Introduction
Due to its location and function, the liver is continually being antigenically challenged by pathogens, toxins, tumor cells, and less harmful antigens such as those acquired with food intake. The bloodstream travels along the hepatic sinusoids which are covered by an endothelial layer devoid of basal membrane and presenting “pores.” This sinusoid structure allows the transmigration of cells towards the hepatic parenchyma. Soon after differentiation and activation, circulating T cells are capable of entering non-lymphoid peripheral organs. Differentiation and activation of T cells within the peripheral lymphoid organs are known to take roughly 48 h. The liver is an exception to the general time course lymphoid trafficking due to its ability to retain mature naïve T cells, mainly CD8+ T cells which, after a 24 h period, are delivered into the peripheral circulation as activated cells (1).
Chronic inflammation of the liver occurs as a consequence of the recruitment and retention of lymphocytes in the tissue. The recruitment of effector and regulatory T cells (Tregs) from the circulation is dependent on interactions between lymphocytes and specific cell surface molecules expressed on endothelial cells (2). Once captured, the retention and positioning of leukocytes within tissue requires certain signals to locate and retain them at sites of target cell damage. These receptors and chemokine signals follow the accepted multistep paradigm of leukocyte adhesion to vascular endothelium, which is relevant to most organ systems although the specific signals involved differ among tissues. Chemokines are critical components of this adhesion cascade and are believed to play two crucial roles: triggering integrin-mediated stable adhesion and directing migration. Chemokines can bind to endothelial glycosaminoglycans and thus allow them to be presented to flowing leukocytes and also provide a mechanism for the paracrine presentation of chemokines secreted by other cells within the microenvironment. Similar mechanisms are believed to be involved in both normal immune surveillance and in inflammatory disease although the chemokines involved differ with constitutive chemokines playing the dominant role in physiological trafficking and inducible inflammatory cytokines involved in inflammation. Chemokines can be classified into four groups based on their amino acid sequence. The two largest groups are the CC chemokines, where conserved cysteine residues lie adjacent to each other, and CXC chemokines, where an amino acid separates the first two cysteine residues. Within the group of CXC chemokines, CXCL10 or interferon (IFN)-inducible protein (IP-10), CXCL9 or monokine induced by IFN-γ (MIG), CXCL11 or IFN-γ - inducible T-cell -chemoattractant (ITAC) display potent lymphocyte chemotactic activity and bind a common receptor and CXCR3, the expression of which is increased on tissue-infiltrating regulatory and Th1-polarized T cells. T cells infiltrating the inflamed liver express high levels of CXCR3. Moreover, CXCR3 ligands are up-regulated on hepatic endothelium at sites of T-cell infiltration in chronic hepatitis, and their presence has been correlated with outcome of inflammatory liver disease (3).
The liver is a residence organ for dendritic cells (DC), whose precursors derive from peripheral blood. The antigenic uptake that occurs within the liver by immature DC promotes their maturation and migration to peripheral lymph nodes, where they act as efficient antigen presenting cells. This is a key process in the initiation of the immune response. Tregs may be attracted to DC residing within the liver to modulate their local function and survival. In secondary lymphoid tissues, Tregs use multiple mechanisms to inhibit DC function and block initiation of autoimmunity. Tregs can suppress effector cell proliferation and function within the liver by secreting immunosuppressive cytokines (IL-10, TGFβ) or through contact-dependent mechanisms to restore immune homeostasis and promote resolution of hepatitis. Effector T cells cause damage to hepatocytes resulting in interface and lobular hepatitis. Infiltrating T cells, macrophages, and stromal cells secrete IFNγ, TNFα, and other proinflammatory cytokines that result in a microenvironment that promotes the persistence of chronic liver inflammation.
Apart from DC, hepatocytes, kupffer cells, and sinusoidal endothelial cells can present antigens to CD8+ T cells. The local presentation of antigens within the liver leads to activation-triggered T cell apoptosis. The balance between the different activation processes that take place within the liver leads to immunological tolerance, which explains the long survival of hepatic grafts and the chronic persistence of hepatotropic viruses (4). The development of intrahepatic tolerance would be due to the low expression of co-stimulatory molecules by hepatocytes (5).
Another feature that makes the liver a specialized immune organ is the type of resident T cells in the parenchyma (6). Fifty percent of intrahepatic lymphocytes (IHL) are non-conventional cells expressing membrane proteins that are typical of T cells and proteins belonging to natural killer cells (CD56, CD161, and/or KIRs, which are receptors that inhibit the cytotoxic response). These cells are termed NKT cells or “natural T cells.” IHLs represent 20% of the total NK cell population, 25% of the T cells, and 5% of the B cells. Classical NKT cells (type I) express only one α chain of the TCR called the invariant chain (iNKT) by which the recognition of antigenic epitopes presented by the non-classical histocompatibility molecule CD1 (7) is accomplished. These cells also express CD45RO, an isoform of the CD45 which features activated lymphocytes and CD161 (NKR-P1A), a co-stimulatory molecule that contributes to the antigenic presentation mediated by CD1.
