Summary
Clinical characteristics.
Dystrophic epidermolysis bullosa (DEB) is a genetic skin disorder affecting skin and nails that usually presents at birth. DEB is divided into two major types depending on inheritance pattern: recessive dystrophic epidermolysis bullosa (RDEB) and dominant dystrophic epidermolysis bullosa (DDEB). Each type is further divided into multiple clinical subtypes. Absence of a known family history of DEB does not preclude the diagnosis.
Clinical findings in severe generalized RDEB include skin fragility manifest by blistering with minimal trauma that heals with milia and scarring. Blistering and erosions affecting the whole body may be present in the neonatal period. Oral involvement may lead to mouth blistering, fusion of the tongue to the floor of the mouth, and progressive diminution of the size of the oral cavity. Esophageal erosions can lead to webs and strictures that can cause severe dysphagia. Consequently, malnutrition and vitamin and mineral deficiency may lead to growth restriction in young children. Corneal erosions can lead to scarring and loss of vision. Blistering of the hands and feet followed by scarring fuses the digits into "mitten" hands and feet, with contractures and pseudosyndactyly. The lifetime risk of aggressive squamous cell carcinoma is higher than 90%.
In contrast, the blistering in the less severe forms of RDEB may be localized to hands, feet, knees, and elbows with or without involvement of flexural areas and the trunk, and without the mutilating scarring seen in severe generalized RDEB.
In DDEB, blistering is often mild and limited to hands, feet, knees, and elbows, but nonetheless heals with scarring. Dystrophic nails, especially toenails, are common and may be the only manifestation of DDEB.
Diagnosis/testing.
The diagnosis of DEB is established in a proband with characteristic clinical findings and the identification of biallelic pathogenic variants (RDEB) or a heterozygous pathogenic variant (DDEB) in COL7A1 by molecular genetic testing. The only gene in which pathogenic variants are known to cause DEB is COL7A1. If molecular genetic testing is not diagnostic, examination of a skin biopsy with direct immunofluorescence (IF) for specific cutaneous markers and/or electron microscopy (EM) may be necessary for diagnosis.
Management.
Treatment of manifestations: New blisters should be lanced, drained, and in most cases dressed with a nonadherent material, covered with padding for stability and protection, and secured with an elastic wrap for integrity. Infants and children with severe generalized RDEB and poor growth require attention to fluid and electrolyte balance and may require nutritional support, including feeding gastrostomy. Anemia is treated with iron supplements and transfusions as needed. Other nutritional supplements may include calcium, vitamin D, selenium, carnitine, and zinc. Occupational therapy may help prevent hand contractures. Surgical release of fingers often needs to be repeated.
Prevention of primary manifestations: If a fetus is known to be affected with any form of DEB, cesarean delivery may reduce trauma to the skin during delivery; age-appropriate play involving activities that cause minimal trauma to the skin is encouraged; dressings and padding are needed to protect bony prominences from blister-inducing impact.
Surveillance: Beginning in the second decade of life, biopsies of abnormal-appearing wounds that do not heal or have exuberant scar tissue are indicated for evidence of squamous cell carcinoma. Suggested regular testing includes screening for anemia and deficiencies of iron, zinc, vitamin D, selenium, and carnitine every 6-12 months. Yearly echocardiograms to identify dilated cardiomyopathy and bone mineral density studies to identify osteoporosis are recommended.
Agents/circumstances to avoid: Poorly fitting or coarse-textured clothing and footwear; activities/bandages that traumatize the skin.
Evaluation of relatives at risk: Evaluating an at-risk newborn for evidence of blistering is appropriate so that trauma to the skin can be avoided as much as possible.
Genetic counseling.
Dystrophic epidermolysis bullosa is inherited in either an autosomal dominant (DDEB) or autosomal recessive (RDEB) manner. Molecular characterization of pathogenic variants is the only accurate method to determine mode of inheritance and recurrence risk; phenotype severity and IF/EM findings alone are not sufficient.
