

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mobley HLT, Mendz GL, Hazell SL, editors. Helicobacter pylori: Physiology and Genetics. Washington (DC): ASM Press; 2001.

In the study of bacterial diseases, research often focuses on virulence factors that distinguish a true pathogen from innocuous organisms inhabiting many body surfaces. The most critical feature of Helicobacter pylori, however, may not be its ability to damage host tissues, but rather its ability to persist for many years within the host. Although some H. pylori strains clearly cause more damage to cells than others, much of the pathogenesis seen in H. pylori infections may be due to misdirection of the host's response to the bacterium rather than any significant toxicity mediated by the bacterium itself. Colonization, therefore, is not synonymous with virulence but rather may refer to the persistence and growth of the organism in a particular site within the host, irrespective of any detrimental effects on the host. This chapter will focus on host, environmental, and bacterial factors that contribute to the establishment and persistence of H. pylori within the stomach rather than on genes for products that specifically contribute to host damage. These topics are covered in other chapters.
The process of colonization consists of four steps: (i) transmission to a new host; (ii) bacterial adherence to a specific niche within the host; (iii) avoidance, subversion, or exploitation of host defense mechanisms; and (iv) acquisition of nutrients resulting in successful replication. The primary difference between colonization and pathogenesis is that mere colonization may not lead to overt tissue damage (105, 106). For example, most Escherichia coli in the gut and staphylococci on the skin never cause damage. Members of the oral flora, such as lactobacilli, staphylococci, and streptococci, are frequently found in the human stomach but cannot truly colonize; they are killed by gastric acid or flushed into the small intestine, only to be "reseeded" with the next swallow. Transmission of H. pylori, however, appears to be infrequent, requiring the organism to take hold quickly and avoid being swept into the intestine. This makes adherence a critical early step in colonization. Colonization can therefore be viewed as the confluence of mechanisms used to persist in the stomach.
The mechanism of H. pylori transmission remains elusive. Many investigators have searched for environmental reservoirs of H. pylori. H. pylori DNA or actively respiring organisms have been detected in well water (135, 152) and saliva (189); however, live organisms have not been cultured. Culture of live H. pylori from stool has only rarely been accomplished (171, 329). It remains possible that H. pylori survives in a dormant state and that current culture methods are inadequate to stimulate growth of this organism.
In the absence of an environmental reservoir, H. pylori is required to transport itself from one stomach to another. The most direct route of such transmission is via contact with vomitus. This may indeed be one route of transmission, as suggested by some investigators (187, 250), but this seems unlikely to be the primary mode of transmission. Epidemiological studies (see chapter 2) indicate transmission occurs most often during early childhood, with the mother as the most common source of infection (93, 201). Children are seldom exposed to the vomitus of adults, however. A more likely mode of transmission would be via saliva. Activities such as kissing and sharing of eating utensils could then promote transmission. H. pylori has been suggested to reside within the oral cavity, perhaps in association with other bacterial species. Circumstantial evidence suggests that, if true, numbers found in the oral cavity are low (20, 351). Natural infection of humans is likely to proceed quite differently from animal infection models, which require large and often repeated inocula.
Following transmission, H. pylori faces a new set of challenges in establishing colonization. H. pylori exhibits both host tropism, colonizing only primates, and tissue tropism, adhering only to the gastric epithelial lining of the antrum or staying in the gastric mucous layer. Over 90% of the H. pylori bacteria are thought to remain in the mucous layer, with the smaller proportion colonizing the gastric surface. H. pylori adheres to the mucus-secreting gastric epithelial cells of the lumen in the upper half of the gastric pit, but not to closely related mucus-secreting neck cells in the lower half of the gastric pit (Fig. 1). H. pylori also does not adhere to chief cells, parietal cells, or endocrine cells in the gastric pit. Although H. pylori adheres almost exclusively to the less acidic antrum region of the stomach, it can occasionally colonize the corpus under conditions of low acid secretion, such as when patients are on long-term acid suppression. Such patients may be at elevated risk for developing gastric cancer (183).
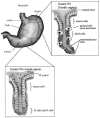
In response to H. pylori colonization of the antral mucosa, G endocrine cells in the antrum are activated to release the hormone gastrin, which stimulates parietal cells in the corpus (body) region of the stomach to hypersecrete acid (154, 209). This increased acid production may lead to gastric metaplasia and damage to the duodenum, predisposing patients to duodenal ulcers. Gastrin release by the G cells inhibits release of somatostatin by the D endocrine cells in the antral mucosa (209).
Adhesion is considered to be necessary for the establishment of H. pylori infection, but many other factors also influence the persistence of H. pylori in the gastric mucosa. In vitro tissue culture models permit the study of adhesion independently from other factors influencing colonization, such as epithelial cell turnover, host immune response, and the impact of gastric contents. For this reason, adhesion will be dealt with separately from colonization, though it is understood to be a key step in the colonization process.
Adherence
Adhesins are bacterial proteins, glycoconjugates, or lipids that are involved in the initial stages of colonization by mediating the interaction between the bacterium and the host cell surface (Table 1). Thus, adhesins are generally regarded as virulence factors for the bacterium. Host cell receptors are composed of lipids, proteins, glycolipids, or glycoproteins. Adherence of bacteria to host cell receptors triggers cellular changes that include signal transduction cascades, leading to infiltration of inflammatory cells (neutrophils and monocytes) and to persistence of the organism. Since the gastric epithelium and mucus are in continual turnover, and peristalsis ensures constant movement of food and cell debris, H. pylori has evolved adherence and colonization mechanisms to maintain itself specifically in the gastric mucosa. In this section, we describe the cellular changes associated with attachment of H. pylori to host cells, the bacterial adhesins themselves, and host cell receptors that have been reported to date.
Table 1
H. pylori adhesins and host cell receptors.
Despite extensive research on H. pylori adherence in vitro, there is little consensus on which H. pylori adhesins are most important in vivo, or even whether adhesins and receptors for the H. pylori-epithelial cell interactions are similar to the H. pylori-phagocytic cell interactions. Numerous problems hinder the unequivocal identification of adhesins and host cell receptors: (i) H. pylori adherence differences among strains; (ii) presence of numerous adhesins in H. pylori; (iii) variable expression of H. pylori adhesins under different culture conditions for a single strain; (iv) variable expression in the coccoid versus bacillary forms of H. pylori; (v) use of nongastric and nonphagocytic cell experimental systems to identify H. pylori adhesins; (vi) autolysis of H. pylori; (vii) variable expression of host cell receptors within a single host upon contact with H. pylori; and (viii) genetic variability in receptor expression in different hosts. Few studies have investigated the effect of H. pylori mutants in specific candidate genes on adherence, and surprisingly, no studies have yet addressed the role of adherence genes in vivo. This may be because H. pylori adhesin-mediated colonization is clearly multifactorial and no single adhesin may be crucial. Regardless, adherence of H. pylori to the gastric epithelium is a necessary early step in colonization and renders H. pylori 100 to 1,000 times more resistant to antibiotics than nonadherent bacteria (211). Furthermore, adherence protects bacteria from being washed away and from the adherence-blocking effects of mucin.
Techniques for Investigating Adherence of H. pylori
There are a number of methods to assess adherence of H. pylori to host cells: microscopy, histology, flow cytometry, enzyme-linked immunosorbent assay, and in situ adherence. A standard method is microscopic counting of bacteria adhering to host cells. However, this method has high experiment-to-experiment variability (132). Histological analysis is useful for investigation of mechanisms of adherence but is not easily quantifiable. Flow cytometry is an excellent tool because it is easily quantifiable and reproducible (132, 194, 368). Logan and colleagues (194) developed a sensitive method for studying adherence using carboxyfluorescein diacetate succinimidyl ester to stain H. pylori and fluorescence-activated cell sorting to sort gastric epithelial cells. In situ assays of H. pylori adherence to human gastric tissue were pioneered by Falk and colleagues (104).
Cellular changes following attachment of H. pylori and role of the cag pathogenicity island
Upon encountering the gastric mucosa, H. pylori swims through the viscous gastric mucus, propelled by its polar flagella, and adheres to gastric epithelial cells. Only a small percentage of the total H. pylori population adheres to the gastric epithelium (<1%); the rest of the population resides in the gastric mucous layer or is cleared by peristalsis or other mechanisms (134, 333). During adherence, H. pylori has a predilection for intercellular junctions of gastric epithelial cells (26, 235). Entrance of H. pylori into gastric epithelial cells (26, 235) or into gastric epithelial cell lines (67) has generally been thought to be rare, although some controversy surrounds this subject (see "Does Invasion into Host Cells Occur?," below).
In the antrum of the human stomach, H. pylori adheres very specifically to mucus-secreting gastric epithelial cells on the luminal surface, the gastric pit surface, within gastric pit pockets, occasionally adjacent to parietal cells, and in between adjacent gastric epithelial cells (42, 62, 104, 274), where they disrupt intercellular tight junctions through the ammonia produced by urease (286) (Fig. 2). Adherence to other cell lineages in the antrum, such as chief, parietal, fibroblasts, or neck cells (104), or to epithelial cells in the duodenum, esophagous, or colon is rare (56, 310). Binding occurs more readily under acidic conditions in vitro (62, 150, 151). Acidity could be a signal for enhanced adhesin transcriptional or translational expression, or could simply favor adhesin-receptor interactions. Despite the specific tropism of H. pylori for the antral mucus-secreting epithelial cells in vivo, H. pylori adheres to a vast range of gastric and nongastric (human and nonhuman) cells in vitro (57). The reasons for this apparent discrepancy and for the specific in vivo tropism of H. pylori are not understood.
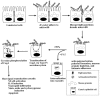
H. pylori probably adheres first to microvilli-containing regions of gastric epithelial cells (Fig. 2); then, by an undetermined mechanism, the microvilli become denuded (6, 26, 28, 35, 57, 62, 84, 125, 136, 140, 174, 230, 235, 272, 274, 284, 310). Upon subsequent tighter adherence, cup-like projections and pedestals form, accompanied by actin polymerization and cytoskeletal rearrangements. Tighter adherence leads to an increase in sialic acid glycoproteins on the host cell surface, depletion of mucous granules within host cells, and cellular degeneration (apoptosis) possibly mediated by the H. pylori vacuolating cytotoxin (VacA). Tyrosine phosphorylation of host and bacterial proteins occurs during intimate attachment (2, 284, 285). Fibrillar or filamentous appendages (variously reported as fimbriae or afimbrial adhesins) at the point of contact between H. pylori and the host cell during adherence have been suggested (35, 98, 140). Many of these properties (microvilli denudement, pedestal formation, actin polymerization, cytoskeletal rearrangements, tyrosine phosphorylation of proteins, and occasional cellular degeneration) are reminiscent of host cellular changes caused by adherence of enteropathogenic E. coli (EPEC) (57, 173). However, the EPEC adhesin called intimin, encoded by the eae gene, has no homolog in the H. pylori genome (7, 84, 332). The bacterial and host genes responsible for pedestal formation with concomitant actin polymerization are unknown, but genes encoded in the cag pathogenicity island may be required (285).
The above observations regarding H. pylori adherence have been made with human and gnotobiotic piglet gastric biopsies, primary human gastric cells, and human gastric cell lines, all with similar general conclusions, with rare exceptions (84, 230). The observation that H. pylori is sometimes found adjacent to acid-secreting parietal cells, coupled with the observation that patients with H. pylori infection suffer achlorhydria, suggests that H. pylori directly disrupts parietal cell function. Recent evidence strongly supports this hypothesis (see below;128,321).
The "cytotoxin-associated gene," cagA, was originally thought to be necessary for VacA vacuolating activity or expression, due to the strong correlation in clinical isolates: cagA + (type I) strains were Tox+, whereas cagA mutant (type II) strains were Tox (63, 337, 363). However, using an isogenic cagA mutant, it has been shown that cagA is not required for expression or vacuolating activity of VacA (338, 363). During the adhesion of H. pylori to gastric epithelial cells, an intense inflammatory response is generated. The cytokine interleukin (IL)-8, a chemotactic factor for human neutrophils, is secreted by gastric epithelial cells (in both gastric biopsies and cell lines) in response to binding of cag+ H. pylori isolates (67, 148, 267). The bacterial factor responsible for IL-8 induction remains a mystery, but genes in the cag pathogenicity island (PAI) are clearly required, based on observations that type I strains (cag+), but not type II strains (cag mutant), induce IL-8 expression and tyrosine phosphorylation of several proteins, including a 145-kDa protein (39, 285). Furthermore, isogenic mutants of numerous genes within the cag PAI abolish IL-8 induction and tyrosine phosphorylation (cagC, cagD, cagE [also known as picB], cagG, cagH, cagI, cagL, cagM, cagX, cagY, and cagβ) (39, 64, 190, 285, 339). Polar effects of one mutant on adjacent genes were not ruled out in these studies. This IL-8 induction is blocked by the protein tyrosine kinase inhibitor herbimycin A, indicating that a tyrosine kinase phosphorylates a protein (bacterial or host) necessary for IL-8 induction (190). Three mutants have no effect on IL-8 induction: cagA, cagF, and cagN (39, 69, 289). Type I strains bind to AGS cells better than type II strains, and the interaction of type I strains with AGS cells may up-regulate one or more host cell receptors triggered by interaction with the bacterium (317). Contact of live, intact H. pylori with gastric epithelial cells is a prerequisite for IL-8 induction (2, 267). IL-8 leads to infiltration of neutrophils and monocytes into the gastric mucosa, causing inflammation and mucosal damage (26, 125, 199). H. pylori adheres to and is phagocytosed by these neutrophils and monocytes in the absence of opsonins.
Further analysis of type I versus type II strains of H. pylori revealed that type I strains carry the 40-kb cag PAI, whereas type II strains do not (39). Nearly all isolates (88 to 100%) from patients with duodenal ulcers are cag+, whereas only 50 to 60% of isolates from patients with uncomplicated gastritis are cag+ (63, 66, 68). These findings indicate that type I (cag+) strains are more virulent than type II (cag mutant) strains, yet the presence of the cag PAI alone is not sufficient to confer full virulence. The organization of the cag PAI from several H. pylori strains has been reported (3, 7, 39, 332). Some of the cag genes are orthologous to the ptl/vir genes encoding a type IV secretion apparatus (e.g., cagE is orthologous to the gene encoding the membrane-associated ATPase, virB4, the VirB4 protein is involved in T-DNA transfer from Agrobacterium tumefaciens to plant cells; reviewed in references 54, 64, 65).
Recent data from four laboratories have shown that CagA is translocated into gastric epithelial cells via the cag PAI-encoded type IV secretion system (18, 238, 283, 313; reviewed in reference 64). Following translocation, CagA (120 to 140 kDa in different strains) is tyrosine phosphorylated. Thus, the 145-kDa tyrosine phosphorylated protein previously observed in host cells and thought to be a host protein is, in fact, the bacterial protein CagA. The cellular tyrosine kinase involved in tyrosine phosphorylation of CagA has not yet been reported. It will be of great interest to understand the role of phosphorylated CagA in perturbing host cell signaling pathways and elucidating the mechanism and signals responsible for translocation of CagA into host cells. Interestingly, a hemolysin mutant of H. pylori (specific gene not specified) cannot induce tyrosine phosphorylation of the 145-kDa protein (probably CagA), but still can induce IL-8 expression from a human gastric epithelial cell line, indicating that tyrosine phosphorylation and IL-8 induction are probably the result of separate signal transduction pathways (285). Part of this pathway may be activation of NF-κB and AP-1, which are known transcriptional activators of the IL-8 gene (2).
Chronic infection modulates the balance between gastric epithelial cell proliferation (cancer) and epithelial cell death (apoptosis) (224). Perturbation of this balance likely results from differences in factors produced by the bacterium, the host, or a combination of the two. H. pylori binds to class II major histocompatibility complex antigens on gastric epithelial cells, and a signal transduction cascade ensues, leading to apoptosis (107). Several groups have shown that the apoptosis cascade utilizes the Fas ligand/antigen pathway (CD95) (146, 163, 273), which fosters down-regulation of Bcl-2 (anti-apoptotic) and up-regulation of Bax (apoptotic) (178). The transcription factor NF-κB, activated by tumor necrosis factor (TNF)-α-mediated phosphorylation of IκB, activates transcription of the genes encoding IL-8, nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2). Several laboratories have recently shown that H. pylori induces iNOS and COX-2 expression in vitro and in vivo (114, 208, 271, 279, 358). It is unclear whether H. pylori-mediated induction of iNOS and COX-2 tilts the balance toward apoptosis or to increased proliferation of gastric epithelial cells leading to metaplasia and cancer. Both lipopolysaccharide (LPS)-dependent and LPS-independent (urease, VacA, cag-mediated protein secretion) H. pylori factors have been implicated (163, 175, 273, 305–308, 358).
The above studies clearly implicate the cag PAI in at least some of the cellular changes occurring immediately after H. pylori adheres to gastric epithelial cells. It will be of great interest to further discern the mechanism of how the cag PAI triggers these host cell signaling events and determine whether the PAI plays a role in apoptosis or proliferation of gastric epithelial cells.
Adherence to other microbes
H. pylori has been shown to adhere to Fusobacterium nucleatum and Fusobacterium periodonticum in vitro in a lactose-dependent manner (11). Data indicating this interaction to be relevant in vivo are not yet available. Fusobacterium spp. are found in human dental plaque. Therefore, dental plaque may serve as a potential reservoir for H. pylori, as has been suggested by other groups (288). This hypothesis is also supported by the finding that H. pylori binds salivary mucins in vitro (350) and that H. pylori adheres to buccal epithelial cells (57). More research is needed to address whether H. pylori can adhere to buccal epithelial cells in vivo and whether this constitutes an important reservoir for person-to-person transmission or for eventual migration of the organism to the gastric mucosa.
H. pylori also adheres via knob-shaped structures (adherence pedestals?) to yeasts from the genus Candida, especially to Candida tropicalis (14). As H. pylori may be found in conjunction with other microbes, including yeasts, in the stomach and possibly in the oral cavity (297), it is presently unclear whether there are interactions of H. pylori with stomach or oral microbial flora that could play an important in vivo role in modulating the pathogenesis and transmission of H. pylori.
Growth-phase-dependent adherence?
H. pylori exists in two major morphologies in vitro and in vivo: the motile, bacillary spiral form and the nonmotile, coccoid form. The coccoid form is possibly a viable but nonculturable state of H. pylori that occurs when the organism encounters stress such as extended stationary phase or nutrient limitation. Whether coccoid organisms are viable, dormant stages or a manifestation of eventual cell death is the subject of much controversy and will be discussed elsewhere (see chapter 6). With this controversy in mind, we will summarize some findings of the effect of coccoid forms and stationary phase (which triggers coccoid formation) on adherence.
In the study by Falk and colleagues, it was observed that stationary phase (containing many coccoid forms) but not exponential phase H. pylori (containing few coccoid forms) adheres to Lewis b antigens in fixed gastric tissue (104). Using the AGS cell line, it was found that coccoid forms of H. pylori adhere to the cells, stimulate cytoskeletal rearrangements, and induce tyrosine phsophorylation of bacterial and host proteins (283, 284). In contrast, other studies found that coccoid forms adhere to KatoIII and AGS cell lines less avidly than bacillary forms (58, 61); adherence to both cell types was Lewis b independent (58). Additionally, coccoid forms induce paltry IL-8 secretion by gastric epithelial cells compared with bacillary forms (61). Differences among these studies include length of culture of H. pylori to yield coccoid forms (range 4 to 20 days) and the type of model used. Nonetheless, from these data taken together it is tempting to speculate that H. pylori may convert from bacillary to coccoid form following attachment to the host gastric epithelium, since coccoid forms are observed routinely in vivo in gastric biopsies. This conversion may alter expression of adhesins. In support for this hypothesis, protein profiles of coccoid forms are quite different from those of bacillary forms (219, 220). Additional evidence for changes in H. pylori adhesins under different in vitro conditions comes from hemagglutination experiments. H. pylori expresses hemagglutinins (putative adhesins) on agar that are absent in broth-grown conditions (100, 218). More research is needed to address this hypothesis and to determine the mechanism of adhesin changes under different in vitro growth conditions.
Bacterial Adhesins and Receptors
Interactions of H. pylori with Mucin
Mucins are a family of high molecular weight oligomeric proteins (>2 million molecular weight) that are sulfated and heavily glycosylated (carbohydrate >80%) and represent a major component of mucus. These glycoproteins serve as host mucosal defenses by preventing colonization of microbes, as well as by protecting the epithelial cells from injury. Mucin contains a wide array of structurally distinct carbohydrate side chains, such as sulfated or nonsulfated sugar moieties, Lewis a, b, X, and/or Y moieties, and sialic acid residues. Because of its lifestyle as a mucosal pathogen, H. pylori has evolved a number of mechanisms to overcome the nonspecific host defenses due to mucin (Fig. 3). First, H. pylori has the ability to bind to gastric and salivary mucin, and optimal binding requires that the mucin be sulfated and/or sialylated (141, 142, 213, 229, 257, 294, 340, 345, 350). Sulfated galactose or sulfated Lewis a within salivary mucin was the likely antigen bound by H. pylori (350), although sialic acids and Lewis b are also possible antigens (104, 213, 236). Mucin binding is elevated by exposure of H. pylori to low pH in vitro, and by the presence of divalent cations (especially nickel and zinc), and is mediated most likely by electrostatic interactions (236, 350). Gastric mucin blocks H. pylori from adhering to the gastric epithelial cells, presumably by competing with the cell surface for H. pylori adhesins (104, 142, 294, 340). Desulfation of mucin by the anti-ulcer compound sulglycotide results in loss of binding of H. pylori to mucin (141, 257), and sialidase treatment of mucin likewise decreases H. pylori binding to gastric mucin and hemagglutination to red blood cells (141, 142, 213, 340). An H. pylori protein that binds mucin has been recently identified as the 16-kDa neutrophil-activating protein (Nap), a protein that binds pleiotropically to sulfated oligosaccharides such as sulfo-Lewis a, sulfogalactose, and sulfo-N-acetylglucosamine found on mucin (229, 327). Nap also binds to Lewis X and sialylated glycoconjugates in vitro (229, 327) and causes enhanced adhesion to human neutrophils by an unknown mechanism (102).

Second, H. pylori possesses several mucinase activities (proteins not yet identified): one desulfates mucin and the other proteolytically cleaves the proteinaceous component of mucin (299, 300, 304). Degradation of mucin allows H. pylori to migrate to and adhere to the gastric epithelial cell surface more quickly. In another study, urease of H. pylori was suggested to disrupt the carbonate-bicarbonate buffer of the stomach through the production of ammonium from urea, thereby leading to breakdown of gastric mucin (293). This was suggested based on the finding that filtrates of H. pylori passaged only once in vitro from gastric biopsies failed to degrade mucin unless urea was present. Whether urease can more directly degrade mucin has not been reported.
Third, H. pylori displays chemotactic activity toward mucin (111; T. L. Testerman, D. J. McGee, and H. L. T. Mobley, unpublished observations), which requires a two-component signal transduction system CheA/Y (111).
Fourth, the H. pylori thioredoxin was shown to reduce mucin such that oligomers of mucin are potentially converted to monomers (359). Reduction of mucin to monomeric form may destroy the ability of mucin to interact with the mucin receptor. Thus, H. pylori may chemotactically move toward mucin, bind mucin, and degrade it. This would decrease the viscosity of the mucous gel and allow H. pylori to facilitate migration to the epithelial cell surface, an event that prevents clearance by peristalsis.
Fifth, H. pylori may occasionally enter the mucin-containing compartment of goblet cells at areas of intestinal metaplasia (a precursor of gastric carcinoma) (117). This could explain the clinical observations that, upon adherence of H. pylori to the gastric epithelium, depletion of the mucin-containing mucous granules occurs (26). The mechanism of depletion of mucin secretion by the glandular epithelium has been further explored in two studies. A study of human biopsies showed that H. pylori infection significantly lowers the amount of mucin contained in cells from antral biopsies, but little effect is seen in cells from gastric body biopsies (170). Rats fed 0.01% ammonia for 4 weeks develop a similar pattern of mucin depletion in antral cells, suggesting that the ammonia produced by H. pylori urease may be responsible for effects on mucin (170). Micots and colleagues used a mucin-secreting cell line to further study the effects of H. pylori on mucus secretion (215). They found that baseline mucin biosynthesis was not impaired over a 24-h period; however, secretion of mucin was impaired at 24 h. Secretion by H. pylori-infected cells was more strongly inhibited following stimulation by forskolin and ionophore A23187, agents that raise intracellular cAMP levels. These studies indicate that H. pylori perturbs the release of mucin, which would facilitate H. pylori adherence to the gastric epithelial cell layer and may contribute to ulcer formation in the antrum. All of these interactions of H. pylori with mucin may also explain the clinical observations that H. pylori is found both free in the mucous layer and attached to gastric epithelial cells and strongly suggest that more research is needed to understand the H. pylori genes involved in these mucin interactions. Also, given the potential for H. pylori LPS to interact with host cell glycoconjugates of the same structure (homotypic interactions) (92, 324), a future area of examination should be determination of whether LPS is involved in adherence of H. pylori to mucin as both LPS and mucin contain Lewis X.