The interaction between antigen presenting cells and NKT cells results in the rapid expression of regulatory/Th2 and proinflammatory (IFN-γ and TNF) cytokines by NKT cells. Activated NKT cells are a source of IFN-γ and IL-4, two cytokines with hepatocytotoxic properties (Figure 1).
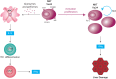
Autoimmune hepatitis (AIH)
Autoimmune hepatitis (AIH) is a chronic and progressive hepatitis of moderate to severe activity. AIH does not cause granulomatous biliary lesions, siderosis, or copper depots (8). The classification of AIH is based on the specificity of the autoantibodies found in the patient’s serum (Table 1). In this chapter, special reference will be made to type I AIH.
Type I AIH
Type I AIH (AIH-I) is a disease of unknown etiology featured by the presence of polyclonal hypergammaglobulinemia, autoantibodies, and a local progressive necroinflammatory response which frequently evolves to cirrhosis and hepatic failure. The actual prevalence of AIH is unknown. The first study to use the International Autoimmune Hepatitis Group scoring system on Alaskan natives reported a prevalence of definite AIH of 35.9 cases per 100,000 (11). A report from the United Kingdom showed that the annual incidence of AIH is 3.0 per 100,000 inhabitants (12). This disease is more frequent in females (70–90%) with a female:male ratio of 3.6:1 (13), and it responds favorably to treatment with immunosuppressant steroids and azathioprine (14,15).
Laboratory findings
AIH-I is featured by the presence of antibodies to actin filaments of the smooth muscle (SMA) and/or antinuclear antibodies (ANA). These antibodies can also be accompanied by perinuclear anti-neutrophil cytoplasm antibodies (p-ANCA). Although these antibodies are useful in the diagnosis of the disease, an immunopathogenic role has not been attributed to them (16). Therefore, no direct correlation exists between the necroinflammatory activity and the presence of such antibodies though their titers are known to decrease upon corticosteroid treatment (15). The activity of aminotransferases and specific IgG titers do not correlate with histological damage and consequently provide limited help with respect to treatment initiation (17). Bilirrubin levels as well as alkaline phosphatase activity are slightly higher in 80% of patients and 75% of them present low serum albumin values (8).
Histological abnormalities
Since aminotransferases and IgG levels do not reflect the extent of histological inflammatory activity, or the presence or absence of cirrhosis, liver biopsy is mandatory to not only confirm the diagnosis but also evaluate the severity of liver damage. The histological evaluation of chronic hepatitis, including AIH-I, consists of the assessment of the necroinflammatory activity (activity degree), the presence of fibrosis, and the disruption of the hepatic parenchyma architecture (stadification). Results of this assessment are expressed as the Knodell’s histological activity index (18, 19). The most frequent histological findings are the presence of interface hepatitis with abundant inflammatory infiltrate of a lymphocytic nature with or without lobular (acinar) and portoportal or portocentral fibrotic bridges accompanied by rosette formation and nodular regeneration.
Clinical manifestations and diagnosis
In individuals with unresolved hepatitis, it is common to find symptoms that are similar to those of acute viral hepatitis. In these patients, a combination of signs and symptoms can be found, e.g. jaundice, acolia, coluria, hepatomegaly, splenomegaly, spider veins, acne, malaise, fatigue, evidence of cirrhosis, or fulminant liver failure. In the first five years after the onset of the disease, 50% of untreated patients die while spontaneous remission is a less frequent phenomenon. Diagnosis of AIH-I is difficult, since clinical, biochemical, or histological findings are not conclusive if taken in isolation. The criteria for the diagnosis of this disease have been established and revised by the International Autoimmune Hepatitis Group (20, 21). More recently, a simplified score intended to be used in clinical practice has been proposed by this group (22). The diagnostic criteria that make it possible to distinguish probable from definite AIH are in Table 2.
Table 2
Diagnostic criteria for autoimmune hepatitis.
The current guidelines for the diagnosis of AIH are the following:
- Diagnosis of AIH requires the assessment of aminotransferase activity, the levels of gammaglobulins, detection of ANA and/or SMA, or the presence of type 1 anti-liver and kidney microsome (anti-LKM-1) antibodies as well as the histological evaluation of a liver biopsy.
- Diagnostic criteria of AIH must be applied to all patients.
- In those cases where the diagnosis of AIH is not conclusive or when the clinical features of hepatitis are atypical, the scoring method must be used (Table 3).
- Even though AIH-I presents histological and serological features that are common to pediatric and adult patients, previous work undertaken in our laboratory have shown that the clinical features and the genetic susceptibility for AIH-I differ between children and adults (Table 4).
Table 3
Differential characteristics between pediatric and adult autoimmune hepatitis.
Table 4
Scoring system for the diagnosis of atypical autoinmune hepatits in adults.
Treatment of AIH-I
AIH-I has been proven to respond favorably to the immunosuppressant treatment provided that there are no clinical signs of acute liver failure, a circumstance under which liver transplantation is mandatory.
The treatment of AIH-I comprises two phases. In newly diagnosed AIH, induction of remission is the main goal. The second phase of therapy is maintenance of remission with the lowest possible dose in order to maintain it while preventing significant side effects. If the diagnosis is correct, and the appropriate therapy is chosen, liver transplantation should be avoidable in patients with AIH.
