

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mendy M, Caboux E, Lawlor RT, et al. Common Minimum Technical Standards and Protocols for Biobanks Dedicated to Cancer Research. Lyon (FR): International Agency for Research on Cancer; 2017. (IARC Technical Report, No. 44.)

Common Minimum Technical Standards and Protocols for Biobanks Dedicated to Cancer Research.
Show details3.1. Ethical, legal, and social issues (ELSI) and governance
This section provides advice on developing an internal governance system for biobanks. It references recommendations and best practices of international organizations, including OECD (2007, 2009), ISBER (2012), GA4GH (2016), and NCI (2016), among others. However, the background of law and guidance is continually developing and should be monitored. For example, the new EU General Data Protection Regulation (European Commission, 2016) has implications for patients’ rights in medical research, CEN norms, and ISO standards.
Governance, in the context of biobanks, is not one-size-fits-all. During the establishment of a biobank, governance systems should be designed to take into account the biobank’s scope and the context in which it operates (Laurie, 2011). A good internal governance system should:
- •
ensure that the biobank remains faithful to its purpose, encouraging trust between the various stakeholders;
- •
be guided by a set of overarching principles when making decisions, including being transparent, accountable, consistent, proportionate, efficient, coordinated, equitable, and fair; and
- •
be dynamic and able to adapt over time.
The internal governance approaches introduced in this section are based on a good governance structure or framework (Section 3.1.1) and documentation on:
- •
informed consent (Section 3.1.2);
- •
data protection, confidentiality, and privacy (Section 3.1.3);
- •
return of results and incidental findings (Section 3.1.4); and
- •
access to and sharing of samples and data (Section 3.1.5; see also Annex 1).
Further sections consider quality (Section 3.4) and records management (Section 3.6).
Good governance includes engaging with the public during the establishment of a biobank and throughout the life-cycle of the biobank. Therefore, the approach to public engagement must be considered from the outset. In addition to engaging with participants, the biobank may need to engage with the scientific community, researchers, patient groups, and/or the wider public using a variety of methods, for example by consultation on study designs and policies, involvement on committees, or publication and outreach. Good biobank governance also includes a strong commitment to researchers, ensuring quality, efficiency, and transparency of service. Therefore, the following recommendations should be put into practice in collaboration with project principal investigators.
3.1.1. Governance framework
A good governance framework should define the organizational structure of the biobank, for daily management and oversight of its strategic policy. This framework usually includes lists and descriptions of the biobank’s personnel, committees, and policies that are required to enable the correct functioning of the biobank. The level of policies and procedures governing the biobank should be scalable to its nature, size, and available resources. For example, smaller biobanks may have more limited policies, whereas larger biobanks will need to develop a detailed protocol and procedures.
The policies are usually stipulated in a governance document that describes the objectives and scope of the biobank, the organizational structure, the scientific and economic strategy of the biobank (which will be articulated in an annually updated business plan), and contingency plans in the event of closure. The governance document also includes policies on data protection and privacy as well as the procedures governing specific operational activities of the biobank.
Defining the structure and mandate of committees and describing policies is an effective way to ensure adherence to proper governance. However, if there are too many committees or policies, or if they are ill-defined, this can impede procedures and cause delays.
3.1.1.1. Governance organization
The biobank should have a structure of committees and appropriately qualified personnel in relevant roles to oversee its governance. The size, type, and number of committees and their composition will vary depending on the size and purpose of the biobank. Careful consideration should be given when participants, patient groups, or public representatives are asked to serve on biobank committees. Their roles on the committee should be clearly communicated, and training should be provided. The following types of committee may be considered (Fig. 1).