Hemagglutination
To investigate the interaction of H. pylori with human cells, hemagglutinating activity of various species of red blood cells (RBCs) by H. pylori has been extensively studied. On the basis of studies from numerous laboratories (10, 17, 47, 48, 51, 95, 100, 147, 149, 151, 160, 161, 184–186, 216, 218, 228, 245, 269, 325), H. pylori exhibits a broad spectrum of hemagglutination. This activity depends on the H. pylori strains used, how long they have been passaged in vitro, how the strains are grown (e.g., broth-grown cells lack sialic acid-dependent hemagglutination activity in contrast with agar-grown bacteria), and the species of red blood cells used in the hemagglutination study. Both cell surface-associated and soluble hemagglutinins exist in H. pylori. Hemagglutinin binding to human RBCs is mediated by at least three specific interactions. Two of the interactions are mediated by distinct sialic acid linkages to carbohydrate side chains of host glycoconjugates, either α2,3- or α2,6-specific for strongly hemagglutinating strains. These interactions are inhibited by pretreatment of the erythrocytes with neuraminidase or by fetuin or mucin, glycoproteins rich in sialylated oligosaccharides, but not by pretreatment with asialofetuin or asialomucin. A third interaction is observed for weaker hemagglutinating strains, and this interaction is sialic acid independent. A protein of 59 kDa has been suggested to be involved in this interaction, but its receptor specificity is unknown (147). In general, hemagglutination activity is inhibited by preincubation of H. pylori at 56°C. This heat treatment does not necessarily alter adherence, however.
In vivo, H. pylori is unlikely to interact with human RBCs. Thus, the physiological role(s) of the hemagglutinins has been addressed more carefully using gastric epithelial cells and human neutrophils and monocytes. Sialic acid-dependent hemagglutinating H. pylori strains appear to resist phagocytosis by human neutrophils and monocytes, in contrast with sialic acid-independent hemagglutinating strains (10, 47, 48, 51). These results suggest a correlation between possession of sialic acid-dependent lectins and protection from phagocytic killing. However, these studies need to be repeated using freshly isolated H. pylori strains and mutants bearing isogenic gene disruptions in candidate genes, such as the gene encoding the putative sialic acid lectin (see below), hpaA, to address what genes are responsible for adhesion and whether they contribute to the virulence of H. pylori.
Several studies have attempted to correlate hemagglutination with interactions of H. pylori strains to KatoIII, AGS, MKN45, or primary gastric epithelial cell lines or to nongastric cell lines. It was found that adherence to some cell lines and primary gastric cells from various animals was sialic acid and heparan sulfate dependent (47, 98, 174). Adherence is inhibited by preincubation of host cells with neuraminidase or by preincubation of H. pylori with the sialic acid-containing proteins fetuin or mucin. Contradictory evidence for a correlation of hemagglutination and adherence to gastric cells has been widely documented (46, 47, 56, 108, 174, 245, 268), although the strain selected and how long it is passaged for each of these studies play a critical role in the type of data obtained (145, 368). Data from various studies suggest H. pylori adherence levels to various cell types as follows: primary gastric cells > HEp-2 (human larynx carcinoma) > AGS > MKN45 cells > KatoIII (174, 234).
Sialic Acid-Binding Adhesins
From the data summarized above on hemagglutination and mucins, there is strong evidence for sialic acid adhesins in H. pylori. Likewise, sialic acid-containing glycoconjugates have been shown to exist on the surface of gastric epithelial cells and on human neutrophils by staining with sialic acid lectins or by conducting binding assays; these sialic acid-containing glycoconjugates are up-regulated upon contact of H. pylori with the host cell (26, 217, 321). Confirmation of the hypothesis for sialic acid adhesins of H. pylori came from Evans and colleagues, who cloned and characterized the first sialic acid adhesin, HpaA (100, 101). The gene encoding the sialic acid adhesin HpaA, hpaA (HP0410), has been cloned, sequenced, and expressed in E. coli (101). The purified protein (~29 kDa) binds to sialoconjugates mainly in an α2,3-specific manner and can be detected on Western blots with antiserum directed against HpaA (100, 101). Antibodies against the putative HpaA sialic acid-binding motif, KRTIQK, inhibit H. pylori sialic acid-dependent hemagglutination, and immunogold labeling shows that HpaA is surface-exposed and is functional (101). However, in E. coli expressing HpaA, no hemagglutination is observed, suggesting that additional genes are necessary for transport, assembly, or regulation of hemagglutination expression in H. pylori. Additionally, HpaA has also been shown to be an inner membrane lipoprotein in E. coli expressing hpaA (246), rather than the expected outer membrane location (101). HpaA has also been observed as a component of the extracellular flagellar sheath (162, 195). An isogenic hpaA mutant of H. pylori still retains sialic acid-dependent hemagglutination activity and adheres normally to five human gastric carcinoma cell lines and to fixed human gastric tissues (162, 246), suggesting that other sialic acid lectins exist and that adherence to epithelial cells is multifactorial. Although these latter studies questioned the relevance of HpaA in H. pylori attachment and virulence, experiments using human neutrophils, monocytes, primary human gastric epithelial cells, and animal studies have not been reported with the hpaA mutant. Simon and colleagues have discovered that 3′-sialyllactose does not inhibit adherence of highly passaged isolates of H. pylori (294), in contrast with lowly passaged isolates, suggesting that high-passage isolates, such as the ones used by O'Toole and colleagues (246), may have lost the expression of certain sialic acid adhesins on the cell surface. Interestingly, the genome sequence of H. pylori predicts an additional HpaA ortholog (HP0492, 30% identical to HpaA at the amino acid level), which may explain some of the perplexing findings (332).
Evidence for the existence of other sialic acid adhesins in H. pylori was provided in several studies. Five candidate sialic acid adhesins of H. pylori of molecular mass of 64, 63, 56, 25, and 20 kDa have been described, but their further characterization has not been reported (147, 184, 185). It is unclear whether the 20-kDa protein may be the same as the 20-kDa conserved antigen reported by Doig and colleagues (75).
Additional evidence for H. pylori binding to sialic acid-containing glycoconjugates comes from transgenic mice lacking parietal cells (321). These mice were developed to mimic the presumed in vivo situation; following H. pylori adherence to the gastric epithelium, parietal cell function rapidly deteriorates (128). In the parietal cell-negative transgenic mice, epithelial multipotent stem cells bearing sialic acid-containing glycoconjugates proliferate; H. pylori specifically attaches to these glycoconjugates in vitro and in vivo (321). This finding confirms the early study of Bode and colleagues (26), who observed up-regulation of sialic acid-containing glycoconjugates following H. pylori adherence to gastric tissue. Other investigators studying normal versus gastric carcinoma gastric tissue have observed elevated sialyltransferase activity (254), leading to increased expression of sialylated glycoconjugates in gastric adenocarcinoma; this is rarely observed in normal gastric mucosa (8, 196, 197, 276). Similarly, drastically elevated sialic acids in the gastric juice of duodenal ulcer patients were observed as compared to normal healthy controls (251, 348), while expression of fucose-rich (Lewis b-positive) glycoproteins is down-regulated in gastric ulcer patients (27). These results prompt the interesting hypothesis that H. pylori directly alters host cell receptor expression and that there is a temporal expression of host cell receptors following H. pylori binding. However, few follow-up experiments have been conducted.
Taken together, the sialic acid adherence studies support two conclusions: (i) H. pylori binds multiple types of sialic acids in vitro and in vivo and (ii) H. pylori adherence to normal gastric epithelium, perhaps first through glycolipids or through the Lewis b antigens (see below), causes the up-regulation of the expression of sialic acid containing glycoconjugates, which allow tighter binding of H. pylori.
Lewis b-Binding Adhesins
An early study by Bode and colleagues (26) suggested the presence of fucose-containing glycoproteins on the surface of gastric epithelial cells. Subsequently, several reports have confirmed that stationary-phase H. pylori can bind to fucosylated glycoconjugates containing Lewis b structures on the surface of gastric epithelial cells within human gastric biopsies (28, 104). Monoclonal antibodies to Lewis b or soluble Lewis b antigens inhibit the binding of H. pylori to fixed gastric tissue (28, 266). In these latter two studies, no evidence for sialic acid-dependent adhesion was obtained. Further support for H. pylori using Lewis b as a receptor comes from transgenic mouse studies. H. pylori binds in a cell lineage-specific manner to the gastric epithelial cells and to the gastric pit in the mucosa of transgenic mice expressing the human α1,3/4 fucosyltransferase gene, in contrast with nontransgenic littermates (103). This enzyme adds a fucose residue to the Lewis b precursor H type 1 antigen. Soluble Lewis b antigens inhibit binding of H. pylori to gastric tissue from these transgenic mice (103). Guruge and colleagues (128) extended this study and found that simultaneous co-inoculation of eight fresh clinical H. pylori isolates resulted in similar colonization levels of both transgenic and nontransgenic littermates, with only one strain predominating (128). H. pylori stayed in the mucous layer in nontransgenic mice, whereas in transgenic animals, H. pylori was found in the mucous layer and adherent to the surface of mucus-secreting gastric epithelial cells, resulting in severe chronic gastritis, parietal cell loss, and production of autoantibodies to Lewis X antigen (found in H. pylori LPS and on parietal cells) (128). Thus, attachment of H. pylori to the gastric epithelial cell surface alters the disease outcome. The fresh H. pylori clinical isolates bound in vitro only to mouse gastric biopsies obtained from the Lewis b transgenic mice, and binding did not depend on presence of flagella (128). Further studies with this transgenic mouse model demonstrated that parietal cell ablation results in amplification of gastric epithelial multipotent stem cells (321). H. pylori attachment to these cells resulted in enhanced cellular and humoral immune responses.
The first H. pylori adhesin responsible for this Lewis b-binding was identified as BabA, which is a member of the large family of paralogous outer membrane proteins (155). The presence of the babA gene in H. pylori correlates with duodenal ulcer and adenocarcinoma and use of this gene marker concomitant with certain vacA and cagA subtypes further strengthens this correlation (118). Presence of babA also correlates well with the ability of H. pylori to bind to Lewis b antigens in vitro (118); babA-deficient clinical isolates always fail to bind Lewis b.
The above studies have contributed much to our understanding of H. pylori adherence to Lewis b receptors. However, there are inconsistencies: (i) not all H. pylori strains bind Lewis b antigens (see below), (ii) Lewis b antigens are widely distributed on epithelial cell types to which H. pylori does not interact and thus does not explain the specific tissue tropism of H. pylori, and (iii) Lewis antigen expression in the host can vary. Thus, host and bacterial strain heterogeneity may play a role in determining bacterium-host cell interactions, and H. pylori can clearly interact with multiple receptors as recently confirmed in vivo using transgenic mice (321).
Recent studies have questioned whether Lewis b serves as the major receptor for H. pylori adherence. Su and colleagues (316) demonstrated that Lewis b antigen-independent adherence to gastric cell lines requires de novo protein synthesis in both host and bacterium. In contrast, Lewis b-dependent adherence occurred to fixed gastric tissue (316), as was originally reported (104). In a second study, using primary gastric cells isolated from gastric biopsies, Clyne and Drumm showed evidence of Lewis a and b on the host cell surface of 18 of 19 samples tested, but three different H. pylori strains bound gastric cells independent of Lewis a or b using quantitative flow cytometry (58). Preincubation of these cells with monoclonal antibodies to Lewis a or b from two different commercial sources did not block binding of H. pylori to these cells. Similar results were obtained using the KatoIII gastric cell line. In other studies, presence of Lewis antigens on gastric cells could not be correlated with Lewis expression on H. pylori LPS (137, 324). Finally, only 4 of 32 H. pylori isolates from infected children expressed the Lewis b binding ability in an in vitro filter binding assay, suggesting that BabA is not expressed in the majority of H. pylori isolates from children (36). BabA is also not expressed in about 50% of H. pylori fresh clinical isolates (118), probably due to phase variation of the babA gene via slipped-strand mispairing (155, 332). Notably, there are also rare babA-positive strains that also fail to bind Lewis b (118).
All H. pylori strains examined to date (n = 49) synthesize a neuraminidase, and 20% of strains produce fucosidase (83), which may cleave host cell sialic acid or fucose residues, respectively, from glycoconjugates. This could result in unmasking of other sugar moieties to which putative H. pylori adhesins could bind and could explain the discrepancies in H. pylori adherence characteristics. However, there is no ortholog of either enzyme in the sequenced genome of H. pylori strain 26695, suggesting that presence of these enzymes is artifactual (143), that H. pylori contains nonhomologous genes, or that only some strains possess these enzymes.
A 61-kDa protein from H. pylori was shown to interact with the H type 2 antigen (the blood group O antigen) and may explain the finding that patients with blood group O are more susceptible to peptic ulcer disease (4). This protein also could bind Lewis a and Lewis b antigens, yet is distinct from BabA.
Taken together, these observations suggest that (i) H. pylori binds to Lewis b in vitro and in vivo, but this is not an absolute requirement for adherence; (ii) adhesins independent of BabA may interact with Lewis b; and (iii) the host cell surface can change its sugar repertoire in response to H. pylori adherence.
Sulfate and Lipid-Binding Adhesins
H. pylori has been shown to interact with a number of sulfated and lipidated compounds in vitro, including heparan sulfate, phosphatidylethanolamine (PE), lactosylceramide, galactosyl- and lactosylceramide sulfate (collectively known as sulfatides), ganglioside GM3, gangliotriaosyl ceramide (Gg3), and gangliotetraosylceramide (Gg4) (13, 19, 49, 50, 123, 124, 144, 167, 191–193, 275, 302). H. pylori also binds to gastric mucin, a sulfated protein that is heavily glycosylated (229, 257, 340). In the study by Kamisago and colleagues, it was observed that adherence of H. pylori to KatoIII gastric epithelial cells is inhibited by sulfated glycoconjugates (167). Lewis b, sialic acid, and GM3 were not involved in this process, but sulfatides were, since a monoclonal antibody to sulfatides markedly reduced H. pylori adherence to KatoIII cells (167). Additionally, bovine milk diluted up to 100-fold inhibits binding of H. pylori to sulfatide by 50% (130).
Adherence of H. pylori to human cells correlates well with the amount of PE present in lipids extracted from these host cells (84, 123). PE, however, does not explain the specific tissue tropism of H. pylori since PE is ubiquitously present in cell membranes throughout the human body. The H. pylori adhesin for Gg3, Gg4, and PE was shown to be related to exoenzyme S of Pseudomonas aeruginosa, since monoclonal antibodies to exoenzyme S block the interaction between H. pylori and these lipids in vitro (124, 191, 193). N-terminal sequence analysis of the purified 63-kDa protein from H. pylori was determined and, based on the complete genome sequence (332), the protein is now known to be catalase, a protein not homologous with exoenzyme S. This result is surprising since bacterial catalases are typically cytosolic, not surface exposed. H. pylori also has a sphingomyelinase, which hydrolyzes sphingomyelin and PE and has hemolytic activity (40). Perhaps after H. pylori interacts with PE, it releases sphingomyelinase to hydrolyze PE receptors, thereby gaining deeper access to the specific cell surface molecules found in the gastric mucosa.
Laminin-Binding Adhesins
H. pylori has been shown in a number of studies to bind to laminin, a basement membrane sialylated glycoprotein (75, 334, 341, 342). A sialic acid glycoconjugate inhibits binding of H. pylori to laminin by 70% and neuraminidase pretreatment of laminin reduces binding by 50%. Heat or protease treatment of H. pylori markedly reduced binding, suggesting that one or more H. pylori proteins are involved in laminin binding (334, 341, 343). Indeed, Doig and colleagues (75, 76) purified a 19.6-kDa laminin-binding protein resembling bacterioferritin from multiple H. pylori strains, and others identified a 25-kDa laminin-binding protein distinct from HpaA that binds in an α2,3 sialic acid-dependent fashion (343). Evidence from several other laboratories suggests that H. pylori LPS is also involved in laminin binding (223, 255, 303, 342). Specifically, a phosphorylated oligosaccharide in the core of H. pylori LPS (in some but not all strains) may be involved in initial binding (342). Remarkable diversity of laminin-binding was observed among distinct H. pylori strains (334).
These data taken together suggest that LPS mediates initial binding of H. pylori to laminin, followed by subsequent tighter binding to laminin by one or more sialic acid lectins, of which the 19.6-kDa and 25-kDa proteins participate. H. pylori binding to laminin may take place in vivo after damage to the epithelial cell layer has exposed the basement membrane.
Role of LPS in Adherence
H. pylori LPS can express antigens that molecularly mimic the Lewis antigens (a, b, X, and Y) found on the surface of gastric epithelial cell glycoconjugates (324). Several studies support the role of LPS as an adherence factor for H. pylori. A monoclonal antibody that inhibits H. pylori adherence to gastric epithelial cells by up to 75% was shown to target the LPS possibly through the Lewis X portion (244). In another study, the core LPS oligosaccharide bound to laminin (342). The O antigen was not involved in binding to laminin, since rough variants (lacking O antigen) still bound laminin. In recent studies, the O antigen LPS side chain, which contains Lewis X antigens and molecularly mimics human cell surface glycoconjugates, was shown to bind to the gastric epithelium in a Lewis X-dependent fashion, and the receptor may be Lewis X itself on the host surface via homotypic interactions (92, 244, 324). Finally, H. pylori LPS interferes with the interaction of gastric mucin with the 97-kDa mucin receptor (256). All of these studies are complicated by the observation that H. pylori LPS content varies from one strain to the next and undergoes phase and antigenic variation within a single strain (15, 16, 222), leading to variable expression of Lewis antigens on H. pylori LPS (262). Despite LPS variation, it is clear that H. pylori LPS plays in important role in adherence and future research should focus on construction of defined LPS mutants to investigate this further.
Role of Heat Shock Proteins in Adherence
Heat shock proteins of H. pylori have surprisingly been shown by numerous laboratories to be surface exposed (150, 365, 366) and involved in adherence (150, 151, 364–367). For example, the intensity of Hsp60 on the H. pylori surface, which is variable among strains, correlates with the degree of adherence (365). Monoclonal or polyclonal antibodies to Hsp60 or Hsp70 block adherence of H. pylori to MKN45 cells, primary gastric epithelial cells, and in an in vitro thin layer chromatography assay (151, 364, 367). However, not all strains of H. pylori could be blocked in adherence by anti-Hsp60 (367). The host receptor for Hsp60 and Hsp70 was found to be sulfatides (150, 151). The Hsp-sulfatide interaction was dependent on a stress induction (exposure to pH 2.5 or 42°C for 5 min). Thus, in the stomach lumen where the pH is 1 to 2, expression of Hsp may be induced so that H. pylori is primed for adhering to the gastric epithelial cell once the organism migrates there.
Role of Outer Membrane Proteins in Adherence
Cotranscribed genes for two outer membrane proteins, AlpA and AlpB, are necessary for Lewis b antigen-independent (and BabA2-independent) adherence of H. pylori to KatoIII gastric epithelial cells and gastric tissue (239, 240). The host receptor for AlpA/B is unknown. HopZ also mediates adherence, since a hopZ isogenic mutant has reduced adherence (253). However, adherence was only qualitatively described and the host receptor is unknown.
AlpA, AlpB, BabA, HopZ, and Hops A–E all belong to a paralogous family of 32 outer membrane proteins (OMPs), 8 of which undergo phase variation by alteration of the number of dinucleotide repeats by slip-strand mispairing (7, 77, 97, 155, 240, 253, 332). That four of these proteins (HopZ, AlpA, AlpB, BabA) are involved in adherence suggests that the entire protein family may have roles in adherence and that the diverse repertoire and phase variation of OMPs allow fine specificity of H. pylori adherence to specific host cell glycoconjugates under different conditions in vivo. This hypothesis may be very difficult to test directly in H. pylori. Rather, an E. coli model that carries one or more H. pylori OMPs could be useful to determine the role and receptor specificity of various OMPs.
Role of Flagellar Secretion Apparatus in Adherence
An isogenic nonmotile mutant in the flagellar secretion apparatus component FliQ results in 30% reduced adherence to AGS cells (112), presumably because the mutation decreases release of a protein involved in adherence that would normally be transported through the apparatus. Since another flagellar, nonmotile mutant (in the flhB gene) adhered similarly to wild type, flagella per se are probably not necessary for adherence (112). However, an open question that still needs to be addressed is whether the flagellar sheath, which contains proteins, including the adhesin HpaA, is involved in adherence. Some of the best studied adhesin-receptor interactions are shown in Fig. 4.
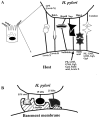
Adherence Antagonists
Adherence of H. pylori to the gastric epithelium is a crucial initial step in colonization, as nonadhering bacteria would be washed away during peristalsis-mediated flushing of the stomach. Therapeutic approaches to inhibit adherence may thus prevent colonization and disease through eradication of H. pylori. Many adherence antagonists have been tested, and some promising candidate adherence antagonists are briefly summarized (Table 2).
Table 2
H. pylori adherence antagonists.
First, administration of 3′-sialyllactose to persistently colonized rhesus monkeys cures or decreases H. pylori colonization in some monkeys (225). 3′-sialyllactose also significantly reduces gastric bacterial load in H. pylori-infected humans (D. Zopf, P. M. Simon, M. Hurley, E. McGuire, and S. Roth, unpublished observation). These data were obtained following promising in vitro studies showing that 3′-sialyllactose inhibits adherence of low-passage isolates of H. pylori to epithelial cells (294). The 3′-sialyllactose presumably binds to and saturates H. pylori sialic acid adhesins, such as HpaA, and prevents binding of these adhesins to host cell sialic acid receptors. This adherence antagonist is safe, is nonimmunogenic, and can potentially be used in combination with other therapeutic agents. Second, the anti-ulcer compound sofalcone inhibits H. pylori adhesion to mucin in a dose-dependent fashion (319), probably by inhibiting the chemotaxis of H. pylori toward mucin (370). Third, the anti-ulcer compounds sucralfate and sulglycotide inhibit adherence by blocking the interaction of H. pylori LPS with laminin, GM3 ganglioside, and lactosylceramide at the gastric epithelium surface (255, 258, 301, 305, 309). These compounds also decrease H. pylori mucinase activity by desulfating mucin (300, 304), and they also block the interaction of H. pylori LPS to the 97-kDa mucin receptor by 90% (256). These activities lead to increased availability of mucin to bind normally to the mucin receptor on the gastric epithelium surface. Fourth, pretreatment of H. pylori with the anti-ulcer compound ecabet sodium inhibits adhesion of H. pylori to gastric epithelial cell lines by increasing stability of mucin to pepsin degradation and also inhibits surface urease activity (133, 172, 291). Fifth, pretreatment of gastric epithelial cells lines MKN-28 and MKN45 with the anti-ulcer compound rebamipide, or derivatives thereof, inhibits adhesion of H. pylori by about 50% (131). Sixth, pretreatment of MKN45 gastric epithelial cells with the commonly used antacid co-magaldrox (Maalox) inhibits adherence of H. pylori by 75% and specifically interferes with IL-8 secretion from the epithelial cells, while reducing surface expression of H. pylori Hsp60 (168). Seventh, the proton pump inhibitor omeprazole inhibits adherence of H. pylori extracts to human neutrophils (320). Eighth, addition of Lactobacillus salivarius, indigenous flora of the mouth, to MKN45 and KatoIII gastric epithelial cells decreases H. pylori adherence by up to 90%, suggesting that lactobacilli could be used as a probiotic agent against H. pylori (166). Lactobacilli are also colonization antagonists in a gnotobiotic murine model (1, 166). Ninth, milk could serve as a potential adherence antagonist by blocking adherence of H. pylori to sulfatide and Lewis b and by preventing cell vacuolation (130). Indeed, human milk inhibits adherence of H. pylori to KatoIII cells by 50 to 70% (59). The inhibitory fraction of milk was shown not to be secretory IgA (59), but rather κ-casein, in a fucose-dependent fashion (315). There is also a casein-independent fraction of human milk that inhibits H. pylori adherence, which could perhaps be the mucin found in milk (59). Tenth, bile acids, such as chenodeoxycholic acid, are effective inhibitors of adherence of certain H. pylori strains by mechanisms that are unclear (205). Finally, fucoidan, a sulfated α1,3 fucan, inhibits both Lewis b and sulfatide-mediated adherence of H. pylori to MKN28 and KatoIII gastric epithelial cells (291). Seven to 10 fucoidan-binding proteins of H. pylori were detected but not identified.
Adherence antagonists are already being tested for efficacy in combination with antibiotic therapy regimens, such as omeprazole with amoxicillin. Another use for adherence antagonists is dissection of molecular mechanisms of adherence. For example, several adhesin-receptor interactions can be blocked so that still additional interactions may be explored (132).
Does Invasion into Host Cells Occur?
A long-standing question in H. pylori research is whether the bacterium can invade gastric epithelial cells and, if so, whether the organisms can persist as viable organisms intracellularly. The answers to these questions, however, are still controversial. The vast majority of researchers agree that invasion is rare (26, 30). However, rare invaders could still be important for intracellular persistence, resistance to antimicrobial therapy, and protection from the human immune response.