The standard treatment consists of the administration of prednisolone with or without azathioprine. With this schedule, remission (normal liver tests) is achieved in more than 90% of the patients (26). In 20% of patients who have undergone remission, drugs can be withdrawn successfully after a few years of treatment, whereas in those patients suffering from AIH-II, the treatment must be maintained for the lifetime. A sustained remission of AIH has been reported in patients undergoing long-term treatment with azathioprine alone (27). Other effective drugs that are employed in the treatment of AIH-I are cyclosporine A and mophetil mycophenolate (28, 29)
Recurrence and de novo appearance of AIH
Autoimmune hepatitis can recur or appear de novo after liver transplantation, resulting in hepatic fibrosis, graft loss, and the need for re-transplantation. Autoimmune hepatitis recurs in 8–12% of transplanted patients at 1 year and in 36–68% at 5 years. Recurrence may be asymptomatic and detected only by surveillance of liver test abnormalities or routine liver tissue examinations. Autoantibodies that characterized the original disease, hypergammaglobulinemia, raised serum immunoglobulin G levels, and characteristic histologic findings typify recurrence. Premature corticosteroid withdrawal and pre-transplant severity of the original disease are possible risk factors.
In patients with AIH, a decrease in the number of peripheral CD4+CD45RO cells has been observed together with the presence of an abundant intrahepatic infiltrate of mononuclear CD45RO cells. These findings have made it possible to suggest that a subset of memory cells could be preferentially migrating towards the liver. This cell subset remains unchanged even after the immunosuppressant treatment is administered and might be associated with occasional relapses of the disease.
A recent study demonstrated that AIH patients in remission have a significantly lower frequency of activated T cells in the peripheral blood, compared to both AIH patients with active disease and healthy subjects (30). De novo autoimmune hepatitis occurs in 1–7% of patients 0.1–9 years after transplantation, especially in children. The appearance of autoantibodies may herald its emergence, and antibodies to glutathione-S-transferase T1 seem to be predictive of the disease. While recurrent disease may reflect recruitment of residual memory T lymphocytes and host-specific genetic predispositions, de novo disease may reflect an allo-antigenic immune response and molecular mimicries that override self-tolerance. Treatment should be appropriate for autoimmune hepatitis and not based on anti-rejection drugs. Corticosteroid therapy alone or combined with azathioprine is the essential treatment (31).
Immunopathogenesis of AIH-I
The establishment of this disease may be considered a consequence of a breakdown of tolerogenic mechanisms which govern the liver milieu. Although the autoantigen/s that trigger AIH-I have not been identified so far, the family of asialoglycoprotein receptors, xenobiotic substances, bacteria, and viruses have been postulated as candidates. Infection with hepatitis A, B, C virus (HAV, HBV, HCV) or measles virus, in particular, can precede the onset of AIH-I.
In Argentina there is a high prevalence of HAV infection. In children, the allele HLA II DRB1*1301 is linked to the development of both chronic HAV hepatitis and AIH-I. Due to this fact, it has been suggested that in children, the development of AIH-I requires the expression of yet unknown genes. The latter hypothesis is in line with the polygenic nature of autoimmune diseases (32). The treatment of chronic B and C hepatitis with IFN-α promotes the synthesis of serum autoantibodies and in some patients, these autoantibodies have been shown to be pathogenic. The neoantigen formation, the molecular mimicry or the induction of proinflammatory cytokines could be considered factors modifying the tolerogenic liver microenvironment that would lead to the development of AIH-I.
It is known that 10% of chronic HCV patients have autoantibodies typical of AIH-II (Table 1). The molecular mimicry between viral proteins and the host’s antigens has been postulated to be the mechanism by which anti-LKM antibodies are generated (33).
Genetic expression studies done on liver biopsies obtained from pediatric patients with AIH-I have demonstrated the over-expression of primary transcripts of IFN-γ, IL-12p40 (a 40 kDa polypeptide), IL-12Rβ2 (the β2 subunit of the IL-12 receptor), IL-18, TGFβ1, IL-4, and Vα24 (34). The increase in the expression of these molecules, some of which were non-detectable in normal livers, has made it possible to infer their role in the mechanisms of liver injury. It is known that IL-2 is capable of inducing the synthesis of the IL-12 receptor chains in naïve T cells. Nevertheless, the rise in the expression of this receptor is dependent on the presence of IL-12, which is released by several antigen-presenting cells. Furthermore, the IL-12-dependent production of IFN-γ is responsible for the maintenance of high levels of IL-12Rβ2 (35, 36), a molecule controlling the differentiation of the Th1 phenotype of T cells (37). One of the effects of the IL-12 is to enhance the expression of the IL-18R present in IFN-γ-producing cells thus attaining a synergistic response between IL-12 and IL-18 to produce IFN-γ (38). The high expression levels of IFN-γ, IL-12p40, and IL-12Rβ2 found in liver biopsies of AIH-I patients is evidence of the role of a Th1-skewed response in this disease.
