant, perhaps because this classification encompasses a wide spectrum of patients because of the generous morphologic criteria for inclusion. The median age tends to be somewhat higher, and there may be an increased incidence of hyperleukocytosis and extramedullary leukemic involvement, as can be seen with monocytic leukemia (see below). There is no particular cytogenetic clustering, and it is impossible to predict reliably short- or long-term outcome in patients with FAB M4.
FAB M4EO (Myelomonocytic Leukemia with Eosinophilia)
Approximately 5% of patients with de novo AML present with typical morphologic features of myelomonocytic leukemia with the presence of variable numbers of dysplastic eosinophils at various stages of maturation. The distinctive eosinophils usually represent only 5 to 10% of the cells of the marrow.95,96 These cells generally contain large basophilic granules in addition to typical eosinophilic granules and are easily detected by experienced morphologists. It is unclear whether these cells are part of the leukemic process or are reactive in nature. Occasional patients with apparent FAB M2 morphology with eosinophilia have also been described.
FAB M4E0 tends to occur in patients of younger age (median age 35–40 years) and is associated with an excellent prognosis. CR rates are high (generally >85%), and failure due to initial drug resistance is unusual. In some series, this variant represents the subtype with the most favorable long-term prognosis. In addition to long initial CRs, second remissions, which are often quite sustained, are generally easier to accomplish in patients with FAB M4E0.97 Older series have suggested that there may be a high rate of central nervous system (CNS) relapse in patients with bone marrow eosinophilia.98 With more intensive regimens using higher doses of cytosine arabinoside (ara-C), CNS relapse in AML is now an unusual occurrence, and this possible predilection in patients with FAB M4E0 does not require prophylactic CNS therapy.
Essentially all patients with FAB M4E0s have a cytogenetic abnormality involving chromosome 16 at band q22. In most patients, the cytogenetic changes involve a pericentric inversion [inv16], although translocations between the two chromosomes 16 with homologous deletions at 16q22 have also been noted.94,95 This can be a very subtle cytogenetic finding, and cytogeneticists should be prompted to look for these changes by the characteristic morphologic changes. This breakpoint has been cloned and involves a fusion between the CBF-β chain and the gene encoding the smooth muscle myosin heavy chain.99 The fusion protein thus generated may recruit nuclear co-repressor activity (in the form of histone deacetylase), which prevents transcription of genes required for myeloid differentiation in a fashion analogous to the CBF-α ETO fusion in t(8; 21) M2 AML. Although it is clear that patients with inv 16 leukemia respond very well to intensive chemotherapy (> 60–70% 3-yr DFS rate in those receiving high-dose ara-C [so-called HIDAC])100, the reason for this is uncertain. One group has suggested that the gene coding for the multidrug resistance protein (MRP) is mutated as a consequence of this translocation, thereby hypothesizing that the enhanced chemosensitivity may be related to a decrease in the normal amounts of this resistance factor.101
FAB M5 (Monocytic Leukemia)
Two variants of monocytic leukemia have been described; in both, >80% of the blasts are of monocytic derivation. Less common is so-called FAB M5a, in which the monocytic blasts usually have round nuclei and small amounts of sometimes deeply basophilic cytoplasm without evidence of morphologic differentiation. In monocytic leukemia with differentiation (FAB M5b), at least 20% of the blasts resemble promonocytes with folding of the nuclei and abundant, lightly granulated cytoplasm, generally without Auer rods. The nuclear folding can often be quite marked with rarification of the nuclear chromatin (Fig. 124.7). Phagocytosis of other hematopoietic elements by these cells is frequently noted in bone marrow preparations. These monocytic elements stain prominently with nonspecific esterase that is inhibited by fluoride.
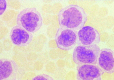
Figure 124.7
FAB M5. Monocytic leukemia. Prominent nuclei filled with nucleoli in some cells, light granulation, and large amounts of lightly basophilic cytoplasm give these cells the appearance of promonocytes.
Although seen in patients of all ages, monocytic leukemias are somewhat more common in older adults. Patients with FAB M5 have higher blast counts at diagnosis, and problems with hyperleukocytosis are most common in this morphologic variant (see “Complications” below).102,103 In addition, the incidence of extramedullary leukemia is highest in FAB M5, particularly those with evidence of morphologic differentiation.104,105 For example, it is common for patients to present to the dentist with gingival hypertrophy, an example of which is seen in Fig. 124.8. Skin infiltration is common both at diagnosis and relapse and frequently represents the initial site of recurrence, often while the bone marrow is still morphologically normal. Other less common areas of extramedullary involvement include the gastrointestinal tract, conjunctivae, and the CNS.106,107 It is likely that the extramedullary infiltration is related to active migration of the leukemic promonocytes to these sites. It has been shown that these partially differentiated cells are capable of migration to skin windows in vivo as well as phagocytosis of microorganisms and adherence to nylon fibers in vitro.108
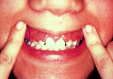
Figure 124.8
FAB M5. Gingival hypertrophy due to infiltration by leukemic cells in acute monocytic leukemia.
Serum levels of lysozyme are elevated in most patients with AML but are generally much higher in patients with monocytic leukemia.109 Lysozyme can affect renal tubular function, and severe, symptomatic hypokalemia can occur in patients with FAB M4 and M5 leukemia. This problem generally resolves with cytoreduction but can also be additive to the hypokalemic side effects of antibiotics and amphotericin B, vomiting, and diarrhea.
In addition to the initial problems presented by complications of hyperleukocytosis, patients with monocytic leukemia have lower complete response rates related to initially drug-resistant disease.105,110,111 In parallel, CR duration tends to be shorter with consequent very low rates of long-term disease-free survival. A variety of cytogenetic abnormalities can be detected, although the most common findings involve abnormalities of chromosome 11 at band q23.111,112 This breakpoint, at what has been termed the MLL gene, has recently been cloned and can be involved in leukemias of myeloid or lymphoid origin as well as in leukemias associated with therapy with epidophyllotoxins and other drugs directed at topoisomerase II.113 The MLL gene, also called All-1 or HRX, may partner with at least 16 different genes in balanced translocation.114 MLL is homologous to a gene important in drosophila development and includes DNA binding elements.115 A relatively common translocation involving the MLL gene, the t(9;11) associated with monocytic leukemia may actually confer a better prognosis than formerly thought,116 with higher initial CR rates. There is an association between M5b and extensive erythrophagocytosis, and the t(8;16) (p11;p13)117,118 a translocation involving that CBP class of translocation factors that are positive regulators of myeloid differentiation.119
FAB M6 (Erythroleukemia)
Erythroleukemia, often termed de Guglielmo’s syndrome in earlier literature, is the variant of AML in which morphologic abnormalities of erythropoiesis are most prominent. Cases of pure erythroleukemia in which the predominant malignant cell is clearly identified as a pronormoblast are extraordinarily rare. Rather, this is almost certainly a disease of the myeloid stem cell with marked dysplastic changes in all three hematopoietic lines with eventual increases in the number of myeloid blasts. Along with the increase in myeloid-appearing blasts, there is persistence of morphologic abnormalities in the erythroid series with profound megablastosis, multinuclearity, karyorrhexis, increased numbers of mitoses, and staining with periodic acid–Schiff, often in a block pattern (Fig. 124.9). Increased iron stores are usually seen, often with ringed sideroblasts.
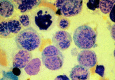
Figure 124.9
FAB M6. Erythroleukemia is characterized by the presence of bizarre megaloblastic and often multinucleated erythroid precursors. Karyorrhexis is seen in some cells. The somewhat arbitrary distinction between FAB M6 and myelodysplastic syndrome with excess (more...)
These changes are morphologically identical to those seen in patients with myelodysplasia, and many observers feel that it is likely that most cases of erythroleukemia are biologically similar, if not identical, to patients with refractory anemia with excess blasts, or refractory anemia with excess blasts in transformation (RAEB or RAEB-T). This contention is supported by the very poor response to therapy in both groups, the tendency for the disease to occur in patients of older age, and the presence of similar cytogenetic abnormalities (complex karyotypic abnormalities with loss of part of all of chromosomes 5 and/or 7 and marker chromosomes).120 Nonetheless, in order to provide some consistency in terms of protocol entry and reports of clinical trials, the FAB group, with a minor recent revision, has somewhat arbitrarily distinguished among FAB M6, myelodysplasia (RAEB or RAEB-T), and other FAB subtypes with significant numbers of erythroblasts by quantification of the number of erythroblasts and myeloblasts.1,38 FAB M6 is defined by the presence of > 30% blasts among nonerythroid cells when > 50% of the marrow nucleated elements are erythroid. Antibodies against glycophorin A are lineage-specific for erythroblasts, but immunologic phenotyping is rarely needed to distinguish among these various morphologic subtypes.
Except for the more advanced age and higher frequency of primary resistance to therapy, there are no distinct clinical or diagnostic karyotypic features associated with FAB M6.121 In occasional patients, transfusion of red blood cells can result in a marked decrease, if not elimination, of the marrow erythroid elements, suggesting the continued response to normal feedback mechanisms.
FAB M7 (Megakaryocytic Leukemia)
Morphologic abnormalities of megakaryocytopoiesis, usually characterized by the presence of mono- or binucleated micromegakaryocytes, are common in many variants of AML and can be particularly prominent in patients with FAB M6 or myelodysplasia. A minority of these patients have thromboyctosis and abnormalities of chromosome 3[inv(3)(q21;q26)], usually in association with other chromosomal deletions, with a variety of primary FAB morphologies (M1, M2, M4).122,123 These patients have a poor response to initial treatment and poor overall survival. Thrombocytosis is not a unique feature of the inv(3) karyotype but can also be present in other patients with AML at the time of diagnosis, particularly those in whom normal numbers of megakaryocytes are present.124 The gene at the chromosome 3q21 breakpoint is associated with the activation of EV11 transcription factor.125
The diagnosis of FAB M7 is reserved for those patients in whom the predominant leukemic cell is of megakaryocytic lineage.37 In some patients, there is obvious evidence of megakaryocytic dysplasia or multinucleated cells that strongly points morphologically toward principal involvement of the megakaryocyte (Fig. 124.10). In many others, however, the leukemia is very undifferentiated morphologically with variable amounts of generally agranular cytoplasm and can sometimes be confused with FAB M1 or even ALL. Sudan black B (Fig. 124.11), peroxidase, and alpha naphthyl butyrate stains are negative, whereas PAS and acid phosphatase may be positive, usually in a diffuse pattern. Histochemical staining is nondiagnostic, however, and the definitive diagnosis depends on either the demonstration of platelet-specific peroxidase by ultrastructural techniques126 or, more commonly, by the demonstration of a variety of platelet antigens (usually glycoprotein IIb/IIIa [CD41] or von Willebrand’s factor) on the surface of the blasts.127 At times, the diagnosis can be quite difficult to confirm, particularly because in most patients there is increased marrow reticulin, rendering the marrow fibrotic and inaspirable. Careful evaluation of peripheral blood blasts is necessary in such patients. It is likely that most patients with what has been termed acute myelosclerosis actually have acute megakaryocytic leukemia.128,129 Acute megakaryocytic leukemia should not be confused with the late stages of myloid metaplasia, and, indeed, prominent splenomegaly is not a clinical feature of FAB M7.
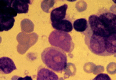
Figure 124.10
FAB M7. Megakaryocytic leukemia. Blasts in this category are often morphologically undifferentiated. The presence of multinucleated cells, dysplastic micromegakaryocytes, and cytoplasmic budding can be helpful diagnostic clues. The diagnosis is confirmed (more...)
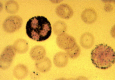
Figure 124.11
Typical granular staining with Sudan black B of a blast and a neutrophil from a patient with FAB M1 AML.
Although an uncommon variant of AML, most series suggest that this subtype is associated with a very poor prognosis. Prolonged aplasia is common following induction chemotherapy, and, because of the marrow fibrosis, it is often difficult to follow the results of therapy with repeated marrow aspirations. There have been relatively few cytogenetic evaluations of this variant, and except for the inv(3) and a few cases of t(1;22) (p13;q13) found in infants,130 no consistent abnormality has been identified to date.
Hybrid Leukemias
Morphologists have long been perplexed by cases that defy easy categorization or seem to have features of both lymphoid and myeloid histology. A variety of terms have been used to describe such cases including hybrid leukemia, mixed lineage leukemia, biclonal leukemia, lineage infidelity or promiscuity, and biphenotypic leukemias.131–133 With the advent of new immunologic and molecular technology, it was hoped that most, e.g., if not all, leukemias could be assigned to a specific lineage. Paradoxically, the serial application of biochemical, immunologic, and molecular biologic techniques has demonstrated that sharing of myeloid and lymphoid characteristics may be a relatively common feature of both myeloid and lymphoid leukemia. The clinical and therapeutic implications of these findings are as yet unclear. There are no accepted diagnostic criteria for hybrid leukemia, and, given the rapid pace of investigation, it may be premature to establish rigorous definitions. Most reports suggest that individual cells co-express myeloid and lymphoid features; it is likely that this pattern is much more common than the simultaneous occurrence of two separate leukemias. The frequency and importance of subpopulation of myeloid only, lymphoid only, null, or mixed Leukemia cells in an individual patient.
Leukemias that co-express features of both lymphoid and myeloid development are generally believed to arise from a relatively less committed stem cell. Such a concept is supported by the finding that the less differentiated myeloid leukemias (e.g., MO AML) have a higher frequency of expression of markers traditionally associated with lymphoid differentiation. TdT, an enzyme found in primitive lymphoid precursors and thymocytes, may be expressed in 20 to 40% of blasts from those with FAB MO AML.53,54
The significance of expression of TdT or other lymphoid markers in those with typical peroxidase positive AML is unclear. Although up to 25% of AML patients may express TdT,134,135 there are conflicting data about whether such a feature confers an adverse prognosis. The importance of T-cell antigen expression or T-cell gene rearrangement in children with AML may136 or may not137 correlate with a higher level of resistance to regimens designed for AML patients.136 In adults with AML treated on CALGB clinical trials, lymphoid antigen expression had no effect on outcome.138 The significance of a T-cell receptor rearrangement, a finding not uncommon in the AML patient,139 is even less clear. In fact, CD7 expression is not uncommon in t(8;21) AML which clearly responds well to chemotherapy.140–142 Patients, generally infants, with a t(4; 11) abnormality frequently display biphenotypia.143
Although the phenotypic features of the leukemic blast are generally constant throughout the patient’s course (e.g., at diagnosis and relapse), examples of lineage switch, usually AML followed by ALL, have been noted.144,145 The optimal therapeutic approach to these hybrid leukemias remains unclear. For patients whose blasts are clearly peroxidase positive and/or nonspecific esterase positive, who express mainly myeloid antigens but who display TdT positivity or express one lymphoid antigen, an AML regimen is appropriate. For those ALL patients whose blasts express one or two myeloid antigens, enrolment onto an ALL protocol is reasonable.55,146,147 For patients who truly have equal myeloid and lymphoid features, an obvious initial approach would be to combine AML and ALL regimens, but there are few data concerning the outcome of this strategy in such patients.
Presenting Signs and Symptoms
Patients with AML generally present initially with symptoms related to complications of pancytopenia including weakness, easy fatigability, infections of variable severity or hemorrhagic findings such as gingival bleeding, ecchymoses, epistaxis, or menorrhagia (Table 124.2). Combinations of these symptoms are common. Occasional patients present because of prominent extramedullary sites of leukemia usually related to either cutaneous or gingival infiltration by leukemia cells. Bone pain is infrequent in adults with AML, although some individuals describe sternal discomfort or tenderness, occasionally with aching in the long bones, particularly of the lower extremities. It is generally difficult to date the onset of AML precisely, at least in part because individuals have different symptomatic thresholds for choosing to seek medical attention. It is likely that most patients have had more subtle evidence of leukemia for weeks, to perhaps months, before diagnosis. This can sometimes make the distinction between de novo leukemia and leukemia associated with prior hematologic disorders arbitrary.
Table 124.2
Initial Diagnostic Evaluation.
The findings on physical examination are variable and generally nonspecific. If fever is present, it is almost always related to infection, and an infectious site must be vigorously sought and generally treated empirically with broad-spectrum antibiotics. A small minority of patients have fever related solely to the underlying leukemia, which abates with appropriate chemotherapy; there is a suggestion that this may be more common in patients with promyelocytic leukemia.93 Examination of the skin can reveal pallor; infiltrative lesions suggestive of leukemic involvement; cutaneous sites of infection, which may be either primary or embolic; or, most commonly, petechiae or ecchymoses related to thrombocytopenia and/or coagulopathy. Examination of the fundus reveals hemorrhages and/or exudates in the majority of patients (see “Ophthalmic Complications” below). The conjunctivae may be pale, according to the magnitude of the anemia. Careful examination of the oropharynx and teeth is important because of the occasional occurrence of leukemia involvement and the value of applying effective dental prophylaxis, if time permits, prior to chemotherapy.148 Palpable adenopathy is uncommon in patients with AML, and significant lymph node enlargement is rare. Similarly, hepatomegaly and splenomegaly are uncommon and, if found, may suggest the possibility of ALL or chronic myeloid leukemia in blast crisis. None of these findings is diagnostic of acute leukemia, and the final diagnosis and categorization depends on appropriate evaluation of peripheral blood and bone marrow.
Because of the rigorous nature of the chemotherapy required for the successful treatment of AML, particular attention should be paid in the history and physical examination to other medical problems that could complicate the patient’s management. A history of congestive heart failure or other heart disease mandates careful monitoring of the large amounts of intravenous fluids that accompany the initial chemotherapy, antibiotics, blood and platelet transfusions, hydration for amphotericin B, and parenteral nutrition during the 3 to 4 weeks of chemotherapeutically induced pancytopenia. Prior transfusion for other disorders or multiple previous pregnancies may presage difficulties with adequate platelet transfusion support or herald the occurrence of transfusion reactions after red blood cell or platelet administration. Careful appraisal for possible drug allergies is critical, since virtually every patient will require antibiotic therapy. A history of prior herpes simplex infections (or the presence of an elevated antibody titer) provides justification for prophylactic administration of acyclovir.149 Menses should be suppressed with estrogens and/or progestational compounds until thrombocytopenia is resolved in premenopausal women. Gonadotropin-releasing hormone antagonists may have a role in menstrual suppression, but the pituitary ovarian axis is not inhibited until one menstrual cycle has passed after the administration of such a drug.150
After the diagnosis is established, the physician and staff must present the goals of therapy and the side effects of treatment to the patient and family. For almost all patients, this discussion can rightfully emphasize the potential benefits of treatment with regard to both the short- and long-term outcome. It is frequently appropriate and necessary to repeat this discussion and counsel later during the patient’s course. Occasionally, intensive treatment with the intent to achieve CR may be less advisable because of advanced patient age, debility, or prior myelodysplasia. Particularly with de novo leukemia, this represents the exception, and the administration of chemotherapy with the intent to achieve CR affords the greatest potential for even short-term benefit for most patients.
Therapy
The therapy of AML has traditionally been divided into stages: induction, postremission therapy of varying intensity and duration, and postrelapse therapy. In newly diagnosed patients with AML, the goal of induction therapy is to achieve what has been termed complete remission (CR), which then permits the administration of subsequent therapy that, in most treatment protocols, is designed to maximize the rate of disease-free survival and cure. Currently, CR is defined primarily on morphologic grounds and includes the development of a morphologically normal bone marrow containing less than 5% blast elements, absence of any signs of extramedullary leukemia, and return of normal neutrophil (> 1,500/μl) and platelet (> 150,000/μl) counts. However, even at the time of CR, and after an estimated 3-log cytoreduction in the leukemic burden, up to approximately 109 residual leukemia cells persist and must be eradicated by subsequent postremission therapy to achieve any chance for prolonged disease-free survival. Even if the bone marrow contains < 5% blasts, patients are not considered to be in remission if distinctive morphologic signs of leukemia, such as Auer rods, are noted. Low hemoglobin levels and the presence of symptoms unrelated to leukemia no longer exclude CR since they are often treatment-related and slow to normalize.1
It has usually been assumed that CR is accomplished because the cytotoxic chemotherapy markedly decreases the number of cells in the leukemic clone, thereby allowing repopulation of the bone marrow by residual, normal progenitors whose proliferation had been suppressed. This explanation is supported by observations that cytogenetic abnormalities present in the original leukemia cells are either not detectable or constitute a small minority of metaphases during remission. However, in some fraction of patients, intensive chemotherapy eliminates the block in differentiation such that the apparently normal cells seen in the bone marrow and peripheral blood during a CR are actually progeny of the leukemic clone.151–153 Studies of female patients in CR using X-chromosome-linked polymorphisms (glucose-6-phosphate dehydrogenase isoenzymes or restriction fragment-length polymorphisms) have suggested that as many as 20% of adult patients in CR have normal hematopoiesis with a monocloncal proliferation of the same genotype as the leukemic clone.151) The frequency of this “clonal CR” (which may be lower in children with AML), its possible association with particular subtypes of AML or remission duration, the mechanism by which this important biologic phenomenon occurs, and a number of important technical issues need to be defined further.154,155
Induction Therapy
General Principles
Induction therapy is designed to produce rapid clearing of leukemic cells from the peripheral blood with subsequent marrow aplasia. Perhaps the only exception to this principle occurs in patients with acute progranulocytic leukemia (FAB M3), in whom, as described earlier, remission can be achieved despite the persistence 2 to 3 weeks later of what appear morphologically to be viable leukemia cells.93,94,156 Approximately 1 week after standard induction therapy is completed (generally 2 weeks after the initiation of treatment), bone marrow aspirates and biopsies are done to evaluate the magnitude of cytoreduction. If indeed the marrow is profoundly hypoplastic, the marrow is repeated at approximately weekly intervals to assess whether there is return of normal hematopoiesis, leukemia cells, or an apparent mixture of both. If the marrow is not hypoplastic and only leukemia cells are noted on the day 14 marrow, then a second course of therapy is generally administered. At times, particularly if the marrow is hypocellular, it can be difficult to distinguish morphologically between residual leukemia cells and normal undifferentiated hematopoietic progenitors. In this instance, it is advisable to delay a few days and perform another marrow aspirate. If there is no evidence of further maturation, then the second course of treatment is indicated. The presence of erythroid precursors, juvenile megakaryocytes, or increases in the peripheral blood platelet or neutrophil counts serve as clues that normal regeneration is occurring and that a second course of treatment should be delayed. With standard regimens, approximately 30% of patients with AML receive two courses of treatment to enter remisssion. It should be noted, however, that despite these guidelines, there remains considerable variability among clinicians in terms of the decision as to when a second course of therapy is indicated. Some clinicians also have advocated performing bone marrow aspirates and biopsies on the day after chemotherapy is completed because of a suggestion that evidence of residual leukemia at this time may be a signal of inadequate cytoreduction and a decreased probability of CR.157,158 Although it has been advocated that further therapy be administered immediately to such patients, there is no conclusive evidence that this approach increases the rate of CR or improves survival; such bone marrows that have been so recently exposed to intensive chemotherapy are also often difficult to interpret.
