

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Brown TA. Genomes. 2nd edition. Oxford: Wiley-Liss; 2002.

Learning outcomes
When you have read Chapter 13, you should be able to:
- State what is meant by the topological problem and explain how DNA topoisomerases solve this problem
- Describe the key experiment that proved that DNA replication occurs by the semiconservative process, and outline the exceptions to semiconservative replication that are known in nature
- Discuss how replication is initiated in bacteria, yeast and mammals
- Give a detailed description of the events occurring at the bacterial replication fork, and indicate how these events differ from those occurring in eukaryotes
- Describe what is currently known about termination of replication in bacteria and eukaryotes
- Explain how telomerase maintains the ends of a chromosomal DNA molecule in eukaryotes, and appraise the possible links between telomere length, cell senescence and cancer
- Describe how genome replication is coordinated with the cell cycle
Genome replication has been studied since Watson and Crick first discovered the double helix structure of DNA back in 1953. In the years since then research has been driven by three related but distinct issues:
- The topological problem was the primary concern in the years from 1953 to 1958. This problem arises from the need to unwind the double helix in order to make copies of its two polynucleotides (see Figure 13.1). The issue assumed center stage in the mid-1950s because it was the main stumbling block to acceptance of the double helix as the correct structure for DNA, but moved into the background in 1958 when Matthew Meselson and Franklin Stahl demonstrated that, despite the perceived difficulties, DNA replication in Escherichia coli occurs by the method predicted by the double helix structure. The Meselson-Stahl experiment enabled research into genome replication to move forward, even though the topological problem itself was not solved until the early 1980s when the mode of action of DNA topoisomerases was first understood (Section 13.1.2).
- The replication process has been studied intensively since 1958. During the 1960s, the enzymes and proteins involved in replication in E. coli were identified and their functions delineated, and in the following years similar progress was made in understanding the details of eukaryotic genome replication. This work is ongoing, with research today centered on topics such as the initiation of replication and the precise modes of action of the proteins active at the replication fork.
- The regulation of genome replication, particularly in the context of the cell cycle, has become the predominant area of research in recent years. This work has shown that initiation is the key control point in genome replication and has begun to explain how replication is synchronized with the cell cycle so that daughter genomes are available when the cell divides.
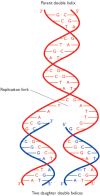
Figure 13.1
DNA replication, as predicted by Watson and Crick. The polynucleotides of the parent double helix are shown in red. Both act as templates for synthesis of new strands of DNA, shown in blue. The sequences of these new strands are determined by base-pairing (more...)
Our study of genome replication will deal with each of these three topics in the order listed above.
13.1. The Topological Problem
In their paper in Nature announcing the discovery of the double helix structure of DNA, Watson and Crick (1953a) made one of the most famous statements in molecular biology:
It has not escaped our notice that the specific pairing we have postulated immediately suggests a possible copying mechanism for the genetic material.
The pairing process that they refer to is one in which each strand of the double helix acts as a template for synthesis of a second complementary strand, the end result being that both of the daughter double helices are identical to the parent molecule (Figure 13.1). The scheme is almost implicit in the double helix structure, but it presents problems, as admitted by Watson and Crick in a second paper published in Nature just a month after the report of the structure. This paper (Watson and Crick, 1953b) describes the postulated replication process in more detail, but points out the difficulties that arise from the need to unwind the double helix. The most trivial of these difficulties is the possibility of the daughter molecules getting tangled up. More critical is the rotation that would accompany the unwinding: with one turn occurring for every 10 bp of the double helix, complete replication of the DNA molecule in human chromosome 1, which is 250 Mb in length, would require 25 million rotations of the chromosomal DNA. It is difficult to imagine how this could occur within the constrained volume of the nucleus, but the unwinding of a linear chromosomal DNA molecule is not physically impossible. In contrast, a circular double-stranded molecule, for example a bacterial or bacteriophage genome, having no free ends, would not be able to rotate in the required manner and so, apparently, could not be replicated by the Watson-Crick scheme. Finding an answer to this dilemma was a major preoccupation of molecular biology during the 1950s.
13.1.1. Experimental proof for the Watson-Crick scheme for DNA replication
The topological problem was considered so serious by some molecular biologists, notably Max Delbrück, that there was initially some resistance to accepting the double helix as the correct structure of DNA (Holmes, 1998). The difficulty relates to the plectonemic nature of the double helix, this being the topological arrangement that prevents the two strands of a coil being separated without unwinding. The problem would therefore be resolved if the double helix was in fact paranemic, because this would mean that the two strands could be separated simply by moving each one sideways without unwinding the molecule. It was suggested that the double helix could be converted into a paranemic structure by supercoiling (see Figure 2.17) in the direction opposite to the turn of the helix itself, or that within a DNA molecule the right-handed helix proposed by Watson and Crick might be ‘balanced’ by equal lengths of a left-handed helical structure. The possibility that double-stranded DNA was not a helix at all, but a side-by-side ribbon structure, was also briefly considered, this idea surprisingly being revived in the late 1970s (e.g. Rodley et al., 1976) and receiving a rather acerbic response from Crick and his colleagues (Crick et al., 1979). Each of these proposed solutions to the topological problem were individually rejected for one reason or another, most of them because they required alterations to the double helix structure, alterations that were not compatible with the X-ray diffraction results and other experimental data pertaining to DNA structure.
The first real progress towards a solution of the topological problem came in 1954 when Delbrück proposed a ‘breakage-and-reunion’ model for separating the strands of the double helix (Holmes, 1998). In this model, the strands are separated not by unwinding the helix with accompanying rotation of the molecule, but by breaking one of the strands, passing the second strand though the gap, and rejoining the first strand. This scheme is in fact very close to the correct solution to the topological problem, being one of the ways in which DNA topoisomerases work (see Figure 13.4A), but unfortunately Delbrück over-complicated the issue by attempting to combine breakage and reunion with the DNA synthesis that occurs during the actual replication process. This led him to a model for DNA replication which results in each polynucleotide in the daughter molecule being made up partly of parental DNA and partly of newly synthesized DNA (Figure 13.2A). This dispersive mode of replication contrasts with the semiconservative system proposed by Watson and Crick (Figure 13.2B). A third possibility is that replication is fully conservative, one of the daughter double helices being made entirely of newly synthesized DNA and the other comprising the two parental strands (Figure 13.2C). Models for conservative replication are difficult to devise, but one can imagine that this type of replication might be accomplished without unwinding the parent helix.

Figure 13.4
The mode of action of Type I and Type II DNA topoisomerases. (A) A Type I topoisomerase makes a nick in one strand of a DNA molecule, passes the intact strand through the nick, and reseals the gap. (B) A Type II topoisomerase makes a double-stranded break (more...)

Figure 13.2
Three possible schemes for DNA replication. For the sake of clarity, the DNA molecules are drawn as ladders rather than helices.
The Meselson-Stahl experiment
Delbrück's breakage-and-reunion model was important because it stimulated experiments designed to test between the three modes of DNA replication illustrated in Figure 13.2. Radioactive isotopes had recently been introduced into molecular biology so attempts were made to use DNA labeling (Technical Note 4.1) to distinguish newly synthesized DNA from the parental polynucleotides. Each mode of replication predicts a different distribution of newly synthesized DNA, and hence of radioactive label, in the double helices resulting after two or more rounds of replication. Analysis of the radioactive contents of these molecules should therefore determine which replication scheme operates in living cells. Unfortunately, it proved impossible to obtain a clearcut result, largely because of the difficulty in measuring the precise amount of radioactivity in the DNA molecules, the analysis being complicated by the rapid decay of the 32P isotope that was used as the label.
The breakthrough was eventually made by Matthew Meselson and Franklin Stahl who, in 1958, carried out the required experiment not with a radioactive label but with 15N, the non-radioactive ‘heavy’ isotope of nitrogen. Now it was possible to analyze the replicated double helices by density gradient centrifugation (Technical Note 2.2), because a DNA molecule labeled with 15N has a higher buoyant density than an unlabeled molecule. Meselson and Stahl (1958) started with a culture of E. coli cells that had been grown with 15NH4Cl and whose DNA molecules therefore contained heavy nitrogen. The cells were transferred to normal medium, and samples taken after 20 minutes and 40 minutes, corresponding to one and two cell divisions, respectively. DNA was extracted from each sample and the molecules examined by density gradient centrifugation (Figure 13.3A). After one round of DNA replication, the daughter molecules synthesized in the presence of normal nitrogen formed a single band in the density gradient, indicating that each double helix was made up of equal amounts of newly synthesized and parental DNA. This result immediately enabled the conservative mode of replication to be discounted, as this predicts that there would be two bands after one round of replication (Figure 13.3B), but did not provide a distinction between Delbrück's dispersive model and the semiconservative process favored by Watson and Crick. The distinction was, however, possible when the DNA molecules resulting from two rounds of replication were examined. Now the density gradient revealed two bands of DNA, the first corresponding to a hybrid composed of equal parts of newly synthesized and old DNA, and the second corresponding to molecules made up entirely of new DNA. This result agrees with the semiconservative scheme but is incompatible with dispersive replication, the latter predicting that after two rounds of replication all molecules would be hybrids.

