

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Amid A, Lal A, Coates TD, et al., editors. Guidelines for the Management of α-Thalassaemia [Internet]. Nicosia (Cyprus): Thalassaemia International Federation; 2023.

Introduction
Haemoglobin (Hb) is a vital respiratory carrier protein that facilitates the transport of oxygen from the lungs to the body’s tissues and the return of carbon dioxide from the tissues to the lungs. Each haemoglobin molecule is structured in a tetrameric form, composed of two α-like and two β-like globin chains. Each globin chain contains a heme group with a ferrous iron atom, which binds with oxygen [1].
The synthesis of globin proteins is controlled by the α-gene cluster on chromosome 16 and the β-gene cluster on chromosome 11 (see Figure 1). The genes within these two clusters are coordinately expressed to ensure an equal amount of α-like and β-like globins are produced. The various globin genes are situated on α- and β-gene loci in the order they are developmentally expressed, leading to the production of different haemoglobins during the stages of human development. In adults, haemoglobin primarily consists of adult haemoglobin (HbA, α2β2), with a smaller proportion of foetal haemoglobin (HbF, α2γ2), and a minor component, HbA2 (α2δ2). The expression of globin genes is regulated by enhancer elements located in the upstream regions of the α-gene and β-gene clusters, known as HS-40 and LCR, respectively.
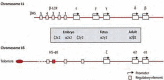
Figure 1
Schematic representation of the chromosomal location of the α- and β-globin clusters on 16p and 11p respectively.
Epidemiology and global burden of α-thalassaemias
Genetic basis of α-thalassaemias
The α-globin gene cluster lies in a 135-155 kb GC-rich, Alu repeat dense and gene-dense genomic DNA region approximately 150 kb from the telomere of chromosome 16 (16p13.3). It contains three functional globin genes, i.e. the embryonic ζ-gene (HBZ) and duplicated foetal/adult α-genes (HBA1 and HBA2), three pseudogenes, i.e. the pseudo ζ (HBZps), the pseudo α1 (HBA1ps), and pseudo α2 (HBM) and the θ (HBQ)-gene of unknown function (see Figure 1). Mutations in the HBA2 gene are associated with more prominent effect on α-globin production than mutations in the HBA1 gene [6].
As previously mentioned, normal haemoglobin consists of two α-globin chains and two β-globin chains. In normal individuals, there are four functional α-globin genes, with two inherited from each parent (αα/αα), and two β-globin genes, with one inherited from each parent (β/β).
α-Thalassaemia primarily arises from large fragment deletions (known as copy number variations or CNV) or point mutations (referred to as single nucleotide variations or SNV) in the regions that encode the α-globin chains. These genetic changes lead to a varying degree of reduced or absent production of α-globin chains. Deletions that result in the loss of duplicated α-genes lead to the absence of α-globin production from that chromosome, and they are called α0-thalassaemia deletions (--), and those that result in the loss of single a gene and decreased production of α-globin from that chromosome are called α+-thalassaemia deletions (-α). Less frequently, α-thalassaemia can occur due to point mutations in either HBA1 or HBA2, resulting in the production of abnormal or unstable variant α-globin chains (αTα or ααT).
The most prevalent deletional α+-thalassaemias result from unequal homologous recombination of duplicated α-genes during meiosis, leading to the -α3.7 and -α4.2 deletions. Conversely, α-gene triplications (ααα anti 3.7 and ααα anti 4.2) represent the complementary events [1, 8]. Among these, -α3.7 is the most common α+-thalassaemia deletion and its compound heterozygosity with α0-thalassaemia deletions is the most common genotype observed in HbH disease.
Larger deletions that eliminate both duplicated α-genes in cis constitute α0-thalassaemias and occur less frequently but may be prevalent in specific populations. If the deletion leaves the HBZ gene intact (e.g. -- SEA deletion), homozygotes develop Hb Bart’s hydrops foetalis syndrome, characterized by development of non-immune hydrops in the second or third trimester (as discussed below). However, when HBZ is also deleted (e.g. --FIL deletion), homozygotes are unlikely to survive even in the earliest developmental stages. The most common α0-deletional type encountered is the --SEA deletion, which is prevalent in Southeast Asia, while the --MED deletion is the most common α0-thalassaemia mutation in the Middle East and Mediterranean region.
