Introduction
Autoimmune thyroid diseases (AITD) are the most prevalent organ-specific autoimmune diseases (ADs) and affect 2 - 5% of the population (1) with great variability between genders (i.e., women 5–15% and men 1–5%) (2). AITD include Graves’ Disease (GD) and Hashimoto Thyroiditis (HT), among others. HT and GD are the major causes of hypothyroidism and hyperthyroidism, respectively (3). They reflect the loss of immunological tolerance and share the presence of cell and humoral immune response against antigens from the thyroid gland with reactive infiltration of T cells and B cells, autoantibody generation and, subsequently, the development of clinical manifestations (4, 5).
The lymphocytic infiltration causes tissue damage and alters the function of the thyroid gland. The injury is caused when the autoantibodies and/or sensitized T cells react with the thyroid cells causing the inflammatory reaction and, in some cases, cell lysis (6). Generally, while T lymphocytes are the main cell type infiltrating the gland in HT, a B cell response predominates and determines the presence of GD (7).
As with other ADs, there is a multifactorial etiology with a complex interaction of environmental factors in genetically susceptible individuals (8) (Figure 1). Some of these genes are specific for GD and HT while others are mutual for both diseases, which indicates a genetic predisposition shared in these processes together. Candidate genes include immunoregulators [e.g., human leukocyte antigen (HLA), cytotoxic T lymphocyte antigen-4 (CTLA-4)] and others specific to the thyroid (e.g., TSH receptors, thyroglobulin, etc.). The main environmental factors are smoking, stress, and iodine consumption (6).

AITD is the most hierarchical AD coexisting in the same individual [e.g., polyautoimmunity and multiple autoimmune syndromes (MAS)] (5,9). In fact, rheumatologists may be consulted for clinical manifestations of AITD that mimic several symptoms of other ADs (3).
Genetics
Genetics play a key role in the pathogenesis of AITD. In fact, a number of immune-related genes have been implicated in genetic susceptibility to AITD. For instance, it is calculated that up to 80% of the susceptibility for the development of GD is secondary to the presence of determined genes (10). These genes include both immunological-synapse genes [e.g., HLA-DR, CTLA-4, CD40, and the protein tyrosine phosphatase, nonreceptor type 22 (PTPN22) gene] and regulatory T–cell genes (e.g., FOXP3, CD25) (3).
The association between HLA alleles and AITD is well known, but the primary etiological variant in this region is still uncertain (1). Thus, the pathological findings for both GD and HT are similar, and are associated with particular HLA-B and HLA-DR, suggesting that inherited risk factors are important in the development of these entities (11,12). HLA-DR locus plays an important role since HLA-DR3 is present in up to 55% of the patients with GD compared to the 30% prevalence in the general population (4). In fact, it has been discovered that HLA-DR3 and HLA-DR5 are linked to HT and provide a greater risk for the disease (4). Moreover, Ban et al. (11) identified arginine at position 74 of the HLA-DRβ1 (DRβ-Arg74) as the critical DR amino acid (a.a) conferring susceptibility to GD, and glutamine at position 74 as a protective a.a. Regarding HT, Menconi et al. (13,14) reported similar findings for this disease. They were able to identify the thyroglobulin peptides that could be presented by HLA-DR pockets containing arginine at position beta 74 to T cells and, therefore, initiate the autoimmune process (14). There are, in turn, studies that suggest alleles from HLA-DQ might be genetic markers that confer resistance to the development of AITD (15). Likewise, atrophic and subacute thyroiditis are strongly associated with HLA-B35 in many ethnic groups while painless thyroiditis is associated with HLA-DR3 (11,12,16).
There are associations with a number of immune-related genes, other than HLA genes, which have also been found in many other ADs and presumably underpin the inherited susceptibility to autoimmunity. Polymorphism in certain alleles of CTLA-4 predispose to GD and HT (17–21). For instance, in a study of 379 patients with GD in the United Kingdom, 42% had a particular allele (G allele) of the CTLA-4 gene compared to 32% of the controls (20). This genetic abnormality also stimulates higher levels of thyroid-specific autoantibodies and clinical thyroid disease when interacting with other loci (4,20–22). Currently, both HLA-DR3 and CTLA-4 are considered to be the main genes associated with the development of AITD.
CD40 is expressed on thyroid follicular cells and on B cells. Polymorphisms of this gene are associated with a 20–30% increase in translation of CD40 mRNA transcripts in patients with AITDs (23).
Polymorphisms of the PTPN22 gene, which encodes a negative regulator of T cell activation, have been associated with AITD and other ADs. A gain of function trait has been identified, but its role in autoimmunity is uncertain (24,25). Finally, abnormalities in the FOXP3 gene (differentiation of T cells into natural Treg cells) have been associated with juvenile GD (26). Changes in the CD25 gene region have also been associated with GD (27).
Epigenetics
Although the link between susceptibility genes and environmental triggers is clear, the mechanisms by which gene variants interact with environmental factors to cause autoimmunity are still unclear. Recent data suggest that epigenetic mechanisms might underlie genetics. Their effects on gene expression have been broadened to include non-DNA-sequence-encoded effects on gene expression that are mitotically stable (28). Epigenetic modulation of gene expression can occur through alterations in DNA methylation, histone modification patterns (e.g., acetylation, deacetylation, and methylation), and RNA interference through microRNAs. Recently, interferon-a (IFN-α) was shown to induce alterations in thyroglobulin (Tg) gene expression through epigenetic changes in histone modifications (29). Since IFN-α is secreted locally during viral infections, this could be an attractive mechanism by which infections can trigger AITD (30). Another epigenetic phenomenon described in the pathogenesis of AITD is the X chromosome inactivation. The degree of X chromosome inactivation is an important contributor to the increased risk females have of developing AITD, as demonstrated by Yin et al. (31).
Environmental factors
There are a number of environmental factors associated with the occurrence of AITD such as low birth weight, iodine excess, selenium deficiency, use of anovulatories, parity, stress, smoking, allergy, radiation exposure, viral or bacterial infections, and fetal microchimerism (32).
Smoking
This habit, as well as cessation, are the main risk factor for AITD (e.g., GD) (33). The increased risk in the onset of AITD with cessation of smoking may be useful in monitoring susceptible patients who stop smoking for the myriad health benefits. For example, cessation of smoking may be associated with weight gain, and hypothyroidism should be considered a possible cause. Cigarette smoke contains cyanide, which is metabolized to thiocyanate and can interfere with iodine concentration in the thyroid and in the lactating breast (34).
Infection
There is evidence suggesting the involvement of infection in the development of this pathology. However, not only pathogenic but also non-pathogenic microorganisms induce pro-inflammatory or regulatory immune responses within the host (e.g., commensal microbiota) (35). Data regarding the role of B. burgdorferi and Y. enterocolitica as triggers of thyroid autoimmunity remain inconclusive. Some reports have suggested molecular mimicry that may explain an association between both of these pathogens and AITD. Additionally, significant homologies had been found for 16 Borrelia spp. proteins and 19 Yersinia spp. proteins. The number of motif copies was found to be greater in the regions of homology of thyroid autoantigens with Yersinia spp. than those with Borrelia spp., with the most common being HLA-DR3, DR-4, and DR-7 (28,36). Other microorganisms implicated in the pathogenesis of AITD include H.pylori, Coxsackie virus, Hepatitis C virus, and retroviruses (28).
Iodine
Although it is essential for normal thyroid function, iodine is one of the most important precipitants of thyroid dysfunction. Thus, while mild iodine deficiency is associated with a lower prevalence of HT, excessive intake is associated with a higher prevalence. Potential mechanisms by which iodine can induce autoimmunity include direct stimulation of immune responses to the thyroid, increased immunogenicity of highly iodinated Tg, and direct toxic effects of iodine on thyrocytes via the generation of reactive oxygen species. However, thyroid autoimmunity associated with iodine might be a transient phenomenom (37-39). Kahaly et al. (37) followed a group of patients with endemic goiter that received iodine for 6 months and another group that received T4. High titers of thyroid autoantibodies were found in 19% of the patients receiving iodine. After iodine was withdrawn, antibodies levels decreased significantly, and after a 4-year follow-up, these levels had normalized in four out of the six patients (66%).
Radiation exposure
Radiation is perhaps the best characterized environmental exposure linked to effects on the thyroid. The most common thyroid manifestation of radiation is hypofunction due to direct destruction of the gland, but stimulation of thyroid autoantibodies may be another mechanism for both hypothyroidism and hyperthyroidism (28). AITD has been linked to therapeutic medical radiation (40) as well as environmental radiation exposure (41). In a study in which 160 people were occupationally exposed to ionizing radiation, 10% of the subjects met criteria for AITD compared to 3.4% of those without exposure. Subjects with more than five years of exposure were considered to be at higher risk. It will be important in future studies to determine if there is a dose-response relationship to radiation exposure (42).
Environmental toxins
Many environmental pollutants have been shown to be toxic to thyroid cells and promote the onset of AITD (43). For instance, a high prevalence of hypothyroidism was observed in individuals exposed to polybrominated biphenyls with an associated elevation in thyroperoxidase antibodies (TPOAb) and Tg antibodies (TgAb). Bisphenol A, commonly used to manufacture plastic products, may bind to the TSH receptor (TSHR) and act as an antagonist to thriiodothyroinine (T3) thus, inhibiting its transcriptional activity (39,43,44).
Medications
Several medications may play a role in the development of AITD. IFN-α, IL-2, lithium, amiodarone, and highly active antiretroviral therapy are the agents most commonly associated with thyroid dysfunction (28,39). For most of these medications, patients at greatest risk of developing AITD are those with previous thyroid autoantibody positivity (28,39,43). Some medications, such as lithium, may not trigger autoimmunity but accelerate the autoimmune process by interfering with thyroid hormone synthesis. Thyroid function testing and measurement of TPOAb should be considered before beginning these medications on patients (32).
Stress
Various types of stress had been linked to GD. The postulated mechanisms include induction of immune suppression by non-antigen-specific mechanisms, perhaps due to the effects of cortisol or corticotropin-releasing hormone on immune cells followed by immune hyperactivity leading to AITD. A mechanism like that might be operative in postpartum thyroiditis. However, there is currently no evidence linking stress to AITD probably because the triggering event occurred years before thyroid gland damage (28,39,43).
Sex steroids and pregnancy
HT is most common in women than in men, suggesting a role for sex steroids. However, older women may be more likely to have HT than younger women, implying that the presence or absence of estrogen may not be the important factor (45). Another possible explanation for female predominance is skewed X-chromosome inactivation that was found in 34% of female twins with AITD and only 11% of controls (31).
During pregnancy, the transition from immune suppression to release from suppression is associated with the onset of a number of ADs (32). Additionally, there is a marked increase in CD4+CD25+ regulatory T cells which leads to diminished function of activated T cells and B cells, causing the rebound from this immunosuppression. Pregnancy immunosuppression is associated with a switch to Th2 cells and a shift in cytokine profiles. Likewise, progesterone produced by the placenta affects cytokine profiles across the whole maternal immune system (46).
Autoantibodies
The TPOAb and TgAb are the most common thyroid autoantibodies present in patients with AITD and are associated with complement - mediated cytotoxicity against thyrocytes (49,50). In fact, the absence of these autoantibodies could exclude a diagnosis of AITD, which means that these tests have high negative predictive value (51). The TSHR antibody (TRAb) is identified in patients with GD (52). There are other less common autoantibodies such as those against the sodium/iodine symporter (NIS) and pendrin, but their clinical utility is limited (53,54) (Table 1 and Figure 2).
Table 1
Antibodies described in AITD, frequency of presentation and detection method.

These autoantibodies may be present in individuals without clinical or laboratory evidence of thyroid dysfunction (55,56). Presence of antibodies in the absence of disease has been related to a greater risk of developing an AITD in the future, especially postpartum thyroiditis, as seen in women with recurrent abortion (57). These antibodies has also been linked to an increased probability of thyroid dysfunction secondary to the use of medication that can potentially alter the thyrocyte function (58). The aging process is, likewise, associated with a greater presence of these antibodies. Moreover, these antibodies have been detected in up to 50% of women with a first degree relative with AITD and in 30% of men in the same situation and thus, suggests a dominant inheritance pattern (19).
Even though the recognition of these antibodies does not generate a direct intervention against them, they allow us to predict a clinical outcome or therapeutic response. It is known that the complete ablation of the thyroid tissue results in complete disappearance of these antibodies, in accordance with the theory that the production of these antibodies is triggered by the presence of autoantigens (59). Another clinical utility of these antibodies is related to thyroid ophthalmopathy. Patients that present with this clinical manifestation have higher titers of TRAb and lower levels of TPOAb (60).