Apart from the classical Th1 and Th2 subsets, an effector T cell subset named Th17 has also been described. Th17 cells are characterized by the production of interleukin-17A (IL- 17A), IL-17F, IL-22, IL-21, IL-6, and tumor necrosis factor α (TNF-α) (39). Although the differentiation and maturation of human Th17 cells is still a matter of controversy, transforming growth factor β (TGF-β), IL-6, IL-21, IL-1β, and IL-23 have been postulated to induce the human Th17 cell lineage (40). IL-23, a heterodimer of the IL-12 family, composed of the p19 and p40 subunits, appears to be essential to maintain and expand Th17 effectors. Meanwhile, IL-27, which is composed of the p28 and EBI3 subunits, suppresses the Th17 differentiation and IL-17 production and promotes Th1 differentiation (41). The presence of Th17 cells has also been reported in inflamed human tissues from patients suffering from a variety of inflammatory and autoimmune disorders (42-44). Only circumstantial signs of Th17 effector functions were found in liver biopsies of patients with AIH-I as deduced from the limited production of IL-17A and IL-17F transcripts (45).
Regulatory T cells in AIH-IN
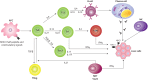
In addition to the Th1, Th2, and Th17 subsets, the functional quartet of CD4+ effectors also includes Tregs constitutively expressing the IL-2 receptor α-chain CD25 and the forkhead/winged helix (FoxP3) master regulatory transcription factor which is highly specific to the CD4+CD25high Treg lineage (46-48). Some studies carried out in mainly pediatric patients have addressed the possible impairment of Treg functions in AIH. These studies have shown that CD4+CD25+ Treg cells in peripheral blood seemed to feature both reduced frequencies and impaired suppressive functions (49, 50). Moreover, earlier studies have suggested that peripheral lymphocytes in autoimmune hepatitis exhibit increased immunosuppressive capability (51, 52). In these studies, however, Treg cells were defined only by staining for CD4 and CD25 since, at that time, it was not possible to distinguish human Tregs from activated effector T cells. In humans, a clear distinction of Tregs from activated conventional effector T cells is difficult since the Treg markers CD25 and Foxp3 can also be expressed transiently by activated effector T cells (53). More recently, a combination of more accurate methods has been developed. Those studies include the methylation status of the FOXP3 locus to search for demethylated FOXP3 TSDR (Treg-specific demethylated region) (54); the analysis of the expression of CD127, which is low in Treg cells and high in activated effector T cells (55); and the analysis of CD39, which seems to be one of the molecules that mediates the suppressive function of Treg cells by producing inhibitory adenosine (56). The results of these studies clearly established that Treg cells belonging to AIH patients are fully functional; Treg frequency is not decreased in AIH patients but rather seems to correlate with disease activity. Another important aspect of this study was the finding that the Treg frequency in the blood of AIH patients is associated with pharmacological immunosuppression, and therefore, not with the remission per se (30).
The complex cell and cytokine environment of the inflamed liver may have profound effects on Treg differentiation, stability, and function as well as alter the susceptibility of effector T cells to suppression. In addition to the above mentioned interaction between Tregs and dendritic cells, local cytokines could reprogram Tregs to express either Tbet which would lead to a Th1 phenotype (57) or RORc and a Th-17 phenotype (58). We have described a higher Vα24, IFN-γ, FoxP3, p28, IL-12p40, and IL-21 expression at the moment of diagnosis as well as a positive correlation between IL-21 and aminotransferase levels. It is interesting that only IFN-γ and FoxP3 decreased during biochemical remission following immunosuppressive treatment (AIH-Ir). An “AIH-I phenotype” described as “high Vα24, IFN-γ, and FoxP3 expression” was observed in a low percentage of AIH-Ir children but not in healthy, age-matched controls. Overall, the presence of a local deregulation of the innate (i.e., NKT cells) and adaptive arms of the immune responses (i.e., Th1, Treg cells) was described (45). In addition, IL-21 was highlighted as a mediator of liver injury. Together with Th17 cells, iNKT cells are a source of IL-21. Since a seemingly limited presence of a Th17 subpopulation and an amplified expression of Vα24+ transcripts were detected, it has been suggested that iNKT cells may be the main source for IL-21 in the livers of patients with AIH-I.
IL-21, together with IL-2 and IL-15 are known to induce the proliferation of NKT cells (59). Apart from its role as a differentiation factor for Th17 cells from naïve cells, the inhibitory role of IL-21 on the differentiation of Treg FoxP3+ cells from the same precursor cells as those of Th17 cells is also noteworthy. The same is true of the ability of this cytokine to confer CD4+ cell resistance to the suppressing effect of Treg cells (60). The high levels of IL-21 found in the AIH-I inflammatory scenario but not in other inflammatory liver diseases (e.g. non-alcoholic fatty liver disease and HCV infection) make it possible to suggest that the immunoregulation of Treg and NKT cell subsets would constitute one of the most probable roles of IL-21 in the pathogenesis of AIH-I. The latter hypothesis implies that the high levels of FoxP3 found in the livers of pediatric patients reflects an increase in the number of Treg cells and that the prevailing function of IL-21 would be to confer resistance to the suppressant effector of Treg cells. Thus, and in spite of their local increase, FoxP3+ cells would not be able to suppress the autoimmune response. An increase in the recruitment of Treg cells from the peripheral compartment to the liver would constitute an attempt to suppress the local immune response. Treatment of AIH will fail if adoptively transferred Treg cells are inhibited or pushed into an effector differentiation pathway by local factors in the liver. Consequently, the need to shift the balance from damage towards resolution of inflammation emphasizes the importance of understanding the effects of the local inflammatory microenvironment (61). The fact that Treg cells are detected at high frequencies in the liver of patients with AIH suggests that suppression of function locally rather than a basic defect in Treg generation or function underlies the persistence of chronic autoimmune hepatitis.
