Clinical Description
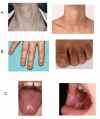
The classic dyskeratosis congenita (DC) triad of abnormal fingernails and toenails, lacy, reticular pigmentation of the neck and upper chest, and oral leukoplakia is diagnostic (); however, these features are not present in all individuals with DC and related telomere biology disorders (DC/TBD) and may or may not develop over time after the appearance of other complications listed here [Savage & Bertuch 2010, Dokal 2011, Ward et al 2018, Niewisch et al 2022]. The time of onset for these medical complications varies considerably among individuals even within the same family and thus the manifestations of DC/TBD do not progress in a predictable pattern. The spectrum ranges from individuals who develop bone marrow failure (BMF) first and then years later develop other classic findings such as nail abnormalities, to others who have severe nail problems and abnormalities of skin pigmentation but normal bone marrow function.
Three forms of DC/TBD with more severe manifestations have been identified: Hoyeraal Hreidarsson syndrome, Revesz syndrome, and Coats plus syndrome (see Severe Forms of DC/TBD).
Dermatologic. Lacy, reticular pigmentation primarily of the neck and chest may be subtle or diffuse hyper- or hypopigmentation. Changes in skin pigmentation may not be present at time of diagnosis, but may develop or become more pronounced over time.
Dysplastic fingernails and toenails may worsen significantly over time and nails may eventually "disappear."
Additional common skin findings in DC/TBD include epiphora, loss of dermatoglyphics, early graying, palmoplantar hyperkeratosis, eyelash loss, and hair loss from scalp [Ward et al 2018]. Dermatoglyphics may be lost with age.
Hyperhidrosis is noted infrequently in some individuals.
Ears, nose, and throat. Oral leukoplakia is part of the diagnostic triad. It may be a presenting sign found in childhood or may develop over time.
Deafness has been reported but is rare.
Ophthalmic. Epiphora caused by stenosis of the lacrimal drainage system can result in blepharitis.
Abnormal eyelash growth includes sparse eyelashes, ectropion, entropion, and trichiasis, which can lead to corneal abrasions, scarring, or infection if not treated.
Bilateral exudative retinopathy seen in Revesz syndrome or Coats plus syndrome can lead to blindness.
Dental. Decreased root-to-crown ratio is attributed to abnormal tooth development.
Taurodontism (enlarged pulp chambers of the teeth) may be noted on dental x-ray.
Growth and development. Short stature has been reported but height is variable.
Intrauterine growth restriction (IUGR) has been noted in children with the more severe Coats plus syndrome, Hoyeraal Hreidarsson syndrome, or Revesz syndrome.
Developmental delay may be present in some. It can be more pronounced in persons with Hoyeraal Hreidarsson syndrome, Revesz syndrome, or Coats plus syndrome.
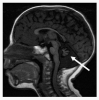
Neurologic. Although most persons with DC/TBD have normal psychomotor development and normal neurologic function, significant developmental delay / intellectual disability is present in Hoyeraal Hreidarsson syndrome and Revesz syndrome. Cerebellar hypoplasia is present in Hoyeraal Hreidarsson syndrome (), and intracranial calcifications have been reported in Revesz syndrome and Coats plus syndrome. Microcephaly has also been reported in some persons with DC/TBD, most commonly in Hoyeraal Hreidarsson syndrome and Revesz syndrome.
MRI of cerebellar hypoplasia in an individual with Hoyeraal Hreidarsson syndrome. Arrow indicates the hypoplastic cerebellum.
Psychiatric. Clinically significant psychiatric diagnosis was reported in 27% of 44 individuals with DC/TBD in a recent retrospective study. The psychiatric diagnoses in adults included depression and/or anxiety/panic attacks, autism spectrum disorder, and bipolar disorder [Bhala et al 2019]. However, the true prevalence of psychiatric disorders is unknown.
Endocrine. Hypogonadism has been noted in a small number of severely affected males.
Gastrointestinal. Esophageal stenosis has been reported in several persons with DC/TBD and may worsen over time.
Enteropathy, which may result in poor growth, has been reported, specifically in Hoyeraal Hreidarsson syndrome.
Liver disease, including nodular regenerative hyperplasia, fibrosis, cirrhosis, portal hypertension, and hepatopulmonary syndrome, is increasingly recognized as a potential severe complication of DC/TBD and may lead to liver transplant [Gorgy et al 2015, Kapuria et al 2019, Niewisch et al 2022]. Individuals who have undergone hematopoetic cell transplant (HCT) need close monitoring for liver disease.
Gastrointestinal telangiectasias and bleeding may occur in the context of portal hypertension or independently.
Genitourinary. Urethral stenosis in males may be present at diagnosis or develop over time.
Musculoskeletal. Osteoporosis and osteopenia have been reported. The contribution of prior treatment and comorbid conditions to these complications is not known. Some individuals have reported bone fractures after minor trauma.
Avascular necrosis of the hips and shoulders can result in pain and reduced function. Several individuals have required hip replacement surgery at young ages.
Cardiovascular. Rare reported congenital heart defects include atrial and ventricular septal defects, myocardial fibrosis, and dilated cardiomyopathy.
Hematologic. Bone marrow failure is a common presenting sign, may develop at any age, and may progress over time. Approximately one half of individuals with DC/TBD develop some degree of bone marrow failure by age 40 years.
Individuals with DC/TBD are at increased risk for leukemia (see Cancer).
Immunologic. Immunodeficiency of variable severity has been reported in DC/TBD. It has not been fully characterized, but it appears that some individuals may have reduced numbers of B cells, T cells, and/or NK cells.
Cancer. Persons with DC/TBD are at high risk for leukemia and squamous cell cancer of the head and neck or anogenital region. Elevated risk of cervical squamous cell cancer has also been reported in DC/TBD.
Investigators at the National Cancer Institute analyzed data from 15 years of follow up in a longitudinal cohort study to further quantify cancer risk in individuals with DC/TBD (n=197). The median age of onset for all cancers was 38 years (range 18-63 years), with an O/E (observed deaths to expected deaths) ratio of 4.2 compared to the normal population. The most frequent solid tumors were head and neck squamous cell carcinomas (with an O/E ratio of 74), followed by leukemia, non-Hodgkin lymphoma, and anorectal carcinoma. Of note, the risk of tongue cancer was increased by 216-fold over the normal population. In this study, myelodysplastic syndrome (MDS) was observed at a 578-fold increased risk, and the median age of MDS onset was 31 years (range 4-73 years).
Respiratory. Pulmonary fibrosis may be a presenting sign or may develop over time. It may be more common in individuals who have had HCT. Pulmonary fibrosis is manifest as bibasilar reticular abnormalities, ground glass opacities, or diffuse nodular lesions on high-resolution computed tomography and abnormal pulmonary function studies that include evidence of restriction (reduced vital capacity with an increase in FEV1/FVC ratio) and/or impaired gas exchange (increased P(A-a)O2 with rest or exercise or decreased diffusion capacity of the lung for carbon monoxide).
Pulmonary arteriovenous malformations have recently been reported in individuals with DC/TBD [Khincha et al 2017]. They may be present in individuals with hypoxia in the absence of pulmonary fibrosis and can be diagnosed by bubble echocardiography.
Clinical Findings of X-Linked DC/TBD in Females
Clinical manifestations in females heterozygous for DKC1 pathogenic variants have been reported, including skin pigmentation abnormalities, nail dysplasia, and bone marrow failure [Vulliamy et al 2006, Xu et al 2016, Hirvonen et al 2019].