Thyroglobulin antibodies
The Tg is the main component of thyroid follicular colloid (51) (Figure 2). It is a large glycoprotein dimer, synthesized by follicular cells, and secreted into the lumen (i.e., colloid) (61). Tg plays an essential role in the storage of iodine and synthesis of the thyroid hormones (51). Normally, no more than 25% of the Tg is iodinated (51). Along with posttranslational modifications (e.g., glycosylation), the extent of the iodination is the most important determinant of Tg immunogenicity. Highly iodinated Tg has been found to be more antigenic (51,62,63).
TgAb are primarily IgG with the main subclasses being IgG2, found in HT, and IgG4, which predominate in GD (64). TgAb is mainly used, together with TPOAb, for the diagnosis of HT. They are found in 50–90% of the patients with HT and less frequently in GD (i.e., 20–40%) (65). TgAb are also present in differentiated thyroid cancer, other ADs, and in up to 20% of the euthyroid population (51,66) (Table 1). Another clinical use of TgAb is in the follow - up of patients with differentiated thyroid cancer post-thyroidectomy and radioactive iodine ablation (51,67).
Thyroid peroxidase antibodies
The TPO is a heme containing oxidoreductase and a membrane-spanning glycoprotein located on the apical surface of thyrocytes (51,68). Its function is to catalyze the iodination of tyrosine residues of Tg to form monoiodotyrosine and diiodotyrosine (Figure 2) (51). The thyroid microsomal antibody was one of the first TPOAb identified (69).
There is circumstantial evidence that TPOAb are responsible for thyroid failure either by inhibiting the TPO or by causing direct damage to the thyrocytes (51). Most of TPOAb are in the IgG1 subclass, which is a complement activator. Moreover, TPOAb can bind through their Fc region to Natural killer cells that, in turn, cause cytotoxic damage to thyrocytes (51,70).
TPOAb are considered diagnostic of AITD (3). They are present in the majority of both HT (i.e., >90%) and GD (i.e., 40–70%) (3). The annual risk for progression to overt hypothyroidism in women with TPOAb is 2.1%, and this is correlated with the titer of the antibody, whether it is weakly, moderately, or strongly positive (23%, 33%, and 53%, respectively) (51). TPOAb also increases the risk of postpartum thyroiditis (discussed below) as the presence of these antibodies during the first trimester is associated with a probability of 30–52% for developing the disease (71). Like TgAb, TPOAb antibodies are found in other ADs and in the general population (51,55,72).
Thyroid - stimulating hormone receptor antibodies
The TSHR is a G-protein coupled receptor (73,74) and the main regulator of the thyroid gland. Signaling through the TSHR via the stimulation of cytosolic second messengers promotes the synthesis of thyroid hormones and the growth of the thyroid follicular cell (51).
The natural ligand of the TSHR is the TSH which binds to multiple sites in the extracellular domain (51). However, there are other hormones with the ability to bind to the receptor (e.g., luteinizing hormone and human chorionic gonadotropin) (51). This receptor is expressed predominantly on the surface of thyroid cells. Nevertheless, TSHR mRNA is also found in other tissues, e.g., adipocytes, cardiac muscle cells, pituitary cells, bone cells, and fibroblasts, though its exact function on these cells is not fully understood (75).
TRAb are usually part of the IgG1 subclass (51). The binding site of the TRAb is the region of the receptor to which TSH binds, which consists of a binding pocket-encompassing leucine-rich repeat region (76). They can be functionally classified into three categories: stimulating, blocking, and neutral.
Stimulating autoantibodies. These antibodies were first identified by their prolonged thyroid – stimulating activity when serum from GD patients was transferred into animals. Initially, this finding was called long – acting thyroid stimulators (77). Stimulating antibodies are those that bind to the TSHR and induce conformational changes that activate cytosolic second messengers and promote the synthesis of thyroid hormones and thyroid growth. By mimicking the action of the TSH, these autoantibodies compete with the hormone for the binding site on the receptor. Stimulating antibodies constitutes the hallmark of GD pathophysiology (3,51).
Blocking autoantibodies. These are antibodies that bind to the TSH receptor but do not induce a conformational change nor promote hormone synthesis. Furthermore, they impede the binding of the TSH to its receptor (3,51). As a consequence, they decrease thyroid function. Blocking antibodies are usually found in patients with HT and in some with GD (51).
Neutral autoantibodies. These antibodies bind to the TSHR but neither induce a conformational change nor block its function. Their pathophysiological or clinical relevance remains unclear (51).
TRAb are useful for the diagnosis of GD as they are present in >95% of the patients (3,51,78). These antibodies are specific but not very sensitive, so a negative result is not conclusive. This sensitivity is due to the fact that TRAb are present in the serum at very low concentrations (51).
Other Antibodies. Antibodies against specific non – thyroid antigens, e.g., antinuclear antibodies (ANA), have been described in patients with AITD. The prevalence of ANA varies from 9–35% and is much higher when TPOAb and TgAb are present (75% and 69%, respectively) (56). Anti-double stranded DNA antibodies are also found in GD and HT in patients without clinical evidence of systemic lupus erythematosus (SLE) (79).
Autoimmune thyroid diseases
The clinical entities found in AITDs are diverse and vary depending on whether a state of hypothyroidism (HT), hyperthyroidism (GD), or both [Painless thyroiditis (PT), Postpartum thyroiditis (PPT), and Subacute thyroiditis (SAT)] predominate in the patient (7) (Table 2).
Table 2
Main characteristics of autoimmune thyroid diseases.
The term thyroiditis encompasses a varied group of disorders characterized by some form of thyroid inflammation. They include conditions that cause acute illness with severe thyroid pain and others in which there is no clinically evident inflammation, and the illness is manifested primarily by thyroid dysfunction or goiter. PT, PPT, HT, and GD all have an autoimmune basis (6).
Hashimoto thyroiditis
HT (chronic autoimmune thyroiditis or chronic lymphocytic thyroiditis) is the prototypic example of organ-specific AD and the most common cause of hypothyroidism in iodine sufficient areas of the world (3,80,81). It was first defined in 1912 by surgeon Hakaru Hashimoto who described four women with a condition he initially denominated struma lymphomatosa (82).
Epidemiology. Its prevalence is about 1 in 1,000 people and increases with age, affecting up to 40% of elderly women (80,81). Thyroid failure is seen in up to 10% of the population. The incidence of HT is higher in countries where there is excess iodine in the diet, approximately 1.3%, compared to 1% in countries that are iodine sufficient (83). It affects more women than men with a female to male ratio of 18:1 (82). The sibling recurrence risk is >20 (21). There is a peak frequency during the fourth decade, and the mean age of presentation is 35 years (82). The disease clusters in families, sometimes alone and sometimes in combination with GD (see below) (84). Although thyroid lymphoma is a rather rare condition, there is an increased risk for this disease in patients with HT with a RR of 67 (85).
Pathogenesis. HT is characterized by gradual loss of thyroid function, goiter, or both due to autoimmune–mediated destruction of the thyroid gland through apoptosis of the thyrocytes (82). Several mechanisms have been suggested to explain the pathogenesis of HT. These include first, molecular mimicry. Here HT is thought to be caused by an immune reaction against an antigen that shares structural similarities with endogenous proteins (86). Second, there is bystander activation. In this case, a virus reaching the gland or activation of non–specific lymphocytes within the thyroid by virus may cause the release of cytokines which, in turn, activate local thyroid–specific T cells and favor an inflammatory reaction (i.e., thyroiditis) (87). Third is thyroid cell expression of HLA antigens. As previously discussed, thyroid follicular cells in patients with HT express HLA-II. The expression of these molecules can be induced by IFN-γ and other products from activated T cells or by virus directly. Thyroid cells expressing HLA-II, in turn, become non-professional antigen presenting cells (88,89). Fourth is thyroid cell apoptosis. In autoimmune thyroiditis, cytokine (e.g., IL–1) production from antigen presenting cells (APC) and Th1 cells induces the expression of Fas and Fas ligand on thyrocytes, thereby causing self–apoptosis (90).
Clinical Manifestations. The two major clinical forms of HT are goitrous autoimmune thyroiditis and atrophic autoimmune thyroiditis (82). The decrease in circulating thyroid hormones causes a negative effect on the metabolic rate and on multiple organ systems (91). The deposit of glycosaminoglycans secondary to an increase in the synthesis of hyaluronic acid and the decrease in the metabolic rate explains most of the clinical manifestations of patients with hypothyroidism (91).
Clinically, the signs and symptoms of hypothyroidism that characterize HT are shown in the Figure 3. These symptoms are usually insidious and are unrecognized by the patient for a prolonged time (82). The most typical clinical manifestations include:
- Skin: findings on the skin depend on the degree of the hypothyroidism and the ethnicity of the patient (91). Usually there is xeroderma, thickening of the skin, cold intolerance, livedo reticularis, and loss of lateral eyebrows (Queen Anne sign) (82,91). The face is swollen, and the tongue is thickened. Patients complain of hair loss and the nails are frail (82).
- Musculoskeletal: fatigue, carpal tunnel syndrome, myopathy, and arthritis may be present (91). Myopathic symptoms consist of proximal muscular weakness accompanied by an increase in serum creatine kinase (91,92). The association between arthritis and hypothyroidism is well-known (93,94). Bland et al. (95) had previously described the arthritis that accompanied patients with hypothyroidism as characterized by affected knees, metacarpophalangeal joints, proximal interphalangeal joints, and metatarsophalangeal joints without the presence of sinovitis. It is thought to be a TSH-dependent increase in hyaluronic acid and proteoglycan synthesis in this subgroup of hypothyroid patients. This observation is supported by the symptomatic response to thyroid hormone replacement with concomitant TSH suppression (93,95).
- Other findings: patients with hypothyroidism may present with hyponatremia secondary to plasma dilution due to decreased free water clearance. This finding together with accumulation of mucopolysaccharides, reduced glomerular filtration rate, and low cardiac output results in edema (100). Hypothyroidism is characterized by abnormal lipid values; more than 90% of patients present with increased low–densitiy lipoproteins (LDL) and apolipoprotein B because of reduced hepatic clearance secondary to a decrease in hepatic LDL receptors (101).
Figure 3
Clinical manifestations of hypothyroidism and hyperthyroidism. GI: gastrointestinal tract; CNS: central nervous system.
Some patients with HT, especially those with the goitrous form, might not present with the classic symptoms of overt hypothyroidism, but with symptoms secondary to the presence of a mass on the neck. The main compression symptoms are dyspnea, dysphagia, and dysphonia (82).
Laboratory Findings. Even though the clinical manifestations are very sensitive, their specificity is rather low (102). This is the reason the laboratory findings constitute the cornerstone for the diagnosis of thyroid dysfunction.
Secretion of the thyroid hormones, thyroxine (T4) and T3, is regulated by the TSH. In turn, TSH secretion is controlled by thyroid hormones. There is a negative relationship between free T4 (fT4) and TSH, where small changes in fT4 concentration induce very large reciprocal changes in TSH concentration (103). This means that serum levels of TSH are best for assessing the thyroid function. However, there is considerable controversy regarding the normal upper limit of serum TSH. Several authors have addressed this issue but, currently, there is no consensus (104–107). Even though most laboratories have used values of 5.0 IU/mL concurrent with the American Academy of Clinical Endocrinology (AACE) (106), an article published by the National Academy of Clinical Biochemistry argues that the upper limit should be reduced to 2.5 IU/mL based on their results where 95% of the euthyroid volunteers had serum values between 0.4 – 2.5 IU/mL (107). Jensen et al. and Hamilton et al. (104,105) found a normal upper TSH level of 4.1 IU/mL, which is more clinically acceptable for initiating therapy. Regarding T4 and T3, the free hormone hypothesis states that the unbound or free hormone is the one available for uptake into cells and interaction with nuclear receptors. The bound hormone, on the other hand, represents a circulating storage pool that is not immediately available for cell uptake. Free T3 (fT3) and fT4 are usually measured rather than serum total T3 and T4 concentrations.
The most common tests done in HT are TSH, fT4, TPOAb, and TgAb. The usual findings in patients with hypothyroidism include high TSH and low fT4 levels. Patients with high TSH but normal fT4 have a condition known as subclinical hypothyroidism (91). High serum TPOAb concentrations are present in 90% of the patients, and high serum TgAb are found in 50–90% of these patients. As discussed earlier, some patients with HT might have TRAb levels (i.e., up to 10%), but their role in the diagnosis is uncertain (91). The diagnosis of HT can be made in patients with high TSH levels, low fT4, and positive TPOAb.
Other tests that may be done include thyroid gland ultrasound (US) that will reveal an enlarged or shrunken gland depending on the clinical presentation of the HT (82). Radioiodine uptake (RaIU) over 24 hours does not provide useful information for diagnosis as it might be normal, reduced, or high depending on the functional phase of the disease (6,82). The fine needle aspiration (FNAB) should be done on every patient who presents with a dominant thyroid nodule and HT since, as discussed previously, lymphoma and thyroid carcinoma must be ruled out. A FNAB positive for HT will reveal the classical pathological findings that include a marked lymphocytic infiltration and the Hürthle cells (6,82).
