Introduction
The nucleotide sequence of the Streptococcus pyogenes genome was first determined in 2001, which inferred a proteome of 1,752 proteins (Ferretti, et al., 2001). The genomic sequencing of additional isolates showed that all streptococcal chromosomes are poly-lysogenized with bacteriophages (Sumby, Whitney, Graviss, DeLeo, & Musser, 2006), which account for approximately 10% of their genome content (Ferretti, et al., 2001). The number and types of prophages that are present in the genomes of different clinical isolates varies significantly, which suggests a stochastic process of co-evolution (Canchaya, Fournous, & Brüssow, 2004). Horizontally transmitted integrative conjugative elements, transposons, and insertion sequences also contribute to genomic variation within the species. Due to the extent of horizontal DNA transmission, S. pyogenes is thought to have a theoretically infinite pan-genome and—by extension—a theoretically infinite proteome (Desiere, McShan, van Sinderen, Ferretti, & Brüssow, 2001). Moreover, multiple protein isoforms can be derived from a single open reading frame (ORF) following post-translational modifications, such as truncation or phosphorylation, and each isoform may have a unique function. As a result, the complexity of an organism’s proteome is estimated to be at least two to three orders of magnitude greater than that of the genome () (Matthiesen & Jensen, 2008; Cain, Solis, & Cordwell, 2014).
The complexity of the Streptococcal proteome is significantly greater than the corresponding core genome due to the acquisition of horizontally transmitted DNA contributing to the pan genome, the regulation of gene expression and post-translational modification (more...)
The availability of the genome sequence of S. pyogenes and advances in mass spectrometry (MS) have greatly enhanced our ability to characterize proteins on a genome-wide scale. Proteomic studies of S. pyogenes have been motivated by the pursuit of answers to fundamental questions, such as: How are proteins trafficked to specific sub-cellular locations? What is the functional significance of protein localization? How does the proteome transition in response to changing conditions encountered in the host? What are the differences between streptococci associated with a localized infection, as compared to those associated with life-threatening invasive infections? What are the functional significances of interactions between streptococcal proteins and human host proteins? What physiologic changes in the proteome occur in response to the presence of antimicrobials? What are correlates of protective immunity? What Streptococcal proteins can be used to vaccinate against disease? What proteins elicit pathogenic antibodies? Answering these questions is important in developing new approaches to mitigate the morbidity and mortality associated with S. pyogenes.
In this chapter, we focus on the advances in our understanding of the S. pyogenes proteome from the perspective shaped by results obtained using proteomics, or studies that have simultaneously characterized a set of proteins. We start by describing the methods that have been used to study the proteome and then discuss how these approaches have led to insights into the pathogen’s response to changing conditions, circumvention of the immune response, and the organization and regulation of extracellular proteins. Finally, we will review advances in identifying proteins that evoke auto- and protective immunity. Given the rapid pace at which various aspects of proteomic investigation are progressing, including instrumentation, workflow strategies, and bioinformatics, the characterization of the S. pyogenes proteome is, in many ways, just beginning.
Sub-cellular proteomes
The term proteome refers to the entire set of proteins that are expressed by an organism at a certain time under specific conditions. Unlike genomics and transcriptomics, proteomics examines compartmentalized sets of proteins. Therefore, protein localization is an important feature for characterizing the proteome. There are four spatially defined sub-proteomes of S. pyogenes: cytoplasmic proteins, cell membrane proteins, cell wall-associated proteins, and soluble exoproteins or culture supernatant proteins (CSPs).
Protein Trafficking
S. pyogenes lacks a twin-arginine translocation pathway (Dilks, Rose, Hartmann, & Pohlschröder, 2003) and most proteins are translocated from the cytoplasm by the general secretion pathway, which is often referred to as the Sec pathway (Schneewind & Missiakas, 2014). These proteins possess an amino-terminal signal peptide that consists of an “N region” of hydrophilic amino acids, an “H region” of approximately 17 hydrophobic amino acids, and a slightly hydrophilic “C region.” During translation, the signal peptide is recognized by the signal recognition particle (SRP), which is composed of the Ffh protein and a small cytoplasmic RNA (Miller, Bernstein, & Walter, 1994). SRP targets the nascent polypeptide to the membrane receptor FtsY and the SecYEG translocon (Schneewind & Missiakas, 2014; Halic, et al., 2006). In S. pyogenes, SecA and other accessory proteins, such as the HtrA chaperone, have been reported to be localized to a single area of the cytoplasmic membrane, or microdomain, which is known as the ExPortal (Rosch & Caparon, 2004), although a separate study has observed SecA throughout the membrane (Carlsson, et al., 2006). Similar to S. mutans (Crowley, Svensäter, Snoep, Bleiweis, & Brady, 2004), the SRP protein Ffh is not required for the viability of S. pyogenes (Rosch, Vega, Beyer, Lin, & Caparon, 2008).
Proteins secreted by the Sec pathway are destined for the cytoplasmic membrane, the cell wall, or for release into the extracellular environment. These proteins are often present in more than one compartment. Proteins covalently attached to the cell wall possess a carboxyl terminal motif LPXTG or a similar motif (Fischetti, Pancholi, & Schneewind, 1990). During export from the cytoplasm, the motif is recognized by the membrane-localized sortase, which cleaves the polypeptide and covalently anchors the protein to peptidoglycan (Mazmanian, Liu, Ton-That, & Schneewind, 1999; Perry, Ton-That, Mazmanian, & Schneewind, 2002). Several proteins that are important to the virulence of S. pyogenes contain the LPXTG motif, including the antiphagocytic M protein, which is often considered to be the single most important virulence factor of S. pyogenes (Cunningham, 2000).
Three sortases have been identified in S. pyogenes. SrtA is a housekeeping sortase that catalyzes the attachment of most cell wall-associated proteins in a two-step reaction. First, the LPXTG motif is cleaved, which leaves the carboxyl group of threonine to react with a free amine present in lipid II and results in the attachment of the protein to the cell wall (Mazmanian, Liu, Ton-That, & Schneewind, 1999; Perry, Ton-That, Mazmanian, & Schneewind, 2002). SrtA anchors the M protein, C5a peptidase (ScpA) protein, G-related α 2M-binding protein (GRAB), protein F (Barnett & Scott, 2002), and probably several more proteins to the cell wall. SrtA is localized to specific foci of the cytoplasmic membrane, mostly the septum (Raz & Fischetti, 2008). A second sortase, SrtB, is encoded in the FCT chromosomal region (so named because it encodes fibronectin, collagen, and T-antigen proteins) and catalyzes the attachment of the T antigen (pilin) to the cell wall (Barnett & Scott, 2002). SrtB recognizes an LPSTG motif. A third sortase, SrtC, is also encoded in the FCT region and anchors the protein encoded by the adjacent gene via a QVPTGV motif (Barnett, Patel, & Scott, 2004).