H. pylori is occasionally localized within endocytic vacuoles in gastric epithelial cells, on the basis of studies using human gastric biopsy specimens or various cell lines (26, 235, 274, 333, 361, 362). Invasion into parietal and chief cells was also observed in patient gastric biopsies in one study (362). Additionally, H. pylori can invade the lamina propria and gain access to the basement membrane (10), where laminin is present. Using a nongastric epithelial cell line (HEp-2) and a gentamicin protection assay, Evans and colleagues observed that invasion of H. pylori was rapid and dependent on the sialic acid adhesin HpaA (99). However, little replication of H. pylori occurred after 24 h, and numerous coccoid forms were observed, suggesting the H. pylori may have died following entry. Indeed, using the same cells and experimental protocol for assessing invasion, Megraud and colleagues failed to recover viable intracellular organisms (211). More recent experiments suggest that the gentamicin protection assay is not useful for accurately assessing invasion of H. pylori because the organism finds extracellular pockets that protect it from the antibiotic, leading to falsely elevated invasion levels (62). HEp-2 cells are also probably a poor model to assess invasion and adherence of H. pylori because it is a nongastric cell line, and since only some strains of H. pylori adhere to these cells (174).
An early elegant study by Bode and colleagues (26) demonstrated internalization of H. pylori into the cytoplasm of mucus-secreting duodenal epithelial cells. This occurred in 10% of the duodenal ulcer patient biopsies examined. This study was subsequently confirmed by Noach and colleagues (235), who used human gastric biopsies in their study and found six samples that were highly suggestive of invasion. H. pylori has also been found inside the mucin compartment of goblet cells in gastric biopsies (117). H. pylori has a predilection for association with intercellular tight junctions (26, 134, 330). Initial attachment of H. pylori to the cell surface may weaken tight junctions between adjacent host cells (26, 235). This weakening may allow deeper penetration of H. pylori between cells, with subsequent internalization (26, 235). However, that coccoid forms of the organism were observed in some of these transmission electron micrographs suggests that the invaders may have been nonviable. Evidence for invasion in the studies mentioned above were by electron microscopy or by gentamicin protection assays and thus "invasion artifacts" cannot unequivocally be ruled out. In contrast, other investigators were unable to show evidence for invasion in gastric biopsies (134, 330) or in cell lines (62, 263).
Whether internalized H. pylori is viable was still not clear until more recently, when invasion of H. pylori was confirmed by immunohistochemical staining for bacterial Hsp60, which was found intracellularly in gastric biopsy specimens (96). Treatment of patients with standard antimicrobial triple therapy for 2 weeks resulted in clearance of the bacteria by day 14, but 5 of the 10 patients in the study relapsed by one month posttherapy, providing the first suggestive evidence for antimicrobial therapy failure due to persistent (and thus, viable) intracellular H. pylori (96). If H. pylori lives in an intracellular environment, then poorly penetrating antibiotics, such as the widely used amoxicillin, would fail and may explain occasional clinical failures of antimicrobial regimens.
Using a tissue culture-based system, H. pylori was shown to invade AGS cells using a gentamicin protection assay (317). Type I (cag+) strains invaded better than type II strains (cag mutant). Invasion was blocked by cytochalasin D, which inhibits actin polymerization, or by a tyrosine phosphatase. Interestingly, a 125- to 130-kDa protein that is almost certainly CagA, based on more recent studies (18, 64, 238, 283, 313), was tyrosine phosphorylated. Invasion was markedly enhanced by transfection of AGS cells with the β1 integrin gene, suggesting that integrins might serve as receptors for H. pylori in this system (317).
A highly sensitive method of assessing invasion of 18 clinical isolates of H. pylori into HEp-2 cells was recently reported by Wilkinson and colleagues (355). In this study, acridine orange was used to stain microbes; intracellular viable microbes fluoresce green, whereas nonviable organisms fluoresce red. Fluorescence of extracellular microbes is quenched by a counterstain. This study showed H. pylori invasion frequencies that exceeded those for Shigella flexneri and paralleled those of Yersinia enterocolitica (355). Despite being mostly coccoid in shape, intracellular H. pylori remained viable for at least 6 h after entry, and no significant correlation was observed between invasion frequencies and clinical state from which the H. pylori was isolated (355). It will be of great interest to apply this sensitive method to more relevant cell lines such as KatoIII gastric epithelial cells and to fresh gastric biopsies. One potential problem with assessing H. pylori invasion is that VacA may contribute to cell lysis, leading to disruption of tissue culture monolayers. Thus use of a vacA mutant of H. pylori may help investigate invasion mechanisms more closely (355).
Except for possibly HpaA (in a nongastric cell model), H. pylori invasins have not yet been identified in any study and specific genes need to be disrupted to lend stronger support for the hypothesis that H. pylori can occasionally invade host cells.
Interations of H. pylori with Phagocytic Cells
H. pylori adheres to and enters human monocytes and neutrophils, phagocytic cells that infiltrate the gastric mucosa. Adherence to neutrophils leads to up-regulation of the integrin CD11b, independent of the H. pylori genes cagA, vacA, and cagE (129). Entry of H. pylori into human neutrophils (9, 26), monocytes, and macrophages (5, 9) is opsonin independent, suggesting that H. pylori adhesins are responsible for entry. Whether these phagocytosed organisms remain viable or are killed depends on the presence of the cag PAI. Type I strains have a delay in phagocytosis followed by homotypic phagosome fusion into "megasomes," whereas type II strains are rapidly phagocytosed and killed (5). The megasomes contained viable organisms (5). The reason for survival of type I strains inside phagosomes is unclear, but perhaps catalase and superoxide dismutase produced by the bacterium detoxify reactive oxygen intermediates inside of phagosomes (241, 312). Also of importance is whether strains possess sialic acid hemagglutinins. Those H. pylori strains containing sialic acid hemagglutinins are more resistant to phagocytosis by human neutrophils and monocytes, in contrast to those lacking sialic acid hemagglutinins (9, 46, 48). However, H. pylori adherence, which can be clearly distinguished from phagocytosis, does require sialic acids on the surface of human monocytes (51). Therefore, sialic acid hemagglutinating strains adhere better yet are more resistant to bactericidal killing and phagocytosis compared with nonhemagglutinating strains. Heparan sulfate significantly enhances phagocytosis of H. pylori by human neutrophils (46, 49).
H. pylori activates monocytes by both LPS-dependent and -independent mechanisms, resulting in the production of IL-1 and TNF-α and in expression of major histocompatibility (MHC) antigens and the IL-2 receptor (199). Monocytes could thus be crucial for ingestion of H. pylori and presentation of bacterial antigens on the cell surface in the context of MHC antigens for eventual humoral or cell-mediated immune response. The role of specific bacterial genes in the interaction of H. pylori with phagocytic cells is a largely unexplored area of research.
Summary of Adherence
Assimilation of all these data supports a model whereby H. pylori first encounters gastric mucin and degrades it. Next, H. pylori induces Hsp expression during their transient exposure to acid in vivo, allowing binding to sulfatides. Upon neutralization of acid by urease and movement of H. pylori to the gastric epithelial cell surface (pH neutral) via motility, the Hsps may be down-regulated and H. pylori then can bind to phosphatidylethanolamine. Following cleavage of PE by sphingomyelinase, H. pylori adheres to Lewis b and up-regulates sialic acid-containing glycoconjugates to which H. pylori subsequently binds. After damage to the gastric epithelium, H. pylori adheres to the basement membrane protein laminin. Hence, H. pylori is a very sticky bacterium that has evolved numerous adhesins to bind to host cell surfaces. Temporal regulation of adhesin and host cell receptor expression is probably very important in vivo, but has not been well studied. The real challenge is to temporally dissect these adhesins and receptors to test the ideas in this model and determine which are important in vivo.
Colonization
Host Factors
Even before the discovery of H. pylori, researchers had found significant correlations between certain genetic markers and prevalence of ulcer disease (Table 3). Similar studies have been performed more recently, and there are early indications that some alleles of host genes may predispose one to become colonized in the first place, while others are associated with subsequent development of ulcers or cancer. This section will focus primarily on factors believed to increase susceptibility to infection, rather than disease outcome; however, the present state of knowledge scarcely allows differentiation between susceptibility and prognosis.
Table 3
Factors influencing colonization by H. pylori.
Numerous epidemiological studies have examined a range of populations and environmental factors to determine what constitutes a risk factor for development of chronic H. pylori infection and subsequent pathologies. Although the evidence is circumstantial, it appears that the H. pylori infection rate varies due to both exposure level and intrinsic susceptibility to colonization. Studies have revealed that nursing home patients, endoscopists, and individuals consuming food from street vendors may be more likely to become infected with H. pylori than the general population (24, 122, 207, 264). These studies point toward a higher risk of exposure to the organism. In other studies, however, it is more difficult to imagine why the exposure rates should differ. For instance, comparisons of H. pylori-specific IgG, indicating an established infection, and IgM, suggesting recent exposure, reveal that women are more than twice as likely to become infected with H. pylori while pregnant than nonpregnant controls (181). Since pregnancy is known to result in many immunological changes, it is possible that host immune status could render the host less able to combat the initial infection with H. pylori. Although numerous studies have shown that acquisition often occurs during childhood (93, 201), at least one study has also demonstrated a high rate of spontaneous clearance among children before age 11 (331). Thus, it is possible that children are both more likely to be exposed to H. pylori and more likely to clear the infection as their immune systems mature, whereas adults are less likely to clear an established infection but are infrequently exposed to the organism.
Many investigators have accumulated evidence that factors specific to an individual host influence colonization by H. pylori. Studies of twins revealed that monozygotic twins were 81% concordant for infection, compared with 63% concordance in dizygotic twins (200). That is, 81% of identical twins were either both infected or neither infected. There were few cases in which one twin was infected and the other twin was not. This difference was maintained regardless of whether the twins were reared together or apart. In a multiethnic Malaysian city, a study of 1,060 consecutive endoscopy patients revealed that, overall, the prevalence of H. pylori infection measured by rapid urease tests of biopsies was 49%; however, only 16.4% of ethnic Malay patients were infected, whereas 48.5% of Chinese and 61.8% of Indian patients were H. pylori positive (121). Another study in a multiethnic region also revealed differences in H. pylori positivity related to ethnicity. Even when socioeconomic status was accounted for, Europeans were significantly less likely to be seropositive than Maori or Pacific Islanders (113). Neither research group could rule out dietary factors or cultural practices that may increase risk of infection; however, Campbell and colleagues noted that Maori and Pacific Islander populations interact more frequently with Europeans than with each other (34). This group also found that strains isolated from Europeans were almost three times more likely to express a biologically inactive VacA cytotoxin than strains isolated from Polynesians. This could mean that VacA confers an advantage to H. pylori only in certain hosts, while it is detrimental for the bacterium in others. Specific host characteristics could therefore influence the ability of H. pylori to colonize in the first place, and subsequently exert adaptive pressure leading to measurable differences in the colonizing strain.
A single encounter with a given strain of H. pylori may not be sufficient for lifelong infection. Ethnic correlations among inhabitants of the same geographic region merit further investigation to determine whether identifiable host markers predispose certain individuals to infection. Polynesians are known to express the Lewis b antigen at a lower frequency than Europeans, but this has not yet been correlated to risk of infection or strain type likely to be present (138, 139). Data regarding the identities or mechanisms of such host factors influencing H. pylori growth are sparse, due to the inherent difficulties in conducting controlled studies. Nonetheless, one study performed in rhesus monkeys has yielded some interesting data regarding host specificity, and apparent changes in selective pressure that occur as colonization progresses. A group of four monkeys that had been previously cured of their natural H. pylori infections was inoculated with a mixture of seven fresh clinical isolates (81). By 10 months postinfection, randomly amplified polymorphic DNA (RAPD) fingerprinting revealed that clinical isolate J166 predominated in all four animals, even though this strain had been detected in only one animal at the earliest timepoint. Two other strains were found early in infection in all four animals, but both had nearly reached undetectable levels by 10 months. Although only a small number of animals were used, this study strongly suggests that either the gastric environment changes as infection becomes established, and that this change affects the relative survival of H. pylori strains, or that strains differ in their relative competence for infection and persistence. It is possible that, had only one strain been used as an inoculum, the monkeys would have been only transiently infected.
Thus far, little is known about specific host factors that influence H. pylori infection. The HLA types that appear to be correlated with susceptibility to helicobacter infection are HLA DR and HLA DQ*0301. HLA DQA1*0301 and HLA DQ5 are frequently associated with more severe disease but may not predispose an individual to becoming infected in the first place. HLA-DQA1*0102 appears to confer partial protection to H. pylori-related atrophic gastritis and intestinal-type gastric adenocarcinoma (21). The identification of additional host cell markers relevant to H. pylori may provide important clues for understanding H. pylori infection and developing treatment strategies.
Immune Factors
The quest for a fully protective H. pylori vaccine has been frustrating, but there is ample evidence that innate and elicited immunity serves to modify and/or partially control H. pylori infection (120, 163, 213). One study revealed a difference between infections of germ-free athymic mice and their euthymic counterparts (169). Although both groups of mice were persistently infected, the athymic mice had colony counts approximately eightfold higher than euthymic mice from week four until the conclusion of the study. Bacterial counts increased rapidly in euthymic germ-free mice during the first week postinfection but subsequently declined until reaching a stable level by week three. In contrast, the bacterial counts in the athymic germ-free group continued to increase until at least week four. Interestingly, the germ-free euthymic group consistently had less inflammation than the athymic group, suggesting that infiltration of inflammatory cells alone is insufficient to reduce H. pylori numbers. Although the authors did not measure antibody titers or other markers of immune response, H. pylori numbers declined after week one, when antibody titers are expected to be rising.
Since H. pylori infection induces a polarized Th1 response, characterized by increased IFN-γ, it was hypothesized that directing the immune system toward a Th2-type response might reduce infection. An initial investigation of IL-4−/− mice, which exhibit a strong Th1 response, infected with H. felis showed increased colonization and inflammation, supporting this hypothesis (221). Chen and colleagues (41) did not find similar increases in colonization or immune response when they compared H. pylori-infected IL-4−/− mice to wild-type mice. Nonetheless, convincing evidence for the role of the Th2 response in control of H. pylori infection is provided by a study utilizing adoptive transfer of splenocytes (221). Mice injected with an H. pylori-specific Th2-cell line showed dramatic reductions in bacterial burden. Mice injected with splenocytes from immunized mice also harbored fewer bacteria, while no difference was noted when splenocytes were transferred from mice that had been infected with H. pylori but not previously immunized. Reductions in bacterial burden correlated significantly with elevated serum IgG1 levels (a marker of the Th2 response) in the recipient mice.
Gastrin is a polypeptide hormone produced by G cells in the antrum of the stomach that stimulates secretion of gastric acid (Fig. 1). H. pylori-positive individuals frequently have elevated gastrin levels, both fasting and following a meal, and often have increased acid output in response to gastrin (33). Both gastrin levels and acid secretion return to normal following eradication of H. pylori (94, 127). Gastrin has been shown to augment growth of H. pylori and may aid in colonization. When grown in brain-heart infusion or Brucella broth medium supplemented with 10% fetal calf serum, H. pylori showed a dose-dependent response to gastrin, with a decreased lag phase and higher final bacterial density, while the gastric peptides somastatin and epidermal growth factor had no effect (53). Gastrin had no effect on growth of Campylobacter jejuni or E. coli. Experiments with 125I-labeled gastrin demonstrated that gastrin is specifically bound and taken up by H. pylori (53). The structurally similar peptides pentagastrin and cholecystokinin octapeptide were able to compete with radiolabeled gastrin for uptake but did not similarly stimulate H. pylori growth. Uptake of radiolabeled gastrin, as measured by autoradiography, was inhibited at 4°C, suggesting an energy-dependent internalization mechanism. Several mechanisms were initially proposed to explain H. pylori-mediated increases in gastrin output. H. pylori might reduce secretion of the G-cell inhibitor somastatin, which is produced by nearby D cells; H. pylori products could directly stimulate G-cell activity (353); or H. pylori could mediate effects on gastrin indirectly via other host cells. Reduced D-cell numbers, somatostatin concentrations, and response to cholecystokinin seem to support the first hypothesis (33, 177); however, other experimental data indicate that the last hypothesis is correct. The proinflammatory cytokines IL-1β and TNF-α produced by monocytic cells have been shown to increase gastrin secretion by endocrine cells and by antral G cells (353).
Thus, there are two interactions between H. pylori and gastrin: (i) H. pylori induces elevated gastrin release, and (ii) H. pylori uses gastrin as a growth factor. H. pylori proteins involved in this response have not yet been identified. Nor is it known what advantage is conferred by H. pylori's uptake and growth response to gastrin. Given the specificity of the response, the simple use of gastrin for nutritional purposes seems unlikely. The utility of using gastrin as a homing signal directing H. pylori toward the gastric mucosa makes intuitive sense; however, it is less clear whether the host side effects of H. pylori colonization are also beneficial to the bacteria. As one of the host's primary innate defenses, an increase in acid secretion following bacterial insult and mucosal inflammation may be beneficial. Moreover, a link has previously been shown between the immune system and gastrin production. Following immunization, reexposure of animals to the immunogen caused an increase in gastrin secretion (326). H. pylori, however, has adapted to cope with the resulting acid load, and perhaps gastrin secretion somehow aids in the perpetuation of H. pylori colonization.
Dietary, Drug, and Environmental Factors
Following colonization, H. pylori is envisioned to reside in a protected niche, safe from stomach acid and many components of the immune system. But how accurate is this picture? Some elements of the immune system are able to cross the mucosal epithelium. Antibodies are secreted, and macrophage-like cells may be able to traverse the gastrointestinal epithelium (349). The gastric mucous layer effectively prevents back-diffusion of hydrochloric acid but does allow diffusion of other solutes, including some over 30 kDa (73, 354). Thus, H. pylori could be exposed to many elements of the host diet. Dietary factors may have a larger influence during and immediately following infection, as H. pylori struggles to colonize the gastric mucosa.
Dietary factors
Excessive salt intake is a dietary factor common to Japanese and to many Americans. A high-salt diet has previously been correlated with gastritis and increased cancer risk but may in fact influence H. pylori colonization as well (336). Mice fed 7.5% salt prior to and following experimental infection with H. pylori SS1 became colonized at a higher level than mice fed a normal diet (110). Salt alone causes atrophy of parietal cells and increases the rate of N-methyl-N′-nitro-N-nitrosoguanidine-induced tumor formation in rodents (352). Perhaps these changes produce a more favorable environment for H. pylori replication. Furthermore, gastric cell proliferation rates correlate with salt intake only when the subject is infected with H. pylori (72, 357), which could make the high salt-H. pylori combination particularly carcinogenic.
Dietary vitamin C (ascorbic acid), on the other hand, is epidemiologically linked to lower rates of gastric carcinoma (31, 295). There is evidence that lower risks may be partially attributable to scavenging of reactive oxygen radicals in the stomach by ascorbate (80), but more specific effects of vitamin C on H. pylori may also play a role. Researchers have shown inhibition of H. pylori, both in vitro and in vivo by vitamin C (373). Vitamin E, another antioxidant, had no effect on growth, even at high concentrations. Furthermore, this inhibitory effect appears to be specific for H. pylori and the closely related Campylobacter species, as various E. coli strains, Salmonella, and Vibrio species were unaffected. Vitamin C is bactericidal for helicobacter in vitro, but only partially inhibitory in vivo. Gerbils dosed orally with 10 mg of vitamin C daily following H. pylori infection had 100-fold fewer CFU per stomach than control mice. The attainable concentrations of vitamin C near the mucosal epithelium are not known, and it is possible that in vivo levels are not high enough for full suppression or that H. pylori is otherwise partially protected in its gastric niche. Additionally, H. pylori may inhibit the secretion of ascorbic acid from the circulation, lowering concentrations in the colonized stomach (280). Vitamin C inhibits urease activity in vitro (231); however, Zhang and colleagues (373) report that growth of urease-negative mutants is similarly inhibited by vitamin C in vitro, suggesting urease-independent mechanisms of growth inhibition. Given the accepted importance of urease for colonization, it is difficult to differentiate between urease-dependent and -independent effects of vitamin C.
In related research, another antioxidant, astaxanthin, was found to reduce both the bacterial load and gastric inflammation in H. pylori-infected mice (25). Astaxanthin, a carotenoid present in high levels in the algae Haematococcus pluvialis, has previously been studied as an anti-cancer agent (45, 323). Astaxanthin may mediate both chemoprotection and amelioration of H. pylori infection via its effects on the immune system. Dietary astaxanthin resulted in increased phytohemagglutinin-induce ed lymphocyte proliferation and increased cytotoxic activity (45). Additionally, astaxanthin suppresses interferon-γ (IFN-γ) production in vitro (165) and increases IL-4 production in vivo, suggesting that astaxanthin promotes a shift from a predominantly Th1 response to a mixed Th1/Th2 response (25). Since IFN-γ activates phagocytic cells, which can result in tissue damage, suppression of IFN-γ may decrease tissue pathology. Thus, astaxanthin may protect the mucosal surface both by decreasing phagocyte activation and via its intrinsic antioxidant activity.
Reports that certain Chinese and Italian populations consuming large amounts of garlic are at decreased risk for gastric cancer have prompted investigations of the antimicrobial effects of garlic (32, 372). Garlic tablets or extracts have demonstrated in vitro growth inhibitory activity against H. pylori in several studies (38, 55, 164, 242, 296). In two studies, a synergistic effect between omeprazole and garlic was observed (38, 164). Unfortunately, human trials have not yet demonstrated amelioration of H. pylori infection (126).
Other dietary components have been tested and discarded as potential risk factors for helicobacter infection. Some early data suggested a role for vitamin E and beta-carotene in prevention of gastric cancer (78, 176); however, a 5-year clinical trial involving male smokers with atrophic gastritis failed to show any protective effect of alpha-tocopherol (vitamin E) or beta-carotene supplementation (347). Likewise, a study of polyunsaturated fatty acid intake showed no decrease in colonization or inflammation. The authors believe that earlier correlations were spurious, and possibly due to confounding factors such as affluence, social class, or smoking (82).
Probiotics
Lactobacilli can also influence levels of colonization by helicobacter. Although gastric acid is famous for its ability to kill ingested organisms, the stomach routinely contains live organisms in the mucus and associated with the gastric wall. Sampling of gastric juice, both fasting and postprandial, routinely produced Viridans streptococci, Neisseria spp., Candida spp., and Lactobacillus spp. (311). One possible reason for the failure of most H. pylori strains to colonize mice may be the higher level of Lactobacillus colonization observed in mice. Isogai et al. hypothesized that increasing the amount of time H. pylori is able to contact the gastric mucosa may give H. pylori a better chance at competing with the normal flora of the mouse (156). They were able to transiently infect conventional mice by temporarily clamping the duodenum for 30 min, thus preventing the H. pylori inoculum from passing through the stomach. They noted, however, that the numbers of lactobacilli in the stomach fell following surgical treatment and H. pylori inoculation. Subsequently, Lactobacillus numbers rose as H. pylori numbers fell. In a more controlled study of the effects of lactobacilli, Kabir et al. infected gnotobiotic BALB/c mice in the presence or absence of L. salivarius, a species frequently found in the human stomach and oral cavity (166). Germ-free animals could be infected by all H. pylori strains tested in the study, including NCTC 11637, which had failed to infect conventional animals in previous studies. Furthermore, the infection was sustained at about 105 CFU per g of tissue for 9 weeks. When the germ-free animals were infected with L. salivarius one week prior to H. pylori inoculation, H. pylori was unable to colonize. Likewise, infecting previously H. pylori-colonized mice with L. salivarius reduced both the level of H. pylori colonization and the anti-H. pylori IgG titers. In contrast, two other organisms found in the mouse stomach, Enterococcus faecalis and Staphylococcus aureus, were unable to prevent H. pylori colonization. This suggests that the inhibitory effects of lactobacilli are due to more than simple competition.
Several investigators have observed that Lactobacillus spp. inhibit adherence of H. pylori in vitro (60, 166), but there is evidence of other inhibitory mechanisms used by at least some lactobacillus strains. A whey-based culture supernatant of Lactobacillus acidophilus (johnsonii) La1 administered daily for 2 weeks was able to decrease urease activity, as measured by 13C-urease breath test (13C-UBT), in H. pylori-infected human volunteers (214). Four weeks after cessation of therapy, 13C-UBT values remained significantly below pretreatment levels. In vitro studies performed using the same supernatants and those from a related strain, L. acidophilus La10, showed that strain La1 was able to inhibit H. pylori growth in vitro, whereas strain La10 could not. The two strains produce similar amounts of lactic acid, implying that lactic acid was not responsible for the effect. In a similar study involving mice, the Servin group examined the effects of human-derived L. acidophilus strain LB on H. pylori adhering to cultured mucus-secreting cells and on mice infected with H. felis. Culture supernatants from strain LB caused a significantly greater decrease in H. pylori viability than did a similar supernatant from L. acidophilus strain GC (60). The activity of both supernatants could be partially abrogated by heating, suggesting a heat-labile antagonist. Lactic acid alone, at a similar concentration and pH to that found in supernatants, had no effect on H. pylori viability. Thus, H. felis is presumably resistant to products produced by naturally occurring lactobacilli in the conventional BALB/c mouse stomach but is sensitive to L. acidophilus LB. Although the in vivo studies were not performed with H. pylori, the experiments demonstrate that addition of a new Lactobacillus strain to the existing flora can alter the state of the Helicobacter infection and may reduce gastric pathology. Lactobacilli are known to produce hydrogen peroxide and bacteriocins, which may be responsible for some of the H. pylori inhibition (159, 344). The above data in conjunction with mouse data suggest that consumption of L. acidophilus or dairy products fermented with these organisms may prevent colonization from occurring or reduce symptoms in previously infected individuals.