Patients with hypothyroidism must be assessed on their cardiovascular risk as severe hypothyroidism leads to hypercholesterolemia and hypertriglyceridemia with a higher risk of atherosclerosis and acute coronary syndrome (101). Lipid profile tests should be done periodically on these patients. Patients with dyslipidemia, in turn, should always be screened for hypothyroidism (91).
Treatment. Thyroid hormone replacement therapy constitutes the main therapeutic strategy for patients with hypothyroidism (82,108). The treatment of choice is oral supplementation with synthetic thyroxine (levo–thyroxine, L-T4). L-T4 is a prohormone with very little intrinsic action. It is, in turn, peripherally deiodinated into T3, the active thyroid hormone. As the L-T4 has a prolonged half–life (i.e., approximately 7 days), once a day treatment ensures steady concentrations of T4 and T3 (109).
The goals of the treatment are improvement of the symptoms and normalization of TSH secretion. In addition, in patients with the goitrous form of presentation, a decrease in the size of the goiter is also considered a therapeutic objective (110). These goals are usually achieved with an average replacement dose of 1.6 mcg/kg/day (111). Elderly patients with coronary disease or multiple coronary risk factors should be treated conservatively since thyroid hormones increase myocardial oxygen demand which can induce angina, arrhythmias, or even myocardial infarction (82). The starting dose for these patients should be 50 mcg/day, and those with a prior history of coronary heart disease must be treated with 25 mcg/day (82).
Patients with replacement therapy must be monitored by assessing serum TSH levels. Although symptoms may begin to resolve a couple of weeks after initiating the treatment, steady–state TSH concentrations are not achieved for at least six weeks. Thus, these patients should be reevaluated with serum TSH in six weeks (112). If the TSH level persists at higher than the normal upper limit, the dose must be increased 12.5–25 mcg per day. Serum fT4 measurements are very insensitive for assessing the appropriateness of the dose (112). In the case of subclinical hypothyroidism, the decision to initiate replacement treatment depends on serum TSH levels. If the TSH level is >10 IU/mL, treatment with L–T4 is indicated to prevent progression to overt hypothyroidism. Patients with TSH levels that are between 4–10 IU/mL who also present with goiter, nonspecific symptoms of hypothyroidism, or high titers of TPOAb must be started on replacement therapy (113). Other therapeutic strategies such as the use of immunosuppressive agents [e.g., glucocorticoids (GCs)] are not required since the lifelong administration of L–T4 is enough for these patients (82).
Painless thyroiditis
PT, also called silent thyroiditis, is characterized by hyperthyroidism, followed by hypothyroidism, and finally, recovery to a euthyroid state (6,114). It is considered a variant form of HT, suggesting that it is part of the spectrum of AITD (114,115). There are many similarities with PPT and SAT (6,114,115).
The incidence of PT is not well-delineated and accounts for 1–5% of the cases of hyperthyroidism. It seems to be more prevalent in areas of higher iodine intake (117). Women are affected more commonly than men at a ratio of 4:1. The mean age of presentation is between 30–40 years (115). It is associated with a specific HLA, most often HLA–DR3, which suggests an inherited susceptibility. However, this association is weaker compared with HLA–B35 and SAT (16).
Factors postulated to initiate PT include excess iodine intake and various cytokines (115,116). It has been reported in patients following cessation of GCs and in external radiation of the neck for Hodgkin lymphoma (117–120). The resulting thyroid inflammation damages thyroid follicles and activates proteolysis of the Tg stored in the colloid. This causes an unregulated release of T4 and T3 into the circulation and results in hyperthyroidism. Once the Tg is exhausted, hormone synthesis ceases and a state of hypothyroidism may develop. As the inflammation subsides, the repaired thyroid follicles resume normal synthesis and secretion of thyroid hormone (6,115).
Approximately 5% to 20% of the patients with PT present with the triphasic course of hyperthyroidism, hypothyroidism, and restoration to normal thyroid function (Figure 4). This clinical course is also described for PPT and SAT (115). The signs and symptoms that characterize hyperthyroidism and hypothyroidism are shown in Figure 3. The hypothyroid phase is recognized and diagnosed more often compared to the hyperthyroid phase (121). The thyroid gland is not painful, non-tender, but it is usually minimally enlarged sometimes firm in texture upon palpation (115).
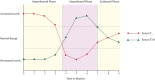
As expected, thyroid function tests vary during the clinical course of the PT, and changes in serum TSH usually lag behind those in fT4 and fT3 (114,115) as explained in Figure 4. Thyroid function tests should be done every four to eight weeks to confirm resolution of hyperthyroidism and detect the development of hypothyroidism (115). Up to 50% of the patients with PT have increased titers of TPOAb but not to the same extent as those with HT (122). Other laboratory results as well as common imaging results are described in Table 2.
Most of the patients do not require any treatment at all since the thyroid dysfunction is not severe enough to make them symptomatic. However, patients who develop clinical manifestations do need treatment based on the phase of the clinical course they are in. During the hyperthyroid phase, the treatment of choice is beta-blockers (BB) (Table 3). In the hypothyroid phase, the replacement therapy is L-T4. Based on the results of the thyroid function tests, the course of the supplementation therapy must be defined (115). Although most patients recover full thyroid function, up to 20% will develop permanent hypothyroidism (123).
Table 3
Beta-blockers used for management of symptomatic hyperthyroidism.
Postpartum thyroiditis
Postpartum thyroiditis (PPT) is a destructive thyroiditis induced by an autoimmune mechanism within a year of parturition. It can also occur after spontaneous or induced abortion. Like PT, PPT is considered a variant form of HT (57,124–126).
The prevalence of PPT varies globally and ranges from 8 - 11% (71). Several risk factors have been identified that have a higher probability of developing PPT. For instance, women with positive TPOAb at the end of the first trimester have a 30–50% higher risk of progression to PPT. Patients with a prior history of thyroid dysfunction have a 40% risk, and for those with type 1 diabetes mellitus (T1DM) or family history of thyroid disease, the probability is about 20% (71,127). PPT has been related to HLA-B and HLA-D, suggesting an inherited pattern (128).
The presentation of PPT can be identical to painless thyroiditis but with a more variable course. Around 20–30% of the patients have the characteristic triphasic course with hyperthyroidism, hypothyroidism, and euthyroidism that lasts 12 months, 20–40% present with just hyperthyroidism, and 40–50% with overt hypothyroidism (125,129). Many of these symptoms are usually attributed to breastfeeding or the stress of having a newborn, making the recognition of PPT difficult at times. Patients with PPT may have a mildly enlarged, diffuse, non-tender thyroid gland that typically disappears with recovery of thyroid function (115).
The thyroid function test results are described in Figure 4. Positive TPOAb are found in 60–85% of the patients (78,124). Table 2 shows the common results of other laboratory tests and imaging studies.
The management of PPT is similar to the treatment of PT with some exceptions. When using BB for hyperthyroidism, propranolol is indicated given its low concentration in breast milk. Regarding hypothyroidism, the replacement should be halved after 6–12 months, unless the woman is pregnant, attempting pregnancy or breastfeeding (130,131). Up to 30% of the patients never recover from the hypothyroid phase and thus, require lifelong supplementation (127). Progression to permanent hypothyroidism may be related to higher initial TSH concentrations and TPOAb titer, maternal age, and female sex of the infant (71).
Subacute thyroiditis
This type of thyroiditis, which is also known as De Quervain thyroiditis in honor of the Swiss surgeon who formally described the disease in 1904, is the main cause of thyroid pain (115,132). It is a self-limiting inflammatory disorder secondary to viral infections with a predictable clinical course of thyroid function evolution as seen in PT and PPT (133).
The estimated incidence is 3 cases per 100,000/year, and it affects four times more women than men. It occurs between 40 – 50 years of age (132–134) and decreases with age (132). The disease was thought to have a seasonal incidence, especially during summer, and clusters have been associated with a number of viruses (135). There is an association between SAT and HLA–B35 in many ethnic groups (11).
The pathogenesis mechanism is the same as the one described for PT. However, the main trigger for the autoimmune disease is a viral infection as the majority of the patients have a prior history of upper respiratory tract infection days before the SAT (133). The main viral microorganisms identified are Echovirus and Coxsackevirus groups A and B, but other reports include measles, mumps, Epstein–Barr virus, and adenovirus.
The clinical course includes the triphasic phase described for PT and PPT. One of the main differences found in SAT is that thyroid pain represents the presenting symptom in up to 96% of the patients. The onset may be sudden or gradual and is usually radiated to the upper neck, jaw, throat, or upper chest. It is exacerbated by coughing or neck movements (136). On physical examination, the thyroid gland may be enlarged with tenderness upon palpation. Up to 50% of the patients have an initial thyrotoxicosis phase with the typical symptoms of hyperthyroidism (115). Another clinical variation found in patients with SAT is the appearance of a prodromic phase during the subclinical viral infection characterized by nonspecific symptoms such as myalgias, pharyngitis, low-grade fever, and fatigue (136).
The most relevant laboratory results and imagining studies are shown in Table 2 (137–140). The main goals in the treatment of SAT are pain relief and management of hyperthyroidism symptoms (115,132). Pain control strategies involve the use of both non-steroidal anti-inflammatories (NSAIDs) and/or systemic GCs (132). First line therapy is NSAIDs, so the patient is usually given ibuprofen or naproxen initially. If after two to three days there is no improvement in the symptoms, the NSAID should be discontinued and prednisone initiated at a dose of 40 mg/day. When the pain is controlled, a tapering dose of 5–10 mg per week should be encouraged to discontinue the medication within 4–8 weeks (132). About 4% of the patients have a recurrence of SAT (141).
Graves’ disease
Graves’ disease (GD), named after the physician Robert Graves who first described the association in 1835, is clinically characterized by the presence of hyperthyroidism, diffuse goiter, ophthalmopathy (GO), and dermopathy (142,143).
Epidemiology. GD is the most common cause of hyperthyroidism, representing up to 80% of the cases (144). The annual incidence of GD is 20–25/100,000 in Caucasian populations. It is lower among people with African ancestry (142). It affects ten times more women than men. A peak frequency between the ages of 40–60 years has been reported (143,144). The concordance rate for GD between monozygotic twins is 38% (10). A family history of thyroid disease, especially among maternal relatives, is associated with increased risk of GD and a younger age of onset (145).
Pathogenesis. GD is the result of the presence of circulating TRAb that bind and activate the TSH receptor, thus stimulating follicle hypertrophy and hyperplasia as well as increasing the synthesis of thyroid hormones and the fraction of T3 relative to T4 (74,142). In GD, as discussed previously, there is an environmental factor that triggers an autoimmune response to the thyroid either by molecular mimicry or by bystander activation (142). Thyroid cells expressing HLA-R molecules may act as APC activating local T cells through the expression of co-stimulating molecules. As a result, there is a production of cytokines which in turn, activates thyroid–specific T cells and B cells to produce TRAb (142).
Clinical Manifestations. The clinical manifestations can be classified into those secondary to the excess of circulating thyroid hormones (hyperthyroidism) and those specific to GD (142). Typical signs and symptoms of hyperthyroidism are shown in Figure 3 (143,145). Some signs and symptoms are highly specific to GD including the following:
- Goiter: diffuse enlargement of the thyroid gland that can vary in size (142).
- Ophthalmopathy: approximately 20–25% of patients with GD have clinically obvious GO (33). The estimated incidence of GO is 16/100,000 persons in women and 3/100,000 in men (146). It appears before the onset of hyperthyroidism in 20% of the patients, concurrently in 40%, and six months after diagnosis in about 20% (147). The characteristics signs are proptosis and periorbital edema. The major symptoms include sense of irritation in the eyes, excessive tearing, retroorbital discomfort or pain, blurring of vision, diplopia, and occasionally, loss of vision (148). Most of the patients have mild non–progressive ocular involvement, but in 5–10% of the cases, there is severe ophthalmopathy associated with significant visual impairment that can lead to visual loss (142). The pathogenesis of GO is a result of a cross-reaction between the TRAb and TSHR found on orbital fibroblasts, which induces the production of glycosaminoglycans, especially hyaluronic acid. (33).
- Thyroid acropachy: clubbing caused by soft tissue swelling and periosteal bone changes in the fingers and toes. This sign represents a rare manifestation of GD (142).
Table 4
Diagnostic criteria for Graves’ disease.
Treatment. The treatment of GD is based upon one of three therapeutic strategies: medical management, using medication to suppress the hyperthyroid symptoms (i.e., BB), and blocking thyroid hormone synthesis with antithyroid medication (i.e., PTU, MMI); radioactive iodine therapy; or surgical approach with total thyroidectomy (149). The decision to choose one over the other depends on each individual and is based on the severity of the thyrotoxicosis, the presence of goiter and ophthalmopathy, and the patient’s preference (149) (Table 5).