Patients can fail to achieve remission for a number of reasons. Until recently, most papers reported results as complete response or no response. It is, however, more helpful in investigations of prognostic factors or assessment of the cytotoxic activity of different regimens to classify more rigorously the causes of failure to achieve remission. One such classification divides nonresponders into those with apparent chemotherapy-resistant leukemia, those who die with aplastic bone marrows in whom the response to chemotherapy cannot be determined, and those in whom either early death or failure to obtain adequate bone marrow studies prior to death preclude determination of whether persistent leukemia was present.159 Patients with drug-resistant leukemia include those patients who survive treatment and those who die but have morphologic evidence of leukemia in the bone marrow or blood antemortem or at autopsy. This categorization is relatively simple to perform in most patients and can be helpful in distinguishing between failure due to ineffective chemotherapy and failure related to inadequacies of supportive care.
The overall rate of CR in large cooperative group studies is approximately 65%. This CR rate is related to a number of prognostic factors, many of which are discussed below. Patient age is the most critical clinical variable. Comparisons among studies can be difficult because of imbalances in prognostic factors. In addition, many reports do not stipulate whether patients with secondary leukemias or prior myelodysplasia were included in the trial. Exclusion of such patients, who represent approximately 5 to 10% of adult patients diagnosed with AML, produces apparently improved results. It is preferable to present results in these patients and in patients with de novo leukemia separately.160 The achievement of CR is highly dependent on the ability to administer appropriate supportive care to prevent hemorrhagic and infectious deaths during the approximately 3 weeks of aplasia. The criteria for CR are generally reached a median of 30 to 35 days after treatment has begun. Perhaps more importantly, patients develop adequate levels of circulating neutrophils (> 500/μl) and no longer require platelet transfusion (at counts of approximately 10–20,000/μl), at least 7 to 10 days earlier.
Although there have been gradual overall improvements in the CR rates worldwide during the past 10 to 15 years, much of this can be attributed to more widespread availability of sophisticated supportive care and not to changes in therapy. With the exception of the use of ATRA for patients with APL (see below), relatively few changes in therapy have been made since the introduction of combined therapy with daunorubicin and ara-C, the so-called “7 and 3” regimen.161–163 This two-drug combination was derived from observations of single agent activity with either compound.164 Daunorubicin is generally administered by intravenous push at doses of 30 to 50 mg/m2/d for 3 days; ara-C is now generally administered at doses of 100 to 200 mg/m2/d by continuous infusion for 7 days (Table 124.3). A series of randomized studies159,162,165–168 by the Cancer and Leukemia Group B (CALGB) demonstrated that (a) results were superior using the 7 and 3 regimen compared with 5 days of ara-C and two doses of daunorubicin; (b) the addition of oral 6-thioguanine (DAT regimen) to the 7 and 3 program did not increase CR rate; (c) continuous infusions of ara-C produced slightly better outcome than twice-daily short intravenous infusions of ara-C when combined with daunorubicin; (d) results were not improved when 10 days of ara-C by continuous infusion were administered compared with 7 days; (e) substitution of Adriamycin for daunorubicin produced almost identical CR rates, although mucosal toxicity was greater with Adriamycin; and (f) there was no overall benefit from doubling the dose of ara-C from 100 mg/m2 to 200 mg/m2/d.
Table 124.3
Representative Regimens for AML.
Other attempts to improve the 7 and 3 induction regimen have also been relatively disappointing. Although there was a suggestion that older patients more frequently achieved CR with a single course of treatment using mitoxantrone in place of daunorubicin, this did not result in enhanced overall patient survival. Alternative anthracyclines or other agents such as rubidizone, aclacinomycin, amsacrine, mitoxantrone, and idarubicin have been used in several trials.5,169–172 The largest reported experience has been with idarubicin where three trials have demonstrated that induction results are at least equivalent to results achieved with daunorubicin and ara-C.169,171,172 None of these studies demonstrated a survival or disease-free survival advantage with these different agents, perhaps because most are relatively similar in structure and mechanism of action and hence become susceptible to the same mechanisms of resistance. Moreover, the duration of myelosuppression was greater in the idarubicin cohorts, calling into question the equitoxicity of the arms. Therefore, it is unlikely that the introduction of these compounds will have a major impact on the long-term disease-free survival rate of patients with AML.
Another recent trial from Australia added etoposide (VP-16) to the 7 and 3 regimen.173 There were similar complete response rates in older patients when compared with daunorubicin and ara-C alone and a possible slight increase in CR rate and modest prolongation of CR duration in younger patients receiving the etoposide. This was a relatively small study and requires confirmation, since the overall disease-free survival in the group receiving the etoposide during both remission induction and postremission therapy was similar to other larger studies using daunorubicin and ara-C alone. Insofar as HIDAC is a beneficial postremission therapy (see below), several groups have tested the concept of using this approach during induction. Studies by the Southwest Oncology Group,174 the Australia Leukemia Group,175 and UCLA176 compared standard 7 and 3 to daunorubicin plus intermediate- or high-dose ara-C (2 g/m2 for 8–12 doses). These studies failed to show an improved CR rate for the recipients of higher-dose ara-C, although the Australian study documented a more prolonged duration of CR (but no change in overall survival) in the patients randomized to HIDAC. The addition of HIDAC to standard daunorubicin/ara-C during induction has also been studied. Although a limited access trial demonstrated an 87% remission rate in patients under 60 years old,177 a cooperative group trial,178 failed to confirm those positive results. Topotecan-based regimens are now being evaluated179; unfortunately, there are few convincing data to suggest that new drugs or alternative induction regimens are likely to make a significant difference in either the CR or cure rates.
The reasons for treatment failure vary according to patient age. With the availability of sophisticated supportive care, it is relatively uncommon for patients less than 50 years of age to die from complications of treatment; most of the approximately 25% induction failure rate is a consequence of drug-resistant leukemia, and most patients survive initial therapy. In contrast, the CR rate in patients >60 years of age is approximately 50%,180 with failures divided equally between drug-resistant leukemia and deaths occurring during marrow aplasia as a consequence of reduced end organ tolerance. Thus, whereas younger patients with poor prognostic factors would benefit from entirely new approaches to achieve CR, including the possible use of new chemotherapeutic agents, older individuals would also benefit from methods by which the period of aplasia can be shortened.
Some observers debate whether older patients or patients with secondary leukemias or prior myelodysplasia benefit from intensive induction approaches at all. In particular, there has been interest in using very low-dose ara-C (10–20 mg/m2/d) in these patients to avoid the acute side effects associated with daunorubicin and even conventional-dose ara-C. Although responses can be seen with low-dose ara-C, many weeks of therapy can be required with consequent pancytopenic complications equivalent to those seen with higher-dose treatment.181 In addition, the CR rate is lower, and although median survivals may be similar using the two different dosage approaches, the prospect for long-term disease-free survival is greater, when, the intent of treatment is to achieve CR.182 Another study documented a survival benefit for the prompt initiation of induction therapy at the time of diagnosis compared with waiting and administering chemotherapy only on development of symptoms.183 However, the degree of benefit was small. To answer the question of whether a 1- to 2-month aggregate survival advantage justifies the side effects associated with induction therapy, a quality of life component in those types of trials is critical.
CRs can be achieved in approximately 20 to 30% of patients whose leukemia followed treatment for another cancer. Responses may be better in patients who do not have an extended pancytopenic or myelodysplastic period prior to developing frank leukemia. Although responses tend to be short in such patients, intensive induction should be offered, particularly to younger patients in whom subsequent bone marrow transplantation (BMT) may be an option.160,183,184 Some younger patients with apparent secondary leukemia have been found to have cytogenetic findings associated with de novo AML of good prognosis such as t(8;21) or t(15;17) and may have a more favorable clinical course than expected.185,186 These patients should be treated in the same fashion as those with de novo disease. It can also sometimes be difficult to distinguish morphologically between FAB M2 with t(8;21) and patients whose marrows contain < 30% blasts who would be classified as RAEB-T according to strict FAB criteria. Cytogenetic studies should therefore be done in all such patients.
Whether to treat patients with well-documented prior MDS and leukemia evolving from RAEB or RAEB-T can be more problematic. Complete response rates are lower, sustained responses are unusual, and prolonged periods of aplasia are common. In addition, these patients are usually older and less able to tolerate pancytopenic complications. However, data from M.D. Anderson Cancer Center suggest that the absolute number of marrow blasts (e.g., whether a patient has RAEB-T or AML) is not of prognostic significance.187 Instead, cytogenetic status and a truly prolonged history of MDS are critical factors. It is possible, however, to define patients with AML who have such poor prognostic factors that enrollment on phase I/II studies using new agents would be appropriate. If a histocompatible donor is available, allogeneic BMT should be considered for younger patients.188–190
Postremission Therapy
Although CR is defined as the absence of morphologically detectable leukemia on bone marrow aspirate, morphologic assessments are subjective and relatively insensitive. It is estimated that as many as 109 leukemia cells may still be present in patients in apparent CR. Rare patients can remain in CR for 1 to 2 years without further treatment, but it has been generally accepted that some form of therapy after CR is required to achieve long-term disease-free survival. Data to support this contention are based on two randomized trials conducted well over a decade ago. In a trial conducted by the German Cooperative Leukemia Group, a subset of 37 patients did not receive postremission therapy for a variety of protocol and medical reasons; all of these individuals relapsed.191 The Eastern Cooperative Oncology Groups (ECOG) reported a randomized study in which patients achieving CR were randomized to either no therapy, lower-dose maintenance therapy, or an intensive postremission program.192 All of the patients in the no-treatment arm relapsed rapidly, with a median CR duration of 4 months, resulting in early termination of this arm of the trial. Similar results were noted in a smaller randomized study reported by Embury and colleagues.193 Although timed sequential therapy in which patients receive additional chemotherapy during the early postinduction recovery phase has been associated with prolonged remissions in the absence of postremission chemotherapy,194 it remains standard practice to administer chemotherapy after remission is achieved.
A variety of different approaches have been evaluated as postremission therapy. Considerable uncertainty and controversy remain about which approach is preferable. These different approaches can be generally categorized according to the intensity of the therapy administered. The terms consolidation and intensification have generally been used to describe therapies that are of equal and greater intensity, respectively, than the regimens used in initial induction therapy.195 It should be noted, however, that these phrases have been used rather loosely in the literature, and readers are advised to assess carefully the doses stipulated in various regimens, the doses actually delivered, and the periods of aplasia experienced by the recipients. The term maintenance has generally referred to lower-dose therapy administered on an intermittent basis for months to years, often on an outpatient schedule, and frequently at doses that do not produce significant myelosuppression. Again, however, the ability to deliver such regimens as scheduled varies considerably. In addition, there have been combinations of consolidation and maintenance approaches, including the use of late intensification treatment for patients who have already had sustained remissions.196 A wide variety of antineoplastic agents have been used including the agents successfully administered in initial induction, sometimes in different doses or schedules, as well as different classes of compounds, some of which have proven activity in AML, others of which may have had only limited activity.
From a historical perspective, earlier postremission approaches tended to focus on maintenance-type therapy administered on a monthly or a bimonthly basis for up to 2 to 3 years.197–199 This is largely derived from the model of maintenance treatment successfully used in childhood ALL. In more recent years, in part based on some of the positive results reported following BMT,200,201 the emphasis has shifted toward the use of intensive postremission consolidation with a particular focus on HIDAC.202–208 There has been considerable variation in the doses and numbers of courses of HIDAC administered, and newer programs are being initiated assessing high-dose therapy with agents shown to be active in phase II trials but that have not previously been used extensively in nonrelapsed patients. Lastly, both allogeneic and autologous BMT have been advocated for selected patients in first remission.200
Although many nonrandomized studies purport to demonstrate better results, using different postremission approaches, these are invariably small studies with potentially selected, generally younger patient populations. Since a variety of prognostic factors significantly affect ultimate outcome, independent of the type of therapy administered, small studies that may reflect inadvertent patient selection, single or oligoinstitutional superiority, or even random luck must ultimately be confirmed by multi-institutional applicability.
Overall, in adults, it can be expected that the administration of some sort of postremission therapy will result in a median CR duration of 12 to 18 months with approximately 20 to 25% of complete responders remaining as long-term disease-free survivors. A review of approximately 1,700 CALGB patients who achieved CR demonstrated a relatively constant relapse rate of 4.7% per month during the first 6 months following CR.209 The overall failure rate decreased in subsequent 6-month intervals (3.5% per month in months 7–12 and 2.4% per month in months 13–18), with a further flattening of the curves after this point. It is likely that patients relapsing earlier have leukemia that is more drug-resistant than those who relapse late.160 Many clinical/laboratory correlative studies that evaluate prognostic factors and mechanisms of drug resistance now combine individuals with very short CR durations together with individuals with primarily resistant leukemia because of probable similarities in the underlying biology of their disease.
Earlier randomized trials of postremission therapy conducted by the CALGB162,165,166,199 have failed to demonstrate long-term benefits from (a) an alternate-month compared with a monthly schedule of maintenance therapy; (b) 3 years of relatively low-dose maintenance therapy compared with 8 months of a similar program (indeed, there was a modest survival advantage for patients randomized to stop therapy after 8 months); (c) doubling of the dose of ara-C from 100 to 200 mg/m2 during maintenance therapy; and (d) addition of nonspecific immunotherapy in the form of methanol-extractable residue of BCG (MER) to maintenance therapy. An earlier ECOG study demonstrated no benefit from the addition of 2 years of maintenance therapy following two courses of postremission treatment with DAT.203 In contrast, results from Germany reported by Buchner and colleagues191 suggest a modest prolongation of CR duration, although with a questionable effect on long-term survival, when long-term maintenance therapy was administered after a single course of DAT. It is unclear whether the same results would have been achieved if more intensive postremission consolidation had been used before the maintenance.
Two important randomized trials have shown that HIDAC in the post-remission setting is better than lower doses of the drug. In an ECOG study, patients randomized to receive one course of very intensive postremission consolidation with a HIDAC-type regimen had a longer median duration of remission than patients receiving 2 years of lower-dose maintenance therapy.210 A CALGB study provides additional evidence of the value of more dose-intensive postremission treatment.211 Five hundred ninety-six AML patients achieving CR were randomized to receive four courses of ara-C administered at three different dose levels (100 mg/m2 by continuous IV infusion [CIV] for 5 days, 400 mg/m2 CIV for 5 days, 3 g/m2 IV for 3 hours q12h on days 1, 3, and 5 [total 6 doses/course]). There was no benefit from the higher-dose arms in patients > 60 years of age (see Fig. 124.1, bottom) with a median duration of CR of approximately 13 months and with only 10 to 12% long-term DFS. In addition, there was a substantial incidence of CNS neurotoxicity in the older patients manifested primarily as cerebellar dysfunction. In contrast, patients younger than 60 years of age benefited substantially from the HIDAC regimen in terms of both relapse-free and overall survival (see Fig. 124.1, top). The long-term results in patients < 40 years of age were similar to those reported with autologous or allogeneic BMT.
The CALGB and ECOG studies strongly support the use of HIDAC-based consolidation programs in younger patients with AML. It is not known how many courses of such therapy are needed, how to most safely and effectively administer HIDAC, and whether the addition of other active agents will improve on these results. Analysis of the CALGB data suggests that the benefit from HIDAC was most pronounced in patients with favorable cytogenetic findings [t(8;21); and inv16] with much less effect in patients with unfavorable karyotypes typically associated with drug resistance.212 When medically feasible, most clinicians attempt to administer at least two to three courses of reasonably intensive HIDAC-based therapy postremission regimens. Programs that use HIDAC in the range of 2 to 3 g/m2 for 8 to 12 doses often are only able to administer one to two courses of such treatment. Obviously, the decision about the type of postremission therapy must take into consideration the patient’s medical condition and the possible persistence of infection (particularly with fungal organisms) acquired during induction, as well as the ability to provide adequate platelet transfusion therapy, in an attempt to balance the risk of intensive postremission approaches with the potential benefit. Depending on the intensity of the consolidation program, a 5% mortality rate in CR is to be expected and must be carefully explained to the patient (see Table 124.3).
In addition, a number of clinical trials suggest that between 5 and 15% of patients achieving CR do not go on to receive intensive postremission programs because of physicians’ concerns about the patient’s ability to tolerate and survive such therapy. Such patients should receive some sort of lower-dose treatment if possible, and we frequently use ara-C (100–200 mg/m2/d by continuous infusion for 5 days or sometimes on a b.i.d. subcutaneous schedule), with or without 1 to 2 doses of an anthracycline for 2 to 4 courses, depending on patient tolerance. It is difficult to recommend a particular postremission approach in older patients not participating in clinical trials. Intensive chemotherapy has not proved beneficial compared with standard doses211,213; some form of therapy, even low-dose ara-C,214 is required, however, to yield any chance at long-term disease-free survival. Since there is no proven role for HIDAC-based treatment in the older adult in remission, we generally use lower-dose regimens as described above. In view of the lack of proven benefit and the expense and morbidity associated with long-term maintenance therapy, prolonged treatment cannot be recommended.
Bone Marrow Transplantation
BMT represents the other major therapeutic option available as postremission therapy. Syngeneic BMT, using identical twins as donors, and allogeneic BMT, using HLA identical siblings, were first evaluated in patients with advanced, refractory AML.215,216 After demonstrating an approximately 10% rate of long-term disease-free survival in this refractory group of patients and a 40 to 50% disease-free survival in patients in first remission receiving syngeneic transplants, studies of allogeneic transplantation in first remission AML were begun.215,217 Early small series were promising, demonstrating a low rate of relapse with most of the approximately 50% mortality being related to complications of acute and chronic graft-versus-host disease (GVHD). Following publication of these initial results, the suggestion was made that all suitable patients with available donors should be considered for allogeneic transplant as the treatment of choice. For years, contentious debate ensued and still continues, despite several large prospective trials designed to address the role of allogeneic as well as the appropriateness of autologous BMT relative to chemotherapy in the postremission setting.218–221
At least in part because of difficulties in identifying suitable donors for many patients as well as the complications of GVHD. considerable attention has been focused on the use of autologous BMT. It has been demonstrated that administration of cryopreserved autologous bone marrow can rapidly reconstitute heavily treated recipients, although marrow recovery, and particularly recovery of platelet count, may be delayed compared with allogeneic BMT recipients.222 Recent experience using cytokine-mobilized peripheral blood stem cells (PBSC) suggests that the duration of neutropenia and particularly thrombocytopenia can be shortened further compared with the use of bone marrow.223 Because of the absence of GVHD, the procedure is much better tolerated and can be readily applied to patients up to 65 years of age and perhaps older. The major theoretical disadvantages of this approach include the absence of a graft-versus-leukemia effect, as well as concern that viable leukemic progenitors will be administered with the autologous stem cells. A number of purging techniques have been used including incubation with monoclonal antibodies directed against myeloid blasts,224,225 as well as incubation with very high concentrations of cytotoxic agents that rather remarkably spare hematopoietic progenitors, although delayed marrow reconstitution is not uncommon.222 It is unknown whether these in vitro manipulations are necessary, and randomized trials have not been reported. When autologous transplantation is performed early in CR, many centers use some type of purging technique. In studies in which autologous BMT was harvested after many months of CR, or after the administration of high-dose consolidation therapy, retrospective comparisons do not suggest an increased rate of relapse after the unpurged transplant, perhaps as a consequence of the therapy that had been administered during these intervening months.220,226,227
A number of centers and groups have reported relapse-free survival rates in excess of 40% in recipients of autologous BMT in first CR.222,224,227–230 Historically, syngeneic transplant experience in this situation demonstrated relapse rates of approximately 50%, which is probably the best that can be expected with the autologous approach. Perhaps the strongest evidence of the potential of this approach consists of reports of apparent cure rates of 20% in patients transplanted in second and third remissions of AML.222 The potential criticisms with regard to patient selection noted above are also applicable to most of the autologous BMT studies.
The controversy about whether a patient in first remission should receive chemotherapy, an autologous BMT, or allogeneic BMT has prompted prospective comparative trials dating back to the early 1980s. Several of the initial trials,231–233 which compared the outcome in patients assigned to allogeneic BMT on the basis of having a histocompatible sibling to those receiving chemotherapy alone, documented a benefit for the transplant. However, the situation in recent times is more complex. Four recently available prospective trials each compared results in first remission patients genetically assigned to allogeneic BMT (usually only in those 45 years old or less) to autologous BMT versus chemotherapy. The latter comparison was based on randomized assignment. In the first of these trials, conducted in Europe,218 422 patients < 45 years of age (median 33 years) were either allocated to allogeneic BMT if an HLA-identical sibling was available or randomized to autologous BMT or a second course of consolidation chemotherapy with intermediate-dose ara-C and m-amsacrine. Disease-free survival was similar in patients undergoing allogeneic BMT (55% projected at 4 years) and autologous BMT (48%) and superior to the chemotherapy group (30%). Overall survival was similar, however, because many patients relapsing after chemotherapy could be successfully reinduced and undergo BMT in second CR. Many patients did not undergo treatment as randomized, and no information was provided about responses to the three treatments in different cytogenetic or FAB risk groups.