Recent studies suggest that additional information is contained within the signal peptide than was previously known. For example, M6 and the fibronectin binding protein PrtF.2 both possess a signal peptide and LPXTG motif. M6 is secreted at the septum and is localized throughout the cell wall, while PrtF.2 is concentrated at the poles. Swapping the signal sequences between the proteins reverses the pattern of localization, which indicates that the signal peptides determine the final location of the proteins (Carlsson, et al., 2006). In a related study, the localization of four proteins (M protein; SPN, an NAD(+)-glycohydrolase; SLO, a secreted cytolysin; and SpeB, a secreted cysteine protease) that possess signal peptides was analyzed in an Ffh mutant (lacking a functional SRP) (Rosch, Vega, Beyer, Lin, & Caparon, 2008). M protein localization to the cell wall was not affected by the absence of SRP (the Ffh mutant). In contrast, both SPN and SLO were not secreted in the mutant strain, which indicates that their export requires SRP. Notably, SpeB was not secreted by the mutant, unless glucose was added to the media. The studies indicate that more remains to be learned about the sorting and localization of proteins to the cell wall and extracellular milieu, as well as the functional consequences of the arrangement of proteins within these compartments.
Many studies have shown the importance of secreted proteins to virulence. In support of those findings, an SRP-deficient mutant of S. pyogenes is less virulent in animal models of infection (Rosch, Vega, Beyer, Lin, & Caparon, 2008), which suggests that therapeutic strategies designed to inhibit SRP-mediated protein localization could be effective in mitigating disease.
Fractionation techniques
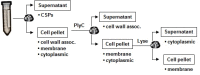
The streptococcal proteome is partitioned during analyses and is often discussed in terms of these fractions, or sub-proteomes, which include CSPs, cell wall-associated proteins, cytoplasmic membrane proteins, and cytoplasmic proteins. summarizes the typical workflow used for fractionating these proteins. The use of media devoid of peptides or proteins, such as chemically defined media or rich media that has been filtered to remove proteins and peptides, can simplify the characterization of the CSPs, because all the proteins and peptides present are derived from S. pyogenes. Following the centrifugation of bacterial cultures and the collection of CSPs, the cell pellet is suspended in a buffer, and the proteins are further fractionated by separating the cell wall-associated proteins from membrane and cytoplasmic proteins. This is done by treating the cells with enzymes that degrade peptidoglycan, thereby releasing proteins attached to the cell wall into solution. Mutanolysin and lysozyme have often been used; however, a proportion of the resulting protoplasts typically lyse during enzymatic treatment. As a result, the cell wall fraction is contaminated with cytoplasmic proteins; this occurs even when using an osmoprotective buffer. One method to improve these results is through the use of a bacteriophage derived N-acetylmuramoyl-L-alanine amidase to hydrolyse the N-acetylmuramic acid, L-alanine bond of the cell wall (Nelson, Loomis, & Fischetti, 2001). The enzyme, known as PlyC, was validated for this purpose and was found to decrease contamination with cytoplasmic proteins (Köller, et al., 2008). Following amidase treatment, centrifugation will separate the cell wall-associated proteins present in the supernatant fraction from the protoplasts that contain the cytoplasmic and membrane localized proteins. These fractions are separated by lysing the protoplasts and separating the membrane proteins (the pellet) and cytoplasmic proteins (supernatant) by centrifugation. The comparative complexity of the cytoplasmic protein fraction and the technical challenges of working with cell membrane proteins have hindered the characterization of these fractions. Nonetheless, cell membrane proteins are present at the host-pathogen interface and are essential to cellular homeostasis, including communication between the host environment and the cytoplasm. As a result, further characterization of this sub-proteome is an important area for future investigation.
Fractionation of the S. pyogenes proteome. CSPs are obtained following centrifugation of the bacterial culture. The bacterial pellet is suspended in buffer and treated with PlcY (or mutanolysin) to release cell-wall–associated proteins covalently (more...)
Separating proteins by gel electrophoresis and liquid chromatography
Streptococcal proteins are usually separated by either gel electrophoresis or liquid chromatography (LC), and proteins of interest are identified with mass spectrometry. Much of the investigation into the S. pyogenes proteome has used two-dimensional gel electrophoresis (2-DE) to resolve complex protein mixtures (O'Farrell, 1975). While it is remarkably powerful in its ability to separate very similar proteins, 2-DE has several limitations. First, to visualize the proteins, they must be stained, and the sensitivity and the dynamic range of protein detection are both dependent on the staining process. Although progress has been made in the past decade to develop more sensitive stains that are compatible with mass spectrometry, protein detection is still limited to relatively abundant proteins. This means that less abundant proteins that are present and that may be of interest, such as regulatory proteins, are below the limits of detection. In addition, not all proteins can be simultaneously analyzed by 2-DE, due to differences in their solubility, size, and other biochemical characteristics. Finally, the 2-DE process is relatively laborious and time consuming. Because of these limitations, the use of gel-free methods, such as multi-dimensional liquid chromatography, has attracted considerable attention, and is being used in many studies on the S. pyogenes proteome. This approach is also referred to as “shotgun” proteomics, due to its similarity to shotgun DNA sequencing. A key feature of this strategy is that a mixture of proteins is first digested with trypsin (or another protease with a specific recognition site) to facilitate identification by mass spectrometry. The peptides are then resolved by liquid chromatography. While protein digestion increases the number of molecules present in the sample, since multiple peptides are derived from each protein, the biochemical homogeneity of peptides simplifies their characterization, as compared to proteins. Nonetheless, because so many peptides are contained in samples, multi-dimensional LC is necessary to resolve the peptides. MuDPIT is one example of such multi-dimensional protein identification technology. In this case, peptides are initially separated by cation exchange chromatography, with the eluting peptides further separated by reverse phase liquid chromatography. The chromatography steps can either be done separately, or more commonly, by sequential analysis. This analysis is achieved by connecting two resolving columns so that peptides separated with the first column (1st dimension) elute directly into a second column (2nd dimension). Advances in the chemistries used for sequential separation and the development of LC pumps that can produce reliable gradients at nanoliters per minute flow rates have greatly contributed to the ability to use multi-dimensional liquid chromatography (Zhang, Fonslow, Shan, Baek, & Yates, 2013). Typically, the peptides from the 2nd dimension column elute directly into a mass spectrometer for characterization and protein identification.
Protein quantification
The quantification of proteins separated by either 1 or 2-dimensional gel electrophoresis is most often (and most easily) accomplished by staining the proteins and then scanning the stained gel. The sum of the number of pixels comprising the protein band, or spot, and the intensity of each pixel is proportional to the amount of protein within the band or spot. The majority of studies have used R-250 or G-250 Coomassie Blue, modified silver staining protocols compatible with mass spectrometry; or one of the many fluorescent stains available, such as SYPRO Ruby, Deep Purple, or LavaPurple. The fluorescent stains are generally more sensitive and have a greater dynamic range, as compared to silver staining methods that are compatible with mass spectrometry. Because of limitations in sensitivity and other drawbacks associated with 2-DE, more studies using gel-free methods to separate proteins have been developed to quantitate proteins using the mass spectrometer.