Point mutations, small deletions, or insertions that affect sequences controlling gene expression fall under the category of non-deletional α+-thalassaemias (αTα or ααT) and are less common than their deletional counterparts. However, some of these non-deletion types can lead to a more pronounced reduction in α-globin synthesis compared to single α-gene deletions. These mutations have been documented to affect gene expression, mRNA splicing, and globin chain stability. Important examples of these mutations are Hb Constant Spring and the IVS I-1 (-5 bp) (HBA2:c.95+2_95+6delTG) [7].
Classification of α-thalassaemia
α-Thalassaemia is highly heterogeneous at both clinical and molecular levels. The clinical course of α-thalassaemia is generally correlated with the number of affected α-globin genes. There are four primary clinical types of α-thalassaemia syndromes (see Figure 4).:
![Figure 4. Classification of α-thalassaemia defects (reproduced with permission from Piel FB and Weatherall DJ. N Engl J Med. 2014 [13]).](/books/NBK602225/bin/chapter1_f4.gif)
Figure 4
Classification of α-thalassaemia defects (reproduced with permission from Piel FB and Weatherall DJ. N Engl J Med. 2014 [13]).
- Silent carrier: This type is defined as heterozygous α+-thalassaemia (-α/αα) resulting from the deletion or dysfunction of one of the four normal α-globin genes. Individuals with this condition are generally healthy and exhibit a normal hematological profile.
- α-Thalassaemia trait: This includes two subtypes:
- Heterozygous α0-thalassaemia (--/αα), resulting from the deletion of two α-genes in cis.
- Homozygous α+-thalassaemia (-α/-α), resulting from deletion of two α-genes in trans.Individuals with α-thalassaemia trait are healthy and asymptomatic, although they may experience mild anaemia or microcytosis. Identification of individuals with α0 trait (--/αα genotype) is a critical step in a screening and prevention programme as inheritance of these deletions is necessary for more severe forms of α-thalassaemia disease.
- HbH disease: This category encompasses compound heterozygous α0 and α+-thalassaemia mutations (--/-α or --/αTα). Additionally, individuals homozygous for non-deletional α+-thalassaemia mutations (αTα/αTα) can also be classified within this group. The severity of haemolytic anaemia and ineffective erythropoiesis varies depending on the specific mutations involved.
- Hb Bart’s hydrops foetalis: This type is defined by homozygous α0-thalassaemia (--/--) with the complete absence of functioning α-genes.
Haemoglobin H disease
Haemoglobin H (HbH) disease is a clinical condition that arises when only one residual functioning α-globin gene is present, resulting in genotypes of (--/-α) or (--/αTα). Consequently, there is a relative excess of β-globin chains, which combine to form β4 tetramers known as HbH. These HbH molecules typically constitute 3-30% of the total haemoglobin in patients with HbH disease.
Conversely, patients with non-deletional HbH disease experience a more severe clinical phenotype. This severity arises from the production of abnormal or unstable α-globin variants, which, when combined with the presence of unstable HbH due to globin chain imbalance, contributes to additional red cell pathobiology. This combined abnormality can lead to increased peripheral hemolysis and ineffective erythropoiesis, resulting in a more pronounced clinical phenotype.
Clinically, a wide variety of phenotypes is observed as a result of homozygosity for non-deletional α+-thalassaemia mutations. For instance, homozygous poly(A) mutations are a common cause of HbH disease, particularly among populations in the Middle East and Central Asia. In contrast, homozygosity for Hb Constant Spring or Hb Koya Dora mutations tends to result in milder thalassaemia syndromes.
Rarely, the combination of specific non-deletional α+-thalassaemia mutations and α0-thalassaemia deletions can give rise to an exceptionally severe phenotype, akin to that of β-thalassaemia major. In its most severe forms, this condition can lead to hydrops foetalis (HbH hydrops foetalis).
Haemoglobin Bart’s hydrops foetalis syndrome
Hb Bart’s hydrops foetalis syndrome represents the most severe form of α-thalassaemia and is characterized by the absence of all four α-globin genes (--/--), as depicted in Figure 4. In the absence of α-globin production, γ-globin chains combine to form Hb Bart’s (γ4) during the foetal period, which switches to HbH (β4) after birth, both of with are non-functional haemoglobins [1]. Foetuses with Hb Bart’s hydrops foetalis (homozygous α°-thalassaemia) typically exhibit 80–90% Hb Bart’s [9].
These affected foetuses experience hypoxia, heart failure, and hydrops foetalis, often succumbing in utero during the second or third trimester of gestation, or they may pass away within hours after birth. This condition is notably the most common cause of hydrops in Southeast Asia. In recent decades, due to population migrations, there has been an increase in the prevalence of this syndrome in other parts of the world [10]. With improvement of antenatal and prenatal care and availability of intrauterine transfusion, an increasing number of long-term survivors with this condition are being reported.