Figure 13.3
The Meselson-Stahl experiment. (A) The experiment carried out by Meselson and Stahl involved growing a culture of Escherichia coli in a medium containing 15NH4Cl (ammonium chloride labeled with the heavy isotope of nitrogen). Cells were then transferred (more...)
13.1.2. DNA topoisomerases provide a solution to the topological problem
The Meselson-Stahl experiment proved that DNA replication in living cells follows the semiconservative scheme proposed by Watson and Crick, and hence indicated that the cell must have a solution to the topological problem. This solution was not understood by molecular biologists until some 25 years later, when the activities of the groups of enzymes called DNA topoisomerases were characterized.
DNA topoisomerases are enzymes that carry out breakage-and-reunion reactions similar but not identical to that envisaged by Delbrück. Three types of DNA topoisomerase are recognized (Table 13.1; Champoux, 2001):
- Type IA topoisomerases introduce a break in one polynucleotide and pass the second polynucleotide through the gap that is formed (Figure 13.4A). The two ends of the broken strand are then re-ligated (Lima et al., 1994). This mode of action results in the linking number (the number of times one strand crosses the other in a circular molecule) being changed by one.
- Type IB topoisomerases act in a similar way to the Type IA enzymes, although the detailed mechanism is different (Redinbo et al., 1998; Stewart et al., 1998). Type IA and IB topoisomerases probably evolved separately.
- Type II topoisomerases break both strands of the double helix, creating a ‘gate’ through which a second segment of the helix is passed (Figure 13.4B; Berger et al., 1996; Cabral et al., 1997). This changes the linking number by two.
Table 13.1
DNA topoisomerases.
DNA topoisomerases do not themselves unwind the double helix. Instead they solve the topological problem by counteracting the overwinding that otherwise would be introduced into the molecule by the progression of the replication fork. The result is that the helix can be ‘unzipped’, with the two strands pulled apart sideways without the molecule having to rotate (Figure 13.5).

Figure 13.5
Unzipping the double helix. During replication, the double helix is ‘unzipped’ as a result of the action of DNA topoisomerases. The replication fork is therefore able to proceed along the molecule without the helix having to rotate.
Replication is not the only activity that is complicated by the topology of the double helix, and it is becoming increasingly clear that DNA topoisomerases have equally important roles during transcription, recombination and other processes that can result in over- or underwinding of DNA. In eukaryotes, topoisomerases form a major part of the nuclear matrix, the scaffold-like network that permeates the nucleus (Section 8.1.1), and are responsible for maintaining chromatin structure and unlinking DNA molecules during chromosome division. Most topoisomerases are only able to relax DNA, but prokaryotic Type II enzymes, such as the bacterial DNA gyrase and the archaeal reverse gyrase, can carry out the reverse reaction and introduce supercoils into DNA molecules.
13.1.3. Variations on the semiconservative theme
No exceptions to the semiconservative mode of DNA replication are known but there are several variations on this basic theme. DNA copying via a replication fork, as shown in Figure 13.1, is the predominant system, being used by chromosomal DNA molecules in eukaryotes and by the circular genomes of prokaryotes. Some smaller circular molecules, such as the human mitochondrial genome (Section 2.2.2), use a slightly different process called displacement replication, which involves continuous copying of one strand of the helix, the second strand being displaced and subsequently copied after synthesis of the first daughter genome has been completed (Figure 13.6A). The advantage of displacement replication as performed by human mitochondrial DNA is not clear. In contrast, the special type of displacement process called rolling circle replication is an efficient mechanism for the rapid synthesis of multiple copies of a circular genome (Novick, 1998). Rolling circle replication, which is used by λ and various other bacteriophages, initiates at a nick which is made in one of the parent polynucleotides. The free 3′ end that results is extended, displacing the 5′ end of the polynucleotide. Continued DNA synthesis ‘rolls off’ a complete copy of the genome, and further synthesis eventually results in a series of genomes linked head to tail (Figure 13.6B). These genomes are single stranded and linear, but can easily be converted to double-stranded circular molecules by complementary strand synthesis followed by cleavage at the junction points between genomes and circularization of the resulting segments.
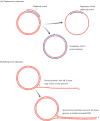
Figure 13.6
DNA replication systems used with small circular DNA molecules. (A) Displacement replication, as displayed by the human mitochondrial genome. (B) Rolling circle replication, used by various bacteriophages.
13.2. The Replication Process
As with many processes in molecular biology, we conventionally look on genome replication as being made up of three phases - initiation, elongation and termination:
- Initiation (Section 13.2.1) involves recognition of the position(s) on a DNA molecule where replication will begin.
- Elongation (Section 13.2.2) concerns the events occurring at the replication fork, where the parent polynucleotides are copied.
- Termination (Section 13.2.3), which in general is only vaguely understood, occurs when the parent molecule has been completely replicated.
As well as these three stages in replication, one additional topic demands attention. This relates to a limitation in the replication process that, if uncorrected, would lead to linear double-stranded DNA molecules getting shorter each time they are replicated (see Figure 13.24). The solution to this problem, which concerns the structure and synthesis of the telomeres at the ends of chromosomes (Section 2.2.1), is described in Section 13.2.4.

Figure 13.24
Two of the reasons why linear DNA molecules could become shorter after DNA replication. In both examples, the parent molecule is replicated in the normal way. A complete copy is made of its leading strand, but in (A) the lagging-strand copy is incomplete (more...)
13.2.1. Initiation of genome replication
Initiation of replication is not a random process and always begins at the same position or positions on a DNA molecule, these points being called the origins of replication. Once initiated, two replication forks can emerge from the origin and progress in opposite directions along the DNA: replication is therefore bidirectional with most genomes (Figure 13.7). A circular bacterial genome has a single origin of replication, meaning that several thousand kb of DNA are copied by each replication fork. This situation differs from that seen with eukaryotic chromosomes, which have multiple origins and whose replication forks progress for shorter distances. The yeast Saccharomyces cerevisiae, for example, has about 300 origins, corresponding to 1 per 40 kb of DNA, and humans have some 20 000 origins, or 1 for every 150 kb.

Figure 13.7
Bidirectional DNA replication of (A) a circular bacterial chromosome and (B) a linear eukaryotic chromosome.
Initiation at the E. coli origin of replication
We know substantially more about initiation of replication in bacteria than in eukaryotes. The E. coli origin of replication is referred to as oriC. By transferring segments of DNA from the oriC region into plasmids that lack their own origins, it has been estimated that the E. coli origin of replication spans approximately 245 bp of DNA. Sequence analysis of this segment shows that it contains two short repeat motifs, one of nine nucleotides and the other of 13 nucleotides (Figure 13.8A). The nine-nucleotide repeat, five copies of which are dispersed throughout oriC, is the binding site for a protein called DnaA. With five copies of the binding sequence, it might be imagined that five copies of DnaA attach to the origin, but in fact bound DnaA proteins cooperate with unbound molecules until some 30 are associated with the origin. Attachment occurs only when the DNA is negatively supercoiled, as is the normal situation for the E. coli chromosome (Section 2.3.1).
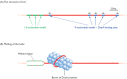
Figure 13.8
The Escherichia coli origin of replication. (A) The E. coli origin of replication is called oriC and is approximately 245 bp in length. It contains three copies of a 13-nucleotide repeat motif, consensus sequence 5′-GATCTNTTNTTTT-3′ where (more...)
The result of DnaA binding is that the double helix opens up (‘melts’) within the tandem array of three AT-rich, 13-nucleotide repeats located at one end of the oriC sequence (Figure 13.8B). The exact mechanism is unknown but DnaA does not appear to possess the enzymatic activity needed to break base pairs, and it is therefore assumed that the helix is melted by torsional stresses introduced by attachment of the DnaA proteins. An attractive model imagines that the DnaA proteins form a barrel-like structure around which the helix is wound. Melting the helix is promoted by HU, the most abundant of the DNA packaging proteins of E. coli (Section 2.3.1).
Melting of the helix initiates a series of events that culminates in the start of the elongation phase of replication. The first step is attachment of a complex of two proteins, DnaBC, forming the prepriming complex. DnaC has a transitory role and is released from the complex soon after it is formed, its function probably being simply to aid the attachment of DnaB. The latter is a helicase, an enzyme which can break base pairs (see Section 13.2.2). DnaB begins to increase the single-stranded region within the origin, enabling the enzymes involved in the elongation phase of genome replication to attach. This represents the end of the initiation phase of replication in E. coli as the replication forks now start to progress away from the origin and DNA copying begins.
Origins of replication in yeast have been clearly defined
The technique used to delineate the E. coli oriC sequence, involving transfer of DNA segments into a non-replicating plasmid, has also proved valuable in identifying origins of replication in the yeast Saccharomyces cerevisiae. Origins identified in this way are called autonomously replicating sequences or ARSs. A typical yeast ARS is shorter than the E. coli origin, usually less than 200 bp in length, but, like the E. coli origin, contains discrete segments with different functional roles, these ‘subdomains’ having similar sequences in different ARSs (Figure 13.9A). Four subdomains are recognized. Two of these - subdomains A and B1 - make up the origin recognition sequence, a stretch of some 40 bp in total that is the binding site for the origin recognition complex (ORC), a set of six proteins that attach to the ARS (Figure 13.9B). ORCs have been described as yeast versions of the E. coli DnaA proteins (Kelman, 2000), but this interpretation is probably not strictly correct because ORCs appear to remain attached to yeast origins throughout the cell cycle (Bell and Stillman, 1992; Diffley and Cocker, 1992). Rather than being genuine initiator proteins, it is more likely that ORCs are involved in the regulation of genome replication, acting as mediators between replication origins and the regulatory signals that coordinate the initiation of DNA replication with the cell cycle (Section 13.3; Stillman, 1996).