Difference in-gel electrophoresis to compare protein abundance
Protocols have been developed to quantitatively compare proteins separated by 2-DE between two or more samples without using separate gels, which helps to avoid problems associated with trying match protein spots among gels. Difference in-gel electrophoresis (DIGE) uses a combination of fluorescent stains or probes with different excitation and emission wavelengths to quantitatively compare proteins from more than one sample (Unlü, Morgan, & Minden, 1997). For example, proteins isolated from a wild-type S. pyogenes isolate can be labeled via lysine residues with succinimidyl esters of propyl-Cy3, and those from a mutant derivative can be similarly labeled with a methyl-Cy5 ester. The two cyanine family fluorophores have the same masses and charges, but have different emission wavelengths. The labeled proteins from each sample are mixed prior to their separation by electrophoresis. Because the samples are separated simultaneously in a single gel, the differentially labeled proteins will migrate to exactly the same place within the gel. The gel is then scanned to measure the intensity of the individual flours. One of the limitations of this technique is that the fluorescent labels are less sensitive than SYPRO Ruby and silver staining (Matthiesen & Amorim, 2010).
Label-based protein quantification with mass spectrometry
Label-based strategies for quantitating proteins with mass spectrometry use either stable isotopes or the mass tag labeling of proteins or peptides. The advantage of these techniques as compared to quantifying stained proteins is that quantitation is done using the mass spectrometer, which is far more sensitive than the currently available protein dyes or flourophores . With these methods, proteins from two, or more, samples are covalently labeled with isotopically distinguishable elements, such as 14N and 15N, and are then analyzed with mass spectrometry. The labeling can be performed before or after protein digestion. After labeling, the samples are combined and the masses of tryptic peptides are determined. The relative difference in protein abundance between the two samples is determined by comparing the ratios of ion peak intensities from the differentially labeled samples (either peptides labeled directly or peptides derived from a labeled protein). For example, the intensity of a peptide from a wild-type sample can be directly compared to that from a mutant, because the specific peptide ions will differ by exactly one mass unit (when labeled with14N and 15N).
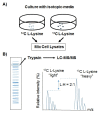
Proteins can also be labeled in vivo by growing streptococci in media containing isotopic precursors, most commonly amino acids. The process is known as stable isotope labeling by amino acids in culture (SILAC) (Ong, et al., 2002). For example, a wild-type strain can be cultured with media that contains 12C-lysine and a mutant derivative similarly cultured with 13C-lysine. When the peptides obtained from the two strains are simultaneously analyzed by mass spectrometry, they will differ by exactly one atomic mass unit, and the intensities of the isotopic ion peaks will be proportional to the abundance of the cognate protein ().
Stable isotope labeling by amino acids in culture (SILAC) to quantitatively compare the abundance of proteins. A) Two strains (wild type and mutant) are grown with either a 13C or 12C amino acid such as lysine. Following culture, cell lysates are obtained (more...)
In vitro protocols use a post-biosynthetic labeling strategy that can be applied to any set of protein samples obtained from streptococci. To do so, proteins are labeled with isotope-coded affinity tags (ICAT) before protein digestion, or with isobaric mass tags (isobaric tags for relative and absolute quantification, or iTRAQ) after protein digestion. ICAT uses biotinylated derivatives of iodoacetamide, which react with the cysteine residues of denatured proteins. The biotin labeling permits cysteine-containing peptides to be isolated from complex peptide mixtures, through their affinity for streptavidin. This strategy reduces the number of peptides introduced into the mass spectrometer, which can enhance both sensitivity and mass accuracy. To determine the relative amounts of proteins from two samples, such as from a wild-type and mutant strain, two isotopic biotin-containing tags are used (12C and 13C). The peptide ion peak intensities of each sample are then compared to determine the relative amounts of the corresponding protein in each sample. Some proteins may not be detected using this method if the cysteine residues are not present or reactive (Shiio & Aebersold, 2006).
The iTRAQ method involves labeling peptides from different sources, such as from a wild-type and mutant strain, after protein digestion with chemical groups that have the same mass. The label contains an amine-specific reactive group, a balancer group, and a reporter mass group. Because the mass of the tagged peptides is identical in both samples, potential differences in the efficiency of ionization associated with peptides that have different mass tags is eliminated. A decrease in ionization efficiency would decrease ion intensity, which is assumed to correlate with protein abundance. With this method, a single parental peptide mass from both samples is measured in MS1, because the peptides from the two samples have tags with identical masses; however, fragmentation of the tryptic peptides during MS2 analysis will release reporter ions with different masses that are unique to each sample (wild-type and mutant). Again, the intensities of the fragment ions correlate to the abundance of the parent protein. The advantage of this procedure is that it eliminates the possibility that a difference in the molecular weight of an isotopically labeled protein will change the efficiency at which tryptic peptides ionize. iTRAQ is the most widely used protocol for isobaric mass tag protein quantitation, and can be used to simultaneously compare up to eight samples (Ross, et al., 2004).
Label-free protein quantitation with mass spectrometry
Three different strategies can be used to determine the quantities of proteins without labeling them. One strategy is based on the assumption that the ion peak intensities of peptides correlate with the abundance of their parent protein in complex samples, which is known as the protein abundance index method (PAI). The other two methods are targeted counting using either selected ion monitoring (SIM) or selected reaction monitoring (SRM).
The PAI method has two main features. First the number of tryptic peptide masses (assuming that trypsin is used in the digestion step) measured for a particular protein is compared to all the theoretical peptides that are predicted from in silico digestion of that particular protein. The assumption is that the number of peptides that are detected, as compared to the theoretical number of peptides, directly correlates to the abundance of the parent protein. When using this approach, it’s important to validate the results by determining the normalized spectral abundance factor (NSAF). This validation is performed by calculating the ratio of the number of MS/MS spectra obtained for each protein (Wasinger, Zeng, & Yau, 2013) to the total number of spectra obtained from all of the proteins detected in the sample (Zhu, Smith, & Huang, 2010). Malmstrom et al. used this label-free method to examine how the intracellular homeostasis of S.
pyogenes is influenced by exposure to human plasma, as discussed in more detail below (Malmström, et al., 2012).
The SIM method can be used with scanning mass spectrometers (those instruments capable of detecting ions throughout a defined mass range, such as quadrupoles and Q-ToFs) by restricting the acquisition mass range to only the ion(s) of interest. In other words, a mass spectrometer is programmed to detect only the masses that correspond to peptides that are derived from the proteins of interest. The narrower the mass range, the more specific the SIM assay is to a particular protein. SIM is highly sensitive because by focusing on a particular mass, and ignoring all the others in the sample, more time is devoted by the instrument to measuring the mass of interest, which improves the overall signal intensity and accuracy of mass determination (Lange, et al., 2008).