Unusual forms of α-thalassaemia
ATR-16 is a rare genetic condition that arises from large chromosomal abnormalities at the telomere end of chromosome 16, which encompasses the α-globin genes. Affected individuals demonstrate an unusual association of α-thalassaemia, cognitive impairment and dysmorphic features [11].
ATR-X syndrome is another rare genetic condition that is associated with a distinct and recognizable dysmorphic appearance in boys that is also associated with α-thalassaemia with severe mental cognitive impairment. It is inherited due to deletions or mutations of ATRX gene located on chromosome X (X-linked). ATRX plays an important role in the incorporation of the histone variant H3.3 into telomere and pericentromeric DNA. When the gene is mutated, among many other effects, this leads to down-regulation of expression of the α-globin genes on the telomere end of chromosome 16 [11].
α-Thalassaemia myelodysplastic syndrome (ATMDS) is an acquired form of α-thalassaemia that results from somatic mutations in ATRX gene or very rarely from acquired loss of telomere end of chromosome 16 during myelodysplastic syndrome or haematological malignancies [11].
Laboratory and genetic diagnosis of α-thalassaemias
Diagnosis of α-thalassaemias requires a combination of laboratory tests, including the measurement of red blood cell indices using automatic haematology analyzers, haemoglobin analysis, and quantification of HbA2 and HbF. Two widely adopted automatic methods for this purpose are High-Performance Liquid Chromatography (HPLC) and Capillary Zone Electrophoresis (CE). These systems provide both qualitative and quantitative analyses of haemoglobin components with high precision and reproducibility, enabling both prenatal and postnatal thalassaemia diagnoses within minutes.
Haematology and haemoglobin analysis
Initial laboratory testing to identify α-thalassaemia carriers involves a complete blood count (CBC), with determination of mean corpuscular volume (MCV) and mean corpuscular haemoglobin (MCH). Carriers with genotypes -α/-α and --/αα typically exhibit reduced MCV and MCH values. In contrast, -α/αα carriers may have normal red cell indices or only slightly reduced MCV and MCH levels.
Patients with HbH disease display significant variations in haemoglobin, MCV, and MCH values, which can vary among individuals (see Table 1).
Table 1
Clinical and haematologic manifestation of deletional and non-deletional forms of haemoglobin H (reproduced with permission from Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT), 2nd edition)
Automatic haemoglobin analyzers
High-performance liquid chromatography: The HPLC system is cation exchange and use two dual piston pumps to set gradient sodium phosphate buffers of increasing ionic strength to pass through a column spherical cation exchange resin during a 6.5 minutes. Hemolysate samples are determined by spectrophotometer that read double wavelengths at 415 and 690 nm. The resulting chromatograms are separated in retention time (RT).
Capillary electrophoresis: The CE system is based on capillary electrophoresis in free solution from cathode to anode. Haemoglobin components are separated in silica capillaries by their electroosmotic flow and at a high voltage (9,800 V) in electrophoretic mobility in an alkaline buffer. The photometry at an absorbance wavelength 415 nm is used to directly detect Hb fractions.
Automatic haemoglobin analyzers are unable to reliably identify individuals who are heterozygous carriers for α-thalassaemia deletions, although they can identify great majority of β-haemoglobinopathies or carriers for some non-deletional α+-thalassaemia mutations.
Patients with HbH disease have reduced (<2%) HbA2, but the characteristic finding is the presence of variable amounts (up to 30%) of HbH. HbH is easily detected as an early-eluting peak in HPLC (Figure 5). In neonates, those with HbH disease can be detected through haemoglobin electrophoresis due to elevated levels (approximately 25% or greater) of Hb Bart’s, making this feature useful for neonatal screening.
![Figure 5. High-performance liquid chromatography (HPLC) and capillary haemoglobin electrophoresis patterns of an adult with HbH (adapted from Harteveld, CL and Higgs DR, Orphanet Journal of Rare Diseases, 2010 [1], with permission).](/books/NBK602225/bin/chapter1_f5.gif)
Figure 5
High-performance liquid chromatography (HPLC) and capillary haemoglobin electrophoresis patterns of an adult with HbH (adapted from Harteveld, CL and Higgs DR, Orphanet Journal of Rare Diseases, 2010 [1], with permission).