Figure 13.9
Structure of a yeast origin of replication. (A) Structure of ARS1, a typical autonomously replicating sequence (ARS) that acts as an origin of replication in Saccharomyces cerevisiae. The relative positions of the functional sequences A, B1, B2 and B3 (more...)
We must therefore look elsewhere in yeast ARSs for sequences with functions strictly equivalent to that of oriC of E. coli. This leads us to the two other conserved sequences in the typical yeast ARS, subdomains B2 and B3 (see Figure 13.9A). Our current understanding suggests that these two subdomains function in a manner similar to the E. coli origin. Subdomain B2 appears to correspond to the 13-nucleotide repeat array of the E. coli origin, being the position at which the two strands of the helix are first separated. This melting is induced by torsional stress introduced by attachment of a DNA-binding protein, ARS binding factor 1 (ABF1), which attaches to subdomain B3 (see Figure 13.9B). As in E. coli, melting of the helix within a yeast replication origin is followed by attachment of the helicase and other replication enzymes to the DNA, completing the initiation process and enabling the replication forks to begin their progress along the DNA, as described in Section 13.2.2.
Replication origins in higher eukaryotes have been less easy to identify
Attempts to identify replication origins in humans and other higher eukaryotes have, until recently, been less successful (Gilbert, 2001). Initiation regions (parts of the chromosomal DNA where replication initiates) were delineated by various biochemical methods, for example by allowing replication to initiate in the presence of labeled nucleotides, then arresting the process, purifying the newly synthesized DNA, and determining the positions of these nascent strands in the genome. These experiments suggested that there are specific regions in mammalian chromosomes where replication begins, but some researchers were doubtful whether these regions contained replication origins equivalent to yeast ARSs. One alternative hypothesis was that replication is initiated by protein structures that have specific positions in the nucleus, the chromosome initiation regions simply being those DNA segments located close to these protein structures in the three-dimensional organization of the nucleus.
Doubts about mammalian replication origins were increased by the failure of mammalian initiation regions to confer replicative ability on replication-deficient plasmids, although these experiments were not considered conclusive because it was recognized that a mammalian origin might be too long to be cloned in a plasmid or might function only when activated by distant sites in the chromosomal DNA. The breakthrough eventually came when an 8 kb segment of a human initiation region was transferred to the monkey genome, where it still directed replication despite being removed from any hypothetical protein structure in the human nucleus (Aladjem et al., 1998). Analysis of this transferred initiation region showed that there are primary sites within the region where initiation occurs at high frequency, surrounded by secondary sites, spanning the entire 8 kb region, at which replication initiates with lower frequency. The presence of discrete functional domains within the initiation region could also be demonstrated by examining the effects of deletions of parts of the region on the efficiency of replication initiation.
The demonstration that the human genome contains replication origins equivalent to those in yeast raises the question of whether mammals possess an equivalent of the yeast ORC. The answer appears to be yes, as several genes whose protein products have similar sequences to the yeast ORC proteins have been identified in higher eukaryotes and some of these have been shown to be able to replace the equivalent yeast protein in the yeast ORC (Carpenter et al., 1996). These results indicate that initiation of replication in yeast is a good model for events occurring in mammals, a conclusion that is very relevant to studies of the control of replication initiation, as we will see in Section 13.3.
13.2.2. The elongation phase of replication
Once replication has been initiated, the replication forks progress along the DNA and participate in the central activity of genome replication - the synthesis of new strands of DNA that are complementary to the parent polynucleotides. At the chemical level the template-dependent synthesis of DNA (Figure 13.10) is very similar to the template-dependent synthesis of RNA that occurs during transcription (compare with Figure 3.5). However, this similarity should not mislead us into making an extensive analogy between transcription and replication. The mechanics of the two processes are quite different, replication being complicated by two factors that do not apply to transcription:
- During DNA replication both strands of the double helix must be copied. This is an important complication because, as noted in Section 1.1.2, DNA polymerase enzymes are only able to synthesize DNA in the 5′→3′ direction. This means that one strand of the parent double helix, called the leading strand, can be copied in a continuous manner, but replication of the lagging strand has to be carried out in a discontinuous fashion, resulting in a series of short segments that must be ligated together to produce the intact daughter strand (Figure 13.11).
- The second complication arises because template-dependent DNA polymerases cannot initiate DNA synthesis on a molecule that is entirely single-stranded: there must be a short double-stranded region to provide a 3′ end onto which the enzyme can add new nucleotides. This means that primers are needed, one to initiate complementary strand synthesis on the leading polynucleotide, and one for every segment of discontinuous DNA synthesized on the lagging strand (Figure 13.11).

Figure 13.10
Template-dependent synthesis of DNA. Compare this reaction with template-dependent synthesis of RNA, shown in Figure 3.5.

Figure 13.11
Complications with DNA replication. Two complications have to be solved when double-stranded DNA is replicated. First, only the leading strand can be continuously replicated by 5′→3′ DNA synthesis; replication of the lagging strand (more...)
Before dealing with these two complications we will first examine the DNA polymerase enzymes themselves.
The DNA polymerases of bacteria and eukaryotes
The principal chemical reaction catalyzed by a DNA polymerase is the 5′→3′ synthesis of a DNA polynucleotide, as shown in Figure 13.10. We learnt in Section 4.1.1 that some DNA polymerases combine this function with at least one exonuclease activity, which means that these enzymes can degrade polynucleotides as well as synthesize them (see Figure 4.7):
- A 3′→5′ exonuclease is possessed by many bacterial and eukaryotic template-dependent DNA polymerases (Table 13.2). This activity enables the enzyme to remove nucleotides from the 3′ end of the strand that it has just synthesized. It is looked on as a proofreading activity whose function is to correct the occasional base-pairing error that might occur during strand synthesis (see Section 14.1.1).
- A 5′→3′ exonuclease activity is less common but is possessed by some polymerases whose function in replication requires that they must be able to remove at least part of a polynucleotide that is already attached to the template strand that the polymerase is copying. This activity is utilized during the process that joins together the discontinuous DNA fragments synthesized on the lagging strand during bacterial DNA replication (see Figure 13.17).
Table 13.2
DNA polymerases involved in replication of bacterial and eukaryotic genomes.
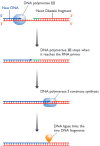
Figure 13.17
The series of events involved in joining up adjacent Okazaki fragments during DNA replication in Escherichia coli. DNA polymerase III lacks a 5′→3′ exonuclease activity and so stops making DNA when it reaches the RNA primer of (more...)
The search for DNA polymerases began in the mid-1950s, as soon as it was realized that DNA synthesis was the key to replication of genes. It was thought that bacteria would probably have just a single DNA polymerase, and when the enzyme now called DNA polymerase I was isolated by Arthur Kornberg in 1957 there was a widespread assumption that this was the main replicating enzyme. The discovery that inactivation of the E. coli polA gene, which codes for DNA polymerase I, was not lethal (cells were still able to replicate their genomes) therefore came as something of a surprise, especially when a similar result was obtained with inactivation of polB, coding for a second enzyme, DNA polymerase II, which we now know is mainly involved in repair of damaged DNA rather than genome replication (Section 14.2.5). It was not until 1972 that the main replicating polymerase of E. coli, DNA polymerase III, was eventually isolated. Both DNA polymerases I and III are involved in genome replication, as we will see in the next section.
The properties of the two E. coli DNA polymerases involved in genome replication are described in Table 13.2. DNA polymerases I and II are single polypeptides but DNA polymerase III, befitting its role as the main replicating enzyme, is multi-subunit, with a molecular mass of approximately 900 kDa. The three main subunits, which form the core enzyme, are called α, ε and θ, with the polymerase activity specified by the α subunit and the 3′→5′ exonuclease by ε. The function of θ is not clear: it may have a purely structural role in bringing together the other two core subunits and in assembling the various accessory subunits. The latter include τ and γ, both coded by the same gene, with synthesis of γ involving translational frameshifting (Section 11.2.3), β, which acts as a ‘sliding clamp’ and holds the polymerase complex tightly to the template, δ, δ′, χ and ψ.
Eukaryotes have at least nine DNA polymerases (Hübscher et al., 2000), which in mammals are distinguished by Greek suffices (α, β, γ, δ, etc.), an unfortunate choice of nomenclature as it tempts confusion with the identically named subunits of E. coli DNA polymerase III. The main replicating enzyme is DNA polymerase δ (Table 13.2), which has two subunits (three according to some researchers) and works in conjunction with an accessory protein called the proliferating cell nuclear antigen (PCNA). PCNA is the functional equivalent of the β subunit of E. coli DNA polymerase III and holds the enzyme tightly to the template. DNA polymerase α also has an important function in DNA synthesis, being the enzyme that primes eukaryotic replication (see Figure 13.12B). DNA polymerase γ, although coded by a nuclear gene, is responsible for replicating the mitochondrial genome.