Fig. 1
Sample committee structure for internal biobank governance.
Executive committee or steering group
All biobanks should have an executive committee or steering group. The responsibilities of this committee may include overall management, defining strategic objectives, monitoring progress, revising and/or adopting policies, and developing a communications strategy. This committee may also conduct an annual review meeting to consider the QMS.
Ethics oversight (or advisory) committee
The ethics oversight committee advises the executive committee on strategy, developments, and procedures relating to ethical oversight, including legal and policy issues. The committee could include, for example, ethicists, scientific researchers, medical experts, lawyers, social scientists, and members of the public or participant organizations. In some cases, this committee may be part of a larger infrastructure, such as a local hospital ethics committee. In some countries, this committee may be a legal requirement.
Laboratory safety and biosecurity committee
All biobanks should establish, or have access to, a committee on laboratory safety, which may also consider general health, safety, and security issues.
Data and sample access committee
Biobanks should consider establishing a data and sample access committee, to oversee access requests, monitor related procedures, and ensure that participants’ interests are protected and biobank protocols are followed. In some cases, this committee is external to the biobank and is composed of independent members.
Operations or management committee
The role of the operations or management committee is to support the executive committee for the strategic decisions of the biobank and to provide expertise in all aspects of biobanking operations (e.g. safety; quality and efficiency, including processing, storage, and distribution of biospecimens).
Larger biobanks may require additional committees, such as the following.
Scientific oversight (or advisory) committee
This committee would provide scientific feedback to the executive committee, advise on scientific strategy and current developments, consider the pertinence of new collections, or advise on procedures. Membership should include relevant professionals. In some biobanks, this committee could be combined with an ethics oversight committee. In some countries, this committee may be a legal requirement.
Public engagement committee
This committee could help biobank personnel and associated researchers to better understand public opinion. For some larger biobanks, advisory panels of study participants meet regularly and provide feedback on new projects and review study materials, newsletters, and questionnaires. Examples are the Avon Longitudinal Study of Parents and Children (ALSPAC) teenage advisory panel (UK Biobank Ethics and Governance Council, 2009) and the NIH Precision Medicine Initiative Cohort Program subcommittee, which has significant participant representation (Precision Medicine Initiative Working Group, 2015).
In terms of personnel, the biobank should have clear reporting lines and accountability, with documented levels of authority and responsibility associated with each role. Clear responsibilities for staff members enable the biobank management to ensure that the biobank’s activities comply with ethical and legal requirements (OECD, 2009). An organizational chart and list of staff members and their responsibilities should be developed, alongside an organizational plan, which defines the organization and management of the biobank and its relationship to external parties. Roles and responsibilities should be clearly defined, to establish who has legal responsibility in relation to the biobank, who has day-to-day operational responsibility, and who is acting as the custodian of the resources.
Specific roles within the biobank will depend on the institutional context but may include the following.
- •
A designated director, who is responsible for implementing biobank policies. The roles and responsibilities of this person in their institution should be clearly defined.
- •
A biobank coordinator or manager, who reports directly to the steering group. To eliminate conflicts of interest, it is recommended that the biobank manager is not an active investigator or a biobank user. The biobank manager may also be designated the custodian of the resource, with the following responsibilities:
- o.
establishing procedures;
- o.
ensuring that ethical guidelines are adopted and respected;
- o.
implementing the decisions of the relevant committees in relation to the control, access, and use of the material;
- o.
maintaining close collaborations with principal investigators;
- o.
distributing information about the biobank and related research; and
- o.
other responsibilities, which should be defined in advance.
- •
A biobank quality manager, who is responsible for the QMS and for periodic review of all SOPs, and has overall responsibility for quality control (QC) and quality assurance (QA).
- •
A data steward, who is responsible for data protection and privacy.
3.1.1.2. Documentation: plans, policies, and procedures
Documentation requirements include plans, policies, and specific SOPs. The documents should be compatible with international standards such as the ISO standards for biobanking and the CEN norms that articulate how activities of the biobank are to be performed (see Section 3.4 for more details about ISO and CEN). Some general principles in relation to these documents should be followed:
- •
Documents should be developed in the context of a QMS, which includes document version control.
- •
Documents should be developed in the context of an up-to-date risk assessment undertaken alongside a procedure that takes into account risks to the health and safety of people.
- •
Documents should include a time frame for review and revision; 2 years is a recommended time frame (NCI, 2016).
- •
To facilitate cooperation between biobanks, documents should adhere to internationally accepted technological standards and norms, ensuring that these are clearly referenced (OECD, 2009).
- •
High-level policies, including data and sample access policies and terms of reference of committees, should be publicly and freely available, for example on a website.
- •
Biobanks should consider implementing monitoring strategies with scheduled audits to ensure that policies and procedures are followed.
- •
Researchers should submit reports annually and at the end of their projects, including information on publications and patent applications (OECD, 2009).
- •
The biobank should maintain a system for reporting adverse events, anomalies, and non-compliance with the QMS; this reporting system supports corrective and preventive actions and enables any relevant documents to be updated (CCB, 2014).
A critical document is the biobank programme document (or biobank protocol), which contains information about the scientific rationale, scope, design, and strategy for the biobank. Other biobank plans, policies, and procedures should be developed in line with this protocol. The biobank’s mission should be clearly outlined in terms of its purpose, the types of research or other users supported (scope), and the types of samples and data collected. Additional considerations include which services are provided (e.g. specific research assays, storage of samples) and whether legacy samples can be incorporated into the biobank. This protocol and associated key biobank documents should be approved by a research ethics committee, and renewed approval should be required if the documents are amended (OECD, 2009).
An annually updated business and continuity plan or model is essential, especially if the biobank is planning to charge for use of the resources (Vaught et al., 2011). The business plan should include a strategy for both medium-term and long-term sustainability. A budgeting or costing exercise will assist in the development of such a plan.
All biobanks should also develop a quality management policy and a policy on access to samples and data from the biobank (see Annex 1).
Additional policies to consider include:
- •
a governance policy, containing information about the biobank’s governance structure and the responsibilities of management (OECD, 2009);
- •
a retention policy, covering biospecimen availability and whether collections can be shared or destroyed;
- •
a policy on storage options;
- •
a safety policy for staff and visitors;
- •
a policy on transportation of material;
- •
a policy on disposal of material and biosafety and biosecurity;
- •
policies covering ethical issues, including information on the protection of the confidentiality and privacy of participants (see Section 3.1.3), informed consent, return of results and incidental findings, and so on;
- •
a policy on the intellectual property generated from the use of the resources and research results;
- •
a publication policy, governing publications arising from the use of the biobank; and
- •
policies on how the termination of the biobank would be handled.
Guidelines recommend that accompanying SOPs should be put in place to govern all biobank activities: recruitment; consent; staff training; biosafety; the collection, receipt, processing, and storage of samples; sample QC, laboratory QA; participant de-identification; data collection, recording, storage, and management; data protection; the monitoring, calibration, maintenance, backup, and repair of equipment; the procurement and monitoring of supplies (disposables and reagents); the distribution and tracking of samples; records and documentation; reporting of non-conformity and complaints; and disaster management.
All staff members should be trained in the procedures at the biobank, and this should be documented.
Box
Key points: biobank governance.
3.1.2. Informed consent
The approach to informed consent is a key consideration when establishing a new biobank, and a policy should be developed (see Section 3.1.1.2). Requesting appropriate informed consent has become a cornerstone for the collection of samples and data for use in research, and is supported by relevant guidance and legislation. This section presents recommendations to assist a biobank in developing a consent policy and associated documentation, and covers the following areas (see also Annex 2):
- •
types of consent;
- •
what information to provide to potential participants;
- •
potentially ethically or legally challenging issues;
- •
what to consider during the process of requesting consent;
- •
what to consider if the country or the research area would benefit from community engagement in relation to the consent process;
- •
what to do if the potential participant does not fully understand the language of the researcher who is administering the consent;
- •
what to do if the potential participants do not have the legal capacity to consent for themselves;
- •
considerations when including samples or data from deceased participants in the biobank;
- •
the continuing nature of consent;
- •
when participants might need to be re-contacted to request new or updated consent; and
- •
how to approach withdrawal of consent.
3.1.2.1. Types of consent
Many biobanks use a broad consent, which allows patients or research participants to consent to a broad range of uses of their data and samples. Although broad consent allows for a broad range of research activities, it is regarded by research ethics committees as specific enough to be considered “informed”, because guidance is provided on the nature of the future undetermined research uses (e.g. research on breast cancer and associated conditions). It is important to note that broad consent forms usually contain a series of statements specific to the biobank, and not statements related to specific research projects. Table 2 outlines the different types of consent associated with biobanks and sample collections, together with key points about each approach and notes on information to be provided to participants.
Table 2
Types of consent and key considerations.
Further guidance on designing and implementing a broad informed consent is provided in Annex 2.
3.1.2.2. What information to provide to potential participants
Information about the biobank and biobanking activities is usually provided to potential participants using a participant information sheet in conjunction with a consent form (in some cases, these two documents together are called the informed consent form). The information provided will vary depending on the nature of the biobank and the type of consent requested. Further details are provided in Table 2 and Annex 2.
3.1.2.3. Potentially ethically or legally challenging issues
The following potential uses of samples and data are examples of issues that may be considered ethically or legally challenging:
- •
transfer of samples or data across national borders; it is important to be aware of regional and national regulations, such as the EU Data Protection Directive of 1995 (Article 26) (European Commission, 1995) and the EU-U.S. Privacy Shield adopted by the EU Commission on 12 July 2016 (http://ec
.europa.eu/justice /data-protection /international-transfers /eu-us-privacy-shield/index_en .htm); - •
use of samples in experiments involving animals;
- •
creation of cell lines from the samples, including stem cell lines;
- •
use of samples and data by commercial researchers;
- •
research linked to reproduction, including use of embryos;
- •
return of individual research results and incidental findings (this is an important topic that warrants extensive discussion, which is beyond the scope of this document); and
- •
research into high-penetrance genes linked to disease.
When information sheets and consent forms are being developed, the biobank should consider whether it may be involved in any potentially challenging uses and include information about these in the information sheet and/or the consent form as appropriate, in addition to the core elements usually included as part of a consent form. The consent form should provide the participant with a means to opt out of uses that the participant feels are ethically questionable. The consent form may also require specific opt-in provisions if they are legally required by national or regional laws; an example is transfer of data outside of Europe, according to the EU Data Protection Directive (European Commission, 1995). A means to opt out of certain uses should be provided by the biobank only if this is recordable (i.e. in a database), actionable (i.e. when distributing the samples for research), and practicable (e.g. given the number of participants or the number of samples distributed).
3.1.2.4. What to consider during the process of requesting consent
It is important to consider the following aspects during the consent process.
- •
Where possible, the information sheet should be distributed ahead of the meeting with the potential participant.
- •
The potential participant must be given adequate time to read and consider the information sheet and should be offered the option to decide and give consent at a later visit if required.
- •
The person administering the consent process must be convinced of the capacity of the potential participant to give consent. If they are not convinced, Section 3.1.2.6 (on participants without the legal capacity to consent) may be applicable. Alternatively, the potential participant may require assistance in reading the form.
- •
The person requesting consent should not coerce the potential participant in any way.
- •
The person requesting consent should encourage open discussion and give the potential participant the opportunity to ask questions.
- •
The person requesting consent should ensure that the potential participant is informed about their right to withdraw consent, about the risks and benefits of participating in the project, and about any other important issues.
3.1.2.5. What to consider if the country or the research area would benefit from community engagement in relation to the consent process
The consent process needs to be appropriate for the local cultural context, and in some cases this means that wider community engagement is appropriate (H3Africa, 2013). In cases where wider community engagement is needed, consultation on the biobank’s consent processes and documents should take place with the wider community, local leaders, and professionals. The requirement for community involvement in the consent process may also be specified in local ethical or legal guidance. In some of these cases, prior consent, assent, or permission may need to be obtained from community, tribal, or family leaders (Nuffield Council on Bioethics, 2002). In all cases, the potential research participant themselves must be approached for consent and must have the right to refuse participation.
3.1.2.6. What to do if the potential participants do not have the legal capacity to consent for themselves
This category of participant is often called “vulnerable people”, and careful consideration must be given to how recruitment will be conducted (WMA, 2013). Recruitment of “vulnerable” participants must respect the requirements in the Declaration of Helsinki that all vulnerable groups and individuals should receive specifically considered protection, that the research is responsive to the health needs of this group and cannot be carried out in a non-vulnerable group, and that this group will benefit from the research.
Three types of participant commonly identified as vulnerable are:
- •
mentally incapacitated adults,
- •
adults in emergency care situations, and
- •
minors/children.
In the case of mentally incapacitated adults, a legally authorized representative can provide consent on their behalf. If the participant made legally approved provisions about research participation before they were incapacitated or if they assigned a legal representative, these should be respected. The participant should be involved as much as possible in the decision to participate, and any resistance or objections should be respected.
In the case of research taking place in an emergency clinical situation where consent has not been obtained, requirements will differ by country and may include:
- •
the local ethics committee explicitly approving the recruitment pathway;
- •
another medical professional authorizing the involvement of the participant;
- •
the known wishes or objections of the participant being respected; and
- •
a maximum time limit being imposed for participant involvement without consent.
Consent should be requested from the participant if the participant regains legal capacity.
In the case of research involving children, they should take part in the consent process in accordance with their age and maturity, and assent to participate (rather than consent). Information and consent materials can be designed for different age groups to aid understanding of the research. Any objection from the child should be respected (Hens et al., 2011). The assent process is based on the age and maturity of the child and on any applicable local laws or ethical guidance on the matter. In addition to the assent of the child, the child’s parent(s) or an appropriate legal representative must provide consent on the child’s behalf. It is also good practice to re-contact child participants once they reach the local legal age of maturity, to request consent, if possible (CIOMS, 2002; Hens et al., 2011).
3.1.2.7. Considerations when including samples or data from deceased participants in the biobank
Consent requirements will vary depending on whether the biobank intends to request consent for samples and data from potential participants before their death (for example, for a brain biobank) or request the samples and data after the participants’ death. Local legislation will dictate the applicable consent and legal requirements. Some legal or practical constraints may exist for biobanks accessing medical records after the participants’ death. Uses of the samples and data, collected before or after death, that fall outside the scope of the original consent will require approval by a research ethics committee (Tassé, 2011).
Where samples are to be collected for research after death, local institutional, ethical, and legal guidelines to obtain consent must be followed, and the individual’s wishes expressed before death, if they are known, should be respected. The consent procedure may be built into existing procedures for postmortems or for clinical use of postmortem samples for organ or tissue transplants, which may differ markedly from country to country.
3.1.2.8. The continuing nature of consent
If the participant has given prior written consent, they should always be asked to confirm (verbally and/or tacitly, as appropriate) their agreement to donate samples to the biobank, and they should always have the opportunity to ask questions, before additional sampling (e.g. blood, biopsy, aspirate, bronchial brushings) or data collection is performed. Where possible, the verbal consent should be recorded electronically or noted and stored with the original consent form. The participant may decline to provide further samples or data at any time. This does not invalidate the consent to use any previous samples or data given to the biobank, unless notice of withdrawal of consent is given.