DDEB. About 70% of individuals diagnosed with DDEB are reported to have an affected parent. If a parent of a
proband with DDEB is affected, the risk to the sibs is 50%. Each child of an individual with DDEB has a 50% chance of inheriting the
pathogenic variant.
RDEB. Each sib of an affected individual whose parents are both carriers has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic
carrier, and a 25% chance of being unaffected and not a carrier.
Once the COL7A1 pathogenic variant(s) have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Diagnosis
Dystrophic epidermolysis bullosa (DEB) is a genetic disorder affecting skin and nails that usually presents at birth. Currently, the classification of DEB is based on the publication of the consensus meeting of 2013 [Fine et al 2014]. Diagnosis is based on clinical suspicion in an individual with fragile skin, a family history of DEB, and diagnostic testing. Molecular genetic analysis is the most definitive test, but direct immunofluorescence (IF) and/or transmission electron microscopy (EM) may be helpful especially in classifying subtypes.
DEB is divided into two major types depending on inheritance pattern: recessive dystrophic epidermolysis bullosa (RDEB) and dominant dystrophic epidermolysis bullosa (DDEB). Each type is further divided into multiple clinical subtypes (see Nomenclature). Absence of a known family history of DEB does not preclude the diagnosis.
Suggestive Findings
Dystrophic epidermolysis bullosa (DEB) should be suspected in individuals with the following clinical findings:
Fragility of the skin, manifest by blistering with minimal trauma that heals with milia and scarring
Blistering and erosions that may:
Lead to aplasia cutis congenita at birth (absence of skin, especially on extremities)
Be present in the neonatal period
Affect the whole body including mucous membranes (most severe forms) or primarily the hands, feet, knees, and elbows (milder forms)
Lead to mutilating pseudosyndactyly of the hands and feet (severe forms)
Lead to oral and/or esophageal scarring and strictures
Lead to corneal erosions with resulting scarring leading to loss of vision
Predispose to squamous cell carcinoma
Dystrophic or absent nails, especially toenails
Establishing the Diagnosis
The diagnosis of DEB is established in a proband with characteristic clinical findings and either biallelic pathogenic (or likely pathogenic) variants (RDEB) or a heterozygous pathogenic (or likely pathogenic) variant (DDEB) in COL7A1 identified on molecular genetic testing (see Table 1). If molecular genetic testing is not diagnostic, examination of a skin biopsy (see Skin Biopsy) with direct IF for specific cutaneous markers and/or EM may be necessary. Routine histology is not useful.
Note: Per ACMG variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants.
It should be noted that not all clinicians have access to the diagnostic tools described in this section (molecular genetic testing, specialized tests on a skin biopsy). A recent study compared a matrix of clinical findings with genetic confirmation in 74 cases and found a high concordance to type and subtype of EB. This technique may be useful in developing countries [Yenamandra et al 2017].
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, concurrent or serial single-gene testing, multigene panel) and comprehensive
genomic testing (chromosomal microarray analysis, exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of DEB is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with epidermolysis bullosa (EB), or presenting at birth or in the neonatal period before more advanced disease progression has occurred, are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of DEB molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
Single-gene testing. Sequence analysis of
COL7A1 detects small intragenic deletions/insertions and
missense,
nonsense, and
splice site variants; typically,
exon or whole-gene deletions/duplications are not detected.
An epidermolysis bullosa multigene panel that includes
COL7A1 and other genes of interest (see
Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see
Table 1).
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by epidermolysis bullosa, comprehensive
genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
Exome array (when clinically available) may be considered if exome sequencing is not diagnostic.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Dystrophic Epidermolysis Bullosa
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
| COL7A1 3 | Sequence analysis 4 | 95% 5 |
| Gene-targeted deletion/duplication analysis 6 | <2% 7 |
- 1.
- 2.
- 3.
