

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Gene Help [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2005-.

Introduction
Gene supplies gene-specific connections in the nexus of map, sequence, expression, structure, function, citation, and homology data. Unique identifiers are assigned to genes with defining sequences, genes with known map positions, and genes inferred from phenotypic information. These gene identifiers are used throughout NCBI's databases and tracked through updates of annotation. Gene includes genomes represented by NCBI Reference Sequences (or RefSeqs) and is integrated for indexing and query and retrieval from NCBI's Entrez and E-Utilities systems. Gene comprises sequences from thousands of distinct taxonomic identifiers, ranging from viruses to bacteria to eukaryotes. It represents chromosomes, organelles, plasmids, viruses, transcripts, and millions of proteins.
Quick Starttips
Gene is accessed like any other Entrez database, namely by
- querying on any word,
- restricting the query term to a certain field, or
- applying filters or properties
Here are some representative queries:
|
Find genes by... |
Search text |
|
free text | |
|
partial name and multiple species |
transporter[title] AND ("Drosophila melanogaster"[orgn] OR "Mus musculus"[orgn]) |
|
chromosome and symbol | |
|
associated sequence accession number | |
|
gene name (symbol) | |
|
publication (PubMed ID) | |
|
Gene Ontology (GO) terms or identifiers | |
|
genes with variants of medical interest | |
|
chromosome and species | |
|
Enzyme Commission (EC) numbers |
When you look at the URLs that underlie these links, you will see that they are constructed by combining ‘http://www.ncbi.nlm.nih.gov/gene/?term=’ with a query term qualified by field names (in square brackets).
How Data Are Maintained
New Records
Records are added to Gene if any of the following conditions is met:
- A RefSeq is created for a completely sequenced genome and that record contains annotated genes. In the case of prokaryotes, only reference genomes and representative genomes from well-sampled species are currently added to Gene. In the case of RNA viruses with polyprotein precursors, annotated proteins may be treated as equivalent to a ”gene”.
- A recognized genome-specific database provides information about genes (preferably with defining sequence) or mapped phenotypes.
- The NCBI Genome Annotation Pipeline reports model genes.
- A model organism is scheduled for sequencing, and representative sequences are identified to characterize known genes.
The minimum set of data necessary for a gene record, therefore, is: a unique identifier, or GeneID, assigned by NCBI; a preferred symbol; and either defining sequence information, map information, or official nomenclature from an authority list.
Gene records are not created for genomes which are incompletely represented by whole genome shotgun (WGS) assemblies. In terms of RefSeqs accessions, this means that genes annotated on accessions of the pattern NZ_ABCD12345678 are not submitted to Gene. Although not all existing records have been removed, loci defined by repetitive elements, endogenous retroviruses not named by nomenclature authorities, and loci identified by single transcripts with no other supporting data also are not in scope for Gene.
Numbering system
A unique GeneID is assigned to each new record. There are currently two number generators being used by Gene; one that is assigning values in the range of 7,000,000 – 99,999,999 and another that is assigning values > 100,000,000. Thus the sequence of GeneIDs is expected to have gaps.
Updates
Records are updated when new information is received. For some genomes, this may occur when a genome is re-annotated and the corresponding RefSeqs are updated. For other genomes, this may occur when any information attached to a single gene record is altered. Updates are processed daily.
Some components of the Gene record are updated automatically from other resources. Table 1 summarizes these data elements, their sources, and the update frequency. For example, GeneRIFs are processed independently of the Gene record. Most GeneRIFs are provided by the staff of the National Library of Medicine's Index Section and are integrated weekly. Those are available with the first update to Gene of the week. Public users are also invited to submit GeneRIFs, via the 'New GeneRIF' link in the Bibliography section of a Gene report.
Table 1.
Data sources for Gene.
When any change is made to a record, the modification date is changed. This includes changes in GeneRIFs. The modification date, therefore, is the later of any update to Gene or supplemental information.
About two days are required for an update to be reflected in all reports from Gene. In some cases, the full report may be more up-to-date than the ftp site because the ftp files are regenerated after a re-index of the database, a process that may lag a day behind the update to the database itself. The last modification date is available in the ftp files.
Suppressed Records
Gene will suppress a record for several reasons:
- Review by NCBI staff and/or collaborators indicates that a record is no longer supported or in scope for Gene. An explanation for the suppression is provided by RefSeq staff.
- Review by NCBI staff and/or collaborators indicates that the original record defined only part of what is now understood to be the functional gene unit. In that event, one record is made secondary to another, and the URL to the current record is provided.
- The molecular basis for a Gene record that was previously only a mapped phenotype is discovered, and there was already a record for the causative locus or loci. The record for the mapped phenotype is made secondary to one of the causative loci and added to the phenotype section of all.
By default, all records, i.e., current and suppressed, are retrieved by a query submitted with no restrictions. You can, however, restrict your results to current records. For example,
- click on Current only from the list filters in the Results filter sidebar at the left of your results display, or
- qualify your query with the phrase “AND alive[property]“
Query Tips provides additional details.
How content is selected
The content of a Gene record depends on availability of information and curatorial decisions. If you have suggestions about types of information that should be included in general, or for a specific record, please let us know by using our update form. More details about maintenance of certain types of information are provided in Gene’s FAQ.
How Data Are Displayed (Display Settings/Format)
NCBI's Entrez system supports multiple display options for each of its databases. The options available can be browsed by clicking on Display Settings (Figure 1). The options depend on whether you are viewing a set of results, or just one record. When viewing a set of results, in Tabular or Summary format for example, Display settings also provides choices for controlling the number of items to display, and their order. Additional customization of display formats and filtering options is possible by configuring your My NCBI preferences.
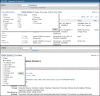
Figure 1.
Display Settings.There are two types of options to configure your display: from a query result (top) or from a record-specific display (bottom). The latter has no need to offer controls on the number of records to display or how to sort them.
Gene provides the following categories of formats:
Tabular
When you process a query, the results are displayed by default in the Tabular format (Figure 2). You can see that this is the Tabular format by noting the word Tabular at the top of the results section to the right of Display Settings.
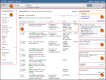
In the Tabular format, a check box is provided at the left of each record. The check box enables you to select which of the records in the retrieval set that you want to review in another format, according to your selection in Display Settings. If none is checked, all are displayed in the selected format.
The Tabular format includes the preferred gene symbol and unique identifier (the GeneID), the Description (including the complete gene name and species), the location on a genomic RefSeq for the reference assembly chromosome (if known), RefSeq accession.version, coordinates, orientation), other symbols and names, and for human only, the Mendelian Inheritance in Man (MIM) number for the gene. If a gene is not annotated on the reference assembly, a location is not reported. Also note that if a gene is on a named plasmid, then the plasmid name is given as the location.
The order of preference for displaying a symbol as preferred is:
- Official symbol
- Locus tag
- First symbol in the set of aliases
The Tabular display is also available as tab-delimited text that includes additional columns in a more parsable format. Gene aliases and other designations are also provided. In Display Settings, select the Tabular (text) format. There is an upper limit of 200 records that can be returned by this mechanism; to download a complete result set in Tabular(text) format, use the Send to: option at the upper right, select File, and Format Tabular (text).
Summary
The functions allowed from the Summary format (also known as the ‘docsum’) are similar to those described for the Tabular format. The Summary format contains similar information as the Tabular(text) format, including gene aliases and other designations.
UI List
This option displays only the unique identifiers (UIs) or GeneIDS for the records retrieved by your query (without the functions supported by Entrez).
Sort by
When the Tabular or Summary options are selected, the Display Settings menu also allows you to reorder the results. The options are:
- Relevance (the current default). Relevance is calculated from Gene's assessment of what fields are the most important by which to find search results. For example, Gene assigns more value to results that match a term in the 'Gene Name' (symbol) field compared to a match in free text such as the RefSeq or GeneRIF summary. Thus if your query is the single term 'cat', then records with symbols of 'cat' will be sorted ahead of records with the term cat only elsewhere in the record.
- Gene Weight. Gene Weight is calculated from multiple lines of evidence geared toward evaluating how well a gene has been characterized. These lines include:
- 1.