3.1.2.9. When participants might need to be re-contacted to request new or updated consent
Several situations may arise where the biobank may need to re-contact the participants to request new or updated consent, apart from contact to request additional data and/or samples for the purposes of the same project. These situations may include children reaching the age of maturity and temporarily incapacitated participants regaining legal capacity (see Section 3.1.2.6), and in such cases full informed consent should be requested (Burke and Diekema, 2006). Another situation is when the information provided in the initial consent form and information sheets is modified or updated (e.g. if the scope of the biobank changes). In this case, re-contact of participants to request an updated consent may be required, given the changing conditions of their participation (Wallace et al., 2016). An appropriate research ethics committee should decide whether re-consent is required or whether a waiver can be applied.
A general guideline is that before re-contact is established, a participant’s options with respect to re-contact should be checked, because participants should be given the option not to be re-contacted (see Annex 3).
3.1.2.10. How to approach withdrawal of consent
At any time, participants can withdraw consent for the biobanking and use of their samples or data without giving a reason. It is crucial for the biobank to present withdrawal options to participants in the consent form or information sheets. This may not mean guaranteeing to destroy all samples and data; for example, the withdrawal options may stipulate that samples and data already released or used in analyses are not retrievable. Examples of withdrawal options include the following.
- •
“No further use” option: the biobank will destroy all samples and data from the participant and will not contact the participant again.
- •
“No further contact” option: the biobank will no longer contact the participant directly by any means but can continue to use samples and data already collected and can continue to access the participant’s medical records if necessary.
- •
“No further access” option: the biobank will not contact the participant or access the participant’s medical records but can continue to use samples and data already collected.
The biobank should also clearly communicate to participants when it is impossible to destroy parts of the samples or data. Examples include being unable to destroy:
- •
samples and data already distributed for research or used in analyses, and
- •
data needed for audit purposes or already archived.
Box
Key points: informed consent.
3.1.3. Data protection, confidentiality, and privacy
This section briefly outlines recommendations about the protection of biobanks’ data and about the confidentiality and privacy of the participants’ data. Legislation and guidance on these issues vary between countries and, where they exist, may also be complemented by local site requirements.
In the EU, the General Data Protection Regulation (European Commission, 2016) replaces the Data Protection Directive (European Commission, 1995) and unifies data protection for individuals in the EU. The processing of personal data outside the EU is also an important component of EU privacy and human rights law.
Other examples of privacy and security policies are those of the Confederation of Cancer Biobanks (CCB, 2014) and GA4GH (GA4GH, 2015b).
At a European level, the changing data protection regulations and requirements in relation to the EU-U.S. Privacy Shield should be taken into account. The biobank must develop a strategy and have an IT framework and policies in place for managing data collected alongside the samples in line with the commitment undertaken with participants.
Methods to ensure data protection, confidentiality, and privacy are discussed in Section 3.3.4 and Section 3.6. The biobank should consider using de-identification methods, such as coding or pseudo-anonymization associated with a procedure to store codes. Explanations of these terms are provided in Section 1 and in Appendix 1 of the privacy and security policy of GA4GH (GA4GH, 2015b).
Examples of particular issues for data protection and privacy for biobanks include the following.
- •
Access to medical records. When possible, staff members should be bound by a professional code of practice with high standards of ethical behaviour. The participant should be asked for permission to access their medical records and should be informed where these data are held in an encrypted, non-identifiable format at the biobank (CCB, 2014).
- •
Data protection mechanisms when biobanks share data with other biobanks or provide data for translational research. Privacy and confidentiality of data must be guaranteed, while facilitating access.
- •
Participants should also be informed, in the informed consent, about what data will be shared and how data will be shared, and how their privacy and the confidentiality of the data will be protected.
- •
Research involving genetic data and next-generation sequencing data may lead to concerns about (i) whether data can identify individuals and/or family members, and (ii) whether to return results from this type of analysis (see Section 3.1.4).
- •
The inclusion of medical images in biobanks (e.g. scans and histology slides) poses specific challenges for de-identification, because identifying data are usually embedded within the images, which by themselves may not be identifying.
In terms of confidentiality, all staff members with access to confidential data should have a duty of professional secrecy (OECD, 2007). Staff members, consultants, or committee members without such a duty must be asked to sign a confidentiality agreement. Access to personal data should be limited, and access to any data should be restricted to those data needed for the research project or other use. Data can be separated into different databases according to type. The biobank should keep specimen metadata in a linked but separate database from the patients’ medical records and demographic information, to keep data safe and confidential. Regular audits of the data systems must be implemented.
Box
Key points: data protection and privacy.
3.1.4. Return of results and incidental findings
This section presents the issues with respect to research results: summary results, individual results from baseline assessment, and individual research results. The principal investigator and the biobank have collective responsibility for deciding whether to return research results to participants. The decision-makers should consider the balance between the duty of care to participants, the ethical and legal requirements to return results, and the logistical and technical ability of the biobank to return results in an appropriate manner. Factors to consider include whether the biobank no longer has contact with the patient, the ease of re-identifying the participants and finding out whether they have chosen to be informed, the potential cost implications of providing re-testing or counselling to participants, if required, and whose responsibility this is.
One toolkit is the framework on the feedback of health-related findings in research (Medical Research Council and Wellcome Trust, 2014).
3.1.4.1. Generalized, non-individual study results
According to best practices, research results can be published on the biobank website or via a newsletter (CCB, 2014). These are summary results of research using samples and/or data from the biobank and cannot be connected back to a specific individual.
The participant should be given the option during the consent process to receive these publications, and researchers using samples and data should commit in the MTA to providing the report of research results to the biobank.
3.1.4.2. Individual results from tests conducted during sign-up or registration
Some biobank studies request participants, after they consent, to undergo initial testing (baseline assessments), such as blood pressure measurements, lung function tests, and vision tests. In addition, biological samples may be routinely tested for infectious diseases, such as HIV, hepatitis B, and hepatitis C, before storage in the biobank. Biobanks should consider whether they will return the results of these tests, and this should be clearly indicated in the participant consent materials. These tests should not be used as incentives for the participants to sign up, and it should be made clear that they are not part of health checks.
3.1.4.3. Individual research results
Individual research results fall into two categories.
- •
Results that can be anticipated because they are in line with the aims of the research. The method for handling these can be considered during the evaluation of the sample request.
- •
Incidental findings that are not linked to the aims of the research. The method for returning these to the biobank can be addressed in the MTA.
In both cases, the biobank will need procedures for evaluating the validity of such results, as well as the period of validity, and for returning these results to the participants, including re-identification and re-contact of the participant if the participant has indicated in the consent form that they wish to be re-contacted (UK Biobank Ethics and Governance Council, 2015). The scope of the results to be returned and how they are to be returned should be defined in advance with relevant experts and the ethics committee (Thorogood et al., 2014), taking into account that biobank participants have the right to choose not to know research results. The ideal time to ask about an individual’s preferences is during the consent process. The information sheet and consent form should provide potential study participants with information and give them the opportunity to choose whether they wish to receive individual research results. The protocol must provide a way for participants to later change their preferences.
3.1.4.4. Results of genetic tests and next-generation sequencing
Returning the results of genetic tests or next-generation sequencing should involve offering a clinical-grade validation and making available clinical expertise or genetic counselling. Recommendations about which genetic test results to return are provided, for example, in the American College of Medical Genetics and Genomics (ACMG) recommendations for reporting of incidental findings in clinical exome and genome sequencing (Green et al., 2013) and in the Genomics England project (https://www.genomicsengland.co.uk/taking-part/results/).
3.1.4.5. Results of imaging studies and scans
Imaging biobanks are defined by the European Society of Radiology as “organised databases of medical images and associated imaging biomarkers (radiology and beyond) shared among multiple researchers, and linked to other biorepositories” (European Society of Radiology, 2015). Specialized facilities and biobanks that also conduct imaging studies such as scans are particularly likely to discover incidental findings. The Royal College of Radiologists provides guidance on the management of incidental findings detected during research imaging (RCR, 2011).
3.1.4.6. Results about children
If the biobank includes biological samples from children, the biobank needs to evaluate whether it has a stronger responsibility to return results in this case because the participants are children (Hens et al., 2011). It must also consider what action to take if the results become available after the child comes of age, because the original consent was not the child’s, and whether the parents have the right to receive all information about the child (see Section 3.1.2.6).
Box
Key points: return of results.
3.1.5. Access to and sharing of samples and data
A catalogue of biological material should be published, to optimize the use of resources and ensure the transparency of biobank activities. Optimally, each sample should be listed, with associated access conditions and consent elements (OECD, 2009).
In some cases, funders may require specific data sharing policies to be designed and implemented (Kaye and Hawkins, 2014; Kosseim et al., 2014). Intended sample and data sharing should be included on the participant information sheets and consent forms (GA4GH, 2015a).
To facilitate data and sample sharing, BBMRI-ERIC has created one of the largest directories for biobanks in Europe. The directory currently contains more than 6 million samples with associated data, which can be accessed for collaboration (BBMRI-ERIC, 2016).
Researchers requesting access to the biobank’s resources should do so by applying to the biobank and following its access policy and access procedures (see Section 3.1.5.1). Furthermore, the biobank should assess and review the type of data requested by the researcher. For example, the biobank cannot, without explicit prior consent, disclose participant-identifiable data to researchers. This should be addressed in any agreement between the researcher and the biobank (such as an MTA or Data Transfer Agreement [DTA]; see Section 3.1.5.5). In all cases, a section stipulating that the researcher may not attempt to re-identify any participants should be included. In general, the policy on disclosing data must consider which identifiers will be removed from the participant’s record to ensure that privacy is protected. Particular care should be taken with regard to data that may not directly lead to re-identification but, in combination with other data, could do so.
3.1.5.1. Access to stored materials and data for research purposes
The biobank should develop an access policy and access procedures, in line with its protocol (Section 3.1.1). The policy and procedures should describe the administrative and approvals process for applying for and obtaining access to samples or data, comprising an overview of applicable restrictions and obligations. A procedure should exist to ensure that the applicants are bona fide researchers, and the policy should be publicly available (CCB, 2014). See Annex 1 for the IARC access policy; other examples include those of the National Cancer Research Institute (NCRI, 2009) and P3G (Harris et al., 2012).
Evaluation of requests should be based on the notion of proportionality, balancing risks against benefits, and ensuring that the intended use follows the biobank’s protocol and priorities and the consent provided by the participants (Mallette et al., 2013). In general, samples should be shared in a fair, transparent, and equitable manner (Chen and Pang, 2015). In the case of scarce samples, a decision could be made to provide samples to projects more closely aligned with the aims and strategy of the biobank. The researcher should sign an MTA/DTA (see Section 3.1.5.5), which will include the obligations of the researcher, before receiving the samples and/or data.
3.1.5.2. Principles for international specimen exchanges
The legal aspects of sample sharing vary between countries, and an assessment should be made to ensure that the relevant legal regimes are compatible with those of the biobank and the consent. Where applicable, the participant should also have given consent for the transfer of data between countries. Examples of legal requirements are the EU-U.S. Privacy Shield principles, which deal specifically with transfer of data from Europe to the USA (see Section 3.1.2.3), and the new EU General Data Protection Regulation (see Section 3.1.3).
Finally, if there are any doubts in relation to privacy implications when samples or data are to be transferred internationally, a data privacy impact assessment can be performed before such transfer (GA4GH, 2015b). For further information on the legal requirements related to international sample sharing, researchers can use the Human Sample Exchange Regulation Navigator (hSERN) tool, available at http://www.hsern.eu/.
Special attention should be given to the transfer of samples to or from countries with regulatory frameworks that are poor or absent (Chen and Pang, 2015).
3.1.5.3. Collaboration with the private sector
Collaboration with the private sector must adhere to the same requirements and obligations with respect to data and sample sharing. It is important for the possibility of sharing samples and data with the private sector to be specifically mentioned in the informed consent and information sheet (European Commission, 2012a).
3.1.5.4. Intellectual property and ownership
Intellectual property policies vary across institutions, but the biobank should define an intellectual property policy. Aspects of this policy should be defined in the MTA/DTA (see Section 3.1.5.5), as well as ownership of biological samples.
Box
Key points: data and sample sharing.
3.1.5.5. Material Transfer Agreement (MTA)/Data Transfer Agreement (DTA)
An MTA, a DTA, or a similar agreement should be put in place before the transfer of samples and/or data between organizations (ISBER, 2012). An MTA/DTA is a legally binding document that governs the conditions under which the samples and/or data can be used (see Annex 4).
The MTA/DTA outlines the type of samples and/or data to be transferred, the purpose of the transfer, and all restrictions or obligations that relate to the use of the samples and data (NCI, 2011; ISBER, 2012; NCI, 2016). These restrictions and obligations must be in line with the conditions of the informed consent, ethics approval, and biobank governance attached to the samples and/or data. The agreement may include a statement that the samples and data have received appropriate ethics approval and consent.
The agreements should include specific aspects relating to the biobank’s policies and provisions that bind the researcher to:
- •
use the samples and data in line with the biobank access approval given;
- •
adhere to applicable laws, regulations, and guidance;
- •
not further distribute the samples or data;
- •
dispose of, or return, the samples and data after use;
- •
guarantee confidentiality and data protection;
- •
not attempt to re-identify participants;
- •
inform the biobank of any issues with the data or samples;
- •
provide traceability of samples;
- •
return research results in the form of individual results, raw data, an interim/final report, relevant publications, or patent applications;
- •
cite or acknowledge the biobank in publications, patents, or other documents, or include a citation in any published work to a specific publication describing the biobank; and
- •
respect intellectual property terms.
An example of an MTA is provided in Annex 4. Other examples include those of Knoppers et al. (2013) (online supplementary material), NCI (NCI, 2016), the National Cancer Research Institute (NCRI, 2009), the Association of Research Managers and Administrators (ARMA, 2016), and Belgian Co-ordinated Collections of Micro-organisms (BCCM, 2016).
3.2. General safety precautions required for working in a biobank
The primary, basic requirement of a biobank is general safety. This includes protection of people and of the environment against biological and chemical hazards. The management of these risks should be based on a general implementation of a precautionary principle similar to those used in laboratories and clinical settings, and should be embodied in a general safety management plan.
3.2.1. General laboratory safety
In addition to biosafety, biobanks must follow strict general safety regulations and procedures in relation to chemical, physical, and electrical safety. The use of liquid gases such as liquid nitrogen (LN2) for cryopreservation poses a serious source of hazard. Where LN2 refrigeration is used, an adequate supply of refrigerant must be maintained. The supply maintained on-site should be at least 20% more than the normal refill use, to allow for emergency situations.
Handling LN2 has serious safety implications. Skin contact with LN2 can cause severe frostbite.
When bulk storage and piping systems are used, blockage of relief valves and/or overpressure may lead to simultaneous leakage of LN2 from several relief valves, causing white-out conditions in a matter of a few seconds. This leads to a drop in visibility to almost zero, and the oxygen level in the area decreases below what is necessary to sustain life. Personnel must evacuate immediately.
Oxygen-level sensors should always be used when LN2 containers are used in a biobank. LN2 expands to 650 times its original volume at room temperature, causing a form of explosion hazard if evaporation is restricted. Storage areas must be well ventilated. Plastic and glass containers can easily explode if liquid is trapped when the container is removed from the LN2.
Protective safety equipment must be worn when handling LN2. Heavy gloves, a face shield, and a protective garment should always be worn (Fig. 2). Protective shoes are also recommended. Safety notices and protocols must be clearly displayed in the biobank area. Appropriate training on the risks of LN2, including safe handling and means of protection, must be given to personnel before they work in a biobank, and should be repeated on a regular basis.