Some pathogenic variants in COL7A1 have been described in both recessive and dominant inheritance patterns [Almaani et al 2011]. If two variants in COL7A1 are found, parental testing may be necessary to establish that the variants are biallelic.
- 4.
- 5.
- 6.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 7.
Skin Biopsy
A definitive diagnosis of EB is made most directly by molecular genetic analysis. In the past, when only single-gene sequencing was available, it was imperative to perform a biopsy first to determine which single gene(s) to analyze. However, now that multigene panels are available [Lucky et al 2018], some clinicians prefer to avoid biopsy unless genetic analysis fails to yield a diagnosis [Pfendner 2015, Tenedini et al 2015].
If a biopsy is determined to be necessary for diagnosis, it should be taken from the leading edge of a fresh blister (<12 hours old) or of a mechanically induced blister and should include some normal adjacent skin; older blisters undergo changes that may obscure the diagnostic morphology. Elliptic or shave excisions are often used. Although a punch biopsy can introduce confusing artifact, careful use of the punch can avoid loss of the epidermis [Intong & Murrell 2010].
Light microscopy is inadequate and unacceptable for the accurate diagnosis of epidermolysis bullosa.
Immunofluorescence (IF). Examination of a skin biopsy by IF antibody/antigen mapping is an appropriate way to establish the diagnosis of DEB. Direct IF may reveal the level of clefting in the skin and help establish the broad category of EB type. Even without a split, presence or absence of specific proteins in the skin may also determine the type of EB. IF also has the advantage of a rapid turnaround time [Pohla-Gubo et al 2010, Meester et al 2018].
Characteristic findings:
Staining of collagen VII using antibodies is diminished or absent.
In milder forms of RDEB and in DDEB, staining for collagen VII may appear normal, but cleavage planes are below the lamina densa.
Normal staining for other antigens (e.g., laminin 332, collagen XVII, plectin, α6β4 integrin, and keratins 5 and 14) helps to confirm the diagnosis of DEB.
Transmission electron microscopy (TEM). Sometimes, especially in milder forms of EB, direct IF studies are not sufficient to make the diagnosis because near-normal antigen levels may be detected and no cleavage plane is observed. In such cases, TEM examination of the skin biopsy can be helpful in examining cellular structures [Eady & Dopping-Hepenstal 2010].
Characteristic findings:
All DEB. Cleavage is observed below the lamina densa of the basement membrane zone.
Recessive DEB (RDEB) severe generalized. Anchoring fibrils are markedly reduced, absent, or abnormal in morphology.
Dominant DEB (DDEB), RDEB-gen and -loc
Anchoring fibrils may appear reduced in number and/or show altered morphology.
Intracellular retention of collagen VII can be observed in some individuals.
Collagen VII may be retained intracellularly within the basal keratinocytes instead of being transported to the basement membrane zone in some individuals who have transient blistering in the newborn period.
Clinical Characteristics
Clinical Description
Before the molecular basis of dystrophic epidermolysis bullosa (DEB) was understood, types and subtypes were identified based primarily on clinical features, mode of inheritance, and the presence or absence of collagen VII and anchoring fibrils detected on skin biopsy. The current classification system is based on inheritance pattern (autosomal dominant DEB [DDEB] vs autosomal recessive DEB [RDEB]) and is further stratified by collagen VII staining and the specific COL7A1 pathogenic variant that is identified in a given affected individual (see Nomenclature) [Fine et al 2014]. For the purposes of this GeneReview the terms "recessive DEB severe generalized" (RDEB-sev gen), "recessive DEB generalized and localized" (which includes several further subtypes), and "dominant DEB" (DDEB) (which also includes further subtypes) have been used and are discussed below.
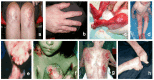
See .
Common findings of dystrophic epidermolysis bullosa: a, b. Scarring on knees and hands and dystrophic nails found in dominant DEB in an adult
Recessive DEB Severe Generalized (RDEB-sev gen)
In this classic severe form of RDEB, blisters are present at birth or become apparent in the neonatal period. Medical consequences of RDEB-sev gen have been recently reviewed [Fine & Mellerio 2009a, Fine & Mellerio 2009b, Murrell 2010, Li et al 2017].