Another similarly designed trial performed in Europe by a French group failed to show a benefit for either type of BMT compared to patients receiving intensive postremission chemotherapy.219 The MRC 10 trial conducted in Great Britain220 appeared to show that autologous BMT was beneficial. All patients not assigned to allogeneic BMT received three cycles of post-CR chemotherapy and were then randomized to autologous BMT (nonpurged) or observation. However, there was no significant difference in overall survival among the two groups. Allogeneic BMT was not significantly better than autologous BMT; in no cytogenetic risk group was one modality clearly better than another. The most recently published trial conducted by the North American Intergroup221 actually found that chemotherapy was at least as good as autologous or allogeneic BMT in leading to cure, at least based on the intent-to-treat analysis. Many patients assigned to autologous BMT did not receive this therapy for a variety of reasons. Although the study was not powered to do so, an attempt was made to review the result according to cytogenetic status at diagnosis.234 In this preliminary analysis, both the favorable and unfavorable risk groups benefited from BMT.
Based on the available evidence, no firm conclusion can be drawn. However, a few general points can be made concerning the role of allogeneic BMT in the management of patients with AML in first CR. (a) The applicability of the technique is severely limited by patient age. Mortality from GVHD increases in a step-wise fashion from decade to decade, and most centers do not perform allogeneic transplantation in patients older than 60 years, thereby excluding most patients with AML. Whether so-called mini-allogeneic transplants (nonmyeloablative conditioning regimens)235 will change this is unclear. (b) Less than one-third of potential recipients have suitable HLA-matched family donors. Although alternatives include the use of partially mismatched donors or matched unrelated donors,236–238 there is a considerable increase in the incidence of severe GVHD with both approaches and appreciable administrative delays in rapidly identifying suitable donors despite the available thousands of HLA-typed donors throughout the world. This is particularly true for patients with less common HLA types.239 (c) Although the relapse rate is lower, and allogeneic BMT can cure some patients with chemotherapy-resistant disease, there remains an appreciable relapse rate post-BMT, with at least some of the factors predictive of drug resistance and relapse after chemotherapy also applying to BMT recipients.240,241 (d) A substantial component of the antileukemic effect following BMT correlates with the occurrence and severity of GVHD, presumably as a result of a graft-versus-leukemia effect. Attempts to attenuate GVHD with a variety of immunosuppressive approaches (T-cell depletion of the infused marrow, cyclosporin) are associated with a decrease in GVHD but also an increase in the relapse rate,242,243 although selective T-cell depletion approaches hold promise in making BMT safer without an increased relapse risk.244,245 (e) Although important advances in supportive care have been made, the consequences of the immunosuppression and direct organ sequelae of GVHD still represent formidable barriers, particularly with regard to infections with cytomegalovirus and interstitial pneumonia. (f) As many as 10 to 20% of surviving patients may have significant symptoms and impairment of performance status because of chronic GVHD. There is also a small increase in the development of secondary tumors in long-term survivors.246
It is hoped that additional ongoing studies, perhaps permitting (or eventually requiring) meta-analysis of results, will clarify many of these issues, particularly with regard to optimal treatments for different risk subgroups of patients. It would appear, however, that all approaches are in need of considerable improvement. Although the causes of failure are different (high rate of relapse with chemotherapy, lower rate of relapse but high mortality due to GVHD for BMT), historic results suggest that the outcomes may be similar with the inevitable conclusion that improved cytoreductive approaches are needed in both arenas. The greater the improvement in chemotherapy, the less will be the need for transplantation.
Therapy of Relapsed and Refractory AML
There are a number of agents available that have clear-cut activity when used alone or in combination in patients with relapsed or refractory AML, including amsacrine, mitoxantrone, diaziquone, idarubicin, fludarabine, 2-clorodeoxyadenosine, etoposide, homoharringtonine, topotecan, and carboplatin.247–258 The pros and cons of these and other individual drugs or therapeutic regimens have been reviewed.251,259,260 There is considerable heterogeneity among this group of patients, and a number of issues, in addition to the specific drugs used, influence the outcome of treatment. Most phase I/II trials enlisting new agents contain relatively small numbers of patients and frequently do not adequately describe their clinical characteristics. Some consistent trends are found in most such studies; however, (a) response rates are uniformly very low in patients with primarily refractory leukemia; (b) patients with short initial CR durations or who have relapsed while receiving postremission chemotherapy also tend to fare poorly with reinduction therapy; (c) secondary leukemias, or leukemias that evolved from a prior hematologic disorder, are particularly resistant to further therapy; and (d) patients in second and subsequent relapse tend to have a poorer prognosis than patients in first relapse, and the durations of subsequent remissions tend to decrease progressively.
Results from the Medical Research Council (MRC) in Britain, which prospectively followed all patients entered on an induction AML trial, are supportive of these conclusions.260 Overall, 155 of 531 relapsed patients (29%) achieved second CRs. The response rate with a variety of different reinduction regimens was directly related to the duration of the first remission (Table 124.4) and patient age, emphasizing the enormous influence of patient selection on the results of phase II trials. In the absence of comparative trials, differing results are more likely the consequence of patient selection than the superiority of a particular regimen. Others249,261 have also reported results essentially identical to the MRC. In one study, patients whose initial remission was greater than 18 months had a reinduction rate of 64% (37/58) compared with a 29% CR rate in 278 patients with shorter initial remissions. The duration of second CR was also longer in the former group (8 months vs. 3 months), with three patients remaining in remission for more than 4 years. In addition to the duration of first CR, favorable karyotypes [inv16; t(8;21), or t(15;17)] were more common in patients with longer first remissions. Such patients tend to be younger, perhaps accounting for the improved results in younger patients noted by the MRC.
Table 124.4
Treatment of AML in Relapse*.
The type and timing of therapy for relapsed patients should therefore be individualized. Patients with other medical problems who have had poor responses to initial therapy have a small likelihood of sustained benefit from reinduction therapy and, indeed, may have life shortened by intensive therapy. Some such patients can be supported for many months with maintenance of a reasonable quality of life, with a more conservative approach using red blood cell and platelet transfusions and oral hydroxyurea to control elevated white blood cell counts or symptoms such as bone pain. However, there is no potential for long-term benefit. Conversely, patients with longer initial responses may derive prolonged benefit from intensive reinduction therapy.
Recurrence of leukemia can be detected in a variety of clinical circumstances. It is common to identify relapse at a time when the patient is asymptomatic, blood counts are normal, and there is modest marrow infiltration by blasts. Although it is logical to begin reinduction therapy at the earliest sign of relapse, when tumor burden is presumably the lowest, there are no data to demonstrate the validity of this approach except possibly for allogeneic BMT.262 The drawback to early treatment is that reinduction therapy is often unsuccessful and may result in excessive early morbidity and premature patient death. Our general policy is to observe such patients until there has been a pattern of deterioration in blood counts but to initiate therapy while adequate numbers of circulating granulocytes and platelets are still present, prior to the development of symptoms or infection. Some patients have relapse diagnosed when bone marrows are done because of falls in blood counts. Such patients usually require reinduction therapy more immediately. In occasional patients, relapse develops in an explosive fashion with high circulating blast counts, fever, and infection. Although there are few prospective data available, it is likely that the outcome in this group of patients is poorer.
There are essentially no comparative trials to provide therapeutic guidelines. The initial decision can generally be outlined as a choice between reuse of drugs that have previously been effective in a given patient, compared with the use of new drugs, some of which may be investigational. If a patient has relapsed while receiving chemotherapy, it makes little sense to use these same agents for reinduction. The oft-quoted exception to this guideline is the use of HIDAC (2–3 g/m2 q12h for 8–12 doses), even in patients who had recently received more conventional doses of this agent.205 Although CR can be achieved in this setting, the duration of CR appears to be short, and it is not clear that the HIDAC approach is of long-term value in such patients.
In contrast, in patients who have longer first remissions, there is logic to reusing initial treatment. There are no clear-cut guidelines as to what constitutes a satisfactory initial first remission. Many centers have arbitrarily felt that patients relapsing more than a year after completion of therapy should again receive the standard treatment. Although the CR rate in such patients is probably in excess of 50% when the original drugs are reused, it is not clear whether the same results could have been achieved in this favorable group using a variety of different tactics, including new investigational agents.
There is no proof of the benefit of postremission therapy in patients achieving second or third remissions. The obvious drawback of such an approach is that one is potentially costing patients time when they would be asymptomatic and out of hospital by the administration of therapy of variable toxicity but no proven long-term efficacy. At the moment, most reinduction studies do not specify that patients in second or later remission receive either maintenance or consolidation therapy.
Although some relapsed patients may have long-term benefit from chemotherapy, it is likely that BMT offers a greater chance of long-term survival than does chemotherapy alone. What is unclear is whether transplantation should be offered at the time of initial relapse or whether patients should be reinduced and transplanted when a second remission is achieved. Unfortunately, a large percentage of patients do not achieve second remission, and a significant fraction of patients develop medical problems as a consequence of reinduction therapy that preclude subsequent transplantation. Enthusiasm for transplantation in relapse derives largely from reports from Seattle,262 indicating that the results of allogeneic transplantation in early relapse were equivalent to or better than those achieved in patients transplanted in second or subsequent remission. It is often a practical problem both to identify patients during early relapse and to be able to refer such patients for transplantation rapidly; the waiting lists at most transplant centers can preclude this approach. Nonetheless, it would probably be best to consider allogeneic transplantation for patients in early relapse if a suitable donor is available and the patient is under the age of 55 to 60 years. Similarly, allogeneic BMT can produce long-term survival in some patients with primary refractory leukemia, and it is advisable to HLA-type patients and their families at diagnosis to allow for this possibility. It is more difficult to use nonrelated donors for these purposes because of delays caused by tissue typing, identification of donors, and marrow procurement.
Patients who achieve second remission probably have a greater chance of long-term survival if transplanted in second remission. Data using both allogeneic and autologous approaches are promising in this regard, although it should be kept in mind that all such reports include highly selected patient populations, most of whom have had second remissions lasting a few months prior to transplantation. These patients have thereby identified themselves as having more chemotherapy-sensitive disease. Some centers now obtain and cryopreserve bone marrow from patients in first CR for use in either early relapse or as consolidation in second CR. Although this is an expensive, administratively cumbersome approach, it merits study in investigational settings, particularly in high-risk patients, prior to its more widespread application.224 Lastly, some selected patients who relapse following BMT can derive benefit from a second BMT and, in general, can receive and tolerate further chemotherapy.263
New Approaches
Hematopoietic Growth Factors
Because the rate of myelosuppression-associated mortality is so high, particularly after induction therapy is administered to older adults with AML, the cloning of a number of hematopoietic growth factors (HGFs) held promise to aid in amelioration of such side effects. Granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF),264 and interleukin-3 have been evaluated extensively; the potentially thrombopoetic factors MGDF and IL-11 are also in clinical development. The use of these agents in AML lagged behind that in solid tumors because of the concern that pharmacologic administration would stimulate the endogenous HGF receptors on the blasts, leading to proliferation (a phenomenon well recognized in the laboratory265) and a poor clinical outcome. Although fears regarding this clinical problem have been unfounded, the HGFs have not lived up to their promise for other reasons.
The first two available studies suggested that G-CSF265 and GM-CSF266 were safe and possibly effective in patients with poor-risk AML. In a pioneering study conducted in Germany, GM-CSF was administered to patients with AML shortly after completion of standard induction chemotherapy. Clinically overt stimulation of leukemia cell growth occurred very infrequently, and duration of neutropenia was slightly shortened compared with historical controls.
The growth factors were therefore considered for use in two circumstances: as hematopoietic support (to shorten neutropenic duration and decrease infectious deaths) and as “priming” (to stimulate leukemic cells into S-phase, thereby making them more sensitive to chemotherapeutic agents such as ara-C). In older patients, complications related to prolonged neutropenia are the major causes of failure to achieve remission. Randomized studies have recently been completed in which older patients received an HGF or placebo beginning on the day following the completion of initial induction therapy with the goals of assessing the duration of severe myelosuppression and the influence on complete response rate.267–273 In addition, it was hoped that if leukemia proliferation was not a significant problem with growth factor given after cytoreductive therapy to patients with known leukemia, then the growth factor should also be usable with safety to attenuate neutropenia in patients in remission receiving intensive consolidation therapy.
Two large placebo-controlled randomized trials using different preparations of GM-CSF given after the completion of standard induction therapy in older patients with AML have been reported. Both trials noted a small decrease in the number of days of neutropenia. In a study of 121 patients reported by the ECOG, there was a significant reduction in infectious deaths and a trend toward a higher CR rate in the patients randomized to GM-CSF.267 There was no difference in the duration of neutropenia in the two groups following intensive consolidation therapy. A larger CALGB study evaluated 365 patients >60 years of age and failed to show a difference in the incidence of severe infection, deaths from infections, CR rate, duration of hospitalization, or survival.268 Other smaller studies suggest a modest shortening of the duration of severe neutropenia, but with variable or no effect on CR rate and infectious morbidity.272,273 One published randomized trial using G-CSF after induction therapy for older patients with AML showed an improved CR rate for the G-CSF recipients.271 Somewhat unexpectedly, the infectious mortality was similar in the two groups of patients, and the reason for the increased CR rate is unclear. There was no difference in overall survival. Other trials269,270 evaluating G-CSF as a supportive agent showed a consistent effect on decreasing the duration of neutropenia by 5 d. Although one of these studies documented a reduction in the duration of hospitalization, there was no effect on CR rate, DFS, or infectious deaths. In aggregate, it would appear that the benefits of growth factors administered after the completion of induction therapy are likely to be modest at best. In contrast, those of G-CSF following intensive consolidation therapy have produced more marked decreases in the duration of neutropenia, albeit without effects on CR duration or survival. Because of the potential for eliminating the need for hospitalization,269 the use of HGF can be recommended following consolidation therapy. Preliminary studies with pegylated thrombopoietin274,275 failed to demonstrate its ability to reduce the need for platelet transfusions during the induction treatment of AML.
Although there is in vitro evidence suggesting that the HGFs can increase the cytotoxic effects of ara-C, at least in part by increasing the fraction of cells in S-phase,276–280 the clinical results with the priming strategy have been disappointing. An early study from the M.D. Anderson group suggested that GM-CSF administered before and during chemotherapy produced lower response rates and survival than seen in a historical control group treated with chemotherapy alone.281 Studies in which patients have been randomized to receive GM-CSF or placebo before/during/after chemotherapy272,273 also generally fail to show an advantage for patients primed with growth factor before initial induction therapy. Some of these studies used HIDAC, whereas others evaluated more conventional continuous-infusion schedules. Two recently presented abstracts suggest that GM-CSF priming does not have a role in older patients with newly diagnosed AML282 or those with relapsed disease.283 Despite all of the relatively uninspiring results with HGFs, they are still widely used, especially during consolidation therapy,284 are safe, and may have a more important future role.
Immune Modulation
A growing body of evidence suggests that a critical component of the therapeutic benefit associated with allogeneic BMT is related to the graft-versus-leukemia effect.285 Although it is likely that this is a complex, multifactorial set of events, further studies may enable the rational application of newly cloned lymphokines to stimulate the appropriate cells in patients treated with chemotherapy. Circulating natural killer (NK) cells can be detected both after autologous and allogeneic transplantation but not chemotherapy, which can be cytotoxic to leukemia cell lines and autologous leukemia cells in vitro.286 The numbers of these killer cells can be expanded by the post-transplant administration of IL-2.287 IL-2 itself has also been shown to have antileukemic effects in patients with AML in relapse.288,289 Pilot trials have documented the feasibility of administering IL-2 in the postchemotherapy setting; randomized trials are now underway. Another important aspect of the immunotherapeutic approach to AML includes the administration of monoclonal antibodies directed against AML blasts coupled to cellular toxins.290 Radio-immunoconjugates are also being developed.291 However, the most promising agent for rapid development is CMA-676, a humanized anti-CD33 antibody linked to the calcheamycin toxin. Phase I and II trials with this agent in patients with relapsed AML have shown a significant (20%) CR rate without extramedullary toxicity.292,293 Vaccine-based approaches using dendritic cell fusions to present leukemia-associated agents,294 or transducing AML cells to express proteins capable of stimulation of immune response, are under development.295
Circumvention of Drug Resistance
The sum of complex and interacting mechanisms determine whether sufficient cytoreduction occurs in a given patient. The pharmacokinetics of antineoplastic agents vary widely among patients receiving the same calculated dose of drug, resulting in marked differences in drug exposure. Cell cycle kinetics also differ widely, potentially modulating the effectiveness of agents such as ara-C that are largely S-phase-specific.296 Similarly, there are potential differences in drug transport across leukemia cell membranes and variations among patients, and among individual leukemia cells within a given patient, in the cellular pharmacology of different antineoplastic agents. The ability of individual leukemia cells to repair sublethal damage is a critical variable as well. It is also likely that clinical drug resistance is conferred by properties present in a small fraction of leukemia cells, presumably that fraction capable of efficient self-renewal. Investigations of these phenomena have been hampered by difficulties in reliably and repeatedly cloning leukemic cells from individual patients to allow serial studies to be performed. In addition, a potential sampling bias exists in that the in vitro results are only a reflection of the characteristics of cells obtained at a single point in time that can grow in an artificial environment.
Despite these problems, there have been many important studies done in the area of leukemia drug resistance. In brief, these evaluations have focused on the pharmacology and metabolism of ara-C,297–299 with a more recent emphasis on studies of the multidrug resistance (MDR) phenotype, which is associated with simultaneous resistance to a number of different classes of anticancer drugs. The ara-C studies have focused on changes in ara-C dose and schedule as well as the administration of modifiers of nucleotide metabolism or cell cycle such as thymidine, l-asparaginase, and fludarabine300 as a means of increasing the amount and rate of incorporation of ara-C into DNA, which appears to be the ultimate lethal event.297 Except for the apparent beneficial effects of very high-dose ara-C, these maneuvers have not clearly resulted in unequivocal improvement in the therapeutic index.
The MDR phenotype is associated in most cases with increased amounts of a membrane glycoprotein (p-170), which serves as a pump accelerating the efflux of a wide variety of agents, including the anthracycline antibiotics, vincristine and mitoxantrone.301,302 Preliminary screens of a wide assortment of human tumors, including acute leukemia, suggest that the levels of p-170, as can be assessed by immunoperoxidase studies of individual cells, Western blots of cell preparations, and indirectly by mRNA levels and gene expression, are increased in patients refractory to chemotherapy.303–306 Preliminary evidence suggests that evidence of the MDR phenotype is infrequent at the time of diagnosis but may be present to a significant extent in patients with relapsed or refractory disease.307 Moreover, older patients,308 and those with adverse prognostic factors and destined to fare poorly, have a higher level of MDR expression.
The exciting implications of this finding relate to the ability, at least in vitro, to reverse the effect of the p-170 by exposure to a wide variety of compounds including calcium channel blockers (verapamil), quinine, and cyclosporine A.309 Verapamil has been used in conjunction with chemotherapy in patients with refractory multiple myeloma and lymphoma with suggestive evidence of benefit for some patients.304 Cardiotoxicity occurs at the doses of verapamil required to produce this effect. Cyclosporine can reverse the MDR phenotype in vitro at drug levels that are likely to be clinically acceptable in terms of toxicity.310 Clinical studies using these and other agents such as the potentially more potent nonimmunosuppressive cyclosporine analogue, PSC833, in conjunction with the anthracyclines, have been completed311 and represent attempts to increase the effectiveness of the available agents against AML and other types of leukemia. Preliminary results of a randomized trial of the addition of cyclosporin A to an ara-C plus continuous-infusion daunorubicin regimen for patients with relapsed AML suggested an overall survival benefit for adults in the experimental arm.312 Because of their effects on normal tissues such as the liver and kidney, these modulators also affect the pharmacokinetics of antineoplastic agents, such that the dosage of anthracyclines or etoposide may have to be attenuated if given with an MDR modulator.313
P-glycoprotein is unlikely to represent the only mechanism of drug resistance in patients with AML. Thus, preliminary evidence suggests overexpression of the multidrug-resistant protein in the cells from some patients with AML, particularly in samples from patients in relapse.314–316 Thus, intervention to circumvent resistance might best be applied during initial treatment prior to the enrichment of subpopulations resistant by other mechanisms.
Differentiating Agents/Acute Progranulocytic Leukemia
The HL-60 human leukemia cell line can be induced to undergo differentiation in vitro by an assortment of compounds including retinoids, vitamin D3, phorbol esters, dimethyl sulfoxide, ara-C, gamma interferon, and hexamethylenebisacetamide.4 In theory, if leukemic progenitors could be forced to undergo terminal differentiation in vivo, then the leukemic clone could lose the capacity for self-renewal and be eliminated, with fewer side effects than are currently associated with intensive cytotoxic chemotherapy. With the exception of blasts obtained from patients with APL, it has been difficult to induce reproducibly fresh leukemia cells to differentiate in vitro.