Informative Gene-PubMed links. Informativeness is inversely proportional to the number of Gene records connected to a PubMed record.
- 2.
Informative symbols or full names. A gene with a symbol constructed as LOC+GeneID is weighted less, for example, than a gene with the symbol 'ABCA1'. A gene with a description that starts with the word 'hypothetical' is weighted less than one with a description that starts with 'cystic fibrosis'.
- 3.
Inclusion in HomoloGene or Protein Clusters. Genes (or their products) that are known to be conserved are weighted more highly.
- 4.
Inclusion in OMIM or Books.
- Name. Results are sorted alphabetically (case insensitive) by the symbol of the gene.
- Chromosome. Results are sorted in the following order:
- 1.
Alphabetically by organism name
- 2.
Numerically by chromosome
- 3.
Numerically by the start position on the chromosome
For example, suppose that the search results include genes for Homo sapiens (human) and Mus musculus (mouse). The human genes will all appear before those for mouse. Within the set of human genes in the results, those that are placed on chromosome 1 will appear first, followed by those placed on chromosome 2, and so on. Finally, within a chromosome, genes will be sorted according to their start positions on the chromosome. Genes that are not placed on a chromosome will appear at the end of the results. Genes that are placed on multiple chromosomes will be sorted according to the first such chromosome.
Subset of data content
Gene Table
The Gene Table display represents the gene structure as annotated on the indicated genomic RefSeq. The default report is based on the reference assembly, but the selection menu in the top box (Figure 3) allows you to generate reports from other RefSeq genomic sequences.
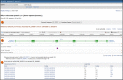
The report provides information about the intron/exon organization of each transcript, and, if an mRNA, the region of each exon that contains coding sequence. It does this in two ways:
- graphically, by repeating the display included in the Full Report
- in a table, by reporting the position of any exon or coding region, and reporting the length of exons, coding regions, and introns
The Gene Table display supports retrieval of gene-related sequence, as summarized in Table 2.
Table 2.
Access to Gene-specific sequence information from Gene.
Please note that Gene Table is not supported when the gene has not (yet) been annotated on any of NCBI's Genomic RefSeqs.
The sequence being retrieved is from the indicated genomic sequence, not the RNA. This means that the length of any non-aligning nucleotides, including a poly(A) tail or vector sequence, is not included in the GeneTable report.
Unaligned tails can be displayed graphically in the Sequence Viewer; follow the Open Full View link on Gene’s Full Report, click Configure on the right side of the Graphical Panel, and add RefSeq Alignments from the Alignments Track tab. Unaligned tails are displayed as boxes with the number of aligned bases shown above. Note that RefSeq transcripts with perfect alignments (excluding poly(A) tail) are NOT displayed in the RefSeq Alignment track. More information on how features are rendered in the Sequence Viewer is available from the Graphical Panel Legend section of the Sequence Viewer Help document.
When following a link from GeneTable to the sequence-specific nucleotide or protein record, use the Display Settings options there to generate the format you prefer (e.g. GenBank).
Because Gene Table reflects the annotation on the current genomic sequence, for bulk access you may prefer to use one of the General Feature Format (GFF, version 3) files in the species-specific GFF subdirectory. For example:
Please note that RefSeq may update annotation on sequences representing a genome less frequently than updates to gene-specific RefSeqs. This means that if the version of a RefSeq RNA has changed, or if the number of transcript variants has changed, the GeneTable display will be out of date with respect to the Reference Sequences section of the full Gene report. Please check also the Reference Sequences section of the Gene record to determine whether updates have occurred (new versions and/or more variants and/or suppression resulting from review).
Please see Table 2 for a summary of how to access gene-specific sequence information via Gene.
GeneRIF
The GeneRIF display for a Gene can also be accessed by a URL constructed as:
http://www.ncbi.nlm.nih.gov/gene/GENEID/?report=generif, where a GeneID replaces GENEID.
Example for GeneID 1059: http://www.ncbi.nlm.nih.gov/gene/1059/?report=generif
This display lists the text of the GeneRIF (which anchors a link to PubMed), the title of the paper, and the authors.
The PubMed (GeneRIF) display provides a listing of all the PubMed uids that are associated with GeneRIFs AND interaction data for a GeneID. Thus the count of GeneRIFs displayed for a gene may differ from the number of results in PubMed when the PubMed (GeneRIF) link is used.
Full Reports
All of the content that Gene provides is defined by the ASN.1 file. The Full Report display is of the HTML transformation of that ASN.1 and includes navigation tools (Table of contents and Related information), discovery elements, diagrams, and text. Some gene-specific information is not maintained in Gene but is maintained in more specialized databases such as GEO and HomoloGene. Access to the additional information maintained in other resources within NCBI or external to NCBI is provided by the listings under Related information (on the right beneath the Table of contents) and by other HTML anchors within the page.
The Full Report display is divided into the gray Search bar (explained in Query tips), navigation and discovery functions at the right, and content elements divided by horizontal separators that display or hide that subsection.
Navigation/Discovery column
The menu at the right of the Gene report supports navigation to multiple sites of interest. Each submenu can be expanded and compressed by clicking on the down ( ) or up (
) or up ( ) arrows, respectively.
) arrows, respectively.
Table of contents
lists the subcategories (or content elements) of information available for a gene. Clicking on a subcategory name takes you to that portion of the gene record.
Genome Browsers
provides links to NCBI and non-NCBI genome browsers, such as Genome Data Viewer, Map Viewer, Variation Viewer, 1000 Genomes Browser, the Ensembl browser, and the UCSC browser.
Related information
indicates other Entrez databases (or report types) that reference Gene. Each line anchors a link to gene-specific data in those databases/reports (Figure 4).
Links to other resources
names sites or files outside of the Entrez system that contain information specific to a gene record. When the identifier for that database needs to be displayed, the link may be repeated in other sections of the full report, such as the Summary or Additional Links sections.
General information
enumerates resources that may help you find and understand the information in Gene. The Help link goes to the default help document. The default help document is also accessed by the question marks ( ) in the horizontal section separators.
) in the horizontal section separators.
Related sites
provides links to home pages of a subset of Entrez databases likely of interest to users of Gene.
Feedback
enumerates several sites where you can comment on or add data to Gene and/or RefSeq.
Subscription
provides a link where you can subscribe to a mailing list to receive announcements about updates to RefSeq.
Recent activity
displays your recent database searches and document views. You can click on any to return to the results of that query or that document.
Content elements
Each content element is divided by a horizontal separator. The arrow at the left end of the separator allows you to open () or close () the display of that section. The arrows pointing up at the right end of the separator ( ) will return you to the top of the page should you want to make a different selection from the Table of contents. A link to this Help document is also provided ().
) will return you to the top of the page should you want to make a different selection from the Table of contents. A link to this Help document is also provided ().
Title
The section immediately below Display Settings/Send to: (Figure 5) provides the preferred symbol and descriptive name in bold font, followed by the italicized binomial in brackets. If there is a recognized authority for the gene nomenclature of a species, then that authority is the source for these values.
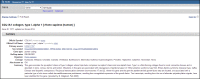
Figure 5.
Representative Title and Summary sections of a Full Report.
The second line of this section contains the NCBI GeneID and the last date a record was changed. The date is in the format day-month-year. Change is defined as any modification to the content of the record, including ancillary changes such as the URL for a displayed link. If a record was merged or discontinued, that information is provided also.
Summary
The section (Figure 5) may include several categories of information, namely:
Official Symbol: and Name: Nomenclature provided by the named external authority.
Primary source: Identifier and link to the major resource outside of NCBI that provided information about this gene. For some taxa, this resource may be the nomenclature authority; in other taxa it may be the group that defines genes and submits annotation to public sequence databases.
Locus tag corresponds to the systematic feature qualifier used by the international sequence collaboration (INSDC, DDBJ/EMBL/GenBank) and can be assigned by sequence submitters as a unique, systematic gene descriptor. When such a value is not available from submitted sequence, the identifier from a collaborating model organism database is used. Locus tag is often used to anchor a link to a database other than Gene. Locus tag may also be used as the preferred symbol if an official symbol has not been identified for a gene.
See related: A listing of other identifiers for this gene, provided as database name/value pairs.