Similarly, the SRM method uses a triple quadrupole (QQQ) mass spectrometer to detect one peptide derived from the protein of interest. Moreover, the mass of a specific fragment ion of the peptide of interest is also targeted for measurement. The peptide and fragment ion are referred to as a transition pair. Similar to SIM, the instrument dedicates its analysis to the measurement of only a few very specific masses while ignoring the other ions in the sample. A QQQ is particularly well suited to this application, because two quadrupoles (Q1 and Q3) are configured as mass filters that monitor ions to measure the masses of interest. The Q1 quadrupole creates a mass gate, in which only ions with approximately the mass of the tryptic peptide of interest are measured. After measuring the mass, the ions are then fragmented in Q2, and the fragmented mass of interest is measured in Q3. To determine the absolute quantity of the protein in a sample, a selected peptide derived from the protein of interest is biochemically synthesized in an isotopic form (13C) and is added to samples at different concentrations. When the samples are analyzed, a standard curve can be created, based on the ion intensity of known amounts of the isotopic peptide. The intensity of the peptide of interest is then used with the standard curve to determine the absolute concentration of the protein of interest. When SRM is applied to multiple proteins, it is called multiple reaction monitoring (MRM) (Lange, et al., 2008). In MRM, the instrument is configured to detect several transition pairs of interest. Lange et al. have used MRM to quantitatively measure streptococcal virulence factors (Lange, et al., 2008). One of the challenges to performing SRM and MRM is in the selection of transition pairs, which has traditionally been a tedious, time-consuming process; however, the development of in silico tools has greatly simplified this process. Moreover, Karlsson et al. have validated over 10,000 transition pairs to quantitate streptococcal proteins by SRM (Karlsson, Malmström, Aebersold, & Malmström, 2012).
Characterization of the S. pyogenes proteome
In the following sections, we review studies that have used proteomic approaches to characterize the streptococcal proteome. includes a summary of these studies, which includes the subproteome investigated, the methods used, and their major findings.
Major studies characterizing the group A Streptococcus proteome.
Proteome changes in response to different environmental conditions
Identifying changes in gene expression is most conveniently done at the transcript level. This is partially due to the availability of highly sensitive methods to measure transcripts, including quantitative RT-PCR, and the biochemical homogeneity of RNA. Nonetheless, because proteins mediate the majority of cellular functions, and because post-transcriptional changes can greatly impact protein abundance and function, it is also important to characterize the changes that occur at the protein level in response to different environmental conditions.
The study of differences in protein abundance between the exponential and post-exponential phases of growth is an experimentally convenient way to characterize the microbes’ response to changes in cell density, the accumulation of metabolites (quorum sensing), nutrient depletion, decreasing pH values, and other stresses. Many of these are relevant to the changing conditions that occur during infection. Cytoplasmic proteins from samples obtained during the exponential and post-exponential phases of growth were compared (Chaussee, Callegari, & Chaussee, 2004). The use of 2-DE resolved 527 proteins; 125 of these were identified, which corresponded to 78 genes, since multiple isomers of a single ORF were often identified. The most abundant cytoplasmic proteins were enolase and GAPDH. In addition to glycolytic enzymes, many other catabolic enzymes were detected, including those involved in pyruvate metabolism (AcoB, Spy_1028; AcoL, Spy_1031), which convert pyruvate to acetyl CoA and CO2; L-lactate dehydrogenase; and the enzymes of the arginine deiminase pathway encoded by the ArcABC operon, which convert arginine to citrulline, NH3, and ATP. The analysis was coupled with transcriptome characterization. Together, the results identified several growth phase associated changes mediated at the post-transcriptional level, including changes in the expression of lactate dehydrogenase and a cell division initiation protein (DivIAV). In addition, the abundance of several protein isoforms varied in a growth-phase–dependent manner, which indicates that regulation at the post-translational level is also an important (though poorly understood) means of adapting to changing environmental conditions.
Other conditions thought to be important to different stages of infection include the concentrations of hyaluronic acid and iron. Iron is essential for bacterial growth, and iron availability has a significant impact on the streptococcal proteome. Iron supplementation decreased the abundance of 17 exoproteins, including several established virulence factors (Sic, SpeF, and Ska (Nakamura, et al., 2004)). Hyaluronic acid, in addition to being the capsule produced by S. pyogenes, is a non-sulfated glycosaminoglycan that is widely distributed in human tissues, including the skin. The addition of hyaluronic acid to cultures increased the abundance of several virulence factors, including the M protein, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a collagen-like surface protein, and two hypothetical proteins (Zhang, et al., 2007). One interpretation of these results is that hyaluronic acid promotes the production of exoproteins that facilitate adherence.
S. pyogenes is often exposed to antimicrobials during the course of infections. High doses of clindamycin and penicillin are recommended for the treatment of invasive diseases, including toxic shock syndrome; in part because clindamycin (an inhibitor of ribosome translocation) reduces the production of exoproteins that contribute to disease (Mascini, Jansze, Schouls, Verhoef, & Van Dijk, 2001; Coyle, Cha, & Rybak, 2003). An examination of the influence of sub-inhibitory levels of clindamycin on the secretome of S. pyogenes by using 2-DE showed that exposure decreased the abundance of the extracellular cysteine protease (SpeB) (Sawai, et al., 2007); however, the abundance of several other exoproteins increased when clindamycin was added to cultures at the beginning or early exponential phase of growth. Many of these proteins are known to be degraded by SpeB (Aziz, et al., 2004), which suggests that the increase in various exoproteins was due to decreased degradation by SpeB. Surprisingly, however, the increases in SLO, NAD-glycohydrolase, and the streptococcal inhibitor of the complement (SIC) were found to be due to increased transcription of the genes (Minami, et al., 2010). In contrast, the addition of clindamycin during the mid-exponential phase of growth decreased the abundance of exoproteins, which suggests that the effect of the antibiotic varies depending on the growth phase of the bacteria (Sawai, et al., 2007). It’s noteworthy that sub-inhibitory concentrations were used in this study, which may occur in micro niches of the infected host—especially if the vasculature at the site of infection is compromised.
Penicillin is the antibiotic of choice for patients with pharyngitis who are not allergic to the drug. Since the 1950s, the incidence of the failure of penicillin to eradicate S. pyogenes has increased, even though S. pyogenes remains universally susceptible to penicillin ex vivo (Pichichero, 1991). Several explanations have been proposed to explain this observation, including the idea that some isolates can be tolerant, but not resistant, to penicillin. To explore changes in the proteome that are associated with phenotypic tolerance to penicillin, cytoplasmic proteins were compared between cultures of S. pyogenes exposed to penicillin, or not, by using 2-DE and tandem mass spectrometry (Chaussee, McDowell, Rieck, Callegari, & Chaussee, 2006). Changes in the abundance of proteins associated with fatty acid biosynthesis, glycolysis, and various stress responsive proteins were identified. In addition, the proteome of a penicillin-tolerant strain of S. pyogenes was characterized and compared to that of the non-tolerant parental strain in both the presence and absence of penicillin. Following penicillin exposure, the proteome of the parental strain was similar to that of the tolerant strain that had not been exposed to penicillin. This finding suggests that the wild-type bacteria respond to penicillin in a manner that reflects the pattern of gene expression in the tolerant strain. The implication is that tolerance is induced by penicillin exposure (at least in a fraction of the bacterial cells in the culture), which might be relevant to clinical treatment failure (Chaussee, McDowell, Rieck, Callegari, & Chaussee, 2006).