Older limited clinical trials had produced disappointing results except for individual patients with APL who derived transient benefit from treatment with cis-retinoic acid.317 More recent reports from China, France, and New York have demonstrated dramatic responses in patients with APL using ATRA administered orally for 30 to 90 days.318–320 CRs were seen in > 80% of both relapsed and previously untreated individuals. Marrow aplasia did not occur, and serial bone marrows demonstrated what appeared to be maturation of the abnormal, hypergranulated promyelocytes. Disseminated intravascular coagulation resolves promptly with a profound reduction in the requirement for platelet transfusions following ATRA treatment. Although the hemorrhagic complications are decreased, approximately 20 to 30% of patients treated with ATRA develop significant side effects that can include fever, rapidly evolving pulmonary insufficiency, pericarditis, and pleurisy. This syndrome can occur independently of the leukocytosis frequently observed with ATRA therapy and can be fatal. The optimal management remains unclear, although amelioration with corticosteroids and/or the initiation of chemotherapy has been noted.321,322 A randomized study conducted in Europe first demonstrated superiority of ATRA with or without the addition of chemotherapy as needed to chemotherapy alone in newly diagnosed patients with APL both in terms of response to induction therapy and disease-free survival.323,324 A large North American trial compared initial ATRA therapy versus chemotherapy as well as ATRA versus observation in the late postremission period.325 Patients who were never randomized to receive ATRA did very poorly compared to patients receiving ATRA during induction or maintenance. Subsequent studies have suggested that optimal results are achieved when ATRA and anthracycline-based chemotherapy are administered concurrently during induction.326,327
Resistance to ATRA eventually develops, and virtually all patients treated with ATRA alone relapse despite ATRA maintenance therapy. Possible pharmacokinetic mechanisms include induction of more rapid metabolism of the ATRA and induction of cytoplasmic retinoic acid binding protein in normal tissues that bind the ATRA, thereby decreasing the exposure of residual APL cells to the drug.328 Most clinical trials incorporate postremission anthracycline-based consolidation because of the unique sensitivity of APL to this class of compounds. A recently published study supported the role of maintenance therapy with ATRA plus methotrexate and 6-mercaptopurine.327 The most exciting new development in APL has been the demonstration that arsenic trioxide (another agent used for years in China) is a safe, effective, and powerful treatment.329,330 Major tumor burden reduction (four to five orders of magnitude based on PCR testing) have been noted even in patients with advanced multiply relapsed disease.
These exciting findings and those related to our understanding of the molecular mechanism accounting for the block in differentiation in APL and the t(8; 21) and inv 16 leukemias have rekindled interest in treatment with other potential differentiating agents. Although the effect of ATRA may be largely limited to APL presumably related to the unique mutation in the nuclear retinoic acid receptor characteristic of this disorder, other specific small molecules (e.g., histone deacytalase inhibitors [which also have a role in APL] and hypomethylating agents such as 5-azacytidine) may be useful in other AML subtypes.
Minimal Residual Disease
A variety of techniques of varying sensitivity are being evaluated to detect residual leukemia cells in patients in morphologic CR, including (in approximate descending order of sensitivity) conventional cytogenetics, Southern blotting for known gene rearrangements, fluorescent in situ hybridization (FISH), multiparameter flow cytometry, and RT-PCR.331,332 Technical problems abound, including the requirement for an already cloned abnormality when applying molecular techniques and the potential for changes in antigen expression when using immunologic monitoring. There are few large prospective studies available. Although preliminary data tend to be compatible with the logical premise that persistence of detectable disease presages eventual relapse, false-positive [see comments on t(8;21) above]66 and false-negative rates may be appreciable and could result in incorrect decisions about further treatment. Serial monitoring is cumbersome, and it may be that detection of MRD at a particular time point after completion of therapy will be most convenient and prognostically significant. This appears to be true of persistence of the t(15;17) in APL after ATRA therapy.323,333 A recent report from Italy suggests that a positive PCR test after all chemotherapy is completed is an ominous sign.334 The key question, of course, is whether earlier intervention with further therapy or allogeneic BMT will be of value if applied earlier, prior to gross relapse. There are no data addressing this issue. Because of the presence of residual disease, autologous collection and high-dose therapy may be predicted to be of less value in this circumstance.
Prognostic Factors
A number of clinical and laboratory factors have been described that can help predict the probability of attaining complete remission, the duration of CR, and, by extension, the probability of disease-free survival. It should be emphasized, however, that, with few exceptions, prognostic factors provide only relative guidelines to be used when discussing the probabilities of treatment outcome with individual patients. Conversely, they are of critical importance in focusing clinical and laboratory research efforts directed toward improving outcome. Most clinical factors are associated with decreased response to initial chemotherapy because they result in poor tolerance of the complications of prolonged pancytopenia. Advanced age (and particularly age > 70 years), renal insufficiency, congestive heart failure, concurrent infection, decreased serum albumin, and history of alloimmunization to blood products are examples.335 Many can be summed in the assessment of the patient’s performance status at the time of presentation.
As supportive care improves, some of these issues become less critical, and the intrinsic biologic characteristics of the patient’s leukemia become the dominant factors. Because the major cause of treatment failure tends to be associated with drug resistance, these biologic predictive factors usually are associated with both decreased complete response rates as well as short durations of remission should CR be achieved. A number of large multivariate analyses have consistently demonstrated that patients with hyperleukocytosis and/or FAB M5 morphology, increases in serum lactate dehydrogenase (which is probably simply a reflection of the two previously mentioned factors), prior history of myelodysplasia or other hematologic disorder, and FAB M6 or M7 and probably M0 morphology are associated with poor initial response rates.335 More advanced age is associated with decreased CR rates because of an increased rate of primarily resistant leukemia as well as difficulties with supportive care. There is evidence suggesting that age greater than 60 years may also presage shorter CR durations, probably because of association with less favorable cytogenetic findings.336 In contrast to patients with ALL, immunologic phenotyping using currently available reagents does not appear to identify distinct prognostic groups, except for the negative influence of CD34 positivity,41,43,44 and the conflicting data about whether patients with AML and associated lymphoid antigens may have an improved or impaired outlook.139 Studies have suggested that overexpression of bcl-2, with a presumed inhibitory effect on apoptosis341 or underexpression of Rb, a tumor suppressor gene,342 may be negative prognostic factors.
Major attention has focused on the prognostic impact of karyotypic abnormalities. A large number of studies demonstrate a high frequency of nonrandom cytogenetic abnormalities in the blasts from patients with AML.339–342 The overall conclusions from these studies are quite consistent. In brief, clonal abnormalities can be identified in 60 to 75% of patients, although some studies maintain that abnormalities can be detected in greater than 95% and, indeed, perhaps in all patients with AML.339 Certainly, it is not unreasonable to assume that genetic abnormalities are present in all patients and that morphologic evaluation of chromosomal changes represents a relatively crude technique compared with molecular approaches. It is also clear that certain recurring abnormalities have a very tight relationship with both morphology and treatment outcome (see Table 124.1). The balanced translocations t(8;21), inv 16, and t(15;17) tend to occur in younger patients and area associated with a high incidence of initial complete response to treatment with a very low incidence of initial failure related to drug-resistant leukemia342 and a better rate of overall disease-free survival. In contrast, multiple abnormalities or deletions of chromosomes 5 and/or 7 are rarely associated with cure. Those with normal chromosomes, trisomy 8, and abnormalities of chromosome 11q23, and inv3 are associated with an intermediate prognosis342,343 (Fig. 124.12). In almost all studies, cytogenetic findings are an independent variable predicting outcome, and in some studies, they represent the major prognostic factor other than patient age. These findings have many intriguing biologic implications and have produced considerable interest in the possibility of specific therapy directed according to different risk groups. Although it is tempting to apply the knowledge of karyotypes to therapeutic decisions, a number of important and unanswered biologic and clinical questions remain.
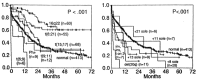
Figure 124.12
Overall survival according to cytogenic findings in patients with de novo AML treated with intensive induction and postremission chemotherapy treated on CALGB studies.
Because of the close association between certain cytogenetic changes and morphology, the same prognostic information can frequently be gleaned by a careful assessment of the FAB classification or, as has been done in the past, by evaluation of patient age, performance status, and initial blast count. In order for the cytogenetic findings to represent more than a simple addition to this descriptive list, basic investigations into the mechanisms by which the specific cytogenetic changes are associated with these clinically recognizable effects are required. With the extraordinary pace of development of molecular biologic and immunologic techniques in the past few years, it is this aspect of the impact of cytogenetics that is potentially most exciting. These chromosomal alterations result in changes in the level or function of normal or mutated protein products, which result in the halt in differentiation that is morphologically recognized as acute leukemia as well as the associated cell kinetic and/or biochemical changes that may be correlated with resistance to therapy. Although it may be naive to assume that single DNA abnormalities are at the root of the biologically complex leukemic process, pharmacologic or molecular therapies, such as antisense fragments, might eventually become available for rational therapeutic attacks in specific chromosomal abnormalities.
Although cytogenetic analyses are not as yet routinely used to guide induction therapy, such data are used, perhaps with insufficient support, to guide the BMT versus chemotherapy postremission decisions. With regard to influencing decisions about induction, for example, it is possible to screen for gross deletions or additions of whole chromosomes or parts of chromosomes by performing Southern blots using appropriate molecular probes with densitometry to quantify the signal and estimate the number of alleles expressed.151 This technique depends on a relatively pure preparation of blasts as well as the assumption that the same finding is present in all of the leukemia cells. Another approach uses in situ hybridization with probes coupled to compounds that are easily visible on cell smears when viewed under fluorescence microscopy. This technique has been used to identify additions or deletions of individual chromosomes and translocation breakpoints for which probes have been adequately cloned.344 This technique permits correlation with cell morphology, can assess the frequency with which abnormalities are found within a given cell population, and can provide information within 48 to 72 hours of patients referral. Either approach requires the serial application of different probes directed against the most common and clinically relevant cytogenetic abnormalities, a tedious task potentially applicable to at most one half of patients, even as new probes are developed. In addition, these techniques are unlikely to replace standard cytogenetics because they do not easily detect other changes that are likely to be biologically and clinically relevant. Nonetheless, molecular procedures can be more sensitive both at the time of diagnosis and for the detection of minimal residual disease and are more readily adaptable to serial evaluations once the cytogenetic abnormality has been defined. Even if the technologies described above become readily available, it is likely that cytogenetic findings could correctly classify fewer than 50% of patients into prognostic groups. A significant fraction of patients have pseudodiploid karyotypes of uncertain prognostic impact, with others having miscellaneous hyper- or hypodiploid karyotypes. In addition, about one-third of patients have apparently normal karyotypes using current cytogenetic techniques, although it is probable that improved cell culture or molecular techniques will somewhat decrease this fraction of normals.
The real utility of adequate, complete, and rapidly available cytogenetic analysis will be in (1) further definition of genes at cytogenetic breakpoints to define pathophysiology and provide therapeutic clues, (2) assign prognosis, if in fact other cytogenetic abnormalities are shown to have independent importance, and (3) determine therapy if prospective evaluations demonstrate a benefit of one form of therapy relative to another for specific cytogenetic subgroup.
Complications
Hyperleukocytosis
Leukemic blasts are considerably less deformable than mature myeloid cells345,346 and are “stickier” than lymphoblasts due to expression of cell surface adhesion molecules. With increasing blast counts, usually at levels greater than 100,000/μl in the myeloid leukemias, blood flow in the microcirculation can be impeded by plugs of these more rigid cells. Local hypoxemia may be exacerbated by the high metabolic activity of the dividing blasts, with endothelial damage and hemorrhage. The situation can be worsened by red blood cell transfusions that rapidly increase whole-blood viscosity in the presence of hyperleukocytosis, thereby further compromising local blood flow. Red blood cell transfusions should therefore be administered very slowly or, if possible, withheld until the blast count is reduced. Lastly, coagulation abnormalities, including disseminated intravascular coagulation, further increase the risk of local hemorrhage. Liberal use of platelet transfusions is recommended, particularly since the platelet count is frequently overestimated because of the presence of fragments of blasts on blood smears,347 which can be mistakenly counted as platelets by automated blood cell counters.
Although pathologic evidence of leukostasis can be found in most organs in patients with extremely high white cell counts, clinical symptomatology is usually related to CNS and pulmonary involvement.348 Occasionally, pulmonary symptomatology with worsening hypoxemia can occur with therapy and lysis of trapped leukemic cells. Spurious elevation of serum potassium can occur because of release from white blood cells during clotting, and it is sometimes necessary to measure potassium levels on heparinized plasma. Similarly, pO2 can appear falsely decreased because of the enhanced metabolic activity of the high level of white blood cells, even when the specimen is appropriately placed on ice during transport to the laboratory. Pulse oximetry provides an accurate assessment of O2 saturation in such circumstances. Hyperleukocytosis is more common in patients with myelomonocytic or monocytic leukemia,102 and it is possible that the clinical manifestations are exacerbated by the migration of leukemic promonocytes into tissue where further proliferation occurs.104
The initial mortality rate for patients with AML and symptomatic hyperleukocytosis is high.103,349 If patients survive the initial period, they tend to have somewhat lower remission rates. Remission durations are also shorter, possibly because of larger initial tumor mass, but more likely related to the biology and intrinsic chemoresistance of their leukemia.103,349 Thus, symptomatic hyperleukocytosis in AML (and rarely in ALL) constitutes a medical emergency, and efforts should be made to lower the white blood cell count rapidly. In most patients, rapid cytoreduction can be achieved by chemotherapy, with either standard induction agents or with high doses of hydroxyurea (3 g/m2/d).350 Some centers also advocate low-dose cranial irradiation, including the retina, in order to prevent further proliferation of leukemic cells in CNS sites where drug delivery may theoretically be compromised. This treatment is well tolerated, although there are no comparative studies to determine whether the results are superior to chemotherapy alone. In some patients, however, the response to chemotherapy can be somewhat delayed, whereas in others, it is impossible to initiate chemotherapy immediately because of renal insufficiency, metabolic problems, delays in initiating allopurinol therapy so as to prevent hyperuricemia, or similar considerations. In such patients, emergency leukapheresis has been used to lower or stabilize the white count.351,352 Although intensive leukapheresis, with procedure times often lasting many hours, can produce rapid improvement of pulmonary and CNS symptomatology, there are theoretic and practical limitations to its benefits. It is difficult, for example, for leukapheresis to affect already established vascular plugs, particularly if vascular invasion has taken place. In such cases, chemotherapy and possibly radiotherapy remain the primary modalities, although, theoretically, leukapheresis could decrease further accumulation of leukocytes at these sites. Furthermore, it is precisely the patient in whom leukostasis is most likely to occur, that is, the patient with high and rapidly rising blasts counts, in whom the technical limitations of leukapheresis are relevant. It is often difficult, even with highly efficient cell separators, to reduce the rapidly rising count. In such patients with a high proliferative thrust, cycle-specific chemotherapeutic agents are more likely to be most rapidly effective, however. Leukapheresis is often of modest benefit to patients who develop pulmonary problems during cytotoxic treatment, since in some such patients the symptoms are related at least in part to a local inflammatory response following leukocyte lysis. Leukapheresis can clearly be of value to the occasional patient to whom chemotherapy cannot be administered immediately.
Central Nervous System Leukemia
Involvement of the CNS is considerably less common in patients with AML than in both adults and children with ALL.353–356 Most clinicians have the impression that the incidence has decreased even further in recent years, perhaps related to the use of higher doses of ara-C, which can penetrate into the CNS, during both initial induction and postremission therapies. It is therefore somewhat difficult to provide a precise estimate of the incidence of CNS leukemia in AML, although it has occurred in less than 5% of large clinical trials in recent years. Because of this low frequency, diagnostic lumbar punctures are not routinely indicated in the absence of CNS symptoms, and chemotherapeutic approaches do not include CNS prophylaxis. The most common presenting symptoms are consequences of increased intracranial pressure and usually consist of a constant headache, sometimes associated with lethargy or other mental changes. Cranial nerve signs (most commonly cranial nerves III or VI) and occasionally peripheral nerve manifestations are secondary to nerve root involvement and can be accompanied by headaches or occur alone.
The diagnosis is usually easily confirmed by examination of cytocentrifuge preparations of cerebrospinal fluid (CSF) after lumbar puncture. Cell counts can vary from as few as 5/μl to > 1,000. Most patients have moderate elevations in CSF protein with a moderate decrease in glucose. After the administration of intrathecal therapy, cytocentrifuge preparations can demonstrate reactive ependymal cells that, particularly if the leukocyte count is low, can be difficult to distinguish from leukemia cells. Treatment consists of the administration of intrathecal chemotherapy, with the addition of cranial radiation (usually 2,400 cGy), to patients who do not respond fully to chemotherapy or in whom cranial nerve involvement is present.357 Either methotrexate (12 mg/dose) or ara-C (50 mg/dose) can be used as initial therapy, with a cross-over to the other agents in the event of refractoriness or relapse. A typical schedule includes treatment 2 to 3 times a week until the CSF has cleared, generally occurring after a few injections. Treatment is then given at weekly intervals for two more doses to be followed by monthly administration, for a total of a year. An Omaya reservoir to permit intraventricular drug administration is frequently needed either because of difficulties in performing repeated lumbar punctures or because of concern that in some individuals, the CSF flow does not deliver sufficient amounts of the drug from the lumbar space to the entire CNS. Successful therapy with systemically administered agents that penetrate the CNS such as diaziquone (AZQ), high-dose methotrexate, or ara-C have also been reported.357 Unfortunately, the relapse rate, even after initially rapidly successful therapy, is high, either in association with bone marrow relapse or independently.
It has been suggested that the incidence of CNS leukemia is higher in patients with FAB M4 E0 morphology and possibly in patients with monocytic leukemia and high circulating blast counts.358 This may no longer be the case using more contemporary treatment regimens, and prophylactic therapy is not indicated in such individuals.
Ophthalmic Complications
Essentially every ocular structure has been described to be involved in the leukemias, sometimes dominating the clinical picture in the prechemotherapy era.359 Leukemia cells can infiltrate the conjunctiva and lacrimal glands, producing obvious masses that may require treatment with radiation therapy. Involvement of the choroid and retina is most common, however. A comprehensive, prospective study of 53 newly diagnosed adults with AML documented retinal or optic nerve (3 patients) abnormalities in 64% of patients.360 Hemorrhage and cotton wool spots (a consequence of nerve fiber ischemia) were most frequent, and the occurrence of these findings was unrelated to patient age, FAB type, white blood cell count, or hematocrit. Initial platelet counts were lower in patients with retinopathy. Ten patients had decreased visual acuity, including five with macular hemorrhages. It was felt that many of the cotton wool spots were either a consequence of or exacerbated by ischemia due to anemia. Definite leukemic infiltrate of the retina could not be confirmed. All patients received aggressive chemotherapy and platelet transfusion support; no patient received cranial or ocular irradiation. All ocular findings resolved in patients achieving CR, and there was no residual visual deficit in any patient. Infectious ocular problems were not noted and seem to be uncommon, perhaps because the rapid, empiric use of antibacterial antibiotics (and more recently of amphotericin B) has decreased the possibility of hematogenous spread of infections to the eye.
Pregnancy
AML is occasionally diagnosed during pregnancy, either because of clinical manifestations or as an incidental finding during blood count checks. If detected during the first trimester, termination of pregnancy followed by treatment of the leukemia is advisable. The management of patients diagnosed later in pregnancy, such as late in the second trimester or during the third trimester, is more problematic. In the latter group of patients, if the leukemia is relatively indolent, it is sometimes possible to manage patients conservatively with leukapheresis and/or transfusion with induction of labor and delivery of the fetus as soon as possible.361 There have also been many reports of patients treated with chemotherapy later in their pregnancy.362,363 The majority of these women have not aborted, and there have been no reports of leukemia occuring in the children, nor an increased incidence of fetal abnormalities in infants who have been exposed to AML chemotherapy during the later stages of gestation.
Metabolic Abnormalities
Patients receiving treatment for AML can experience a wide range of metabolic problems as a consequence of vomiting, diarrhea, impaired nutrition, or renal dysfunction, usually due to side effects from antibiotics or amphotericin B. Some metabolic disorders are related to the leukemic process itself. Hyperuricemia, occasionally accompanied by urate nephropathy with renal insufficiency, is the most frequent metabolic accompaniment of AML. All patients should receive allopurinol, 300 mg/d or more, as soon as the diagnosis of acute leukemia is established, so that the chemotherapy can be administered as soon as is medically appropriate. In most patients, the urate nephropathy can be avoided or ameliorated with vigorous hydration and urinary alkalization with systemic or oral administration of sodium bicarbonate. Allopurinol can usually be discontinued within a day or two after chemotherapy is completed.
Tumor lysis syndrome occurs more frequently in patients with ALL, although some AML patients experience hyperphosphatemia, hypocalcemia, hyperkalemia, and renal insufficiency as a consequence of massive leukemic cell death.364 The inciting cause seems to be release of large amounts of phosphate from lysed blasts, which co-precipitates with calcium in the kidneys, leading to hypocalcemia and sometimes to oliguric renal failure. Hyperuricemia further contributes to this problem, which is usually self-limited and responds to judicious hydration. Despite markedly hypercellular marrows, hypercalcemia is extremely unusual in patients with AML. Severe, occasionally symptomatic, hypokalemia is not infrequent, particularly in patients with monocytic leukemias. The mechanism appears to be renal potassium loss because of tubular damage induced by the high levels of lysozyme often noted in these patients.108 Aggressive replacement with parenteral potassium is required; the syndrome usually abates after cytoreduction by chemotherapy. Lastly, rare patients have been described in whom lactic acidosis has been a constant metabolic accompaniment of the leukemia both at the time of presentation and relapse.365 The mechanism is unclear, although anaerobic metabolism by the leukemic cells at sites of leukostasis has been postulated.