A common theme appears to be emerging from research on dietary influences on H. pylori infection. Agents, which in themselves promote tissue damage or cellular proliferation, appear to increase the risk of helicobacter infection and eventual cancerous lesions. Tissue damage may give H. pylori an advantage as it establishes colonization of the gastric mucosa.
Dietary supplements and alteration may at least form part of a rational treatment regimen, in combination with drug therapy. Dietary interventions may be less expensive than drugs; this is useful for developing countries or other populations lacking access to advanced health care. Extracts of commonly consumed plants, such as garlic, or bacteria may also prove safer and less likely to cause side effects, an important consideration when treating children and pregnant women. Lack of correlation between epidemiological evidence and in vitro and in vivo data warrants caution when investigating dietary supplements. Correlations based on epidemiology may be spurious, and confounding host factors may limit in vivo efficacy. Therefore, carefully designed human and/or animal studies are required.
Non-antibiotic drugs
Bismuth, which has antidiarrheal, antibacterial, and anti-inflammatory properties (109), has been used to treat gastrointestinal disorders for at least 300 years (180, 249), yet its mechanism of action is only now being uncovered. Bismuth-containing compounds appear to have several activities relevant to inhibition of H. pylori. Colloidal bismuth subcitrate inhibits activity of phospholipases 2 and C (PLA2, PLA-C) derived from H. pylori lysates or culture supernatants (247, 248). There are conflicting reports regarding whether bismuth is competing with calcium ions for binding to PLA2 (247, 248). It has been suggested that PLA activity damages the hydrophobic barrier that protects the gastric mucosa against damage by acid (79). Inhibition of phospholipases may help preserve the integrity of the gastric mucosa, thereby interfering with H. pylori colonization by restricting access to the gastric epithelium.
Bismuth subsalicylate also appears to scavenge reactive oxygen species. Damage to normal human gastric tissue removed by biopsy was assessed following exposure to ethanol, hydrochloric acid, or sodium hydroxide (22). Increases in superoxide production, hydroxyl radical production, and DNA fragmentation were measured following exposure to each of the above agents. Bismuth subsalicylate measurably reduced these forms of damage in a dose-dependent manner. Although these experiments did not involve helicobacter, other lines of evidence support the notion that H. pylori infection results in increased reactive oxygen species within tissues. Both increased reactive oxygen species and increased DNA damage have been detected in H. pylori-infected patients (23, 71, 204). This tissue damage was initially thought to be due to activation of neutrophils; however, Obst and colleagues (237) demonstrated that DNA damage induced by H. pylori extracts can be inhibited by various radical scavengers.
The antioxidant activity of bismuth has been shown to interfere with at least one H. pylori enzyme, alcohol dehydrogenase (270). The relevance of alcohol dehydrogenase inhibition in terms of colonization ability is uncertain; however, several hypotheses have been proposed. Acetaldehyde, produced by the action of alcohol dehydrogenase on ethanol, is toxic to mucosal cells (232) and inhibits proliferation of gastric cell lines in vitro (206). Thus, acetaldehyde may impair the ability of the mucosal layer to repair damage. Alternatively, it has been proposed that the reverse reaction may occur under microaerobic conditions, resulting in ethanol production and energy production (278). In support of the notion that fermentation may be an important means of energy production, a potent alcohol dehydrogenase-inhibitor, 4-methylpyrazole, also inhibited growth of H. pylori in culture (277). Reduction in damage may make the gastric epithelium more resistant to colonization. Therefore, antioxidant activities of bismuth or other compounds may comprise one facet of the complex set of interactions leading to clearance of H. pylori or long-term colonization.
Bismuth may have other direct inhibitory effects on H. pylori. Investigators have reported disruptions in the H. pylori cell wall, loss of motility, and decreased viability following treatment with various bismuth compounds (314, 360). Cytotoxicity associated with cell wall disturbances was also inhibited by divalent cations (314). Thus, displacement of required divalent cations may be a common mechanism of bismuth activity.
The anti-ulcer drug, ecabet, has two reported activities that may interfere with H. pylori colonization. First, ecabet has a high affinity for gastric mucus and decreases degradation of mucous glycoproteins by pepsin (172). Second, ecabet inhibits urease activity (158). In experiments with jack bean urease, ecabet-mediated urease inhibition was shown to be irreversible (157). The authors of this study suggest that permanent inhibition is due to specific binding and denaturation of urease. H. pylori is able to survive in buffer at pH 3 if 10 mM urea is present. Addition of ecabet to the buffered urea results in decreased viability, similar to survival seen in the absence of urea (158). No evidence has been presented to address the question of whether ecabet is taken up by H. pylori, where it could suppress activity of intracellular urease. Nonetheless, several clinical studies have demonstrated improved clearance of H. pylori infection when ecabet is given in combination with antibiotics, suggesting that the drug's anti-urease activity is relevant to infection (243, 287, 292). Improvement of mucin stability by ecabet may therefore force H. pylori to spend longer in the lumen of the stomach, where it is exposed to the lowest pH values, while ecabet simultaneously deprives H. pylori of its ability to neutralize acid via urease activity.
Bacterial Factors
Since H. pylori has no known animal reservoir and is not believed to replicate outside the host, all naturally occurring strains of H. pylori are presumed able to infect some subset of the population. The search for genes that are not required for viability but do influence colonization is progressing rapidly, with the assistance of two fully sequenced H. pylori genomes. One strategy is to examine properties that are common to all H. pylori strains but absent in other bacteria. Genome sequences also facilitate the search for homologs of genes known to be critical to other pathogens. Some genes may influence pathogenesis, without having an effect on colonization. For example, the vacuolating cytotoxin VacA (see chapter 9) has profound effects on host cells, but insertional deletion of the vacA gene does not reduce colonization (87). This section summarizes current knowledge on genes that may influence gastric colonization by H. pylori.
Urease was the first colonization factor identified in H. pylori, and continuing experimentation has uncovered new roles for this enzyme (see chapter 16). Urease was initially thought to be important solely for neutralization of gastric acid, yet it is required for colonization even in achlorhydric animals (88). Research has uncovered potential roles for urease in nitrogen assimilation and provision of energy for flagellar rotation (356, 371). Several drugs used to combat H. pylori infection, including bismuth, sucralfate, and sulglycotide, exert at least some of their effects through inhibition of urease activity.
The first urease mutant of H. pylori was generated by mutation with nitrosoguanidine (85). This mutant was unable to colonize gnotobiotic piglets. Due to the undefined nature of the original mutants, questions remained regarding possible second site mutations. The Eaton group later reported the use of an insertional deletion urease mutant and repeated the colonization study, with similar findings (88). Other groups found similar results with urease mutants tested in normochlorhydric nude mice, ferrets, and cynomolgus monkeys (12, 322, 335). The insertional deletion mutant persisted at low numbers for at least 5 days in some piglets rendered achlorhydric by treatment with omeprezole. Fewer than 100 CFU per g of tissue were recovered, however, from two of four animals at 5 days postinfection. In contrast, eradication therapy with the urease inhibitor flurofamide failed to eradicate H. mustelae infection in ferrets, even when given in combination with amoxycillin (260). These data suggest that acid neutralization is one important function of urease, but that urease also plays other roles during the infection process. Furthermore, urease may not be absolutely required once infection is established.
H. pylori has mechanisms other than urease for protection against acid. These include arginase, an aliphatic amidase, and amino acid deamidases (210, 212, 298). The rocF gene encoding arginase protects H. pylori against acid in the presence of arginine by converting it to ornithine and urea (210). The rocF mutant is 1,000 times more sensitive to acid than the wild-type strain in the absence of urea. One rocF mutant examined had a 2-log reduction in the ability to colonize mice, suggesting that arginase could be an important bacterial factor in vivo.
γ-Glutamyl transpeptidase (GGT) is constitutively expressed by all strains of H. pylori but has only been found in a few other bacteria (44). This enzyme catalyzes the transfer of glutamyl groups between compounds such as glutathione and amino acids. In mammalian cells, GGT influences glutathione metabolism. Its role in bacteria is unknown, but GGT has been hypothesized to play a role in the transport of amino acids. A ggt isogenic mutant of the mouse-virulent SS1 H. pylori strain was convincingly shown to be unable to colonize mice (44). Additionally, the mutant strain was unable to infect in the presence of wild-type H. pylori. Further studies on the H. pylori ggt mutant have not yet been performed, however, and an E. coli ggt mutant was found to be sensitive to hydrogen peroxide and chlorine compounds, and suppression of GGT activity resulted in leakage of glutathione from the bacterial cells (43, 227). Although the gene is constitutively expressed, the ggt mutant strain demonstrated no obvious deficiencies when grown in vitro. Elucidation of the role played by H. pylori GGT in vivo may provide clues regarding the relevant stresses faced by H. pylori during colonization.
Phospholipases are believed to play roles in the virulence of several organisms, including Clostridium perfringens, Listeria monocytogenes, and P. aeruginosa. H. pylori has PLA1, PLA2, and PLC activities (247). As mentioned above, the anti-ulcer drug bismuth inhibits phospholipase activity. To test the importance of phospholipase activity in vivo, Dorrell and coworkers (79) constructed a mutation in pldA, the only H. pylori phospholipase gene identified to date. The resulting mutant H. pylori strain had no detectable PLA2 activity, no zone of clearing on egg yolk agar, and markedly reduced hemolytic activity. This mutant was not detected in mice at 2 or 8 weeks following infection, suggesting an inability to colonize. Nonetheless, infected mice generated H. pylori-specific antibody titers. Although titers were significantly lower than those elicited by wild-type H. pylori, this may indicate that the pldA mutant is able to persist within the mouse for a short period. PLA2 levels and lysolecithin, the product of cellular phospholipid hydrolysis, have been found to be higher in H. pylori-infected patients than in uninfected controls (182). This indicates that PLA2 could contribute to cellular damage in vivo and thus may play an active role in pathogenesis.
Iron is required for growth of nearly all pathogens. H. pylori actively acquires iron from the host and may, in fact, contribute to anemia in some patients (52, 252). Human transferrin is the protein used to sequester iron in the systemic circulation, while lactoferrin is used in the mucosa, including tears, saliva, and milk (70). Other bacterial species are known to utilize transferrin and lactoferrin as iron sources, and specificity for the human or bovine proteins is believed to contribute to species specificity of Neisseria gonorrhoeae and Pasteurella haemolytica, respectively (281, 282). H. pylori produces a 70-kDa outer membrane protein, Lbp, which specifically binds human lactoferrin (74). Bovine lactoferrin partially inhibits interaction with human lactoferrin; however, bovine lactoferrin cannot be effectively used by H. pylori for growth in iron-deficient medium (153). Lactoferrin may be a particularly important source of iron for bacteria residing in the gastric mucosa, as it retains binding capabilities even at pH extremes (153). Serum antibodies specific for H. pylori Lbp were found in patients infected with H. pylori but not in control patients, suggesting that this protein is expressed by H. pylori in vivo (74).
Other bacterial colonization factors have not yet been precisely identified. Van Doorn and colleagues (346) found that a spontaneous mutation within strain SPM326 affected colonization in BALB/c and C57Bl/6 mice. The wild-type strain induced strong IL-8 production in KATOIII cells, whereas the spontaneous mutant did not induce IL-8. The wild-type strain, however, was unable to colonize BALB/c mice, whereas the IL-8 mutant strain could infect these animals. Both strains were able to colonize C57Bl/6 mice, but the wild-type stain colonized at a significantly lower level, and with a further decrease in bacterial load by 12 weeks postinfection. Wild-type strain SPM326 is able to persistently colonize other mouse strains (119), suggesting that host factors play a role as well. The site of the spontaneous mutation is not known; however, mutations in certain genes in the cag pathogenicity island influence IL-8 production (3, 39, 339). The authors speculate that the wild-type SPM326 strain may be more susceptible to host factors than strain SS1, which readily infects both BALB/c and C57Bl/6 mice. Perhaps the mutant deficient in IL-8 induction elicits a weaker immune response than the parent strain. In three of seven SPM326-infected C57Bl/6 mice, increases were noted in the number of T cells in the mucosa, and increased MHC class II antigen expression was not confined to small cells located between the gastric glands, as it was in control mice (346). The mechanism of these colonization and immunological effects will need to be elucidated to determine whether similar gene disruptions influence H. pylori colonization in humans.
One significant impediment to studying bacterial colonization factors is the lack of data regarding the natural route of transmission and the physiological state of the organism at the time of infection. Since H. pylori is capable of converting to a coccoid morphology, and this conversion takes place after prolonged in vitro culture, it is possible that coccoid organisms are responsible for naturally acquired infections. The literature contains conflicting reports regarding the capacity of coccoid H. pylori to infect animals. Cellini and colleagues were able to demonstrate colonization of BALB/c mice by culture, histology, and serology following administration of coccoid organisms (37). A similar attempt to infect gnotobiotic piglets with coccoid H. pylori failed to produce any evidence of even transient infection (86). Neither group attempted to assess viability of coccoid organisms using any measure other than the formation of colonies. Therefore, one cannot infer what proportion of each inoculum was dormant or truly dead. Far more research on the conversion to—and possible reversion from—the coccoid state is needed to resolve the question of whether coccoid H. pylori is infectious.
Motility and Chemotaxis
To avoid being continually bathed in acid, and eventually flushed from the stomach entirely, H. pylori must be motile. H. pylori flagella are composed of flagellin, encoded by flaA and flaB (179, 188, 318), and are surrounded by a membranous sheath containing LPS and protein (116, 195). Aflagellate H. pylori mutants are unable to colonize (89, 90) but still generate an immune response, indicating survival at least for a short time (91). Repeated in vivo passage of H. pylori resulted in greater colonization efficiency (203). Further investigation revealed that bacteria from later passages expressed more flaA mRNA and flagellin protein than did earlier passage isolates.
The flagella of H. pylori possess unique bulb structures at their tips, called terminal bulbs. The function of the terminal bulbs and the genes controlling their development are not yet known. These structures may aid in adherence or in propelling the bacteria through the viscous mucous layer. Indeed, viscosity has been shown to stimulate chemotaxis (369). The panorama of bismuth's antihelicobacter activities may include reduction in motility. Inhibition of urease activity with various inhibitors abolished the motility and the chemotactic response in a viscous solution, in spite of the presence of flagella and the ability to move through a nonviscous solution (226, 369). The energy source for flagellar movement is derived from the proton motive force; hydrolysis of urea may generate the additional energy required for movement through a viscous environment (371).
The ability to swim may not be sufficient to maintain the H. pylori population in the stomach. Some evidence has arisen indicating that the capacity for directed motility, or chemotaxis, may be crucial. Analysis of the H. pylori genome revealed genes homologous to those encoding the known chemotaxis regulators, CheA and CheY. The H. pylori homologs were designated cheY1 and cheAY2. Mutations in these genes, which abolished chemotaxis, also rendered the mutants unable to colonize mice, although significant antibody responses were detected (111).
Conclusions
We are only beginning to understand the complex mechanisms used by H. pylori to establish infection and to maintain colonization in the same host over a period of many years. Penetration of the mucous layer likely involves motility, phospholipase activity, and mucinase activity. H. pylori-mediated reduction of mucus synthesis or secretion may also assist the bacterium in gaining access to the epithelium and persisting at this location. Adherence allows the bacteria to anchor themselves to the epithelial layer, but bacteria that remain attached to epithelial cells will eventually be swept away as these cells die and are exfoliated. Thus, a proportion of the H. pylori population exists in the nonadherent state. H. pylori must also contend with antibodies and phagocytic cells as the host mounts an immune response.
Colonization by H. pylori has only been confirmed in the gastric mucosa. Limitation to this site could be due to competition by other organisms in the mouth and small intestine. Other Helicobacter spp. have been isolated from blood and deep tissues, but H. pylori has not. Perhaps the exquisite sensitivity of H. pylori to complement-mediated lysis (261, 265) renders it incapable of causing extragastric disease; however, circumstantial evidence suggests that H. pylori may be present in the liver (233, 259).
Preventing the establishment of long-term colonization, or even curing an established H. pylori infection, may be achieved either by killing all organisms or by antagonizing one or more of the persistence mechanisms and suppressing growth until the combination of host immune response, exfoliation, and movement of gastric contents purge the last bacterium from the stomach. Data in humans on prevention of colonization or spontaneous cure are sparse; however, there is some evidence for both phenomena (29, 115, 198, 202, 328, 372). Widespread treatment of H. pylori infection with multidrug regimens is problematic even in industrialized nations. Thus, until an effective vaccine is developed to prevent H. pylori infection, improved sanitation and public education may be the most practical means of limiting infection.
At present, dissecting the individual contributions of the factors discussed in this chapter is exceedingly difficult. The development of new research tools, such as the completed genome sequence and superior in vivo and in vitro models will no doubt facilitate future studies of H. pylori adherence and colonization.
References
- 1.
- Alba Y., Suzuki N., Kabir A. M., Takagi A., Koga Y. Lactic acid-mediated suppression of Helicobacter pylori by the oral administration of Lactobacillus salivarius as a probiotic in a gnotobiotic murine model. Am. J. Gastroenterol. 1998;93:2097–2101. [PubMed: 9820379]
- 2.
- Aihara M., Tsuchimoto D., Takizawa H., Azuma A., Wakebe H., Ohmoto Y., Imagawa K., Kikuchi M., Mukaida N., Matsushima K. Mechanisms involved in Helicobacter pylori-induced interleukin-8 production by a gastric cancer cell line, MKN45. Infect. Immun. 1997;65:3218–3224. [PMC free article: PMC175455] [PubMed: 9234778]
- 3.
- Akopyants N. S., Clifton S. W., Kersulyte D., Crabtree J. E., Youree B. E., Reece C. A., Bukanov N. O., Drazek E. S., Roe B. A., Berg D. E. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol. Microbiol. 1998;28:37–53. [PubMed: 9593295]
- 4.
- Alkout A. M., Blackwell C. C., Weir D. M., Poxton I. R., Elton R. A., Luman W., Palmer K. Isolation of a cell surface component of Helicobacter pylori that binds H type 2, Lewis(a), and Lewis(b) antigens. Gastroenterology. 1997;112:1179–1187. [PubMed: 9098001]
- 5.
- Allen L. A., Schlesinger L. S., Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J. Exp. Med. 2000;191:115–128. [PMC free article: PMC2195807] [PubMed: 10620610]
- 6.
- Allen L. A. H. Intracellular niches for extracellular bacteria: lessons from Helicobacter pylori. J. Leuk. Biol. 1999;66:753–756. [PubMed: 10577505]
- 7.
- Alm R., Ling L. S., Moir D. T., King B. L., Brown E. D., Doig P. C., Smith D. R., Noonan B., Guild B. C., deJonge B. L., Carmel G., Tummino P. J., Caruso A., Uria-Nickelsen M., Mills D. M., Ives C., Gibson R., Merberg D., Mills S. D., Jiang Q., Taylor D. E., Vovis G. F., Trust T. J. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. [PubMed: 9923682]
- 8.
- Amado M., Carniero F., Seixas M., Clausen H., Sobrinho-Simones M. Dimeric sialyl/Lex expression in gastric carcinoma correlates with venous invasion and poor outcome. Gastroenterology. 1998;114:462–470. [PubMed: 9496936]
- 9.
- Andersen L. P., Blom J., Nielsen H. Survival and ultrastructural changes of Helicobacter pylori after phagocytosis by human polymorphonuclear leukocytes and monocytes. APMIS. 1993;101:61–72. [PubMed: 8457327]
- 10.
- Andersen L. P., Holck S. Possible evidence of invasiveness of Helicobacter (Campylobacter) pylori. Eur. J. Clin. Microbiol. Infect. Dis. 1990;9:135–138. [PubMed: 2318218]
- 11.
- Andersen R. N., Ganeshkumar N., Kolenbrander P. E. Helicobacter pylori adheres selectively to Fusobacterium spp. Oral Microbiol. Immunol. 1998;13:51–54. [PubMed: 9573823]
- 12.
- Andrutis K. A., Fox J. G., Schauer D. B., Marini R. P., Murphy J. C., Yan L., Solnick J. V. Inability of an isogenic urease-negative mutant stain of Helicobacter mustelae to colonize the ferret stomach. Infect. Immun. 1995;63:3722–3725. [PMC free article: PMC173518] [PubMed: 7642314]
- 13.
- Ångstrom J., Teneberg S., Milh M. A., Larsson T., Leonardsson I., Olsson B. M., Halvarsson M. O., Danielsson D., Näslund I., Ljungh Å., Wadström T., Karlsson K.-A. The lactosylceramide binding specificity of Helicobacter pylori. Glycobiology. 1998;8:297–309. [PubMed: 9499377]
- 14.
- Ansorg R., Schmid E. N. Adhesion of Helicobacter pylori to yeast cells. Zentralbl. Bakteriol. 1998;288:501–508. [PubMed: 9987188]
- 15.
- Appelmelk B. J., Shiberu B., Trinks C., Tapsi N., Zheng P. Y., Verboom T., Maaskant J., Hokke C. H., Schiphorst W. E., Blanchard D., Simoons-Smit I. M., van den Eijnden D. H., Vandenbroucke-Grauls C. M. Phase variation in Helicobacter pylori lipopolysaccharide. Infect. Immun. 1998;66:70–76. [PMC free article: PMC107860] [PubMed: 9423841]
- 16.
- Appelmelk B. J., Simoons-Smit I., Negrini R., Moran A. P., Aspinall G. O., Forte J. G., deVries T., Quan H., Verboom T., Maaskant J. J., Ghiara P., Kuipers E. J., Bloemena E., Tadema T. M., Townsend R. R., Tyagarajan K., Crothers J. M., Monteiro M. A., Savio A., deGraaff J. J. Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in auto-immunity. Infect. Immun. 1996;64:2031–2040. [PMC free article: PMC174033] [PubMed: 8675304]
- 17.
- Armstrong J. A., Cooper M., Goodwin C. S., Robinson J., Wee S. H., Burton M., Burke V. Influence of soluble haemagglutinins on adherence of Helicobacter pylori to HEp-2 cells. J. Med. Microbiol. 1991;34:181–187. [PubMed: 2010909]
- 18.
- Asahi M., Azuma T., Ito S., Ito Y., Suto M., Nagai Y., Tsubokawa M., Tohyama Y., Maeda S., Omata M., Suzuki T., Sasakawa C. Helicobacter pylori CagA protein can be tyosine phosphorylated in gastric epithelial cells. J. Exp. Med. 2000;191:593–602. [PMC free article: PMC2195829] [PubMed: 10684851]
- 19.
- Ascencio F., Franson L. A., Wadström T. Affinity of the gastric pathogen Helicobacter pylori for the N-sulphated glycosaminoglycan heparan sulphate. J. Med. Microbiol. 1993;38:240–244. [PubMed: 7682621]
- 20.
- Asikainen S., Chen C., Slots J. Absence of Helicobacter pylori in subgingival samples determined by polymerase chain reaction. Oral Microbiol. Immunol. 1994;9:318–320. [PubMed: 7808777]
- 21.
- Azuma T., Ito S., Sato F., Yamazaki Y., Miyaji H., Ito Y., Suto H., Kuriyama M., Kato T., Kohli Y. The role of the HLA-DQA1 gene in resistance to atrophic gastritis and gastric adenocarcinoma induced by Helicobacter pylori infection. Cancer. 1998;82:1013–1018. [PubMed: 9506344]
- 22.
- Bagchi D., McGinn T. R., Ye X., Balmoori J., Bagchi M., Stohs S. J., Kuszynski C. A., Carryl O. R., Mitra S. Mechanism of gastroprotection by bismuth subsalicylate against chemically induced oxidative stress in cultured human gastric mucosal cells. Dig. Dis. Sci. 1999;44:2419–2428. [PubMed: 10630491]
- 23.
- Baik S. C., Youn H. S., Chung M. H., Lee W. K., Cho M. J., Ko G. H., Park C. K., Kasai H., Rhee K. H. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res. 1996;56:1279–1282. [PubMed: 8640814]
- 24.
- Begue R. E., Gonzales J. L., Correa-Gracian H., Tang S. C. Dietary risk factors associated with the transmission of Helicobacter pylori in Lima, Peru. Am. J. Trop. Med. Hyg. 1998;59:637–640. [PubMed: 9790444]
- 25.
- Bennedsen M., Wang X., Willen R., Wadstrom T., Andersen L. P. Treatment of H. pylori infected mice with antioxidant astaxanthin reduces gastric inflammation, bacterial load and modulates cytokine release by splenocytes. Immunol. Lett. 1999;70:185–189. [PubMed: 10656672]
- 26.
- Bode G., Malfertheiner P., Ditschuneit H. Pathogenic implications of ultrastructural findings in Campylobacter pylori related gastroduodenal disease. Scand. J. Gastroenterol. 1988;142:25–39. [PubMed: 3166531]
- 27.
- Bonvicini F. M., Maltarello C., Versura P., Bianchi D., Gasbarrini G., Laschi R. Correlative scanning electron microscopy in the study of human gastric mucosa. Scanning Electron Microsc. 1986;2:687–702. [PubMed: 3099375]
- 28.