Figure 13.12
Priming of DNA synthesis in (A) bacteria and (B) eukaryotes. In eukaryotes the primase forms a complex with DNA polymerase α, which is shown synthesizing the RNA primer followed by the first few nucleotides of DNA.
Discontinuous strand synthesis and the priming problem
The limitation that DNA polymerases can synthesize polynucleotides only in the 5′→3′ direction means that the lagging strand of the parent molecule must be copied in a discontinuous fashion, as shown in Figure 13.11. The implication of this model - that the initial products of lagging-strand replication are short segments of polynucleotide - was confirmed in 1969 when Okazaki fragments, as these segments are now called, were first isolated from E. coli (Okazaki and Okazaki, 1969). In bacteria, Okazaki fragments are 1000–2000 nucleotides in length, but in eukaryotes the equivalent fragments appear to be much shorter, perhaps less than 200 nucleotides. This is an interesting observation that might indicate that each round of discontinuous synthesis replicates the DNA associated with a single nucleosome (140 and 150 bp wound around the core particle plus 50–70 bp of linker DNA: Section 2.2.1).
The second difficulty illustrated in Figure 13.11 is the need for a primer to initiate synthesis of each new polynucleotide. It is not known for certain why DNA polymerases cannot begin synthesis on an entirely single-stranded template, but it may relate to the proofreading activity of these enzymes, which is essential for the accuracy of replication. As described in Section 14.1.1, a nucleotide that has been inserted incorrectly at the extreme 3′ end of a growing DNA strand, and hence is not base-paired to the template polynucleotide, can be removed by the 3′→5′ exonuclease activity of a DNA polymerase. This means that the 3′→5′ exonuclease activity must be more effective than the 5′→3′ polymerase activity when the 3′ nucleotide is not base-paired to the template. The implication is that the polymerase can extend a polynucleotide efficiently only if its 3′ nucleotide is base-paired, which in turn could be the reason why an entirely single-stranded template, which by definition lacks a base-paired 3′ nucleotide, cannot be used by a DNA polymerase.
Whatever the reason, priming is a necessity in DNA replication but does not present too much of a problem. Although DNA polymerases cannot deal with an entirely single-stranded template, RNA polymerases have no difficulty in this respect, so the primers for DNA replication are made of RNA. In bacteria, primers are synthesized by primase, a special RNA polymerase unrelated to the transcribing enzyme, with each primer 4–15 nucleotides in length (Frick and Richardson, 2001). Once the primer has been completed, strand synthesis is continued by DNA polymerase III (Figure 13.12A). In eukaryotes the situation is slightly more complex because the primase is tightly bound to DNA polymerase α, and cooperates with this enzyme in synthesis of the first few nucleotides of a new polynucleotide. This primase synthesizes an RNA primer of 8–12 nucleotides, and then hands over to DNA polymerase α, which extends the RNA primer by adding about 20 nucleotides of DNA. This DNA stretch often has a few ribonucleotides mixed in, but it is not clear if these are incorporated by DNA polymerase α or by intermittent activity of the primase. After completion of the RNA-DNA primer, DNA synthesis is continued by the main replicative enzyme, DNA polymerase δ (Figure 13.12B).
Priming needs to occur just once on the leading strand, within the replication origin, because once primed, the leading-strand copy is synthesized continuously until replication is completed. On the lagging strand, priming is a repeated process that must occur every time a new Okazaki fragment is initiated. In E. coli, which makes Okazaki fragments of 1000–2000 nucleotides in length, approximately 4000 priming events are needed every time the genome is replicated. In eukaryotes the Okazaki fragments are much shorter and priming is a highly repetitive event.
Events at the bacterial replication fork
Now that we have considered the complications introduced by discontinuous strand synthesis and the priming problem, we can move on to study the combination of events occurring at the replication fork during the elongation phase of genome replication.
In Section 13.2.1 we identified attachment of the DnaB helicase, followed by extension of the melted region of the replication origin, as representing the end of the initiation phase of replication in E. coli. To a large extent, the division between initiation and elongation is artificial, the two processes running seamlessly one into the other. After the helicase has bound to the origin to form the prepriming complex, the primase is recruited, resulting in the primosome, which initiates replication of the leading strand. It does this by synthesizing the RNA primer that DNA polymerase III needs in order to begin copying the template.
DnaB is the main helicase involved in genome replication in E. coli, but it is by no means the only helicase that this bacterium possesses: in fact there were eleven at the last count (van Brabant et al., 2000). The size of the collection reflects the fact that DNA unwinding is required not only during replication but also during diverse processes such as transcription, recombination and DNA repair. The mode of action of a typical helicase has not been precisely defined, but it is thought that these enzymes bind to single-stranded rather than double-stranded DNA, and migrate along the polynucleotide in either the 5′→3′ or 3′→5′ direction, depending on the specificity of the helicase. Breakage of base pairs in advance of the helicase requires energy, which is generated by hydrolysis of ATP. According to this model, a single DnaB helicase could migrate along the lagging strand (DnaB is a 5′→3′ helicase), unzipping the helix and generating the replication fork, the torsional stress generated by the unwinding activity being relieved by DNA topoisomerase action (Figure 13.13). This model is probably a good approximation of what actually happens, although it does not provide a function for the two other E. coli helicases thought to be involved in genome replication. Both of these, PriA and Rep, are 3′→5′ helicases and so could conceivably complement DnaB activity by migrating along the leading strand, but they may have lesser roles. The involvement of Rep in DNA replication might in fact be limited to participation in the rolling circle process used by λ and a few other E. coli bacteriophages (Section 13.1.3).

Figure 13.13
The role of the DnaB helicase during DNA replication in Escherichia coli. DnaB is a 5′→3′ helicase and so migrates along the lagging strand, breaking base pairs as it goes. It works in conjunction with a DNA topoisomerase (see (more...)
Single-stranded DNA is naturally ‘sticky’ and the two separated polynucleotides produced by helicase action would immediately reform base pairs after the enzyme has passed, if allowed to. The single strands are also highly susceptible to nuclease attack and are likely to be degraded if not protected in some way. To avoid these unwanted outcomes, single-strand binding proteins (SSBs) attach to the polynucleotides and prevent them from reassociating or being degraded (Figure 13.14A). The E. coli SSB is made up of four identical subunits and probably works in a similar way to the major eukaryotic SSB, called replication protein A (RPA), by enclosing the polynucleotide in a channel formed by a series of SSBs attached side by side on the strand (Figure 13.14B; Bochkarev et al., 1997). Detachment of the SSBs, which must occur when the replication complex arrives to copy the single strands, is brought about by a second set of proteins called replication mediator proteins (RMPs; Beernick and Morrical, 1999). As with helicases, SSBs have diverse roles in different processes involving DNA unwinding.

Figure 13.14
The role of single-strand binding proteins (SSBs) during DNA replication. (A) SSBs attach to the unpaired polynucleotides produced by helicase action and prevent the strands from base-pairing with one another or being degraded by single-strand-specific (more...)
After 1000–2000 nucleotides of the leading strand have been replicated, the first round of discontinuous strand synthesis on the lagging strand can begin. The primase, which is still associated with the DnaB helicase in the primosome, makes an RNA primer which is then extended by DNA polymerase III (Figure 13.15). This is the same DNA polymerase III complex that is synthesizing the leading-strand copy, the complex comprising, in effect, two copies of the polymerase. It is not two complete copies because there is only a single γ complex, containing subunit γ in association with δ, δ′, χ and ψ. The main function of the γ complex is to interact with the β subunit (the ‘sliding clamp’) and hence control the attachment and removal of the enzyme from the template, a function that is required primarily during lagging-strand replication when the enzyme has to attach and detach repeatedly at the start and end of each Okazaki fragment. Some models of the DNA polymerase III complex place the two enzymes in opposite orientations to reflect the different directions in which DNA synthesis occurs, towards the replication fork on the leading strand and away from it on the lagging strand. It is more likely, however, that the pair of enzymes face the same direction and the lagging strand forms a loop, so that DNA synthesis can proceed in parallel as the polymerase complex moves forwards in pace with the progress of the replication fork (Figure 13.16).
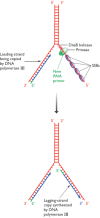
Figure 13.15
Priming and synthesis of the lagging-strand copy during DNA replication in Escherichia coli.

Figure 13.16
A model for parallel synthesis of the leading- and lagging-strand copies by a dimer of DNA polymerase III enzymes. It is thought that the lagging strand loops through its copy of the DNA polymerase III enzyme, in the manner shown, so that both the leading (more...)
The combination of the DNA polymerase III dimer and the primosome, migrating along the parent DNA and carrying out most of the replicative functions, is called the replisome. After its passage, the replication process must be completed by joining up the individual Okazaki fragments. This is not a trivial event because one member of each pair of adjacent Okazaki fragments still has its RNA primer attached at the point where ligation should take place (Figure 13.17). Table 13.2 shows us that this primer cannot be removed by DNA polymerase III, because this enzyme lacks the required 5′→3′ exonuclease activity. At this point, DNA polymerase III releases the lagging strand and its place is taken by DNA polymerase I, which does have a 5′→3′ exonuclease and so removes the primer, and usually the start of the DNA component of the Okazaki fragment as well, extending the 3′ end of the adjacent fragment into the region of the template that is exposed. The two Okazaki fragments now abut, with the terminal regions of both composed entirely of DNA. All that remains is for the missing phosphodiester bond to be put in place by a DNA ligase, linking the two fragments and completing replication of this region of the lagging strand.
The eukaryotic replication fork: variations on the bacterial theme
The elongation phase of genome replication is similar in bacteria and eukaryotes, although the details differ. The progress of the replication fork in eukaryotes is maintained by helicase activity, although which of the several eukaryotic helicases that have been identified are primarily responsible for DNA unwinding during replication has not been established. The separated polynucleotides are prevented from reattaching by single-strand binding proteins, the main one of these in eukaryotes being RPA.
We begin to encounter unique features of the eukaryotic replication process when we examine the method used to prime DNA synthesis. As described on page 397, the eukaryotic DNA polymerase α cooperates with the primase enzyme to put in place the RNA-DNA primers at the start of the leading-strand copy and at the beginning of each Okazaki fragment. However, DNA polymerase α is not capable of lengthy DNA synthesis, presumably because it lacks the stabilizing effect of a sliding clamp equivalent to the β subunit of E. coli DNA polymerase III or the PCNA accessory protein that aids the eukaryotic DNA polymerase δ. This means that although DNA polymerase α can extend the initial RNA primer with about 20 nucleotides of DNA, it must then be replaced by the main replicative enzyme, DNA polymerase δ (see Figure 13.12B).
The DNA polymerase enzymes that copy the leading and lagging strands in eukaryotes do not associate into a dimeric complex equivalent to the one formed by DNA polymerase III during replication in E. coli. Instead, the two copies of the polymerase remain separate. The function performed by the γ complex of the E. coli polymerase - controlling attachment and detachment of the enzyme from the lagging strand - appears to be carried out by a multi-subunit accessory protein called replication factor C (RFC).
As in E. coli, completion of lagging-strand synthesis requires removal of the RNA primer from each Okazaki fragment. There appears to be no eukaryotic DNA polymerase with the 5′→3′ exonuclease needed for this purpose and the process is therefore very different to that described for bacterial cells. The central player is the ‘flap endonuclease’, FEN1 (previously called MF1), which associates with the DNA polymerase δ complex at the 3′ end of an Okazaki fragment, in order to degrade the primer from the 5′ end of the adjacent fragment. Understanding exactly how this occurs is complicated by the inability of FEN1 to initiate primer degradation because it is unable to remove the ribonucleotide at the extreme 5′ end of the primer, because this ribonucleotide carries a 5′-triphosphate group which blocks FEN1 activity (Figure 13.18). Two alternative models to circumvent this problem have been proposed (Waga and Stillman, 1998):
- The first possibility is that a helicase breaks the base pairs holding the primer to the template strand, enabling the primer to be pushed aside by DNA polymerase δ as it extends the adjacent Okazaki fragment into the region thus exposed (Figure 13.19A). The flap that results can be cut off by FEN1, whose endonuclease activity can cleave the phosphodiester bond at the branch point where the displaced region attaches to the part of the fragment that is still base-paired.
- Alternatively, most of the RNA component of the primer could be removed by RNase H, which can degrade the RNA part of a base-paired RNA-DNA hybrid, but cannot cleave the phosphodiester bond between the last ribonucleotide and the first deoxyribonucleotide. However, this ribonucleotide will carry a 5′-monophosphate rather than triphosphate and so can be removed by FEN1 (Figure 13.19B).