Summary
Largely due to improvements in supportive care and the anticipatory management of complications of the treatment and underlying disease, the overall outlook for adults with AML has improved throughout the world in the past 10 to 15 years. It is equally clear, however, that results have leveled off and that new approaches are needed to increase the fraction of patients cured. Dramatic advances in molecular biology and molecular genetics offer promise in this regard both because of increased understanding of leukemia biology and the production of small molecules, monoclonal antibodies, cytokines, and HGF with potential clinical utility. Further clarification of mechanisms of drug resistance with the possibility of enhancement of the effectiveness of currently available drugs is also an exciting and achievable prospect. These strategies, as well as clinical trials designed to assess the appropriate use of BMT and newer chemotherapeutic drugs, provide the hope that therapy for AML will be both more successful and less empiric in the future.366
References
- 1.
- Cheson B D, Cassileth P A, Head D R. et al. Report of the NCI-sponsored workshop on definitions of diagnosis and response in acute myeloid leukemia. J Clin Oncol. 1990;8:813. [PubMed: 2185339]
- 2.
- Pizzo P A. Fever in immunocompromised patients. N Engl J Med. 1999;341:893. [PubMed: 10486422]
- 3.
- Wade J C. Management of infection in patients with acute leukemia. Hematol Oncol Clin North Am. 1993;7:293. [PubMed: 8449863]
- 4.
- Collin S J. The HL-60 promyelocytic leukemia cell line: proliferation, differentiation, and cellular oncogene expression. Blood. 1987;70:1233. [PubMed: 3311197]
- 5.
- Arlin Z, Case D C Jr, Moore J. et al. and the Lederle Cooperative Group. Randomized multicenter trial of cytosine arabinoside with mitoxantrone or daunorubicin in previously untreated adult patients with acute nonlymphocytic leukemia (ANLL). Leukemia. 1990;4:177. [PubMed: 2179638]
- 6.
- Bloom G E, Warner S, Gerald P S, Diamond L K. Chromosome abnormalities in constitutional aplastic anemia. N Engl J Med. 1966;274:8. [PubMed: 5901871]
- 7.
- Head D R. Revised classification of acute myeloid leukemia. Leukemia. 1996;10:1826. [PubMed: 8892688]
- 8.
- Nichols J, Nimer S D. Transcription factors, translocations, and leukemia. Blood. 1992;80:2953. [PubMed: 1361370]
- 9.
- Toksoz D, Farr C J, Marshall C J. Ras genes and acute myeloid leukemia. Br J Haematol. 1989;71:1. [PubMed: 2644966]
- 10.
- van Kamp H, dePijper C, Verlaan-de Vries M. et al. Longitudinal analysis of point mutations of the N-ras proto-oncogene in patients with myelodysplasia using archived blood smears. Blood. 1992;79:1266. [PubMed: 1536950]
- 11.
- Rosnet O, Schiff C, Pébusque M J, Marcheto S, Tonnelle C, Toiron Y, Birg F, Birnbaum D. Human FLT 3/FLK gene: cDNA cloning and expression in hematopoietic cells. Blood. 1993;82:1110. [PubMed: 8394751]
- 12.
- Kornlau S M, Xu H -J, del Giglio A. et al. Clinical complications of decreased retinoblastoma protein expression in acute myelogenous leukemia. Cancer Res. 1992;52:4587. [PubMed: 1511426]
- 13.
- Sugimoto K, Hirano N, Toyoshima H. et al. Mutations of the p53 gene in myelodysplastic syndrome (MDS) and MDS-derived leukemia. Blood. 1993;81:3022. [PubMed: 8499637]
- 14.
- Look A T. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059. [PubMed: 9353180]
- 15.
- Bizzozero O J Jr, Johnson K G, Ciocco A. et al. Radiation-related leukemia in Hiroshima and Nagasaki. I. Distribution, incidence, and appearance time. N Engl J Med. 1966;274:1095. [PubMed: 5932020]
- 16.
- Darby S C, Doll R, Gill S K, Smith P G. Long term mortality after a single treatment course with X-rays in patients treated for ankylosing spondylitis. Br J Cancer. 1987;55:179. [PMC free article: PMC2002095] [PubMed: 3814487]
- 17.
- Jacobs A. Benzene and leukemia. Br J Haematol. 1989;72:119. [PubMed: 2667621]
- 18.
- Austin A, Delzell E, Cole P. Benzene and leukemia: a review of the literature and a risk assessment. Am J Epidemiol. 1988;127:419. [PubMed: 3277397]
- 19.
- Andrieu J -M, Ifrah N, Payen C. et al. Increased risk of secondary acute nonlymphocytic leukemia after extended-field radiation therapy combined with MOPP chemotherapy for Hodgkin’s disease. J Clin Oncol. 1990;8:1148. [PubMed: 2193118]
- 20.
- Pui C -H, Ribeiro R C, Hancock M L. et al. Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N Engl J Med. 1991;325:1682. [PubMed: 1944468]
- 21.
- Crane M M, Keating M J, Trujillo J M, LaBarthe D R, Frankowski R F. Environmental exposures in cytogenetically defined subsets of acute nonlymphocytic leukemia. JAMA. 1989;262:634. [PubMed: 2746815]
- 22.
- Kinlen L J, Rogot E. Leukaemia and smoking habits among United States veterans. BMJ. 1988;297:657. [PMC free article: PMC1834341] [PubMed: 3179546]
- 23.
- Brownson R C, Novotny T E, Perry M C. Cigarette smoking and adult leukemia. Arch Intern Med. 1993;153:469. [PubMed: 8435026]
- 24.
- Ahilbom A. A review of the epidemiologic literature on magnetic fields and cancer. Scand J Work Environ Health. 1988;14:337. [PubMed: 3062769]
- 25.
- Won O, Raabe G K. Critical review of cancer epidemiology in petroleum industry employees, with a quantitative meta-analysis by cancer site. Am J Ind Med. 1989;15:283. [PubMed: 2929617]
- 26.
- Wongsrichanalai C, Delzell E, Cole P. Mortality for leukemia and other diseases among workers at a petroleum refinery. J Occup Med. 1989;31:106. [PubMed: 2709160]
- 27.
- Chaganti R S K, Miller D R, Meyers P A, German J. Cytogenetic evidence of the intrauterine origin of acute leukemia in monozygotic twins. N Engl J Med. 1979;300:1032. [PubMed: 285335]
- 28.
- Clarkson B D, Boyse E A. Possible explanation of the high concordance for acute leukemia in monozygous twins. Lancet. 1971;1:699. [PubMed: 4101637]
- 29.
- Davidson R J L, Walker W, Watt J L, Page B M. Familial erythroleukemia: a cytogenetic and haematological study. Scand J Haematol. 1978;20:351. [PubMed: 274807]
- 30.
- Gunz F W, Gunz J P, Vincent P C. et al. Thirteen cases of leukemia in a family. J Natl Cancer Inst. 1978;60:1243. [PubMed: 274587]
- 31.
- Heath C W Jr, Moloney W C. Familial leukemia: five cases of acute leukemia in three generations. N Engl J Med. 1964;272:882. [PubMed: 14274439]
- 32.
- Ho C Y, Otterund B, Legare R D. et al. Linkage of a familial platelet disorder with a propensity to develop myeloid malignancies to human chromosome 21q22.1-22.2. Blood. 1996;87:5218. [PubMed: 8652836]
- 33.
- Bortin M M, D’Amaro J, Bach F H, Rimm A A, Roos J J V. HLA associations with leukemia. Blood. 1987;70:227. [PubMed: 3474040]
- 34.
- Daly P A, Simon R, Schiffer C A. et al. A study of HLA antigens and haplotypes in a population of Caucasians with acute non-lymphocytic leukemia. Leuk Res. 1979;3:75. [PubMed: 449392]
- 35.
- Bennett J M, Catovsky D, Daniel M T. et al. Proposals for the classification of the acute leukaemias. Br J Haematol. 1976;33:451. [PubMed: 188440]
- 36.
- Head D R, Savage R A, Cerezo L. et al. Reproducibility of the French-American-British classification of acute leukemia: the Southwest Oncology Group experience. Am J Hematol. 1985;18:47. [PubMed: 2981467]
- 37.
- Bennett J M, Catovsky D, Daniel M T. et al. Criteria for the diagnosis of acute leukemia of megakaryocyte lineage (M7). Ann Intern Med. 1985;103:460. [PubMed: 2411180]
- 38.
- Bennett J M, Catovsky D, Daniel M T. et al. Proposed revised criteria for the classification of acute myeloid leukemia. Ann Intern Med. 1985;103:626. [PubMed: 3862359]
- 39.
- Bennett J M, Catovsky D, Daniel M -T. et al. Proposal for the recognition of minimally differentiated acute myeloid leukaemia (AML-MO). Br J Haematol. 1991;78:325. [PubMed: 1651754]
- 40.
- Harris N L, Jaffe E S, Diebold J. et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835. [PubMed: 10577857]
- 41.
- Bradstock K, Matthews J, Benson E, Page F, Bishop J. Australian Leukaemia Study Group. Prognostic value of immunophenotyping in acute myeloid leukemia. Blood. 1994;84:1220. [PubMed: 8049437]
- 42.
- Civin C I, Strauss I C, Brovall C. et al. A hematopoietic progenitor cell surface antigen defined by a monoclonal antibody raised against KG-Ia cells. J Immunol. 1984;133:157. [PubMed: 6586833]
- 43.
- Borowitz M J, Gockerman J P, Moore J O. et al. Clinicopathologic and cytogenetic features of CD34 (My 10)-positive acute nonlymphocytic leukemia. Am J Clin Pathol. 1989;91:1265. [PubMed: 2646903]
- 44.
- Campos L, Guyotat D, Archimbaud E. et al. Surface marker expression in adult acute myeloid leukemia: correlations with initial characteristics, morphology and response to therapy. Br J Haematol. 1989;72:161. [PubMed: 2757962]
- 45.
- Geller R B, Zahurak M, Hurwitz C A. et al. Prognostic importance of immunophenotyping in adults with acute myelocytic leukaemia: the significance of the stem-cell glycoprotein CD34 (My10). Br J Haematol. 1990;76:340. [PubMed: 1702006]
- 46.
- Raspadori D, Lauria F, Vetura M A. et al. Incidence and prognostic relevance of CD34 expression in acute myeloblastic leukemia: analysis of 141 cases. Leuk Res. 1997;21:603. [PubMed: 9301680]
- 47.
- Bernstein I D, Singer J W, Smith F O. et al. Differences in the frequency of normal and clonal precursors of colony-forming cells in chronic myelogenous leukemia and acute myelogenous leukemia. Blood. 1992;79:1811. [PubMed: 1373089]
- 48.
- Kibbelaar R E, Mulder J W R, Dreef E J. et al. Detection of monosomy 7 and trisomy 8 in myeloid neoplasia: a comparison of banding and fluorescence in situ hybridization. Blood. 1993;82:904. [PubMed: 8338953]
- 49.
- Fialkow P J, Singer J W, Adamson J W. et al. Acute nonlymphocytic leukemia: heterogeneity of stem cell origin. Blood. 1981;57:1068. [PubMed: 6939452]
- 50.
- Keinanen M, Griffin J D, Bloomfield C D, Machnicki J, de la Chapelle A. Clonal chromosomal abnormalities showing multiple-cell-lineage involvement in acute myeloid leukemia. N Engl J Med. 1988;318:1153. [PubMed: 3163100]
- 51.
- Kibbelaar R E, van Kamp H, Dreef E J. et al. Combined immunophenotyping and DNA in situ hybridization to study lineage involvement in patients with myelodysplastic syndromes. Blood. 1992;79:1823. [PubMed: 1558974]
- 52.
- Koeffler H P. Syndromes of acute nonlymphocytic leukemia. Ann Intern Med. 1987;107:748. [PubMed: 3310797]
- 53.
- Cuneo A, Ferrant A, Michaux J L. et al. Cytogenetic profile of minimally differentiated (FAB MO) acute myeloid leukemia: correlation with clinicobiologic findings. Blood. 1995;85:3688. [PubMed: 7780152]
- 54.
- Stasi R, Del Poeta G, Venditti A. et al. Analysis of treatment in patients with minimally differentiated acute myeloid leukemia (AML-MO). Blood. 1994;83:1619. [PubMed: 8123853]
- 55.
- Larson R A, Dodge R K, Burns C P. et al. A five-drug remission induction regimen with intensive consolidation for adults with acute lymphoblastic leukemia: Cancer and Leukemia Group B study 8811. Blood. 1995;85:2025. [PubMed: 7718875]
- 56.
- Dohner H, Arthur D C, Ball E D. et al. Trisomy 13—a new recurring chromosome abnormality in acute leukemia. Blood. 1990;76:1614. [PubMed: 1698482]
- 57.
- Matsuo T, Cox C, Bennett J M. Prognostic significance of myeloperxidase positivity of blast cells in acute myeloblastic leukemia without maturation (FAB: M1): an ECOG study. Hematol Pathol. 1989;3:153. [PubMed: 2560775]
- 58.
- Berger R, Bernheim A, Ochoae-Noguera M E. et al. Prognostic significance of chromosomal abnormalities in acute nonlymphocytic leukemia: a study of 343 patients. Cancer Genet Cytogenet. 1987;28:293. [PubMed: 3621141]
- 59.
- Keating M J, Cork A, Broach T. et al. Toward a clinically relevant cytogenetic classification of acute myelogenous leukemia. Leuk Res. 1987;11:119. [PubMed: 3469482]
- 60.
- Keating M J, Smith T L, Kantarjian H. et al. Cytogenetic pattern in acute myelogenous leukemia: a major reproducible determinant of outcome. Leukemia. 1988;2:403. [PubMed: 3164797]
- 61.
- Tallman M S, Hakimian D, Shaw J M. et al. Granulocytic sarcoma is associated with the 8;21 translocation in acute myeloid leukemia. J Clin Oncol. 1993;11:690. [PubMed: 8478662]
- 62.
- Downing J R, Head D R, Curcio-Brint A M. et al. An AML1/ETO fusion transcript is consistently detected by RNA-based polymerase chain reaction in acute myelogenous leukemia containing the (8;21)(q22;q22) translocation. Blood. 1993;81:2860. [PubMed: 8499624]
- 63.
- Liu P P, Tarle S A, Hajre A. et al. Fusion between transcription factor CBFβ/PEBP2β and myosin heavy chain in acute myeloid leukemia. Science. 1993;261:1041. [PubMed: 8351518]
- 64.
- Okuda T, van Deursen J, Hiebert S W, Grosveld G, Downing J R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321. [PubMed: 8565077]
- 65.
- Gelmetti V, Zhang J, Fanelli M. et al. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia partner ETO. Mol Cell Biol. 1998;18:7185. [PMC free article: PMC109300] [PubMed: 9819405]
- 66.
- Nucifora G, Birn D J, Erickson P. et al. Detection of DNA rearrangements in the AML1 and ETO loci and of an AML1/ETO fusion mRNA in patients with t(8;21) acute myeloid leukemia. Blood. 1993;81:883. [PubMed: 8427996]
- 67.
- Cunningham I, Gee T S, Reich L M. et al. Acute promyelocytic leukemia: treatment results during a decade at Memorial Hospital. Blood. 1989;73:1116. [PubMed: 2930837]
- 68.
- Stone R M, Mayer R J. The unique aspects of acute promyelocytic leukemia. J Clin Oncol. 1990;8:1913. [PubMed: 2230879]
- 69.
- Warrell R P, de The H, Wang Z -Y. et al. Acute promyelocytic leukemia. J Clin Oncol. 1990;8:1913. [PubMed: 2230879]
- 70.
- Grignani F, Fagioli M, Alcalay M. et al. Acute promyelocytic leukemia: from genetics to treatment. Blood. 1994;83:10. [PubMed: 8274729]
- 71.
- Golomb H M, Rowley J D, Vardiman J W, Testa J R, Butler A. Microgranular acute promyelocytic leukemia: a distinct clinical, ultrastructural, and cytogenetic entity. Blood. 1980;55:253. [PubMed: 6928105]
- 72.
- Griffin J D, Davis R, Nelson D A. et al. Use of surface marker analysis to predict outcome of adult acute myeloblastic leukemia. Blood. 1986;68:1232. [PubMed: 2946331]
- 73.
- Davey F R, Davis R B, MacCallum J M. et al. Morphologic and cytochemical characteristics of promyelocytic leukemia. Am J Hematol. 1989;30:221. [PubMed: 2539013]
- 74.
- Douer D, Preston-Martin S, Chang E. et al. High frequency of acute promyelocytic leukemia among latinos with acute myeloid leukemia. Blood. 1996;87:308. [PubMed: 8547657]
- 75.
- Estey E, Thall P, Kantarjian H. et al. Association between increased body mass index and a diagnosis of acute promyelocytic leukemia in patients with acute myeloid leukemia. Leukemia. 1997;11:1661. [PubMed: 9324286]
- 76.
- Grainick H R, Sultan C. Acute promyelocytic leukemia. Haemorrhagic manifestation and morphologic criteria. Br J Haematol. 1975;29:373. [PubMed: 1059471]
- 77.
- Gralnick H R, Abrell E. Studies of the procoagulant and fibrinolytic activity of promyelocytes in acute promyelocytic leukaemia. Br J Haematol. 1973;24:89. [PubMed: 4577065]
- 78.
- Sakata Y, Murakami T, Noro A, Mori K, Matsuda M. The specific activity of plasminogen activator inhibitor-12 in disseminated intravascular coagulation with acute promyelocytic leukemia. Blood. 1991;77:1949. [PubMed: 1708294]
- 79.
- Goldberg M A, Ginsburg D, Mayer R J. et al. Is heparin administration necessary during induction chemotherapy for patients with acute promyelocytic leukemia? Blood. 1987;69:187. [PubMed: 3466655]
- 80.
- Gralnick H R, Bagley J, Abrell E. Heparin treatment for the hemorrhagic diathesis of acute promyelocytic leukemia. Am J Med. 1972;52:167. [PubMed: 4500954]
- 81.
- Avvisati G, Wouterten Cate J, Buller H R, Mandelli F. Tranexamic acid for control of hemorrhage in acute promyleocytic leukemia. Lancet. 1989;2:122. [PubMed: 2567893]
- 82.
- Degos L, Dombret H, Chomienne C. et al. All-trans-retinoic acid as a differentiating agent in the treatment of acute promyelocytic leukemia. Blood. 1995;85:2643. [PubMed: 7742522]
- 83.
- Larson R A, Kondo K, Vardiman J W. et al. Evidence of a 15;17 translocation in every patient with acute promyelocytic leukemia. Am J Med. 1984;76:827. [PubMed: 6586073]
- 84.
- Slack J L, Arthur D C, Lawrence D. et al. Secondary cytogenetic changes in acute promyelocytic leukemia-prognostic importance in patients treated with chemotherapy alone and association with the nitron 3 breakpoint of the PML gene: a Cancer and Leukemia Group B study. J Clin Oncol. 1997;15:1786. [PubMed: 9164186]
- 85.
- Borrow J, Goddard A D, Sheer D, Solomon E. Molecular analysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science. 1990;249:1577. [PubMed: 2218500]
- 86.
- de The H, Chomienne C, Lanotte M, Degos L, Dejean A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature. 1990;347:558. [PubMed: 2170850]
- 87.
- de The H, Lavau C, Marchio A. et al. The PML-RARa fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 1991;66:675. [PubMed: 1652369]
- 88.
- Diverio D, Rossi V, Avvisati G. et al. Early detection of relapse by 7 prospective reverse transcriptase-polymerase chain reaction analysis of the PML/RAR alpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter “AIDA” trial. GIMEMA-AIEOP Multicenter “AIDA” Trial. Blood. 1998;92:784. [PubMed: 9680345]
- 89.
- Grignani F, De Matteis S, Nervi C. et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukemia. Nature. 1998;391:815. [PubMed: 9486655]
- 90.
- Lin R J, Nagy L, Inoue S. et al. Role of the histone deacetylase complex in acute promyelocytic leukemia. Nature. 1998;391:811. [PubMed: 9486654]
- 91.
- Melnick A, Licht J D. Deconstructing a disease: RARα, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999;93:3167. [PubMed: 10233871]
- 92.
- He L Z, Guidez F, Tribioli C. et al. Distinct interactions of PML-RARalpha and PLZF-RARalpha with co-repressors determine differential responses to RA in APL. Nat Genet. 1998;18:126. [PubMed: 9462740]
- 93.
- Daly P A, Schiffer C A, Wiernik P H. Acute promyelocytic leukemia: clinical management of 15 patients. Am J Hematol. 1980;8:347. [PubMed: 6932177]
- 94.
- Kantarjian H M, Keatin M J, McCredie K B. et al. A characteristic pattern of leukemic cell differentiation without cytoreduction during remission induction in acute promyelocytic leukemia. J Clin Oncol. 1985;3:793. [PubMed: 3859586]
- 95.
- Arthur D C, Bloomfield C D. Partial deletion of the long arm of chromosome 16 and bone marrow eosinophilia in acute nonlymphocytic leukemia: a new association. Blood. 1983;61:994. [PubMed: 6831056]
- 96.
- Hogge D E, Misawa S, Parsa N Z, Pollak A, Testa J R. Abnormalities of chromosome 16 in association with acute myelomonocytic leukemia and dysplastic bone marrow eosinophils. J Clin Oncol. 1984;2:550. [PubMed: 6587018]
- 97.
- Kantarjian H M, Keating M J, Walters R S, McCredie K B, Freireich E J. The characteristics and outcome of patients with late relapse acute myelogeneous leukemia. J Clin Oncol. 1988;6:232. [PubMed: 3276823]
- 98.
- Holmes R, Keating M J, Cork A. et al. A unique pattern of central nervous system leukemia in acute myelomonocytic leukemia associated with inv(16)(p13q22). Blood. 1985;65:1071. [PubMed: 3857943]
- 99.
- Claxton D F, Liu P, Marlton P. et al. Detection of fusion transcripts generated by the inversion 16 chromosome in acute myelogenous leukemia. Blood. 1994;83:1750. [PubMed: 8142642]
- 100.