Fig. 2
Equipment for safe handling of liquid nitrogen: (a) individual oxygen detector, (b) knitted gloves, (c) cryogenic gloves, and (d) face shield.
There are also risks associated with the use of chemical fixatives and solvents used in tissue processing. In addition, electrical safety is an important concern. Freezers must be properly wired to adequate sources of electrical supply, and grounded.
Work in a biobank also entails several occupational hazards typical of the laboratory environment. These risks must be taken into account before setting up a biobank, and their prevention must be integrated into all aspects of the SOPs of the biobank.
3.2.2. Biological hazards
Laboratory biosafety requires the implementation of good laboratory practices and procedures as well as the proper use of safety equipment and facilities, to prevent unintentional exposure to microorganisms and toxins, or their accidental release.
All biological specimens should be considered as potentially infectious. They should always be handled with great care to avoid potential exposure. Their collection and processing represents a source of hazard both for the person who is the source of the specimens and for the staff members involved in these processes. It is recommended that potentially infectious samples should be handled under a biological safety hood to minimize exposure of laboratory staff. The risk group of the samples held in a biobank should be determined, and the biobank should comply with the biosafety levels corresponding to the risk group of the samples.
Immunization of biobank staff members is recommended when appropriate vaccines are available. In particular, immunization against hepatitis B virus (HBV) is mandatory for staff members involved in collecting and processing human blood or tissues. Other significant risks are posed by exposure to hepatitis C virus (HCV) and HIV as well as to the prion that causes Creutzfeldt–Jakob disease. Other pathogens can also represent a serious hazard.
Further sources of biological risk have been identified. Recommendations for laboratory practices in a safe working environment have been provided by the United States Centers for Disease Control and Prevention (CDC) in Guidelines for Safe Work Practices in Human and Animal Medical Diagnostic Laboratories (Miller et al., 2012).
3.2.3. Biosecurity
Laboratory biosecurity describes the protection of, control of, and accountability for valuable biological materials, to prevent their unauthorized access, loss, misuse, theft, or intentional release.
The scope of laboratory biosecurity is broadened by addressing the safekeeping of all valuable biological materials, including not only pathogens and toxins but also scientifically, historically, and economically important biological materials, such as collections and reference strains, pathogens and toxins, vaccines and other pharmaceutical products, food products, genetically modified organisms, non-pathogenic microorganisms, extraterrestrial samples, cellular components, and genetic elements.
Biosecurity can also refer to precautions that should be taken to prevent the use of pathogens or toxins for bioterrorism or biological warfare. Securing pathogens and toxins at research and diagnostic laboratories cannot prevent bioterrorism but can make it more difficult for potential terrorists to divert material from a legitimate facility so as to build a biological weapon (OECD, 2007). In 2006, the World Health Organization developed the publication Biorisk Management: Laboratory Biosecurity Guidance, which defines the scope and applicability of “laboratory biosecurity” recommendations, narrowing them strictly to human, veterinary, and agricultural laboratory environments (WHO, 2006).
Laboratory biosecurity measures should be based on a comprehensive programme of accountability for valuable biological material that includes:
- •
assessment of biosecurity risks,
- •
restricted and controlled access,
- •
containment-in-containment architecture,
- •
regularly updated inventories with storage locations,
- •
identification and selection of personnel with access,
- •
plan of use of valuable biological material,
- •
clearance and approval processes, and
- •
documentation of internal and external transfers within and between facilities and of any inactivation and/or disposal of the material.
Institutional laboratory biosecurity protocols should include how to handle breaches in laboratory biosecurity, including:
- •
incident notification,
- •
reporting protocols,
- •
investigation reports, and
- •
recommendations and remedies.
Adoption of these security requirements is important for biobanks that store pathogenic or toxic biospecimens.
3.3. Infrastructure and storage facilities
The biobank infrastructure and storage system depend on the type of material being stored, the required storage conditions, the anticipated period of storage, the intended use of the materials, and the resources available for purchasing the storage equipment. The storage infrastructure also depends on the available resources and support to the biobank (Mendy et al., 2013). The storage system is fundamental to maintaining high sample quality.
The data and databases related to biospecimen annotation, quality, storage location, and use, including the patients’ clinical and epidemiological information, are important attributes of biobank infrastructure. The collection, storage, uses, and management of data linked to biospecimens are discussed in Section 3.6 and Section 3.8.2.
Box
Key points: creating a biobank.
3.3.1. Storage conditions
Biospecimens should be stored under stabilized conditions to meet the requirements of potential future use in research. In selecting the biospecimen storage temperature, it is essential to consider the type of biospecimen, the intended period of storage, the frequency of use of biospecimens, the biomolecules and analyses of interest, the intended purpose of the sample, and whether the goals include preserving viable cells. Other factors that should be considered include the humidity level, the light intensity in the facilities, access to a continuous power supply, and backup systems in case of freezer breakdowns, loss of power, and other emergencies.
3.3.1.1. Cryopreservation
Cryopreservation is the recommended standard for preservation of human biological samples for a wide range of research applications. The challenge of tissue preservation is to be able to block, or at least slow down, intracellular functions and enzymatic reactions while at the same time preserving the physicochemical structures on which these functions depend.
Cryopreservation is a process in which cells or whole tissues are preserved by cooling to ultra-low subzero temperatures, typically −80 °C (freezer) or −196 °C (LN2 phase). At these low temperatures, most biological activity is effectively stopped, including the biochemical reactions that would lead to cell autolysis. However, due to the particular physical properties of water, the process of cryopreservation may damage cells and tissue by thermal stress, dehydration and increase in salt concentration, and formation of water crystals. Specific applications (e.g. proteomics or storage of primary cell cultures) may require more complex cryopreservation procedures. General information is available on the principles of cryopreservation (Cryo Bio System, 2013) and on the optimal temperature for selected biomarkers and metabolites (Hubel et al., 2014). The process of thawing may also influence cellular structure or metabolite analyses.
Specimen freezing can be performed, for example, by placing the specimen in a sealed (but not airtight) container and immersing the container in the freezing medium. The ideal medium for rapid freezing is isopentane that has been cooled to its freezing point (−160 °C). To achieve this, the vessel containing the isopentane should be placed in a container of LN2. The freezing point approximately corresponds to the moment when opaque drops begin to appear in the isopentane. Direct contact of the specimen with LN2 should be avoided because this can damage tissue structure.
3.3.1.2. Other fixation and preservation methods
Formalin or alcohol fixation and paraffin embedding is the best method for morphological analysis, morphology-related methods, and immunohistochemistry. It may also be used as an alternative method to preserve tissues at relatively low cost when adequate freezing procedures and storage facilities are not available. Paraffin blocks and histological slides may be stored in light- and humidity-controlled facilities at 22 °C (Figs. 3 and 4).