Gene type: Possible values are tRNA, rRNA, snRNA, scRNA, snoRNA, miscRNA, ncRNA, protein coding, pseudo, other, and unknown. These are indexed as properties of a gene. Descriptions of these gene types are detailed at properties.
Feature type(s): Feature types annotated on RefSeq(s) associated with genes with a biological region Gene type. Annotated INSDC features are listed along with feature classes or controlled vocabularies in the feature_type: feature_class or feature_type: controlled vocabulary, where each INSDC feature_type is listed on a separate line, and multiple feature_class or controlled_vocabulary terms associated with each feature_type are provided in a comma-separated list following the colon, e.g.:
misc_feature: conserved_region
regulatory: TATA box, locus_control_region, promoter, transcriptional_cis_regulatory_region
These are indexed as properties of a gene, and related feattype properties are listed in the Properties section below.
RefSeq status: Any of the set of status descriptions defined by RefSeq. The aim is to describe the gene-level curation status for a given locus, defined as the best RefSeq status found on any of the RefSeq records (NM_, NR_, NG_, XM_, XR_ accession records) associated with the gene, ranked in the order: reviewed > validated > provisional > inferred > predicted > model. In particular, note that an individual locus may be represented by both known (NM_, NR_) and model (XM_, XR_) RefSeq records, and the Gene RefSeq status is based on the known RefSeq records. In this case, the models are provided as supplemental information. Further information about RefSeq statuses and record curation is available on the RefSeq site.
Organism: The binomial, and strain when appropriate, with a link to NCBI’s Taxonomy resource.
Lineage: Binomial and lineage from the Taxonomy database.
Also known as: Unofficial symbols and descriptions that have been used for this gene and its products. If there is no official symbol, and no locus_tag, the symbol at the top of the display is repeated in this section. These names are integrated from several sources, including model organism databases, annotation on sequence records, and interactive curation from the published literature.
Annotation information: Information about annotation oddities for a gene on the reference assembly. May be a report from NCBI’s genome annotation pipeline or a comment written by a RefSeq curator to explain how a gene is (or is not) represented in NCBI’s annotation. Not provided if the RefSeq group does not provide annotation for a genome or if there are no problems in the annotation.
Summary: Descriptive text about the gene, its cellular localization, its function, its expression, and its effect on phenotype. Records with a summary section can be retrieved by use of the property has_summary (Table 3).
Table 3.
Other properties in Gene (excluding those related to genetype, rnatype, source, srcdb refseq, and feattype).
Expression: A teaser sentence briefly describing tissue-specific expression of the gene, based on data in the Expression section (see below). A gene is considered to be expressed in a particular sample if it is at a level >=5% of the expression seen in the most strongly expressing sample. Please note that for organisms with expression data from multiple projects, the teaser sentence and indexing are only available on data in the primary expression dataset.
Orthologs: Orthologous genes as determined from genome annotation pipeline data. These are also reported in the file gene_orthologs.gz available by FTP.
Genomic Context
The Genomic Context section (Figure 6) reports the location of the gene on the chromosome in non-sequence coordinates. The section also provides information on the primary sequence location of the gene, which is the location(s) on the primary assembly of the current reference assembly, or the location(s) on alternate loci or alternate assemblies if the gene is not annotated on the reference primary assembly. For many genes, including those annotated using NCBI’s Eukaryotic Genomic Annotation Pipeline, the sequence location information is provided as a table that includes the Annotation Release number and Assembly name. Note that the Assembly column includes a link, by accession, to NCBI’s Assembly database.
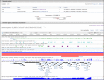
To facilitate working with previous assembly versions, the sequence coordinates from the last annotation of the previous assembly version may also be listed. This feature is currently limited to human, where the location on the GRCh37.p13 assembly is provided, but will be expanded to more organisms with future assembly updates. A link to NCBI's Genome Data Viewer resource is provided in the upper right hand corner of this section.
If the gene is included in the current genome annotation, the section also diagrams neighboring genes and indicates their orientations. If the name of a gene is too long to use for a label, it is truncated and marked with an ellipsis (...). The gene being shown on the diagram is in maroon. All other diagrams and labels anchor links to specific Gene pages, supporting quick navigation to review neighboring genes by clicking in the area of the symbol/arrow.
The diagram shows the gene’s placement on any and all chromosomes in the current genome annotation. Otherwise, the diagram will show another genomic placement in the current genome annotation in this order of precedence: reference contig; reference genomic region (NG); alternate assembly chromosome; alternate assembly contig. The location information for all current placements will be provided in the ASN.1 of the record and in the Reference Sequences Section. If a gene is not included in the current genome annotation, no diagram is provided.
Genomic Regions, Transcripts, and Products
The Genomic Regions, Transcripts, and Products section (Figure 6) is provided when a gene has been annotated on a genomic RefSeq, in other words, when the intron/exon/coding region information, or the position of a pseudogene, is available in some genomic coordinate system. The display in this section is generated from NCBI’s Sequence Viewer, the same software that drives the Graphics sequence display option available from the sequence databases, and provides some of the navigation features. A legend describes how annotated features are rendered in this display, and a link in the top right hand corner of the sequence panel provides complete Help documentation.
Depending on the data that are available, you can add data tracks to the display using the Configure button in the top right hand corner of the graphical panel to:
- view the intron/exon/coding region organization of a gene and its RNA product(s), or the placement of a pseudogene, on a genomic RefSeq
- identify the RefSeqs that correspond to any RNA or protein product and see an overview of the exons they represent
- evaluate expression under different experimental conditions by adding RNA-seq tracks
- explore differences between genome assemblies
You may also:
- alter the zoom level of the display (more…)
- hover over a feature to display information about it via a tool tip
- move upstream and downstream of the sequence being displayed (more…)
- navigate to a full display of the genomic context via the link to Graphics
- navigate to the genomic sequence of the gene in FASTA format
- navigate to the genomic sequence of the gene in GenBank format
- change the display of the genomic sequence on which the gene is annotated. The default display is the chromosome of the reference assembly; for some taxa there are alternate assemblies. For human, the RefSeqGene can also be selected
Each position of a gene product, when represented by a RefSeq RNA and/or protein, is provided relative to the genomic accession on which it is annotated. For some species, including human and other vertebrates, the genomic RefSeqs are updated independently of the annotated product RNAs, with the latter being updated more frequently. This means that several kinds of discrepancies between the diagram and the current RefSeq RNAs may result.
- The diagram may be labeled with an mRNA accession for a predicted model transcript (of the format XM_123456 or XM_123456789), yet display of that accession from Entrez Nucleotide indicates that the accession is no longer primary. That means that a curated mRNA (accession of the format NM_123456 or NM_123456789) has been generated to replace the previous model accession. The new "NM" accession will be reported in the Reference Sequences section of Gene.
- The diagram may be labeled with curated RNA accession numbers (of the format NM_123456 or NM_123456789 or NR_123456) different from those listed in the Reference Sequences section. This will result if curation after the submission of the annotated genome identified more transcript variants, which therefore are listed only in the Reference Sequence section and not in the diagram. It will also result if curation after submission of the annotated genome identified an error in the annotated product, and the accession for that product was suppressed. In that case, the Genomic regions, transcripts and products section will indicate a transcript not listed in the Reference Sequences section of the Gene report.
- The diagram may be labeled with a curated RNA accession number that represents a previous version of the accession. A version number change (e.g., NM_321321.1 -> NM_321321.2) occurs to a RefSeq record when there is any update to the sequence of that record. Sequence updates include the alteration, addition, or removal of nucleotides or amino acids from a record. Older RefSeq records (NM_321321.1) may be labeled on the diagram but updated RefSeq records (NM_321321.2) will be reported in the Reference Sequences section of Gene. The diagram shows the RefSeq records that were annotated in the last release while the Reference Sequences section shows the current version of the RefSeq records. The diagram is updated upon a new annotation release. Between releases, BLAST2SEQ can be used to determine sequence differences between older and newer RefSeq records.
Changing the zoom level in the display
- Select and display only a subsequence. Left click in the white section with the coordinates and ruler, and drag to select your region of interest. Then, right click, select zoom on range, and the display will refresh to provide the region of interest.