Figure 2 depicts the interaction between genetic susceptibility, an antigenic peptide, and the production of different cytokines favoring the autoimmune attack (62).
Oxidative stress
Reactive oxygen species (ROS) are highly reactive small molecules that are generated as by-products of oxygen metabolism during intracellular physiological processes. The production of ROS is finely regulated by enzymatic systems and reducing species that are able to clear these intermediates. Under physiological conditions, they play a crucial role in cell signaling pathways. However, when an imbalance between pro-oxidant and homeostatic anti-oxidant mechanisms occurs, a status known as oxidative stress arises. Under these circumstances, the ROS are cytotoxic due to their ability to react with most macromolecules. Thus, they induce the inactivation of enzymes, DNA damage, produce post-translational protein modifications, lipoperoxidation, and induce cell death (63).
Oxidative stress is not disease-specific, and its demonstration in animal models and humans with diverse liver diseases suggests that it has a complementary role in the pathogenesis of possibly all forms of acute and chronic hepatitis. It has been demonstrated that the livers of primary biliary chirrosis and AIH patients possess a higher expression of inducible nitric oxide synthase as well as nitrotyrosine (a result of an irreversible reaction between oxidant peroxynitrites and cell proteins) accumulation. It is likely that these findings suggest that oxidative damage to cell structures may play a role in the pathogenesis of AIH (64) and that complementary antioxidant therapy could be of value in selected patients (65).
Animal models
Due to the fact that AIH is often recognized during the late course of disease, it is difficult to know the immunological mechanisms responsible for initiation of the disease. Current AIH models have been helpful for understanding and modulating liver immune responses, but are not suitable for studying mechanisms in chronic AIH or for developing new therapies. The experimental AIH-I murine model induced by Concanavalin A has been useful for demonstrating that NKT cells are necessary for the development of the disease. Furthermore, the production of IL-4 by these cells plays an important role since this cytokine increases the autocrine cytotoxic activity by inducing the expression of granzime B and FasL (66).
While transgenic AIH models deal with short-term hepatitis, models with natural antigens are either self-limited or have unknown target antigens. Therefore, novel animal models with defined onset of AIH and a standard course of the disease are essential for a better understanding of the disease and its pathophysiology. To obtain a preclinical platform for new therapeutic approaches or to be able to prevent the onset of AIH, a positive impact of conventional, standard, therapeutic interventions in the model would be helpful. For decades, AIH research has lacked such a reliable preclinical model with chronic immune response against the liver. Initial results in breaking tolerance to hepatocytes have only led to mild and transient hepatitis. Transgenic models were helpful in understanding different aspects of hepatic immune regulation. At present, the fate of T cells, especially CD8+ T cells, is the focus of research. Ignorance, anergy, deletion, or TCR downregulation of T cells especially are the mechanisms of tolerance against hepatic antigens. Furthermore, the importance of professional antigen-presenting cells, and particularly liver sinusoidal cells, in liver tolerance has been demonstrated in many studies. Other models have shown the mechanism of interaction of adaptive and innate immune cells in the liver. Recently, approaches have been made to establish AIH models reflecting the situation in AIH patients. This will allow new studies in the field to be done and will provide an opportunity to study the onset and pathophysiology of AIH. Furthermore, new options for therapeutic approaches will be tried out in these models, and options to prevent onset of disease may be shown (67).
Conclusions
Due to the high incidence in many geographical areas as well as to the high percentage of liver transplantations due to autoimmune hepatitis, it is important to encourage more insightful studies to be done. Experimental evidence allows us to conclude that the liver damage in AIH-I would be a consequence of not only a Th-1-biased response but also the deregulation of hepatic Treg cell function. The molecular species responsible for the development of the different types of this disease in both children and adults remains to be determined. Knowing the autoantigen that triggers these processes is of paramount importance for it would broaden the frontiers for the development of more accurate methodologies of diagnosis, prognosis, and treatment of this pathology.
Follow-up studies are necessary to elucidate the mechanisms of the immune response that remain active despite the treatment and whether those pathways would be amenable to further therapeutic intervention.
References
- 1.
- Mehal WZ, Juedes AE, Crispe IN. Selective retention of activatedCD8+ T cells by the normal liver. J Immunol. 1999;163:3202–10. [PubMed: 10477588]
- 2.
- Oo YH, Weston CJ, Lalor PF, et al. Distinct roles for CCR4 and CXCR3 in the recruitment and positioning of regulatory T cells in the inflamed human liver. J Immunol. 2010;184:2886–98. [PubMed: 20164417]
- 3.