Table 5
Treatment strategies for Graves’ Disease.
Polyautoimmunity
Several clinical signs and symptoms that are shared among ADs –physiopathological mechanism and risk factors– indicate that they have a common origin, which has been called the autoimmune tautology (5,8). The clinical evidence of the autoimmune tautology highlights the co-occurrence of more than one AD in a single patient, namely polyautoimmunity, or MAS, a form of polyautoimmunity which corresponds to the coexistence of two or more well-defined ADs (5,9). The importance of these terms is due to the fact that patients with polyautoimmunity or MAS may have a modified disease course (i.e., with a worse prognosis or a better one) and a modified clinical presentation (9).
AITD has been described as the most prevalent AD as well as being associated with other organ-specific and non-organ specific ADs (150). In a study in which we analyzed 1,083 patients, polyautoimmunity was observed in 34.4% of the cases, and AITD was the most frequent polyautoimmunity found (9). This finding was supported by systemic literature review where three basic, large clusters were found. According to the analysis, the most hierarchical (i.e., chaperon) AD in the MAS cases is represented by AITD. It was associated with sclerodema (SSc) in 23% of the cases, rheumatoid arthritis (RA) in 21%, SLE in 17.9%, and multiple sclerosis (MS) in 9.1%. Female gender was a shared factor that was significantly associated with polyautoimmunity in the four ADs (9). Possible explanations for the relationship of these AD include the immunomodulatory effects of antithyroid antibodies, molecular mimicry between thyroid and disease-specific epitopes, and a genetic link between thyroid autoimmunity and the susceptibility to AD. Other studies have demonstrated the association between AITD and non–organ specific AD, particularly with RA, SLE, and Sjögren’s syndrome (SS) (150-152) (Table 6).
Table 6
Polyautoimmunity and AITD.
Familial autoimmunity (FA) is defined as the presence of any AD in first-degree relatives (FDRs) of the proband, which are at increased risk of developing an AD (1,4–6). Recently, we found FA to be strongly associated with AITD (69). For instance, if the proband has AITD, the most common AD in the FDRs are: Addison’s disease (AdD), celiac disease (CD), inflammatory bowel disease (IBD), myasthenia gravis, MS, pernicious anemia (PA), RA, SLE, T1DM, vitiligo (VIT), localized SSc, and discoid lupus erythematosus. On the other hand, if AITD is the AD in FDRs, the probands are predisposed to T1DM, SLE, RA, MS, SS, SSc, IBD, VIT, juvenile rheumatoid arthritis, juvenile lupus erythematous, inflammatory idiopathic myositis, CD, and alopecia areata. These finding shows that there is familial clustering of AITD in FDRs, particularly in female relatives (7).
Boelaert et al. (153) described FA among probands with HT or GD. Both ADs were significantly associated with the presence of T1DM, RA, PA, SLE, CD, VIT, and MS. Only GD was associated with AdD and IBD. Compared to the general population, FA in GD probands disclosed PA as the strongest association (RR:14.1) followed by RA (RR:13.5). Hemminki et al. (154) assessed FA only in probands with GD from Sweden. To calculate familial risk within a large community based cohort, they calculated standardized incidence ratios (SIR) as the ratio between the observed and the expected frequency for each disease. A value over one indicates a higher frequency of what is expected whereas a value below one indicates a decreased frequency. The analysis was stratified based on the FDR involved. For a single parent affected, HT, PA, and RA were the only diseases significantly associated as they had a SIR of 2.04, 1.82, and 1.48 respectively and thus, showed a frequency that was higher than what was expected. Significant associations for singleton siblings were found for T1DM, discoid lupus, and localized SSc if a parent and a sibling were affected with the same AD. The significant association was between HT with a SIR of 37.41 and SLE with a SIR of 14.33 (154). In another study, FA was significantly associated with polyautoimmunity in SLE and SSc patients (9), while Walker et al. (155) found an excess risk for AITD in RA multicase families compared to the general population.
AITD and rheumatoid arthritis
For several decades, an increasing occurrence of thyroid disorders in patients suffering from RA has been documented, both autoimmune and non-autoimmune in nature (94,156–158). In addition, rheumatological and non-rheumatological manifestations of AITD have been described (55). Within these manifestations, it is noteworthy that the most common symptoms are polyarthralgia and unclassified arthritis, which are the main features of RA. Genetic background is an important aspect in autoimmunity. Genetic risk factors shared among diseases have been described in AITD and RA (55,159,160).
The prevalence of AITD in RA cases has ranged from 0.5% in Morocco (161) to 27% in Slovakia (78). It varies between regions and ethnicities [i.e., 1% in Germany (162), 2.1–9.8% in North America (163-166)]. For more details, see Figure 5. In Latin America, the prevalence of polyautoimmunity was reported to be 9.8% in a cross sectional analytical study of Colombian RA patients. This association, adjusted by gender and RA duration, was related to the presence of diabetes, thrombosis, and abnormal body mass index. Furthermore, a lower AITD frequency was demonstrated for the lowest educational level than for the highest one. This is also true when antimalarials are used (150).

Figure 5
Prevalence of AITD and thyroid antibodies worldwide. Tg: thyroglobulin; TPO: thyroperoxidase.
It is widely accepted that, among the thyroid antibodies, the most frequent is TPOAb compared to TgAb (80). The prevalence for TgAb ranged from 5% in men from the UK (167) and 6% regardless of gender in Egypt (168), to 31% in RA patients from Japan (169). The prevalence for TPOAb was within the range of 5% in Egypt (170) to 37% in Italy (151). Cárdenas-Roldán et al. (150) in a recent cross-sectional analysis of 800 RA patients found that the presence of antibodies was 37.8% for TPOAb and 20.8% for TgAb. In contrast, Ruggeri et al. (171) show the assessment of thyroid antibodies at three points in time. There were more patients with TPOAb and TgAb than with a clinical diagnosis of AITD. Considering the idea that autoantibodies are predictors of disease (45,172), it is important to remain vigilant in following the clinical course of these patients. TPOAb and TgAb are known to predict AITD. This was demonstrated in the Wickham cohort (173). Patients within the accepted TSH reference range and who had the above mentioned autoantibodies had a greater risk of developing overt hypothyroidism. A careful assessment of those patients with a normal range of TSH but presenting specific antibodies should be done. For more details, see Figure 5.
The association between AITD and extra-articular manifestations (EAM) seems to be linked to CVD (174), and it is considered a major predictor of poor prognosis (175). In fact, Raterman et al. (156) agreed that the presence of hypothyroidism, including HT, is a risk factor for CVD in patients with RA. Moreover, McCoy et al. (165) found that L-T4 supplementation was significantly associated with CVD, which supports the fact that the administration of this medication does not decrease the occurrence of this outcome. Autoimmunity itself may be an independent risk factor for CVD. As both diseases increase inflammatory parameters and cytokines and cause endothelial dysfunction, a relationship between polyautoimmunity and the occurrence of CVD is not surprising.
With respect to RA severity, the literature is scarce. Charles et al. (176) did not find a relationship between the presence of thyroid antibodies and the occurrence of anti-cyclic citrullinated peptide antibody (anti-CCP), although they did the analysis with the PTPN22 R620W allele. Likewise, another cohort did not find a correlation between AITD and proxy variables for RA severity such as erosions, biological agent use, the presence of anti-CCP, and EAM (150). One cannot but hypothesize that many of these studies are cross-sectional in nature and because the importance of DAS28 and HAQ is along a timeline, it is not relevant to include these variables in the analysis.
AITD and systemic lupus erythematous
White et al. (177) and Hijmans et al. (158) first described the association between SLE and AITD by showing an increased presence of thyroid dysfunction in patients with SLE compared to the general population. There are several studies that have suggested an increased prevalence of AITD in SLE (178–181). However, this is a subject of controversy to some authors (182).
Thyroid dysfunction in SLE may have various pathogenic mechanisms, but the underlying processes are unknown (183). For instance, genetic influence might play a role as suggested by Namjou et al. (184) in a study in which they found a gene of susceptibility identified in 5q14.3–q15, a major locus of susceptibility for SLE also found in AITD. This result highlights a potential genetic link between these diseases.
The most common thyroid function abnormality found in patients with SLE is hypothyroidism with a prevalence of 4–14% (178–181,183), which is significantly higher than the general population prevalence of 1%. Additionally, subclinical hypothyroidism is present in up to 12% of the patients with SLE (178,179).
Whether hyperthyroidism is more prevalent in SLE than in the background population is still debatable (180,183). While the prevalence in different studies is up to 1.7%, the frequency in the general population is approximately 1.9%, which suggests that there is no significant difference (180).
When assessing the thyroid function of patients with SLE, some factors should be considered such as age, use of immunosuppressant, and disease activity (183). Kumar et al. (178) found a significantly higher prevalence of thyroid disease in the SLE cohort when compared to age and sex matched controls (36% vs. 8%). The studies assessing the relationship between disease activity and thyroid dysfunction are controversial with non–conclusive results (183). However, patients with greater clinical activity and severity of SLE have significant changes in the hypothalamus – pituitary – thyroid axis even with no evidence of thyroid disease (179). There is a subgroup of patients in which the disease activity affects the thyroid function, and are those with the “euthyroid sick syndrome.” These patients had a SLEDAI that was statistically significant different from those without thyroid dysfunction, implying the effect of disease activity on this type of thyroid abnormality in SLE patients (178,185). Disease duration, in turn, has no statistically significant effect on thyroid function abnormalities (178).
Both TgAb and TPOAb are found with greater frequency in patients with SLE than in the general population even in those with no thyroid function abnormalities (178,183). In a recent study, 30% of the patients were positive for antithyroid antibodies, and 12% of them did not have thyroid function abnormality (178). Overall, there is a trend toward TPOAb being found more frequently than TgAb. Antonelli et al. (179) reported a 27.6% prevalence of TPOAb in patients with SLE as opposed to 15.4% of TgAb.
Thyroid involvement being non-life threatening in SLE compared to organ involvement, it can be undetected for a long time while contributing to the morbidity of the illness. Symptoms of thyroid disease can be confused with those of SLE. Therefore, it is necessary to identify the thyroid dysfunction in these patients and treat them accordingly (183).
AITD and sjögren’s syndrome
The most common association reported between an endocrine and rheumatological autoimmune disease is that found between SS and AITD (183). However, the controlled studies looking at this association have been few and the results have not been uniform thus, making them highly variable (186).
The lacrimal, salivary, and thyroid glands have a number of histological and functional similarities including the uptake and concentration of iodine by these glands (187). From the histopathological point of view, they share three characteristics (183,188). First, focal or diffuse infiltration by activated T lymphocytes similar to those from HT are found in patients with SS, which suggests that the same autoimmune response is directed towards the thyrocytes and salivary gland epithelium (183,187). Second, the participation of HLA of the haplotypes HLA-B8 and DR3 in both SS and AITD is demonstrated by the higher frequency of those haplotypes in Caucasian patients with these diseases (189). Third is clonal B cell expansion (183,189). These observations may suggest a common pathogenic mechanism in both diseases. It is possible that antigens shared by the salivary and thyroid glands are responsible for the associated autoimmunity directed toward each organ (187). In addition, Hansen et al. (190) reported five cases of focal autoimmune sialadenitis in 19 patients with AITD. Thus, it is sometimes hard to establish whether the salivary and eye involvement represent an extra-thyroidal manifestation of AITD, or rather, an extra-exocrine manifestation of SS (183).
The prevalence in the association between AITD and SS varies between studies (186,187,189,191) and ranges from 10–18%. The major cause of thyroid disease is hypothyroidism. Lazarus et al. (192) reported that the most common AD developed by patients with SS in their cohort, approximately 16%, was hypothyroidism. Moreover, Foster et al. (193) showed a high prevalence of thyroid disease and anti-thyroid antibodies in female relatives of 42 patients with SS (13.7% vs. 3.3% in female controls). SS is ten times more frequent in patients with AITD, and HT is nine times more frequent in SS as compared to the general population (183). In a study of 479 patients with SS, the frequency of HT was greater than that in the general population, 6.25% and 1–2%, respectively (194). Ramos-Casals et al. (187), in turn, found that the most frequent hormonal profile observed was subclinical hypothyroidism, detected in 51% of their cohort.
Only a few studies have focused on the prevalence of hyperthyroidism in patients with SS, which is why it is thought to be infrequent (187,191,192). The prevalence in these studies is as low as 1.8% (192) and as high as 18% (187); thus, further studies are needed regarding this specific association.
The frequency of anti-thyroid antibodies in SS is approximately 11% (186). Punzi et al. (191) reported a 17.6% increase in the frequency of TPOAb and of 13.4% in TgAb in 119 female patients with SS, compared to 199 female controls.