In addition to examining the response to antimicrobials that are currently in use, proteomics has been used to investigate the mechanism of action of compounds that are being developed as therapeutic agents. Rhodomyrtron is a phytochemical isolated from Rhodomyrtus tomentosa that inhibits the growth of S. pyogenes and other Gram-positive bacteria. Using a gel-based approach, cytoplasmic and CSPs were compared between cultures grown in the presence or absence of sub-inhibitory concentrations of rhodomyrtron in an attempt to determine the mechanism of inhibition. Changes in metabolic enzymes, as well as secreted virulence factors (CAMP factor and SpeC), were identified (Limsuwan, Hesseling-Meinders, Voravuthikunchai, van Dijl, & Kayser, 2011). While the results seem to indicate that the compound perturbs the proteome, additional study is needed to determine how exactly these changes influence the growth of S. pyogenes.
Transcriptional regulation of sub-proteomes
Proteins localized to the cell wall or the extracellular milieu directly influence the outcome of host-pathogen interactions. As a result, the characterization of these sub-proteomes can be useful in identifying the molecular bases for differences in virulence. For example, proteomics have helped to identify at least part of the basis for the virulence attenuation associated with a perR mutant (Wen, et al., 2011). PerR is a Fur-like transcriptional regulator that contributes to iron homeostasis, the oxidative stress response, and virulence (Ricci, Janulczyk, & Björck, 2002). CSPs produced by wild-type strain A20 and a perR mutant were separated with 2-DE and compared. Thirty-eight differences in protein abundance were identified, including a difference in the production of MF-3, a secreted nuclease encoded by bacteriophage (Wen, et al., 2011). Follow-up studies confirmed that there was less MF-3 produced by the perR mutant; showed that PerR bound to the promoter region of the mf-3 gene; and showed that decreased expression of mf-3 in the parental A20 strain decreased virulence. Notably, the growth rate and yield of the perR mutant was less when DNA was the sole carbon source present in the media, which suggests that DNA was being utilized as a catabolic substrate; presumably following hydrolysis by extracellular nucleases, including MF-3 (Wen, et al., 2011).
Another study on using proteomics to characterize regulons focused on Nra, a transcriptional regulator in the RALP family that represses the expression of virulence factors localized primarily to the cell wall, including a fibronectin-binding protein (SfbX), serum opacity factor (SOF), C5a peptidase (ScpA), M protein, and others (Kreikemeyer, et al., 2007). Proteomics was used, in conjunction with DNA microarrays, to identify other members of the Nra regulon. Both culture supernatant and cell wall-associated proteins were obtained from wild-type and nra mutant strains and were separated with 2-DE. In this study, phage lysin C was used to release the cell wall-associated proteins. A total of 67 proteins were identified as being differentially expressed (Kreikemeyer, et al., 2007). The largest differences in protein abundance among CSPs and the cell wall-associated proteins were PrtF.2 (a fibronectin binding protein) and SclA (a collagen-like protein), respectively. With some exceptions, the changes identified with proteomics correlated to the results obtained by DNA microarrays.
A systems approach was used to investigate a serotype M89 isolate from an invasive infection (Tsatsaronis, et al., 2013). The isolate possessed a naturally acquired mutation in the csrS/covS gene, which is the histidine kinase component of the two component regulator csrRS/covRS. The secretomes of the clinical isolate and a derivative that possessed a functional CsrS/CovS protein were compared. SclA and Gls24 were identified in samples from the clinical isolate, but were absent from the secretome of the complemented isolate that possessed a functional csrS/covS gene. Gls24 is a stress-responsive protein that is required for virulence in Entercoccus faecalis (Teng, Nannini, & Murray, 2005). Subsequent investigation revealed that both SclA and Gls24 are required for the survival of S. pyogenes in human blood. As a result, proteomics has discovered a novel role for SclA and Gls24 in streptococcal pathogenesis.
Identifying the host-acquired proteome
S. pyogenes surface proteins bind human proteins, thereby acquiring a surface composition partially derived from humans; the so called “host-acquired proteome.” Decorating the cell surface with human proteins confers several advantages to the microbe, including adherence, internalization, evasion of the host immune response, and pathogen dissemination.
Human proteins that regulate complement activation accumulate on the surfaces of pathogens and contribute to virulence. The cell wall-associated M protein is the best characterized protein involved in recruiting human proteins to the surface. M proteins typically bind to Factor H, fibrinogen (Fg), C4b-binding protein (C4BP), plasminogen, collagen, and albumin, although the binding specificity and affinity varies among the different serotypes (Smeesters, McMillan, & Sriprakash, 2010). Factor H and C4BP are large glycoproteins that are present in human plasma. They down-regulate complement activation on human cells. Fg binding to the surface of several S. pyogenes serotypes decreases complement deposition and activation (Smeesters, McMillan, & Sriprakash, 2010). SRM MS was used to measure the binding of IgG subclasses and complement proteins present in pooled human plasma and saliva to the surface of S. pyogenes (Nordenfelt, et al., 2012). The results showed a clear distinction between the fluids. After incubation with plasma, the host proteins bound to the bacterial surface were predominately IgG1 and IgG3, as well as proteins associated with both the classical and alternative complement pathways. In contrast, the predominant host proteins identified on the bacterial surface following incubation with saliva were IgG1 and IgG2, as well as complement factor H, which inhibits complement activation (Nordenfelt, et al., 2012). The results highlight the dynamic nature of the bacterial surface protein composition during the colonization of different niches within the human host.
Mga is a global transcriptional regulator that is required for the expression of several extracellular proteins, including the M protein. SRM was used to compare the binding of human plasma proteins to the surfaces of both a wild-type M1 strain and an mga mutant derivative (Sjöholm, Karlsson, Linder, & Malmström, 2014). Plasma proteins partially mimic the environment to which the pathogen is exposed during both bacteremia and localized infections, where the inflammatory response causes vascular leakage and an increase in plasma proteins at the site of infection. The results showed differences in the binding of more than 28 plasma proteins. The mga mutant bound significantly less Fg, C4BP, and S proteins to the pathogen surface, which is as expected, due to the absence of M protein. The results also showed an increase in the binding of human complement proteins associated with the membrane attack complex to the bacterial surface of the mga mutant strain. The results re-emphasized the importance of the Mga regulon in regulating host-protein recruitment to the surface of the pathogen.
An alternate strategy to characterize a host-acquired proteome used protein chips to analyze multiple streptococcal proteins for their ability to bind various ligands, including host proteins. A chip consisting of 106 recombinant streptococcal proteins predicted to be surface localized was created by expressing the genes in E. coli, purifying the epitope tagged proteins, and arraying the proteins on nitrocellulose coated slides. Using the chip, Margarit et al. identified proteins with the capacity to bind Fg, fibronectin, and C4BP. In addition to confirming the previously described interactions, the study discovered some new interactions. Specifically, two streptococcal membrane proteins were identified that bind to C4BP (Spy_1037 and Spy_1326), and a novel Fg binding protein (Spy_0591) was discovered (Margarit, et al., 2009).