Fig. 3
Paraffin-embedded tissues.

Fig. 4
Histological slides.
Tissue fixed according to strict protocols may be used for DNA extraction. The DNA is usually fragmented but remains suitable for polymerase chain reaction (PCR)-based analysis of short DNA fragments. However, fixed tissue is of limited usefulness for RNA extraction.
RNAlater is a commercial aqueous, non-toxic tissue storage reagent that rapidly permeates tissues to stabilize and protect cellular RNA at room temperature. RNAlater eliminates the need to immediately process tissue samples or to freeze samples in LN2 for later processing. Tissue pieces can be harvested and submerged in RNAlater for storage for specific periods without jeopardizing the quality or quantity of RNA obtained after subsequent RNA isolation.
PAXgene tissue fixation is increasingly used for tissue preservation. PAXgene tissue systems are formalin-free solutions for the simultaneous preservation of histomorphology and biomolecules and the purification of high-quality RNA, DNA, microRNA (miRNA), proteins, and phosphoproteins from the same sample. Tissue specimens are collected, fixed, and stabilized with the PAXgene tissue fixation and stabilization products. PAXgene-fixed tissue can be processed and embedded in paraffin similarly to formalin-fixed tissue, and biomolecules can be extracted (Gündisch et al., 2014).
3.3.2. Biospecimen storage infrastructure
Two types of storage systems are used for biospecimen storage: ultra-low-temperature (or low-temperature) storage systems and ambient-temperature storage systems. “Ultra-low temperature” can be defined as temperatures below −80 °C (e.g. LN2), and “low temperature” as temperatures between 0 °C and −80 °C.
3.3.2.1. Liquid nitrogen storage
LN2 facilities contain LN2 in liquid-phase tanks (Fig. 5) and vapour-phase containers (Fig. 6). Cryogenic storage using LN2 is an effective long-term storage system, because its extreme ultra-low temperatures slow down most biological, chemical, and physical reactions that may cause biospecimens to deteriorate.

Fig. 5
Liquid nitrogen facility with LN2 tanks.

Fig. 6
Tank for storage in vapour-phase liquid nitrogen.
LN2 vapour-phase containers with LN2 in the base of the tank can maintain samples below Tg (the critical glass-transition temperature, i.e. −132 °C), and submersion in LN2 guarantees a stable −196 °C temperature environment for all samples. Vapour-phase storage is preferred over liquid-phase storage, because it avoids some of the safety hazards inherent in liquid-phase storage, including the risk of transmission of contaminating agents (Fig. 6). The design of the tank is critical to maintain a sufficient amount of LN2 in the vapour phase.
Liquid-phase storage needs less frequent resupply of LN2 and thus affords better security in case of a crisis in LN2 supply. Closed LN2 tanks can maintain samples at below −130 °C for several weeks without the need to refill the LN2 tank. The initial investment and the availability and cost of LN2 can be major drawbacks. Also, safety hazards inherent in the use of LN2, such as burning or oxygen deficit risks, should be managed. When LN2 tanks are used, oxygen-level sensors must be used, and they should be calibrated every few years. The use of protective equipment – in particular, face shields, cryogenic gloves, and individual oxygen detectors – should be mandatory, and this equipment should be easily accessible (Fig. 2). Appropriate training in the safe handling of LN2 must be provided, and this should be included in an SOP describing the potential health hazards and required safety precautions.
3.3.2.2. Mechanical freezers
Mechanical freezers are used for a variety of storage systems with temperatures ranging from low-temperature to ultra-low-temperature conditions, including −20 °C, −40 °C, −70 °C to −80 °C, and −150 °C, and come in a wide range of sizes and configurations (Figs. 7 and 8).

Fig. 7
Freezer facility.

Fig. 8
Freezer racks.
Ice crystals may form in biological samples at temperatures between 0 °C and −40 °C, and protein activity may persist until −70 °C or −80 °C; therefore, freezer temperatures should preferably be below −80 °C. Cascade compressor technologies may produce temperatures as low as −140 °C. Mechanical freezers, which generally require a lower initial investment than LN2 tanks and provide easy access to biospecimens, can be installed if appropriate electrical power is available. However, the compressor technology requires constant electrical power to maintain subzero temperatures, so a backup power system and an emergency response plan are needed. Whether samples warm up significantly during power outages or freezer breakdowns depends on the temperature, type, and volume of the stored biospecimen, the ambient conditions of the environment where the freezers are stored, and the design and maintenance of the freezer.
Ambient temperature and humidity influence temperature stability considerably if doors are left open for prolonged periods, for example for sample loading, or if frost forms in the freezer, racks, or samples. Overheating of compressors may shorten their lives. Mechanical freezers and refrigerators should be positioned with sufficient air flow around the units and preferably in rooms that are air-conditioned or have equipment for extraction of the hot air generated by the compressors. Regular cleaning and maintenance of freezers should be planned; this should consist, at a minimum, of cleaning filters and removing ice around the door and seals. Freezers should be equipped with alarms set at about 20 °C warmer than the nominal operating temperature of the unit. An independent temperature monitoring system should be in place (Figs. 9 and 10), to prevent freezer failure or breakdown and/or to alert personnel in case this happens. Some of the freezers (approximately 10%) should be kept empty and cool to be used as a backup system.

Fig. 9
Temperature monitoring system.

Fig. 10
Graph of temperature log obtained from monitoring system.
3.3.2.3. Refrigerators
Refrigerators are commonly used for samples that can be maintained at ambient temperature. However, the longevity of biospecimens being stored is enhanced if they are stored below ambient temperature, due to biomolecular degradation that can occur at high ambient temperatures. Storage at 4 °C can be a temporary storage solution as an intermediate step before preparation for ultra-low-temperature storage or before sample processing. For refrigerators, as for mechanical freezers, it is important to maintain and monitor the temperature in the required operating range and to have a backup power system.
3.3.2.4. Ambient-temperature storage
If a biobank does not have mechanical freezers or cryogenic storage equipment, because of practical or financial reasons, then specific biological storage matrices may be used for long-term maintenance of some biological components at room temperature. Formalin-, PAXgene-, or ethanol-fixed, paraffin-embedded tissues and lyophilized samples can be stored at ambient temperatures. Dried samples, such as blood spots on filter paper, can be stored at ambient temperature (Figs. 11 and 12). There are also some new techniques for storage of DNA at ambient temperature, for example in mini-capsules after dehydration. A mini-capsule consists of a glass vial containing the sample, enclosed in a stainless steel shell with a cap. The mini-capsule is sealed by a laser, which welds the junction between the shell and the cap under an anhydrous and anoxic inert atmosphere.

Fig. 11
Room-temperature storage facility.

Fig. 12
Box of samples stored at room temperature.
Biological storage matrices should be evaluated before use to ensure that they are appropriate for downstream applications. Temperature, humidity, and oxygen levels should be controlled to avoid mould growth and microbial contamination.
3.3.3. Storage services, access, and security
Biobanks should have dedicated storage facilities that are not shared with other activities, for the safety and security of biospecimen collections. Sufficient air conditioning must be provided for air circulation and to maintain the ambient temperature at 22 °C or below, to prevent excess freezer wear and early failure. Rooms that contain LN2 tanks should be equipped with appropriate air flow systems to avoid the accumulation of N2 in case of leakage, coupled with an oxygen-level alarm system, to monitor N2 release from the tanks. In general, storage facilities and equipment should be monitored by appropriate alarm systems (Figs. 13 and 14).

Fig. 13
A tank fitted with a monitoring device, which shows the level of liquid nitrogen inside the tank.

Fig. 14
Video monitoring in liquid nitrogen facilities.
Biobanks should be equipped with a system that adequately limits access to authorized staff members and protects against intrusion by unauthorized individuals (Fig. 15). In principle, only people assigned to biobank activities should have access to the storage facility and biospecimens, and all materials added or withdrawn should be documented. The documentation of sample movement is important for traceability and for the updating of biobank catalogues.

Fig. 15
Controlled access in a secured room.
All biobanks require a constant source of electrical power. Given that the commercial electrical power grid is likely to fail at some point, a backup power system is required. This backup system should operate independently from the grid and from any other facilities. The most common type of backup power is a diesel generator. Such a system should have the capacity to run for a sufficient time to allow the restoration of the power supply (typically 48–72 hours) and should be tested regularly (Fig. 16). Enough fuel should be available on-site to run the generator for several days. The fuel should also be tested to ensure its quality.

Fig. 16
Power generator.
Biobanks with LN2 facilities should have an LN2 supply stock from which to refill the LN2 tank (Figs. 17 and 18). Adequate backup capacity for low-temperature units must be maintained. The total amount of backup storage required for large biobanks must be determined empirically but will typically be 10% of the total freezer capacity for mechanical freezer storage. Because LN2 storage is safer than using mechanical freezers, the backup capacity for LN2 storage could be less than 10% (ISBER, 2012).

Fig. 17
Liquid nitrogen supply stock tank.