Dermatologic and skin cancer risk
Aplasia cutis congenita, especially of the extremities, which can occur in any type of EB, may be found in the newborn period.
Blisters can affect the whole body including the skin, oral mucosa, esophageal mucosa, and corneas as early as the newborn period. Chronic nonhealing wounds and secondary infection are common, often with Staphylococcus, Pseudomonas, and Streptococcus.
Blistering continues throughout life with scarring that may lead to disfigurement and orthopedic issues (see Orthopedic below).
Many individuals develop large irregular brown patches that histologically comprise collections of nevus cells and are called EB nevi [
Lanschuetzer et al 2010]. No instances of melanoma arising in these nevi have been reported to date.
The lifetime risk of aggressive squamous cell carcinoma (SCC) is greater than 90% with significant metastatic potential [
Fine et al 2009]. SCC usually appears in the third decade but can appear as early as the second decade [
Ayman et al 2002]. Affected individuals usually succumb to aggressive metastatic SCC [
Mellerio et al 2016].
Oral, gastrointestinal, growth, and nutritional issues
Oral involvement may lead to fusion of the tongue to the floor of the mouth (ankyloglossia) and progressive diminution of the size of the oral cavity and mouth opening (microstomia), which, along with poor dental hygiene and caries, impairs food intake and ultimately nutrition [
Krämer et al 2012].
Esophageal blisters and erosions as well as webs and strictures can cause severe dysphagia with resultant poor nutrition [
Azizkhan et al 2006,
Mortell & Azizkhan 2010]. Rarely, affected individuals can have esophageal disease with few or no skin manifestations. Gastroesophageal reflux disease is also common.
Anal erosions, poor intake of fluid and fiber, and use of opioid analgesics contribute to frequent severe constipation.
Malnutrition caused by poor intake and an increased nutritional demand for tissue healing can result in growth restriction in young children and absent or delayed puberty in older children.
Vitamin and mineral deficiencies can occur especially with iron, zinc, carnitine, selenium, and vitamin D [
Haynes 2010].
Anemia results from poor iron intake and the anemia of chronic disease with bone marrow suppression.
Zinc deficiency may impede proper healing of skin wounds.
Carnitine and selenium deficiencies have been associated with cardiomyopathy in other conditions and may contribute to this finding in DEB.
Osteopenia and osteoporosis is often associated with vitamin D deficiency, and results from poor nutrition, lack of exposure to adequate sunlight, and inactivity [
Martinez & Mellerio 2010,
Rodari et al 2017].
Ocular. Corneal erosions can lead to scarring and loss of vision [Matsumoto et al 2005].
Cardiac. Dilated cardiomyopathy, sometimes associated with selenium and carnitine deficiency, has been reported in RDEB and can be fatal in some cases [Lara-Corrales et al 2010, Ryan et al 2016].
Urologic/renal. Urethral erosions, strictures, bladder dysfunction, and glomerulonephritis can occur, sometimes leading to renal failure [Fine et al 2004].
Orthopedic. Individuals with RDEB tend to get contractures and pseudosyndactyly of the fingers resulting in a "mitten" or "cocoon" hand and consequent impairment of function and decreased quality of life [Eismann et al 2014]. Although fusion of the toes is not detrimental to function, painful blistering and progressive contractures of the foot and ankle as well as the larger joints (knees, hips, neck) can interfere with ambulation and function.
Psychosocial. Severe stress may affect the affected individual and family because of the complications of this disorder and the chronic pain endured by most individuals with EB. Quality of life can be decreased and psychosocial disorders including anxiety, depression, and drug dependence/abuse may occur in older persons [Frew & Murrell 2010] – although a recent study showed that pain in individuals with DEB is not correlated with anxiety or depression [Fortuna et al 2016].