Figure 13.18
The ‘flap endonuclease’ FEN1 cannot initiate primer degradation because its activity is blocked by the triphosphate group present at the 5′ end of the primer.

Figure 13.19
Two models for completion of lagging strand replication in eukaryotes. See the text for details. The new DNA (blue strand) is synthesized by DNA polymerase δ but this enzyme is not shown in order to increase the clarity of the diagrams.
Both schemes are made attractive by the possibility that both the RNA primer and all of the DNA originally synthesized by DNA polymerase α are removed. This is because DNA polymerase α has no 3′→5′ proofreading activity (see Table 13.2) and therefore synthesizes DNA in a relatively error-prone manner. To prevent these errors from becoming permanent features of the daughter double helix, this region of DNA might be degraded and resynthesized by DNA polymerase δ, which does have a proofreading activity and so makes a highly accurate copy of the template. At present this possibility remains speculative.
The final difference between replication in bacteria and eukaryotes is that in eukaryotes there is no equivalent of the bacterial replisome. Instead, the enzymes and proteins involved in replication form sizeable structures within the nucleus, each containing hundreds or thousands of individual replication complexes. These structures are immobile because of attachments with the nuclear matrix, so DNA molecules are threaded through the complexes as they are replicated. The structures are referred to as replication factories (Figure 13.20) and may in fact also be features of the replication process in at least some bacteria (Lemon and Grossman, 1998; Cook, 1999).

Figure 13.20
Replication factories in a eukaryotic nucleus. Equivalent transcription factories are responsible for RNA synthesis. Reproduced with permission from Nakamura H et al. (1986) Exp. Cell Res., 165, 291–297, Academic Press, Inc., Orlando, FL.
13.2.3. Termination of replication
Replication forks proceed along linear genomes, or around circular ones, generally unimpeded except when a region that is being transcribed is encountered. DNA synthesis occurs at approximately five times the rate of RNA synthesis, so the replication complex can easily overtake an RNA polymerase, but this probably does not happen: instead it is thought that the replication fork pauses behind the RNA polymerase, proceeding only when the transcript has been completed (Deshpande and Newlon, 1996).
Eventually the replication fork reaches the end of the molecule or meets a second replication fork moving in the opposite direction. What happens next is one of the least understood aspects of genome replication.
Replication of the E. coli genome terminates within a defined region
Bacterial genomes are replicated bidirectionally from a single point (see Figure 13.7), which means that the two replication forks should meet at a position diametrically opposite the origin of replication on the genome map. However, if one fork is delayed, possibly because it has to replicate extensive regions where transcription is occurring, then it might be possible for the other fork to overshoot the halfway point and continue replication on the ‘other side’ of the genome (Figure 13.21). It is not immediately apparent why this should be undesirable, the daughter molecules presumably being unaffected, but it is not allowed to happen because of the presence of terminator sequences. Seven of these have been identified in the E. coli genome (Figure 13.22A), each one acting as the recognition site for a sequence-specific DNA-binding protein called Tus.
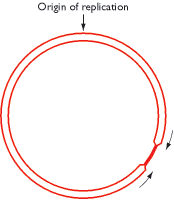
Figure 13.21
A situation that is not allowed to occur during replication of the circular Escherichia coli genome. One of the replication forks has proceeded some distance past the halfway point. This does not happen during DNA replication in E. coli because of the (more...)

Figure 13.22
The role of terminator sequences during DNA replication in Escherichia coli. (A) The positions of the six terminator sequences on the E. coli genome are shown, with the arrowheads indicating the direction that each terminator sequence can be passed by (more...)
The mode of action of Tus is quite unusual. When bound to a terminator sequence, a Tus protein allows a replication fork to pass if the fork is moving in one direction, but blocks progress if the fork is moving in the opposite direction around the genome. The directionality is set by the orientation of the Tus protein on the double helix. When approached from one direction, Tus blocks the passage of the DnaB helicase, which is responsible for progression of the replication fork, because the helicase is faced with a ‘wall’ of β-strands which it is unable to penetrate. But when approaching from the other direction, DnaB is able to disrupt the structure of the Tus protein, probably because of the effect that unwinding of the double helix has on Tus, and so is able to pass by (Figure 13.22B; Kamada et al., 1996).
The orientation of the termination sequences, and hence of the bound Tus proteins, in the E. coli genome is such that both replication forks become trapped within a relatively short region on the opposite side of the genome to the origin (see Figure 13.22A). This ensures that termination always occurs at or near the same position. Exactly what happens when the two replication forks meet is unknown, but the event is followed by disassembly of the replisomes, either spontaneously or in a controlled fashion. The result is two interlinked daughter molecules, which are separated by topoisomerase IV.
Little is known about termination of replication in eukaryotes
No sequences equivalent to bacterial terminators are known in eukaryotes, and proteins similar to Tus have not been identified. Quite possibly, replication forks meet at random positions and termination simply involves ligation of the ends of the new polynucleotides. We do know that the replication complexes do not break down, because these factories are permanent features of the nucleus (see Figure 13.20).
Rather than concentrating on the molecular events occurring when replication forks meet, attention has been focused on the difficult question of how the daughter DNA molecules produced in a eukaryotic nucleus do not become impossibly tangled up. Although DNA topoisomerases have the ability to untangle DNA molecules, it is generally assumed that tangling is kept to a minimum so that extensive breakage-and-reunion reactions, as catalyzed by topoisomerases (see Figure 13.4), can be avoided. Various models have been proposed to solve this problem (Falaschi, 2000). One of these (Cook, 1998; Wei et al., 1998) suggests that a eukaryotic genome is not randomly packed into the nucleus, but is ordered around the replication factories, which appear to be present in only limited numbers. It is envisaged that each factory replicates a single region of the DNA, maintaining the daughter molecules in a specific arrangement that avoids their entanglement. Initially, the two daughter molecules are held together by cohesin proteins, which are attached immediately after passage of the replication fork by a process that appears to involve DNA polymerase κ (Takahashi and Yanagida, 2000), an enigmatic enzyme that is essential for replication but whose only known role does not obviously require a DNA polymerase activity. The cohesins maintain the alignment of the sister chromatids until the anaphase stage of nuclear division, when they are cleaved by cutting proteins, enabling the daughter chromosomes to separate (Figure 13.23; Murray, 1999).