- Use the in/out zoom functions. Right click, and select either zoom in or zoom out. The display will refresh and change the region displayed by a factor of 2.
Move upstream and downstream
- A single left click anywhere in the display other than the ruler section, followed by a drag, results in a shift to display upstream and downstream sequence.
Expression
Expression data for some genes are now being displayed graphically after the Genomic regions, transcripts, and products section. Records with an expression data section can be retrieved by use of the property “has expression data”, while other properties exist to look for different expression patterns (Table 3). The data are computed from RNA-seq alignments compared to the most recent RefSeq gene models on the reference genome, and then normalized by RPKM (Reads Per Kilobase of transcript per Million mapped reads).
Because methodologies differ between RNA-seq projects, the data are binned by specific BioProject to reduce variability. If more than one BioProject is available, they can be accessed separately using the toggle bar at the top of the section.
Many BioProjects include a number of replicates per sample. In these cases, the data are shown per sample and are reported as an average \+/- standard deviation. However, the full data are provided in a table accessible by going to the See details link at the top right corner of the Expression section. The table lists the data by sample, and each sample has a + button that can be used to display the replicate data for each sample.
The data for the single BioProject shown can be downloaded by selecting the Download button in the top right corner of the See details page. The full dataset in XML format is available from the Gene FTP site.
Note: NCBI's staff have chosen studies which sample a range of representative tissues, with internal replicates of each tissue where possible. The expression profiles displayed provide a reliable starting point for assessing the expression of genes in the body. For interpretation of the data, researchers should consider examining several independent relevant studies rather than relying on any single assay. Please see PMID: 26996076 for a discussion of the challenges of making RNA-seq expression data relevant to the clinical setting.
Bibliography
The Bibliography section (Figure 7) may have two components:
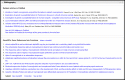
Figure 7.
Representative Bibliography section displaying articles in PubMed and GeneRIFs. If the number of citations exceeds 5 (PubMed) or (10) GeneRIFs, the first 5 or 10 are displayed, along with the total count and a link to the display of all records.
- A.
An embedded display of a subset of PubMed citations.
- B.
An embedded display of a subset of GeneRIFs.
The approach in both components is to display a limited number of records within the full display (5 for PubMed, 10 for GeneRIF), provide a count of the total records available, and support links to a display of all records. The GeneRIFs component also provides a link to submit a new GeneRIF for the gene, or to submit a request to the RefSeq curators to review information in the record.
What is a GeneRIF?
A GeneRIF is a concise phrase describing a function or functions of a gene, with the PubMed citation supporting that assertion. The majority of GeneRIFs have been provided by a collaboration between the NLM's Index Section and NCBI. There is no constraint on the number of independent submissions of GeneRIFs per PubMed id, although those from non-NLM sources are reviewed by RefSeq staff. The GeneRIF homepage provides more information about the project, including how general users can make submissions. If more than one GeneRIF for a gene has the same text but a different citation, the link to PubMed (icon at the left) will result in a display of all citations.
Each species has a GeneID with the symbol NEWENTRY. When staff of the NLM indexer sections cannot identify the gene to which a publication belongs, the GeneRIF is connected to the NEWENTRY, which is a placeholder for all the 'unconnected' GeneRIFs for a species. The GeneRIF text remains associated with the NEWENTRY GeneID until a RefSeq curator can identify or create the specific gene or genes to which the submission should be connected.
The full display of GeneRIFs for a gene can be generated at any time by selecting GeneRIF as the format from Display Settings.
Phenotypes
This section reports the effect of the gene on phenotype, especially disease. For human genes (Figure 8), the first row links to the NIH Genetic Testing Registry (GTR), a central location for genetic test information that is submitted voluntarily by test providers. The second row links to the Phenotype-Genotype Integrator, (PheGenI, pronounced FEE-GEE-NEE), a web portal providing a tabular display of genome-wide association study results relating the gene and/or its expression to a phenotype. PheGenI includes links to Genotype-Tissue Expression (GTex) results and viewers to display the relationships among genetic variants at the nucleotide level. Subsequent rows of the Phenotypes section may display the following:
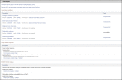
Professional guidelines: As professional practice guidelines, position statements, and recommendations are identified that relate to a disorder, gene, or variation, staff at NCBI connect them to the appropriate records. An alphabetical list of many of these guidelines can be found here: MedGen summary of professional guidelines
You can also identify all conditions associated with guidelines via this URL: http://www.ncbi.nlm.nih.gov/medgen?term="has%20guideline"[Properties]
Associated conditions: each row of a named phenotype provides links to more information, as available. In the case of human disease, this may include links to MedGen, OMIM, and GeneReviews; a link to the NIH Genetic Testing Registry (GTR) comparing laboratories offering the test may also be provided.
Copy number response: provides evidence of dosage sensitivity (either haploinsufficiency or triplosensitivity) as determined by the ClinGen group (https://www.clinicalgenome.org/).
NHGRI GWAS Catalog: provides a link to the SNP-trait associations reported in the NHGRI Catalog of Genome-Wide Association Studies, and the associated PubMed citation.
Variation
The section is designed to make it easier to navigate to gene-specific reports of sequence variation in NCBI's major variation resources, namely (1) dbSNP for variations of length less than approximately 50 bp, (2) dbVar for longer variations, including complex rearrangements, and (3) ClinVar, for the subset of both types of variation that may have medical relevance. ClinVar is available only for human. For human genes where variation may be related to a condition, and as practice guidelines, position statements, and recommendations are developed, links to Professional guidelines may be provided in the Phenotypes section.
The links that are provided to ClinVar and dbVar are equivalent to the links provided to those resources in the Related information section at the right.
To view, search, and navigate the human variations in dbSNP, dbVar, and ClinVar in a genomic context, follow the links to 'See Variation Viewer …'. Links to the GRCh38 and GRCh37.p13 assemblies are available.
There are several types of links provided for data in dbSNP:
- See SNP Geneview Report is equivalent to the link named SNP: GeneView in the Related information section. It displays by default only the variants in the coding region (note that cSNP is checked). To see all variations, select 'in gene region' instead. Note that this page also supports downloads.
- See SNP Genotype Report is equivalent to the link named SNP: Genotype in the Related information section. It displays information about populations and submitters of genotype data in the region of gene. An LD plot is also provided.
- See SNP Variation Viewer report is equivalent to the link named SNP: VarView in the Related information section and is available only for human. This display makes it easier to display both medically relevant and all short variations submitted to dbSNP in the region of a gene.
HIV-1 interactions
This subcategory is divided further into Replication interactions and Protein interactions.
Replication interactions
This section reports human proteins shown to be required for HIV-1 infectivity and replication. The interaction data are provided by the Southern Research Institute (https://southernresearch.org/) based on published whole genome screens that used small interfering RNAs. The data is provided without review by Gene staff; if you identify an incorrect or missing interaction, please contact the external source directly for correction. The display on human records reports:
- a concise description of the interaction
- links to papers in PubMed that support the described interaction
Protein interactions
The HIV-1, Human Protein Interaction Database is funded by the Division of Acquired Immunodeficiency Syndrome (DAIDS) of the National Institute of Allergy and Infectious Diseases (NIAID). As the title indicates, this project focuses on the human proteins that have been shown to interact with proteins from HIV-1. Interaction data is provided solely by the HIV-1, Human Protein Interaction Database without review by Gene staff; if you identify an incorrect or missing interaction, please contact the external source directly for correction. The format of this section is different for the human and HIV-1 gene reports. For human, the display consists of:
- the HIV-1 protein, linked to the sequence record in the Protein database
- the HIV-1 gene, linked to the Gene record for that gene product
- a concise description of the interaction
- links to papers in PubMed that support the described interaction
For HIV-1, the display is subdivided by peptide name and includes:
- a key word categorizing the interaction
- the full name of the human gene, linked to the Gene record
- links to papers in PubMed that support the described interaction
Please note that there are separate reports from this section that are available for download, both from the HIV-1, Human Protein Interaction Database homepage and the GeneRIF subdirectory of the Gene FTP site.