Using a combination of shotgun proteomics and SRM, Sjoholm (Sjöholm, Karlsson, Linder, & Malmström, 2014) examined several aspects of host protein binding to the bacterial surface. The most interesting results came from quantitating the abundance of plasma proteins bound to the streptococcal surface. The most abundant human proteins on the surface were Fg, immunoglobulins, C4BP, albumin, and protein S. Protein S is a glycoprotein that inhibits coagulation, among other functions. Much of the protein S in human plasma is associated with C4BP (Dahlbäck & Stenflo, 1981), which perhaps indicates that protein S-C4BP complexes bind to the bacterial surface, rather than through sequential binding of each protein, or the binding of each protein to distinct bacterial surface proteins.
Many of the human proteins recruited to the bacterial cell surface regulate different aspects of the immune response or mediate adherence and internalization; however, a unique function was discovered by comparing the abundance of streptococcal cytoplasmic proteins during culture with varying concentrations of human plasma. The presence of plasma altered the abundance of over 200 streptococcal proteins, as determined by using label free LC MS/MS. The most prominent changes included a decrease in the abundance of enzymes involved in fatty acid biosynthesis. Malmstrom et al. further investigated this finding and discovered that the plasma component responsible for repression was human serum albumin (HSA) bound to fatty acids (Malmström, et al., 2012). While it has been known for years that streptococcal M (and M-like) proteins bind HSA (Wagner, Schmidt, Wagner, & Köhler, 1986; Retnoningrum & Cleary, 1994), it was not known that binding resulted in the acquisition of fatty acids and a subsequent decrease in the expression of fatty acid biosynthetic enzymes. Thus, in addition to this new information, these results provide an example of how using an unbiased proteomics approach can lead to novel insights.
Additional human proteins are likely to be bound to the S. pyogenes surface, either in amounts below the current limits of detection or as a result of indirect binding through other human proteins. For example, several cell wall-associated streptococcal proteins bind human fibronectin (Schwarz-Linek, Höök, & Potts, 2006). By using LC-MS/MS, over 30 different human plasma proteins were identified that bind to the amino terminal region of human fibronectin. As a result of fibronectin binding to the bacterial cell surface, dozens of other human proteins may also be recruited to the streptococcal surface (Moussavi-Harami, et al., 2013). Thus, the bacterial surface composition (surfome) is likely to be quite different in various micro-niches (including blood, pharynx, subcutaneous tissue, or lung, among others) occupied by S. pyogenes, depending on the expression of cell wall-associated proteins, posttranslational changes to cell surface proteins (discussed below), and the availability of different human proteins for recruitment to the surface.
Protein-protein interactions at the cell surface
While a great deal of progress has been made in identifying proteins associated with the bacterial cell surface, little is known about their spatial organization, which is likely to influence their functions. Proteomics can be used as an alternative to two-hybrid type experiments to identify protein-protein interactions. One such study explored the protein-protein interactions of S. pyogenes surface proteins using protein chips that consisted of 83 purified streptococcal proteins, which were selected based on their possession of a signal peptide, a lipoprotein motif, or the presence of an LPXTG motif. The capacity of each of these immobilized proteins to bind to other extracellular proteins was then assessed by incubating the array with soluble, biotinylated derivatives of each protein. Protein-protein interactions were further characterized by surface plasma resonance and confocal microscopy (Galeotti, et al., 2012). Among the highlights of the study was the finding that a surface associated protein annotated as Mur1.2 binds to several proteins that are known to be important to virulence, including hyaluronidase (HylA), SKA, SLO, a likely superantigen (SpeI), ScpA, and a fibronectin binding protein (SpyM3_0104). The primary structure of Mur1.2 is similar to N-acetyl muramidases, such as FlgJ. Among some Gram-negative species, FlgJ remodels peptidoglycan and binds to other extracellular proteins to facilitate the assembly of flagella. While speculative, the results suggest that Mur1.2 is involved in the spatial organization of streptococcal cell wall-associated proteins. The future use of proteomics, in combination with microscopy, is likely to aid in determining the architecture of the bacterial cell surface and contribute to our understanding of the ways in which the organization of extracellular proteins influences the function of these proteins.
Culture supernatant proteins
One might predict that CSPs would only include proteins that possess a type II signal sequence for transport across the cytoplasmic membrane and that lack a cell wall anchoring motif. Moreover, given the amino acid conservation of these motifs, one might also suspect the straightforward prediction of CSPs, based on inferences of the genome sequence. Experimental results obtained from proteomics have shown this is not the case. Initial reports that the glycolytic enzymes glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Pancholi & Fischetti, 1992) and enolase (Pancholi & Fischetti, 1998) were localized to the cell wall and culture supernatant were confirmed by a proteomic analysis that showed the presence of nearly all the glycolytic enzymes (with the exception being glucose phosphate isomerase) in culture supernatant fractions (Lei, Mackie, Lukomski, & Musser, 2000). Moreover, the results showed that approximately half of all the CSPs have no discernable type II secretion signal, and that many are also associated with the bacterial cell surface.
The canonical cytoplasmic proteins (such as enolase) possess different functions when localized extracellularly, as compared to when they are cytoplasmic. As a result, they are often referred to as “moonlighting” proteins., Most of these proteins are involved in metabolism when localized to the cytoplasm and virulence when localized extracellularly (Henderson & Martin, 2011). For example, surface-localized GAPDH, which binds to human plasminogen (Pancholi & Fischetti, 1992), mediates adherence to pharyngeal cells and is antiphagocytic (Boël, Jin, & Pancholi, 2005). Similarly, surface-localized enolase binds to human plasminogen (Pancholi & Fischetti, 1998) with an even greater affinity than that of GAPDH. Plasminogen can subsequently be activated by SKA to plasmin, which degrades host tissue and promotes pathogen dissemination. The presence of moonlighting proteins on the surface of microbes has been described in several other bacterial species. Moreover, similar to S. pyogenes, many of the moonlighting extracellular glycolytic enzymes contribute to virulence (Henderson & Martin, 2011). The paradigm even extends to other kingdoms, including mammalian cells, where normally cytosolic glycolytic enzymes (hexokinase, lactate dehydrogenase and glyceraldehyde-3-phosphate dehydrogenase) also localize to the nucleus and function in transcriptional regulation. Others (such as glucose-6-phosphate isomerase) influence motility and apoptosis (such as glucokinase, glyceraldehyde-3-phosphate dehydrogenase, and hexokinase) (Kim & Dang, 2005). As a result, protein localization is a critical component to the understanding of protein function.
The trafficking of moonlighting proteins has been, and continues to be, an enigma. In mammalian cells, the phosphorylation of lactate dehydrogenase is thought to be the signal for localization to the nucleus (Henderson & Martin, 2011). In S. pyogenes, one theory is that the extracellular localization of proteins that are usually considered to be cytoplasmic proteins is simply the result of autolysis and the coincidental affinity of the proteins for other proteins localized to the cell wall. This idea is attractive, because the most abundant extracellular moonlighting proteins are also the most abundant cytoplasmic proteins. However, the discovery that adding a twelve amino-acid peptide to the carboxyl terminus of GAPDH diminished extracellular localization indicates that autolysis may not be involved, and implies that an unidentified transport system specifically targets these proteins to the extracellular milieu (Boël, Jin, & Pancholi, 2005). While the mechanism of localization is not resolved, there is nonetheless a great deal of evidence that the glycolytic proteins are present on the cell surface and that they significantly influence host-pathogen interactions.