Fig. 18
Liquid nitrogen piping.
Every facility should use basic security systems; these must be monitored and alarms must be responded to 24 hours a day, 7 days a week by people who can take the necessary action to respond to an alarm within a time frame that prevents or minimizes loss or damage to biospecimen collections. Systems should allow for calls to other key staff members from a list of staff telephone numbers if the first person fails to acknowledge the alarm.
Whenever possible, it is recommended to consider splitting stored biospecimen collections into two sets of aliquots, with each set stored in a different location. This strategy will enable the preservation of a set of samples in the case of adverse events in one location. For multicentre studies, it is recommended that each recruitment centre retain a set of aliquots at the place of collection, with the second set transported to a central location that is accessible to all recruitment centres.
3.3.4. Basic informatics infrastructure
The biobank informatics infrastructure needs to contain hardware and software that are sufficient to address the functional requirements of the biobank, record and store the information acquired during each biobank process (see Section 3.8), and provide an electronic method for records management (see Section 3.6). It is important that the hardware and software infrastructure is designed in such a way that it not only meets these capacity and traceability requirements but also meets the requirements for security, data protection, and privacy (see Section 3.1.3).
One challenge that persists for IT solutions is importing specimen-associated clinical data (Section 3.8) that have been inputted into a separate system. Although it is not essential for the specimen-associated data to be in the same database as the biobank-specific data, it is important for the clinical data to be easily accessible via a link or a regular import. There may be logistic concerns in directly accessing hospital IT systems, and careful attention should be given to this during the planning of the biobank IT infrastructure.
The IT infrastructure should also be part of the QMS, and the records stored in the system should be checked for veracity. QC checks should include the verification of biospecimen locations to assess the concordance between physical storage and database location.
3.3.4.1. IT functionality
Biobank management software must guarantee the management of different functions and data related to biobanking activities (see Section 3.8). It is fundamentally important that there is a method to track each sample throughout the biobank process and to document the actions that have been carried out on the sample.
Documentation related to sample collection (informed consent, participant information sheet, sample collection protocol), sample processing, sample sharing (MTA and DTA), and shipment (proof of shipment and delivery) must be appropriately referenced in the IT system (see Section 3.6). The IT system requires a backup process to ensure that all data recorded in the IT system are correctly tracked and maintained, and are recoverable in the event of erroneous modifications.
Semantic interoperability, in particular, presents a significant challenge for biobanking and IT support.
The specific location of every stored aliquot relating to a sample should be tracked. The biobank database containing this information should be updated in real time as a biospecimen is moved within or out of the biobank.
In addition to IT software to record the information at each point of the biobanking process, there need to be software solutions to document information about monitoring of storage infrastructure and to report alarms about adverse events.
It is also recommended that the biobank software record information about operations and operators. This should include information about the standard and regular measures taken to calibrate and repair biobank instruments.
The management of these functions is fundamental to provide high-quality samples.
3.3.4.2. Software solutions
As biobanking evolves in terms of the types of samples that are collected, archived, and stored and the downstream use of the samples, there continues to be a need to develop informatics tools for the management of biobanks. Different options may be considered depending on the needs, financial resources, and IT resources of the specific biobank. For the rapidly growing field of biobanking, commercial software solutions are increasingly available. Recently, open-source systems have emerged, and some have been selected by European biobanks (Kersting et al., 2015). However, commercial and open-source solutions mainly cover particular aspects and require adaptation to respond to the requirements of the individual biobank.
An alternative to commercial and open-source systems may be the development of a dedicated in-house system, noting that the internal cost of maintaining a development team for modifications and maintenance can be considerable (Voegele et al., 2010). In a larger scope, a laboratory information management system (LIMS) enables the management not only of the biobank but also of the entire sample life-cycle workflow. Electronic laboratory notebooks are also a solution for the management of procedures performed in a laboratory, and can be used by scientists, engineers, and technicians (Voegele et al., 2013).
3.3.4.3. Data management and informatics security
Biobank management systems must permit access to sample data in order to stimulate collaboration, but they must also guarantee the confidentiality of sample records.
Data security systems should be adequate to ensure confidentiality and safety. Electronic records should be adequately protected through regular backups on appropriate media. Intrusion-proof management systems should include solutions such as dedicated servers, secure networks, firewalls, data encryption, and user authentication through verification of user names and passwords.
All computers used by biobank personnel should be password-protected and have automatic timeout mechanisms. The biobank management software should also be password-protected and should have different user profiles to permit different levels of access. Each biobank staff member should have an individual user ID, to provide complete traceability of all actions performed on biobank data.
The protection of personal information and individual data associated with specimen collection is a fundamental requirement of a biobank. This should be achieved through the use of safe, structured bioinformatics systems. Personal identifiers should be replaced by codes, and all individual data stored in the biobank management system should be protected with the same stringency as patient clinical files. This also applies to data that are considered to be sensitive. Communication to third parties of data files containing personal information and identifiers should be strictly prohibited unless it is required by law or explicit permission to do so was granted. Examples of methods of coding are provided in Appendix 2 of the privacy and security policy of GA4GH (GA4GH, 2015b).
3.3.4.4. Biobank networking infrastructure
The facilitation of scientific networking is an important aspect of IT infrastructure. Networking can increase biobank use and therefore is an important element of biobank sustainability. Publication of data on the Internet can greatly increase the visibility of the biobank and its ability to participate in biobank networks. It is recommended that a biobank develop a website to present its operations to the scientific community, in addition to an online catalogue with information on the nature, characteristics, and quality of its biological samples. Networking to facilitate exchange and access to an increased number of samples requires that biobanks adhere to standards for use of samples and data to ensure semantic interoperability between different systems and different biobanks, and this in particular presents a significant challenge for biobanking and IT support (see Section 3.8, Section 3.5, and Section 2.1.4)
Box
Key points: IT systems.
3.3.5. Basic storage disaster recovery – monitoring, backup, and additional storage
Biobanks require a disaster recovery (DR) plan to protect their assets, biological material, and associated data. The ability to respond to a disaster and protect the integrity of the samples and data directly affects their quality.
In its most simple terms, DR entails taking whatever steps are needed to ensure that, in the event of a disaster, the loss caused by the disaster is kept to an acceptable level and operations can return to normal as smoothly and as quickly as possible. DR encompasses all processes, policies, and procedures for recovery or continuation of infrastructure operation after a natural or human-caused disaster, and must include planning for key personnel, facilities, and data recovery. DR plans are not a one-size-fits-all solution; in order for DR plans to work, they need to address the needs of the specific biobank.
The best possible DR planning for biological materials and data is to ensure that there are duplicated aliquots stored in two or more locations. The more distinct these locations are in terms of geographical area and reliance on the same utilities (power, generator, LN2, carbon dioxide [CO2] supply, ambient-temperature control, and other elements that pertain to the functioning of the biobank storage infrastructure), the better the ability of one of the locations to withstand a particular disaster. Although this strategy will avoid unnecessary loss in case of adverse events in one location, this approach has three difficulties.
- •
There needs to be enough of the original specimen to produce two identical aliquots (in the presence of tumour heterogeneity, this is questionable for tumour tissue samples).
- •
Additional logistics are involved in regular transportation of the fresh and/or frozen samples to ensure that the biobank has the same content at each location.
- •
Retrieval of samples from the duplicate locations involves increased costs and time delays.
Retrieval of samples from duplicate locations is hampered by increased distances (preferable for the DR plan) and transportation facilities (couriers, transportation infrastructure). Preventive measures such as different methods of storage or reducing the specimen to its basic derivative components, such as nucleic acids, will provide opportunities for innovative storage methods. Also, nucleic acids are more stable once extracted from tissue, and therefore are more resistant to temperature fluctuations and permit longer response times.
A similar situation relates to biobank data: saving the data contemporaneously at two distinct sites would guarantee the same safeguards to the data. This is potentially more feasible than storing samples in separate locations, because duplicating and transferring data is an easier task and does not necessarily require physical transportation. However, where continual data transfer is not feasible, periodic backups should be carried out, with backups stored off-site to reduce loss of data.
Each biobank infrastructure DR plan should contain an evaluation of the events and elements that can affect the biobank, the probability of these occurring, and the means to address them. These can be either natural events (e.g. earthquakes, hurricanes, storms, floods, fires, plane crashes, excess temperatures and humidity) or human-caused events (e.g. breakdown of a single freezer, breakdown of multiple freezers, power outage or power fluctuations, CO2 outage, air conditioning breakdown, air extraction breakdown, inaccessible room due to gas leak). Only those elements that affect the biobank, either directly or indirectly, should be considered in the individual plan.
Apart from faults with a single container (caused by blown fuses, battery discharges, blocked refill valves, broken compressors, broken covers or doors, or worn seals), external events will affect each biobank to a different extent.
- •
Biobanks with −80 °C freezers are most affected by electricity supply, CO2 supply, biobank room temperature, dusty conditions, humidity, and air conditioner faults.
- •
LN2 biobanks are most affected by LN2 supply.
- •
Automatic LN2 filling systems are most affected by faulty sensors and faulty transfer pipes.
- •
Monitoring systems are most affected by electricity supply, Internet connectivity, wireless connections, and telephone lines.
Events such as a power outage or power fluctuations can have low priority if there is a way to mitigate or avoid the problem by providing either an uninterrupted power supply or a backup diesel generator. The backup generator should be able to start automatically, needs to have the capacity to run for a sufficient time to allow the restoration of the power supply (typically 48–72 hours), and should not be affected by the adverse event that caused the power outage.
It is always important to consider the cascading effect of a single event. An example is a fault in the air conditioning system that causes the biobank’s ambient temperature to rise. This temperature rise, in turn, causes the mechanical freezers to need CO2 to maintain their temperature of −80 °C. If the CO2 supply is depleted by the time the ambient temperature returns to an acceptable level, then the temperature of the mechanical freezers will also rise, potentially leading to damage of the samples they contain.
A complete DR plan requires the following steps.
- •
Categorize the stored samples in order of priority. In case of an emergency, high-priority samples will be moved to an external facility before lower-priority samples.
- •
Evaluate the acceptable downtime (the time during which the biobank is inaccessible).
- •
Evaluate the acceptable loss (the number of samples and their associated data that can be lost). The acceptable loss should be considered in terms of delays to research, and by evaluating how much time and money would be needed to collect the lost material again, considering the availability of such samples and their associated data as well as the effort required by personnel, the equipment to be used, and the consumables. Samples from longitudinal studies acquire more value over time as their associated samples and data accumulate, and therefore the cost of replacing them increases with time.
- •
Carry out a risk analysis to evaluate the potential disasters, the probability of each type of disastrous event occurring, and the impact each event would have on the biobank. The risk analysis makes it possible to prioritize the events that need to be addressed. Then, based on the resources available, it can be decided how to address each event.
- •
Calculate the response times in the event of a disaster. Response times are critical because they directly affect the potential loss of samples, and they must be calculated based on the acceptable loss and the time needed to either return to normal functioning or mitigate the problem caused by the adverse event.
- •
For each type of disaster, calculate a maximum response time to ensure the integrity of the conserved samples, such as either fixing a broken freezer so that it returns to its desired temperature before it has reached critical temperature, or moving the samples to a different location before their quality is compromised. This calculation must take into consideration the different reactions of the containers and the different effects of temperature change on each sample type stored (tissue, blood, plasma, serum, DNA, RNA).
- •
Assign people to be on call to respond to any alarms at all times (24 hours a day, 7 days a week); it is essential that they are able to respond and carry out the DR plan within the allocated time.
- •
Carry out simulation exercises to ensure effective training of the people assigned to respond.
- •
Specify methods for transporting samples without affecting their quality and integrity, in case the samples need to be moved.
- •
Ensure that backup facilities are available, in case samples need to be moved from the current biobank or freezer. Adequate backup capacity for low-temperature units must be maintained (10% is the best practice). The total amount of backup storage required for large biobanks must be determined empirically. Typically, the minimum should be the capacity of a single container (where there are different sizes, this should be calculated based on the capacity of the largest freezer) or, for large biobanks, 10% of the total container capacity (NCI, 2016).
- •
Prepare a detailed list of actions for each evaluated event. These lists must be presented in the form of SOPs, so that in the event of a disaster, action can be taken immediately.
The DR plan should be part of the QMS and should be reviewed annually to guarantee that it responds adequately to the biobank’s evolution. The DR plan must be tested as extensively as possible using simulated scenarios and should be updated regularly as the biobank infrastructure changes.
Box
Key points: disaster recovery plan.
3.4. Quality
Biobanks are key for the development of clinically useful biomarkers of disease and disease progression, for discovering and monitoring new drugs, and for understanding the mechanisms of cancer and related diseases. All of these possibilities are underpinned by the availability of high-quality, well-annotated samples of diseased and control tissue, blood, and other biological materials and associated data.
The availability of high-quality samples is also important to demonstrate to funders of biobanks and to the research community that the facility provides a good return on their investments in sample and data collection, which will accelerate progress in cancer research.
The scientific and technical management of the biobank infrastructure and resources – such as storage facilities, pre-analytical processing tools, trained personnel, robust governance, and policy management – is central to maintaining quality and determines the relevance and success of a biobank.
The key components that can affect the quality of samples and data are presented in Fig. 19.
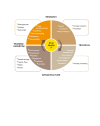
Fig. 19
Overview of the key issues related to quality in biobanking.
In general, biobanks should implement systems that specify QC and QA for sample collection, processing, storage, shipment, and disposition. Such systems are essential for maintaining a fit-for-purpose biobank for cancer research. The ISO 15189 standard currently referred to by biobanks (ISO, 2012) is based on ISO/IEC 17025 and ISO 9001, which provide general requirements for the competence of testing and calibration laboratories and for the QMS, respectively. They are not specific for biobanking processes and procedures. However, in 2015 CEN published a series of Technical Specifications for molecular in vitro diagnostic examinations – Specifications for pre-examination processes, which are relevant for diagnostic laboratories as well as biobanks. See Table 3 for a list of CEN Technical Specifications. It is recommended that these standards are used.
Table 3
CEN Technical Specifications for molecular in vitro diagnostic examinations.
In addition, ISO is developing standards for biobanks and bioresources. The Technical Committee of ISO 276 (standardization in the field of biotechnology) will include:
- •
biobanking terms and definitions;
- •
biobanks and bioresources;
- •
analytical methods;
- •
bioprocessing; and
- •
data processing, including annotation, analysis, validation, comparability, and data integration (Furuta and Schacter, 2015).
Biobanks should have appropriate QA and QC programmes with respect to equipment maintenance and repair, staff training, data management and record-keeping, and adherence to principles of good laboratory practice. All biobank operations must be subject to regular audits. The timing, scope, and outcome of these audits should be documented.
3.4.1. Biospecimen quality biomarkers
Biospecimen quality biomarkers are useful to assess the quality of material before it is included in experimental platforms and to avoid the unnecessary use of biospecimens. In 2013, the ISBER Biospecimen Science Working Group reported on the identification of evidence-based biospecimen QC markers (Betsou et al., 2013). The findings are summarized in Table 4. Although the report provided evidence for several quality biomarkers, their level of applicability and accessibility varies. In Table 4, only markers that scored highly for applicability and accessibility are included. These markers provide QC tools for assessing biospecimens in relation to pre-analytical conditions. NCI’s Biorepositories and Biospecimen Research Branch has initiated the Biospecimen Research Network (http://biospecimens.cancer.gov/researchnetwork/projects/default.asp), which aims to stimulate original research and disseminate available data in biospecimen science.
Table 4
Identified biospecimen molecular diagnostic biomarkers, with QC scope and evaluation.
3.4.2. Quality of tissue and derivatives
The procurement of tissue, both diseased (neoplastic, pre-neoplastic, and inflammatory) and normal, must always be carried out by, or under the supervision of, a pathologist. This permits a more accurate macroscopic sampling followed by a microscopic confirmation. This is standard QA for tissue procurement (Hainaut et al., 2009).
3.4.2.1. Quality control of tissue (e.g. frozen section)
QC should be done during sampling in the grossing room on surgical samples using the frozen section method. This is done by sampling the area of suspected cancer macroscopically, performing a routine frozen section, preparing a stained slide, and documenting the review data on the sample collection sheet. The following information should be provided, which defines the quality of the tissue sample:
- •
frozen section performed (yes or no);
- •
pathologist who performed frozen section review;
- •
tumour confirmed;
- •
percentage of tumour cells;
- •
percentage of stromal and inflammatory cells;
- •
percentage of surface occupied by necrosis; and
- •
other comments.
3.4.2.2. Methods for quality control of tissue sections for DNA/RNA extraction
Regardless of whether a frozen section is performed at the time of sampling, microscopic pathology review should be performed on the tissue sections taken for nucleic acid extraction. It is recommended that this is performed every 20 sections of 5 µm, because of the potential heterogeneity in the sample. This is also recommended for sections taken from formalin-fixed, paraffin-embedded (FFPE) blocks.
3.4.3. Quality control of nucleic acids from tissue
The quality of a nucleic acid is based on quantity, concentration, purity, and integrity.
Concentration is calculated for DNA, RNA, and proteins using the ultraviolet (UV) absorbance reading at a wavelength of 260 nm and a conversion factor based on the extinction coefficient for each nucleic acid (A260 of 1.0 = 50 µg/mL for double-stranded DNA [dsDNA], 40 µg/mL for RNA, and 33 µg/mL for single-stranded DNA [ssDNA]).
Purity is calculated from the ratio of the absorbance contributed by the nucleic acid to the absorbance of the contaminants. Aromatic amino acids absorb light at 280 nm, so absorbance measurements at that wavelength are used to estimate the amount of protein in the sample. Measurements at 230 nm are used to determine the amount of other contaminants that may be present in the samples, such as guanidine thiocyanate, which is common in nucleic acid purification kits. Typical requirements for A260/A280 ratios are 1.8–2.2; A260/A280 of pure DNA is ~1.8, and A260/A280 of pure RNA is ~2. Requirements for A260/A230 ratios are generally > 1.7. The A260/A230 ratio may also predict sample amplifiability (the ability of the extracted sample to be amplified by PCR).
Acceptable ratios for purity vary with the downstream application. A230 is often constant for nucleic acid purified using a specific kit, whereas the amount of nucleic acid can vary depending on the sample source. Thus, the A260/A230 ratio often decreases when small amounts of nucleic acids are isolated.
Integrity represents intactness or state of degradation. This is often presented as the DNA integrity number (DIN) and the RNA integrity number (RIN). The higher the RIN value, the better the integrity of the RNA. RNA is considered to be of high quality when the RIN value is ³ 7. RNA with RIN values of 5 and 6 may be considered acceptable. Care must be taken when using instruments to determine these values, because the concentration of the sample can affect the resulting value.
The quality of a nucleic acid extracted from tissue can vary depending on the sample source and the extraction method applied. Quality requirements can be very different depending on the downstream application. Nucleic acids that are unsuitable for one application may provide perfectly acceptable results in another application. The qualification process consists of the quantification of dsDNA and the assessment of its suitability for downstream applications, such as high-throughput next-generation sequencing. Microarray experiments may require nucleic acid samples with specific values of concentration, purity, and integrity, whereas quantitative PCR (qPCR)-based assays may accept samples with lower quality scores because the amplicons are small (typically < 100 bp). Correctly interpreting data obtained from quantification and QC analysis is essential.
3.4.3.1. Methods for evaluating quality of nucleic acids from tissue
Spectrophotometers can measure absorbance and provide values for wavelengths of 260 nm, 280 nm, and 230 nm. However, they lack the sensitivity to measure small quantities of DNA. All nucleic acids (dsDNA, RNA, and ssDNA) absorb at 260 nm, and this method cannot distinguish between the various forms of nucleic acid. For example, the amount of genomic DNA (gDNA) present in an RNA preparation or the amount of RNA present in a gDNA sample cannot be determined. These contaminants contribute to the absorbance value, resulting in an overestimation of nucleic acid concentration. In addition, if samples are degraded, single nucleotides will also contribute to the 260 nm reading, and thus the nucleic acid concentration will be overestimated.
Fluorescent dye-based quantification uses dyes that only fluoresce when bound to specific molecules, dsDNA, ssDNA, or RNA, and thus the concentration of the specific molecule can be measured. This makes the measurement more accurate for samples that contain nucleic acid contaminants or samples that are partially degraded. Although this method provides a more accurate concentration of the sample for the molecule of interest, it does not give an indication of the contamination of the sample.
Gel electrophoresis verifies the integrity of DNA and RNA molecules by separating their fragments based on size and charge and thus estimating the size of DNA and RNA fragments.
The RIN is an algorithm for assigning integrity values to RNA measurements. The integrity of RNA is a major concern for gene expression studies and traditionally has been evaluated using the 28S/18S ribosomal RNA (rRNA) ratio. The RIN algorithm is applied to electrophoretic RNA measurements and is based on a combination of different features that contribute information about the RNA integrity to provide a robust universal measure. If RNA is purified from FFPE samples, the 28S/18S rRNA ratio and the RIN value are not useful for assessing RNA quality.
The DIN determines the level of sample degradation using an algorithm to evaluate the entire electrophoretic trace. The higher the DIN value, the better the integrity of the gDNA sample.
qPCR can be a useful technique in the QC of gDNA for downstream sequencing, because it simultaneously assesses DNA concentration and suitability for PCR amplification. However, this technique is labour-intensive and has higher costs.
Sequential use of spectrophotometric and fluorescence-based methodologies permits the cost-effective assessment of DNA quality for high-throughput downstream applications. This combination also enables the detection of impurities, and thus their removal from samples. This is particularly useful for samples such as FFPE samples that are available in limited amounts.
3.5. Contents of standard operating procedures (SOPs)
Biobanks should develop, document, and regularly update policies and procedures in a standardized written format incorporated into an SOP manual that is readily available to all laboratory personnel. The SOP manual is a key part of the overall QMS of the biobank, is important to the success of biobanking, and is a major contributor to the development of biomedical practice worldwide.
The SOP manual should specifically include:
- •
procedures for obtaining informed consent and withdrawal of consent from participants;
- •
records management policies, including access control, a backup system, clinical annotation, and document maintenance and archiving;
- •
policies and procedures for specimen handling, including supplies, methods, and equipment;
- •
laboratory procedures for specimen processing (e.g. collection, transportation, processing, aliquoting, tests, storage, and QC);
- •
procedures for sharing and transferring specimens (access policy, MTA);
- •
procedures for a business model and cost recovery, when applicable;
- •
policies and procedures for shipping and receiving specimens;
- •
QA and QC policies and procedures for supplies, equipment, instruments, reagents, labels, and processes used in sample retrieval and processing;
- •
procedures for security in biobank facilities;
- •
policies and procedures related to emergencies and safety, including reporting of staff injuries and exposure to potential pathogens;
- •
policies and procedures for the investigation, documentation, and reporting of accidents, errors, complaints, and adverse events;
- •
policies, procedures, and schedules for equipment inspection, maintenance, repair, and calibration;
- •
emergency procedures in case of failure of a refrigerator, freezer, or LN2 tank;
- •
procedures for disposal of medical waste and other hazardous waste; and
- •
policies and procedures describing the requirements of recruitment and training programmes for biobank staff.
3.6. Records management
The importance of an adequate records management strategy cannot be overstated.
Documentation related to sample collection, sample processing, sharing of samples (MTA and DTA), and shipment of samples (proof of shipment and delivery) must be appropriately maintained and archived in a traceable and secure manner. A backup system must be implemented to guarantee appropriate maintenance of all documents.
All documents and documentation must be kept centrally and should include:
- •
the SOP manual
- •
quality certifications
- •
personnel training records
- •
templates of forms and spreadsheets
- •
documentation of biobank audits
- •
documentation of adverse events
- •
instrument calibration records
- •
maintenance and repair records
- •
signed informed consents
- •
signed collaboration agreements
- •
sample request forms
- •
signed MTAs and DTAs
- •
shipping notes.
Similarly to SOPs, each form should have a unique number and title. All changes made to forms should be noted, dated, and signed to provide a trace of all modifications.
All hard copies of records must be archived in a secure manner, to be accessed only by authorized personnel. All stored records should be stored in a manner that provides easy access for inspection by authorized personnel.
Each container, tank, freezer, refrigerator, or room-temperature storage cabinet should have a unique identifier. The hierarchy of each storage unit should be clearly defined, to enable stored samples to be located easily. A convention should be established for numbering shelves, racks, and boxes as well as each location within the container.
An IT solution (see Section 3.3.4) can provide a centralized system to maintain traceable records of samples. Where possible, hard copies of records should be scanned into an IT system to provide a backup.
All records should be archived for a period in line with institutional or local regulations, where they exist. Where there are no such regulations, the biobank should decide the period for record retention depending on the type of record. Records pertaining to samples that no longer exist may be destroyed if the records are considered to no longer be valuable. Records pertaining to samples that were withdrawn should be destroyed in a secure manner. Records pertaining to instruments may be destroyed once the instrument has been retired. The destruction of records should be carried out in a manner in line with the security requirements of the record.
Box
Key points: records management system.
3.7. Specimen collection, processing, and storage
Many types of biological material can be stored for cancer research purposes. The methods used to collect biospecimens will vary depending on what the intended end use is and how the specimens will be processed.
The recommendations presented here are derived from multiple sources, such as international publications and articles (Eiseman et al., 2003), including the biorepository protocols of the Australasian Biospecimen Network. Although this book focuses on cancer research, the research community realizes that samples may also be used in other areas of research; the key issue is the importance of harmonization of techniques and practices to facilitate multidisciplinary collaboration.
This section provides general advice about the collection of:
- •
whole blood and derivatives
- •
solid tissues
- •
urine
- •
buccal cells and saliva
- •
bronchoalveolar lavage
- •
bone marrow aspirate
- •
cerebrospinal fluid
- •
semen
- •
cervical and urethral swabs
- •
hair
- •
nails.
3.7.1. Collection of blood or blood-derived products
Detailed instructions and protocols for the collection of blood or blood derivatives are provided in Section 4.
3.7.1.1. Blood
The following general guidelines should be considered.
- •
All blood should be treated as potentially infectious. Blood samples for research purposes should be collected at the same time as routine clinical blood samples, so as to limit discomfort to individuals. Blood should be collected from fasting individuals (i.e. after abstinence from food, alcohol, and caffeine-containing beverages for 8–12 hours).
- •
Blood should not be collected after prolonged venous occlusion.
- •
Tubes into which the blood is collected should be clearly labelled (Fig. 20).
- •
For blood collection, the recommended order of draw is the following (Calam and Cooper, 1982; CLSI, 2007):
- 1.
blood culture tube;
- 2.
coagulation tube;
- 3.
serum tube with or without clot activator, with or without gel;
- 4.
heparin tube with or without plasma separating gel;
- 5.
ethylenediaminetetraacetic acid (EDTA) tube with or without separating gel;
- 6.
glycolytic inhibitor.
- •
For the preparation of plasma, blood may be collected into an EDTA tube, an acid citrate dextrose (ACD) tube, or a lithium heparin tube.
- •
Ideally, blood should be processed within 1 hour of collection. After that time, cell viability decreases rapidly, resulting in poor cell structure and degradation of proteins and nucleic acids.
- •
Lithium heparin is generally used if cytology studies will be performed, but it is not recommended for proteomics work.
- •
PCR was clearly interfered with when heparinized blood (heparin 16 U/mL blood) was used as a source of template DNA (Yokota et al., 1999).
- •
Either EDTA or ACD tubes can be used if DNA will be extracted or lymphoblastoid cell lines will be derived. Lithium heparin is not recommended for establishment of lymphoblastoid cell lines.
- •
EDTA tubes are recommended if protein studies will be performed. The use of EDTA tubes results in less proteolytic cleavage compared with the use of heparin tubes or ACD tubes.
- •
For the preparation of plasma, the blood should be centrifuged as soon as possible. For the preparation of serum, the blood should be processed within 1 hour of collection.
- •
The amount of blood collected should be justified when applying for ethical clearance.
- •
A reduced volume of blood in a tube containing additives should be recorded to avoid confounding the results.
- •
The time and date of blood collection and the time of freezing should be recorded, as well as any deviations from the standard processing protocol.
- •
Blood should be transported at room temperature or on melting ice depending on the particular applications. Samples to be used for proteomics assays should be processed immediately at room temperature, because cool temperatures can activate platelets and release peptides into the sample ex vivo.
- •
Blood spot collection should be considered as an alternative to whole blood for validated techniques when conditions necessitate easier collection and cheap room-temperature storage. Different types of collection cards are available (e.g. Guthrie cards, “fast transient analysis” [FTA] cards, IsoCode cards) (see Section 4).