Figure 13.23
Cohesins. Cohesin proteins attach immediately after passage of the replication fork and hold the daughter molecules together until anaphase. During anaphase, the cohesins are cleaved, enabling the replicated chromosomes to separate prior to their distribution (more...)
13.2.4. Maintaining the ends of a linear DNA molecule
There is one final problem that we must consider before leaving the replication process. This concerns the steps that have to be taken to prevent the ends of a linear double-stranded molecule from gradually getting shorter during successive rounds of chromosomal DNA replication. There are two ways in which this shortening might occur:
- The extreme 3′ end of the lagging strand might not be copied because the final Okazaki fragment cannot be primed, the natural position for the priming site being beyond the end of the template (Figure 13.24A). The absence of this Okazaki fragment means that the lagging-strand copy is shorter than it should be. If the copy remains this length then when it acts as a parental polynucleotide in the next round of replication the resulting daughter molecule will be shorter than its grandparent.
- If the primer for the last Okazaki fragment is placed at the extreme 3′ end of the lagging strand, then shortening will still occur, although to a lesser extent, because this terminal RNA primer cannot be converted into DNA by the standard processes for primer removal (Figure 13.24B). This is because the methods for primer removal (as illustrated in Figure 13.17 for bacteria and Figure 13.19 for eukaryotes) require extension of the 3′ end of an adjacent Okazaki fragment, which cannot occur at the very end of the molecule.
Once this problem had been recognized, attention was directed at the telomeres, the unusual DNA sequences at the ends of eukaryotic chromosomes. We noted in Section 2.2.1 that telomeric DNA is made up of a type of minisatellite sequence, being comprised of multiple copies of a short repeat motif, 5′-TTAGGG-3′ in most higher eukaryotes, a few hundred copies of this sequence occurring in tandem repeats at each end of every chromosome. The solution to the end-shortening problem lies with the way in which this telomeric DNA is synthesized.
Telomeric DNA is synthesized by the telomerase enzyme
Most of the telomeric DNA is copied in the normal fashion during DNA replication but this is not the only way in which it can be synthesized. To compensate for the limitations of the replication process, telomeres can be extended by an independent mechanism catalyzed by the enzyme telomerase. This is an unusual enzyme in that it consists of both protein and RNA. In the human enzyme the RNA component is 450 nucleotides in length and contains near its 5′ end the sequence 5′-CUAACCCUAAC-3′, whose central region is the reverse complement of the human telomere repeat sequence 5′-TTAGGG-3′ (Feng et al., 1995). This enables telomerase to extend the telomeric DNA at the 3′ end of a polynucleotide by the copying mechanism shown in Figure 13.25, in which the telomerase RNA is used as a template for each extension step, the DNA synthesis being carried out by the protein component of the enzyme, which is a reverse transcriptase (Lingner et al., 1997). The correctness of this model is indicated by comparisons between telomere repeat sequences and the telomerase RNAs of other species (Table 13.3): in all organisms that have been looked at, the telomerase RNA contains a sequence that enables it to make copies of the repeat motif present at the organism's telomeres. An interesting feature is that in all organisms the strand synthesized by telomerase has a preponderance of G nucleotides; it is therefore referred to as the G-rich strand.
Figure 13.25
Extension of the end of a human chromosome by telomerase. The 3′ end of a human chromosomal DNA molecule is shown. The sequence comprises repeats of the human telomere motif 5′-TTAGGG-3′. The telomerase RNA base-pairs to the end (more...)
Table 13.3
Sequences of telomere repeats and telomerase RNAs in various organisms.
Telomerase can only synthesize this G-rich strand. It is not clear how the other polynucleotide - the C-rich strand - is extended, but it is presumed that when the G-rich strand is long enough, the primase-DNA polymerase α complex attaches at its end and initiates synthesis of complementary DNA in the normal way (Figure 13.26). This requires the use of a new RNA primer, so the C-rich strand will still be shorter than the G-rich one, but the important point is that the overall length of the chromosomal DNA has not been reduced.

Figure 13.26
Completion of the extension process at the end of a chromosome. It is believed that after telomerase has extended the 3′ end by a sufficient amount, as shown in Figure 13.25, a new Okazaki fragment is primed and synthesized, converting the 3′ (more...)
Telomere length is implicated in senescence and cancer
Perhaps surprisingly, telomerase is not active in all mammalian cells. The enzyme is functional in the early embryo, but after birth is active only in the reproductive and stem cells. The latter are progenitor cells that divide continually throughout the lifetime of an organism, producing new cells to maintain organs and tissues in a functioning state. The best-studied examples are the hemopoietic stem cells of the bone marrow, which generate new blood cells.
Cells that lack telomerase activity undergo chromosome shortening every time they divide. Eventually, after many cell divisions, the chromosome ends could become so truncated that essential genes are lost, but this is unlikely to be a major cause of the defects that can occur in cells lacking telomerase activity. Instead, the critical factor is the need to maintain a protein ‘cap’ on each chromosome end, to protect these ends from the effects of the DNA repair enzymes that join together the uncapped ends that are produced by accidental breakage of a chromosome (Section 2.2.1). The proteins that form this protective cap, such as TRF2 in humans, recognize the telomere repeats as their binding sequences, and so have no attachment points after the telomeres have been deleted. If these proteins are absent then the repair enzymes can make inappropriate linkages between the ends of intact, although shortened, chromosomes; it is this that is probably the underlying cause of the disruption to the cell cycle that results from telomere shortening.
Telomere shortening will therefore lead to the termination of a cell lineage. For several years biologists have attempted to link this process with cell senescence, a phenomenon originally observed in cell cultures. All normal cell cultures have a limited lifetime: after a certain number of divisions the cells enter a senescent state in which they remain alive but cannot divide (Figure 13.27). With some mammalian cell lines, notably fibroblast cultures (connective tissue cells), senescence can be delayed by engineering the cells so that they synthesize active telomerase (Reddel, 1998). These experiments suggest a clear relationship between telomere shortening and senescence, but the exactness of the link has been questioned (Blackburn, 2000), and any extrapolation from cell senescence to aging of the organism is fraught with difficulties (Kipling and Faragher, 1999).

Figure 13.27
Cultured cells become senescent after multiple cell divisions.
Not all cell lines display senescence. Cancerous cells are able to divide continuously in culture, their immortality being looked upon as analogous to tumor growth in an intact organism. With several types of cancer, this absence of senescence is associated with activation of telomerase, sometimes to the extent that telomere length is maintained through multiple cell divisions, but often in such a way that the telomeres become longer than normal because the telomerase is overactive. It is not clear if telomerase activation is a cause or an effect of cancer, although the former seems more likely because at least one type of cancer, dyskeratosis congenita, appears to result from a mutation in the gene specifying the RNA component of human telomerase (Marciniak and Guarente, 2001). The question is critical to understanding the etiology of the cancer but is less relevant to the therapeutic issue, which centers on whether telomerase could be a target for drugs designed to combat the cancer. Such a therapy could be successful even if telomerase activation is an effect of the cancer, because inactivation by drugs would induce senescence of the cancer cells and hence prevent their proliferation.
13.3. Regulation of Eukaryotic Genome Replication
Genome replication in eukaryotic cells is regulated at two levels:
- 1.
Replication is coordinated with the cell cycle so that two copies of the genome are available when the cell divides.
- 2.
The replication process itself can be arrested under certain circumstances, for example if the DNA is damaged and must be repaired before copying can be completed.
We will end this chapter by looking at these regulatory mechanisms.
13.3.1. Coordination of genome replication and cell division
The concept of a cell cycle emerged from light microscopy studies carried out by the early cell biologists. Their observations showed that dividing cells pass through repeated cycles of mitosis (see Figure 5.14) - the period when nuclear and cell division occurs - and interphase, a less dramatic period when few dynamic changes can be detected with the light microscope. It was understood that chromosomes divide during interphase, so when DNA was identified as the genetic material, interphase took on a new importance as the period when genome replication must take place. This led to a re-interpretation of the cell cycle as a four-stage process (Figure 13.28), comprising:
- Gap 1 or G1 phase, an interval when transcription, translation and other general cellular activities occur;
- Synthesis or S phase, when the genome is replicated;
- Gap 2 or G2 phase, a second interval period.
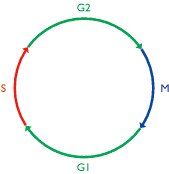
Figure 13.28
The cell cycle. The lengths of the individual phases vary in different cells. Abbreviations: G1 and G2, gap phases; M, mitosis; S synthesis phase.
It is clearly important that the S and M phases are coordinated so that the genome is completely replicated, but replicated only once, before mitosis occurs. The periods immediately before entry into S and M phases are looked upon as key cell cycle checkpoints, and it is at one of these two points that the cycle becomes arrested if critical genes involved in cell-cycle control are mutated, or if the cell undergoes trauma such as extensive DNA damage. Attempts to understand how genome replication is coordinated with mitosis have therefore concentrated on these two checkpoints, especially the pre-S checkpoint, the period immediately before replication.
Establishment of the pre-replication complex enables genome replication to commence
Studies primarily with Saccharomyces cerevisiae have led to a model for controlling the timing of S phase which postulates that genome replication requires construction of pre-replication complexes (pre-RCs) at origins of replication, these pre-RCs being converted to post-RCs as replication proceeds. A post-RC is unable to initiate replication and so cannot accidentally re-copy a piece of the genome before mitosis has occurred (Stillman, 1996). The ORC, the complex of six proteins that is assembled onto domains A and B1 of a yeast ARS (see Figure 13.9B), was an early contender for the pre-RC but is probably not a central component because ORCs are present at origins of replication at all stages of the cell cycle. Instead, the ORC is looked on as the ‘landing pad’ on which the pre-RC is constructed.
Various types of protein have been implicated as components of the pre-RC. The first is Cdc6p, which was originally identified in yeast and subsequently shown to have homologs in higher eukaryotes. Yeast Cdc6p is synthesized at the end of G2, as the cell enters mitosis, and becomes associated with chromatin in early G1 before disappearing at the end of G1, when replication begins (Figure 13.29). The involvement of Cdc6p in the pre-RC is suggested by experiments in which its gene is repressed, which results in an absence of pre-RCs, and other experiments in which Cdc6p is over-produced, which leads to multiple genome replications in the absence of mitosis. There is also biochemical evidence for a direct interaction between Cdc6p and yeast ORCs.
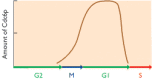
Figure 13.29
Graph showing the amount of Cdc6p in the nucleus at different stages of the cell cycle.
A second component of the pre-RC is thought to be the group of proteins called replication licensing factors (RLFs). As with Cdc6p, the first examples of these proteins were identified in yeast (the MCM family of proteins; Tye, 1999) with homologs in higher eukaryotes discovered at a later date. RLFs become bound to chromatin towards the end of M phase and remain in place until the start of S phase, after which they are gradually removed from the DNA as it is replicated. Their removal may be the key event that converts a pre-RC into a post-RC and so prevents re-initiation of replication at an origin that has already directed a round of replication (Blow and Tada, 2000).
Regulation of pre-RC assembly
Identification of the components of the pre-RC takes us some distance towards understanding how genome replication is initiated, but still leaves open the question of how replication is coordinated with other events in the cell cycle. Cell cycle control is a complex process, mediated largely by protein kinases which phosphorylate and activate enzymes and other proteins that have specific functions during the cell cycle. The same protein kinases are present in the nucleus throughout the cell cycle, so they must themselves be subject to control. This control is exerted partly by proteins called cyclins (whose abundance varies at different stages of the cell cycle), partly by other protein kinases that activate the cyclin-dependent protein kinases, and partly by inhibitory proteins. Even before we start looking for regulators of pre-RC assembly we can anticipate that the control system will be convoluted.
A number of cyclins have been linked with activation of genome replication and prevention of pre-RC reassembly after replication has been completed (Stillman, 1996). These include the mitotic cyclins, whose main function was originally thought to be activation of mitosis but which also repress genome replication. When the effects of these cyclins are blocked by, for example, overproduction of proteins that inhibit their activity, the cell is not only incapable of entering M phase but also undergoes repeated genome replication. There are also more specific S-phase cyclins, such as Clb5p and Clb6p in S. cerevisiae, inactivation of which delays or prevents genome replication, and other mitotic cyclins that are active during G2 phase and prevent the assembly of pre-RCs in the period after genome replication and before cell division (Figure 13.30).