- Borén T., Falk P., Roth K. A., Larson G., Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. [PubMed: 8018146]
- 29.
- Brenner H., Berg G., Lappus N., Kliebsch U., Bode G., Boeing H. Alcohol consumption and Helicobacter pylori infection: results from the German National Health and Nutrition Survey. Epidemiology. 1999;10:214–218. [PubMed: 10230827]
- 30.
- Buck G. E., Gourley W. K., Lee W. K., Subramanyam K., Latimer J. M., DiNuzzo A. R. Relationship of Campylobacter pyloridis to gastritis and peptic ulcer. J. Infect. Dis. 1986;153:664–669. [PubMed: 3950448]
- 31.
- Buiatti E., Palli D., Decarli A., Amadori D., Avellini C., Bianchi S., Bonaguri C., Cipriani F., Cocco P., Giacosa A. A case-control study of gastric cancer and diet in Italy: II. Association with nutrients. Int. J. Cancer. 1990;45:896–901. [PubMed: 2335393]
- 32.
- Buiatti E., Palli D., Decarli A., Amadori D., Avellini C., Bianchi S., Biserni R., Cipriani F., Cocco P., Giacosa A., Marubini E., Puntoni R., Vindigni C., Fraumeni J., Blot W. A case-control study of gastric cancer and diet in Italy. Int. J. Cancer. 1989;44:611–616. [PubMed: 2793233]
- 33.
- Calam J. Helicobacter pylori, acid and gastrin. Eur. J. Gastroenterol. Hepatol. 1995;7:310–317. [PubMed: 7600135]
- 34.
- Campbell S., Fraser A., Holliss B., Schmid J., O'Toole P. W. Evidence for ethnic tropism of Helicobacter pylori. Infect. Immun. 1997;65:3708–3712. [PMC free article: PMC175528] [PubMed: 9284141]
- 35.
- Caselli M., Figura N., Trevisani L., Pazzi P., Guglielmetti P., Bovolenta M. R., Stabellini G. Patterns of physical modes of contact between Campylobacter pylori and gastric epithelium: implications about the bacterial pathogenicity. Am. J. Gastroenterol. 1989;84:511–513. [PubMed: 2719007]
- 36.
- Çelik J., Su B., Tirén U., Finkel Y., Thoresson A.-C., Engstrand L., Sandstedt B., Berander S., Normark S. Virulence and colonization-associated properties of Helicobacter pylori isolated from children and adolescents. J. Infect. Dis. 1998;177:247–252. [PubMed: 9419200]
- 37.
- Cellini L., Allocati N., Angelucci D., Lezzi T., Di Campli E., Marzio L., Dainelli B. Coccoid Helicobacter pylori not culturable in vitro reverts in mice. Microbiol. Immunol. 1994;38:843–850. [PubMed: 7898382]
- 38.
- Cellini L., Di Campli E., Masulli M., Di Bartolomeo S., Allocati N. Inhibition of Helicobacter pylori by garlic extract (Allium sativum). FEMS Immunol. Med. Microbiol. 1996;13:273–277. [PubMed: 8739190]
- 39.
- Censini S., Lange C., Xiang Z., Crabtree J. E., Ghiara P., Borodovsky M., Rappuoli R., Covacci A. cag, a pathogenicity island of Helicobacter pylori, encodes type-I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA. 1996;93:14648–14653. [PMC free article: PMC26189] [PubMed: 8962108]
- 40.
- Chan E. C., Chang C. C., Li Y. S., Chang C. A., Chiou C. C., Wu T. Z. Purification and characterization of neutral sphingomyelinase from Helicobacter pylori. Biochemistry. 2000;39:4838–4845. [PubMed: 10769141]
- 41.
- Chen W., Shu D., Chadwick V. S. Helicobacter pylori infection in interleukin-4-deficient and transgenic mice. Scand. J. Gastroenterol. 1999;34:987–992. [PubMed: 10563668]
- 42.
- Chen X. G., Correa P., Offerhaus J., Rodriguez E., Janney F., Hoffmann E., Fox J., Hunter F., Diavolitsis S. Ultrastructure of the gastric mucosa harboring Campylobacter-like organisms. Am. J. Clin. Pathol. 1986;86:575–582. [PubMed: 2430450]
- 43.
- Chesney J. A., Eaton J. W., Mahoney, Jr J. R. Bacterial glutathione: a sacrificial defense against chlorine compounds. J. Bacteriol. 1996;178:2131–2135. [PMC free article: PMC177915] [PubMed: 8606194]
- 44.
- Chevalier C., Thiberge J. M., Ferrero R. L., Labigne A. Essential role of Helicobacter pylori gamma-glutamyltranspeptidase for the colonization of the gastric mucosa of mice. Mol. Microbiol. 1999;31:1359–1372. [PubMed: 10200957]
- 45.
- Chew B. P., Park J. S., Wong M. W., Wong T. S. A comparison of the anticancer activities of dietary beta-carotene, canthaxanthin and astaxanthin in mice in vivo. Anticancer Res. 1999;19:1849–1853. [PubMed: 10470126]
- 46.
- Chmiela M., Czkwianianc E., Wadström T., Rudnicka W. Role of Helicobacter pylori surface structures in bacterial interaction with macrophages. Gut. 1997;40:20–24. [PMC free article: PMC1027002] [PubMed: 9155570]
- 47.
- Chmiela M., Lawnik M., Czkwianianc E., Rechcinski T., Planeta-Malecka I., Wadström T., Rudnicka W. Attachment of Helicobacter pylori strains to human epithelial cells. J. Physiol. Pharmacol. 1997;48:393–404. [PubMed: 9376622]
- 48.
- Chmiela M., Lelwala-Guruge J., Wadström T. Interaction of cells of Helicobacter pylori with human polymorphonuclear leucocytes: possible role of haemagglutinins. FEMS Immunol. Med. Microbiol. 1994;9:41–48. [PubMed: 7522735]
- 49.
- Chmiela M., Ljungh Å., Rudnicka W., Wadström T. Phagocytosis of Helicobacter pylori bacteria differing in the heparan sulfate binding by human polymorphonuclear leukocytes. Zentralbl. Bakteriol. 1996;283:346–350. [PubMed: 8861873]
- 50.
- Chmiela M., Paziak-Domanska B., Rudnicka W., Wadström T. The role of heparan sulphate-binding activity of Helicobacter pylori bacteria in their adhesion to murine macrophages. APMIS. 1995;103:469–474. [PubMed: 7546650]
- 51.
- Chmiela M., Paziak-Domanska B., Wadström T. Attachment, ingestion and intracellular killing of Helicobacter pylori by human peripheral blood mononuclear leucocytes and mouse peritoneal inflammatory macrophages. FEMS Immunol. Med. Microbiol. 1995;10:307–316. [PubMed: 7773248]
- 52.
- Choe Y. H., Kim S. K., Son B. K., Lee D. H., Hong Y. C., Pai S. H. Randomized placebo-controlled trial of Helicobacter pylori eradication for iron-deficiency anemia in preadolescent children and adolescents. Helicobacter. 1999;4:135–139. [PubMed: 10382128]
- 53.
- Chowers M. Y., Keller N., Tal R., Barshack I., Lang R., Bar-Meir S., Chowers Y. Human gastrin: a Helicobacter pylori-specific growth factor. Gastroenterology. 1999;117:1113–1118. [PubMed: 10535874]
- 54.
- Christie P. J. The Agrobacterium tumefaciens T-complex transport apparatus: a paradigm for a new family of multifunctional transporters in eubacteria. J. Bacteriol. 1997;179:3085–3094. [PMC free article: PMC179082] [PubMed: 9150199]
- 55.
- Chung J. G., Chen G. W., Wu L. T., Chang H. L., Lin J. G., Yeh C. C., Wang T. F. Effects of garlic compounds diallyl sulfide and diallyl disulfide on arylamine N-acetyltransferase activity in strains of Helicobacter pylori from peptic ulcer patients. Am. J. Chin. Med. 1998;26:353–364. [PubMed: 9862023]
- 56.
- Clyne M., Drumm B. Adherence of Helicobacter pylori to primary human gastrointestinal cells. Infect. Immun. 1993;61:4051–4057. [PMC free article: PMC281123] [PubMed: 8406792]
- 57.
- Clyne M., Drumm B. Cell envelope characteristics of Helicobacter pylori: their role in adherence to mucosal surfaces and virulence. FEMS Immunol. Med. Microbiol. 1996;16:141–155. [PubMed: 8988394]
- 58.
- Clyne M., Drumm B. Absence of Lewis A and Lewis B expression on adherence of H. pylori to human gastric cells. Gastroenterology. 1997;113:72–80. [PubMed: 9207264]
- 59.
- Clyne M., Thomas J., Weaver L., Drumm B. In vitro evaluation of the role of antibodies against Helicobacter pylori in inhibiting adherence of the organism to gastric cells. Gut. 1997;40:731–738. [PMC free article: PMC1027197] [PubMed: 9245926]
- 60.
- Coconnier M. H., Lievin V., Hemery E., Servin A. L. Antagonistic activity against Helicobacter infection in vitro and in vivo by the human Lactobacillus acidophilus strain LB. Appl. Environ. Microbiol. 1998;64:4573–4580. [PMC free article: PMC106686] [PubMed: 9797324]
- 61.
- Cole S. P., Cirillo D., Kagnoff M. F., Guiney D. G., Eckmann L. Coccoid and spiral Helicobacter pylori differ in their abilities to adhere to gastric epithelial cells and induce interleukin-8 secretion. Infect. Immun. 1997;65:843–846. [PMC free article: PMC176138] [PubMed: 9009355]
- 62.
- Corthésy-Theulaz I., Porta N., Pringault E., Racine L., Bogdanova A., Kraehenbuhl J.-P., Blum A. L., Michetti P. Adhesion of Helicobacter pylori to polarized T84 human intestinal cell monolayers is pH dependent. Infect. Immun. 1996;64:3827–3832. [PMC free article: PMC174299] [PubMed: 8751935]
- 63.
- Covacci A., Censini S., Bugnoli M., Petracca R., Burroni D., Macchia G., Massone A., Papini E., Xiang Z., Figura N., Rappuoli R. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA. 1993;90:5791–5795. [PMC free article: PMC46808] [PubMed: 8516329]
- 64.
- Covacci A., Rappuoli R. Tyrosine-phosphorylated bacterial proteins: Trojan horses for the host cell. J. Exp. Med. 2000;191:587–592. [PMC free article: PMC2195833] [PubMed: 10684850]
- 65.
- Covacci A., Telford J. L., Del Giudice G., Parsonnet J., Rappuoli R. Helicobacter pylori, virulence and genetic geography. Science. 1999;284:1328–1333. [PubMed: 10334982]
- 66.
- Cover T. L., Dooley C. P., Blaser M. J. Characterization of and human serologic response to proteins in Helicobacter pylori broth culture supernatants with vacuolizing cytotoxin activity. Infect. Immun. 1990;58:603–610. [PMC free article: PMC258508] [PubMed: 2307514]
- 67.
- Crabtree J. E., Farmery S. M., Lindley I. J. D., Figura N., Peichl P., Tompkins D. S. CagA/cytotoxic strains of Helicobacter pylori and interleukin-8 in gastric epithelial cell lines. J. Clin. Pathol. 1994;47:945–950. [PMC free article: PMC502181] [PubMed: 7962609]
- 68.
- Crabtree J. E., Taylor J. D., Wyatt J. I., Heatley R. V., Shallcross T. M., Tompkins D. S., Rathbone B. J. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration, and gastric pathology. Lancet. 1991;338:332–335. [PubMed: 1677696]
- 69.
- Crabtree J. E., Xiang Z., Lindley I. J., Tompkins D. S., Rappuoli R. Induction of interleukin-8 secretion from gastric epithelial cells by a cagA isogenic mutant of Helicobacter pylori. J. Clin. Pathol. 1995;48:967–969. [PMC free article: PMC502959] [PubMed: 8537502]
- 70.
- Crichton R. R., Charloteaux-Wauters M. Iron transport and storage. Eur. J. Biochem. 1987;164:485–506. [PubMed: 3032619]
- 71.
- Davies G. R., Banatvala N., Collins C. E., Sheaff M. T., Abdi Y., Clements L., Rampton D. S. Relationship between infective load of Helicobacter pylori and reactive oxygen metabolite production in antral mucosa. Scand. J. Gastroenterol. 1994;29:419–424. [PubMed: 8036457]
- 72.
- De Koster E., Buset M., Fernandes E., Deltenre M. Helicobacter pylori: the link with gastric cancer. Eur. J. Cancer Prev. 1994;3:247–257. [PubMed: 8061590]
- 73.
- Desai M. A., Mutlu M., Vadgama P. A study of macromolecular diffusion through native porcine mucus. Experientia. 1992;48:22–26. [PubMed: 1737572]
- 74.
- Dhaenens L., Szczebara F., Husson M. O. Identification, characterization, and immunogenicity of the lactoferrin-binding protein from Helicobacter pylori. Infect. Immun. 1997;65:514–518. [PMC free article: PMC176089] [PubMed: 9009306]
- 75.
- Doig P., Austin J. W., Kostrzynska M., Trust T. J. Production of a conserved adhesin by the gastroduodenal pathogen Helicobacter pylori. J. Bacteriol. 1992;174:2539–2547. [PMC free article: PMC205892] [PubMed: 1556073]
- 76.
- Doig P., Austin J. W., Trust T. J. The Helicobacter pylori 19.6-kilodalton protein is an iron-containing protein resembling ferritin. J. Bacteriol. 1993;175:557–560. [PMC free article: PMC196173] [PubMed: 8419304]
- 77.
- Doig P., Exner M. M., Hancock R. E., Trust T. J. Isolation and characterization of a conserved porin protein from Helicobacter pylori. J. Bacteriol. 1995;177:5447–5452. [PMC free article: PMC177350] [PubMed: 7559328]
- 78.
- Dorgan J. F., Schatzkin A. Antioxidant micronutrients in cancer prevention. Hematol. Oncol. Clin. North Am. 1991;5:43–68. [PubMed: 2026568]
- 79.
- Dorrell N., Martino M. C., Stabler R. A., Ward S. J., Zhang Z. W., McColm A. A., Farthing M. J., Wren B. W. Characterization of Helicobacter pylori PldA, a phospholipase with a role in colonization of the gastric mucosa. Gastroenterology. 1999;117:1098–1104. [PubMed: 10535872]
- 80.
- Drake I. M., Davies M. J., Mapstone N. P., Dixon M. F., Schorah C. J., White K. L., Chalmers D. M., Axon A. T. Ascorbic acid may protect against human gastric cancer by scavenging mucosal oxygen radicals. Carcinogenesis. 1999;17:559–562. [PubMed: 8631145]
- 81.
- Dubois A., Berg D. E., Incecik E. T., Fiala N., Heman-Ackah L. M., Del Valle J., Yang M., Wirth H. P., Perez-Perez G. I., Blaser M. J. Host specificity of Helicobacter pylori strains and host responses in experimentally challenged nonhuman primates. Gastroenterology. 1999;116:90–96. [PubMed: 9869606]
- 82.
- Duggan A. E., Atherton J. C., Cockayne A., Balsitis M., Evison S., Hale T., Hawkey C. J., Spiller R. C. Clarification of the link between polyunsaturated fatty acids and Helicobacter pylori-associated duodenal ulcer disease: a dietary intervention study. Br. J. Nutr. 1997;78:515–522. [PubMed: 9389880]
- 83.
- Dwarakanath A. D., Tsai H. H., Sunderland D., Hart C. A., Figura N., Crabtree J. E., Rhodes J. M. The production of neuraminidase and fucosidase by Helicobacter pylori: their possible relationship to pathogenicity. FEMS Immunol. Med. Microbiol. 1995;12:213–216. [PubMed: 8745005]
- 84.
- Dytoc M., Gold B., Louie M., Huesca M., Fedorko L., Crowe S., Lingwood C., Brunton J., Sherman P. Comparison of Helicobacter pylori and attaching-and-effacing Escherichia coli adhesion to eukaryotic cells. Infect. Immun. 1993;61:448–456. [PMC free article: PMC302749] [PubMed: 8380793]
- 85.
- Eaton K. A., Brooks C. L., Morgan D. R., Krakowka S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 1991;59:2470–2475. [PMC free article: PMC258033] [PubMed: 2050411]
- 86.
- Eaton K. A., Catrenich C. E., Makin K. M., Krakowka S. Virulence of coccoid and bacillary forms of Helicobacter pylori in gnotobiotic piglets. J. Infect. Dis. 1995;171:459–462. [PubMed: 7844390]
- 87.
- Eaton K. A., Cover T. L., Tummuru M. K. R., Blaser M. J., Krakowka S. Role of vacuolating cytotoxin in gastritis due to Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 1997;65:3462–3464. [PMC free article: PMC175490] [PubMed: 9234813]
- 88.
- Eaton K. A., Krakowka S. Effect of gastric pH on urease-dependent colonization of gnotobiotic piglets by Helicobacter pylori. Infect. Immun. 1994;62:3604–3607. [PMC free article: PMC303008] [PubMed: 8063376]
- 89.
- Eaton K. A., Morgan D. R., Krakowka S. Campylobacter virulence factors in gnotobiotic piglets. Infect. Immun. 1989;57:1119–1125. [PMC free article: PMC313239] [PubMed: 2925243]
- 90.
- Eaton K. A., Morgan D. R., Krakowka S. Motility as a factor in the colonization of gnotobiotic piglets by Helicobacter pylori. J. Med. Microbiol. 1992;37:123–127. [PubMed: 1629897]
- 91.
- Eaton K. A., Suerbaum S., Josenhans C., Krakowka S. Colonization of gnotobiotic piglets by Helicobacter pylori deficient in two flagellin genes. Infect. Immun. 1996;64:2445–2448. [PMC free article: PMC174096] [PubMed: 8698465]
- 92.
- Edwards N. J., Monteiro M. A., Faller G., Walsh E. J., Moran A. P., Roberts I. S., High N. J. Lewis X structures in the O antigen side-chain promote adhesion of Helicobacter pylori to the gastric epithelium. Mol. Microbiol. 2000;35:1530–1539. [PubMed: 10760152]
- 93.
- Elitsur Y., Adkins L., Saeed D., Neace C. Helicobacter pylori antibody profile in household members of children with H. pylori infection. J. Clin. Gastroenterol. 1999;29:178–182. [PubMed: 10478881]
- 94.
- El-Omar E., Penman I., Dorrian C. A., Ardill J. E., McColl K. E. Eradicating Helicobacter pylori infection lowers gastrin mediated acid secretion by two thirds in patients with duodenal ulcer. Gut. 1993;34:1060–1065. [PMC free article: PMC1374354] [PubMed: 8174954]
- 95.
- Emödy L., Carlsson Å., Ljungh Å., Wadström T. Mannose-resistant haemagglutinin by Campylobacter pylori. Scand. J. Infect. Dis. 1988;20:353–354. [PubMed: 3406678]
- 96.
- Engstrand L., Graham D. Y., Scheynius A., Genta R. M., El-Zaatari F. Is the sanctuary where Helicobacter pylori avoids antibacterial treatment intracellular? Am. J. Clin. Pathol. 1997;108:504–509. [PubMed: 9353088]
- 97.
- Exner M. M., Doig P., Trust T. J., Hancock R. E. Isolation and characterization of a family of porin proteins from Helicobacter pylori. Infect. Immun. 1995;63:1567–1572. [PMC free article: PMC173190] [PubMed: 7534278]
- 98.
- Evans D. G., Evans D. J., Graham D. Y. Receptor-mediated adherence of Campylobacter pylori to mouse Y-1 adrenal cell monolayers. Infect. Immun. 1989;57:2272–2278. [PMC free article: PMC313441] [PubMed: 2473033]
- 99.
- Evans D. G., Evans D. J., Graham D. Y. Adherence and internalization of Helicobacter pylori by HEp-2 cells. Gastroenterology. 1992;102:1557–1567. [PubMed: 1568565]
- 100.
- Evans D. G., Evans D. J., Mould J. J., Graham D. Y. N-acetylneuraminyllactose-binding fibrillar hemagglutinin of Campylobacter pylori: a putative colonization factor antigen. Infect. Immun. 1988;56:2896–2906. [PMC free article: PMC259668] [PubMed: 2459065]
- 101.
- Evans D. G., Karjalainen T. K., Evans D. J., Graham D. Y., Lee C.-H. Cloning, nucleotide sequence, and expression of a gene encoding an adhesin subunit protein of Helicobacter pylori. J. Bacteriol. 1993;175:674–683. [PMC free article: PMC196205] [PubMed: 7678592]
- 102.
- Evans D. J., Evans D. G., Takemura T., Nakano H., Lampert H. C., Graham D. Y., Granger D. N., Kvietys P. R. Characterization of a Helicobacter pylori neutrophil-activating protein. Infect. Immun. 1995;63:2213–2220. [PMC free article: PMC173288] [PubMed: 7768601]
- 103.
- Falk P. G., Bry L., Holgersson J., Gordon J. I. Expression of a human alpha-1,3/4-fucosyltransferase in the pit cell lineage of FVB/N mouse stomach results in production of Lewis b-containing glycoconjugates: a potential transgenic mouse model for studying Helicobacter pylori infection. Proc. Natl. Acad. Sci. USA. 1995;92:1515–1519. [PMC free article: PMC42550] [PubMed: 7878011]
- 104.
- Falk P., Roth K. A., Borén T., Westblom T. U., Gordon J. I., Normark S. An in vitro adherence assay reveals that Helicobacter pylori exhibits cell lineage-specific tropism in the human gastric epithelium. Proc. Acad. Natl. Sci. USA. 1993;90:2035–2039. [PMC free article: PMC46015] [PubMed: 8383333]
- 105.
- Falkow S. Bacterial entry into eukaryotic cells. Cell. 1991;65:1099–1102. [PubMed: 1905978]
- 106.
- Falkow S. What is a pathogen? ASM News. 1997;63:359–365.
- 107.
- Fan X., Crowe S. E., Behar S., Gunasena H., Ye G., Haeberle H., Van Houten N., Gourley W. K., Ernst P. B., Reyes V. E. The effect of class II major histocompatibility complex expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: a mechanism for T helper cell type I-mediated damage. J. Exp. Med. 1998;187:1659–1669. [PMC free article: PMC2212295] [PubMed: 9584144]
- 108.
- Fauchère J.-L., Blaser M. J. Adherence of Helicobacter pylori cells and their surface components to HeLa cell membranes. Microb. Pathog. 1990;9:427–439. [PubMed: 2097496]
- 109.
- Fine K. D., Lee E. L. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology. 1998;114:29–36. [PubMed: 9428215]
- 110.
- Fox J. G., Dangler C. A., Taylor N. S., King A., Koh T. J., Wang T. C. High-salt diet induces gastric epithelial hyperplasia and parietal cell loss, and enhances Helicobacter pylori colonization in C57BL/6 mice. Cancer Res. 1999;59:4823–4828. [PubMed: 10519391]
- 111.
- Foynes S., N., Dorrell, Ward S. J., Stabler R. A., McColm A. A., Rycroft A. N., Wren B. W. Helicobacter pylori possesses two CheY response regulators and a histidine kinase sensor, CheA, which are essential for chemotaxis and colonization of the gastric mucosa. Infect. Immun. 2000;68:2016–2023. [PMC free article: PMC97381] [PubMed: 10722597]
- 112.
- Foynes S., N., Dorrell, Ward S. J., Zhang Z. W., McColm A. A., Farthing M. J. G., Wren B. W. Functional analysis of the roles of FliQ and FlhB in flagellar expression in Helicobacter pylori. FEMS Microbiol. Lett. 1999;174:33–39. [PubMed: 10234819]
- 113.
- Fraser A. G., Scragg R., Metcalf P., McCullough S., Yeates N. J. Prevalence of Helicobacter pylori infection in different ethnic groups in New Zealand children and adults. Aust. N. Z. J. Med. 1996;26:646–651. [PubMed: 8958359]
- 114.
- Fu S., Ramaanujam K. S., Wong A., Fantry G. T., Drachenberg C. B., James S. J., Meltzer S. J., Wilson K. T. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology. 1999;116:1319–1329. [PubMed: 10348815]
- 115.
- Ganga-Zandzou P. S., Michaud L., Vincent P., Husson M. O., Wizla-Derambure N., Delassalle E. M., Turck D., Gottrand F. Natural outcome of Helicobacter pylori infection in asymptomatic children: a two-year follow-up study. Pediatrics. 1999;104:216–221. [PubMed: 10428997]
- 116.
- Geis G., Suerbaum S., Forsthoff B., Leying H., Opferkuch W. Ultrastructure and biochemical studies of the flagellar sheath of Helicobacter pylori. J. Med. Microbiol. 1993;38:371–377. [PubMed: 8487294]
- 117.
- Genta R. M., Gürer I. E., Graham D. Y., Krishnan B., Segura A. M., Gutierrez O., Kim J. G., Burchette, Jr J. L. Adherence of Helicobacter pylori to areas of incomplete intestinal metaplasia in the gastric mucosa. Gastroenterology. 1996;111:1206–1211. [PubMed: 8898634]
- 118.
- Gerhard M., Lehn N., Neumayer N., Borén T., Rad R., Schepp W., Miehlke S., Classen M., Prinz C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc. Natl. Acad. Sci. USA. 1999;96:12778–12783. [PMC free article: PMC23096] [PubMed: 10535999]
- 119.