Fig. 20
Blood sample.
3.7.1.2. Blood spots
Guthrie cards (Schleicher & Schuell 903 filter paper) are used to collect heel-stick blood from newborns for metabolic disease screening. The 903 paper is manufactured from 100% pure cotton linters with no wet-strength additives. The critical parameters for collection of newborn screening samples are blood absorbency, serum uptake, and circle size for a specified volume of blood. Blood spots archived for as long as 17 years, sometimes at room temperature, have also provided valuable sources of amplifiable DNA (Makowski et al., 1996) (Fig. 21).

Fig. 21
Card with dried blood spots.
Modified cards (IsoCode cards or FTA cards) have been developed. These consist of filter paper impregnated with a proprietary mix of chemicals that serves to lyse cells, to denature proteins, to prevent growth of bacteria and other microorganisms, and to protect nucleic acids from nucleases, oxidation, and UV damage. Room-temperature transportation of such cards in folders or envelopes (by hand or by mail) has been common for years. The papers protect DNA in the samples for some years under ambient conditions. The main variable is expected to be the quality of the storage atmosphere, particularly the content of acid gases and free-radical-generating pollutants, although FTA paper can protect against such conditions (Smith and Burgoyne, 2004). Sample integrity is optimized when FTA cards are stored in a multi-barrier pouch with a desiccant pack. Whatman Protein Saver Cards are commonly used to collect dried blood spots for molecular and genomic studies and for the isolation of viruses (Mendy et al., 2005) and bacteria (see Section 4).
3.7.1.3. Buffy coat
For DNA testing, if DNA cannot be extracted from blood within 3 days of collection, the buffy coat may be isolated and stored at −70 °C or below before DNA isolation. Buffy coat specimens that are being used for immortalization by Epstein–Barr virus should be transported frozen on dry ice (solid-phase CO2). RNA should be isolated from buffy coat within 1–4 hours of specimen collection; alternatively, RNA stabilization solution (e.g. RNAlater) should be used (see Section 4).
3.7.2. Collection of solid tissues
Solid tissues for research are collected by biopsy or after surgical excision. Detailed procedures are presented in Section 4.
The following important points should be considered when planning tissue collection for research.
- •
The collection of samples should be carefully planned with surgeons, clinical staff, and pathologists. Collection of solid tissue for research from surgically excised tissue should always occur in the grossing room unless the standard procedure for clinical care permits collection in the operating theatre or nearby pathology suite.
- •
The collection of samples for research should never compromise the diagnostic integrity of a specimen. Only tissue that is excess to diagnostic purposes should be collected for research. It is the responsibility of the pathologist to decide this.
- •
The intact surgical specimen or biopsy sample should be sent to pathology.
- •
Tissue bank staff members must be present in pathology, to collect, freeze, or fix the tissue as quickly as possible.
- •
All materials and instruments should be prepared in advance. If a fresh sample is to be obtained, transport medium (RPMI 1640, 10% fetal bovine serum [FBS], 100 U/mL penicillin/streptomycin, 100 U/mL amphotericin) should be prepared. If a sample is to be vitally cryopreserved, cryopreserving solution should be prepared (RPMI 1640, 10% dimethyl sulfoxide [DMSO], 20% FBS).
- •
A pathologist should supervise the procurement of the tissue for research purposes. The pathologist will examine the sample and, allowing adequate tissue for diagnostic purposes, will remove portions of the tumour and adjacent apparently normal tissue and other areas of interest. Where possible, two or more samples of the tumour tissue should be taken, representing different areas, i.e. different macroscopic patterns in the body of the tumour. Normal tissue can be taken from a non-diseased resected organ, but where the normal tissue is required for use as matched control, it should be taken preferably > 10 mm from the diseased tissue.
- •
If applicable, involved lymph nodes and metastases will also be collected. Tissues must be sliced with sterile forceps and scalpel blades, and staff members must use sterile gloves. The use of the same scalpel blade for normal and neoplastic areas should be avoided. If this is not possible, normal tissue should be collected before tissue from tumour areas.
- •
Standard diagnostic processes usually place surgical specimens in formalin after excision. Where fresh, vitally cryopreserved, or fresh frozen samples are required, samples must be transferred as fresh specimens. In this case, fresh specimens should be placed in a closed container in a sterile cloth on wet ice for transportation from surgery to pathology. An alternative, which also permits a delay in the need for immediate processing, is to vacuum-pack the tissue.
- •
Transfer of specimens on wet ice must be carried out as soon as possible, to minimize the effect of hypoxia on gene expression and degradation of RNA, proteins, and other tissue components. Transfer of vacuum-packed specimens is less time-critical; the samples may be stored for up to 115 hours in a 4 °C refrigerator before and/or after transportation from the operating theatre, until processing. The temperature of the specimen during transfer should be documented.
- •
It is recommended that surgical specimens or biopsy samples be preserved within 1 hour of excision. However, tissue subject to a delay up to 2 hours should still be collected (Eiseman et al., 2003). Detailed records should be kept of the timing of events from excision (or arterial clamping, in the case of larger specimens) to fixation or freezing.
- •
All tissue should be treated as potentially infectious; the collection process should be carried out under the most aseptic conditions possible.
- •
Each specimen collection receptacle must be clearly labelled when multiple samples are being collected for the biobank.
- •
Fresh tissue required for xenografting or for creation of cultures or cell lines must be placed in transport medium (RPMI 1640, 10% FBS, 100 U/mL penicillin/streptomycin, 100 U/mL amphotericin). If this tissue is to be vitally cryopreserved, it should be placed in freezing medium (RPMI 1640, 10% DMSO, 20% FBS). Because DMSO requires slow freezing, the tissue can be placed into a household −20 °C freezer for 30 minutes and then placed into −80 °C storage overnight before final storage in LN2. A specific system to reduce the temperature of the tissue by 1 °C per minute can also be used before the tissue is transferred.
- •
Tissue required for expression profiling and other molecular profiling, such as whole-genome sequencing or epigenetic studies, must be snap-frozen. Each tissue sample should be placed on card and covered with optimal cutting temperature (OCT) compound before vapour-freezing the sample by holding it over LN2. The sample can also be frozen by placing it into a container immersed in freezing medium (e.g. precooled isopentane). Tissue should never be immersed directly in LN2, because of the potential formation of cryo-artefacts. When dry ice or LN2 is not readily available, tissue collection in RNAlater is a good alternative, provided that this tissue is not required for diagnostic purposes and that permission has been given by the pathologist. Alternatively, PAXgene can be used as a fixative that preserves nucleic acids and morphology for histopathological analyses (Viertler et al., 2012).
- •
Where possible, it is advisable for a cryostat section to be taken, to prepare a haematoxylin and eosin (H&E)-stained slide for review by the pathologist for confirmation and QC of the tissue sample being conserved. An indication of the cancer cellularity is important for tissue banking because it predetermines the need for microdissection of tissue for nucleic acid extraction in next-generation sequencing.
- •
FFPE tissue can be used for targeted immunohistochemistry, fluorescence in situ hybridization (FISH), and next-generation sequencing and validation studies. RNA can also be extracted from FFPE tissue for gene fusion studies, next-generation sequencing, or quantitative reverse transcription PCR (RT-PCR). The same procedure as for diagnostic tissue may be followed, with the samples placed in containers of different colours to identify them as samples for research purposes.
- •
Care should be taken in the evaluation of biopsy material for research, because the sample has a much smaller quantity of tissue and most of it may be needed for diagnostic purposes. When needed for diagnosis, the biopsy sample should follow the standard diagnostic process and be formalin-fixed and paraffin-embedded. After the diagnostic process, any leftover material can be recovered for research.
- •
Each specimen conservation receptacle (tube) must be clearly labelled before it is placed in the biobank (see Figs. 24–26).

Fig. 24
Printed linear barcode.

Fig. 26
Pre-labelled tube with two-dimensional barcode.
3.7.3. Collection of other specimens
3.7.3.1. Urine
Urine is easy to collect and is a suitable source of proteins, DNA, and metabolites. Urine should be routinely stored at −80 °C. Ambient-temperature storage before freezing should be kept to a minimum (see Section 4).
3.7.3.2. Buccal cells
The collection of buccal cells is not difficult and does not require highly trained staff. Buccal cell collection is considered when non-invasive, self-administered, or mailed collection protocols are required for DNA analysis (Steinberg et al., 2002). However, buccal cells will yield only limited amounts of DNA compared with blood. Different methods of self-collection are available, depending on the end-points and the analyses to be performed (Mulot et al., 2005).
Cytobrush
With this method, buccal cells are collected on a sterile cytobrush by twirling it on the inner cheek for 15 seconds. The operation is repeated three times, on the two cheeks. The swabs are separated from the stick with scissors and transferred to a cryotube. The duration of the collection can influence the DNA yield. García-Closas et al. (2001) reported that cytobrushes produce DNA with good quality. However, King et al. (2002) concluded that the mouthwash method of collecting buccal cells is superior for reactions that require long fragments.
Mouthwash
With this method, buccal cells are collected by rinsing the mouth for 10 seconds with 10 mL of sterile water and expectorating the rinse into a 50 mL centrifuge tube. This operation is repeated three times. The effect of lag time of storage at room temperature is observed for mouthwashes, whereas cytobrushes are less sensitive to the lag time at room temperature.
Cytobrushes and mouthwashes are generally considered unsuitable for children, because cytobrushes are abrasive. Mouthwashes require participants to expectorate and may be aspirated or swallowed.
3.7.3.3. Saliva
Saliva is used as a biological fluid for the detection of different biomarkers, such as proteins, drugs, and antibodies. Saliva meets the demand for a non-invasive, accessible, and highly efficient diagnostic medium. The collection of saliva is non-invasive (and thus not painful), and a sample can easily be collected without a need for various devices. Whole saliva is collected by expectoration into a provided tube, whereas for the collection of submandibular saliva and sublingual saliva, different ducts need to be blocked by cotton gauze. For the collection of parotid saliva, a parotid cup should be used (see Section 4).
Treated cards
These cards are treated to inhibit the growth of bacteria and kill viruses, thereby minimizing degradation of nucleic acids. Saliva is expectorated into a sterile cup. The tip of the triangle of treated card is placed into the saliva, which is wicked onto the matrix. The treated card is air-dried and placed in a bag with a desiccant pack. Treated cards correspond to the lowest efficiency for DNA yield, because of the small quantity of collected saliva. Moreover, some proteins are left in the solution of extracted DNA. Therefore, the DNA cannot be kept for long-term conservation. However, an advantage of this method of saliva collection is its low cost, because of the absence of an extraction step.
3.7.3.4. Bronchoalveolar lavage
The airways, and particularly the alveoli, are covered with a thin layer of epithelial lining fluid, which is a rich source of many different cells and of soluble components of the lung that help protect the lung from infections and preserve its gas-exchange capacity. Bronchoalveolar lavage performed during fibre-optic bronchoscopy is the most common way to obtain samples of epithelial lining fluid (Reynolds, 2000). The cellular and protein composition of the epithelial lining fluid reflects the effects of the external factors that affect the lung, and changes in this composition are of primary importance in the early diagnosis, assessment, and characterization of lung disorders as well as in the search for disease markers (Griese, 1999).
Bronchoalveolar lavage is classically performed by instillation of buffered saline solution divided into three or four aliquots (typically a total volume of 100–150 mL) through a flexible fibre-optic bronchoscope, after local anaesthesia. The first 10 mL should be processed separately and is denoted as bronchial lavage. The rest of the lavage, denoted as bronchoalveolar lavage, should be pooled into a sterile siliconized bottle and immediately transported on ice to the laboratory. At the laboratory, the total volume of the lavage is measured, and cells and proteins are separated by centrifugation. The lavage fluid should be frozen and stored at −80 °C until use.
3.7.3.5. Bone marrow aspirate
The following paragraphs on bone marrow aspirate and cerebrospinal fluid are derived from the Australasian Biospecimen Network recommendations (see Table 1) and the publication Guidelines on Standard Operating Procedures for Microbiology (Kanagasabapathy and Kumari, 2000).
Bone marrow is the soft tissue found in the hollow interior of bones. In adults, the marrow in large bones produces new blood cells. There are two types of bone marrow: red marrow (also known as myeloid tissue) and yellow marrow. In cancer research, red bone marrow from the crest of the ilium is typically examined.
Bone marrow should be collected by a doctor who is well trained in this procedure. Bone marrow should be aspirated by sterile percutaneous aspiration into a syringe containing an EDTA anticoagulant, and the specimens should be chilled immediately. Heparin is not recommended as an anticoagulant for molecular testing. If a specimen contains erythrocytes, it should be processed to remove the erythrocytes before freezing. The bone marrow samples should be fresh frozen and stored at −80 °C.
3.7.3.6. Cerebrospinal fluid (CSF)
Cerebrospinal fluid (CSF) originates from the blood. The choroid plexuses in the first, second, and third ventricles of the brain are the sites of CSF production. CSF is formed from plasma by the filtering and secretory activities of the choroid plexus and the lateral ventricles. CSF circulates around the brain and the spinal cord. It nourishes the tissues of the central nervous system and helps to protect the brain and the spinal cord from injury. It primarily acts as a water shock absorber. It totally surrounds the brain and the spinal cord, and thus absorbs any blow to the brain. CSF also acts as a carrier of nutrients and waste products between the blood and the central nervous system.
CSF is a very delicate biological material. Often, only small volumes of CSF are available for analysis, because of the difficulty of collecting CSF, and therefore it should be handled with care. Only a physician or a specially trained nurse should collect the specimen. After collection, the specimen should be transferred into a clean penicillin vial containing about 8 mg of a mixture of EDTA and sodium fluoride in the ratio of 1:2. Centrifuging CSF is recommended before freezing if the sample contains red blood cells or particulate matter. The specimen should be frozen and stored at −80 °C or in LN2. Do not delay freezing the CSF, because cells are rapidly lysed once the CSF is removed from the body.
3.7.3.7. Semen
Seminal fluid, which is the liquid component of sperm, provides a safe surrounding for spermatozoa. At pH 7.35–7.50, it has buffering properties, protecting spermatozoa from the acidic environment of the vagina. Seminal fluid contains a high concentration of fructose, which is a major nutriment source for spermatozoa during their journey in the female reproductive tract. The complex content of seminal plasma is designed to ensure the successful fertilization of the oocyte by one of the spermatozoa present in the ejaculate. Seminal plasma is a mixture of secretions from several male accessory glands, including the prostate gland, seminal vesicles, epididymis, and Cowper’s glands (Pilch and Mann, 2006).
After collection, the fresh ejaculate should immediately be spun down at 4 °C to separate the seminal fluid from the spermatozoa. Protease inhibitors should then be added to the sample, to avoid digestion by powerful proteases present in seminal fluid. To ensure complete separation of cell debris or occasional spermatozoa from seminal plasma, the sample can be centrifuged a second time. The sample should be stored at −80 °C.
3.7.3.8. Cervical and urethral swabs
The quality of collected cervical and urethral specimens depends on appropriate collection methods. Swabs, brushes, or other collection devices should be placed in a transport medium, or transported dry in a sealed tube and resuspended in the transport medium upon arrival. The transport fluid may either be stored at −70 °C or below or immediately centrifuged, and the pellet processed for DNA or RNA extraction (see Section 4).
3.7.3.9. Hair
Currently, hair analysis is used for purposes of assessing environmental exposures, such as exposure to mercury from eating fish. Hair analysis is also used to test for illegal drug use and to conduct criminal investigations (see Section 4). Hair should be kept in sealable plastic bag, stored in the dark at room temperature (Fig. 22).

Fig. 22
Hair sample.