Interactions
The general interactions in this section are provided, without review by Gene staff, by the external sources listed in ftp://ftp.ncbi.nlm.nih.gov/gene/GeneRIF/interaction_sources. If you identify an incorrect or missing interaction, please contact the external source directly for correction. Interactions are reported as pairs where the product of the gene that is part of the interaction is given in the first column. If there are more than 25 pairs, pagination is provided. Depending on the type of interaction, the rest of the display may report:
- the other interactant, anchoring a link to more information
- the gene name of the other interactant, anchoring a link to that record in Gene
- the complex to which the interactant(s) belongs
- the source of these data, anchoring a link to the record at that source
- links to papers in PubMed that support the described interaction
- a concise description of the interaction, if available
General Gene Information
This section includes several subcategories of information, including:
Homology: a partial listing of orthologs in other species reported from different sources, including HomoloGene. NCBI's Eukaryotic Genome Annotation Pipeline provides the Orthologs from Annotation Pipeline report calculated using a combination of protein sequence similarity and local synteny information. Orthology is determined between a genome being annotated and a reference genome, typically human, and the set of pairwise orthologs tracked as a group and reported here. Links to a comparative display in Genome Data Viewer and to the OrthoDB catalog of eukaryotic orthologs also may be provided.
GeneOntology (GO): Specific GO terms provided by the Gene Ontology Annotation Database and listed by category and term, with evidence information and links to supporting publications. Each GO term supports a link to the AmiGO browser. Abbreviations in the Evidence Code column indicate the level of support for assigning a GO term to a gene. Explanations for these abbreviations are provided by the Gene Ontology website.
Gene does not alter the associations provided by a model organism database, nor does Gene recapitulate the directed acyclic graph structure provided by GO. Thus, Gene does not support retrieval of all genes associated with a specific GO term based on that term's parent. If you identify a GO term that is inappropriate for a gene, please contact the model organism database directly. ftp.ncbi.nlm.nih.gov/gene/DATA/go_process.xml documents the authorities Gene uses to connect GO terms to GeneIDs.
Genotypes: Links to various reports from dbSNP about allele frequencies in one or more populations, all variations for a gene, or disease-associated variations.
Markers: An enumeration of the markers that are related to this gene. The relationship is reported based on direct reports. Links are provided to the NCBI Probe database.
Readthrough: Information about genes that are sometimes transcribed with others. More information about readthrough transcription and how these events are represented in Gene are described in a FAQ.
Related gene/pseudogenes: If a gene, provides a link to view the records of pseudogenes related to the functional gene. If a pseudogene, provides a link to the functional gene.
Related region gene/members: Region records in Gene define officially named loci that are composed of multiple parts or represent clusters of related genes. If the record defines a region, provides a link to all members of the region. If a member of the region, provides a link to the region record.
Relationships: This section reports some of the public sequences that were used to support the prediction of the indicated RefSeq model. The report is not comprehensive and is provided only for those genomes for which NCBI calculates annotation, and only for those genes where there is not a supporting curated RefSeq.
The above relationships between two or more genes are reported in the file gene_group.gz available by FTP.
General Protein Information
This section applies only to genes that encode proteins. It reports the name or names that have been assigned to proteins encoded by the gene and provides other descriptive text. The names are as annotated on the RefSeq protein, when that protein is available. The sources of these names include model organism databases, annotation on public sequence databases, and curation by RefSeq staff.
NCBI Reference Sequences (RefSeqs)
This section describes the gene-specific NCBI reference sequences (RefSeqs) that have been established for this gene. In addition to enumerating the accession numbers and providing links to the appropriate Entrez sequence database, this section may also include descriptions of each transcript variant, accession numbers of the public sequences used to support any transcript, links to matching related Ensembl transcripts and proteins, and a listing of computed domains in an encoded protein. The text provided in this section therefore supports retrieving gene records based on descriptions of conserved domains.
The Reference Sequence group uses several approaches in maintaining information. These can be broadly categorized as:
- 1.
RefSeqs maintained independently of Annotated Genomes (Figure 9). RefSeqGene and RefSeq RNA and protein sequences are updated continuously, independently of any comprehensive reannotation of a genome. Because these reference sequences are curated independently of the genome annotation cycle, their versions may not match the RefSeq versions in the current genome build. You can identify updates by comparing versions in this section to versions in the Genomic regions, transcripts, and products section. GenBank and FASTA and Sequence Viewer (Graphics) anchor links to sequence in the given formats
- 2.
RefSeqs of Annotated Genomes (Figure 10). This section reports genomic RefSeqs from all assemblies on which this gene is annotated, such as RefSeqs for chromosomes and scaffolds (contigs) from both reference and alternate assemblies. The position and strand of the gene feature is provided (offset 1). GenBank and FASTA and Sequence Viewer (Graphics) anchor links to sequence in the given formats. Model RNAs and proteins are also reported here.
- 3.
Genome Annotation. RefSeq RNA and protein sequence are provided only through the process of genome/chromosome annotation.
- 4.
Suppressed Reference Sequence(s). Accession numbers listed in this section were suppressed for the cited reason(s). Suppressed RefSeqs do not appear in BLAST databases, related sequence links, or BLAST links (BLink) but may still be retrieved by from the Nucleotide or Protein databases, and by clicking on the hyperlinked accession.version.
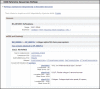
Figure 10.
Representative subsection RefSeqs of Annotated Genomes in the NCBI Reference Sequences (RefSeq) section of a Full Report display. This subsection follows RefSeqs maintained independently of Annotated Genomes (Figure 9). It includes the accession numbers (more...)
Related Sequences
This section has two subsections, one in which the nucleotide sequence is primary and one for protein sequences only (GenPept or UniProtKB). It contains sequence accessions that are related to the gene and provides links to the appropriate sequence record in Entrez Nucleotide, Entrez Protein or UniProtKB. It is not intended to be a comprehensive list of all sequences related to any gene; such sequences can more explicitly be found by using BLAST to query sequence databases or by using pre-calculated reports of related sequences via Entrez Nucleotide, Entrez Protein, or BLink. The sequence accessions in this section are provided in a tab-delimited format in the gene2accession.gz file in the DATA directory of the Gene FTP site.
Depending on the genome of the gene being reported, the sequences included may or may not be restricted to the same subspecies or strain.
Gene purposely lists protein accessions on records represented as not protein-coding. The intent is to make the connection between sequence annotation and Gene's current representation of the type of gene. For example, a nomenclature group may call a gene protein-coding or UniProt may create a sequence record for a protein based on an open reading frame, but RefSeq staff may judge the evidence to be weak based on a lack of cross-species homology or experimental support. Gene will report the protein sequences derived from the locus but will represent the gene as not protein-coding consistent with the RefSeq curation decision. Records of this type are reviewed periodically as new evidence is made available.
Users with evidence indicating that the Gene record should be reviewed are encouraged to contact RefSeq staff.
Accessions are reported as related sequences based on several criteria:
- mRNAs with unique best placement on a genome coinciding with an annotated gene
- cDNA/cDNA sequence relatedness (calculated based on criteria of identity, length of overlap to known accessions, and coverage of the novel accession)
- submissions from model organism databases or nomenclature authorities
- identification of proteins with identical sequences
- curation by RefSeq staff
- annotated GeneIDs from the ORFeome Collaboration or Celera
Additional Links
This section provides a view of a subset of links to information both within and external to NCBI. Some of these links overlap those included in the Related information menu. The intent of this section is to provide a printable report of, for example, MIM numbers, and gene- or gene family-specific websites.
Gene LinkOut
LinkOut provides easy access to relevant online resources outside of the Entrez system. These connections, and their groupings, are maintained by the external database.
ASN.1
The ASN.1 display provides gene records structured according to the Gene specification. An XML transformation of the ASN.1 is also available. Detailed information about the specification is provided in the Tips for Programmers section.
XML
Any record or selected set of records can be displayed in XML format. The XML is generated automatically from the ASN.1 record that is used to support the display, with the names of the tags defined by the ASN.1 specification. Detailed information about the specification is provided in the Tips for Programmers section.