Post-translational regulation of the extracellular proteome
Proteins secreted to the extracellular milieu are important for the adaptation of S. pyogenes to various niches occupied by the pathogen during infection. The composition of proteins associated with the cell wall, and those that are freely soluble, is regulated both at the transcriptional level and post-translationally. In general, genes that encode proteins with the LPXTG motif are transcribed in the exponential phase of growth, while those with type II secretion peptides, but no LPXTG motif, are transcribed in the post-exponential phase of growth.
The secreted cysteine protease SpeB is the most important post-translational regulator of the extracellular proteome. The transcriptional regulation of speB expression is complex and was recently reviewed (Carroll & Musser, 2011). In general, speB is expressed in the post-exponential phase of growth following activation of expression by Rgg1 in response to a variety of signals, including glycolytic flux (Lyon, Gibson, & Caparon, 1998; Loughman & Caparon, 2006). In addition to transcriptional regulation, SpeB is secreted as zymogen, and specific environmental conditions are required for conversion of the zymogen to the enzymatically active protease. Proteins involved in the secretion and maturation of SpeB include RopA, PrsA, and HtrA (Lyon, Gibson, & Caparon, 1998; Lyon & Caparon, 2004). A proteomic study examined the effects of deleting the htrA gene on proteins localized to both the cell wall and the culture supernatant by using 2-DE, and indicated that HtrA indirectly enhances SpeB maturation, but is not essential for conversion of the zymogen to the active protease (Cole, et al., 2007). The diminished amounts of proteolytically active SpeB in the htrA mutant were associated with the presence of many proteins in cell wall and culture supernatant fractions that were likely degraded by SpeB in wild-type fractions. In addition, the results indicated that essentially all the proteolytic activity in culture supernatant fractions isolated from strain HSC5 could be attributed to SpeB (Cole, et al., 2007).
In 1945, Elliott reported that the extracellular SpeB protease (referred to then as streptococcal proteinase) degrades the cell wall-associated M protein (Elliott, 1945). This also results in the release of host proteins bound to M, such as Fg and immunoglobulin (Nelson, Garbe, & Collin, 2011). Subsequent studies showed that the protease can cleave, or degrade, a plethora of additional streptococcal and host proteins (Nelson, Garbe, & Collin, 2011). In fact, results obtained using proteomics showed that SpeB degrades nearly the entire secretome of an invasive serotype M1 isolate (Aziz, et al., 2004). As a result, SpeB can significantly alter the protein environment of the pathogen, which is comprised of both bacterial and host proteins.
SpeB-mediated remodeling of the streptococcal surface proteome significantly influences host-pathogen interactions and has been associated with pathogen dissemination. For example, when the bacterial cell density at the site of infection reaches a critical level, SpeB is expressed, secreted, and activated. Proteolysis results in both the enzymatic degradation of streptococcal proteins that mediate adherence and internalization (Chaussee, Cole, & van Putten, 2000), and the degradation of host extracellular matrix proteins (Kapur, et al., 1993). As a result, SpeB has been proposed to be a spreading factor that promotes pathogen dissemination.
In contrast, other results suggest that SpeB production is associated with localized infections. All clinical isolates possess the speB gene; however, not all of these isolates synthesize the protein. This can be the result of naturally selected mutations in either the csrS/covS gene or the rgg1 gene. Mutations in these loci typically increase the expression of many virulence-associated exoproteins and decrease, or abrogate, the expression of SpeB (Chaussee, et al., 2002; Treviño, et al., 2009). Remarkably, isolates from patients with invasive diseases are more likely to have mutations in either csrS/covS or rgg1, as compared to isolates from localized pharyngeal infections (Sumby, Whitney, Graviss, DeLeo, & Musser, 2006; Ikebe, et al., 2010; Carroll, et al., 2011).
Two complementary mechanisms have been identified to explain the ways in which a loss of SpeB production (as occurs with csrS/covS and rgg1 mutants) can increase pathogen dissemination. First, SpeB degrades an extracellular DNase (Sda1) that is important in mediating a pathogen’s escape from neutrophil extracellular traps (NETs) (Walker, et al., 2007). Thus, when SpeB is active, there is insufficient Sda1 to mediate the escape from NETs and the infection tends to remain localized. On the other hand, in the absence of SpeB, Sda1 is active, and as a result, the pathogen is more likely to escape NETs and disseminate. Second, SpeB degrades SKA (an activator of human plasminogen) (Cole, et al., 2006). Therefore, in the absence of SpeB, SKA can convert human plasminogen to plasmin, which accumulates on the bacterial surface through binding to the M-like protein PAM, as well as the moonlighting proteins enolase and GAPDH. Because plasmin degrades many human proteins, including fibrin, fibronectin, thrombospondin, laminin, and others, the pathogen is able to invade tissues and disseminate.
From a practical standpoint, the results show the importance of accounting for SpeB protease activity when analyzing extracellular proteins. SpeB-mediated changes in the proteome can be experimentally controlled in several ways: first, by using speB mutants, which fail to produce either the protein (knock-out mutation) or site-specific mutants that lack the active site cysteine residue that is required for protease activity; second, by maintaining culture conditions that inhibit activation of the zymogen or by harvesting proteins prior to SpeB activation and subsequent protein degradation; and third, by including cysteine protease inhibitors in the media. In this instance, the epoxide cysteine protease inhibitor E64 is typically used.
Discovering immunogenic streptococcal proteins
Characterizing immunogenic streptococcal proteins is necessary to identify those that elicit cross-reactive antibodies involved in post-infection sequelae, as well as those that elicit protective antibodies, which may be used to vaccinate against S. pyogenes.
Streptococcal induced autoimmunity
Previous studies have identified antigens that contribute to the pathogenesis of post-infection sequelae. Cross-reactive antibodies directed at the M protein and the role they play in acute rheumatic fever (ARF) are of particular importance to this process. Neurological complications, including Syndeham’s chorea, are well-known symptoms of ARF; however, in recent decades, a broader collection of neurological symptoms, including obsessive-compulsive disorders and tics, have been recognized as sequelae of S. pyogenes infections (Snider & Swedo, 2004). To identify streptococcal antigens that elicit neuropathogenic antibodies, Bombaci et al. used a protein chip that consists of over one hundred recombinant S. pyogenes proteins arrayed on glass slides to measure reactivity with sera obtained from 335 children, including 61 with neuropsychiatric symptoms (Bombaci, et al., 2009). The results indicated that symptomatic patients had elevated antibody titers to multiple streptococcal proteins, as opposed to strong reactivity to just a few proteins (such as the M protein). The continued development and use of protein chips is likely to yield important new information into the pathogenesis of post-infection sequelae.