- Bloomfield C D, Lawrence D, Byrd J C. et al. Frequency of prolonged remission duration after high dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998;58:4173. [PubMed: 9751631]
- 101.
- Kuss B J, Deeley R G, Cole S P C. et al. Deletion of the gene for the multidrug resistance associated protein (MRP) is associated with good prognosis in acute myeloid leukemia with the inversion of chromosome 16. Lancet. 1994;343:1531. [PubMed: 7911871]
- 102.
- Cuttner J, Conjalka M S, Reilly M. et al. Association of monocytic leukemia in patients with extreme leukocytosis. Am J Med. 1980;69:555. [PubMed: 6932815]
- 103.
- Dutcher J P, Schiffer C A, Wiernik P H. Hyperleukocytosis in adult acute nonlymphocytic leukemia: impact on remission rate and duration, and survival. J Clin Oncol. 1987;5:1364. [PubMed: 3625254]
- 104.
- Straus D J, Mertelsmann R, Koziner B. et al. The acute monocytic leukemias: multidisciplinary studies in 45 patients. Medicine (Baltimore). 1980;59:409. [PubMed: 7003298]
- 105.
- Scott C S, Strak A N, Limbert J H. et al. Diagnostic and prognostic factors in acute monocytic leukemia: an analysis of 51 cases. Br J Haematol. 1988;69:247. [PubMed: 3291931]
- 106.
- Eshghabadi M, Shojania A M, Carr I. Isolated granulocytic sarcoma: report of a case and review of literature. J Clin Oncol. 1986;4:912. [PubMed: 2423654]
- 107.
- Neiman R S, Barcos M, Berard C. et al. Granulocytic sarcoma: a clinopathologic study of 61 biopsied cases. Cancer. 1981;48:1426. [PubMed: 7023656]
- 108.
- Schiffer C A, Wiernik P H. Functional evaluation of circulating leukemic cells in acute non-lymphocytic leukemia. Leuk Res. 1977;1:271.
- 109.
- Sexton C, Buss D, Powell B. et al. Usefulness and limitations of serum and urine lysozyme levels in the classification of acute myeloid leukemia: an analysis of 208 cases. Leuk Res. 1996;20:467. [PubMed: 8709618]
- 110.
- Tobelman G, Jacquillat C, Chastang C. et al. Acute monoblastic leukemia: a clinical and biologic study of 74 cases. Blood. 1980;55:71. [PubMed: 6927957]
- 111.
- Fourth International Workshop on Chromosomes in Leukemia 1982. Clinical significance of chromosomal abnormalities in acute nonlymphoblastic leukemia. Cancer Genet Cytogenet. 1984;11:332. [PubMed: 6704938]
- 112.
- Kaneko Y, Maseki N, Takasaki N. et al. Clinical and hematologic characteristics in acute leukemia with 11q23 translocation. Blood. 1986;67:484. [PubMed: 3942833]
- 113.
- Rubnitz J E, Behm F G, Pui C H. et al. Genetic studies of childhood acute lymphoblastic leukemia with emphasis on p16, MLL, and ETV6 gene abnormalities: results of St. Jude Total Therapy Study XII. Leukemia. 1997;11:1201. [PubMed: 9264370]
- 114.
- Rubnitz J E, Behm F, Downing J R. 11q23 rearrangements in acute leukemia. Leukemia. 1996;10:74. [PubMed: 8558942]
- 115.
- Hunger S P, Cleary M L. What significance should be attributed to the detection of MLL fusion transcripts? Blood. 1998;92:709. [PubMed: 9680337]
- 116.
- Mrozek K, Heinonen K, Lawrence D. et al. Adult patients with de novo acute myeloid leukemia and t(9;11)(p22;q23) have a superior outcome to patients with other translocations involving band 11q23: a Cancer and Leukemia Group B study. Blood. 1997;90:4532. [PubMed: 9373264]
- 117.
- Heim S, Avanzi G C, Billstrom R. et al. A new specific chromosomal rearrangement t(8;16) (p11,p13) in acute monocytic leukemia. Br J Haematol. 1987;66:323. [PubMed: 3476150]
- 118.
- Giles R H, Dauwerse J G, Higgins C. et al. Detection of CBP rearrangements in acute myelogeous leukemia with t(8;16). Leukemia. 1997;11:2087. [PubMed: 9447825]
- 119.
- Boyes J, Byfield P, Nakatani Y, Ogryzko V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature. 1998;396:594. [PubMed: 9859997]
- 120.
- Olopade O I, Thangavelu M, Larson R A. et al. Clinical, morphologic, and cytogenetic characteristics of 26 patients with acute erythroblastic leukemia. Blood. 1992;80:2873. [PubMed: 1450412]
- 121.
- Nakamura H. Cytogenetic heterogeneity in erythroleukemia defined as M6 by the French-American-British (FAB) Cooperative Group Criteria. Leukemia. 1990;4:517. [PubMed: 2927179]
- 122.
- Bernstein R, Pinto M R, Beher A, Mendelow B. Chromosome 3 abnormalities in acute nonlymphocytic leukemia (ANLL) with abnormal thrombopoiesis: report of three patients with a new inversion anomaly and a further case of homologous translocation. Blood. 1982;60:613. [PubMed: 7104489]
- 123.
- Bitter M A, Neilly M E, Le Beau M M, Pearson M G, Rowley J D. Rearrangements of chromosome 3 involving bands 3q21 and 3q26 are associated with normal or elevated platelet counts in acute nonlymphocytic leukemia. Blood. 1985;66:1362. [PubMed: 4063525]
- 124.
- Lee E J, Schiffer C A, Tomiyasu T, Testa J R. Clinical and cytogenetic correlations of abnormal megakaryocytopoiesis in patients with acute leukemia and chronic myelogenous leukemia in blast crisis. Leukemia. 1990;4:350. [PubMed: 2388480]
- 125.
- Suzukawa K, Parganas E, Gajjar A. et al. Identification of a breakpoint cluster region 3’ of the ribophorin I gene at 3q21 associated with the transcriptional activation of the EV11 gene in acute myelogenous leukemias with inv(3)(q21q26). Blood. 1994;84:2681. [PubMed: 7919381]
- 126.
- Breton-Gorius J, Reyes F, Duhamel G, Najma S, Gorin N C. Megakaryoblastic acute leukemia: identification by the ultrastructural demonstration of platelet peroxidase. Blood. 1978;51:45. [PubMed: 201318]
- 127.
- Huang M, Li C Y, Nichols W L, Young J H, Katzmann J A. Acute leukemia with megakaryocytic differentiation: a study of 12 cases identified immunocytochemically. Blood. 1984;64:427. [PubMed: 6378277]
- 128.
- Bain B J, Catovsky D, O’Brien M. et al. Megakaryoblastic leukemia presenting as acute myelofibrosis: a study of four cases with the platelet-peroxidase reaction. Blood. 1981;58:206. [PubMed: 6264993]
- 129.
- San Miguel F J, Gonzalez M, Canizo M O. et al. Leukemias with megakaryoblastic involvement: clinical, hematologic, and immunologic characteristics. Blood. 1988;72:402. [PubMed: 3165292]
- 130.
- Carroll A, Civin C, Schneider N. et al. The t(1;22)(p13;q13) is nonrandom and restricted to infants with acute megakaryoblastic leukemia: a Pediatric Oncology Group study. Blood. 1991;78:748. [PubMed: 1859887]
- 131.
- Hoffbrand A V, Leber B F, Browett P J, Norton J D. Mixed acute leukaemias. Blood Rev. 1988;2:9. [PubMed: 3289656]
- 132.
- Mirro J, Zipf T F, Pui C H. et al. Acute mixed lineage leukemia: clinicopathologic correlations and prognostic significance. Blood. 1985;66:115. [PubMed: 3931724]
- 133.
- Kantarjian H M, Hirsch-Ginsberg C, Yee G. et al. Mixed-lineage leukemia revisited: acute lymphocytic leukemia with myeloperoxidase-positive blasts by electron microscopy. Blood. 1990;76:808. [PubMed: 2166608]
- 134.
- Benedetto P, Mertelsmann R, Szatrowski T H. et al. Prognostic significance of terminal deoxynucleotidyl transferase activity in acute nonlymphoblastic leukemia. J Clin Oncol. 1986;4:489. [PubMed: 3958762]
- 135.
- Schachner J, Kantarjian H, Dalton W. et al. Cytogenetic association and prognostic significance of bone marrow blast cell terminal transferase in patients with acute myeloblastic leukemia. Leukemia. 1988;2:667. [PubMed: 3172842]
- 136.
- Parreira A, Pombo de Oliveira M S, Matutes E. et al. Terminal deoxynucleotidyl transferase positive acute myeloid leukaemia: an association with immature myeloblastic leukaemia. Br J Haematol. 1988;69:219. [PubMed: 3164631]
- 137.
- Creutzig U, Harbott J, Sperling C. et al. Clinical significance of surface antigen expression in children with acute myeloid leukemia: results of study AML-BFM-87. Blood. 1995;86:3097. [PubMed: 7579404]
- 138.
- Cross A H, Goorha R M, Nuss R. et al. Acute myeloid leukemia with T-lymphoid features: a distinct biologic and clinical entity. Blood. 1988;72:579. [PubMed: 3261183]
- 139.
- Ball E D, Davis R, Griffin J D. et al. Prognostic value of lymphocyte surface markers in acute myeloid leukemia (AML): a Cancer and Leukemia Group B (CALGB) Study. Blood. 1991;77:2242. [PubMed: 1709379]
- 140.
- Saxena A, Sheridan D P, Card R T. et al. Biologic and clinical significance CD7 expression in acte myeloid leukemia. Am J Hematol. 1998;58:278. [PubMed: 9692390]
- 141.
- Jensen A W, Hokland M, Jorgensen H. et al. Solitary expression of CD7 among T-cell antigens in acute myeloid leukemia: identification of a group of patients with similar T-cell receptor β and γ rearrangements and course of disease suggestive of poor prognosis. Blood. 1991;78:1292. [PubMed: 1715222]
- 142.
- Kita K, Miwa H, Nakase K. et al. Clinical importance of CD7 expression in acute myelocytic leukemia. Blood. 1993;81:2399. [PubMed: 7683218]
- 143.
- Chen C S, Sorensen P H, Domer P H. Molecular rearrangements on chromosome 11q23 predominate in infant acute lymphoblastic leukemia and are associated with specific biologic variables and poor outcome. Blood. 1993;81:2386. [PubMed: 8481519]
- 144.
- Pui C H, Dahl G V, Melvin S. et al. Acute leukaemia with mixed lymphoid and myeloid phenotype. Br J Haematol. 1984;56:121. [PubMed: 6584167]
- 145.
- Stass S A, Mirro J. Lineage heterogeneity in acute leukemia: acute mixed lineage leukemia and lineage switch. Clin Haematol. 1986;15:811. [PubMed: 3536242]
- 146.
- Hoelzer D, Thiel E, Loffler H. et al. Prognostic factors in multicenter study for treatment of acute lymphoblastic leukemia in adults. Blood. 1998;71:123. [PubMed: 3422030]
- 147.
- Larson R A, Dodge R K, Linker C A. et al. A randomized controlled trial of filgrastim during remission induction and consolidation chemotherapy for adults with acute lymphoblastic leukemia: CALGB study 9111. Blood. 1998;92:1556. [PubMed: 9716583]
- 148.
- Williford S K, Salisburg P L III, Peacock J E. et al. The safety of dental extractions in patients with hematologic malignancies. J Clin Oncol. 1989;7:798. [PubMed: 2523958]
- 149.
- Saral R, Arminder R F, Burns W H. et al. Acyclovir prophylaxis against herpes simplex virus infection in patients with leukemia: a randomized, double-blind, placebo-controlled study. Ann Intern Med. 1983;99:773. [PubMed: 6359995]
- 150.
- Carr BR, Bradshaw KD. Disorders of the ovary and female reproductive tract. In Harrison’s Principles of Internal Medicine. 14th Ed. Edited by AS Fauci, E Braunwald, KJ Isselbacher. New York: McGraw-Hill, 1998; p 2097.
- 151.
- Fearon E R, Burke P J, Schiffer C A, Zehnbauer B A, Vogelstein B. Evidence for differentiation of leukemia cells to polymorphonuclear leukocytes in patients with acute non-lymphocytic leukemia. N Engl J Med. 1986;315:15. [PubMed: 3086723]
- 152.
- Hittelman W, Agbor P, Petkovic I. et al. Detection of leukemic clone maturation in vivo by premature chromosome condensation. Blood. 1988;72:1950. [PubMed: 3196873]
- 153.
- Jacobson R J, Temple M J, Singer J W. et al. A clonal complete remission in a patient with acute nonlymphocytic leukemia originating in multipotent stem cell. N Engl J Med. 1984;310:1513. [PubMed: 6717542]
- 154.
- Busque L, Gilliland D G. Clonal evolution in acute myeloid leukemia. Blood. 1993;82:337. [PubMed: 8329693]
- 155.
- Gale R E, Wheadon H, Goldstone A H, Burnett A K, Linch D C. Frequency of clonal remission in acute myeloid leukaemia. Lancet. 1993;341:138–142. [PubMed: 8093744]
- 156.
- Stone R M, Maguire M, Goldberg M A. et al. Complete remission in acute promyelocytic leukemia despite persistence of abnormal bone marrow promyelocytes during induction therapy: experience in 34 patients. Blood. 1988;71:690. [PubMed: 3422828]
- 157.
- Bowman G, Preisler H, Raza A. et al. Use of the day 6 bone marrow to alter remission induction therapy in patients with acute myeloid leukaemia: a leukemia intergroup study. Br J Haematol. 1989;71:493. [PubMed: 2653407]
- 158.
- Cassileth P A, Gerson S L, Bonner H. et al. Identification of early relapsing patients with adult acute nonlymphocytic leukemia by bone marrow biopsy after initial induction chemotherapy. J Clin Oncol. 1984;2:107. [PubMed: 6699661]
- 159.
- Preisler H, Bjornsson S, Henderson E S. et al. Remission induction in acute nonlymphocytic leukemia: comparison of a seven-day infusion of cytosine arabinoside in combination with adriamycin. Med Pediatr Oncol. 1979;7:269. [PubMed: 542191]
- 160.
- Gajewski J L, Ho W G, Nimer S D. et al. Efficacy of intensive chemotherapy for acute myelogenous leukemia associated with a preleukemic syndrome. J Clin Oncol. 1989;7:1637. [PubMed: 2809680]
- 161.
- Stone R M, Mayer R J. Treatment of the newly diagnosed adult with de novo acute myeloid leukemia. Hematol Oncol Clin North Am. 1993;7:47. [PubMed: 8449864]
- 162.
- Rai K R, Holland J F, Glidewell O J. et al. Treatment of acute myelocytic leukemia: a study by Cancer and Leukemia Group B. Blood. 1981;58:1203. [PubMed: 6946847]
- 163.
- Yates J W, Wallace J Jr, Ellison R R, Holland J F. Cytosine arabinoside (MSC A63878) and daunorubicin (NSC A83142) in acute non-lymphocytic leukemia. Cancer Chemother Rep. 1973;57:485. [PubMed: 4586956]
- 164.
- Weil M, Glidewell O J, Jacquillat C. et al. Daunorubicin in the therapy of acute granulocytic leukemia. Cancer Res. 1973;33:921–928. [PubMed: 4512953]
- 165.
- Dillman R O, Davis R B, Green M R. et al. Comparative study of two remission induction regimens of ara-c for acute myelocytic leukemia (AML). Blood. 1991;78:2520. [PubMed: 1824249]
- 166.
- Wiernik P H, Glidewell O J, Hoagland C. et al. A comparative trial of daunorubicin, cytosine, arabinoside, and thioquanine, and a combination of the three agents for the treatment of acute myelocytic leukemia. Med Pediatr Oncol. 1979;6:261. [PubMed: 381887]
- 167.
- Preisler H D, Anderson K, Rai K. et al. Cancer And Acute Leukemia Group B. The frequency of long-term remission in patients with acute myelogenous leukemia treated with conventional maintenance chemotherapy: a study of 760 patients with a minimal follow-up time of 6 years. Br J Haematol. 1989;71:189. [PubMed: 2923805]
- 168.
- Yates J, Glidewell O, Wiernik P. et al. Cytosine arabinoside with daunorubicin or Adriamycin for therapy for acute myelocytic leukemia: a CALGB study. Blood. 1982;60:454. [PubMed: 6953986]
- 169.
- Berman E, Heller G, Santorsa J. et al. Results of a randomized trial comparing idarubicin and cytosine arabinoside with daunorubicin and cytosine arabinoside in adult patients with newly diagnosed acute nonlymphocytic leukemia. Blood. 1991;77:1666. [PubMed: 2015395]
- 170.
- Clarkson B D, Gee T, Mertelsman R. et al. Current status of treatment of acute leukemia in adults: an overview of the Memorial experience and review of the literature. Crit Rev Oncol Hematol. 1986;4:221. [PubMed: 3513984]
- 171.
- Keating M J, Gehan E A, Smith T L. et al. A strategy for evaluation of new treatments in untreated patients: application to a clinical trial of AMSA for acute leukemia. J Clin Oncol. 1987;5:710. [PubMed: 3553435]
- 172.
- Vogler W R, Velez-Garcia E, Weiner R S. et al. A phase-three trial comparing idarubicin and daunorubicin in combination with cytarabine in acute myelogenous leukemia. A Southeaster Cancer Group Study. J Clin Oncol. 1992;10:1103. [PubMed: 1607916]
- 173.
- Bishop J F, Lowenthal R M, Josua D E. et al. A randomized trial of cytosine arabinoside, daunorubicin 1 VP 160213 (73 VS 737) as initial induction in adults with acute non-lymphocytic leukemia. Blood. 1990;75:27.
- 174.
- Weick J K, Kopecky K J, Appelbaum F R. et al. A randomized investigation of high-dose versus standard-dose cytosine arabinoside with daunorubicin in patients with previously untreated acute myeloid leukemia: a Southwest Oncology Group Study. Blood. 1996;88:2841. [PubMed: 8874180]
- 175.
- Bishop J F, Matthews J P, Young G A. et al. Randomized study of high-dose cytarabine in induction in acute myeloid leukemia. Blood. 1996;87:1710. [PubMed: 8634416]
- 176.
- Schiller G, Gajewski J, Nimer S. et al. A randomized study of intermediate versus conventional-dose cytarabine as intensive induction for acute myelogenous leukemia. Br J Haematol. 1992;81:170. [PubMed: 1643014]
- 177.
- Mitus A J, Miller K B, Schenkein D P. et al. Improved survival for patients with acute myelogenous leukemia. J Clin Oncol. 1995;13:560. [PubMed: 7884416]
- 178.
- Petersdorf S, Rankin C, Terebolo H. et al. A phase II study of standard dose daunomycin and cytosine arabinoside (ara-C) with high dose ara-C induction therapy followed by sequential high dose ara-C consolidation for adults with previously untreated acute myelogenous leukemia: a Southwest Oncology Group study (SWOG 9500). Proc ASCO. 1998;17:55a.
- 179.
- Beran M, Kantarijian H. Topotecan in the treatment of hematologic malignancies. Semin Hematol. 1998;35(Suppl 4):26. [PubMed: 9779879]
- 180.
- Latagliata R, Petti M C, Mandelli F. Acute myeloid leukemia in the elderly: ‘per aspera ad astra’? Leuk Res. 1999;23:603. [PubMed: 10400181]
- 181.
- Bolwell B J, Cassileth P A, Gale R P. Low dose cytosine arabinoside in myelodysplasia and acute myelogenous leukemia: a review. Leukemia. 1987;1:575. [PubMed: 3312846]
- 182.
- Tilly H, Castaigne S, Bordessoule D. et al. Low-dose cytarabine versus intensive chemotherapy in the treatment of acute nonlymphocytic leukemia in the elderly. J Clin Oncol. 1990;8:272. [PubMed: 2299370]
- 183.
- Larson R A, Wernli M, Le Baeu M. et al. Short remission duration in the therapy-related leukemia despite cytogenetic complete responses to high dose cytarabine. J Clin Oncol. 1988;6:495. [PubMed: 3167210]
- 184.
- Preisler H D, Raza A, Barcos M. et al. High-dose cytosine arabinoside as the initial treatment of poor-risk patients with acute nonlymphocytic leukemia: a Leukemia Intergroup study. J Clin Oncol. 1987;5:75. [PubMed: 3806162]
- 185.
- Kantarjian H M, Keating M J, Walters R S. et al. The association of specific favorable cytogenetic abnormalities with secondary leukemia. Cancer. 1986;58:924. [PubMed: 3719557]
- 186.
- Fenaux P, Detuumignies L. Therapy-related acute promyelocytic leukemia. Br J Haematol. 1994;87:445. [PubMed: 7947301]
- 187.
- Estey E, Thall P, Beran M. et al. Effect of diagnosis (refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, or acute myeloid leukemia (AML) on the outcome of AML-type chemotherapy. Blood. 1997;90:2969. [PubMed: 9376577]
- 188.
- Appelbaum F R, Barrall J, Storb R. et al. Bone marrow transplantation for patients with myelodysplasia. Ann Intern Med. 1990;112:590. [PubMed: 2183666]
- 189.
- Anderson J E, Appelbaum F R, Fisher L D. et al. Allogenic bone marrow transplantation for 93 patients with myelodysplastic syndrome. Blood. 1993;82:677. [PubMed: 8329721]
- 190.
- DeWitte T, Zwaan F, Hermans J. et al. Allogeneic bone marrow transplantation for secondary leukaemia and myelodysplastic syndrome: a survey by the Leukemia Working Party of the European one Marrow Transplant Group (EBMTG). Br J Haematol. 1990;74:151. [PubMed: 2180469]
- 191.