- Ghiara P., Marchetti M., Blaser M. J., Tummuru M. K., Cover T. L., Segal E. D., Tompkins L. S., Rappuoli R. Role of the Helicobacter pylori virulence factors vacuolating cytotoxin, CagA, and urease in a mouse model of disease. Infect. Immun. 1995;63:4154–4160. [PMC free article: PMC173584] [PubMed: 7558333]
- 120.
- Ghiara P., Rossi M., Marchetti M., Di Tommaso A., Vindigni C., Ciampolini F., Covacci A., Telford J. L., De Magistris M. T., Pizza M., Rappuoli R., Del Giudice G. Therapeutic intragastric vaccination against Helicobacter pylori in mice eradicates an otherwise chronic infection and confers protection against reinfection. Infect. Immun. 1997;65:4996–5002. [PMC free article: PMC175721] [PubMed: 9393788]
- 121.
- Goh K. L. Prevalence of and risk factors for Helicobacter pylori infection in a multi-racial dyspeptic Malaysian population undergoing endoscopy. J. Gastroenterol. Hepatol. 1997;12:S29–S35. [PubMed: 9195409]
- 122.
- Goh K. L., Parasakthi N., Ong K. K. Prevalence of Helicobacter pylori infection in endoscopy and non-endoscopy personnel: results of field survey with serology and 14C-urea breath test. Am. J. Gastroenterol. 1996;91:268–270. [PubMed: 8607491]
- 123.
- Gold B. D., Dytoc M., Huesca M., Philpott D., Kuksis A., Czinn S., Lingwood C. A., Sherman P. M. Comparison of Helicobacter mustelae and Helicobacter pylori adhesion to eukaryotic cells in vitro. Gastroenterology. 1995;109:692–700. [PubMed: 7657097]
- 124.
- Gold B. D., Huesca M., Sherman P. M., Lingwood C. A. Helicobacter mustelae and Helicobacter pylori bind to common lipid receptors in vitro. Infect. Immun. 1993;61:2632–2638. [PMC free article: PMC280894] [PubMed: 8500901]
- 125.
- Goodwin C. S., Armstrong J. A., Marshall B. J. Campylobacter pyloridis, gastritis, and peptic ulceration. J. Clin. Pathol. 1986;39:353–365. [PMC free article: PMC499829] [PubMed: 3517070]
- 126.
- Graham D. Y., Anderson S. Y., Lang T. Garlic or jalapeno peppers for treatment of Helicobacter pylori infection. Am. J. Gastroenterol. 1999;94:1200–1202. [PubMed: 10235193]
- 127.
- Graham D. Y., Opekun A., Lew G. M., Evans, Jr D. J., Klein P. D., Evans D. G. Ablation of exaggerated meal-stimulated gastrin release in duodenal ulcer patients after clearance of Helicobacter (Campylobacter) pylori infection. Am. J. Gastroenterol. 1990;85:394–398. [PubMed: 2327380]
- 128.
- Guruge J. L., Falk P. G., Lorenz R. G., Dans M., Wirth H.-P., Blaser M. J., Berg D. E., Gordon J. I. Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc. Natl. Acad. Sci. USA. 1998;95:3925–3930. [PMC free article: PMC19939] [PubMed: 9520469]
- 129.
- Hansen P. S., Go M. F., Varming K., Andersen L. P., Graham D. Y., Nielsen H. Proinflammatory activation of neutrophils and monocytes by Helicobacter pylori is not associated with cagA, vacA or picB genotypes. APMIS. 1999;107:1117–1123. [PubMed: 10660142]
- 130.
- Hata Y., Kita T., Murakami M. Bovine milk inhibits both adhesion of Helicobacter pylori to sulfatide and Helicobacter pylori-induced vacuolation of Vero cells. Dig. Dis. Sci. 1999;44:1696–1702. [PubMed: 10492155]
- 131.
- Hayashi S., Sugiyama T., Amano K.-I., Isogai H., Isogai E., Alhara M., Kikuchi M., Asaka M., Yokota K., Oguma K., Fugii N., Hirai Y. Effect of rebamipide, a novel antiulcer agent, on Helicobacter pylori adhesion to gastric epithelial cells. Antimicrob. Agents Chemother. 1998;42:1895–1899. [PMC free article: PMC105706] [PubMed: 9687380]
- 132.
- Hayashi S., Sugiyama T., Asaka M., Yokota K., Oguma K., Hirai Y. Modification of Helicobacter pylori adhesion to human gastric epithelial cells by antiadhesion agents. Dig. Dis. Sci. 1998;43(Suppl.):56S–60S. [PubMed: 9753227]
- 133.
- Hayashi S., Sugiyama T., Yachi A., Yokota K., Hirai Y., Oguma K., Fugii N. Effect of ecabet sodium on Helicobacter pylori adhesion to gastric epithelial cells. J. Gastroenterol. 1997;32:593–597. [PubMed: 9349983]
- 134.
- Hazell S. L., Lee A., Brady L., Hennessy W. Campylobacter pyloridis and gastritis: association with intracellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. J. Infect. Dis. 1986;153:658–663. [PubMed: 3950447]
- 135.
- Hegarty J. P., Dowd M. T., Baker K. H. Occurrence of Helicobacter pylori in surface water in the United States. J. Appl. Microbiol. 1999;87:697–701. [PubMed: 10594710]
- 136.
- Hemalatha S. G., Drumm B., Sherman P. Adherence of Helicobacter pylori to human gastric epithelial cells in vitro. J. Med. Microbiol. 1991;35:197–202. [PubMed: 1941988]
- 137.
- Heneghan M. A., McCarthy C. F., Moran A. P. Relationship of blood group determinants on Helicobacter pylori lipopolysaccharide with host Lewis phenotype and inflammatory response. Infect. Immun. 2000;68:937–941. [PMC free article: PMC97226] [PubMed: 10639467]
- 138.
- Henry S. M., Benny A. G., Woodfield D. G. Investigation of Lewis phenotypes in Polynesians: evidence of a weak secretor phenotype. Vox Sang. 1990;58:61–66. [PubMed: 2316213]
- 139.
- Henry S. M., Woodfield D. G., Samuelsson B. E., Oriol R. Plasma and red-cell glycolipid patterns of Le(a + b +) and Le(a + b −) Polynesians as further evidence of the weak secretor gene Se(w). Vox Sang. 1993;65:62–69. [PubMed: 8362517]
- 140.
- Hessey S. J., Spencer J., Wyatt J. I., Sobala G., Rathbone B. J., Axon A. T., Dixon M. F. Bacterial adhesion and disease activity in Helicobacter associated chronic gastritis. Gut. 1990;31:134–138. [PMC free article: PMC1378366] [PubMed: 2311970]
- 141.
- Hirmo S., Artursson E., Puu G., Wadström T., Nilsson B. Helicobacter pylori interactions with human gastric mucin studied with a resonant mirror biosensor. J. Microbiol. Methods. 1999;37:177–182. [PubMed: 10445316]
- 142.
- Hirmo S., Kelm S., Iwersen M., Hotta K., Goso Y., Ishihara K., Suguri T., Morita M., Wadström T. Inhibition of Helicobacter pylori sialic acid-specific haemagglutination by human gastrointestinal mucins and milk glycoproteins. FEMS Immunol. Med. Microbiol. 1998;20:275–281. [PMC free article: PMC7135180] [PubMed: 9626932]
- 143.
- Hirmo S., Kelm S., Wadström T., Schauer R. Lack of evidence for sialdase activity in Helicobacter pylori. FEMS Immunol. Med. Microbiol. 1997;17:67–72. [PubMed: 9061351]
- 144.
- Hirmo S., Utt M., Ringner M., Wadström T. Inhibition of heparan sulphate and other glycosaminoglycans binding to Helicobacter pylori by various polysulphated carbohydrates. FEMS Immunol Med. Microbiol. 1995;10:301–306. [PubMed: 7539671]
- 145.
- Hopkins R. J., Morris J. G., Papadimitriou J. C., Drachenberg C., Smoot D. T., James S. P., Panigrahi P. Loss of Helicobacter pylori hemagglutination with serial laboratory passage and correlation of hemagglutination with gastric epithelial cell adherence. Pathobiology. 1996;64:247–254. [PubMed: 9068007]
- 146.
- Houghton J., Korah R. M., Condon M. R., Kim K. H. Apoptosis in Helicobacter pylori-associated gastric and duodenal ulcer disease is mediated via the Fas antigen pathway. Dig. Dis. Sci. 1999;44:465–473. [PubMed: 10080136]
- 147.
- Huang J., Keeling P. W. N., Smith C. J. Identification of erythrocyte-binding antigens in Helicobacter pylori. J. Gen. Microbiol. 1992;138:1503–1513. [PubMed: 1512579]
- 148.
- Huang J., O'Toole P. W., Doig P., Trust T. J. Stimulation of interleukin-8 production in epithelial cell lines by Helicobacter pylori. Infect. Immun. 1995;63:1732–1738. [PMC free article: PMC173217] [PubMed: 7729879]
- 149.
- Huang J., Smyth C. J., Kennedy N. P., Arbuthnott J. P., Keeling P. W. N. Haemagglutinating activity of Campylobacter pylori. FEMS Microbiol. Lett. 1988;56:109–112.
- 150.
- Huesca M., Borgia S., Hoffman P., Lingwood C. A. Acidic pH changes receptor binding specificity of Helicobacter pylori: a binary adhesion model in which surface heat shock (stress) proteins mediate sulfatide recognition in gastric colonization. Infect. Immun. 1996;64:2643–2648. [PMC free article: PMC174121] [PubMed: 8698490]
- 151.
- Huesca M., Goodwin A., Bhagwansingh A., Hoffman P., Lingwood C. A. Characterization of an acidic-pH-inducible stress protein (hsp70), a putative sulfatide binding adhesin, from Helicobacter pylori. Infect. Immun. 1998;66:4061–4067. [PMC free article: PMC108486] [PubMed: 9712748]
- 152.
- Hulten K., Han S. W., Enroth H., Klein P. D., Opekun A. R., Gilman R. H., Evans D. G., Engstrand L., Graham D. Y., El-Zaatari F. A. Helicobacter pylori in the drinking water in Peru. Gastroenterology. 1996;110:1031–1035. [PubMed: 8612990]
- 153.
- Husson M. O., Legrand D., Spik G., Leclerc H. Iron acquisition by Helicobacter pylori: importance of human lactoferrin. Infect Immun. 1993;61:2694–2697. [PMC free article: PMC280902] [PubMed: 8500909]
- 154.
- Ilver, D., A. Arnqvist, J. Ögren, and T. Borén. 1997. Adherence of Helicobacter pylori: mechanisms and relevance to pathogenesis, p. 16–27. In A. P. Moran and C. A. O'Morain (ed.), Pathogenesis and Host Response in Helicobacter pylori Infections. Normed Verlag, Englewood, N.J.
- 155.
- Ilver D., Arnqvist A., Ögren J., Frick I.-M., Kersulyte D., Incecik E. T., Berg D. E., Covacci A., Engstrand L., Borén T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. [PubMed: 9430586]
- 156.
- Isogai H., Isogai E., Hayashi S., Kimura K., Kubota T., Fujii N., Oguma K. Experimental Helicobacter pylori infection in association with other bacteria. Microbiol. Immunol. 1997;41:361–365. [PubMed: 9159411]
- 157.
- Ito Y., Hongo A., Kinoshita M., Tamaki H. Mechanism of antiurease action by the anti-ulcer drug ecabet sodium. Biol. Pharm. Bull. 1995;18:850–853. [PubMed: 7550119]
- 158.
- Ito Y., Shibata K., Hongo A., Kinoshita M. Ecabet sodium, a locally acting antiulcer drug, inhibits urease activity of Helicobacter pylori. Eur. J. Pharmacol. 1998;345:193–198. [PubMed: 9600637]
- 159.
- Jack R. W., Tagg J. R., Ray B. Bacteriocins of gram-positive bacteria. Microbiol. Rev. 1995;59:171–200. [PMC free article: PMC239359] [PubMed: 7603408]
- 160.
- Johansson L., Johansson P., Miller-Podraza H. Neu5Ac—3Gal is part of the Helicobacter pylori binding epitope in polyglycosylceramides of human erythrocytes. Eur. J. Biochem. 1999;266:559–565. [PubMed: 10561598]
- 161.
- Johansson L., Karlsson K.-A. Selective binding by Helicobacter pylori of leucocyte gangliosides with 3-linked sialic acid, as identified by a new approach of linkage analysis. Glycoconj. J. 1998;15:713–721. [PubMed: 9881777]
- 162.
- Jones A. C., Logan R. P. H., Foynes S., Cockayne A., Wren B. W., Penn C. W. A flagellar sheath protein of Helicobacter pylori is identical to HpaA, a putative N-acetyl-neuraminyllactose-binding hemagglutinin, but is not an adhesin for AGS cells. J. Bacteriol. 1997;179:5643–5647. [PMC free article: PMC179448] [PubMed: 9287032]
- 163.
- Jones N. L., Day A. S., Jennings H. A., Sherman P. M. Helicobacter pylori induces gastric epithelial cell apoptosis in association with increased Fas receptor expression. Infect. Immun. 1999;67:4237–4242. [PMC free article: PMC96730] [PubMed: 10417197]
- 164.
- Jonkers D., van den Broek E., van Dooren I., Thijs C., Dorant E., Hageman G., Stobberingh E. Antibacterial effect of garlic and omeprazole on Helicobacter pylori. J. Antimicrob. Chemother. 1999;43:837–839. [PubMed: 10404325]
- 165.
- Jyonouchi H., Sun S., Mizokami M., Gross M. D. Effects of various carotenoids on cloned, effector-stage T-helper cell activity. Nutr. Cancer. 1996;26:313–324. [PubMed: 8910913]
- 166.
- Kabir A. M. A., Aiba Y., Takagi A., Kamiya S., Miwa T., Koga Y. Prevention of Helicobacter pylori infection by lactobacilli in a gnotobiotic murine model. Gut. 1997;41:49–55. [PMC free article: PMC1027227] [PubMed: 9274471]
- 167.
- Kamisago S., Iwamori M., Tai T., Mitamura K., Yazaki Y., Sugano K. Role of sulfatides in adhesion of Helicobacter pylori to gastric cancer cells. Infect. Immun. 1996;64:624–628. [PMC free article: PMC173811] [PubMed: 8550217]
- 168.
- Kamiya S., Yamaguchi H., Osaki T., Taguchi H., Fukuda M., Kawakami H., Hirano H. Effect of an aluminum hydroxide-magnesium hydroxide combination drug on adhesion, IL-8 inducibility, and expression of Hsp60 by Helicobacter pylori. Scand. J. Gastroenterol. 1999;34:663–670. [PubMed: 10466876]
- 169.
- Karita M., Li Q., Cantero D., Okita K. Establishment of a small animal model for human Helicobacter pylori infection using germ-free mouse. Am. J. Gastroenterol. 1994;89:208–213. [PubMed: 8304305]
- 170.
- Kawano S., Tsujii M., Nagano K., Ogihara T., Tanimura H., Hayashi N., Ito T., Sato N., Kamada T., Tamura K., Tanaka M. Different effect of Helicobacter pylori on the human gastric antral and body mucosal intracellular mucin. Scand. J. Gastroenterol. 1990;25:997–1003. [PubMed: 1702231]
- 171.
- Kelly S. M., Pitcher M. C., Farmery S. M., Gibson G. R. Isolation of Helicobacter pylori from feces of patients with dyspepsia in the United Kingdom. Gastroenterology. 1994;107:1671–1674. [PubMed: 7958677]
- 172.
- Kinoshita M., Endo M., Yasoshima A., Saito N., Yamasaki K., Chishima S., Narita H. Ecabet sodium, a novel locally-acting anti-ulcer agent, protects the integrity of the gastric mucosal gel layer from pepsin-induced disruption in the rat. Aliment. Pharmacol. Ther. 1999;13:687–694. [PubMed: 10233194]
- 173.
- Knutton S., Lloyd D. R., McNeish A. S. Adhesion of enteropathogenic E. coli to human intestinal epithelial enterocytes and cultured human intestinal mucosa. Infect. Immun. 1987;55:67–69. [PMC free article: PMC260281] [PubMed: 3539808]
- 174.
- Kobayashi Y., Okazaki K.-I., Murakami K. Adhesion of Helicobacter pylori to gastric epithelial cells in primary cultures obtained from stomachs of various animals. Infect. Immun. 1993;61:4058–4063. [PMC free article: PMC281124] [PubMed: 7691743]
- 175.
- Kohda K., Tanaka K., Aiba Y., Yasuda M., Miwa T., Koga Y. Role of apoptosis induced by Helicobacter pylori infection in the development of duodenal ulcer. Gut. 1999;44:456–462. [PMC free article: PMC1727459] [PubMed: 10075950]
- 176.
- Kono S., Hirohata T. Nutrition and stomach cancer. Cancer Causes Control. 1996;7:41–55. [PubMed: 8850434]
- 177.
- Konturek J. W., Gillessen A., Konturek S. J., Domschke W. Eradication of Helicobacter pylori restores the inhibitory effect of cholecystokinin on postprandial gastrin release in duodenal ulcer patients. Gut. 1995;37:482–487. [PMC free article: PMC1382897] [PubMed: 7489932]
- 178.
- Konturek P. C., Pierzchalski P., Konturek S. J., Meixner H., Faller G., Kirchner T., Hahn E. G. Helicobacter pylori induces apoptosis in gastric mucosa through an up regulation of Bax expression in humans. Scand. J. Gastroenterol. 1999;34:375–383. [PubMed: 10365897]
- 179.
- Kostrzynska M., Betts J. D., Austin J. W., Trust T. J. Identification, characterization, and spatial localization of two flagellin species in Helicobacter pylori flagella. J. Bacteriol. 1991;173:937–946. [PMC free article: PMC207209] [PubMed: 1704004]
- 180.
- Lambert J. R. Pharmacology of bismuth-containing compounds. Rev. Infect. Dis. 1991;13:S691–S695. [PubMed: 1925310]
- 181.
- Lanciers S., Despinasse B., Mehta D. I., Blecker U. Increased susceptibility to Helicobacter pylori infection in pregnancy. Infect. Dis. Obstet. Gynecol. 1999;7:195–198. [PMC free article: PMC1784742] [PubMed: 10449268]
- 182.
- Langton S. R., Cesareo S. D. Helicobacter pylori associated phospholipase A2 activity: a factor in peptic ulcer production? J. Clin. Pathol. 1992;45:221–224. [PMC free article: PMC495476] [PubMed: 1556229]
- 183.
- Lee A., Dixon M. F., Danon S. J., Kuipers E., Megraud F., Larsson H., Mellgard B. Local acid production and Helicobacter pylori: a unifying hypothesis of gastroduodenal disease. Eur. J. Gastroenterol. 1995;7:461–465. [PubMed: 7614109]
- 184.
- Lelwala-Guruge J., Ascencio F., Kreger A. S., Ljungh Å., Wadström T. Isolation of a sialic acid-specific surface haemagglutinin of Helicobacter pylori strain NCTC 11637. Zentralbl. Bakteriol. 1993;280:93–106. [PubMed: 7506596]
- 185.
- Lelwala-Guruge J., Ascencio F., Ljungh Å., Wadström T. Rapid detection and characterization of sialic acid-specific lectings of Helicobacter pylori. APMIS. 1993;101:695–702. [PubMed: 7694599]
- 186.
- Lelwala-Guruge J., Ljungh Å., Wadström T. Haemagglutination patterns of Helicobacter pylori. APMIS. 1992;100:908–913. [PubMed: 1445697]
- 187.
- Leung W. K., Siu K. L., Kwok C. K., Chan S. Y., Sung R., Sung J. J. Isolation of Helicobacter pylori from vomitus in children and its implication in gastro-oral transmission. Am. J. Gastroenterol. 1999;94:2881–2884. [PubMed: 10520837]
- 188.
- Leying H., Suerbaum S., Geis G., Haas R. Cloning and genetic characterization of Helicobacter pylori flagellin gene. Mol. Microbiol. 1992;6:2863–2874. [PubMed: 1435261]
- 189.
- Li C., Ha T., Ferguson, Jr D. A., Chi D. S., Zhao R., Patel N. R., Krishnaswamy G., Thomas E. A newly developed PCR assay of H. pylori in gastric biopsy, saliva, and feces. Evidence of high prevalence of H. pylori in saliva supports oral transmission. Dig. Dis. Sci. 1996;41:2142–2149. [PubMed: 8943965]
- 190.
- Li S. D., Kersulyte D., Lindley I. J. D., Neelam B., Berg D. E., Crabtree J. E. Multiple genes in the left half of the cag pathogenicity island of Helicobacter pylori are required for tyrosine kinase-dependent transcription of interleukin-8 in gastric epithelial cells. Infect. Immun. 1999;67:3893–3899. [PMC free article: PMC96669] [PubMed: 10417153]
- 191.
- Lingwood C. A., Huesca M., Kuksis A. The glycerolipid receptor for Helicobacter pylori (and Exoenzyme S) is phosphatidylethanolamine. Infect. Immun. 1992;60:2470–2474. [PMC free article: PMC257183] [PubMed: 1587616]
- 192.
- Lingwood C. A., Pellizzari A., Law H., Sherman P., Drumm B. Gastric glycerolipid as a receptor for Campylobacter pylori. Lancet. 1989;2:238–240. [PubMed: 2569053]
- 193.
- Lingwood C. A., Wasfy G., Han H., Huesca M. Receptor affinity purification of a lipid-binding adhesin from Helicobacter pylori. Infect. Immun. 1993;61:2474–2478. [PMC free article: PMC280871] [PubMed: 8500882]
- 194.
- Logan R. P. H., Robins A., Turner G. A., Cockayne A., Borriello S. P., Hawkey C. J. A novel flow cytometric assay for quantitating adherence of Helicobacter pylori to gastric epithelial cells. J. Immunol. Methods. 1998;213:19–30. [PubMed: 9671122]
- 195.
- Luke C. J., Penn C. W. Identification of a 29 kDa flagellar sheath protein in Helicobacter pylori using a murine monoclonal antibody. Microbiology. 1995;141:597–604. [PubMed: 7711897]
- 196.
- Macartney J. C. Lectin histochemistry of galactose and N-acetyl-galactosamine glycoconjugates in normal gastric mucosa and gastric cancer and the relationship with ABO and secretor status. J. Pathol. 1986;150:135–144. [PubMed: 3794866]
- 197.
- Magnani J. L., Nilsson B., Brockhaus M., Zopf D., Steplewski Z., Koprowski H., Ginsburg V. A monoclonal antibody-defined antigen associated with gastrointestinal cancer in a ganglioside containing sialylated lacto-N-fuco-pentaose. J. Biol. Chem. 1982;257:14365–14369. [PubMed: 7142214]
- 198.
- Mahalanabis D., Rahman M. M., Sarker S. A., Bardhan P. K., Hildebrand P., Beglinger C., Gyr K. Helicobacter pylori infection in the young in Bangladesh: prevalence, socioeconomic and nutritional aspects. Int. J. Epidemiol. 1996;25:894–898. [PubMed: 8921472]
- 199.
- Mai U. E. H., Perez-Perez G. I., Wahl L. M., Wahl S. M., Blaser M. J., Smith P. D. Soluble surface proteins from Helicobacter pylori activate monocytes/macrophages by lipopolysaccharide-independent mechanism. J. Clin. Invest. 1991;87:894–900. [PMC free article: PMC329879] [PubMed: 1847939]
- 200.
- Malaty H. M., Engstrand L., Pedersen N. L., Graham D. Y. Helicobacter pylori infection: genetic and environmental influences. A study of twins. Ann. Intern. Med. 1994;120:982–986. [PubMed: 8185146]
- 201.
- Malaty H. M., Graham D. Y., Klein P. D., Evans D. G., Adam E., Evans D. J. Transmission of Helicobacter pylori infection. Studies in families of healthy individuals. Scand. J. Gastroenterol. 1991;26:927–932. [PubMed: 1947784]
- 202.
- Manes G., Dominguez-Munoz J. E., Uomo G., Labenz J., Hackelsberger A., Malfertheiner P. Increased risk for Helicobacter pylori recurrence by continuous acid suppression: a randomized controlled study. Ital. J. Gastroenterol. Hepatol. 1998;30:28–33. [PubMed: 9615260]
- 203.
- Mankoski R., Hoepf T., Krakowka S., Eaton K. A. flaA mRNA transcription level correlates with Helicobacter pylori colonisation efficiency in gnotobiotic piglets. J. Med. Microbiol. 1999;48:395–399. [PubMed: 10509483]
- 204.
- Mannick E. E., Bravo L. E., Zarama G., Realpe J. L., Zhang X. J., Ruiz B., Fontham E. T., Mera R., Miller M. J., Correa P. Inducible nitric oxide synthase, nitrotyrosine, and apoptosis in Helicobacter pylori gastritis: effect of antibiotics and antioxidants. Cancer Res. 1996;56:3238–3243. [PubMed: 8764115]
- 205.
- Mathai E., Arora A., Cafferkey M., Keane C. T., O'Morain C. The effect of bile acids on the growth and adherence of Helicobacter pylori. Aliment. Pharmacol. Ther. 1991;5:653–658. [PubMed: 1782308]
- 206.
