Clinical Description
Classic Ehlers-Danlos syndrome (cEDS) is a heritable connective tissue disorder characterized by skin hyperextensibility, abnormal wound healing, and generalized joint hypermobility.
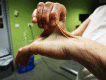
Skin. Skin is hyperextensible; it extends easily and snaps back after release (unlike lax, redundant skin, as in cutis laxa).
The skin is soft, velvety, or doughy to the touch.
The skin is fragile, as manifested by splitting of the dermis following relatively minor trauma, especially over pressure points (knees, elbows) and areas prone to trauma (shins, forehead, chin). Skin fragility may cause dehiscence of sutured incisions in skin or mucosa.
Wound healing is poor, and stretching, thinning, and pigmentation of scars is characteristic. Scars become wide, with a "cigarette-paper"-like (papyraceous) appearance, also referred to as atrophic and/or hemosiderotic scars.
Other dermatologic features in cEDS:
Molluscoid pseudotumors
Subcutaneous spheroids
Piezogenic papules: small, painful, reversible herniations of underlying adipose tissue globules through the fascia into the dermis, such as on medial and lateral aspects of the feet upon standing
Elastosis perforans serpiginosa: characterized by skin-colored to erythematous keratotic papules, some enlarging outward in serpiginous or arcuate configurations, leaving slightly atrophic centers
Acrocyanosis: painless constriction or narrowing of the small blood vessels in the skin (affecting mainly the hands) in which the affected areas turn blue and become cold and sweaty; localized swelling may also occur
Chilblains: cold injuries, characterized by red, swollen skin that is tender and hot to the touch and may itch; can develop in less than two hours in skin exposed to cold
Tissue fragility. Manifestations of generalized tissue extensibility and fragility can be observed in multiple organs:
Cervical insufficiency during pregnancy
Inguinal and umbilical hernia
Hiatal and incisional hernia
Recurrent rectal prolapse in early childhood
Joints. Complications of joint hypermobility including dislocations/subluxations of large and small joints (e.g., shoulder, patella, digits, hip, radius, and clavicle) may occur and usually resolve spontaneously or are easily managed by the affected individual. Many individuals with cEDS experience chronic joint and limb pain, despite normal skeletal radiographs.
Other problems related to joint hypermobility are joint instability, foot deformities such as congenital clubfoot or pes planus, and temporomandibular joint dysfunction [Bowen et al 2017].
Neurologic features. Primary muscular hypotonia may occur and may cause delayed motor development, problems with ambulation, and mild motor disturbance in children with cEDS. Fatigue and muscle cramps are relatively frequent.
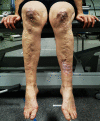
Easy bruising. Easy bruising is a common finding and manifests as spontaneous ecchymoses, frequently recurring in the same areas and causing a characteristic brownish discoloration of the skin, especially in exposed areas such as shins and knees. There is a tendency toward prolonged bleeding (e.g., following brushing of the teeth) in spite of a normal coagulation status.
Cardiovascular. Structural cardiac malformations are uncommon in cEDS.
Mitral valve prolapse and (less frequently) tricuspid valve prolapse may occur. Stringent criteria should be used for the diagnosis of mitral valve prolapse. When it does occur, mitral valve prolapse tends to be of little clinical consequence [Atzinger et al 2011].
Aortic root dilatation has been reported in individuals with cEDS [Wenstrup et al 2002, McDonnell et al 2006, Atzinger et al 2011]. It appears to be more common in young individuals and rarely progresses [Bowen et al 2017].
Individuals with cEDS are at risk for spontaneous rupture of large arteries, although the prevalence of such complications is much lower than in those with vascular EDS.
Dental. Classic EDS may be associated with abnormal dentinogenesis resulting in localized root anomalies, with shortened or bulbous roots, that lead to loosening of the teeth. Calcification of the pulp is another common finding in cEDS. This is usually of little clinical significance but may complicate root canal treatment [Lepperdinger et al 2021].
Pregnancy. Pregnancy in a woman with cEDS places both the newborn and the mother at risk for complications (see Pregnancy Management). As a whole, the complications are more frequent than in the normal population, although it is difficult to quantitate the incidence of each complication in affected individuals because no good studies exist [Kang et al 2020].
Premature rupture of the membranes and prematurity are reported to be twice as common in fetuses with cEDS born to healthy mothers, compared with fetuses without EDS born to a mother with cEDS.
Because of hypotonia, breech presentation is more frequent if the baby is affected and may lead to dislocation of the hips or shoulder of the newborn.
Intrauterine growth restriction may occur.
Perineal tearing, postpartum hemorrhage, pelvic prolapse, and incontinence following delivery may occur.