Query Tips: How to submit detailed queries, and more…
Gene uses functions common to other NCBI databases. Most functions of the Entrez indexing and query engine are used by Gene. This section summarizes only how to use the tools in the context of the Gene database. Entrez Help and PubMed Help provide general information on how to save searches, use the Clipboard, history, and Advanced Search. For general information about Entrez, see Entrez Help.
Each Entrez database provides a query bar where you can select a database to interrogate, and enter a search term or terms. If a simple query is not powerful enough, there are options available to construct Advanced search queries.
Advanced search
The Advanced Search Builder (Figure 11) accessed from the query bar or http://www.ncbi.nlm.nih.gov/gene/advanced/ is a powerful resource to construct useful queries and to view terms that have been indexed under any field name. Table 5 describes the fields used in indexing the records and provides some representative queries using those fields. This section describes:
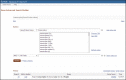
Figure 11.
Advanced Search. Shown is an advanced search in which the user had queried Gene using the word transcription. Then, given the large number of results, whose counts are displayed in the History section, the user decided to explore the indices (by clicking (more...)
Table 5.
Fields used to categorize information in Gene records.
- filters in general and how they can be used to search Gene for records of interest
- properties assigned to Gene records with examples of how to use them
- text phrases and how they differ from text words
Filter
The term filter is used in this context to describe categories of records that are grouped according to their relationship either to other Entrez databases or to external resources that have submitted LinkOut connections. If the former, the filter is named according to the pattern “gene other_Entrez_database”, such as “gene protein”. If the latter, the first two letters of the filter's name are ”lo”, for LinkOut. For a comprehensive listing of filters valid for the Gene database and the number of records in each, follow these steps:
- 1.
Click on the Advanced Search on the query bar.
- 2.
Use the pull-down menu named All Fields and select Filter.
- 3.
Click on Show Index under the open box to show the names of filter and the number of instances of each.
Filters are powerful tools to retrieve records of interest. For example, to retrieve all records for human genes that are associated with OMIM (i.e., have connections to OMIM) and have links to Entrez GEO, use the “AND” operator with both “gene omim” and “gene geo”. Table 4 provides a partial list of filters for Gene; the complete list is available here.
Table 4.
Filter sets (partial).
Properties
In general, properties are assigned to Gene records based on content rather than relationship to other database records, which is the role of filters (see Filter). There is however a small amount of redundancy between properties and filters. Many of the properties assigned to Gene records fall into these major categories:
- Type of gene: Property named as genetype name_of_type.
- Type of RNA: Property named as rnatype name_of_type.
- Source of the gene: Property named as source name_of_source.
- Type of RefSeq provided for the gene: Property named as srcdb refseq type_of_refseq.
- Type of feature annotation associated with the gene: Property named as feattype name_of_feattype.
The genetype option follows the conventions for mol_type used in the feature table of the International Nucleotide Sequence Databases (INSDC). The values should be self-explanatory, except perhaps for miscrna, other, and unknown. The genetype miscrna (misc_rna, miscellaneous RNA) is assigned to any gene that encodes an RNA product not included specifically at ncRNA vocab. The genetype other property is applied to loci of known type, but a specific category has not yet been applied in the Gene data model (e.g., immunoglobulin and TCR gene segments). The genetype unknown property is applied to probable genes for which the type is still under review. This category is frequently used when the defining sequence has uncertain coding propensity. We appreciate your suggestions for any improvements.
To summarize, the genetype property values are:
- genetype biological region (experimentally validated non-genic genomic regions that are in scope for RefSeq Functional Elements representation, including gene regulatory elements, known structural elements, and well-characterized DNA replication origins, DNA recombination regions, and sites of genomic instability)
- genetype miscrna (miscellaneous RNA)
- genetype ncrna (non-coding RNA; includes all ncRNA classes except for snRNA, snoRNA, and scRNA [which have their own gene types]. The largest counts are from miRNA and lncRNA. ncRNA classes are documented at ncRNA vocab.)
- genetype other (when the type is known, but there is no specific enumeration for it; includes immunoglobulin and TCR gene segments, repetitive elements, and regions)
- genetype protein coding
- genetype pseudo (pseudogene)
- genetype rrna (ribosomal RNA)
- genetype scrna (small cytoplasmic RNA)
- genetype snorna (small nucleolar RNA)
- genetype snrna (small nuclear RNA)
- genetype trna (transfer RNA)
- genetype unknown (when the type of gene is uncertain)
The rnatype property values identify the types of RNAs that are represented on the gene:
- rnatype mirna (micro RNA)
- rnatype miscrna (miscellaneous RNA)
- rnatype mrna (messenger RNA)
- rnatype ncrna (non-coding RNA)
- rnatype other
- rnatype other genetic
- rnatype pre rna
- rnatype rnase p rna
- rnatype rrna (ribosomal RNA)
- rnatype snorna (small nucleolar RNA)
- rnatype snrna (small nuclear RNA)
- rnatype srp rna
The source property values should be self-explanatory, with the exception of source other used where a specific category has not yet been applied in the Gene data model. Values are:
- source extrachromosomal
- source genomic
- source mitochondrion
- source organelle
- source other
- source plasmid
- source plastid
- source proviral
- source virion
The srcdb refseq values are as enumerated by RefSeq and will not be duplicated here.
The feattype property values are derived from feature annotation on RefSeq(s) associated with the gene, predominately genes with a genetype biological region. Further information about these feattype values can be found in the Feature Annotation Glossary for RefSeq Functional Elements, including links to INSDC feature specifications and controlled vocabularies, as well as links to equivalent terms in the Sequence Ontology. The values are:
- feattype caat signal
- feattype cage cluster
- feattype chromosome breakpoint
- feattype conserved region
- feattype dnase i hypersensitive site
- feattype enhancer
- feattype enhancer blocking element
- feattype epigenetically modified region
- feattype gc signal
- feattype imprinting control region
- feattype insulator
- feattype locus control region
- feattype matrix attachment region
- feattype meiotic
- feattype micrococcal nuclease hypersensitive site
- feattype misc feature
- feattype misc recomb
- feattype misc structure
- feattype mitotic
- feattype mobile element
- feattype non allelic homologous
- feattype nucleotide cleavage site
- feattype nucleotide motif
- feattype promoter
- feattype protein bind
- feattype recombination hotspot
- feattype regulatory
- feattype rep origin
- feattype repeat instability region
- feattype repeat region
- feattype replication regulatory region
- feattype replication start site
- feattype response element
- feattype sequence alteration
- feattype sequence comparison
- feattype sequence feature
- feattype silencer
- feattype stem loop
- feattype tata box
- feattype transcription start site
- feattype transcriptional cis regulatory region
Other properties used to categorize Gene records are explained in Table 3.
Text Phrases
A text phrase is a special type of text search that uses two or more words to form a phrase. An ordinary text search of two or more words will find gene records that contain all of the specified words anywhere in the gene record. By contrast, a text phrase search will find gene records that contain all of the specified words together and in the specified order.
A text phrase search is constructed by placing double quotes around the phrase. A list of certain phrases that can be used to find records of interest in gene is in Table 6.
Table 6.
Text phrases.
Finding subsets of your results; the ‘Results filter sidebar’ and ‘Filter your results’ options
When reviewing a query result in HTML format (not text), there are two options that allow you to display only a subset of the results:
Using the ‘Results filter sidebar’
The Results filter sidebar (Figure 2) is displayed to the left of your search results and is used to narrow the search results. Clicking a sidebar filter activates that filter, and all subsequent searches will be filtered until the selected filter(s) is cleared.
A check mark is located next to an active filter and the Filters activated: message is displayed above the Results table. The Search details box on the right side displays the updated query. Selecting more than one filter narrows the search further (equivalent to using a Boolean AND). A search can be expanded by replacing AND with OR in the Search details box.
Turn off the sidebar filters in any of these ways:
- Use the ‘Clear all’ link at the top
- Use the ‘clear’ link next to a filter group to clear the filters within that group
- Click on a check mark to clear an individual filter
Sidebar filter groups (described below) include Gene sources, Categories, Sequence content, Status, Chromosome locations, and Search fields. Within a filter group, only filter options valid for the current search results are listed. Use the ‘Show additional filters’ link to add or remove a filter group from the sidebar. A filter group with a greyed check mark in the ‘Additional filters’ menu cannot be removed.