- Curbishley SM, Eksteen B, Gladue RP, Lalor P, Adams DH. CXCR 3 activationpromotes lymphocyte transendothelial migration across human hepatic endothelium under fluid flow. Am J Pathol. 2005;167:887–99. [PMC free article: PMC1698725] [PubMed: 16127166]
- 4.
- Leithauser WMJ, Reimann J. The hepatic immune system. Crit RevImmunol. 2002;22:47–103. [PubMed: 12186188]
- 5.
- Knolle PA, Gerken G. Local control of the immune response in the liver. Immunol Rev. 2000;174:21–34. [PubMed: 10807504]
- 6.
- Doherty DG, Norris S, Madrigal-Estebas L, et al. The human liver contains multiple populations of NK Cells, T Cells, and CD3+CD56+ Natural T cells with distinct citotoxic activities and Th1, Th2, and Th0 cytokine secretion patterns. J Immunol. 1999;163:2314–21. [PubMed: 10438977]
- 7.
- Porcelli EM, Furman M, García J, Balk S. CD161 (NKR-P1A) costimulation of CD1d-dependent activation of human T cells expressing invariant Va24JaQ T cell receptor a cAIHns. J Exp Med. 1998;188:867–76. [PMC free article: PMC2213391] [PubMed: 9730888]
- 8.
- Cuarterolo M, Dávila M, Roig A, Mondiglio C, Sassone A, Ciocca M. Hepatitis autoinmune 8 años de experiencia. Medicina Infantil. 1995;2:237–41.
- 9.
- Palioura S, Sherrer RL, Steitz TA, Soll D, Simonovic M. The human SepSecStRNASec complex reveals the mechanism of selenocysteine formation. Science. 2009;325:321–5. [PMC free article: PMC2857584] [PubMed: 19608919]
- 10.
- Zachou K, Rigopoulou E, Dalekos GN. Autoantibodies and autoantigens in autoimmune hepatitis: important tools in clinical practice and to study pathogenesis of the disease. J Autoimm Dis. 2004;1:2. [PMC free article: PMC544946] [PubMed: 15679907]
- 11.
- Hurlburt KJ, McMahon BJ, Deubner H, Hsu-Trawinski B, Williams JL, Kowdley KV. Prevalence of autoimmune liver disease in Alaska Natives. Am J Gastroenterol. 2002;97:2402–7. [PubMed: 12358264]
- 12.
- Whalley S, Puvanachandra P, Desai A, Kennedy H. Hepatology outpatient service provision in secondary care: a study of liver disease incidence and resource costs. Clin Med. 2007;7:119–24. [PMC free article: PMC4951824] [PubMed: 17491498]
- 13.
- Diamantis I, Boumpas DT. Autoimmune hepatitis: evolving concepts. Autoimm Rev. 2004;3:207–14. [PubMed: 15110233]
- 14.
- Czaja AJ, Manns MP, McFarlane IG, Hoofnagle JH. Autoimmune hepatitis: The investigational and clinical challenges. Hepatology. 2000;31:1194–200. [PubMed: 10796898]
- 15.
- Obermayer-Straub P, Strannburg CP, Manns MP. Autoimmune hepatitis. J Hepatol. 2000;32:181–97. [PubMed: 10728804]
- 16.
- Mieli-Vergani G, Vergani D. Immunological liver disease in children. Semin Liver Dis. 1998;18:271–9. [PubMed: 9773427]
- 17.
- Krawitt EL. Clinical features and management of autoimmune hepatitis. World J Gastroenterol. 2008;14:3301–5. [PMC free article: PMC2716584] [PubMed: 18528927]
- 18.
- Knodell R, Ishak KG, Black WC, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1:431–5. [PubMed: 7308988]
- 19.
- Desmet VJ, Gerber M, Hoofnagle JH, Manns M, Scheuer PJ. Classification of chronic hepatitis: diagnosis, grading and staging. Hepatology. 1984;19:1513–20. [PubMed: 8188183]
- 20.
- Johnson PJ, McFarlane IG. Meeting report: International Autoimmune Hepatitis Group. Hepatology. 1993;18:998–1005. [PubMed: 8406375]
- 21.
- Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group report: review of criteria for diagnosis of autoimmnune hepatitis. J Hepatol. 1999;31:929–38. [PubMed: 10580593]
- 22.
- Hennes EM, Zeniya M, Czaja AJ, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology. 2008;48:169–76. [PubMed: 18537184]
- 23.
- Fainboim L, Marcos Y, Pando M, et al. Chronic active autoimmune hepatitis in children: Strong association with a particular HLA-DR6 (HLA-DRB1*1301) haplotype. Hum Immunol. 1994;41:146–50. [PubMed: 7860360]
- 24.
- Pando M, Larriba J, Fernández G, et al. Pediatric and adult forms of type I autoimmune hepatitis in Argentina: evidence for differential genetic predisposition. Hepatology. 1999;30:1374–80. [PubMed: 10573514]
- 25.
- Marcos Y, Fainboim HA, Capucchio M, et al. Two-locus involvement in the association of human leukocyte antigen with the extrahepatic manifestations of autoimmune chronic active hepatitis. Hepatology. 1994;19:1371–4. [PubMed: 8188167]
- 26.