Currently, there are no robust studies reporting the clinical manifestations of patients with AITD that will develop SS. Petri et al. (195), looking at the reverse association, found that there was no increase in the rheumatological symptoms in patients with AITD. However, it has been found that around 32% of patients with HT present with conjunctivitis sicca and xerostomia (194). According to this, it is justifiable to periodically screen patients with SS for AITD, especially HT, given the frequency of this association apart from the presence of symptomatology.
Conclusions
AITD is a term used to bring together a group of pathologies that include thyroid dysfunction and an autoimmune response to the thyroid gland. Even though the prevalence of this disease in the general population varies between countries, AITD can be regarded as the most common autoimmune endocrine disease. It includes a group of AD clustered together with a diverse clinical presentation that depends on whether it causes hypothyroidism, hyperthyroidism, or both. An international consensus to accurately diagnose AITD is warranted.
AITD is frequently associated with other organ specific and non-organ specific AD, most commonly RA, SLE, and SS. AITD is clinically important in the context of autoimmunity, and it is mandatory to screen patients with hypothyroidism or hyperthyroidism symptoms for the autoimmune etiology when there is suspicion of the coexistence of AITD (i.e. polyautoimmunity).
Routine screening for CVD among these patients should be considered. These results may help to further the study of the common mechanisms of AD, to improve patient outcome, and to define public health policies, especially for RA patients.
Abbreviation list
- AACE:
American Academy of Endocrinology
- AD:
autoimmune diseases
- AdD:
Addison Disease
- AITD:
autoimmune thyroid disease
- ANA:
antinuclear antibodies
- ANCA:
anti-neutrophil cytoplasmic antibodies
- APC:
antigen presenting cell
- ATD:
anti-thyroid drugs
- BB:
beta-blockers
- BID:
two-times per day
- CBC:
complete blood count
- CRP:
C-reactive protein
- CTLA-4:
cytotoxic T lymphocyte antigen-4
- CVD:
cardiovascular disease
- EAM:
extra-articular manifestations
- ELISA:
enzyme-linked immunosorbent assay
- ESR:
erythrocyte sedimentation rate
- FA:
familial autoimmunity
- fT3:
free-fraction triiodothyronine
- fT4:
free-fraction thyroxine
- FNAB:
fine-needle aspiration biopsy
- GCs:
glucocorticoids
- GD:
Graves’ disease
- GO:
Graves’ ophthalmopathy
- HLA:
human leukocyte antigen
- HT:
Hashimoto thyroiditis
- IBD:
inflammatory bowel disease
- IFN:
interferon
- L-T4:
levothyroxine.
- MAS:
multiple autoimmune syndrome
- MMI:
methimazole
- MS:
multiple sclerosis
- NIS:
sodium/iodine symporter
- NSAIDs:
non-steroidal anti-inflammatory drugs
- PA:
pernicious anemia
- PPT:
postpartum thyroiditis
- PTPN22:
protein tyrosine phosphatase, nonreceptor type 22
- PT:
painless thyroiditis
- PTU:
propylthiouracyl
- QD:
once per day
- QID:
four-times per day
- RA:
rheumatoid arthritis
- RaIU:
radioactive iodine uptake
- SAT:
subacute thyroiditis
- SLE:
systemic lupus erythematosus
- SLEDAI:
systemic lupus erythematosus disease activity index
- SS:
Sjögren syndrome.
- SSc:
scleroderma
- T1DM:
type 1 diabetes mellitus
- T3:
triiodothyrosine
- T4:
thryoxine
- Tg:
thyroglobulin
- TgAb:
thyroglobulin antibodies
- TID:
three-times per day
- TPO:
thyroperoxidase
- TPOAb:
thyroperoxidase antibodies
- TRAb:
thyroid stimulating hormone receptor antibodies
- TSH:
thyroid stimulating hormone
- TSHR:
thyroid stimulating hormone receptor
- US:
ultrasonography
- VIT:
vitiligo
References
- 1.
- Simmonds MJ, Gough SC. Unravelling the genetic complexity of autoimmune thyroid disease: HLA, CTLA-4 and beyond. Clin Exp Immunol. 2004;136:1–10. [PMC free article: PMC1808990] [PubMed: 15030506]
- 2.
- Dayan CM, Daniels GH. Chronic autoimmune thyroiditis. N Engl J Med. 1996;335:99–107. [PubMed: 8649497]
- 3.
- Stathatos N, Daniels GH. Autoimmune thyroid disease. Curr Opin Rheumatol. 2012;24:70–5. [PubMed: 22157414]
- 4.
- Tomer Y, Davies TF. Searching for the autoimmune thyroid disease susceptibility genes: from gene mapping to gene function. Endocr Rev. 2003;24:694–717. [PubMed: 14570752]
- 5.
- Anaya JM, Castiblanco J, Rojas-Villarraga A, et al. The multiple autoimmune syndromes. A clue for the autoimmune tautology. Clin Rev Allergy Immunol. 2012;43:256–64. [PubMed: 22648455]
- 6.
- Pearce EN, Farwell AP, Braverman LE. Thyroiditis. N Engl J Med. 2003;348:2646–55. [PubMed: 12826640]
- 7.
- Shin J Il, Kim MJ, Lee JS. Graves’ disease, rheumatoid arthritis, and anti-tumor necrosis factor-alpha therapy. J Rheumatol. 2009;36:449–50. [PubMed: 19208577]
- 8.
- Anaya JM, Rojas-Villarraga A, García-Carrasco M. The autoimmune tautology: from polyautoimmunity and familial autoimmunity to the autoimmune genes. Autoimmune Dis. 2012;2012:1–2. [PMC free article: PMC3362807] [PubMed: 22666553]
- 9.
- Rojas-Villarraga A, Amaya-Amaya J, Rodriguez-Rodriguez A, Mantilla RD, Anaya JM. Introducing polyautoimmunity: secondary autoimmune diseases no longer exist. Autoimmune Dis. 2012;2012:1–9.
- 10.
- Brix TH, Kyvik KO, Christensen K, Hegedüs L. Evidence for a major role of heredity in Graves’ disease: a population-based study of two Danish twin cohorts. J Clin Endocrinol Metab. 2001;86:930–4. [PubMed: 11158069]
- 11.
- Ban Y, Davies TF, Greenberg DA, Concepcion ES, Tomer Y. The influence of human leucocyte antigen (HLA) genes on autoimmune thyroid disease (AITD): results of studies in HLA-DR3 positive AITD families. Clin Endocrinol. 2002;57:81–8. [PubMed: 12100074]
- 12.
- Kologlu M, Fung H, Darke C, Richards CJ, Hall R, McGregor AM. Postpartum thyroid dysfunction and HLA status. Eur J Clin Invest. 1990;20:56–60. [PubMed: 2108037]
- 13.
- Menconi F, Monti MC, Greenberg DA, et al. Molecular amino acid signatures in the MHC class II peptide-binding pocket predispose to autoimmune thyroiditis in humans and in mice. Proc Natl Acad Sci U S A. 2008;105:14034–9. [PMC free article: PMC2544574] [PubMed: 18779568]
- 14.
- Jacobson EM, Yang H, Menconi F, et al. Employing a recombinant HLA-DR3 expression system to dissect major histocompatibility complex II-thyroglobulin peptide dynamism: a genetic, biochemical, and reverse immunological perspective. J Biol Chem. 2009;284:34231–43. [PMC free article: PMC2797193] [PubMed: 19776016]
- 15.
- Tamai H, Kimura A, Dong RP, et al. Resistance to autoimmune thyroid disease is associated with HLA-DQ. J Clin Endocrinol Metab. 1994;78:94–7. [PubMed: 8288722]
- 16.
- Farid NR, Hawe BS, Walfish PG. Increased frequency of HLA-DR3 and 5 in the syndromes of painless thyroiditis with transient thyrotoxicosis: evidence for an autoimmune aetiology. Clin Endocrinol. 1983;19:699–704. [PubMed: 6606505]
- 17.
- Kotsa K, Watson PF, Weetman AP. A CTLA-4 gene polymorphism is associated with both Graves disease and autoimmune hypothyroidism. Clin Endocrinol. 1997;46:551–4. [PubMed: 9231050]
- 18.
- Yanagawa T, Hidaka Y, Guimaraes V, Soliman M, DeGroot LJ. CTLA-4 gene polymorphism associated with Graves’ disease in a Caucasian population. J Clin Endocrinol Metab. 1995;80:41–5. [PubMed: 7829637]
- 19.
- Vaidya B, Kendall-Taylor P, Pearce SH. The genetics of autoimmune thyroid disease. J Clin Endocrinol Metab. 2002;87:5385–97. [PubMed: 12466323]
- 20.
- Heward JM, Allahabadia A, Armitage M, et al. The development of Graves’ disease and the CTLA-4 gene on chromosome 2q33. J Clin Endocrinol Metab. 1999;84:2398–401. [PubMed: 10404810]
- 21.
- Villanueva R, Greenberg DA, Davies TF, Tomer Y. Sibling recurrence risk in autoimmune thyroid disease. Thyroid. 2003;13:761–4. [PubMed: 14558919]
- 22.
- Vieland VJ, Huang Y, Bartlett C, Davies TF, Tomer Y. A multilocus model of the genetic architecture of autoimmune thyroid disorder, with clinical implications. Am J Hum Genet. 2008;82:1349–56. [PMC free article: PMC2427261] [PubMed: 18485327]
- 23.
- Jacobson EM, Concepcion E, Oashi T, Tomer Y. A Graves’ disease-associated Kozak sequence single-nucleotide polymorphism enhances the efficiency of CD40 gene translation: a case for translational pathophysiology. Endocrinology. 2005;146:2684–91. [PubMed: 15731360]
- 24.
- Criswell LA, Pfeiffer KA, Lum RF, et al. Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. Am J Hum Genet. 2005;76:561–71. [PMC free article: PMC1199294] [PubMed: 15719322]
- 25.
- Vang T, Congia M, Macis MD, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. 2005;37:1317–9. [PubMed: 16273109]
- 26.
- Tomer Y, Menconi F, Davies TF, et al. Dissecting genetic heterogeneity in autoimmune thyroid diseases by subset analysis. J Autoimmun. 2007;29:69–77. [PubMed: 17644307]
- 27.
- Brand OJ, Lowe CE, Heward JM, et al. Association of the interleukin-2 receptor alpha (IL-2Ralpha)/CD25 gene region with Graves’ disease using a multilocus test and tag SNPs. Clin Endocrinol. 2007;66:508–12. [PubMed: 17371467]
- 28.
- Eschler DC, Hasham A, Tomer Y. Cutting edge: the etiology of autoimmune thyroid diseases. Clin Rev Allergy Immunol. 2011;41:190–7. [PMC free article: PMC3129418] [PubMed: 21234711]
- 29.
- Stefan M, Jacobson EM, Huber AK, et al. Novel variant of thyroglobulin promoter triggers thyroid autoimmunity through an epigenetic interferon alpha-modulated mechanism. J Biol Chem. 2011;286:31168–79. [PMC free article: PMC3173071] [PubMed: 21757724]
- 30.
- Hasham A, Tomer Y. Genetic and epigenetic mechanisms in thyroid autoimmunity. Immunol Res. 2012;54:204–13. [PMC free article: PMC3601048] [PubMed: 22457094]
- 31.
- Yin X, Latif R, Tomer Y, Davies TF. Thyroid epigenetics: X chromosome inactivation in patients with autoimmune thyroid disease. Ann N Y Acad Sci. 2007;1110:193–200. [PubMed: 17911434]
- 32.
- Prummel MF, Strieder T, Wiersinga WM. The environment and autoimmune thyroid diseases. Eur J Endocrinol. 2004;150:605–18. [PubMed: 15132715]
- 33.
- Bahn RS. Graves’ ophthalmopathy. N Engl J Med. 2010;362:726–38. [PMC free article: PMC3902010] [PubMed: 20181974]
- 34.
- Pearce EN, Braverman LE. Environmental pollutants and the thyroid. Best Pract Res Clin Endocrinol Metab. 2009;23:801–13. [PubMed: 19942155]
- 35.
- Mori K, Nakagawa Y, Ozaki H. Does the gut microbiota trigger Hashimoto’s thyroiditis? Discov Med. 2012;14:321–6. [PubMed: 23200063]
- 36.
- Benvenga S, Santarpia L, Trimarchi F, Guarneri F. Human thyroid autoantigens and proteins of Yersinia and Borrelia share amino acid sequence homology that includes binding motifs to HLA-DR molecules and T Cell receptor. Thyroid. 2006;16:225–36. [PubMed: 16571084]
- 37.
- Kahaly GJ, Dienes HP, Beyer J, Hommel G. Iodine induces thyroid autoimmunity in patients with endemic goiter: a randomised, double-blinde, placebo controlled trial. Eur J Endocrinol. 1998;139:290–7. [PubMed: 9758438]
- 38.