- Matysiak-Budnik T., Karkkainen P., Methuen T., Roine R. P., Salaspuro M. Inhibition of gastric cell proliferation by acetaldehyde. J. Pathol. 1995;177:317–322. [PubMed: 8551395]
- 207.
- Matysiak-Budnik T., Megraud F. Epidemiology of Helicobacter pylori infection with special reference to professional risk. J. Physiol. Pharmacol. 1997;48(Suppl. 4):3–17. [PubMed: 9440051]
- 208.
- McCarthy C. J., Crofford L. J., Greeneson J., Scheiman J. M. Cyclooxygenase-2 expression in gastric antral mucosa before and after eradication of Helicobacter pylori infection. Am. J. Gastroenterol. 1999;94:1218–1223. [PubMed: 10235197]
- 209.
- McColl, K. L. E., E. El-Omar, and D. Gillen. 1997. Helicobacter pylori-induced alterations in gastric acid secretion, p. 119–127. In A. P. Moran and C. A. O'Morain (ed.), Pathogenesis and Host Response in Helicobacter pylori Infections. Normed Verlag, Englewood, N.J.
- 210.
- McGee D. J., Radcliff F. J., Mendz G. L., Ferrero R. L., Mobley H. L. Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease activity. J. Bacteriol. 1999;181:7314–7322. [PMC free article: PMC103695] [PubMed: 10572136]
- 211.
- Mégraud F., Trimoulet P., Lamouliatte H., Boyanova L. Bactericidal effect of amoxicillin on Helicobacter pylori in an in vitro model using epithelial cells. Antimicrob. Agents Chemother. 1991;35:869–872. [PMC free article: PMC245121] [PubMed: 1854168]
- 212.
- Mendz G. L., Hazell S. L. Amino acid utilization by Helicobacter pylori. Int. J. Biochem. Cell Biol. 1995;27:1085–1093. [PubMed: 7496998]
- 213.
- Mentis A., Tzouvelekis L., Spiliadis C., Blackwell C. C., Weir D. M. Inhibition of Helicobacter pylori haemagglutination activity by human salivary mucins. FEMS Microbiol. Immunol. 1990;64:125–128. [PubMed: 2257167]
- 214.
- Michetti P., Dorta G., Wiesel P. H., Brassart D., Verdu E., Herranz M., Felley C., Porta N., Rouvet M., Blum A. L., Corthesy-Theulaz I. Effect of whey-based culture supernatant of Lactobacillus acidophilus (johnsonii) La1 on Helicobacter pylori infection in humans. Digestion. 1999;60:203–209. [PubMed: 10343133]
- 215.
- Micots I., Augeron C., Laboisse C. L., Muzeau F., Mégraud F. Mucin exocytosis: a major target for Helicobacter pylori. J. Clin. Pathol. 1993;46:241–245. [PMC free article: PMC501178] [PubMed: 8463418]
- 216.
- Miller-Podraza H., Bergström J., Milh M. A., Karlsson K.-A. Recognition of glycoconjugates by Helicobacter pylori. Comparison of two sialic acid-dependent specificities based on haemmagglutination and binding to human erythrocyte glycoconjugates. Glycoconj. J. 1997;14:467–471. [PubMed: 9249144]
- 217.
- Miller-Podraza H., Bergström J., Teneberg S., Milh M. A., Longard M., Olsson B.-M., Uggla L., Karlsson K.-A. Helicobacter pylori and neutrophils: sialic-acid dependent binding to various isolated glycoconjugates. Infect. Immun. 1999;67:6309–6313. [PMC free article: PMC97034] [PubMed: 10569742]
- 218.
- Miller-Podraza H., Milh M. A., Bergström J., Karlsson K.-A. Recognition of glycoconjugates by Helicobacter pylori: an apparently high-affinity binding of human polyglycosylceramides, a second sialic acid-based specificity. Glycoconj. J. 1996;13:453–460. [PubMed: 8781976]
- 219.
- Mizoguchi H., Fujioka T., Kishi K., Nishizono A., Kodama R., Nasu M. Diversity in protein synthesis and viability of Helicobacter pylori coccoid forms in response to various stimuli. Infect. Immun. 1998;66:5555–5560. [PMC free article: PMC108699] [PubMed: 9784573]
- 220.
- Mizoguchi H., Fujioka T., Nasu M. Evidence for viability of coccoid forms of Helicobacter pylori. J. Gastroenterol. 1999;34(Suppl. 11):32–36. [PubMed: 10616763]
- 221.
- Mohammadi M., Nedrud J., Redline R., Lycke N., Czinn S. J. Murine CD4 T-cell response to Helicobacter infection: TH1 cells enhance gastritis and TH2 cells reduce bacterial load. Gastroenterology. 1997;113:1848–1857. [PubMed: 9394724]
- 222.
- Monteiro M. A., Chan K. H., Rasko D. A., Taylor D. E., Zheng P. Y., Appelmelk B. J., Wirth H.-P., Yang M., Blaser M. J., Hynes S. O., Moran A. P., Perry M. B. Simultaneous expression of type 1 and type 2 Lewis blood group antigens by Helicobacter pylori lipopolysaccharides. Molecular mimicry between H. pylori lipopolysaccharides and human gastric epithelial cell surface glycoforms. J. Biol. Chem. 1998;273:11533–11543. [PubMed: 9565568]
- 223.
- Moran A. P., Kuusela P., Kosunen T. U. Interaction of Helicobacter pylori with extracellular matrix proteins. J. Appl. Bacteriol. 1993;57:187–189.
- 224.
- Moss S. F. Helicobacter pylori and apoptosis. Yale J. Biol. Med. 1998;71:53–61. [PMC free article: PMC2578891] [PubMed: 10378350]
- 225.
- Mysore J. V., Wigginton T., Simon P. M., Zopf D., Heman-Ackah L. M., Dubois A. Treatment of Helicobacter pylori infection in rhesus monkeys using a novel anti-adhesion compound. Gastroenterology. 1999;117:1316–1325. [PubMed: 10579973]
- 226.
- Nakamura H., Yoshiyama H., Takeuchi H., Mizote T., Okita K., Nakazawa T. Urease plays an important role in the chemotactic motility of Helicobacter pylori in a viscous environment. Infect. Immun. 1998;66:4832–4837. [PMC free article: PMC108597] [PubMed: 9746586]
- 227.
- Nakayama R., Kumagai H., Tochikura T. Leakage of glutathione from bacterial cells caused by inhibition of gamma-glutamyltranspeptidase. Appl. Environ. Microbiol. 1984;47:653–657. [PMC free article: PMC239743] [PubMed: 6144289]
- 228.
- Nakazawa T., Ishibashi M., Konishi H., Takemoto T., Shigeeda M., Kochiyama T. Hemagglutinating activity of Campylobacter pylori. Infect. Immun. 1989;57:989–991. [PMC free article: PMC313210] [PubMed: 2917797]
- 229.
- Namavar F., Sparrius M., Veerman E. C. I., Appelmelk B. J., Vandenbroucke-Grauls C. M. J. E. Neutrophil-activating protein mediates adhesion of Helicobacter pylori to sulfated carbohydrates on high-molecular-weight salivary mucin. Infect. Immun. 1998;66:444–447. [PMC free article: PMC107925] [PubMed: 9453593]
- 230.
- Neman-Simha V., Mégraud F. In vitro model for Campylobacter pylori adherence properties. Infect. Immun. 1988;56:3329–3333. [PMC free article: PMC259747] [PubMed: 3182085]
- 231.
- Nilius M., Bode G., Lehnhardt G., Malfertheiner P. In vitro inhibition of Helicobacter pylori urease: biochemical and ultrastructural analysis. Eur. J. Clin. Invest. 1991;21:551–557. [PubMed: 1752295]
- 232.
- Nilius M., Malfertheiner P. Helicobacter pylori enzymes. Aliment. Pharmacol. Ther. 1996;10(Suppl. 1):65–71. [PubMed: 8730261]
- 233.
- Nilsson H. O., Taneera J., Castedal M., Glatz E., Olsson R., Wadstrom T. Identification of Helicobacter pylori and other helicobacter species by PCR, hybridization, and partial DNA sequencing in human liver samples from patients with primary sclerosing cholangitis or primary biliary cirrhosis. J. Clin. Microbiol. 2000;38:1072–1076. [PMC free article: PMC86342] [PubMed: 10698999]
- 234.
- Nishihara K., Nozawa Y., Nomura S., Kitazato K., Miyake H. Analysis of Helicobacter pylori binding site on HEp-2 cells and three cell lines from gastric carcinoma. Fundam. Clin. Pharmacol. 1999;13:555–561. [PubMed: 10520728]
- 235.
- Noach L. A., Rolf T. M., Tytgat G. N. J. Electron microscopic study of association between Helicobacter pylori and gastric and duodenal mucosa. J. Clin. Pathol. 1994;47:699–704. [PMC free article: PMC502139] [PubMed: 7962619]
- 236.
- Nordman H., Boren T., Davies J. R., Engstrand L., Carlstedt I. pH-dependent binding of Helicobacter pylori to pig gastric mucins. FEMS Immunol. Med. Microbiol. 1999;24:174–181. [PubMed: 10378417]
- 237.
- Obst B., Wagner S., Sewing K. F., Beil W. Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis. 2000;21:1111–1115. [PubMed: 10836997]
- 238.
- Odenbreit S., Püls J., Sedlmaier B., Gerland E., Fischer W., Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. [PubMed: 10688800]
- 239.
- Odenbreit S., Till M., Haas R. Optimized BlaM-transposon shuttle mutagenesis of Helicobacter pylori allows the identification of novel genetic loci involved in bacterial virulence. Mol. Microbiol. 1996;20:361–373. [PubMed: 8733234]
- 240.
- Odenbreit S., Till M., Hofreuter D., Faller G., Haas R. Genetic and functional characterization of the alpAB gene locus essential for the adhesion of Helicobacter pylori to human gastric tissue. Mol. Microbiol. 1999;31:1537–1548. [PubMed: 10200971]
- 241.
- Odenbreit S., Wieland B., Haas R. Cloning and genetic characterization of Helicobacter pylori catalase and construction of a catalase-deficient mutant strain. J. Bacteriol. 1996;178:6960–6967. [PMC free article: PMC178599] [PubMed: 8955320]
- 242.
- O'Gara E. A., Hill D. J., Maslin D. J. Activities of garlic oil, garlic powder, and their diallyl constituents against Helicobacter pylori. Appl. Environ. Microbiol. 2000;66:2269–2273. [PMC free article: PMC101489] [PubMed: 10788416]
- 243.
- Ohkusa T., Takashimizu I., Fujiki K., Araki A., Ariake K., Shimoi K., Honda K., Enomoto Y., Sakurazawa T., Horiuchi T., Suzuki S., Ishii K., Ishikura T. Prospective evaluation of a new anti-ulcer agent, ecabet sodium, for the treatment of Helicobacter pylori infection. Aliment. Pharmacol. Ther. 1998;12:457–461. [PubMed: 9663726]
- 244.
- Osaki T., Yamagucki H., Taguchi H., Fukuda M., Kawakami H., Hirano H., Wantanabe S., Takagi A., Kamiya S. Establishment and characterisation of a monoclonal antibody to inhibit adhesion of Helicobacter pylori to gastric epithelial cells. J. Med. Microbiol. 1998;47:505–512. [PubMed: 9879969]
- 245.
- Osaki T., Yamagucki H., Taguchi H., Fukuda M., Kumada J., Ogata S., Kamiya S. Studies on the relationship between adhesive activity and haemagglutination by Helicobacter pylori. J. Med. Microbiol. 1997;46:117–121. [PubMed: 9060870]
- 246.
- O'Toole P. W., Janzon L., Doig P., Huang J., Kostrzynska M., Trust T. J. The putative neuraminyllactose-binding hemagglutinin HpaA of Helicobacter pylori CCUG 17874 is a lipoprotein. J. Bacteriol. 1995;177:6049–6057. [PMC free article: PMC177441] [PubMed: 7592366]
- 247.
- Ottlecz A., Romero J. J., Hazell S. L., Graham D. Y., Lichtenberger L. M. Phospholipase activity of Helicobacter pylori and its inhibition by bismuth salts. Biochemical and biophysical studies. Dig. Dis. Sci. 1993;38:2071–2080. [PubMed: 8223083]
- 248.
- Ottlecz A., Romero J. J., Lichtenberger L. M. Effect of ranitidine bismuth citrate on the phospholipase A2 activity of Naja naja venom and Helicobacter pylori: a biochemical analysis. Aliment. Pharmacol. Ther. 1999;13:875–881. [PubMed: 10383521]
- 249.
- Paradies, H. H. 1990. Bismuth—from the element to the tablet: the general chemistry of bismuth with relevance to pharmacy and medicine, p. 409–426. In P. Malfertheiner and H. Ditsehuneit (ed.), Helicobacter pylori, Gastritis, and Peptic Ulcer. Springer-Verlag, New York, N.Y.
- 250.
- Parsonnet J., Shmuely H., Haggerty T. Fecal and oral shedding of Helicobacter pylori from healthy infected adults. JAMA. 1999;282:2240–2245. [PubMed: 10605976]
- 251.
- Pasquler M. C., Vatier J., Poitevin C., Vallot T., Mignon M. Assessment of mucus glycoprotein erosion by measurement of sialic acid in gastric secretions: pathophysiologic and therapeutic aspects. J. Clin. Gastroent. 1991;13(Suppl. 1):S22–S31. [PubMed: 1940193]
- 252.
- Peach H. G., Bath N. E., Farish S. J. Helicobacter pylori infection: an added stressor on iron status of women in the community. Med. J. Aust. 1998;169:188–190. [PubMed: 9734575]
- 253.
- Peck B., Ortkamp M., Diehl K. D., Hundt E., Knapp B. Conservation, localization and expression of HopZ, a protein involved in adhesion of Helicobacter pylori. Nucleic Acids Res. 1999;27:3325–3333. [PMC free article: PMC148566] [PubMed: 10454640]
- 254.
- Petretti T., Schulze B., Schlag P. M., Kemmner W. Altered mRNA expression of glycosyltransferases in human gastric carcinomas. Biochim. Biophys. Acta. 1999;1428:209–218. [PubMed: 10434038]
- 255.
- Piotrowski J., Czajkowski A., Yotsumoto F., Slomiany A., Slomiany B. L. Helicobacter pylori lipopolysaccharide inhibition of gastric mucosal laminin receptor: effect of sulglycotide. Gen. Pharmacol. 1993;24:1467–1472. [PubMed: 8112522]
- 256.
- Piotrowski J., Majka J., Murty V. L. N., Czajkowski A., Slomiany A., Slomiany B. L. Inhibition of gastric mucosal mucin receptor by Helicobacter pylori lipopolysaccharide: effect of sulglycotide. Gen. Pharmacol. 1994;25:969–976. [PubMed: 7835646]
- 257.
- Piotrowski J., Slomainy A., Murty V. L. N., Fekete Z., Slomiany B. L. Inhibition of Helicobacter pylori colonization by sulfated gastric mucin. Biochem. Int. 1991;24:749–756. [PubMed: 1799374]
- 258.
- Piotrowski J., Yamaki K., Slomiany A., Slomiany B. L. Inhibition of gastric mucosal laminin receptor by Helicobacter pylori lipopolysaccharide: effect of sucralfate. Am. J. Gastroenterol. 1991;86:1756–1760. [PubMed: 1835819]
- 259.
- Ponzetto A., Pellicano R., Leone N., Cutufia M. A., Turrini F., Grigioni W. F., D'Errico A., Mortimer P., Rizzetto M., Silengo L. Helicobacter infection and cirrhosis in hepatitis C virus carriage: is it an innocent bystander or a troublemaker? Med. Hypotheses. 2000;54:275–277. [PubMed: 10790764]
- 260.
- Pope A. J., Toseland C. D., Rushant B., Richardson S., McVey M., Hills J. Effect of potent urease inhibitor, fluorofamide, on Helicobacter sp. in vivo and in vitro. Dig. Dis. Sci. 1998;43:109–119. [PubMed: 9508511]
- 261.
- Pruul H., Lee P. C., Goodwin C. S., McDonald P. J. Interaction of Campylobacter pyloridis with human immune defence mechanisms. J. Med. Microbiol. 1987;23:233–238. [PubMed: 3585959]
- 262.
- Rasko D. A., Wilson T. J., Zopf D., Taylor D. E. Lewis antigen expression and stability in Helicobacter pylori isolated from serial gastric biopsies. J. Infect. Dis. 2000;181:1089–1095. [PubMed: 10720535]
- 263.
- Rautelin H., Kihistrom E., Jurstrand M., Danielsson D. Adhesion to and invasion of HeLa cells by Helicobacter pylori. Int. J. Med. Microbiol. Virol. Parasitol. Infect. Dis. 1995;282:50–53. [PubMed: 7734829]
- 264.
- Regev A., Fraser G. M., Braun M., Maoz E., Leibovici L., Niv Y. Seroprevalence of Helicobacter pylori and length of stay in a nursing home. Helicobacter. 1999;4:89–93. [PubMed: 10382121]
- 265.
- Reina J., Alomar P. Campylobacter pylori's sensitivity to the bactericidal action of human serum. Enferm. Infecc. Microbiol. Clin. 1989;7:8–11. [PubMed: 2490652]
- 266.
- Reinhard J., Basset C., Holton J., Binks M., Youinou P., Vaira D. Image analysis method to assess adhesion of Helicobacter pylori to gastric epithelium using confocal laser scanning microscopy. J. Microbiol. Meth. 2000;39:179–187. [PubMed: 10670764]
- 267.
- Rieder G., Hatz R. A., Moran A. P., Walz A., Stolte M., Enders G. Role of adherence in interleukin-8 induction in Helicobacter pylori-associated gastritis. Infect. Immun. 1997;65:3622–3630. [PMC free article: PMC175515] [PubMed: 9284128]
- 268.
- Ringnér M., Aleljung P., Wadström T. Adherence of haemagglutinating Helicobacter pylori to five cell lines. Zentralbl. Bakteriol. 1993;280:107–112. [PubMed: 8280930]
- 269.
- Robinson J., Goodwin C. S., Cooper M., Burke V., Mee B. J. Soluble and cell-associated haemagglutinins of Helicobacter (Campylobacter) pylori. J. Med. Microbiol. 1990;33:277–284. [PubMed: 2258915]
- 270.
- Roine R. P., Salmela K. S., Hook-Nikanne J., Kosunen T. U., Salaspuro M. Colloidal bismuth subcitrate and omeprazole inhibit alcohol dehydrogenase mediated acetaldehyde production by Helicobacter pylori. Life Sci. 1992;51:L195–L200. [PubMed: 1435071]
- 271.
- Romano M., Ricci V., Memoli A., Tuccillo C., Di Popolo A., Sommi P., Acquaviva A. M., Del Vecchio C. B., Bruni C. B., Zarrilli R. Helicobacter pylori up-regulates cyclooxygenase-2 mRNA expression and prostaglandin E2 synthesis in MKN 28 gastric mucosal cells in vitro. J. Biol. Chem. 1998;273:28560–28563. [PubMed: 9786845]
- 272.
- Rosberg K., Berglingh T., Gustavsson S., Hübinette R., Rolfsen W. Adhesion of Helicobacter pylori to human gastric mucosal biopsy specimens cultivated in vitro. Scand. J. Gastroenterol. 1991;26:1179–1187. [PubMed: 1754854]
- 273.
- Rudi J., Kuck D., Strand S., von Herbay A., Mariani S. M., Krammer P. H., Galle P. R., Stremmel W. Involvement of the CD95 (APO-1/Fas) receptor and ligand system, in Helicobacter pylori-induced gastric epithelial apoptosis. J. Clin. Invest. 1998;102:1506–1514. [PMC free article: PMC509000] [PubMed: 9788963]
- 274.
- Rudmann D. G., Eaton K. A., Krakowka S. Ultrastructural study of Helicobacter pylori adherence properties in gnotobiotic piglets. Infect. Immun. 1992;60:2121–2124. [PMC free article: PMC257125] [PubMed: 1563801]
- 275.
- Saitoh T., Natomi H., Zhao W., Okuzumi K., Sugano K., Iwamori M., Nagai Y. Identification of glycolipid receptors for Helicobacter pylori by TLC-immunostaining. FEBS Lett. 1991;282:385–387. [PubMed: 2037054]
- 276.
- Sakamoto S., Watanabe T., Tokumaru T., Takagi H., Nakazoto H., Floyd K. O. Expression of Lewisa, Lewisb, Lewisx, sialyl-Lewisa, and sialyl-Lewisx blood group antigens in human gastric carcinoma and in normal gastric tissue. Cancer Res. 1989;49:745–752. [PubMed: 2910493]
- 277.
- Salaspuro M. Helicobacter pylori alcohol dehydrogenase. EXS. 1994;71:185–195. [PubMed: 8032149]
- 278.
- Salmela K. S., Roine R. P., Hook-Nikanne J., Kosunen T. U., Salaspuro M. Acetaldehyde and ethanol production by Helicobacter pylori. Scand. J. Gastroenterol. 1994;29:309–312. [PubMed: 8047804]
- 279.
- Sawaoka H., Kawano S., Tsuji S., Tsuiji M., Sun W., Gunawan E. S., Hori M. Helicobacter pylori infection induces cyclooxygenase-2 expression in human gastric mucosa. Prostaglandins Leukot. Essent. Fatty Acids. 1998;59:313–316. [PubMed: 9888205]
- 280.
- Schorah C. J., Sobala G. M., Sanderson M., Collis N., Primrose J. N. Gastric juice ascorbic acid: effects of disease and implications for gastric carcinogenesis. Am. J. Clin. Nutr. 1991;53:287S–293S. [PubMed: 1985400]
- 281.
- Schryvers A. B., Gonzalez G. C. Receptors for transferrin in pathogenic bacteria are specific for the host's protein. Can. J. Microbiol. 1990;36:145–147. [PubMed: 2110858]
- 282.
- Schryvers A. B., Morris L. J. Identification and characterization of the human lactoferrin-binding protein from Neisseria meningitidis. Infect. Immun. 1988;56:1144–1149. [PMC free article: PMC259775] [PubMed: 3128478]
- 283.
- Segal E. D., Cha J., Lo J., Falkow S., Tompkins L. S. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. USA. 1999;96:14559–14564. [PMC free article: PMC24475] [PubMed: 10588744]
- 284.
- Segal E. D., Falkow S., Tompkins L. S. Helicobacter pylori attachment to gastric cells induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins. Proc. Natl. Acad. Sci. USA. 1996;93:1259–1264. [PMC free article: PMC40067] [PubMed: 8577751]
- 285.
- Segal E. D., Lange C., Covacci A., Tompkins L. S., Falkow S. Induction of host signal transduction pathways by Helicobacter pylori. Proc. Natl. Acad. Sci. USA. 1997;94:7595–7599. [PMC free article: PMC23867] [PubMed: 9207137]
- 286.
- Segal E. D., Shon J., Tompkins L. S. Characterization of Helicobacter pylori urease mutants. Infect. Immun. 1992;60:1883–1889. [PMC free article: PMC257089] [PubMed: 1563778]
- 287.
- Sekine H., Ohara S., Iijima K., Kato K. Recurrence rate of H. pylori after successful eradication and second eradication therapy after initial failure of treatment. Nippon Rinsho. 1999;57:116–120. [PubMed: 10036947]
- 288.
- Shames B., Krajden S., Fuksa M., Babida C., Penner J. L. Evidence for the occurrence of the same strain of Campylobacter pylori in the stomach and dental plaque. J. Clin. Microbiol. 1989;12:2849–2850. [PMC free article: PMC267140] [PubMed: 2592545]
- 289.
- Sharma S. A., Tummuru M. K., Miller G. G., Blaser M. J. Interleukin-8 response of gastric epithelial cell lines to Helicobacter pylori stimulation in vitro. Infect. Immun. 1995;63:1681–1687. [PMC free article: PMC173210] [PubMed: 7729872]
- 290.
- Shibata H., Kimura-Takagi I., Nagaoka M., Hashimoto S., Sawada H., Ueyama S., Yokokura T. Inhibitory effect of Cladosiphon fucoidan on the adhesion of Helicobacter pylori to human gastric cells. J. Nutr. Sci. Vitaminol. 1999;45:325–336. [PubMed: 10524351]
- 291.
- Shibata K., Ito Y., Hongo A., Yasoshima A., Endo T., Ohashi M. Bactericidal activity of a new antiulcer agent, ecabet sodium, against Helicobacter pylori under acidic conditions. Antimicrob. Agents Chemother. 1995;39:1295–1299. [PMC free article: PMC162730] [PubMed: 7574519]
- 292.
- Shimoyama T., Fukuda Y., Fukuda S., Munakata A., Yoshida Y., Shimoyama T. Ecabet sodium eradicates Helicobacter pylori infection in gastric ulcer patients. J. Gastroenterol. 1996;31(Suppl. 9):59–62. [PubMed: 8959522]
- 293.
- Sidebotham R. L., Batten J. J., Karim Q. N., Spencer J., Baron J. H. Breakdown of gastric mucus in presence of Helicobacter pylori. J. Clin. Pathol. 1991;44:52–57. [PMC free article: PMC497015] [PubMed: 1997534]
- 294.