- Gregorio GV, Portmann B, Reid F, et al. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology. 1997;25:541–7. [PubMed: 9049195]
- 27.
- Johnson PJ, McFarlane IG, Williams R. Azathioprine for long-term maintenance of remission in autoimmune hepatitis. N EnglJ Med. 1995;333:958–63. [PubMed: 7666914]
- 28.
- Alvarez F, Ciocca M, Canero-Velasco C, et al. Short term cyclosporine induces a remission of autoimmune hepatitis in children. J Hepatol. 1999;30:222–7. [PubMed: 10068099]
- 29.
- Richardson PD, James PD, Ryder SD. Mycophenolate mofetil for maintenance of remission in autoimmune hepatitis in patients resistant to or intolerant of azathioprine. J Hepatol. 2000;33:371–5. [PubMed: 11019991]
- 30.
- Peiseler M, Sebode M, Franke B, et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J Hepatol. 2012;57:125–32. [PubMed: 22425700]
- 31.
- Czaja AJ. Diagnosis, pathogenesis and treatment of autoimmune hepatitis after liver transplantation. Dig Dis Sci. 2012;57:2248–66. [PubMed: 22562533]
- 32.
- Fainboim L, Cañero Velazco MC, Marcos Y, et al. Protracted, but not acute, Hepatitis A virus infection is strongly associated with HLADRB1* 1301, a marker for pediatric autoimmune hepatitis. Hepatology. 2001;36:1512–7. [PubMed: 11391541]
- 33.
- Strassburg CP, Vogel A, Manns MP. Autoimmunity and hepatitis C. Autoimmun Rev. 2003;2:322–31. [PubMed: 14550873]
- 34.
- Cherñavsky AC, Paladino N, Rubio AE, et al. Simultaneous expression of Th1 cytokines and IL-4 conferes severe characteristics to type I autoimmune hepatitis in children. Human Immunol. 2004;65:683–91. [PubMed: 15301856]
- 35.
- Zhang M, Gong J, Presky DH, Xue W, Barnes PF. Expression of the IL-12 receptor beta 1 and beta 2 subunits in human tuberculosis. J Immunol. 1999;162:2441–7. [PubMed: 9973527]
- 36.
- Chang JT, Segal BM, Shevach EM. Role of costimulation in the induction of the IL-12/IL-12 receptor pathway and the development of autoimmunity. J Immunol. 2000;164:100–6. [PubMed: 10604999]
- 37.
- Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–24. [PMC free article: PMC2196166] [PubMed: 9120387]
- 38.
- Okamura H, Kashiwamura S, Tsutsui H, Yoshimoto T, Nakanishi K. Regulation of interferon-gamma production by IL-12 and IL-18. Curr Opin Immunol. 1998;10:259–64. [PubMed: 9638361]
- 39.
- Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67. [PMC free article: PMC3424508] [PubMed: 18400188]
- 40.
- Sallusto F, Lanzavecchia A. Human Th17 cells in infection and autoimmunity. Microbes Infect. 2009;5:620–4. [PubMed: 19371794]
- 41.
- Diveu C, McGeachy MJ, Cua DJ. Cytokines that regulate Autoimmunity. Curr Opin Immunol. 2008;20:63–8. [PubMed: 18834938]
- 42.
- Singh R, Aggarwal A, Misra R. Th1/Th17 cytokine profiles in patients with reactive arthritis/undifferentiated spondyloarthropathy. J Rheumatol. 2007;34:2285–90. [PubMed: 17937463]
- 43.
- Pene J, Chevalier S, Preisser L, et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol. 2008;180:7423–30. [PubMed: 18490742]
- 44.
- Nistala K, Moncrieffe H, Newton KR, Varsani H, Hunter P, Wedderburn LR. Interleukin-17-producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheum. 2008;58:875–87. [PMC free article: PMC2675006] [PubMed: 18311821]
- 45.
- Ferreyra Solari NE, Galoppo C, Cuarterolo M, et al. The simultaneous high expression of Vα24, IFN-γ and FoxP3 characterizes the liver of children with Type I Autoimmune Hepatitis. Clin Immunol. 2010;137:396–405. [PubMed: 20884299]
- 46.
- Shevach EM. CD4+CD25+ suppressor T-cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. [PubMed: 12093005]
- 47.
- Fontenot JD, Gavin MA. FoxP3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. [PubMed: 12612578]
- 48.
- Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–42. [PubMed: 12612581]
- 49.
- Longhi MS, Ma Y, Bogdanos DP, Cheeseman P, Mieli-Vergani G, Vergani D. Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol. 2004;41:31–7. [PubMed: 15246204]
- 50.
- Longhi MS, Hussain MJ, Mitry RR, et al. Functional study of CD4+CD25+ regulatory T cells in health and autoimmune hepatitis. J Immunol. 2006;176:4484–91. [PubMed: 16547287]
- 51.
- Lohse AW, Kögel M, Meyer zum Büschenfelde KH. Evidence for spontaneous immunosuppression in autoimmune hepatitis. Hepatology. 1995;22:381–8. [PubMed: 7635404]
- 52.