To filter by organism, use the ‘Top Organisms’ section at the upper right of the results page. Additional filters are available but are managed through your My NCBI account; see ‘Filter your results’ using My NCBI.
Sidebar filter groups include:
Gene sources
Filter your search results based on the type of gene in the results set.
- Genomic: genes encoded by chromosomes or the major genomic macromolecule for the taxon.
- Mitochondrial: genes encoded by mitochondria.
- Organelles: genes encoded by organelles, including mitochondria, plastids, and macronuclei
- Plastids: genes encoded by plastids.
Categories
Filter your search results based on the existence of alternatively spliced RefSeqs, or on protein-coding capacity. NEWENTRY records support submission of GeneRIFs, by species, for a gene not currently in Gene.
Sequence content
Filter your search results based on these properties:
- CCDS: records that encode a protein sequence belonging to a Consensus CDS (CCDS) set. See http://www.ncbi.nlm.nih.gov/projects/CCDS/.
- Ensembl: records that match Ensembl annotation based on comparison of mRNA and protein features. See Table 3 for more information.
- RefSeq: records with an associated RefSeq record.
- RefSeqGene: records with an associated gene-specific genomic RefSeq in the RefSeqGene class.
Status
Restrict your search results for records that are ‘Current Only’. This is a particularly useful filter that removes discontinued or replaced records from the result set. It is equivalent to submitting a query that contains the expression ‘AND alive[property]’.
Chromosome locations
Restrict your search results by Organism, reference assembly chromosome or organelle, and location.
Search fields
Restrict your search results using any of the listed search fields. Table 5 summarizes these search fields (grouped into sub-categories) used to categorize information in Gene records. The table also provides examples of how to use these entities effectively to retrieve records.
‘Filter your results’ using My NCBI
In addition to the sidebar filters Gene provides by default, you can take advantage of any of the standard filters for Gene available via My NCBI. For example, if you are interested in Gene records that have a record in OMIM, you can use My NCBI to define "Gene records with MIM (Mendelian Inheritance in Man) numbers" as one of your standard filters. These filter results will be shown at the upper right of the query results screen. In addition to the standard filters, My NCBI also provides a button to ‘Create custom filters’. See Working with Filters for more information.
Words Excluded From Queries
Common, but uninformative, words and terms (also known as stopwords) are automatically eliminated from searches. However, a search term that is a stopword will be included if the term is explicitly qualified by a field name. For example, if you want to search for the term was, you could use:
- was [All Fields]
Enclosing the term in double quotes would have the same effect.
A list of stopwords used in Gene is in Table 7.
Table 7.
Stopwords.
Finding Data Related to Gene in Other Databases
The Related information menu, in the right column of the Full Report display, supports the function to retrieve information in other Entrez databases related to your result set. This function is supported by the links provided by NCBI’s Entrez system. The calculation of Entrez links is documented here. If you navigate to that documentation on the web, click on Gene to navigate quickly to the description of gene-specific links. Navigation in the Related information menu is based on the same infrastructure in Entrez that supports navigation to records related to a set of query results. The following provides more details about some of these links.
3D structures
3D structures provide experimentally resolved structures of proteins, RNA, and DNA derived from the Protein Data Bank, and include links to literature, related sequences, and more. To retrieve all records in Gene in this category, try the query "gene structure"[filter] from the Gene Search bar.
Books
An increasing number of Gene records are annotated specifically in books and monographs provided in Bookshelf. One example, restricted to human genes, is the GeneReviews book provided in collaboration with the GeneTests group of the University of Washington. To retrieve all records in Gene in this category, try the query "gene books"[filter] from the Gene Search bar. To retrieve only genes referenced in GeneReviews, use gene_genereviews[filter].
CCDS
Genes for which the protein products were accessioned by the Consensus CDS (CCDS) project. To retrieve all records in Gene in this category, try the query "gene ccds"[filter] from the Gene Search bar.
ClinVar
ClinVar maintains information about the relationships among human variations and phenotypes, including supporting evidence. ClinVar records associated with your Gene search results can be retrieved by using this display option. To retrieve all records in Gene with variations registed in ClinVar, try the query "gene clinvar"[filter] from the Gene Search bar.
Conserved Domains
Protein sequences are routinely compared to canonical sequences for domains in the Conserved Domain Database. Domain records connected to protein associated with records associated with your Gene search results can be retrieved by using this display option. To retrieve all records in Gene in this category, try the query "gene cdd"[filter] from the Gene Search bar.
Gene neighbors
The Genomic Context diagram of the Full Report display shows a gene’s genomic placement along with neighboring genes. Gene Neighbors contains the raw data corresponding to the Genomic Context diagram. To retrieve all records in Gene in this category, try the query "gene gene neighbors"[filter] from the Gene Search bar.
Genome
Genome maintains information about chromosomes and complete genomes. Genome records associated with your Gene search results can be retrieved by using this display option. To retrieve all records in Gene in this category, try the query "gene genome"[filter] from the Gene Search bar.
Genetic Testing Registry (GTR)
The GTR is a central repository for the voluntary submission of genetic test information by test providers. To retrieve all records in Gene in this category, try the query "gene gtr"[filter] from the Gene Search bar.
HomoloGene
HomoloGene compares protein-coding genes in several key genomes to identify homologs. HomoloGene records associated with r Gene search results can be retrieved by using this display option. To retrieve all records in Gene in this category, try the query "gene homologene"[filter] from the Gene Search bar.
NIH cDNA clone
Some of the mRNAs associated with your Gene search results are available from NIH-supported cDNA repositories. Reports of clones in the Nucleotide database associated with your Gene search results can be retrieved by using this display option.
Nucleotide
Nucleotide sequences associated with your Gene search results can be retrieved by using this display option. To retrieve all records in Gene with nucleotide sequence information, try the query "gene nucleotide"[filter] from the Gene Search bar.
MedGen
MedGen is NCBI’s portal to information related to medical genetics, including clinical features, available tests, up-to-date literature, practice guidelines, and consumer resources. To retrieve all records in Gene in this category, try the query “gene medgen diseases”[filter] from the Gene Search bar..
OMIM
OMIM records associated with your Gene search results can be retrieved by using this display option. To retrieve all records in Gene in this category, try the query "gene omim"[filter] from the Gene Search bar.
PubChem BioAssay
Genes with products having screening results reported in the PubChem BioAssay database. To retrieve all records in Gene in this category, try the query "gene pcassay"[filter] from the Gene Search bar.
PubChem Compound
Genes with products having screening results in the PubChem Compound database. To retrieve all records in Gene in this category, try the query "gene pccompound"[filter] from the Gene Search bar.
PubChem Substance
Genes with products having screening results in the PubChem Substance database. To retrieve all records in Gene in this category, try the query "gene pcsubtance"[filter] from the Gene Search bar.
PMC
Publications available as full text from PubMedCentral may include explicit references to Gene. Publications may also be connected to Gene via a PubMed ID. PubMedCentral records associated with your Gene search results can be retrieved by using this display option. To retrieve all records in Gene in this category, try the query "gene pmc"[filter] from the Gene Search bar.
Probe
Probe records, such as those for resequencing primers or RNAi sequences, related to your Gene search results can be retrieved by using this display option. To retrieve all records in Gene in this category, try the query "gene probe"[filter] from the Gene Search bar.
Protein
Protein sequences associated with your Gene search results can be retrieved by using this display option. To retrieve all records in Gene with protein sequence information, try the query "gene protein"[filter] from the Gene Search bar.
PubMed
PubMed citations associated with your Gene search results can be retrieved by using this display option. Those that were generated from GeneRIFs, including interaction data, are indicated by the PubMed (GeneRIF) option. To retrieve all records in Gene with citations in PubMed, try the query "gene pubmed"[filter] from the Gene Search bar.
SNP
Use these display options to navigate to information about variation reported in the dbSNP database for the gene records in your search results. To retrieve all records in Gene with reported variation, try the query "gene snp"[filter] from the Gene Search bar.
Taxonomy
Use this display option to navigate to information about the taxonomy of the genomes in which the gene records in your search results are found.