- Buchner T H, Urbanitz D, Hiddemann W. et al. Intensified induction and consolidation with or without maintenance chemotherapy for acute myeloid leukemia (AML): two multicenter studies of the German AML cooperative group. J Clin Oncol. 1985;3:1583. [PubMed: 3906048]
- 192.
- Cassileth P A, Harrington D P, Hines J D. et al. Maintenance chemotherapy prolongs remission duration in adult acute nonlymphocytic leukemia. J Clin Oncol. 1988;6:583. [PubMed: 3282032]
- 193.
- Embury S H, Elias L, Heller P H. et al. Remission maintenance therapy in acute myelogenous leukemia. West J Med. 1977;126:267. [PMC free article: PMC1237541] [PubMed: 266313]
- 194.
- Archimbaud E, Thomas X, Leblond V. et al. Timed sequential chemotherapy for previously treated patients with acute myeloid leukemia: long-term follow-up of the etoposide, mitoxantrone, and cytarabine-86 trial. J Clin Oncol. 1995;13:11. [PubMed: 7799010]
- 195.
- Bloomfield C D. Postremission therapy in acute myeloid leukemia. J Clin Oncol. 1985;3:1570. [PubMed: 3906047]
- 196.
- Bodey G P, Freireich E J, Gehan E. et al. Late intensification therapy for acute leukemia in remission: chemotherapy and immunotherapy. JAMA. 1976;235:1021. [PubMed: 946184]
- 197.
- Clarkson B D, Gee T, Mertelsman R. et al. Current status of treatment of acute leukemia in adults: an overview of the Memorial experience and review of the literature. Crit Rev Oncol Hematol. 1986;4:221. [PubMed: 3513984]
- 198.
- Preisler H D, Anderson K, Rai K. et al. for Cancer and Acte Leukemia Group B. The frequency of long-term remission in patients with acute myelogenous leukemia treated with conventional maintenance chemotherapy: a study of 760 patients with a minimal follow-up time of 6 years. Br J Haematol. 1989;71:189. [PubMed: 2923805]
- 199.
- Wiernik P H, Glidewell O J, Hoagland C. et al. A comparative trial of daunorubicin, cytosine arabinoside, and thioguanine, and a combination of the tree agents for the treatment of acute myelocytic leukemia. Med Pediatr Oncol. 1979;6:261. [PubMed: 381887]
- 200.
- Santos G W. Marrow transplantation in acute nonlymphocytic leukemia. Blood. 1989;74:901. [PubMed: 2665867]
- 201.
- Thomas E D, Buckner C D, Clift R A. et al. Marrow transplantation for acute non-lymphoblastic leukemia in first remission. N Engl J Med. 1979;301:597. [PubMed: 381925]
- 202.
- Cassileth P A, Begg C B, Bennett J M. et al. A randomized study of the efficacy of consolidation therapy in adult acute nonlymphocytic leukemia. Blood. 1984;63:843. [PubMed: 6704545]
- 203.
- Cassileth P A, Begg C B, Silber R. et al. Prolonged unmaintained remission after intensive consolidation therapy in adult acute nonlymphocytic leukemia. Cancer Treat Rep. 1987;71:137. [PubMed: 3802110]
- 204.
- Champlin R, Gajewski J, Nimer S. et al. Postremission chemotherapy for adults with acute myelogenous leukemia: improved survival with high-dose cytarabine and daunorubicin consolidation treatment. J Clin Oncol. 1990;8:1199. [PubMed: 1694236]
- 205.
- Herzig R H, Lazarus H M, Wolff S N, Phillips G L, Herzig G P. High dose cytosine arabinoside therapy with and without anthracycline antibiotics for remission reinduction of acute nonlymphoblastic leukemia. J Clin Oncol. 1985;3:992. [PubMed: 3894588]
- 206.
- Kufe D W, Spriggs D R. Biochemical and cellular pharmacology of cytosine arabinoside. Semin Oncol. 1985;12:34. [PubMed: 3925560]
- 207.
- Wolff S N, Herzig R H, Fay J W. et al. High-dose cytarabine and daunorubicin as consolidation therapy for acute myeloid leukemia in first remission: long-term follow-up and results. J Clin Oncol. 1989;7:1260. [PubMed: 2769327]
- 208.
- Philips G L, Reece D E, Shepherd J D. et al. High-dose cytarabine and daunorubicin induction and postremission chemotherapy for the treatment of acute myelogenous leukemia in adults. Blood. 1991;77:1429. [PubMed: 2009367]
- 209.
- Schiffer C A, Davis R, Mayer R J, Holland J, Frei E. The rate of early failure following complete remission (CR) in patients with acute non-lymphocytic leukemia (ANLL): potential effect on the interpretation of bone marrow transplantation (BMT) results. Blood. 1988;72:224a.
- 210.
- Cassileth P A, Lynch E, Hines J D. et al. Varying intensity of postremission therapy in acute myeloid leukemia. Blood. 1992;79:1924. [PubMed: 1562720]
- 211.
- Mayer R J, Davis R B, Schiffer C a. et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Blood. 1992;79:1924. [PubMed: 8078551]
- 212.
- Bloomfield C D, Lawrence D, Byrd J C. et al. Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998;58:4173. [PubMed: 9751631]
- 213.
- Stone R M, Berg D T, George S L. et al. Post remission therapy in older patients with de novo acute myeloid leukemia: a randomized trial of mitoxantrone/intermediate dose cytarabine ‘v’ standard dose cytarabine (CALGB 8923). Blood. 1997;90(Suppl 1):2255a.
- 214.
- Lowenberg B, Suciu S, Archimbaud E. et al. Mitoxantrone versus daunorubicin in induction-consolidation chemotherapy-remission, and an assessment of prognostic factors in acute myeloid leukemia in the elderly: final report of the Leukemia Cooperative Group of the European Organization for the Research and Treatment of Cancer and the Dutch-Belgian Hemato-oncology Cooperative HOVON Group randomized phase III study AML-9. J Clin Oncol. 1998;16:872. [PubMed: 9508168]
- 215.
- Clift R A, Buckner C D, Thomas E D. et al. The treatment of acute nonlymphoblastic leukemia by allogeneic marrow transplantation. Bone Marrow Transplant. 1987;2:243. [PubMed: 3332174]
- 216.
- Fefer A, Cheever M A, Thomas E D. et al. Bone marrow transplantation for refractory acute leukemia in 34 patients with identical twins. Blood. 1981;57:421. [PubMed: 7006708]
- 217.
- Blume K G, Bentler E, Bross K J. et al. Bone marrow ablations and allogeneic marrow transplantation in acute leukemia. N Engl J Med. 1980;302:1041. [PubMed: 6245359]
- 218.
- Zittoun R A, Mandelli F, Willemze R. et al. for the European Organization for Research and Treatment of Cancer (EORTC) and the Gruppo Italiano Malattie Ematologiche Maligne Dell’Adulto (GIMEMA). Leukemia Cooperative Groups: autologous or allogeneic bone marrow transplantation compared with intensive chemotherapy in acute myelogenous leukemia. N Engl J Med. 1995;332:217–223. [PubMed: 7808487]
- 219.
- Harousseau J L, Cahn J Y, Pignon B. et al. Comparison of autologous bone marrow transplantation and intensive chemotherapy as postremission therapy in adult acute myeloid leukemia. The Groupe Ouest Leucemies Aigues Myeloblastiques (GOELAM). Blood. 1997;90:2978. [PubMed: 9376578]
- 220.
- Burnett A, Goldstone A H, Stevens R M. et al. Randomized comparison of addition of autologous bone-marrow transplantation to intensive chemotherapy of acute myeloid leukemia in first remission: results of MRC AML 10 trial. UK Medical Research Council Adult and Children’s Leukaemia Working Parties. Lancet. 1998;351:700. [PubMed: 9504514]
- 221.
- Cassileth P A, Harrington D P, Appelbaum F R. et al. Chemotherapy compared with autologous or allogeneic bone marrow transplantation in the management of acute myeloid leukemia in first remission. N Engl J Med. 1998;339:1649. [PubMed: 9834301]
- 222.
- Yeager A M, Kaizer H, Santos G W. et al. Autologous bone marrow transplantation in patients with acute nonlymphocytic leukemia, using ex vivo marrow treatment with 4-hydroperoxycyclophosphamide. N Engl J Med. 1986;315:141. [PubMed: 3523241]
- 223.
- Saz M A, de la Rubia J, Sanz G F. et al. Busulfan plus cyclophosphamide followed by autologous blood stem-cell transplantation for patients with acute myeloblastic leukemia in first complete remission: a report from a single institution. J Clin Oncol. 1993;11:1661. [PubMed: 8102640]
- 224.
- Ball E B, Mills L E, Corwell G G III. et al. Autologous bone marrow transplantation for acute myeloid leukemia using monoclonal antibody-purged bone marrow. Blood. 1990;75:1199. [PubMed: 2306522]
- 225.
- Robertson M J, Soiffer R J, Freedman A S. et al. Human bone marrow depleted of CD33-positive cells mediates delayed but durable reconstitution of hematopoiesis: clinical trial of MY9 monoclonal antibody-purged autografts for the treatment of acute myeloid leukemia. Blood. 1992;79:2229. [PubMed: 1571539]
- 226.
- Gorin N C, Aegerter P, Auvert B. et al. Autologous bone marrow transplantation for acute myelocytic leukemia in first remission: a European survey of the role of marrow purging. Blood. 1990;75:1606. [PubMed: 2328313]
- 227.
- Linker C A, Ries C A, Damon L E, Rugo H S, Wolf J L. Autologous bone marrow transplantation for acute myeloid leukemia using busulfan plus etoposide as a preparative regimen. Blood. 1993;81:311. [PubMed: 8422457]
- 228.
- Gorin N C. Autologous stem cell transplantation in acute myelocytic leukemia. Blood. 1998;92:1073. [PubMed: 9694694]
- 229.
- Cassileth P A, Andersen J, Lazarus H M. et al. Autologous bone marrow transplant in acute myeloid leukemia in first remission. J Clin Oncol. 1993;11:314. [PubMed: 8426209]
- 230.
- Hermans J, Suciu S, Stijnen T H. et al. Treatment of acute myelogenous leukemia. An EBMT EORTC retrospective analysis of chemotherapy versus allogeneic or autologous bone marrow transplantation. Eur J Cancer Clin Oncol. 1989;25:545. [PubMed: 2649378]
- 231.
- Zander A R, Keating M, Dicke K. et al. A comparison of marrow transplantation with chemotherapy for adults with acute leukemia of poor prognosis in first complete remission. J Clin Oncol. 1988;6:1548. [PubMed: 3049949]
- 232.
- Appelbaum F R, Fisher L D, Thomas E D. Chemotherapy versus marrow transplantation with acute nonlymphocytic leukemia. A 5-year follow-up. Blood. 1988;72:179. [PubMed: 3291979]
- 233.
- Champlin R E, Ho W G, Gale R P. et al. Treatment of acute myelogenous leukemia. A prospectively controlled trial of bone marrow transplantation versus consolidation chemotherapy. Ann Intern Med. 1985;102:285. [PubMed: 3882039]
- 234.
- Slovak M L, Kopecky K J, Cassileth P A. et al. Karyotypic analysis predicts outcome of pre- and post-remission therapy in adult acute myeloid leukemia (AML): a SWOG/ECOG Intergroup study. Blood. 1998;92:2795a. [PubMed: 11110676]
- 235.
- Giralt S, Estey E, Albiters et al. Engraftment of allogeneic hematopoietic progenitor cells with purine analog-containing chemotherapy: harnessing graft-versus-leukemia with myeloblative therapy. Blood. 1997;89:4531. [PubMed: 9192777]
- 236.
- Guinan E C, Boussiotis V A, Neuberg D. et al. Transplantation of anergic histoincompatible bone marrow allografts. N Engl J Med. 1999;340:1704. [PubMed: 10352162]
- 237.
- Aversa F, Tabilio A, Velardi A. et al. Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N Engl J Med. 1998;339:1186. [PubMed: 9780338]
- 238.
- Henslee-Downey P J, Abhyankar S H, Parrish R S. et al. Use of partially mismatched related donors extends access to allogeneic marrow transplant. Blood. 1997;89:3864. [PubMed: 9160695]
- 239.
- Beatty P G, Dahlberg S, Mickelson E M. et al. Probability of finding HLA-matched unrelated marrow donors. Transplantation. 1988;45:714. [PubMed: 3282353]
- 240.
- Bostrom B, Brunning R D, McGlave P. et al. Bone marrow transplantation for acute nonlymphocytic leukemia in first remission: analysis of prognostic factors. Blood. 1985;65:1. [PubMed: 3888309]
- 241.
- Tallman M S, Kopecky K J, Amos D. et al. Analysis of prognostic factors for the outcome of marrow transplantation or further chemotherapy for patients with acute nonlymphocytic leukemia in first remission. J Clin Oncol. 1989;7:326. [PubMed: 2645386]
- 242.
- Sullivan K M, Storb R, Buckner C D. et al. Graft-versus-host disease as adoptive immunotherapy in patients with advanced hematologic neoplasms. N Engl J Med. 1989;320:828. [PubMed: 2648143]
- 243.
- Sullivan K M, Weiden P L, Storb R. et al. Influence of acute and chronic graft-versus-host disease on relapse and survival after bone marrow transplantation from HLA-identical siblings as treatment of acute and chronic leukemia. Blood. 1989;73:1720. [PubMed: 2653460]
- 244.
- Soiffer R J, Fairlough D, Robertson M. et al. CD6-depleted allogeneic bone marrow transplantation for acute leukemia in first complete remission. Blood. 1997;89:3039. [PubMed: 9108425]
- 245.
- Papadopoulos E B, Carabasi M H, Castro-Malaspina H. et al. T-cell depleted allogeneic bone marrow transplantation as postremission therapy for acute myelogenous leukemia: Freedom from relapse in the absence of graft-versus-host disease. Blood. 1998;91:1083. [PubMed: 9446672]
- 246.
- Witherspoon R P, Fisher L D, Schoch G. et al. Secondary cancers after bone marrow transplantation for leukemia or aplastic anemia. N Engl J Med. 1989;321:784. [PubMed: 2671734]
- 247.
- Brown R A, Herzig R H, Wolff S N. et al. High-dose etoposide and cyclophosphamide without bone marrow transplantation for resistant hematologic malignancy. Blood. 1990;76:473. [PubMed: 2378980]
- 248.
- Hiddemann W, Kreutzmann H, Straif K. et al. High-dose cytosine arabinoside and mitoxantrone: a highly effective regimen in refractory acute myeloid leukemia. Blood. 1987;69:744. [PubMed: 3469002]
- 249.
- Hiddemann W, Martin W R, Sauerland C M, Heinecke A, Buchner T. Definition of refractoriness against conventional chemotherapy in acute myeloid leukemia: a proposal based on the results of retreatment by thioguanine, cytosine arabinoside and daunorubicin (TAD 9) in 150 patients with relapse after standardized first line therapy. Leukemia. 1990;4:184. [PubMed: 2314117]
- 250.
- Ho A D, Lipp T, Ehninger G. et al. Combination of mitoxantrone and etoposide in refractory acute myelogenous leukemia: an active and well-tolerated regimen. J Clin Oncol. 1988;6:213. [PubMed: 3422260]
- 251.
- Keating M J, Kantarjian H, Smith T L. et al. Response to salvage therapy and survival after relapse in acute myelogenous leukemia. J Clin Oncol. 1989;7:1071. [PubMed: 2666590]
- 252.
- Vahdat L, Wong E T, Wile M J. et al. Therapeutic and neurotoxic effects of 2-chlorodeoyadenosine in adults with acute myeloid leukemia. Blood. 1994;84:3429. [PubMed: 7949097]
- 253.
- Ghandi V, Estey E, Keating M J. et al. Fludarabine potentiates metabolism of cytarabine in patients with acute myelogenous leukemia during therapy. J Clin Oncol. 1993;11:116. [PubMed: 8418222]
- 254.
- Rowinsky E K, Adjei A, Donehower R C. et al. Phase I and pharmacodynamic study of the topoismerase I-inhibitor topotecan in patients with refractory acute leukemia. J Clin Oncol. 1994;12:2193. [PubMed: 7931489]
- 255.
- Amadori S, Arcese W, Isacchi G. et al. Mitoxantrone, etoposide, and intermediate-dose cytarabine: an effective and tolerable regimen for the treatment of refractory acute myeloid leukemia. J Clin Oncol. 1991;9:1210. [PubMed: 2045861]
- 256.
- Lee E J, Egorin M J, Van Echo D A. et al. A phase I and pharmacokinetic trial of carboplatin in refractory adult leukemia. J Natl Cancer Inst. 1988;80:131. [PubMed: 3278122]
- 257.
- Lee E J, Van Echo D A, Egorin M J. et al. Diaziquone given as a continuous infusion is an active agent for relapsed adult acute non-lymphocytic leukemia. Blood. 1986;67:182. [PubMed: 3940546]
- 258.
- Warrell R P Jr, Coonley C J, Gee T S. Homoharringtonine: an effective new drug for remission induction in refractory nonlymphoblastic leukemia. J Clin Oncol. 1985;3:617. [PubMed: 3889229]
- 259.
- Schiffer C A, Lee E J. Approaches to the therapy of relapsed acute myeloid leukemia. Oncology. 1989;3:23. [PubMed: 2519195]
- 260.
- Rees J K H, Swirsky D, Gray R G, Hayhoe F G J. Principal results of the Medical Research Councils 8th acute myeloid leukaemia trial. Lancet. 1986;2:1236. [PubMed: 2878130]
- 261.
- Kantarjian H M, Keating M J, Walters R S, McCredie K B, Freireich E J. The characteristics and outcome of patients with late relapse acute myelogenous leukemia. J Clin Oncol. 1988;6:232. [PubMed: 3276823]
- 262.
- Clift R A, Buckner C D, Appelbaum F R. et al. Allogeneic marrow transplantation during untreated first relapse of acute myeloid leukemia. J Clin Oncol. 1992;10:1723. [PubMed: 1403055]
- 263.
- Kumar L. Leukemia: management of relapse after allogeneic bone marrow transplantation. J Clin Oncol. 1994;12:1710. [PubMed: 8040682]
- 264.
- Vallenga E, Young D C, Wagner K. et al. The effects of GM-CSF and G-CSF in promoting growth of clonogenic cells in acute myeloblastic leukemia. Blood. 1987;69:1771. [PubMed: 3495305]
- 265.
- Ohno R, Tomonaga M, Kobayashi T. et al. Effect of granulocyte colony-stimulating factor after intensive induction therapy in relapsed or refractory acute leukemia. N Engl J Med. 1990;323:871. [PubMed: 1697646]
- 266.
- Buchner T, Hiddemann W, Koenigsmann M. et al. Recombinant human granulocyte-macrophage colony-stimulating factor after chemotherapy in patients with acute myeloid leukemia at higher age or after relapse. Blood. 1991;78:1190–1197. [PubMed: 1878586]
- 267.
- Rowe J, Andersen J, Mazza J J. et al. A randomized placebo-controlled phase II study of granulocyte-macrophage colony-stimulating factor in adult patients (>55 to 70 years of age) with acute myelogenous leukemia: a study of the Eastern Cooperative Oncology Group (E1490). Blood. 1995;86:457. [PubMed: 7605984]
- 268.
- Stone R M, Berg D T, George S L. et al. Granulocyte-macrophage colony-stimulating factor after initial chemotherapy for elderly patients with primary acute myelogenous leukemia. N Engl J Med. 1995;332:1671. [PubMed: 7760868]
- 269.
- Godwin J E, Kopecky K J, Head D R. et al. A double-blind placebo-controlled trial of granulocyte colony-stimulating factor in elderly patients with previously untreated acute myeloid leukemia: a Southwest Oncology Group (9031). Blood. 1998;91:3607. [PubMed: 9572995]
- 270.
- Heil G, Hoelzer D, Sanz M A. et al. A randomized, double-blind, placebo-controlled, phase II study of filgrastim in remission induction and consolidation therapy for adults with de novo acute myeloid leukemia. Blood. 1997;90:4710. [PubMed: 9389686]
- 271.
- Dombret H, Chastang C, Fenaux P. et al. A controlled study of recombinant human granulocyte-stimulating factor in elderly patients after treatment for acute myelogenous leukemia. N Engl J Med. 1995;332:1678. [PubMed: 7539109]
- 272.
- Lowenberg B, Suciu S, Archimbaud Ossenkopple G. et al. Using of recombinant granulocyte-macrophage colony-stimulating factor during and after remission induction chemotherapy in patients 61 years and older with acute myeloid leukemia (AML): final report of AML-11, a phase III randomized study of the Leukemia Cooperative Group of European Organization and Treatment of Cancer (EORTC-LCG) and Dutch Belgian Hemato-oncology Cooperative Group (HOVON). Blood. 1997;90:2952. [PubMed: 9376575]
- 273.
- Witz F, Sadoun A, Perrin M -C. et al. A placebo-controlled study of recombinant human granulocyte-macrophage colony-stimulating factor administered during and after induction treatment for de novo acute myelogenous leukemia in elderly patients. Blood. 1998;91:2722. [PubMed: 9531581]
- 274.
- Schiffer C A, Miller K, Larson R A. et al. A double-blind, placebo-controlled trial of pegylated recombinant human megakaryocyte growth and development factor as an adjunct to induction and consolidation therapy for patients with acute myeloid leukemia. Blood. 2000;95:2530. [PubMed: 10753831]
- 275.
- Archimbaud E, Ottmann O G, Yin J A. et al. A randomized double-blind, placebo-controlled study with pegylated recombinant human megakaryocyte growth and development factor (PEG-rHuMGDF) as an adjunct to chemotherapy for adults with de novo acute myeloid leukemia. Blood. 1999;94:3694. [PubMed: 10572081]
- 276.