- Walsh JP, Ward LC, Burke V, et al. Small changes in thyroxine dosage do not produce measurable changes in hypothyroid symptoms, well-being, or quality of life: results of a double-blind, randomized clinical trial. J Clin Endocrinol Metab. 2006;91:2624–30. [PubMed: 16670161]
- 39.
- Burek CL, Talor M V. Environmental triggers of autoimmune thyroiditis. J Autoimmun. 2009;33:183–9. [PMC free article: PMC2790188] [PubMed: 19818584]
- 40.
- Dunkelmann S, Wolf R, Koch A, Kittner C, Groth P, Schuemichen C. Incidence of radiation-induced Graves’ disease in patients treated with radioiodine for thyroid autonomy before and after introduction of a high-sensitivity TSH receptor antibody assay. Eur J Nucl Med Mol Imaging. 2004;31:1428–34. [PubMed: 15221291]
- 41.
- Agate L, Mariotti S, Elisei R, et al. Thyroid autoantibodies and thyroid function in subjects exposed to Chernobyl fallout during childhood: evidence for a transient radiation-induced elevation of serum thyroid antibodies without an increase in thyroid autoimmune disease. J Clin Endocrinol Metab. 2008;93:2729–36. [PubMed: 18430771]
- 42.
- Völzke H, Werner A, Wallaschofski H, et al. Occupational exposure to ionizing radiation is associated with autoimmune thyroid disease. J Clin Endocrinol Metab. 2005;90:4587–92. [PubMed: 15886237]
- 43.
- Brent GA. Environmental exposures and autoimmune thyroid disease. Thyroid. 2010;20:755–61. [PMC free article: PMC2935336] [PubMed: 20578899]
- 44.
- Barragán-Martínez C, Speck-Hernández CA, Montoya-Ortiz G, Mantilla RD, Anaya JM, Rojas-Villarraga A. Organic solvents as risk factor for autoimmune diseases: a systematic review and meta-analysis. PLoS One. 2012;7(12):1–18.
- 45.
- Scofield RH. Autoantibodies as predictors of disease. Lancet. 2004;363:1544–6. [PubMed: 15135604]
- 46.
- Somerset DA, Zheng Y, Kilby MD, Sansom DM, Drayson MT. Normal human pregnancy is associated with an elevation in the immune suppressive CD25+ CD4+ regulatory T Cell subset. Immunology. 2004;112:38–43. [PMC free article: PMC1782465] [PubMed: 15096182]
- 47.
- Ando T, Davies TF. Clinical Review 160: Postpartum autoimmune thyroid disease: the potential role of fetal microchimerism. J Clin Endocrinol Metab. 2003;88:2965–71. [PubMed: 12843128]
- 48.
- Fugazzola L, Cirello V, Beck-Peccoz P. Microchimerism and endocrine disorders. J Clin Endocrinol Metab. 2012;97:1452–61. [PubMed: 22399520]
- 49.
- Mariotti S, Caturegli P, Piccolo P, Barbesino G, Pinchera A. Antithyroid peroxidase autoantibodies in thyroid diseases. J Clin Endocrinol Metab. 1990;71:661–9. [PubMed: 2168432]
- 50.
- Blanchin S, Estienne V, Durand-Gorde J-M, Carayon P, Ruf J. Complement activation by direct C4 binding to thyroperoxidase in Hashimoto’s thyroiditis. Endocrinology. 2003;144:5422–9. [PubMed: 12960013]
- 51.
- Lynne Burek, Noel Rose PC. Thyroglobulin, Thyroperoxidase, and Thyrotropin-Receptor autoantibodies. In: Eric Gershwin PLM, editor. Yehuda Shoenfeld. Autoantibodies. Second. Oxford, UK: Elsevier; 2007. pp. 403–14.
- 52.
- Michalek K, Morshed SA, Latif R, Davies TF. TSH receptor autoantibodies. Autoimmun Rev. 2009;9:113–6. [PMC free article: PMC3753030] [PubMed: 19332151]
- 53.
- Seissler J, Wagner S, Schott M, et al. Low frequency of autoantibodies to the human Na(+)/I(-) symporter in patients with autoimmune thyroid disease. J Clin Endocrinol Metab. 2000;85:4630–4. [PubMed: 11134119]
- 54.
- Czarnocka B. Thyroperoxidase, thyroglobulin, Na(+)/I(-) symporter, pendrin in thyroid autoimmunity. Front Biosci. 2011;16:783–802. [PubMed: 21196203]
- 55.
- Punzi L, Betterle C. Chronic autoimmune thyroiditis and rheumatic manifestations. Joint Bone Spine. 2004;71:275–83. [PubMed: 15288851]
- 56.
- Soy M, Guldiken S, Arikan E, Altun BU, Tugrul A. Frequency of rheumatic diseases in patients with autoimmune thyroid disease. Rheumatol Int. 2007;27:575–7. [PubMed: 17102943]
- 57.
- Marqusee E, Hill JA, Mandel SJ. Thyroiditis after pregnancy loss. J Clin Endocrinol Metab. 1997;82:2455–7. [PubMed: 9253317]
- 58.
- Schwartzentruber DJ, White DE, Zweig MH, Weintraub BD, Rosenberg SA. Thyroid dysfunction associated with immunotherapy for patients with cancer. Cancer. 1991;68:2384–90. [PubMed: 1933775]
- 59.
- Chiovato L, Latrofa F, Braverman LE, et al. Disappearance of humoral thyroid autoimmunity after complete removal of thyroid antigens. Ann Intern Med. 2003;139:346–51. [PubMed: 12965943]
- 60.
- Goh SY, Ho SC, Seah LL, Fong KS, Khoo DH. Thyroid autoantibody profiles in ophthalmic dominant and thyroid dominant Graves’ disease differ and suggest ophthalmopathy is a multiantigenic disease. Clin Endocrinol. 2004;60:600–7. [PubMed: 15104563]
- 61.
- Gentile F, Conte M, Formisano S. Thyroglobulin as an autoantigen: what can we learn about immunopathogenicity from the correlation of antigenic properties with protein structure? Immunology. 2004;112:13–25. [PMC free article: PMC1782462] [PubMed: 15096179]
- 62.
- Fenouillet E, Fayet G, Hovsepian S, Bahraoui EM, Ronin C. Immunochemical evidence for a role of complex carbohydrate chains in thyroglobulin antigenicity. J Biol Chem. 1986;261:15153–8. [PubMed: 2429965]
- 63.
- Hutchings PR, Cooke A, Dawe K, et al. A thyroxine-containing peptide can induce murine experimental autoimmune thyroiditis. J Exp Med. 1992:175869–72. [PMC free article: PMC2119149] [PubMed: 1740668]
- 64.
- Caturegli P, Kuppers RC, Mariotti S, Burek CL, Pinchera A, Ladenson PW, et al. IgG subclass distribution of thyroglobulin antibodies in patients with thyroid disease. Clin Exp Immunol. 1994;98:464–9. [PMC free article: PMC1534515] [PubMed: 7994910]
- 65.
- Ericsson UB, Christensen SB, Thorell JI. A high prevalence of thyroglobulin autoantibodies in adults with and without thyroid disease as measured with a sensitive solid-phase immunosorbent radioassay. Clin Immunol Immunopathol. 1985;37:154–62. [PubMed: 3930112]
- 66.
- Szyper-Kravitz M, Marai I, Shoenfeld Y. Coexistence of thyroid autoimmunity with other autoimmune diseases: friend or foe? Additional aspects on the mosaic of autoimmunity. Autoimmunity. 2005;38:247–55. [PubMed: 16126513]
- 67.
- Clark PM, Beckett G. Can we measure serum thyroglobulin? Ann Clin Biochem. 2002;39:196–202. [PubMed: 12038593]
- 68.
- Magnusson RP, Chazenbalk GD, Gestautas J, Seto P, Filetti S, DeGroot LJ, et al. Molecular cloning of the complementary deoxyribonucleic acid for human thyroid peroxidase. Mol Endocrinol. 1987;1:856–61. [PubMed: 3153466]
- 69.
- Czarnocka B, Ruf J, Ferrand M, Carayon P, Lissitzky S. Purification of the human thyroid peroxidase and its identification as the microsomal antigen involved in autoimmune thyroid diseases. FEBS Lett. 1985;190:147–52. [PubMed: 2995127]
- 70.
- Weetman AP. Autoimmune thyroid disease. Autoimmunity. 2004:337–40. [PubMed: 15518055]
- 71.
- Nicholson WK, Robinson KA, Smallridge RC, Ladenson PW, Powe NR. Prevalence of postpartum thyroid dysfunction: a quantitative review. Thyroid. 2006;16:573–82. [PubMed: 16839259]
- 72.
- Mavragani CP, Danielides S, Zintzaras E, Vlachoyiannopoulos PG, Moutsopoulos HM. Antithyroid antibodies in antiphospholipid syndrome: prevalence and clinical associations. Lupus. 2009;18:1096–9. [PubMed: 19762385]
- 73.
- Misrahi M, Loosfelt H, Atger M, Sar S, Guiochon-Mantel A, Milgrom E. Cloning, sequencing and expression of human TSH receptor. Biochem Biophys Res Commun. 1990;166:394–403. [PubMed: 2302212]
- 74.
- Davies TF, Ando T, Lin R-Y, Tomer Y, Latif R. Thyrotropin receptor-associated diseases: from adenomata to Graves disease. J Clin Invest. 2005;115:1972–83. [PMC free article: PMC1180562] [PubMed: 16075037]
- 75.
- Abe E, Marians RC, Yu W, et al. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115:151–62. [PubMed: 14567913]
- 76.
- Núñez Miguel R, Sanders J, Chirgadze DY, Furmaniak J, Rees Smith B. Thyroid stimulating autoantibody M22 mimics TSH binding to the TSH receptor leucine rich domain: a comparative structural study of protein-protein interactions. J Mol Endocrinol. 2009;42:381–95. [PubMed: 19221175]
- 77.
- Adams DD. The presence of an abnormal thyroid-stimulating hormone in the serum of some thyrotoxic patients. J Clin Endocrinol Metab. 1958;18:699–712. [PubMed: 13549548]
- 78.
- Lazúrová I, Benhatchi K, Rovenský J, et al. Autoimmune thyroid disease and autoimmune rheumatic disorders: a two-sided analysis. Ann N Y Acad Sci. 2009;1173:211–6. [PubMed: 19758153]
- 79.
- Katakura M, Yamada T, Aizawa T, et al. Presence of antideoxyribonucleic acid antibody in patients with hyperthyroidism of Graves’ disease. J Clin Endocrinol Metab. 1987;64:405–8. [PubMed: 3493254]
- 80.
- Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87:489–99. [PubMed: 11836274]
- 81.
- Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223–43. [PubMed: 9281381]
- 82.
- Rocchi R, Rose NR, Caturegli P. Hahimoto Thyroiditis. In: Shoenfeld Y, Cervera R, Gershwin ME, editors. Diagnostic Criteria in Autoimmune Diseases. Humana Press; 2008. pp. 217–220.
- 83.
- Teng W, Shan Z, Teng X, et al. Effect of iodine intake on thyroid diseases in China. N Engl J Med. 2006;354:2783–93. [PubMed: 16807415]
- 84.
- Tamai H, Ohsako N, Takeno K, et al. Changes in thyroid function in euthyroid subjects with a family history of Graves’ disease: a follow-up study of 69 patients. J Clin Endocrinol Metab. 1980;51:1123–7. [PubMed: 6774999]
- 85.
- Holm LE, Blomgren H, Löwhagen T. Cancer risks in patients with chronic lymphocytic thyroiditis. N Engl J Med. 1985;312:601–4. [PubMed: 3838363]
- 86.
- Heufelder AE, Wenzel BE, Gorman CA, Bahn RS. Detection, cellular localization, and modulation of heat shock proteins in cultured fibroblasts from patients with extrathyroidal manifestations of Graves’ disease. J Clin Endocrinol Metab. 1991;73:739–45. [PubMed: 1890149]
- 87.
- Arata N, Ando T, Unger P, Davies TF. By-stander activation in autoimmune thyroiditis: studies on experimental autoimmune thyroiditis in the GFP+ fluorescent mouse. Clin Immunol. 2006;121:108–17. [PubMed: 16916620]
- 88.
- Neufeld DS, Platzer M, Davies TF. Reovirus induction of MHC class II antigen in rat thyroid cells. Endocrinology. 1989;124:543–5. [PMC free article: PMC7108507] [PubMed: 2491810]
- 89.
- Khoury EL, Pereira L, Greenspan FS. Induction of HLA-DR expression on thyroid follicular cells by cytomegalovirus infection in vitro. Evidence for a dual mechanism of induction. Am J Pathol. 1991;138:1209–23. [PMC free article: PMC1886009] [PubMed: 1850961]
- 90.