3.8. Specimen annotations and data sets
Data associated with the biological specimens provide added value to the samples and increase the types of research for which the biospecimens can be used. Specimen annotations provide basic information such as sample type, quantity, and current form (how the sample was stabilized and conserved), which can be used to evaluate the use of the sample in a specific assay. Specimen annotation also provides parameters that define the quality of the specimen and thus the quality of the downstream assay.
For biobanks wishing to share samples in large studies and for research that requires samples from different sources, it is important that, as with all other elements of a biobank, the data are standardized or at least harmonized to permit effective aggregation.
Information to annotate the sample should be collected at each phase of the biobank/biospecimen processes:
- •
consent;
- •
donor/patient ID, sample ID;
- •
collection (technique, date, time);
- •
processing/stabilization;
- •
conservation/storage;
- •
for tissues: organ of origin;
- •
for tissues: disease features (e.g. tumour, non-neoplastic);
- •
quality parameters;
- •
donor/patient-related data;
- •
distribution/use; and
- •
returned data.
It is important to determine a minimum data set, because this establishes a basic quality of all samples collected in the biobank (see Table 5). This should not, however, compromise the ability to collect additional data for particular sample sets that may be useful in the future. The minimum data set must be as completely defined as possible, indicating values for each of the fields in the data set, to reduce the heterogeneity of sample-associated data and thus improve its quality.
Table 5
Example of IARC minimum data set for a study or collection in a biobank.
One potential problem of data values involves different systems that use different date formats: DD/MM/YYYY or MM/DD/YYYY or YYYY/MM/DD. Another problem for sample annotation is unfilled fields. It is important that a value for “not available” is defined, because blank fields can potentially be interpreted as zero and can provide incorrect evaluations when used in research.
During the creation of a data set, it is important that for data that refer to classification systems, the version to which the value applies is also indicated. As versions change, these values can be correctly interpreted or adjusted. One such example is the staging score used for defining the stage of a cancer. The requirement to indicate the scoring system also applies to values that may be retrieved using different assays. An example of this is the concentration value taken using a spectrophotometer or a fluorescent dye-based quantification. Other fields for which the situation may be similar are units of time (e.g. time to stabilization, time from diagnosis to collection, time from diagnosis to follow-up), which may be recorded in minutes, hours, days, months, or years, as well as units of measurement of size (e.g. millimetres, centimetres) or weight (e.g. nanograms, milligrams).
Systems already exist to address the need to standardize biobank- and sample-associated information.
The SPREC tool, in particular, addresses the pre-analytical data required and contains seven data fields and defined values for the definition of sample type and the processes of collection, stabilization, and storage. There is a SPREC available for fluids and one for solids, to address the different collection and stabilization processes required. However, the SPREC systems will be replaced by CEN norms and ISO standards.
The Biospecimen Reporting for Improved Study Quality (BRISQ) standard covers pre-acquisition, acquisition, stabilization/preservation, storage/transportation, and QA measures (Moore et al., 2011). The elements in the BRISQ list are prioritized into three tiers according to the relative importance of their being reported.
- •
Tier 1 comprises 15 elements of necessary data, covering all sample types and including the manner in which the specimens were collected, stabilized, and preserved. It also contains clinical data associated with the patient.
- •
Tier 2 comprises 19 elements of beneficial data, covering patient demographic information, times and temperatures, and methods of enrichment.
- •
Tier 3 comprises 16 elements of nice-to-have data, pertaining to environmental conditions such as ischaemia, therapy, exposures, disease state, and storage containers and shipping parameters.
Tier 1 is now required for many journals.
MIABIS 2.0 represents the minimum information required to initiate collaborations between biobanks. This standard currently comprises aggregated descriptive data, and not sample-specific data, to permit a harmonized exchange of samples and data among biobanks. The MIABIS standard pertains to descriptive data of a biobank, collection, and study, which include collection types and contact information. It also includes aggregated data such as sex, age range, material type, data categories, and diseases. MIABIS is the only standard to provide indications for this collective type of information. This information is useful when creating biobank catalogues and inventories to provide visibility to available resources.
3.8.1. Annotations on patients/individuals
It is important for the biobank to annotate the consent that is collected for the samples and data that belong to the patient. This information should include whether the consent or a waiver was used to permit the use of the sample or data in research. It is important to indicate the scope of the permission, the type of research that can be performed, information such as what types of entities can access the sample (public, private), and whether there are geographical restrictions (local, national, international) on shipment of samples and data. In the era of personalized medicine and open access, it is also important to record what participant-related data can be associated with the sample (clinical data, pathological data, follow-up data) and, in particular, whether genetic data can be used and whether these data can be placed in public databases (publication or research). It is vital to indicate whether a donor/patient wishes to be re-contacted for further studies, whether they wish to be informed of incidental findings, and what should be done if any findings have hereditary implications.
For all samples, it is vital to have the diagnosis and pathological data. It is preferable that accepted nomenclature is used for diagnosis and classification systems are used for all pathology data, because they provide standard comparable parameters. One such example is the tumour–node–metastasis (TNM) system for classification of malignant tumours.
Participant-associated data are important, to provide additional value because they permit an extended evaluation of downstream assays. The more information can be collected during the patient’s clinical care path, the more valuable the sample becomes. Some of these data categories are:
- •
demographic information;
- •
family history;
- •
environmental exposure;
- •
lifestyle;
- •
diagnosis;
- •
clinical data/medical history;
- •
complete pathology report, including immunohistochemical and molecular biology markers;
- •
treatment;
- •
follow-up/outcome; and
- •
molecular/genetic characterization.
Not all of these data categories of participant-associated data are required for all studies, and even within a single category, different data fields will be required depending on the type of sample being collected and the intended use of the sample.
3.8.2. Annotations on stored specimens
Obtaining and storing information about the stored specimens – in particular, the pre-analytical variables related to the collection, transportation, ischaemia times, stabilization, and conservation of the specimens – is mandatory in CEN and ISO. The data categories to consider for sample-specific annotations are:
- •
pre-acquisition;
- •
collection;
- •
processing/stabilization;
- •
conservation/transportation; and
- •
quality.
3.8.3. General recommendations for data sets
- •
Wherever possible, the biobank should collect its data from existing clinical databases for patient-related data. This avoids the need for duplicate input and reduces the possibility of human error. If the data are collected through a link between the biobank database and the clinical database, this may also permit tracking of additional clinical data over time.
- •
Because it is not always possible to set minimum data sets common to multiple sample or disease types, macro fields should be set to indicate the presence of such data (e.g. clinical data, epidemiological data, follow-up data).
- •
All donor/patient-related data should be associated with each sample collected. This should be done either within the database system functionality or through a parent ID field that relates initially to the donor data. This avoids repeating each donor data annotation for each sample.
- •
It is important to have a method to indicate participants for which different sample types are available. This should be done either within the database system functionality or through a field to indicate the availability of other types of samples related to the same case.
- •
For all classification systems (ontologies) that are used to annotate samples, the version should be indicated.
- •
All numerical values indicated should have associated units of measurement (e.g. months, days, hours, nanograms, microlitres).
- •
All null values (not inserted) should have default values that are not zero.
- •
Values lists for data fields avoid the introduction of error and are preferable to free-text fields.
See Table 5 and Annex 5.
3.8.4. Specimen labelling and aliquoting
Each specimen should be labelled in such a manner that the labelling will survive all potential storage conditions, in particular dry ice and LN2 and potentially water bath.
Ink used on the label should be resistant to all common laboratory solvents. A minimum requirement is to print labels with a barcode (linear or two-dimensional), thus providing a direct link to database software and preventing human error in identification. However, it is also essential to include human-readable indications of contents in case no barcode reader is available (Figs. 24–26). The barcode template should be documented. The software used for labelling should enable data import and export in standard formats and should be able to link with the biobank management system.

Fig. 25
Printed two-dimensional barcode.
Ideally, all specimens should be labelled with at least two human-readable forms of identification without revealing the identity of the donor. The anonymity of the donor must be guaranteed in all cases. Radio-frequency identification (RFID) is another option but is not in widespread use for biobanking.
Information on the label should include the biobank’s unique identifier number, the name of the project, the type of biospecimen, and/or the number of the location within the storage system, with the same information repeated in the barcode if available.
After primary samples are processed, derived products should be stored in appropriate and optimized containers. As technologies for analysing biospecimens improve, smaller volumes of sample are required. Therefore, the volume of aliquots should be adapted to avoid unnecessary freeze–thaw cycles.
A wide range of tubes of different sizes, with or without a pre-printed barcode, is now available and affordable. Coloured caps can be used to distinguish between different types of samples and to facilitate the retrieval of samples. More and more analyses platforms are using robots, and sample storage in SBS format containers is also important to consider, to facilitate downstream analyses. Otherwise, specific boxes must be used for appropriate storage of this SBS format tube, allowing space saving in the biobank storage facility. Consideration and attention should be given to the composition of plastic, potential interaction with some analytes, and resistance to ultra-low storage temperatures.
3.9. Specimen shipping
Human biospecimens are considered to be “dangerous goods”, defined by the International Air Transport Association (IATA) as “articles or substances which are capable of posing a risk to health, safety, property or the environment”. According to United Nations regulations, dangerous goods meet the criteria of one or more of nine United Nations hazard classes (DGI, 2016). The relevant class for biological specimens is Class 6, Division 6.2: Infectious substances (IATA, 2015b).
The shipping and dispatch of biospecimens is subject to international regulations. These regulations, applicable to any mode of transport, are based on the recommendations of the Committee of Experts on the Transport of Dangerous Goods, a committee of the United Nations Economic and Social Council (UNECE, 2015).
The International Civil Aviation Organization (ICAO) Technical Instructions for the Safe Transport of Dangerous Goods by Air (ICAO, 1986) are legally binding international regulations. The Dangerous Goods Regulations incorporate the ICAO provisions and may add further restrictions. The ICAO rules apply on all international flights. For national flights, i.e. flights within one country, national civil aviation authorities apply national legislation. This is usually based on the ICAO provisions but may incorporate variations. State and operator variations are published in the ICAO Technical Instructions and in the IATA Dangerous Goods Regulations (IATA, 2015a; WHO, 2012).
Each person involved in the transportation of biospecimens classified as dangerous goods by IATA should undergo an initial training session followed by a refresher course every 2 years. This training is for staff members involved in the preparation of documentation and also for those involved in packaging biospecimens.
3.9.1. Regulations
Infectious substances fall into two categories: Category A and Category B.
Category A comprises any infectious substance that is transported in a form that, when exposure to it occurs, is capable of causing permanent disability or life-threatening or fatal disease in otherwise healthy humans or animals. Category A specimens include, but are not restricted to, specimens contaminated by highly pathogenic viruses (Ebola, Hantaan, Marburg, Lassa, etc.) or cultures of viruses such as dengue, HIV, or HBV. The proper shipping name for such substances is UN 2814: “Infectious substances affecting humans” or UN 2900: “Infectious substances affecting animals only”.
Category B comprises any infectious substance that does not meet the above-mentioned criteria. Most human specimens, such as blood samples, tissues, saliva, exfoliated cells, or urine not contaminated by highly pathogenic viruses, will fall into Category B. The proper shipping name for such substances is UN 3373: “Biological Substance, Category B”.
Biospecimens or derived products that have been specifically treated to neutralize infectious agents, or for which there is a minimal likelihood that pathogens are present, are not subject to these regulations. The proper shipping name for such substances is “Exempt Human (or Animal) Specimens”.
3.9.2. Packaging
The basic triple packaging system applies to all substances. It consists of three layers, as follows (Fig. 27).

Fig. 27
The triple packaging system.
- •
The primary receptacle is a watertight, leakproof receptacle containing the specimen, packaged with enough absorbent material to absorb all fluid in case of breakage.
- •
The secondary packaging is a durable, watertight, leakproof packaging to enclose and protect the primary receptacle. Several primary receptacles may be placed in one secondary packaging, but sufficient additional absorbent material should be used to absorb all fluid in case of breakage.
- •
The outer packaging is the shipping packaging, made of a suitable cushioning material, to protect the contents from outside influences while the package is in transit.
Appropriate insulation should be used. For example, for 8 °C to −20 °C, use gel packs; for −78.5 °C, use dry ice (Fig. 28); and if samples need to be kept at −150 °C, transport them in a dry shipper containing LN2. Ensure that enough refrigerant is included to allow for a 24-hour delay in shipping.

Fig. 28
Dry ice (−78.5 °C).
In-transit temperature monitoring solutions that feature alarms as well as reporting are commercially available.
The triple packaging system also applies to “Exempt Human Specimens”, such as Guthrie cards (which should be transported in watertight plastic bags) and histopathological slides (which need to be cushioned to prevent breakage). In all cases, desiccants should be used for samples that are sensitive to humidity.
3.9.3. Labelling of parcels
All outer packages must bear a United Nations packaging specification marking, according to the category in which the specimens fall. For Category A, Packaging Instruction P620 applies. For Category B, Packing Instruction P650 applies. Detailed instructions are described in the IATA Dangerous Goods Regulations (IATA, 2015c). All packages must have shipper details and consignee details (name of institute, address, contact name, email, and telephone number).
3.9.4. Constraints
When preparing to transport biospecimens, it is important to consider shipping time, distance, climate, season, method of transportation, and regulations, as well as the type and number of biospecimens to be sent and their intended use. It is also important to confirm with the recipient before the shipment that someone will be available to receive the samples.
When shipping biospecimens internationally, the sender must be aware of the requirements and regulations in the destination country before initiating the shipment, and must ensure that the consignment adheres to these regulations.
It is important to select an appropriate shipping company. Some companies offer more dedicated services for biospecimens, such as refilling of dry ice, handling of customs paperwork, and step-by-step monitoring and tracking.
3.10. Biobank workflow
Fig. 29 shows the sequence and the flow of information, data, and biospecimens, from the study design to final laboratory analyses. This scheme underlines the central role of biobanks as the transfer structure between biospecimen collection and laboratory analysis. It also underlines the fact that, in developing a study protocol, each step in this sequence of events must be clearly defined. The flow of information and biospecimens, as defined by protocols and procedures, will ensure the formation of a collection that contains traceable biospecimens and yields interpretable results. The biobank is an essential source of information and recommendations for the collection of biospecimens and for their annotation, storage, processing, and flow from the participant to the laboratory where they will be analysed.
Fig. 29
Biobank workflow.
- Ethical, legal, and social issues (ELSI) and governance
- General safety precautions required for working in a biobank
- Infrastructure and storage facilities
- Quality
- Contents of standard operating procedures (SOPs)
- Records management
- Specimen collection, processing, and storage
- Specimen annotations and data sets
- Specimen shipping
- Biobank workflow
- Recommendations for biobanks - Common Minimum Technical Standards and Protocols ...Recommendations for biobanks - Common Minimum Technical Standards and Protocols for Biobanks Dedicated to Cancer Research
Your browsing activity is empty.
Activity recording is turned off.
See more...