Red blood cell morphology
Red cell inclusion bodies (precipitated β4 tetramers or H bodies) can often be detected under a microscope in a significant portion of red blood cells. This detection is facilitated by using supravital staining dyes such as methylene blue or brilliant cresyl blue. These inclusion bodies are a characteristic feature of HbH disease and play a key role in its diagnosis.
Molecular analysis
Since these genetic tests can often be costly, it is crucial to accurately characterize hematologic features, such as haemoglobin levels, MCV, and MCH to guide the selection of samples for genetic analysis. While low MCV and MCH are characteristic of thalassemic red blood cells, these indices alone cannot distinguish between thalassaemia trait and iron deficiency and the hematological parameters in these two conditions may closely resemble each other, leading to confusion between α-thalassaemia trait and iron-deficiency anaemia. In such cases, assessing iron status (e.g., serum iron, transferrin saturation, or red blood cell zinc protoporphyrin levels) can be helpful in making an accurate diagnosis although it may not be sufficient in those where the two conditions co-exist.
Molecular testing for α-thalassaemia typically entails a battery of methods aimed at detecting and characterizing both known and unknown mutations, with the majority of these methods relying on polymerase chain reaction (PCR). Amplifying the α-globin gene cluster poses particular challenges due to the sequence homology shared among genes within this cluster, such as HBA1 and HBA2. Therefore, careful attention must be given to assay design to ensure specific amplification and differentiation of these homologous genes.
The gap-PCR (gap polymerase chain reaction) method serves as a simple, rapid and inexpensive technique for identifying common deletions within specific populations. This method employs primers designed to flank known deletion breakpoints, allowing for the targeted detection of deletions. Common single α-globin gene deletions include --MED, -α20.5, -α3.7, -α4.2, --THAI, --SEA, --FIL, as well as the triplication, anti-α3.7 (ααα).
In cases where gap-PCR analysis yields a negative result for individuals showing haematological indications that suggest the presence of a deletional variant, further investigation is warranted using multiplex ligation-dependent probe amplification (MLPA). MLPA is another technique employed to characterize deletions in thalassaemia. This method relies on the ligation of multiple probe-pairs hybridized across the entire locus of interest, facilitating the quantification of gene copy numbers. MLPA represents a valuable alternative or supplementary method to gap-PCR, particularly when examining both known and unknown deletions and duplications underlying α-thalassaemia.
For the detection of single nucleotide variants (SNVs), Sanger sequencing is the most practical method, enabling comprehensive detection of all variants without prior knowledge of family history. However, sequencing α-globin genes presents a unique challenge due to the near-complete homology shared by the two α-globin genes (HBA1 and HBA2). Therefore, specialized design and optimization of PCR conditions are necessary for accurate sequencing. In instances where the specific carrier mutation within a family is known, direct mutation detection methods, such as restriction enzyme digestion PCR (RED-PCR), designed for that particular variation, may be employed.
Nevertheless, this approach can be time-consuming, labour-intensive, and it sometimes carries the risk of not detecting certain variants. The landscape of human globin gene mutation detection methods has significantly evolved with the advent of next generation sequencing (NGS) platforms. NGS has revolutionized genetic diagnosis by enabling rapid, highly multiplexed, and high-throughput detection of genetic variants. To simplify traditional strategies, some molecular testing laboratories have introduced targeted NGS testing for the genetic analysis of thalassaemias. Commercial targeted NGS kits now allow for the comprehensive analysis of the entire spectrum of thalassaemias, detecting variations, SNVs, indels, and copy number variations (CNVs) in HBA1, HBA2, and HBB using a single one-tube NGS assay. This fast, straightforward, and robust NGS workflow replaces complex multi-step protocols and eliminates the need for maintaining multiple thalassaemia assays in your laboratory.
Summary and recommendations
- α-Thalassaemia is one of the most commonly inherited blood conditions. Initially prevalent in areas where malaria was most common, α-thalassaemia is now considered a global health concern due to population migration.
- Clinically significant forms of α-thalassaemia, HbH disease and haemoglobin Bart’s hydrops foetalis result from compound heterozygosity for α0-thalassaemia deletions with other mutations or deletions of α-globin genes. Hence, identification of carriers of α0-thalassaemia deletions is pivotal for population control and prenatal diagnosis.
- HbH disease is the most common clinically significant form of α-thalassaemia, which is most prevalent in Southeast Asia, South China and also in some areas in the Middle East or Mediterranean region.
- HbH disease has a considerable heterogeneity, both genetically and clinically. Individuals with deletional forms of HbH disease have generally a benign course but those who harbour a non-deletional mutation have a higher rate of transfusion requirement and experience more frequent thalassaemia-related complications. As a result, identification of the underlying genetic abnormality has clinical implications.