- Giordano C, Stassi G, De Maria R, Todaro M, Richiusa P, Papoff G, et al. Potential involvement of Fas and its ligand in the pathogenesis of Hashimoto’s thyroiditis. Science. 1997;275:960–3. [PubMed: 9020075]
- 91.
- Almandoz JP, Gharib H. Hypothyroidism: etiology, diagnosis, and management. Med Clin North Am. 2012;96:203–21. [PubMed: 22443971]
- 92.
- Scott KR, Simmons Z, Boyer PJ. Hypothyroid myopathy with a strikingly elevated serum creatine kinase level. Muscle Nerve. 2002;26:141–4. [PubMed: 12115960]
- 93.
- Tagoe CE, Zezon A, Khattri S. Rheumatic manifestations of autoimmune thyroid disease: the other autoimmune disease. J Rheumatol. 2012;39:1125–9. [PubMed: 22505695]
- 94.
- Becker KL, Ferguson RH, McConahey WM. The connective-tissue diseases and symptoms associated with Hashimoto’s thyroiditis. N Engl J Med. 1963;268:277–80. [PubMed: 13970125]
- 95.
- Bland JH, Frymoyer JW, Newberg AH, Revers R, Norman RJ. Rheumatic syndromes in endocrine disease. Semin Arthritis Rheum. 1979;9:23–65. [PubMed: 386520]
- 96.
- Bauer M, Silverman DHS, Schlagenhauf F, London ED, Geist CL, van Herle K, et al. Brain glucose metabolism in hypothyroidism: a positron emission tomography study before and after thyroid hormone replacement therapy. J Clin Endocrinol Metab. 2009;94:2922–9. [PubMed: 19435829]
- 97.
- Ebert EC. The thyroid and the gut. J Clin Gastroenterol. 2010 Jul;44:402–6. [PubMed: 20351569]
- 98.
- Krassas GE, Pontikides N, Kaltsas T, et al. Disturbances of menstruation in hypothyroidism. Clin Endocrinol. 1999;50:655–9. [PubMed: 10468932]
- 99.
- Bhasin S, Enzlin P, Coviello A, Basson R. Sexual dysfunction in men and women with endocrine disorders. Lancet. 2007;369:597–611. [PubMed: 17307107]
- 100.
- Hanna FW, Scanlon MF. Hyponatraemia, hypothyroidism, and role of arginine-vasopressin. Lancet. 1997;350:755–6. [PubMed: 9297992]
- 101.
- Duntas LH. Thyroid disease and lipids. Thyroid. 2002;12:287–93. [PubMed: 12034052]
- 102.
- Zulewski H, Müller B, Exer P, Miserez AR, Staub JJ. Estimation of tissue hypothyroidism by a new clinical score: evaluation of patients with various grades of hypothyroidism and controls. J Clin Endocrinol Metab. 1997;82:771–6. [PubMed: 9062480]
- 103.
- Spencer CA, LoPresti JS, Patel A, et al. Applications of a new chemiluminometric thyrotropin assay to subnormal measurement. J Clin Endocrinol Metab. 1990;70:453–60. [PubMed: 2105333]
- 104.
- Jensen E, Hyltoft Petersen P, Blaabjerg O, et al. Establishment of a serum thyroid stimulating hormone (TSH) reference interval in healthy adults. The importance of environmental factors, including thyroid antibodies. Clin Chem Lab Med. 2004;42:824–32. [PubMed: 15327019]
- 105.
- Hamilton TE, Davis S, Onstad L, Kopecky KJ. Thyrotropin levels in a population with no clinical, autoantibody, or ultrasonographic evidence of thyroid disease: implications for the diagnosis of subclinical hypothyroidism. J Clin Endocrinol Metab. 2008;93:1224–30. [PMC free article: PMC2729186] [PubMed: 18230665]
- 106.
- Baskin HJ, Cobin RH, Duick DS, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. Endocr Pract. 2002;8:457–69. [PubMed: 15260011]
- 107.
- Baloch Z, Carayon P, Conte-Devolx B, et al. Laboratory medicine practice guidelines. Laboratory support for the diagnosis and monitoring of thyroid disease. Thyroid. 2003;13:3–126. [PubMed: 12625976]
- 108.
- Wiersinga WM. Thyroid hormone replacement therapy. Horm Res. 2001;56:74–81. [PubMed: 11786691]
- 109.
- Fish LH, Schwartz HL, Cavanaugh J, Steffes MW, Bantle JP, Oppenheimer JH. Replacement dose, metabolism, and bioavailability of levothyroxine in the treatment of hypothyroidism. Role of triiodothyronine in pituitary feedback in humans. N Engl J Med. 1987;316:764–70. [PubMed: 3821822]
- 110.
- Hegedüs L, Hansen JM, Feldt-Rasmussen U, Hansen BM, Høier-Madsen M. Influence of thyroxine treatment on thyroid size and anti-thyroid peroxidase antibodies in Hashimoto’s thyroiditis. Clin Endocrinol. 1991;35:235–8. [PubMed: 1742880]
- 111.
- Roos A, Linn-Rasker SP, van Domburg RT, Tijssen JP, Berghout A. The starting dose of levothyroxine in primary hypothyroidism treatment: a prospective, randomized, double-blind trial. Arch Intern Med. 2005;165:1714–20. [PubMed: 16087818]
- 112.
- Carr D, McLeod DT, Parry G, Thornes HM. Fine adjustment of thyroxine replacement dosage: comparison of the thyrotrophin releasing hormone test using a sensitive thyrotrophin assay with measurement of free thyroid hormones and clinical assessment. Clin Endocrinol. 1988;28:325–33. [PubMed: 3139338]
- 113.
- Helfand M. Screening for subclinical thyroid dysfunction in nonpregnant adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2004;140:128–41. [PubMed: 14734337]
- 114.
- Bindra A, Braunstein GD. Thyroiditis. Am Fam Physician. 2006;73:1769–76. [PubMed: 16734054]
- 115.
- Samuels MH. Subacute, silent, and postpartum thyroiditis. Med Clin North Am. 2012;96:223–33. [PubMed: 22443972]
- 116.
- Volpé R. Is silent thyroiditis an autoimmune disease? Arch Intern Med. 1988;148:1907–8. [PubMed: 3415401]
- 117.
- Wilkins M, Moe MM. Acute painless thyroiditis with transient thyrotoxicosis during external beam irradiation to non-Hodgkin’s lymphoma of the thyroid gland. Clin Oncol (R Coll Radiol). 2001;13:311. [PubMed: 11554636]
- 118.
- Morita S, Ueda Y, Yokoyama N. Painless thyroiditis induced by the cessation of betamethasone. Intern Med. 2001;40:744–6. [PubMed: 11518115]
- 119.
- Yamakita N, Sakata S, Hayashi H, Maekawa H, Miura K. Case report: silent thyroiditis after adrenalectomy in a patient with Cushing’s syndrome. Am J Med Sci. 1993;305:304–6. [PubMed: 8484389]
- 120.
- Miller KK, Daniels GH. Association between lithium use and thyrotoxicosis caused by silent thyroiditis. Clin Endocrinol. 2001;55:501–8. [PubMed: 11678833]
- 121.
- Nishimaki M, Isozaki O, Yoshihara A, Okubo Y, Takano K. Clinical characteristics of frequently recurring painless thyroiditis: contributions of higher thyroid hormone levels, younger onset, male gender, presence of thyroid autoantibody and absence of goiter to repeated recurrence. Endocr J. 2009;56:391–7. [PubMed: 19225214]
- 122.
- Morita T, Tamai H, Oshima A, et al. The occurrence of thyrotropin binding-inhibiting immunoglobulins and thyroid-stimulating antibodies in patients with silent thyroiditis. J Clin Endocrinol Metab. 1990;71:1051–5. [PubMed: 1976125]
- 123.
- Mittra ES, McDougall IR. Recurrent silent thyroiditis: a report of four patients and review of the literature. Thyroid. 2007;17:671–5. [PubMed: 17696838]
- 124.
- Stagnaro -Green A. Pospartum Thyroiditis. In: Shoenfeld Y, Cervera R, Gershwin ME, editors. Diagnostic Criteria in Autoimmune Diseases. Humana Press; 2008. pp. 237–240.
- 125.
- Stagnaro-Green A. Approach to the patient with postpartum thyroiditis. J Clin Endocrinol Metab. 2012;97:334–42. [PubMed: 22312089]
- 126.
- Stagnaro-Green A, Glinoer D. Thyroid autoimmunity and the risk of miscarriage. Best Pract Res Clin Endocrinol Metab. 2004;18:167–81. [PubMed: 15157834]
- 127.
- Stuckey BGA, Kent GN, Ward LC, Brown SJ, Walsh JP. Postpartum thyroid dysfunction and the long-term risk of hypothyroidism: results from a 12-year follow-up study of women with and without postpartum thyroid dysfunction. Clin Endocrinol. 2010;73:389–95. [PubMed: 20184598]
- 128.
- Tachi J, Amino N, Tamaki H, Aozasa M, Iwatani Y, Miyai K. Long term follow-up and HLA association in patients with postpartum hypothyroidism. J Clin Endocrinol Metab. 1988;66:480–4. [PubMed: 3162458]
- 129.
- Lazarus JH, Hall R, Othman S, Parkes AB, Richards CJ, McCulloch B, et al. The clinical spectrum of postpartum thyroid disease. QJM. 1996;89:429–35. [PubMed: 8758046]
- 130.
- De Groot L, Abalovich M, Alexander EK, et al. Management of thyroid dysfunction during pregnancy and postpartum: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:2543–65. [PubMed: 22869843]
- 131.
- Stagnaro-Green A, Abalovich M, Alexander E, et al. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid. 2011;21:1081–125. [PMC free article: PMC3472679] [PubMed: 21787128]
- 132.
- Atisha -Fregoso Y, García-Carrasco M. Subacute Thyroiditis. In: Shoenfeld Y, Cervera R, Gershwin ME, editors. Diagnostic Criteria in Autoimmune Diseases. Humana Press; 2008. pp. 227–229.
- 133.
- Fatourechi V, Aniszewski JP, Fatourechi GZE, Atkinson EJ, Jacobsen SJ. Clinical features and outcome of subacute thyroiditis in an incidence cohort: Olmsted County, Minnesota, study. J Clin Endocrinol Metab. 2003;88:2100–5. [PubMed: 12727961]
- 134.
- Golden SH, Robinson KA, Saldanha I, Anton B, Ladenson PW. Clinical review: Prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab. 2009;94:1853–78. [PMC free article: PMC5393375] [PubMed: 19494161]
- 135.
- Martino E, Buratti L, Bartalena L, et al. High prevalence of subacute thyroiditis during summer season in Italy. J Endocrinol Invest. 1987;10:321–3. [PubMed: 3624803]
- 136.
- Nishihara E, Ohye H, Amino N, et al. Clinical characteristics of 852 patients with subacute thyroiditis before treatment. Intern Med. 2008;47:725–9. [PubMed: 18421188]
- 137.
- Pearce EN, Bogazzi F, Martino E, et al. The prevalence of elevated serum C-reactive protein levels in inflammatory and noninflammatory thyroid disease. Thyroid. 2003;13:643–8. [PubMed: 12964969]
- 138.
- Matsumoto Y, Amino N, Kubota S, et al. Serial changes in liver function tests in patients with subacute thyroiditis. Thyroid. 2008;18:815–6. [PubMed: 18631018]
- 139.
- 1Hiromatsu Y, Ishibashi M, Miyake I, et al. Color Doppler ultrasonography in patients with subacute thyroiditis. Thyroid. 1999;9:1189–93. [PubMed: 10646657]
- 140.
- Weihl AC, Daniels GH, Ridgway EC, Maloof F. Thyroid function tests during the early phase of subacute thyroiditis. J Clin Endocrinol Metab. 1977;44:1107–14. [PubMed: 874047]
- 141.
- Mizukoshi T, Noguchi S, Murakami T, Futata T, Yamashita H. Evaluation of recurrence in 36 subacute thyroiditis patients managed with prednisolone. Intern Med. 2001;40:292–5. [PubMed: 11334386]
- 142.
- Menconi F, Oppenheim Y, Tomer Y. Graves Disease. In: Shoenfeld Y, Cervera R, Gershwin ME, editors. Diagnostic Criteria in Autoimmune Diseases. Humana Press; 2008. pp. 231–235.
- 143.
- Brent GA. Clinical practice. Graves’ disease. N Engl J Med. 2008;358:2594–605. [PubMed: 18550875]
- 144.
- Weetman AP. Graves’ disease. N Engl J Med. 2000;343:1236–48. [PubMed: 11071676]
- 145.
- Cooper DS. Hyperthyroidism. Lancet. 2003;362:459–68. [PubMed: 12927435]
- 146.
- BBartley GB, Fatourechi V, Kadrmas EF, et al. The incidence of Graves’ ophthalmopathy in Olmsted County, Minnesota. Am J Ophthalmol. 1995;120:511–7. [PubMed: 7573310]
- 147.