Figure 13.30
Cell-cycle control points for cyclins involved in regulation of genome replication. See the text for details.
In addition to these cyclin-dependent control systems, genome replication is also regulated by a cyclinindependent protein kinase, Cdc7p-Dbf4p, found in organisms as diverse as yeasts and mammals. The proteins activated by this kinase have not been identified, separate lines of evidence suggesting that both RLFs and ORCs are targeted. Whatever the mechanism, Cdc7p-Dbf4p activity is a prerequisite for replication, the cyclin-dependent processes on their own being insufficient to push the cell into S phase.
13.3.2. Control within S phase
Regulation of the G1-S transition can be looked upon as the major control process affecting genome regulation, but it is not the only one. The specific events occurring during S phase are also subject to regulation.
Early and late replication origins
Initiation of replication does not occur at the same time at all replication origins, nor is ‘origin firing’ an entirely random process. Some parts of the genome are replicated early in S phase and some later, the pattern of replication being consistent from cell division to cell division (Fangman and Brewer, 1992). The general pattern is that actively transcribed genes and the centromere are replicated early in S phase, and non-transcribed regions of the genome later on. Early-firing origins are therefore tissue specific and reflect the pattern of gene expression occurring in a particular cell.
Understanding what determines the firing time of a replication origin is proving quite difficult. It is not simply the sequence of the origin, because transfer of a DNA segment from its normal position to another site in the same or a different chromosome can result in a change in the firing pattern of origins contained in that segment. This positional effect may be linked with chromatin organization and hence influenced by structures such as locus control regions (Section 8.1.2) that control DNA packaging. The position of the origin in the nucleus may also be important as origins that become active at similar periods within S phase appear to be clustered together, at least in mammals.
Checkpoints within S phase
The final aspect of the regulation of genome replication that we will consider is the function of the checkpoints that exist within S phase. These were first identified when it was shown that one of the responses of yeast cells to DNA damage is a slowing down and possibly a complete halting of the genome replication process (Paulovich and Hartwell, 1995). This is linked with the activation of genes whose products are involved in DNA repair (Section 14.2; Zhou and Elledge, 2000).
As with entry into S phase, cyclin-dependent kinases are implicated in the regulation of S-phase checkpoints. These kinases respond to signals from proteins associated with the replication fork. The identity of these damage-detection proteins has not yet been confirmed, although DNA polymerase ε, which has not been assigned a precise function during DNA synthesis (Table 13.2), is a particularly strong candidate because mutant yeast cells that have abnormal DNA polymerase ε enzymes do not respond to DNA damage in the same way as normal cells. Other replication-fork proteins, including components of the PCNA and the accessory protein RFC, have also been assigned roles in damage detection (Waga and Stillman, 1998). The signals from these proteins are mediated by kinases such as ATM, ATR, Chk1 and Chk2, which elicit the appropriate cellular response. The replication process can be arrested by repressing the firing of origins of replication that are usually activated at later stages in S phase (Santocanale and Diffley, 1998) or by slowing the progression of existing replication forks. If the damage is not excessive then DNA repair processes are activated (Section 14.2); alternatively the cell may be shunted into the pathway of programmed cell death called apoptosis, the death of a single somatic cell as a result of DNA damage usually being less dangerous than allowing that cell to replicate its mutated DNA and possibly give rise to a tumor or other cancerous growth. In mammals, a central player in induction of cell cycle arrest and apoptosis is the protein called p53. This is classified as a tumor-suppressor protein, because when this protein is defective, cells with damaged genomes can avoid the S-phase checkpoints and possibly proliferate into a cancer. p53 is a sequence-specific DNA-binding protein that activates a number of genes thought to be directly responsible for arrest and apoptosis, and also represses expression of others that must be switched off to facilitate these processes.
Study Aids For Chapter 13
Key terms
Give short definitions of the following terms:
- γ complex
- Apoptosis
- Autonomously replicating sequence (ARS)
- Cell cycle
- Cell-cycle checkpoint
- Cell senescence
- Cohesin
- Conservative replication
- Cyclin
- Dispersive replication
- Displacement replication
- DNA ligase
- DNA polymerase α
- DNA polymerase δ
- DNA polymerase γ
- DNA polymerase I
- DNA polymerase II
- DNA polymerase III
- DNA topoisomerase
- FEN1
- G1 phase
- G2 phase
- Helicase
- Initiation region
- Lagging strand
- Leading strand
- M phase
- Meselson-Stahl experiment
- Okazaki fragment
- Origin of replication
- Origin recognition complex (ORC)
- Origin recognition sequence
- Paranemic
- Plectonemic
- Post-replication complex (post-RC)
- Prepriming complex
- Pre-replication complex (pre-RC)
- Primase
- Primer
- Primosome
- Proliferating cell nuclear antigen (PCNA)
- Proofreading
- Replication factor C (RFC)
- Replication factory
- Replication licensing factor (RLF)
- Replication mediator protein (RMP)
- Replication protein A (RPA)
- Replisome
- Rolling circle replication
- S phase
- Semiconservative replication
- Single-strand binding protein (SSB)
- Stem cell
- Telomerase
- Terminator sequence
- Transcription factory
- Transition
- Transversion
- Tus
Self study questions
- 1.
Distinguish between the terms ‘dispersive’, ‘semiconservative’ and ‘conservative’, as applied to DNA replication.
- 2.
Draw a fully annotated diagram illustrating the Meselson-Stahl experiment. What conclusions can be drawn from the results of this experiment?
- 3.
Explain why the discovery of DNA topoisomerases was an important step in the development of knowledge about DNA replication.
- 4.
With the aid of diagrams, indicate how displacement replication and rolling circle replication differ from the semiconservative process.
- 5.
Give a detailed description of the structure of the Escherichia coli origin of replication and outline the role of each component of the origin in the initiation of replication.
- 6.
Compare and contrast the Escherichia coli origin of replication with those of yeast and mammals.
- 7.
What impact does the inability of DNA polymerases to synthesize DNA in the 3′→5′ direction have on DNA replication?
- 8.
Explain why DNA replication must be primed, and describe how the priming problem is solved by Escherichia coli and by eukaryotes.
- 9.
Construct a table listing the DNA polymerases involved in DNA replication in Escherichia coli and in eukaryotes, and summarize the function and key features of each enzyme.
- 10.
Give a detailed description of the events occurring at the replication fork in Escherichia coli.
- 11.
In what ways do events at the eukaryotic replication fork differ from those occurring in Escherichia coli?
- 12.
Outline how replication terminates in Escherichia coli. What is currently known about the termination of replication in eukaryotes?
- 13.
Explain why the ends of a chromosomal DNA molecule could become shortened after repeated rounds of DNA replication, and show how telomerase prevents this from occurring.
- 14.
Discuss the links between telomeres, cell senescence and cancer.
- 15.
Draw a diagram of the eukaryotic cell cycle and indicate the periods that act as cell-cycle checkpoints.
- 16.
Outline our current knowledge concerning the composition of pre-replication complexes, the factors that influence their assembly, and the events that lead to their conversion into post-replication complexes.
- 17.
Describe how genome replication is regulated during S phase.
Problem-based learning
- 1.
Evaluate the status of current research into mammalian replication origins.
- 2.
Why is inactivation of the Escherichia coli polA gene, coding for DNA polymerase I, not lethal?
- 3.
Write an extended report on ‘DNA helicases’.
- 4.
Our current knowledge of genome replication in eukaryotes is biased towards the events occurring at the replication fork. The next challenge is to convert this DNA-centered description of replication into a model that describes how replication is organized within the nucleus, addressing issues such as the role of replication factories and the processes used to avoid tangling of the daughter molecules. Devise a research plan to address one or more of these issues.
- 5.
Explore the links between telomeres, cell senescence and cancer.
References
- Aladjem MI, Rodewald LW, Kolman JL, Wahl GM. Genetic dissection of a mammalian replicator in the human β-globin locus. Science. (1998);281:1005–1009. [PubMed: 9703500]