- Accurate diagnosis of α-thalassaemia syndromes requires application of a range of diagnostic techniques, including complete blood count (CBC) with reticulocyte count, haemolytic panel, peripheral blood smears, automatic haemoglobin analyzers, and different modalities of molecular analysis.
- Knowledge of the patient’s clinical phenotype and the prevalence of specific mutations in the region is essential to strategically select the appropriate molecular analysis.
References
- 1.
- Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13. [PMC free article: PMC2887799] [PubMed: 20507641] [CrossRef]
- 2.
- Modell B., Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–7. [PMC free article: PMC2647473] [PubMed: 18568278] [CrossRef]
- 3.
- Weatherall DJ. Thalassaemia as a global health problem: recent progress toward its control in the developing countries. Ann N Y Acad Sci. 2010;1202:17–23. [PubMed: 20712767] [CrossRef]
- 4.
- Cao A, Kan YW. The prevention of thalassaemia. Cold Spring Harb Perspect Med. 2013;3(2):a011775. [PMC free article: PMC3552345] [PubMed: 23378598] [CrossRef]
- 5.
- Vlok M, Buckley HR, Miszkiewicz JJ, Walker MM, Domett K, Willis A, Trinh HH, Minh TT, Nguyen MHT, Nguyen LC, Matsumura H, Wang T, Nghia HT, Oxenham MF. Forager and farmer evolutionary adaptations to malaria evidenced by 7000 years of thalassaemia in Southeast Asia. Sci Rep. 2021 Mar 11;11(1):e5677. [PMC free article: PMC7952380] [PubMed: 33707498] [CrossRef]
- 6.
- Farashi S, Harteveld CL. Molecular basis of alpha-thalassaemia. Blood Cells Mol Dis. 2018;70:43–53. [PubMed: 29032940] [CrossRef]
- 7.
- Kountouris P, Kousiappa I, Papasavva T, Christopoulos G, Pavlou E, et al. The molecular spectrum and distribution of haemoglobinopathies in Cyprus: a 20-year retrospective study. Sci Rep. 2016 May 20;6:26371. [PMC free article: PMC4873807] [PubMed: 27199182] [CrossRef]
- 8.
- Higgs DR, Hill AV, Bowden DK, Weatherall DJ, Clegg JB. Independent recombination events between the duplicated human alpha globin genes; implications for their concerted evolution. Nucleic Acids Res. 1984 Sep 25;12(18):6965–77. [PMC free article: PMC320136] [PubMed: 6091047] [CrossRef]
- 9.
- Lorey F, Charoenkwan P, Witkowska HE, Lafferty J, Patterson M, Eng B, Waye JS, Finklestein JZ, Chui DH. Hb H hydrops foetalis syndrome: a case report and review of literature. Br J Haematol. 2001 Oct;115(1):72–8. [PubMed: 11722414] [CrossRef]
- 10.
- Chui DH, Wayne JS. Hydrops foetalis caused by alpha-thalassaemia: an emerging health care problem. Blood. 1998;91(7):2213–22. [PubMed: 9516118]
- 11.
- Gibbons RJ. α-Thalassaemia, mental retardation, and myelodysplastic syndrome. Cold Spring Harb Perspect Med. 2012 Oct 1;2(10):a011759. [PMC free article: PMC3475406] [PubMed: 23028133] [CrossRef]
- 12.
- Traeger-Synodinos J, Harteveld CL, Old JM, Petrou M, Galanello R, Giordano P, Angastioniotis M, De La Salle B, Henderson S, May A. EMQN haemoglobinopathies best practice meeting. EMQN Best Practice Guidelines for molecular and haematology methods for carrier identification and prenatal diagnosis of the haemoglobinopathies. Eur J Hum Genet. 2015 Apr;23(4):426–37. [PMC free article: PMC4666573] [PubMed: 25052315] [CrossRef]
- 13.
- Piel FB, Weatherall DJ. The α-thalassaemias. N Engl J Med. 2014 Nov 13;371(20):1908–16. [PubMed: 25390741] [CrossRef]
- EPIDEMIOLOGY, PATHOPHYSIOLOGY AND DIAGNOSIS OF α-THALASSAEMIA - Guidelines for t...EPIDEMIOLOGY, PATHOPHYSIOLOGY AND DIAGNOSIS OF α-THALASSAEMIA - Guidelines for the Management of α-Thalassaemia
Your browsing activity is empty.
Activity recording is turned off.
See more...