- BBartley GB, Fatourechi V, Kadrmas EF, et al. Chronology of Graves’ ophthalmopathy in an incidence cohort. Am J Ophthalmol. 1996;121:426–34. [PubMed: 8604736]
- 148.
- Bartley GB, Fatourechi V, Kadrmas EF, et al. Clinical features of Graves’ ophthalmopathy in an incidence cohort. Am J Ophthalmol. 1996;121:284–90. [PubMed: 8597271]
- 149.
- Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011;17:456–520. [PubMed: 21700562]
- 150.
- Cárdenas Roldán J, Amaya-Amaya J, Castellanos-de la Hoz J, et al. Autoimmune thyroid disease in rheumatoid arthritis: a global perspective. Arthritis. 2012;2012:864907. [PMC free article: PMC3505628] [PubMed: 23209899]
- 151.
- Atzeni F, Doria A, Ghirardello A, et al. Anti-thyroid antibodies and thyroid dysfunction in rheumatoid arthritis: prevalence and clinical value. Autoimmunity. 2008;41:111–5. [PubMed: 18176873]
- 152.
- Biró E, Szekanecz Z, Czirják L, et al. Association of systemic and thyroid autoimmune diseases. Clin Rheumatol. 2006;25:240–5. [PubMed: 16247581]
- 153.
- Boelaert K, Newby PR, Simmonds MJ, et al. Prevalence and relative risk of other autoimmune diseases in subjects with autoimmune thyroid disease. Am J Med. 2010;123:1–9. [PubMed: 20103030]
- 154.
- Hemminki K, Li X, Sundquist J, Sundquist K. The epidemiology of Graves’ disease: evidence of a genetic and an environmental contribution. J Autoimmun. 2010;34:J307–13. [PubMed: 20056533]
- 155.
- Walker DJ, Griffiths M, Griffiths ID. Occurrence of autoimmune diseases and autoantibodies in multicase rheumatoid arthritis families. Ann Rheum Dis. 1986;45:323–6. [PMC free article: PMC1001877] [PubMed: 3458433]
- 156.
- Raterman HG, van Halm VP, Voskuyl AE, Simsek S, Dijkmans BC, Nurmohamed MT. Rheumatoid arthritis is associated with a high prevalence of hypothyroidism that amplifies its cardiovascular risk. Ann Rheum Dis. 2008;67:229–32. [PubMed: 17557891]
- 157.
- Raterman HG, Nielen MMJ, Peters MJL, Verheij RA, Nurmohamed MT, Schellevis FG. Coexistence of hypothyroidism with inflammatory arthritis is associated with cardiovascular disease in women. Ann Rheum Dis. 2012;71:1216–8. [PubMed: 22419774]
- 158.
- Hijmans W, Doniach D, Roitt IM, Holborow EJ. Serological overlap between lupus erythematosus, rheumatoid arthritis, and thyroid auto-immune disease. Br Med J. 1961;2:909–14. [PMC free article: PMC1969944] [PubMed: 13714204]
- 159.
- Wiebolt J, Koeleman BPC, van Haeften TW. Endocrine autoimmune disease: genetics become complex. Eur J Clin Invet. 2010;40:1144–55. [PubMed: 20718847]
- 160.
- Chistiakov DA, Turakulov RI. CTLA-4 and its role in autoimmune thyroid disease. J Mol Endocrinol. 2003;31:21–36. [PubMed: 12914522]
- 161.
- Benamour S, Zeroual B, Fares L, el Kabli H, Bettal S. Rheumatoid arthritis in Morocco. Apropos of 404 observations. Rev Rhum Mal Osteoartic. 1992;59:801–7. [PubMed: 1364064]
- 162.
- Herrmann F, Hambsch K, Müller P, Häntzschel H, Zugehör M. Incidence of goiter and thyroiditis in chronic inflammatory rheumatism. Z Gesamte Inn Med. 1990;45:52–5. [PubMed: 2327135]
- 163.
- Becker K, Titus J, Woolner L, Ferguson R. Thyroiditis and rheumatoid arthritis. Proceedings of the staff meetings. Mayo Clinic. 1963 Mar;38:125–9. [PubMed: 13970126]
- 164.
- Linos A, Worthington JW, Palumbo PJ, O’Fallon WM, Kurland LT. Occurrence of Hashimoto’s thyroiditis and diabetes mellitus in patients with rheumatoid arthritis. J Chronic Dis. 1980;33:73–7. [PubMed: 6965494]
- 165.
- McCoy SS, Crowson CS, Gabriel SE, Matteson EL. Hypothyroidism as a risk factor for development of cardiovascular disease in patients with rheumatoid arthritis. J Rheumatol. 2012;39:954–8. [PMC free article: PMC3518421] [PubMed: 22337246]
- 166.
- Shiroky JB, Cohen M, Ballachey M-L, Neville C. Thyroid dysfunction in rheumatoid arthritis: A controlled prospective survey. Ann Rheum Dis. 1993;52:454–6. [PMC free article: PMC1005071] [PubMed: 8323398]
- 167.
- Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87:489–99. [PubMed: 11836274]
- 168.
- Silman AJ, Ollier WER, Bubel MA. Autoimmune thyroid disease and thyroid autoantibodies in rheumatoid arthritis patients and their families. Br J Rheumatol. 1989;28:18–21. [PubMed: 2645007]
- 169.
- Mousa AA, Ghonem M, Hegazy A, El-Baiomy AA, El-Diasty A. Thyroid Function and Auto-antibodies in Egyptian Patients with Systemic Lupus Erythematosus and Rheumatoid Arthritis. Trends in Medical Research. 2012;7:25–33.
- 170.
- Nakamura H, Usa T, Motomura M, et al. Prevalence of interrelated autoantibodies in thyroid diseases and autoimmune disorders. J Endocrinol Invest. 2008;31:861–5. [PubMed: 19092289]
- 171.
- El-Sherif WT, El Gendi SS, Ashmawy MM, Ahmed HM, Salama MM. Thyroid disorders and autoantibodies in systemic lupus erythematosus and rheumatoid arthritis patients. The Egyptian journal of immunology / Egyptian Association of Immunologists. 2004;11:81–90. [PubMed: 16734120]
- 172.
- Ruggeri RM, Galletti M, Mandolfino MG, et al. Thyroid hormone autoantibodies in primary Sjögren syndrome and rheumatoid arthritis are more prevalent than in autoimmune thyroid disease, becoming progressively more frequent in these diseases. J Endocrinol Invest. 2002;25:447–54. [PubMed: 12035942]
- 173.
- Bizzaro N. The predictive significance of autoantibodies in organ-specific autoimmune diseases. Clin Rev Allergy Immunol. 2008;34:326–31. [PubMed: 18085442]
- 174.
- Vanderpump MP, Tunbridge WM, French JM, et al. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham Survey. Clin Endocrinol. 1995;43:55–68. [PubMed: 7641412]
- 175.
- Ortega-Hernandez OD, Pineda-Tamayo R, Pardo AL, Rojas-Villarraga A, Anaya JM. Cardiovascular disease is associated with extra-articular manifestations in patients with rheumatoid arthritis. Clinical rheumatology. 2009;28:767–75. [PubMed: 19277815]
- 176.
- DeMaria AN. Relative risk of cardiovascular events in patients with rheumatoid arthritis. Am J Cardiol. 2002;89:33D–38D. [PubMed: 11909559]
- 177.
- Charles PJ, Plant D, Chowdhury M, Worthington J, Venables P. Antibodies to thyroglobulin and thyroid peroxidase in rheumatoid arthritis: environmental and genetic associations. Ann Rheum Dis. 2011;70:A88–89.
- 178.
- White RG, Bass BH, Williams E. Lymphadenoid goitre and the syndrome of systemic lupus erythematosus. Lancet. 1961;1:368–73. [PubMed: 13784857]
- 179.
- Kumar K, Kole AK, Karmakar PS, Ghosh A. The spectrum of thyroid disorders in systemic lupus erythematosus. Rheumatol Int. 2012;32:73–8. [PubMed: 20658291]
- 180.
- Antonelli A, Fallahi P, Mosca M, et al. Prevalence of thyroid dysfunctions in systemic lupus erythematosus. Metabolism. 2010;59:896–900. [PubMed: 20005534]
- 181.
- Pyne D, Isenberg DA. Autoimmune thyroid disease in systemic lupus erythematosus. Ann Rheum Dis. 2002;61:70–2. [PMC free article: PMC1753864] [PubMed: 11779764]
- 182.
- Appenzeller S, Pallone AT, Natalin RA, Costallat LTL. Prevalence of thyroid dysfunction in systemic lupus erythematosus. J Clin Rheumatol. 2009;15:117–9. [PubMed: 19300286]
- 183.
- Scofield RH. Autoimmune thyroid disease in systemic lupus erythematosus and Sjögren’s syndrome. Clin Exp Rheumatol. 1996;14:321–30. [PubMed: 8809450]
- 184.
- Robazzi TCMV, Adan LFF. Autoimmune thyroid disease in patients with rheumatic diseases. Rev Bras Reumatol. 2012;52:417–30. [PubMed: 22641595]
- 185.
- Namjou B, Kelly JA, Kilpatrick J, et al. Linkage at 5q14.3–15 in multiplex systemic lupus erythematosus pedigrees stratified by autoimmune thyroid disease. Arthritis Rheum. 2005;52:3646–50. [PubMed: 16255059]
- 186.
- Park DJ, Cho CS, Lee SH, Park SH, Kim HY. Thyroid disorders in Korean patients with systemic lupus erythematosus. Scand J Rheumatol. 1995;24:13–7. [PubMed: 7863271]
- 187.
- Tunc R, Gonen MS, Acbay O, Hamuryudan V, Yazici H. Autoimmune thyroiditis and anti-thyroid antibodies in primary Sjogren’s syndrome: a case-control study. Ann Rheum Dis. 2004;63:575–7. [PMC free article: PMC1755005] [PubMed: 15082490]
- 188.
- Ramos-Casals M, Garcia-Carrasco M, Cervera R, et al. Thyroid disease in primary Sjögren syndrome. Study in a series of 160 patients. Medicine. 2000;79:103–8. [PubMed: 10771708]
- 189.
- Alfaris N, Curiel R, Tabbara S, Irwig MS. Autoimmune thyroid disease and Sjögren syndrome. J Clin Rheumatol. 2010;16:146–7. [PubMed: 20216331]
- 190.
- D’Arbonneau F, Ansart S, Le Berre R, Dueymes M, Youinou P, Pennec Y-L. Thyroid dysfunction in primary Sjögren’s syndrome: a long-term followup study. Arthritis Rheum. 2003;49:804–9. [PubMed: 14673967]
- 191.
- Hansen BU, Lindgren S, Eriksson S, et al. Clinical and immunological features of Sjögren’s syndrome in patients with primary biliary cirrhosis with emphasis on focal sialadenitis. Acta Med Scand. 1988;224:611–9. [PubMed: 3207072]
- 192.
- Punzi L, Ostuni PA, Betterle C, De Sandre P, Botsios C, Gambari PF. Thyroid gland disorders in primary Sjögren’s syndrome. Rev Rhum Engl Ed. 1996;63:809–14. [PubMed: 9010968]
- 193.
- Lazarus MN, Isenberg DA. Development of additional autoimmune diseases in a population of patients with primary Sjögren’s syndrome. Ann Rheum Dis. 2005;64:1062–4. [PMC free article: PMC1755577] [PubMed: 15958760]
- 194.
- Foster H, Fay A, Kelly C, Charles P, Walker D, Griffiths I. Thyroid disease and other autoimmune phenomena in a family study of primary Sjögren’s syndrome. Br J Rheumatol. 1993;32:36–40. [PubMed: 8422556]
- 195.
- Zeher M, Horvath IF, Szanto A, Szodoray P. Autoimmune thyroid diseases in a large group of Hungarian patients with primary Sjögren’s syndrome. Thyroid. 2009 Jan;19:39–45. [PubMed: 19119981]
- 196.
- Petri M, Karlson EW, Cooper DS, Ladenson PW. Autoantibody tests in autoimmune thyroid disease: a case-control study. J Rheumatol. 1991;18:1529–31. [PubMed: 1765977]
Publication Details
Author Information and Affiliations
Authors
Juan-Sebastián Franco, Jenny Amaya-Amaya, and Juan-Manuel Anaya.Copyright
Publisher
El Rosario University Press, Bogota (Colombia)
NLM Citation
Franco JS, Amaya-Amaya J, Anaya JM. Thyroid disease and autoimmune diseases. In: Anaya JM, Shoenfeld Y, Rojas-Villarraga A, et al., editors. Autoimmunity: From Bench to Bedside [Internet]. Bogota (Colombia): El Rosario University Press; 2013 Jul 18. Chapter 30.