Vaccine development
Reverse vaccinology uses genomics (and often functional genomics) to identify proteins that are likely to be good candidates for vaccines. Protein localization to the cell wall or membrane is among the criteria often used to select candidate immunogens because these proteins are likely to be accessible to antibodies. Other criteria include proteins that are well conserved among isolates of S. pyogenes, those that are highly expressed, and those that are known to elicit antibodies during infection of either humans or mice. Proteins that meet these criteria are typically purified and tested in animal models to assess the ability of the protein to confer protective immunity.
Using bioinformatics, Sharma et al. identified between 199 and 237 proteins with a type II signal peptide among eight sequenced genomes. Next, the presence of the corresponding genes in the chromosome of isolates from India was determined (Sharma, et al., 2013). To identify these expressed genes, cell wall and membrane proteins were enriched from M1 and M49 strains and were separated by SDS-PAGE. The gels were then sliced into multiple pieces. The proteins were digested within the gel slices, the peptides were eluted, and the proteins were identified by MS. A total of 128 and 373 proteins were detected in the M1 and M49 strains, respectively; 116 proteins were identified in samples from both strains. As observed with other fractionation methods, many of the proteins identified are canonical cytoplasmic membranes, including a transcriptional regulator (Sharma, et al., 2013). Of all the proteins experimentally identified, 52 possessed a type II signal peptide.
Antibodies to surface proteins can be opsonic and can neutralize important functions related to virulence, such as adherence, which makes them attractive vaccine candidates. CSPs have also been investigated as vaccine targets for two major reasons. First, several CSPs are thought to be critical to virulence, and consequently, their neutralizing antibodies would likely decrease virulence. Second, proteomic studies indicate that many CSPs are also non-covalently associated with the cell wall. Thus, antibodies to CSPs may also be opsonic. The first study to use proteomics to identify CSP vaccine candidates analyzed the proteins collected from both exponential and stationary phase cultures of invasive isolates that represented serotypes M1 and M3. The proteins were separated with 2-DE, and the amino termini of selected proteins was determined through Edman degradation. Inter-strain differences in CSP composition were evident, including differences in the abundance of extracellular DNAse, NAD-glycohydrolase, SLO, and other proteins. CSPs that elicited an antibody response during the course of human infections were identified by 2-DE immunoblotting with patient sera (Lei, Mackie, Lukomski, & Musser, 2000). Many of the proteins that reacted with antibodies were moonlighting proteins that lacked a type II signal sequence. In addition, because Edman degradation (and not mass spectrometry) was used to identify protein spots excised from the gel, the results also showed that some of the protein isoforms separated by 2-DE were derived from truncation of the polypeptide at the amino terminus, presumably post-translationally. For example, two forms of mitogenic factor (a secreted DNase) were identified. The amino terminus of one corresponded to amino acid 43 of the inferred polypeptide, while the other corresponded to amino acid 42. While it is unclear if there are functional differences between the isoforms, the results illustrate the power of 2-DE to separate highly similar proteins and also provides insight into the types of differences that distinguish protein isoforms (Lei, Mackie, Lukomski, & Musser, 2000).
A similar study used human antisera to identify immunogenic cell wall-associated proteins produced by serotypes M69, M53, and M6. Proteins were isolated and separated with 2-DE. 155 protein spots were identified with MALDI-TOF MS, which corresponded to 74 proteins (Cole, et al., 2005). 45% of the identified proteins were antigenic, as determined by using pooled sera obtained from children living in a region of Australia where 70% of the children have impetigo. Cole et al. (Cole, et al., 2005) also exposed S. pyogenes to biotin prior to isolating cell wall proteins, which were then separated with 2-DE. To identify proteins that were surface-accessible (based on their being biotinylated) immunoblotting was done with streptavidin and anti-streptavidin antibodies. About 30% of the proteins reacted with anti-streptavidin, which implies that they are surface exposed. The biotinylated proteins generally also reacted with pooled sera from the children. For proteins that were not biotinylated but that reacted with sera, it is difficult to know if the proteins were actually not surface exposed during biotinylation (or if a reactive amino group necessary for biotinylation was not surface exposed. As with other studies, several metabolic enzymes were identified in the cell wall-associated fraction.
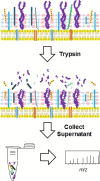
Another strategy to identify surface-exposed proteins for potential use as immunogens involves treating washed intact streptococcal cells with a protease, such as trypsin (Rodríguez-Ortega, et al., 2006; Severin, et al., 2007). The assumption is that trypsin will only cleave those proteins that are surface exposed. Following centrifugation to pellet the bacterial cells, the tryptic peptides present in the supernatant were collected, separated with liquid chromatography, and analyzed with MS/MS to identify the corresponding proteins (Rodríguez-Ortega, et al., 2006; Severin, et al., 2007). This technique is often referred to as “surface shaving” (). Bioinformatics predicted that 72 proteins encoded by the SF370 genome were surface exposed, and by using surface shaving, 95% of these proteins were identified (Rodríguez-Ortega, et al., 2006). The shaving approach should also identify membrane proteins with relatively large extracellular domains; however, only 37 of the estimated 524 membrane proteins encoded in the streptococcal genome were identified in the study (Rodríguez-Ortega, et al., 2006). It’s possible that the cell wall and/or polysaccharide capsules may inhibit trypsin activity near the cytoplasmic membrane surface, thereby reducing the number of membrane proteins identified with this approach (Rodríguez-Ortega, et al., 2006). Rodriguez-Ortega tested 14 surface exposed proteins to determine if any could confer protection in a mouse model against subsequent challenges with S. pyogenes. The results of this work identified a new protective protein, Spy_0416, which is annotated as a peptidase that contains the LPXTG motif (Rodríguez-Ortega, et al., 2006).
Proteolytic shaving to identify surface exposed proteins. Washed S. pyogenes cells are suspended in buffer and incubated with trypsin. Centrifugation is used to pellet the cells and tryptic peptides derived from surface exposed proteins are collected (more...)
Surface proteins of strain SF370 were similarly analyzed during the exponential and post-exponential phases of growth. 79 proteins were identified, including 14 which possessed the LPXTG motif (Severin, et al., 2007). Approximately one-quarter of the proteins were moonlighting proteins—a finding that is similar to results obtained by using cell wall hydrolases to extract surface associated proteins. When combined with results obtained by Rodriquez-Ortega, 118 surface exposed-proteins were identified. Remarkably, despite using similar approaches, only about 30% of the proteins identified were common to both studies.
Bensi et al. (Bensi, et al., 2012) used bioinformatics to identify proteins that are likely to be secreted to the surface. The corresponding genes were then expressed in E. coli, purified, and used to create a protein array. The purified proteins were also used to generate antibodies, which were used to identify highly expressed surface proteins by measuring protein production among a panel of clinical isolates. In addition, sera from patients with pharyngitis were used to identify proteins that were expressed during the course of human infection. Finally, the surface-shaving method was used to identify surface-exposed proteins. Six proteins were identified that were surface exposed, highly expressed, and elicited antibodies during human infection. Notably, two of these proteins, SLO and ScpA, have already been considered as vaccine candidates. The other four were SpyCEP protease (SPy_0416); PrgA (SPy_0269), which is annotated as a surface exclusion protein; SPy0019, which is a surface protein with an amidase motif; and internalin InlA (SPy_2010).