Constructing Powerful Queries
Constructing queries based on free text, filters, and properties can be quite powerful in retrieving records of interest from Gene. Table 8 summarizes some of these approaches by describing:
Table 8.
Constructing queries.
- Scope: The intent of a query.
- Query: How to construct a query that meets that intent.
- Notes: How usage of Gene to retrieve these data may compare to other gene-related resources, namely HomoloGene or Genome Data Viewer.
Although these examples use field restriction (see Table 5 for the comprehensive list of fields used to index the information in Gene records), free text can also be submitted. Gene then weights the retrievals based on the field in which a result was found. For example, if your query matches a gene symbol in one record and arbitrary text in another, the record where the match is on the symbol will be displayed before the other in the results. Thus Gene controls the default order in which results are returned by evaluating what fields are more critical to matching your query. This default sorting order is termed 'relevance'.
Tips for Programmers
The Gene Data Model and DTD
The data model for Gene is documented in the Gene specification. It combines several definitions used by other NCBI databases, such as seqfeat, but also establishes definitions specific to Gene. Of special note is the Gene-commentary, which is used to represent many descriptors of genes. Each Gene-commentary is defined by type and supports specific representation of such elements as sequence database accession numbers (accession, version), citations (refs), external or internal resources defining the data (source), and position information. Heading, label, and text are used for general data, with the choice influenced by display in the Gene viewers.
The DTD for Gene is available from NCBI's DTD directory and is called NCBI Gene.dtd.
Entrez Programming Utilities and Gene
The full power of Entrez Programming Utilities (e-Utils) can be used to extract information from Gene programmatically. The basic strategy is to identify the query that will return the desired records and then submit that query via ESearch. The GeneIDs identified by that search can then be submitted to another function, such as ESummary or EFetch. Examples for Gene are ESummary.
Extracting Gene Summaries and other information from Gene’s Document Summary
The Summary text provided via Gene and on RefSeq records can be extracted by taking advantage of the following:
- the text of the Summary is included in the Document Summary (docsum) from Gene.
- genes with Summary text can be identified by the has_summary property.
In other words:
- 1.
use eSearch to find all GeneIDs with the has_summary property
- 2.
use eSummary to retrieve the Summary text (e.g. http://eutils
.ncbi.nlm .nih.gov/entrez/eutils/esummary .fcgi?db =gene&id=672&retmode =xml) - 3.
Extract the string in the Summary tag.
Table 9 lists the name attributes of Gene’s docsum that can be extracted in a similar manner. An example docsum is provided here:
Table 9.
The Name attributes of Gene’s Document Summary (docsum).
http://eutils.ncbi.nlm.nih.gov/entrez/eutils/esummary.fcgi?db=gene&id=4267&filter=asis
Extracting Gene Neighbors
Gene Neighbors can be queried programmatically using the Entrez Links function of E-utilities. For example, to find all neighbors of GeneID 672, use this:
Note that gene neighbors identified by this method are not associated with a specific genomic placement but with all reported genomic placements. In many cases, a gene's neighbors are the same for all genomic placements. However, in some cases, a gene's neighbors may differ from one genomic placement to another, for example, between the reference assembly and an alternate assembly.
Gene FTP Site
The FTP site for Gene (README) has three major subdirectories: DATA, GeneRIF, and Tools.
DATA
DATA contains files that provide key attributes of genes, including:
- all associated accession numbers, including RefSeqs (gene2accession.gz)
- matching Ensembl annotation (gene2ensembl.gz)
- GO terms (gene2go.gz)
- citations (gene2pubmed.gz)
- associated RefSeq accession numbers (gene2refseq.gz)
- relationships to other genes (gene_group.gz)
- genes that are no longer current (gene_history.gz)
- nomenclature, ID, and map data (gene_info.gz)
- neighboring genes (gene_neighbors)
- MIM numbers and records in MedGen (mim2gene_medgen)
- relationship to UniProtKB proteins (gene_refseq_uniprotkb_collab.gz)
- ortholog data (gene_orthologs.gz)
Details of the construction of these files are reported in the (README) file.
DATA also contains the ASN_BINARY subdirectory. This path contains both a comprehensive extraction from Gene (All_Data.ags.gz), several subsets categorized by source (Organelles, Plasmids), and subdirectories grouped broadly by taxonomy. Records of genes from species that are requested frequently are also provided in species-specific files, for example these mammals. The format of these extractions is compressed binary ASN.1. The program gene2xml is available to convert these files to XML or ASN.1 text. Be aware that the converted files will take approximately 100-fold more space than the original compressed binary ags.gz files.
The GENE_INFO subdirectory of DATA provides subsets of the gene_info file grouped broadly by taxonomy. This directory structure mirrors that of the ASN_BINARY path. Thus if you want the type of information provided in gene_info, but do not want to have to process the complete text, you can use one of the files in the appropriate subdirectory, for example these plants.
GeneRIF
GeneRIF contains files that provide supplemental information about gene functions, either from the GeneRIF pipeline (generifs_basic.gz) or the HIV-1, Human Protein Interaction Database (hiv_interactions.gz). The tab-delimited files are not subdivided by species of interest. All files except the file reporting GeneID/PubMedID relationships (gene2pubmed.gz) have a column with the ID from the NCBI Taxonomy database to facilitate the extraction of a subset of the data from the file by species.
Tools
Gene_tools provides or points to programs and scripts to mine data from Gene. Of particular interest is gene2xml, which can be used to convert the binary ASN.1 in the ASN_BINARY directory to XML or to ASN.1 in text format (README).
Connecting Users of Gene to Your Website
Gene can serve as a gateway to information on your website served from your local database. Users of Gene will discover your website if you participate in our LinkOut system and become a LinkOut provider. Any Entrez database will support LinkOut. Linkout Help’s Information for Other Resource Providers explains the details of this opportunity.
There are many benefits to becoming a LinkOut provider. If you want access to your database to be apparent from Gene, you can control the description of your resource, the update cycle, and the icon to anchor links to your site. In other words, you do not have to wait for NCBI staff to go to your site to obtain and process information and match to Gene records. You know your site best—you can identify which records are related to Gene records and provide the most accurate and informative URL to connect that Gene record to your site. If you already provide LinkOuts to other Entrez databases, such as Nucleotide or Protein, you do not have to re-register as a provider; you need only notify LinkOut staff and start to submit a new resource file.
With the implementation of My NCBI, it is even more advantageous to become a LinkOut provider. One of the options registered users of My NCBI can select is to display the icons for any LinkOut provider at the top of a record. The presence of your familiar logo would invite users of Gene to go to your site.
Connecting your site to Gene
URLs can be constructed to query Gene, or to display a specific record if you know the GeneID. For example, if your site maintains the identifiers (GeneID) used by Gene, you can construct a link from your site to Gene by combining this base
http://www.ncbi.nlm.nih.gov/gene/
with the GeneID. For example, to link to GeneID 1, use this URL:
http://www.ncbi.nlm.nih.gov/gene/1
URLs that query Gene are constructed by adding ?term=[search term]
For example, to find records in Gene containing the phrase ‘immunoglobulin domain’, use this URL
http://www.ncbi.nlm.nih.gov/gene/?term=immunoglobulin_domain
More examples of queries are provided on Gene’s Home page, and general rules for building URLs to query Entrez databases are provided in the Creating a Web Link to the Entrez Databases chapter of this book. The valid display options are also documented in that chapter.
Historical Information about LocusLink
This version of Gene's help document removed detailed information about LocusLink. If you have any question about the history of LocusLink, please use this form.
- Introduction
- Quick Starttips
- How Data Are Maintained
- How content is selected
- How Data Are Displayed (Display Settings/Format)
- Query Tips: How to submit detailed queries, and more…
- Finding subsets of your results; the ‘Results filter sidebar’ and ‘Filter your results’ options
- Words Excluded From Queries
- Finding Data Related to Gene in Other Databases
- Constructing Powerful Queries
- Tips for Programmers
- Historical Information about LocusLink
- Gene Help: Integrated Access to Genes of Genomes in the Reference Sequence Colle...Gene Help: Integrated Access to Genes of Genomes in the Reference Sequence Collection - Gene Help
Your browsing activity is empty.
Activity recording is turned off.
See more...