- Bhalla K, Tang C, Ibrado A M. et al. Granulocyte macrophage colony-stimulating factor/interleukin-3 fusion protein (pIXY 321) enhances high-dose ara-C-induced programmed cell death or apoptosis in human myeloid leukemia cells. Blood. 1992;80:2883. [PubMed: 1450413]
- 277.
- Cannistra S A, Groshek P, Griffin J D. Granulocyte-macrophage colony-stimulating factor enhances the cytotoxic effects of cytosine arabinoside in acute myeloblastic leukemia and in the myeloid blast crisis phase of chronic myeloid leukemia. Leukemia. 1989;3:328. [PubMed: 2654494]
- 278.
- Miyauchi J, Kelleher C A, Wange C, Minkin S, McCulloch E A. Growth factors influence the sensitivity of leukemic stem cells to cytosine arabinoside in culture. Blood. 1989;73:1272. [PubMed: 2784697]
- 279.
- Tafuri A, Lemoli R M, Chen R. et al. Combination of hematopoietic growth factors containing IL-3 induce acute myeloid leukemia cell sensitization by cycle specific and cycle non-specific drugs. Leukemia. 1994;8:749. [PubMed: 7514244]
- 280.
- de Boekhorst P A W, LËwenberg B, Sonneveld P. Hematopoietic growth factor stimulation and cytarabine cytotoxicity in vitro: effects in untreated and relapsed or primary refractory acute myeloid leukemia cells. Leukemia. 1994;8:1480. [PubMed: 7522289]
- 281.
- Estey E H, Dixon D, Kantarjian H M. et al. Treatment of poor-prognosis, newly diagnosed acute myeloid leukemia with ara-C and recombinant human granulocyte-macrophage colony-stimulating factor. Blood. 1990;75:1766. [PubMed: 2184901]
- 282.
- Rowe J M, Neuberg D, Friedenberg W. et al. A phase II study of priming with yeast-derived granulocyte-macrophage colony-stimulating-factor (rhuGM-CSF) for older adult patients (>55 yrs) with acute myelogenous leukemia (AML): a study of the Eastern Cooperative Oncology Group. Blood. 1998;92(Suppl 1):2799a.
- 283.
- Peterson B A, George S L, Bhalla K. et al. A phase II trial with and without GM-CSF administered before and during high dose cytarabine in patients with relapsed refractory acute myelogenous leukemia. Proc ASCO. 1996;15:504a.
- 284.
- Moore J O, Dodge R K, Amrein P C. et al. Granulocyte-colony stimulating factor (Filgrastim) accelerates granulocyte recovery after intensive post-remission chemotherapy for acute myeloid leukemia with aziridinyl benzoquinone and mitoxantrone: Cancer and Leukemia Group B Study 9022. Blood. 1997;89:780. [PubMed: 9028308]
- 285.
- Gale R P, Horowitz M M, Ash R C. et al. Identical-twin bone marrow transplants for leukemia. Ann Intern Med. 1994;120:646. [PubMed: 8135448]
- 286.
- Reittie J E, Gottlieb D, Heslp H E. et al. Endogenously generated activated killer cells circulate after autologous and allogeneic marrow transplantation but not after chemotherapy. Blood. 1989;73:1351. [PubMed: 2495037]
- 287.
- Soiffer R J, Murray C, Gonin R, Ritz J. Effect of low-dose interleukin-2 on disease relapse after T-cell-depleted allogeneic bone marrow transplantation. Blood. 1994;84:964. [PubMed: 8043878]
- 288.
- Foa R. Does interleukin-2 have a role in the management of acute leukemia? J Clin Oncol. 1993;11:1817. [PubMed: 8355048]
- 289.
- Meloni G, Foa R, Vignetti M. et al. Interleukin-2 may induce prolonged remissions in advanced acute myelogenous leukemia. Blood. 1994;84:2158. [PubMed: 7919330]
- 290.
- Caron P C, Scheinberg D A. Immunotherapy for acute leukemias. Curr Opin Oncol. 1994;6:14. [PubMed: 8204688]
- 291.
- Jurcic J G, Divgi C R, McDevitt M R. et al. Potential for myeblation with tritium-90-labeled HuM195 (ANTI-CD33): a phase I trial in advanced myeloid leukemia. Blood. 1998;92(Suppl 1):2526a.
- 292.
- Sievers E L, Appelbaum F R, Spielberger R T. et al. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: a phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood. 1999;93:3678. [PubMed: 10339474]
- 293.
- Sievers E, Larson R A, Estey E. et al. Preliminary results of the efficacy and safety of CMA-676 in patients with AML in first relapse. Proc Am Soc Clin Oncol. 1999;18:7a.
- 294.
- Cignetti A, Bryant E, Allione B. et al. CD34(+) acute myeloid and lymphoid leukemic blasts can be induced to differentiate to dendritic cells. Blood. 1999;94:2048. [PubMed: 10477734]
- 295.
- Dunussi-Joannopoulos K, Dranoff G, Weinstein H J. et al. Gene immunotherapy in murine acute myeloid leukemia: granulocyte-macrophage colony-stimulating factor tumor cell vaccines elicit more potent antitumor immunity compared with B7 family and other cytokine vaccines. Blood. 1998;91:222. [PubMed: 9414288]
- 296.
- Raza A, Maheshwari Y, Preisler H D. Differences in cell cycle characteristic among patients with acute nonlymphocytic leukemia. Blood. 1987;69:1647. [PubMed: 3555651]
- 297.
- Kufe D W, Spriggs D R. Biochemical and cellular pharmacology of cytosine arabinoside. Semin Oncol. 1985;12:34. [PubMed: 3925560]
- 298.
- Plunkett W, Liliemark J O, Adams T M. et al. Saturation of 1-β-d-arabinofuranosylcytosine 5’-triphosphate accumulation in leukemia cells during high-dose 1-β-d-arabinofuranosylcytosine therapy. Cancer Res. 1987;47:3005. [PubMed: 3471322]
- 299.
- Ross D D, Wooten P J, Ton Y. et al. Synergistic reversal of multidrug resistance phenotype in acute myeloid leukemia cells by cyclosporin A cremophor EL. Blood. 1994;83:1337. [PubMed: 8118035]
- 300.
- Estey E, Thall P, Andreef M. et al. Use of granulocyte colony-stimulating factor before, during, and after fludarabine plus cytarabine induction therapy of newly diagnosed acute myelogenous leukemia or myelodysplasia syndromes: comparison with fludarabine plus cytarabine without granulocyte colony-stimulating factor. J Clin Oncol. 1994;12:671–678. [PubMed: 7512125]
- 301.
- McGrath T, Center M S. Mechanisms of multidrug resistance in HL-60 cells: evidence that a surface membrane protein distinct from P-glycoprotein contributes to reduced cellular accumulation of drug. Cancer Res. 1988;48:3959. [PubMed: 2898287]
- 302.
- Moscow J A, Cowan K H. Multidrug resistance. J Natl Cancer Inst. 1987;80:14. [PubMed: 2892943]
- 303.
- Chan H S L, Thorner P S, Haddad G, Ling V. Immunohistochemical detection of p-glycoprotein: prognostic correlation in soft tissue sarcoma of childhood. J Clin Oncol. 1990;8:689. [PubMed: 1968964]
- 304.
- Dalton W S, Grogan T M, Meltzer P S. et al. Drug-resistance in multiple myeloma and non-Hodgkin’s lymphoma: detection of p-glycoprotein and potential and potential circumvention by addition of verapamil to chemotherapy. J Clin Oncol. 1989;7:415. [PubMed: 2564428]
- 305.
- Holmes J, Jacobs A, Carter G, Janowska-Wieczorek A, Padua R A. Multidrug resistance in haemopoietic cell lines, myelodysplastic syndromes and acute myeloblastic leukaemia. Br J Haematol. 1989;72:40. [PubMed: 2736241]
- 306.
- Ma D D F, Davey R A, Harman D H. et al. Detection of a multidrug resistant phenotype in acute nonlymphoblastic leukemia. Lancet. 1987;1:135. [PubMed: 2879973]
- 307.
- Ito Y, Tanimoto M, Kumazawa T. et al. Increased p-glycoprotein expression and multidrug-resistant gene (mdr1) amplification are infrequently found in fresh leukemia cells. Cancer. 1989;63:1534. [PubMed: 2564311]
- 308.
- Leith C P, Kopecky K J, Godwin J. et al. Acute myeloid leukemia in the elderly: assessment of multidrug resistance (MDR) and cytogenetics distinguishes biologic subgroups with remarkably distinct responses to standard chemotherapy. A Southwest Oncology Group study. Blood. 1997;89:3323. [PubMed: 9129038]
- 309.
- Inaba M, Maruyama F. Reversal of resistance of vincristine in P388 leukemia by various polycyclic clinical drugs, with a special emphasis on quinacrine. Cancer Res. 1988;48:2064. [PubMed: 3349478]
- 310.
- Ross D D, Wooten P J, Tong Y. et al. Synergistic reversal of multidrug resistance phenotype in acute myeloid leukemia cells by cyclosporine A and cremophor EL. Blood. 1994;83:1337. [PubMed: 8118035]
- 311.
- Lee E J, George S T, Caligiuri M. et al. Parallel phase I studies of daunorubicin given with cytyosine arabinoside and etoposide with or without the multidrug resistance modulator PSC-833 in previously untreated patients ≥ 60 years of age with acute myloid leukemia: results of cancer and leukemia group B study 9420. J Clin Oncol. 1999;17:2831–2839. [PubMed: 10561359]
- 312.
- List A F, Kopecky K J, Willman C L. et al. Benefit of cyclosporine (CsA) modulation of antracycline resistance in high-risk AML: a Southwest Oncology Group (SWOG) study. Blood. 1998;92(Suppl 1):128a.
- 313.
- Lum B L, Kaubisch S, Yahanda A M. et al. Alteration of etoposide pharmacokinetics and pharmacodynamics by cyclosporine in a phase I trial to modulate multidrug resistance. J Clin Oncol. 1992;10:1635. [PubMed: 1403041]
- 314.
- Hart S M, Ganeshaguru K, Hoffbrand A V, Prentice H G, Mehta A B. Expression of the multidrug resistance-associated protein (MRP) in acute leukaemia. Leukemia. 1994;8:2163. [PubMed: 7808005]
- 315.
- Schneider E, Cowan K H, Bader H. et al. Increased expression of the multidrug resistance-associated protein gene in relapsed acute leukemia. Blood. 1995;85:186. [PubMed: 7528566]
- 316.
- Schuurhuis G J, Broxterman H J, Ossenkoppele G J. et al. Functional multidrug resistance phenotype associated with combined overexpression of Pgp/MDR1 and MRP together with 1-β-d-arabinofuranosylcytosine sensitivity may predict clinical response in acute myeloid leukemia. Clin Cancer Res. 1995;1:81. [PubMed: 9815890]
- 317.
- Fontana J A, Rogers J S II, Durham J P. The role of 13 cis-retinoic acid in the remission induction of a patient with acute promyelocytic leukemia. Cancer. 1986;57:209. [PubMed: 3455835]
- 318.
- Meng-er H, Yu-chen Y, Shu-rong C. et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72:567. [PubMed: 3165295]
- 319.
- Castaigne S, Chomienne C, Daniel M T. et al. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. 1990;76:1704. [PubMed: 2224119]
- 320.
- Warrell R P, Frankel S R, Miller W H. et al. Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans retinoic acid). N Engl J Med. 1991;324:1385. [PubMed: 1850498]
- 321.
- Frankel S R, Eardley A, Lauwers G, Weiss M, Warrell R P Jr. The retinoic acid syndrome in acute promyelocytic leukemia. Ann Intern Med. 1992;117:292. [PubMed: 1637024]
- 322.
- De Botton S, Dombret H, Sanz M. et al. Incidence, clinical features, and outcomes of all trans-retinoic acid syndrome in 413 cases of newly diagnosed acute promyelocytic leukemia. The Europe APL Group. Blood. 1998;92:2712. [PubMed: 9763554]
- 323.
- Degos L, Dombret H, Chomienne C. et al. All-trans-retinoic acid as a differentiating agent in the treatment of acute promyelocytic leukemia. Blood. 1995;85:2643. [PubMed: 7742522]
- 324.
- Fenaux P, Le Deley M C, Castaigne S. et al. the European APL 91 Group. Effect of all trans retinoic acid in newly diagnosed acute promyelocytic leukemia. Results in a multicenter randomized trial. Blood. 1993;82:3241. [PubMed: 8241496]
- 325.
- Tallman M S, Anderen J W, Schiffer C A. et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337:1021. [PubMed: 9321529]
- 326.
- Fenaux P, Chastang C, Chevret S. et al. A randomized comparison of all trans retinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. Blood. 1999;94:1192. [PubMed: 10438706]
- 327.
- Mandelli F, Diverio D, Avvisati G. et al. Molecular remission in PML/RAR alpha-positive acute promyelocytic leukemia by combined all-trans retinoic acid and idarubicin (AIDA) therapy. Gruppo Italiana-Malattie Ematologiche Maligne dell’Adulto and Associazione Italiana di Ematologia de Oncologia Pediatrica Cooperative Groups. Blood. 1997;90:1014. [PubMed: 9242531]
- 328.
- Warrell R P Jr. Retinoid resistance in acute promyelocytic leukemia: new mechanisms, strategies and implications. Blood. 1993;82:1949. [PubMed: 8400247]
- 329.
- Soignet S L, Maslak P, Wang Z G. et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med. 1998;339:1341. [PubMed: 9801394]
- 330.
- Niu C, Yan H, Yu T. et al. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood. 1999;94:3315. [PubMed: 10552940]
- 331.
- Campana D, Pui C -H. Detection of minimal residual disease in acute leukemia: methodologic advances and clinical significance. Blood. 1995;85:1416–1434. [PubMed: 7888664]
- 332.
- Wormann B. Implications of detection of minimal residual disease. Curr Opin Oncol. 1993;5:3. [PubMed: 8427891]
- 333.
- Miller W H Jr, Kakizuk A, Frankel A. et al. Reverse transcription polymerase chain reaction for the rearranged acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 1992;89:2694. [PMC free article: PMC48728] [PubMed: 1372989]
- 334.
- Diverio D, Rossi V, Avvisati G. et al. Early detection of relapse by prospective reverse transcriptase-polymerase chain reaction analysis of the PML/RAR alpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter “AIDA” trial. GIEMEMA-AIEOP Multicenter “AIDA” Trial. Blood. 1998;92:784. [PubMed: 9680345]
- 335.
- Estey E, Smith T L, Keating M J. et al. Prediction of survival during induction therapy in patients with newly diagnosed acute myeloblastic leukemia. Leukemia. 1989;3:257. [PubMed: 2927176]
- 336.
- Samuels B L, Larson R A, Le Beau M M. et al. Specific chromosomal abnormalities in acute nonlymphocytic leukemia correlate with drug susceptibility in vivo. Leukemia. 1988;2:79. [PubMed: 3343866]
- 337.
- Campos L, Rouault J -P, Sabido O. et al. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81:3091–3096. [PubMed: 7684624]
- 338.
- Kornblau S M, Andreeff M, Hu S -X. et al. Low and maximally phosphorylated levels of the retinoblastoma protein confer poor prognosis in newly diagnosed acute myelogenous leukemia: a prospective study. Clin Cancer Res. 1998;4:1955. [PubMed: 9717825]
- 339.
- Yunis J J, Brunning R D, Howe R B, Lobell H. High-resolution chromosomes as an independent prognostic indicator in adult acute nonlymphocytic leukemia. N Engl J Med. 1984;311:812. [PubMed: 6472383]
- 340.
- Keating M J, Cork A, Broach T. et al. Toward a clinically relevant cytogenetic classification of acute myelogenous leukemia. Leuk Res. 1987;11:119. [PubMed: 3469482]
- 341.
- Keating M J, Smith T L, Kantarjian H. et al. Cytogenetic pattern in acute myelogenous leukemia: a major reproducible determinant of outcome. Leukemia. 1988;2:403. [PubMed: 3164797]
- 342.
- Schiffer C A, Lee E J, Tomiyasu T, Wiernik P H, Testa J R. Prognostic impact of cytogenetic abnormalities in patients with de novo acute non-lymphoblastic leukemia. Blood. 1989;73:263. [PubMed: 2910364]
- 343.
- Grimwade D, Walker H, Oliver F. et al. The importance of patients entered into MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukemia Working Parties. Blood. 1998;92:2322. [PubMed: 9746770]
- 344.
- Pinkel D, Landegent J, Collins C. et al. Fluorescence in situ hybridization with human chromosome-specific libraries: detection of trisomy 21 and translocations of chromosome 4. Proc Natl Acad Sci U S A. 1988;85:9138. [PMC free article: PMC282679] [PubMed: 2973607]
- 345.
- Lichtman M A, Weed R I. Peripheral cytoplasmic characteristics of leukocytes in monocytic leukemia: relationship to clinical manifestations. Blood. 1972;40:52. [PubMed: 4504250]
- 346.
- Lichtman M A, Rowe J M. Hyperleukocytic leukemias: rheological, clinical, and therapeutic considerations. Blood. 1982;60:279. [PubMed: 7046844]
- 347.
- Armitage J O, Goeken J A, Feagler J R. Spurious elevation of the platelet count in acute leukemia. JAMA. 1978;239:433. [PubMed: 304490]
- 348.
- Lester T J, Johnson J W, Cuttner J. Pulmonary leukoistasis as the single worst prognostic factor in patients with acute myelocytic leukemia and hyperleukocytosis. Am J Med. 1985;79:43. [PubMed: 3860006]
- 349.
- Vaughan W P, Kimball A W, Karp J E, Dragon L H, Burke P J. Factors affecting survival of patients with acute myelocytic leukemia presenting with high WBC counts. Cancer Treat Rep. 1981;65:1007. [PubMed: 6945910]
- 350.
- Grund F M, Armitage J O, Burns C P. Hydroxyurea in the prevention of the effects of leukostasis in acute leukemia. Arch Intern Med. 1977;137:1246. [PubMed: 268956]
- 351.
- Bunin N J, Kunkel K, Callihan T R. Cytoreductive procedures in the early management in cases of leukemia and hyperleukocytosis in children. Med Pediatr Oncol. 1987;15:232. [PubMed: 3477680]
- 352.
- Cuttner J, Holland J F, Norton L. et al. Therapeutic leukapheresis for hyperleukocytosis in acute myelocytic leukemia. Med Pediatr Oncol. 1983;11:76. [PubMed: 6572782]
- 353.
- Bleyer W A, Poplack D G. Prophylaxis and treatment of leukemia in the central nervous system and other sanctuaries. Semin Oncol. 1985;12:131. [PubMed: 3892694]
- 354.
- Peterson B A, Brunning R D, Bloomfield C D. et al. Central nervous system involvement in acute nonlymphocytic leukemia: a prospective study of adults in remission. Am J Med. 1987;83:464. [PubMed: 3661583]
- 355.
- Stewart D J, Keating M J, McCredie K B. et al. Natural history of central nervous system acute leukemia. Cancer. 1974;33:893. [PubMed: 7459807]
- 356.
- Wolk R W, Masse S R, Conkin R, Freireich E J. The incidence of central nervous system leukemia in adults with acute leukemia. Cancer. 1974;33:863. [PubMed: 4521890]
- 357.
- Frick J, Ritch P S, Hansen R M, Anderson T. Successful treatment of meningeal leukemia using systemic high-dose cytosine arabinoside. J Clin Oncol. 1984;2:365. [PubMed: 6726293]
- 358.
- Cassileth P A, Sylvester L S, Bennett J M, Begg C B. High peripheral blast count in adult acute myelogenous leukemia is a primary risk factor for CNS leukemia. J Clin Oncol. 1988;6:495. [PubMed: 3162514]
- 359.
- Rosenthal A R. Ocular manifestations of leukemia. Ophthalmology. 1983;90:899. [PubMed: 6195573]
- 360.
- Karesh J W, Goldman E J, Reck K. et al. A prospective ophthalmic evaluation of patients with acute myeloid leukemia: correlation of ocular and hematologic findings. J Clin Oncol. 1989;7:1528. [PubMed: 2778482]
- 361.
- Mayer R J, Cuttner J, Truog P, Ambinder E P, Hollan J F. Therapeutic leukopheresis of acute myelomonocytic leukemia in pregnancy. Med Pediatr Oncol. 1978;4:77. [PubMed: 272483]
- 362.
- Bartsch H H, Meyer D, Teichmann A T, Speer C H P. Treatment of promyelocytic leukemia during pregnancy: a case report and review of the literature. Blut. 1988;57:51. [PubMed: 3164640]
- 363.
- Pizzuto J, Aviles A, Noriega L. et al. Treatment of acute leukemia during pregnancy: presentation of nine cases. Cancer Treat Rep. 1980;64:679. [PubMed: 6933005]
- 364.
- Tsokos G C, Balow J E, Spiegel R J, Magrath I T. Renal and metabolic complications of undifferentiated and lymphoblastic lymphomas. Medicine (Baltimore). 1981;60:218. [PubMed: 6894477]
- 365.
- Wainer R A, Wiernik P H, Thompson W L. Metabolic and therapeutic studies of a patient with acute leukemia and severe lactic acidosis of prolonged duration. Am J Med. 1973;55:255. [PubMed: 4722860]
- 366.
- Lowenberg B, Downing J R, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051. [PubMed: 10502596]
- Acute Myeloid Leukemia in Adults - Holland-Frei Cancer MedicineAcute Myeloid Leukemia in Adults - Holland-Frei Cancer Medicine
- Diogenichthys atlanticus[orgn] (0)Genome
- probable inactive lysine-specific demethylase JMJ19 isoform X1 [Brassica rapa]probable inactive lysine-specific demethylase JMJ19 isoform X1 [Brassica rapa]gi|1079413036|ref|XP_018514859.1|Protein
- ranbp2[All Fields] AND 1[has_clinical] (58)dbVar
Your browsing activity is empty.
Activity recording is turned off.
See more...