- Bae S-H, Bae K-H, Kim J-A, Seo Y-S. RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature. (2001);412:456–461. [PubMed: 11473323]
- Beernick HTH, Morrical SW. RMPs: recombination/ replication mediator proteins. Trends Biochem. Sci. (1999);24:385–389. [PubMed: 10500302]
- Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. (1992);357:128–134. [PubMed: 1579162]
- Berger JM, Gamblin SJ, Harrison SC, Wang JC. (1996)Structure and mechanism of DNA topoisomerase II Nature 379225–232. Erratum in: Nature 1996, 380, 179. [PubMed: 8538787]
- Bielinsky A-K, Gerbi SA. Discrete start sites for DNA synthesis in the yeast ARS1 origin. Science. (1998);279:95–98. [PubMed: 9417033]
- Blackburn EH. Telomere states and cell fates. Nature. (2000);408:53–56. [PubMed: 11081503]
- Blow JJ, Tada S. A new check on issuing the licence. Nature. (2000);404:560–561. [PMC free article: PMC3605502] [PubMed: 10766226]
- Bochkarev A, Pfuetzner RA, Edwards AM, Frappier L. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. (1997);385:176–181. [PubMed: 8990123]
- Cabral JHM, Jackson AP, Smith CV, Shikotra N, Maxwell A, Liddington RC. Crystal structure of the breakage-reunion domain of DNA gyrase. Nature. (1997);388:903–906. [PubMed: 9278055]
- Carpenter PB, Mueller PR, Dunphy WG. Role for a Xenopus Orc2-related protein in controlling DNA replication. Nature. (1996);379:357–360. [PubMed: 8552193]
- Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Ann. Rev. Biochem. (2001);70:369–413. [PubMed: 11395412]
- Cook P. Duplicating a tangled genome. Science. (1998);281:1466–1467. [PubMed: 9750117]
- Cook PR. The organization of replication and transcription. Science. (1999);284:1790–1795. [PubMed: 10364545]
- Crick FHC, Wang JC, Bauer WR. Is DNA really a double helix? J. Mol. Biol. (1979);129:449–461. [PubMed: 458852]
- Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science. (1996);272:1030–1033. [PubMed: 8638128]
- Diffley JFX, Cocker JH. Protein-DNA interactions at a yeast replication origin. Nature. (1992);357:169–172. [PubMed: 1579168]
- Eickbush TH. Telomerase and retrotransposons: which came first? Science. (1997);277:911–912. [PubMed: 9281073]
- Falaschi A. Eukaryotic DNA replication: a model for a foxed double replisome. Trends Genet. (2000);16:88–92. [PubMed: 10652536]
- Fangman WL, Brewer BJ. A question of time - replication origins of eukaryotic chromosomes. Cell. (1992);71:363–366. [PubMed: 1423601]
- Feng J, Funk WD, Wang S-S. et al. The RNA component of human telomerase. Science. (1995);269:1236–1241. [PubMed: 7544491]
- Frick DN, Richardson CC. DNA primases. Ann. Rev. Biochem. (2001);70:39–80. [PubMed: 11395402]
- Gilbert DM. Making sense of eukaryotic replication origins. Science. (2001);294:96–100. [PMC free article: PMC1255916] [PubMed: 11588251]
- Greider CW. Telomere length regulation. Ann. Rev. Biochem. (1996);65:337–365. [PubMed: 8811183]
- Holmes FL. The DNA replication problem, 1953-1958. Trends Biochem. Sci. (1998);23:117–120. [PubMed: 9581505]
- Hübscher U, Nasheuer H-P, Syväoja JE. Eukaryotic DNA polymerases: a growing family. Trends Biochem. Sci. (2000);25:143–147. [PubMed: 10694886]
- Kamada K, Horiuchi T, Ohsumi K, Shimamoto N, Morikawa K. Structure of a replicator-terminator protein complexed with DNA. Nature. (1996);383:598–603. [PubMed: 8857533]
- Kelman Z. The replication origin of archaea is finally revealed. Trends Biochem. Sci. (2000);25:521–523. [PubMed: 11084357]
- Kipling D, Faragher RGA. Ageing hard or hardly ageing? Nature. (1999);398:191–193. [PubMed: 10094039]
- Lemon KP, Grossman AD. Localization of bacterial DNA polymerase: evidence for a factory model of replication. Science. (1998);282:1516–1519. [PubMed: 9822387]
- Lima CD, Wang JC, Mondragón A. Three-dimensional structure of the 67K N-terminal fragment of E. coli DNA topoisomerase I. Nature. (1994);367:138–146. [PubMed: 8114910]
- Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. (1997);276:561–567. [PubMed: 9110970]
- Marciniak R, Guarente L. Testing telomerase. Nature. (2001);413:370–373. [PubMed: 11574867]
- Meselson M, Stahl F. The replication of DNA in Escherichia coli. Proc. Natl Acad. Sci. USA. (1958);44:671–682. [PMC free article: PMC528642] [PubMed: 16590258]
- Murray A. A snip separates sisters. Nature. (1999);400:19–21. [PubMed: 10403242]
- Myllykallio H, Lopez P, López-Garcia P. et al. Bacterial mode of replication with eukaryotic-like machinery in a hyperthermophilic archaeon. Science. (2000);288:2212–2215. [PubMed: 10864870]
- Novick RP. Contrasting lifestyles of rolling-circle phages and plasmids. Trends Biochem. Sci. (1998);23:434–438. [PubMed: 9852762]
- Okazaki T, Okazaki R. Mechanisms of DNA chain growth. Proc. Natl Acad. Sci. USA. (1969);64:1242–1248. [PMC free article: PMC223275] [PubMed: 4989398]
- Pardue ML, Danilevskaya ON, Lowenhaupt K, Slot F, Traverse KL. Drosophila telomeres: new views on chromosome evolution. Trends Genet. (1996);12:48–52. [PubMed: 8851970]
- Paulovich AG, Hartwell LH. A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell. (1995);82:841–847. [PubMed: 7671311]
- Reddel RR. A reassessment of the telomere hypothesis of senescence. BioEssays. (1998);20:977–984. [PubMed: 10048297]
- Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WGJ. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science. (1998);279:1504–1513. [PubMed: 9488644]
- Rodley GA, Scobie RS, Bates RHT, Lewitt RM. A possible conformation for double-stranded polynucleotides. Proc. Natl Acad. Sci. USA. (1976);73:2959–2963. [PMC free article: PMC430891] [PubMed: 1067594]
- Santocanale C, Diffley JFX. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. (1998);395:615–618. [PubMed: 9783589]
- Stewart L, Redinbo MR, Qiu X, Hol WGJ, Champoux JJ. A model for the mechanism of human topoisomerase I. Science. (1998);279:1534–1541. [PubMed: 9488652]
- Stillman B. Cell cycle control of DNA replication. Science. (1996);274:1659–1664. [PubMed: 8939847]
- Takahashi K, Yanagida M. Replication meets cohesion. Science. (2000);289:735–736. [PubMed: 10950718]
- Tye BK. MCM proteins in DNA replication. Ann. Rev. Biochem. (1999);68:649–686. [PubMed: 10872463]
- Tzfati Y, Fulton TB, Roy J, Blackburn EH. Template boundary in a yeast telomerase specified by RNA structure. Science. (2000);288:863–867. [PubMed: 10797010]
- van Brabant AJ, Stan R, Ellis NA. DNA helicases, genome instability, and human genetic disease. Annu. Rev. Genomics Hum. Genet. (2000);1:409–459. [PubMed: 11701636]
- Waga S, Stillman B. The DNA replication fork in eukaryotic cells. Ann. Rev. Biochem. (1998);67:721–751. [PubMed: 9759502]
- Watson JD, Crick FHC. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature. (1953a);171:737–738. [PubMed: 13054692]
- Watson JD, Crick FHC. Genetical implications of the structure of deoxyribonucleic acid. Nature. (1953b);171:964–967. [PubMed: 13063483]
- Wei X, Samarabandu J, Devdhar RS, Siegel AJ, Acharya R, Berezney R. Segregation of transcription and replication sites into higher order domains. Science. (1998);281:1502–1505. [PubMed: 9727975]
- Zhou B-BS, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. (2000);408:433–439. [PubMed: 11100718]
Further Reading
- Adams RLP (1991) DNA Replication: In Focus. IRL Press, Oxford. —A concise introduction to the subject.
- Benkovic SJ, Valentine AM, Salinas F. Replisome-mediated DNA replication. Ann. Rev. Biochem. (2001);70:181–208. —Details of events at the bacterial replication fork. [PubMed: 11395406]
- Collins K. Ciliate telomerase biochemistry. Ann. Rev. Biochem. (1999);68:187–218. —Details of telomerase action in ciliates, which are used as model organisms in telomere studies. [PubMed: 10872448]
- DePamphilis ML (ed.) (1996) DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. —A more advanced and detailed treatment.
- Judson HF (1979) The Eighth Day of Creation: Makers of the Revolution in Biology. Penguin Books, London. —Includes an account of the topological problems and the Meselson-Stahl experiment.
- Kelly TJ, Brown GW. Regulation of chromosome replication. Ann. Rev. Biochem. (2000);69:829–880. [PubMed: 10966477]
- Kornberg A (1989) For the Love of Enzymes: The Odyssey of a Biochemist. Harvard University Press, Boston. —A fascinating autobiography by the discoverer of DNA polymerase.
- McEachern MJ, Krauskopf A, Blackburn EH. Telomeres and their control. Ann. Rev. Genet. (2000);34:331–358. —Describes the processes involved in regulation of telomere length. [PubMed: 11092831]
- Nasmyth K, Peters J-M, Uhlmann F. Splitting the chromosome: cutting the ties that bind sister chromatids. Science. (2000);288:1379–1384. —Reviews our current understanding of the role of cohesins during the final stages of DNA replication. [PubMed: 10827941]
- Patel SS, Picha KM. Structure and function of hexameric helicases. Ann. Rev. Biochem. (2000);69:651–697. —A detailed review. [PubMed: 10966472]
- Soultanas P, Wigley DB. Unwinding the ‘Gordian knot’ of helicase action. Trends Biochem. Sci. (2001);26:47–54. —Recent developments in the study of helicase action. [PubMed: 11165517]
- Wang JC. DNA topoisomerases. Ann. Rev. Biochem. (1996);65:635–692. [PubMed: 8811192]
- Genome Replication - GenomesGenome Replication - Genomes
Your browsing activity is empty.
Activity recording is turned off.
See more...