- Simon P. M., Goode P. L., Mobasseri A., Zopf D. Inhibition of Helicobacter pylori binding to gastrointestinal epithelial cells by sialic acid-containing oligosaccharides. Infect. Immun. 1997;65:750–757. [PMC free article: PMC176121] [PubMed: 9009338]
- 295.
- Singh V. N., Gaby S. K. Premalignant lesions: role of antioxidant vitamins and beta-carotene in risk reduction and prevention of malignant transformation. Am. J. Clin. Nutr. 1991;53:386S–390S. [PubMed: 1985417]
- 296.
- Sivam G. P., Lampe J. W., Ulness B., Swanzy S. R., Potter J. D. Helicobacter pylori—in vitro susceptibility to garlic (Allium sativum) extract. Nutr. Cancer. 1997;27:118–121. [PubMed: 9121937]
- 297.
- Sjöstedt S. The upper gastrointestinal microflora in relation to gastric diseases and gastric surgery. Acta Chir. Scand. Suppl. 1989;551:1–57. [PubMed: 2741634]
- 298.
- Skouloubris S., Labigne A., De Reuse H. Identification and characterization of an aliphatic amidase in Helicobacter pylori. Mol. Microbiol. 1997;25:989–998. [PubMed: 9364923]
- 299.
- Slomiany B. L., Bilski J., Serosisk J., Murty V. L. N., Dworkin H., VanHorn K., Zielenski J., Slomiany A. Campylobacter pyloridis degrades mucin and undermines gastric mucosal integrity. Biochem. Biophys. Res. Commun. 1987;14:307–314. [PubMed: 3579908]
- 300.
- Slomiany B. L., Murty V. L., Piotrowski J., Grabska M., Slomiany A. Glycosulfatase activity of H. pylori toward human gastric mucin: effect of sucralfate. Am. J. Gastroenterol. 1992;87:1132–1137. [PubMed: 1381553]
- 301.
- Slomiany B. L., Murty V. L., Piotrowski J., Slomiany A. Gastroprotective agents in mucosal defense against Helicobacter pylori. Gen. Pharmacol. 1994;25:833–841. [PubMed: 7835626]
- 302.
- Slomiany B. L., Piotrowski J., Samanta A., Van Horn K., Murty V. L. N., Slomiany A. Campylobacter pylori colonization factor shows specificity for lactosylceramide sulfate and GM3 ganglioside. Biochem. Int. 1989;19:929–936. [PubMed: 2619758]
- 303.
- Slomiany B. L., Piotrowski J., Sengupts S., Slomiany A. Inhibition of gastric mucosal laminin receptor by Helicobacter pylori lipopolysaccharide. Biochem. Biophys. Res. Commun. 1991;175:963–970. [PubMed: 1827258]
- 304.
- Slomiany B. L., Piotrowski J., Slomiany A. Effect of sucralfate on the degradation of human gastric mucus by Helicobacter pylori protease and lipases. Am. J. Gastroenterol. 1992;87:595–599. [PubMed: 1595646]
- 305.
- Slomiany B. L., Piotrowski J., Slomiany A. Effect of sucralfate on gastric mucosal inflammatory responses induced by Helicobacter pylori lipopolysaccharide. Scand. J. Gastroenterol. 1998;33:916–922. [PubMed: 9759945]
- 306.
- Slomiany B. L., Piotrowski J., Slomiany A. Induction of caspase-3 and nitric oxide synthase-2 during gastric mucosal inflammatory reaction to Helicobacter pylori lipopolysaccharide. Biochem. Mol. Biol. Int. 1998;46:1063–1070. [PubMed: 9861460]
- 307.
- Slomiany B. L., Piotrowski J., Slomiany A. Gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide: down-regulation of nitric oxide synthase-2 and caspase-3 by sulglycotide. Biochem. Biophys. Res. Commun. 1999;261:15–20. [PubMed: 10405316]
- 308.
- Slomiany B. L., Piotrowski J., Slomiany A. Involvement of endothelin-1 in up-regulation of gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide. Biochem. Biophys. Res. Commun. 1999;258:17–20. [PubMed: 10222227]
- 309.
- Slomiany B. L., Piotrowski J., Slomiany A. Suppression of gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide by sulglycopeptide. Gen. Pharmacol. 1999;32:251–257. [PubMed: 10188628]
- 310.
- Smoot D. T., Resau J. H., Naab T., Desborges B. C., Gilliam T., Henry K. B., Curry S. B., Nidiry J., Sewchand J., Mills-Robertson K., Frontin K., Abebe E., Dillon M., Chippendale G. R., Phelps P. C., Scott V. F., Mobley H. L. T. Adherence of Helicobacter pylori to cultured human gastric epithelial cells. Infect. Immun. 1993;61:350–355. [PMC free article: PMC302729] [PubMed: 8418061]
- 311.
- Snepar R., Poporad G. A., Romano J. M., Kobasa W. D., Kaye D. Effect of cimetidine and antacid on gastric microbial flora. Infect. Immun. 1982;36:518–524. [PMC free article: PMC351258] [PubMed: 7085070]
- 312.
- Spiegelhalder C., Gerstenecker B., Kersten A., Schlitz E., Kist M. Purification of Helicobacter pylori superoxide dismutase and sequencing of the gene. Infect. Immun. 1993;61:5315–5325. [PMC free article: PMC281317] [PubMed: 8225605]
- 313.
- Stein M., Rappuoli R., Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc. Natl. Acad. Sci. USA. 2000;97:1263–1268. [PMC free article: PMC15590] [PubMed: 10655519]
- 314.
- Stratton C. W., Warner R. R., Coudron P. E., Lilly N. A. Bismuth-mediated disruption of the glycocalyx-cell wall of Helicobacter pylori: ultrastructural evidence for a mechanism of action for bismuth salts. J. Antimicrob. Chemother. 1999;43:659–666. [PubMed: 10382887]
- 315.
- Stromqvist M., Falk P., Bergstrom S., Hansson L., Lonnerdal B., Normark S., Hernell O. Human milk-casein and inhibition of Helicobacter pylori adhesion to human gastric mucosa. J. Pediatr. Gastroenterol. 1995;21:288–296. [PubMed: 8523212]
- 316.
- Su B., Hellström P. M., Rubio C., Çelik J., Granström M., Normark S. Type I Helicobacter pylori shows Lewisb-independent adherence to gastric cells requiring de novo protein synthesis in both host and bacteria. J. Infect. Dis. 1998;178:1379–1390. [PubMed: 9780259]
- 317.
- Su B., Johansson S., Fällman M., Patarroyo M., Granström M., Normark S. Signal transduction-mediated adherence and entry of Helicobacter pylori into cultured cells. Gastroenterology. 1999;117:595–604. [PubMed: 10464135]
- 318.
- Suerbaum S., Josenhans C., Labigne A. Cloning and genetic characterization of the Helicobacter pylori and Helicobacter mustelae flaB flagellin genes and construction of H. pylori flaA- and flaB-negative mutants by electroporation-mediated allelic exchange. J. Bacteriol. 1993;175:3278–3288. [PMC free article: PMC204724] [PubMed: 8501031]
- 319.
- Sunairi M., Watanabe K., Suzuki T., Tanaka N., Kuwayama H., Nakajima M. Effects of anti-ulcer agents on antibiotic activity against Helicobacter pylori. Eur. J. Gastroenterol. Hepatol. 1994;6:S121–S124. [PubMed: 7735928]
- 320.
- Suzuki M., Mori M., Fukumura D., Suzuki H., Miura S., Ishii H. Omeprazole attenuates neutrophil-endothelial cell adhesive interaction induced by extracts of Helicobacter pylori. J. Gastroenterol. Hepatol. 1999;14:27–31. [PubMed: 10029274]
- 321.
- Syder A. J., Guruge J. L., Li Q., Hu Y., Oleksiewicz C. M., Lorenz R. G., Karam S. M., Falk P. G., Gordon J. I. Helicobacter pylori attaches to NeuAc__2,3Gal__1,4glycoconjugates produced in the stomach of transgenic mice lacking parietal cells. Mol. Cell. 1999;3:263–274. [PubMed: 10198629]
- 322.
- Takahashi S., Igarashi H., Nakamura K., Masubuchi N., Saito S., Aoyagi T., Itoh T., Hirata I. Helicobacter pylori and urease activity—comparative study between urease positive and negative mutant strains. Nippon Rinsho. 1993;51:3149–3153. [PubMed: 8283623]
- 323.
- Tanaka T., Morishita Y., Suzui M., Kojima T., Okumura A., Mori H. Chemoprevention of mouse urinary bladder carcinogenesis by the naturally occurring carotenoid astaxanthin. Carcinogenesis. 1994;15:15–19. [PubMed: 8293542]
- 324.
- Taylor D. E., Rasko D. A., Sherburne R., Ho C., Jewell L. D. Lack of correlation between Lewis antigen expression by Helicobacter pylori and gastric epithelial cells in infected patients. Gastroenterology. 1998;115:1113–1122. [PubMed: 9797366]
- 325.
- Taylor N. S., Hasubski A. T., Fox J. G., Lee A. Haemagglutination profiles of Helicobacter species that cause gastritis in man and animals. J. Med. Microbiol. 1992;37:299–303. [PubMed: 1279175]
- 326.
- Teichmann R. K., Andress H. J., Leibich H., Seifert J., Brendel W. Possible role of la-positive cells in the antrum in gastrin secretion. Eur. Surg. Res. 1984;16:64–65.
- 327.
- Teneberg S., Miller-Podraza H., Lampert H. C., Evans D. J., Evans D. G., Danielsson D., Karlsson K. A. Carbohydrate binding specificity of the neutrophil-activating protein of Helicobacter pylori. J. Biol. Chem. 1997;272:19067–19071. [PubMed: 9228091]
- 328.
- Thomas J. E., Dale A., Harding M., Coward W. A., Cole T. J., Weaver L. T. Helicobacter pylori colonization in early life. Pediatr. Res. 1999;45:218–223. [PubMed: 10022593]
- 329.
- Thomas J. E., Gibson G. R., Darboe M. K., Dale A., Weaver L. T. Isolation of Helicobacter pylori from human faeces. Lancet. 1992;340:1194–1195. [PubMed: 1359263]
- 330.
- Thomsen L. L., Gavin J. B., Tasman-Jones C. Relation of Helicobacter pylori to the human gastric mucosa in chronic gastritis of the antrum. Gut. 1990;31:1230–1236. [PMC free article: PMC1378690] [PubMed: 2253904]
- 331.
- Tindberg Y., Blennow M., Granstrom M. Clinical symptoms and social factors in a cohort of children spontaneously clearing Helicobacter pylori infection. Acta Paediatr. 1999;88:631–635. [PubMed: 10419247]
- 332.
- Tomb J.-F. O. White, Kerlavage A. R., Clayton R. A., Sutton G. G., Fleischmann R. D., Ketchum K. A., Klenk H. P., Gill S., Dougherty B. A., Nelson K., Quackenbush J., Zhou L., Kirkness E. F., Peterson S., Loftus B., Richardson D., Dodson R., Khalak H. G., Glodek A., McKenney K., Fitzgerald L. M., Lee N., Adams M. D., Hickey E. K., Berg D. E., Gocayne J. D., Utterback T. R., Peterson J. D., Kelley J. M., Cotton M. D., Weidman J. M., Fujii C., Bowman C., Watthey L., Wallin E., Hayes W. S., Borodovsky M., Karp P. D., Smith H. O., Fraser C. M., Venter J. C. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. [PubMed: 9252185]
- 333.
- Tricottet V., Bruneval P., Vire O., Camilleri J. P. Campylobacter-like organisms and surface epithelium abnormalities in active, chronic gastritis in humans: an ultrastructural study. Ultrastruct. Pathol. 1986;10:113–122. [PubMed: 3961927]
- 334.
- Trust T. J., Doig P., Emödy L., Kienle Z., Wadström T., O'Toole P. High-affinity binding of the basement membrane proteins collagen Type IV and laminin to the gastric pathogen Helicobacter pylori. Infect. Immun. 1991;59:4398–4404. [PMC free article: PMC259055] [PubMed: 1937798]
- 335.
- Tsuda M., Karita M., Morshed M. G., Okita K., Nakazawa T. A urease-negative mutant of Helicobacter pylori constructed by allelic exchange mutagenesis lacks the ability to colonize the nude mouse stomach. Infect. Immun. 1994;62:3586–3589. [PMC free article: PMC303000] [PubMed: 8039935]
- 336.
- Tsugane S., Tei Y., Takahashi T., Watanabe S., Sugano K. Salty food intake and risk of Helicobacter pylori infection. Jpn. J. Cancer Res. 1994;85:474–478. [PMC free article: PMC5919501] [PubMed: 8014104]
- 337.
- Tummuru M. K., Cover T. L., Blaser M. J. Cloning and expression of a high-molecular-mass antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect. Immun. 1993;61:1799–1809. [PMC free article: PMC280768] [PubMed: 8478069]
- 338.
- Tummuru M. K., Cover T. L., Blaser M. J. Mutation of the cytotoxin-associated cagA gene does not affect vacuolating cytotoxin activity of Helicobacter pylori. Infect. Immun. 1994;62:2609–2613. [PMC free article: PMC186552] [PubMed: 8188385]
- 339.
- Tummuru M. K., Sharma S. A., Blaser M. J. Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secreted protein, is required for induction of IL-8 in gastric epithelial cells. Mol. Microbiol. 1995;18:867–876. [PubMed: 8825091]
- 340.
- Tzouvelekis L. S., Mentis A. F., Makris A. M., Spiliadis C., Blackwell C., Weir D. M. In vitro binding of Helicobacter pylori to human gastric mucin. Infect. Immun. 1991;59:4252–4254. [PMC free article: PMC259024] [PubMed: 1937781]
- 341.
- Valkonen K. H., Ringner M., Ljungh Å., Wadström T. High-affinity binding of laminin by Helicobacter pylori: evidence for a lectin-like interaction. FEMS Immunol. Med. Microbiol. 1993;7:29–38. [PubMed: 8364520]
- 342.
- Valkonen K. H., Wadström T., Moran A. P. Interaction of lipopolysaccharides of Helicobacter pylori with basement membrane protein laminin. Infect. Immun. 1994;62:3640–3648. [PMC free article: PMC303013] [PubMed: 8063380]
- 343.
- Valkonen K. H., Wadström T., Moran A. P. Identification of the N-acetylneuraminyllactose-specific laminin-binding protein of Helicobacter pylori. Infect. Immun. 1997;65:916–923. [PMC free article: PMC175069] [PubMed: 9038297]
- 344.
- Vandenbergh P. A. Lactic acid bacteria, their metabolic products and interference with microbial growth. FEMS Microbiol. Rev. 1993;12:221–238.
- 345.
- Van den Brink G. R., Tytgat K. M., Van der Hulst R. W., Van der Loos C. M., Einerhand A. W., Buller H. A., Dekker J. H. pylori colocalises with MUC5AC in the human stomach. Gut. 2000;46:601–607. [PMC free article: PMC1727935] [PubMed: 10764701]
- 346.
- Van Doorn N. E., Namavar F., Sparrius M., Stoof J., van Rees E. P., van Doorn L. J., Vandenbroucke-Grauls C. M. Helicobacter pylori-associated gastritis in mice is host and strain specific. Infect. Immun. 1999;67:3040–3046. [PMC free article: PMC96618] [PubMed: 10338517]
- 347.
- Varis K., Taylor P. R., Sipponen P., Samloff I. M., Heinonen O. P., Albanes D., Harkonen M., Huttunen J. K., Laxen F., Virtamo J. Gastric cancer and premalignant lesions in atrophic gastritis: a controlled trial on the effect of supplementation with alpha-tocopherol and beta-carotene. The Helsinki Gastritis Study Group. Scand. J. Gastroenterol. 1998;33:294–300. [PubMed: 9548624]
- 348.
- Vatier J., Poitevin C., Mignon M. Sialic acid content and proteolytic activity in gastric juice in humans. An approach for appreciating mucus glycoprotein erosion. Dig. Dis. Sci. 1988;33:144–151. [PubMed: 3123182]
- 349.
- Vazquez-Torres A., Jones-Carson J., Baumler A. J., Falkow S., Valdivia R., Brown W., Le M., Berggren R., Parks W. T., Fang F. C. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature. 1999;401:804–808. [PubMed: 10548107]
- 350.
- Veerman E. C., Bank C. M., Namavar F., Appelmelk B. J., Bolscher J. G., Nieuw Amerongen A. V. Sulfated glycans on oral mucin as receptors for Helicobacter pylori. Glycobiology. 1997;7:737–743. [PubMed: 9376676]
- 351.
- Wahlfors J., Meurman J. H., Toskala J., Korhonen A., Alakuijala P., Janatuinen E., Karkkkainen U. M., Nuutinen P., Janne J. Development of a rapid PCR method for identification of Helicobacter pylori in dental plaque and gastric biopsy specimens. Eur. J. Clin. Microbiol. Infect. Dis. 1995;14:780–786. [PubMed: 8536726]
- 352.
- Watanabe H., Takahashi T., Okamoto T., Ogundigie P. O., Ito A. Effects of sodium chloride and ethanol on stomach tumorigenesis in ACI rats treated with N-methyl-N′-nitro-N-nitrosoguanidine: a quantitative morphometric approach. Jpn. J. Cancer Res. 1992;83:588–593. [PMC free article: PMC5918881] [PubMed: 1644663]
- 353.
- Weigert N., Schaffer K., Schusdziarra V., Classen M., Schepp W. Gastrin secretion from primary cultures of rabbit antral G cells: stimulation by inflammatory cytokines. Gastroenterology. 1996;110:147–154. [PubMed: 8536851]
- 354.
- Werther J. L. The gastric mucosal barrier. Mt. Sinai J. Med. 2000;67:41–53. [PubMed: 10677782]
- 355.
- Wilkinson S. M., Uhl J. R., Kline B. C., Cockerill III F. R. Assessment of invasion frequencies of cultured Hep-2 cells by clinical isolates of Helicobacter pylori using an acridine orange assay. J. Clin. Pathol. 1998;51:127–133. [PMC free article: PMC500507] [PubMed: 9602686]
- 356.
- Williams C. L., Preston T., Hossack M., Slater C., McColl K. E. Helicobacter pylori utilises urea for amino acid synthesis. FEMS Immunol. Med. Microbiol. 1996;13:87–94. [PubMed: 8821403]
- 357.
- Willis P., Lynch D. A., Prescott R., Lamonby S. Cell proliferation in the post-surgical stomach, dietary salt, and the effect of H. pylori eradication. J. Clin. Pathol. 1999;52:665–669. [PMC free article: PMC501541] [PubMed: 10655987]
- 358.
- Wilson K. T., Ramanujam K. S., Mobley H. L. T., Musselman R. F., James S. P., Meltzer S. J. Helicobacter pylori stimulates inducible nitric oxide synthase expression and activity in a murine macrophage cell line. Gastroenterology. 1996;111:1524–1533. [PubMed: 8942731]
- 359.
- Windle H. J., Fox A., Ni Eidhin D., Kelleher D. The thioredoxin system of Helicobacter pylori. J. Biol. Chem. 2000;275:5081–5090. [PubMed: 10671551]
- 360.
- Worku M. L., Sidebotham R. L., Karim Q. N. Effects of ranitidine bismuth citrate on Helicobacter pylori motility, morphology and survival. Aliment. Pharmacol. Ther. 1999;13:753–760. [PubMed: 10383504]
- 361.
- Wyle F. A., Tarnawski A., Dabros W., Gergely H. Campylobacter pylori interactions with gastric cell tissue culture. J. Clin. Gastroenterol. 1990;12:S99–S103. [PubMed: 2212557]
- 362.
- Wyle F. A., Tarnawski A., Schulman D., Dabros W. Evidence for gastric mucosal cell invasion by C. pylori: an ultrastructural study. J. Clin. Gastroenterol. 1990;12:S92–S98. [PubMed: 2212556]
- 363.
- Xiang Z., Censini S., Bayeli P. F., Telford J. L., Figura N., Rappuoli R., Covacci A. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect. Immun. 1995;63:94–98. [PMC free article: PMC172962] [PubMed: 7806390]
- 364.
- Yamaguchi H., Osaki T., Kurihara N., Taguchi H., Hanawa T., Yamamoto T., Kamiya S. Heat-shock protein 60 homologue of Helicobacter pylori is associated with adhesion of H. pylori to human gastric epithelial cells. J. Med. Microbiol. 1997;46:825–831. [PubMed: 9364138]
- 365.
- Yamaguchi H., Osaki T., Taguchi H., Hanawa T., Yamamoto T., Kamiya S. Flow cytometric analysis of the heat shock protein 60 expressed on the cell surface of Helicobacter pylori. J. Med. Microbiol. 1996;45:270–277. [PubMed: 8849701]
- 366.
- Yamaguchi H., Osaki T., Taguchi H., Hanawa T., Yamamoto T., Kamiya S. Induction and epitope analysis of Helicobacter pylori heat shock protein. J. Gastroenterol. 1996;31(Suppl. 9):12–15. [PubMed: 8959511]
- 367.
- Yamaguchi H., Osaki T., Taguchi H., Hanawa T., Yamamoto T., Kamiya S. Relationship between expression of HSP60, urease activity, production of vacuolating cytotoxin, and adherence activity of Helicobacter pylori. J. Gastroenterol. 1998;33(Suppl. 10):6–9. [PubMed: 9840008]
- 368.
- Yamamoto-Osaki T., Yamaguchi H., Taguchi H., Ogata S., Kamiya S. Adherence of Helicobacter pylori to cultured human gastric carcinoma cells. Eur. J. Gastroenterol. Hepatol. 1995;7:S89–S92. [PubMed: 8574746]
- 369.
- Yoshiyama H., Nakamura H., Kimoto M., Okita K., Nakazawa T. Chemotaxis and motility of Helicobacter pylori in a viscous environment. J. Gastroenterol. 1999;34(Suppl. 11):18–23. [PubMed: 10616760]
- 370.
- Yoshiyama H., Nakamura H., Okamoto T., Okita K., Nakazawa T. A novel in vitro effect of the mucosal protective agent sofalcone—inhibition of chemotactic motility in Helicobacter pylori. Aliment. Pharmacol. Ther. 2000;14(Suppl. 1):230–236. [PubMed: 10807429]
- 371.
- Yoshiyama H., Nakazawa T. Unique mechanism of Helicobacter pylori for colonizing the gastric mucus. Microbes Infect. 2000;2:55–60. [PubMed: 10717541]
- 372.
- You W. C., Zhang L., Gail M. H., Ma J. L., Chang Y. S., Blot W. J., Li J. Y., Zhao C. L., Liu W. D., Li H. Q., Hu Y. R., Bravo J. C., Correa P., Xu G. W., Fraumeni, Jr J. F. Helicobacter pylori infection, garlic intake and pre-cancerous lesions in a Chinese population at low risk of gastric cancer. Int. J. Epidemiol. 1998;27:941–944. [PubMed: 10024185]
- 373.
- Zhang H. M., Wakisaka N., Maeda O., Yamamoto T. Vitamin C inhibits the growth of a bacterial risk factor for gastric carcinoma: Helicobacter pylori. Cancer. 1997;80:1897–1903. [PubMed: 9366290]
- Review Immune responses to Helicobacter pylori colonization: mechanisms and clinical outcomes.[Clin Sci (Lond). 2006]Review Immune responses to Helicobacter pylori colonization: mechanisms and clinical outcomes.Portal-Celhay C, Perez-Perez GI. Clin Sci (Lond). 2006 Mar; 110(3):305-14.
- [Intensification of Helicobacter pylori colonization and serological markers of Helicobacter pylori infection and cytotoxicity in blood].[Pol Merkur Lekarski. 2009][Intensification of Helicobacter pylori colonization and serological markers of Helicobacter pylori infection and cytotoxicity in blood].Bak-Romaniszyn L, Kałuzyński A, Chmiela M, Rudnicka W, Kulig A, Płaneta-Małecka I. Pol Merkur Lekarski. 2009 May; 26(155):407-11.
- Review Colonization and infection by Helicobacter pylori in humans.[Helicobacter. 2007]Review Colonization and infection by Helicobacter pylori in humans.Andersen LP. Helicobacter. 2007 Nov; 12 Suppl 2:12-5.
- Role of Helicobacter pylori virulence factors for iron acquisition from gastric epithelial cells of the host and impact on bacterial colonization.[Future Microbiol. 2011]Role of Helicobacter pylori virulence factors for iron acquisition from gastric epithelial cells of the host and impact on bacterial colonization.Boyanova L. Future Microbiol. 2011 Aug; 6(8):843-6.
- Muc1 mucin limits both Helicobacter pylori colonization of the murine gastric mucosa and associated gastritis.[Gastroenterology. 2007]Muc1 mucin limits both Helicobacter pylori colonization of the murine gastric mucosa and associated gastritis.McGuckin MA, Every AL, Skene CD, Linden SK, Chionh YT, Swierczak A, McAuley J, Harbour S, Kaparakis M, Ferrero R, et al. Gastroenterology. 2007 Oct; 133(4):1210-8. Epub 2007 Jul 10.
- Adherence and Colonization - Helicobacter pyloriAdherence and Colonization - Helicobacter pylori
Your browsing activity is empty.
Activity recording is turned off.
See more...