- Lohse AW, Meyer zum Büschenfelde KH. Remission of experimental autoimmune hepatitis is associated with antigen-specific and non-specific immunosuppression. Clin Exp Immunol. 1993;94:163–7. [PMC free article: PMC1534357] [PubMed: 8403500]
- 53.
- Wang J, Ioan-Facsinay A, van der Voort EIH, Huizinga TWJ, Toes REM. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol. 2007;37:129–38. [PubMed: 17154262]
- 54.
- Wieczorek G, Asemissen A, Model F, et al. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res. 2009;69:599–608. [PubMed: 19147574]
- 55.
- Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ Treg cells. J Exp Med. 2006;203:1701–11. [PMC free article: PMC2118339] [PubMed: 16818678]
- 56.
- Ernst PB, Garrison JC, Thompson LF. Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol. 2010;185:1993–8. [PMC free article: PMC3036969] [PubMed: 20686167]
- 57.
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. [PMC free article: PMC2712126] [PubMed: 19412181]
- 58.
- Voo KS, Wang YH, Santori FR, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci. USA. 2009;106:4793–8. [PMC free article: PMC2653560] [PubMed: 19273860]
- 59.
- Davis ID, Skak K, Smyth MJ, Kristjansen PE, Miller DM, Sivakumar PV. Interleukin-21 Signaling: Functions in Cancer and Autoimmunity. Clin Cancer Res. 2007;13:6926–32. [PubMed: 18056166]
- 60.
- Clough LE, Wang CJ, Schmidt EM, et al. Release from regulatory T cell-mediated suppression during the onset of tissue-specific autoimmunity is associated with elevated IL-21. J Immunol. 2008;180:5393–401. [PubMed: 18390721]
- 61.
- Oo YH, Adams DH. Regulatory T cells and autoimmune hepatitis: Defective cells or a hostile environment? J Hepatol. 2012;57:6–8. [PubMed: 22522382]
- 62.
- Liberal R, Grant CR, Mieli-Vergani G, Vergani D. Autoimmune hepatitis: A comprehensive review. J Autoimmun. 2013;41:126–39. [PubMed: 23218932]
- 63.
- Marí M, Colell A, Morales A, von Montfort C, Garcia-Ruiz C, Fernández-Checa JC. Redox control of liver function in health and disease. Antioxid Redox Signal. 2010;12:1295–331. [PMC free article: PMC2864660] [PubMed: 19803748]
- 64.
- Sanz-Cameno P, Medina J, García-Buey L, et al. Enhanced intrahepatic inducible nitric oxide synthase expression and nitrotyrosine accumulation in primary biliary cirrhosis and autoimmune hepatitis. J Hepatol. 2002;37:723–9. [PubMed: 12445411]
- 65.
- Moreno-Otero R. May Oxidative Stress Contribute to Autoimmune Hepatitis Pathogenesis, and Can Antioxidants Be of Value as Adjuvant Therapy for Refractory Patients? Dig Dis Sci. Dig Dis Sci. 2013;58:1440–1. [PubMed: 23504353]
- 66.
- Toyabe S, Seki S, Iiai T, et al. Requirement of IL-4 and liver NK1+ T cells for Concenavalin A-induced hepatic injury in mice. J Immunol. 1997;159:1537–42. [PubMed: 9233653]
- 67.
- Hardtke-Wolenski M, Noyan F, Jaeckel E. Requirements and challenges of a preclinical autoimmune hepatitis mouse model. Dig Dis. 2011;29:402–10. [PubMed: 21894011]
Publication Details
Author Information and Affiliations
Authors
Nazarena Ferreyra Solari and Alejandra Claudia Cherñavsky.Copyright
Publisher
El Rosario University Press, Bogota (Colombia)
NLM Citation
Ferreyra Solari N, Claudia Cherñavsky A. Autoimmune hepatitis. In: Anaya JM, Shoenfeld Y, Rojas-Villarraga A, et al., editors. Autoimmunity: From Bench to Bedside [Internet]. Bogota (Colombia): El Rosario University Press; 2013 Jul 18. Chapter 31.
%20present%20the%20antigen%20to%20NKT%20cells%20within%20the%20context%20of%20CD1%20HLA%20complex%20through%20the%20TCR%20V%003B124.%20During%20antigen%20presentation%2C%20DC%20release%20IL-12%20which%201-%20promotes%20NKT%20cytotoxicity%20and%20regulates%20the%20number%20of%20DC%20(negative%20regulation)%20and%202-%20promotes%20the%20expression%20of%20Fas%20lingand%20(FasL)%20on%20the%20surface%20of%20NKT%20cells.%20The%20latter%20ones%20thus%20acquire%20a%20cytotoxic%20potential%20on%20hepatocytes%20via%20Fas%2FFasL%20and%20granzime%2Fperforin%20pathways.%20IL-12%20is%2C%20in%20turn%2C%20the%20main%20stimulating%20cytokine%20for%20the%20differentiation%20of%20type%201%20helper%20T%20cells%20(Th1%20cells).&p=BOOKS&id=459451_autoimmunehepatitisf1.jpg "Click on image to zoom")