Recessive DEB Generalized and Localized
Multiple clinical phenotypes make up the spectrum of RDEB, many of which are not as severe as RDEB-sev gen. The phenotype may be mild, with blistering localized to hands, feet, knees, and elbows as well as dystrophic nails, or relatively more widespread including flexural areas and trunk, but without the severe, mutilating scarring seen in RDEB-sev gen. Onset of blistering ranges from birth to childhood depending on type.
Some distinctive features of the less common RDEB-gen and -loc variants:
RDEB inversa. Blistering and skin atrophy occurs on the trunk, neck, thighs, and legs while few changes are observed on the hands, feet, elbows, or knees. Otherwise, the
phenotype resembles DEB types with blistering and resulting scarring. Blisters of the hands and feet may be present in infancy.
RDEB pretibial and pruriginosa often affect the shins. Pretibial blisters develop into prurigo-like hyperkeratotic lesions. The lesions occur predominantly on the pretibial areas, sparing the knees and other parts of the skin. Other findings include nail dystrophy, small, white scars (albopapuloid skin lesions), and hypertrophic scars without pretibial predominance.
RDEB generalized intermediate (RDEB-gen intermediate) exhibits widespread blistering with scarring, milia, and nevi. Pseudosyndactyly may occur along with oral lesions and damaged or absent nails. Growth retardation is possible but not as severe as with RDEB-sev gen. Squamous cell carcinoma also develops in some affected individuals.
RDEB localized exhibits blistering with scarring which may be severe but is localized to the hands and feet. Other sites are not affected. The nails are often absent. Growth restriction and systemic illness are also absent. Squamous cell carcinoma has not been reported in individuals who have this subtype.
RDEB centripetalis (RDEB-CE) is apparent at birth and involves the hands, feet, and pretibial areas only. Nails are absent. Growth retardation and systemic illness have not been reported. Squamous cell carcinoma has not been reported in individuals with this subtype.
Dominant DEB (DDEB)
In this milder form of DEB, blistering is often limited to the hands, feet, knees, and elbows. Blistering may be relatively benign but nonetheless heals with scarring. Dystrophic nails, especially toenails, are common and loss of nails may occur. In the mildest forms, dystrophic nails may be the only characteristic noted [Dharma et al 2001, Sato-Matsumura et al 2002, Tosti et al 2003]. Blistering in DDEB often improves somewhat with age, possibly as a result of reduced physical activity. The subtypes of DDEB resemble those of RDEB but may present with milder manifestations. There may be great clinical variability among members of the same family.
DDEB generalized (DDEB-gen) is a milder form of EB in which a single
pathogenic variant in
COL7A1 results in a generalized blistering disease that affects most sites of friction in infancy but often evolves to less severe disease in adulthood. Blisters form with scarring and the nails are often absent. Other systems are generally unaffected and growth retardation and squamous cell carcinoma are rarely reported.
DDEB pretibial and pruriginosa represent the same phenotypes as RDEB pretibial and pruriginosa (see
above); however,
heterozygous pathogenic variants in
COL7A1 lead to an
autosomal dominant pattern of inheritance.
DDEB localized, nails only affects the nails, which are dystrophic and fragile. No skin findings are identified. Other family members, however, may have more severe manifestations.
Penetrance
Until recently, pathogenic variants in COL7A1 were considered to be 100% penetrant when family members were evaluated for mild features of the disease. However, in several families, an individual with DDEB and a known COL7A1 pathogenic variant had relatives with the same variant who had no signs of the disease. Penetrance therefore appears to be less than 100%, at least in DDEB [Almaani et al 2011; Authors, unpublished observations].
Nomenclature
Recessive DEB severe generalized (RDEB-sev gen) was originally called Hallopeau-Siemens type (RDEB-HS).
Recessive DEB generalized intermediate (RDEB-gen intermed) and RDEB localized (RDEB-loc) were originally called non-Hallopeau-Siemens type (RDEB-non-HS).
The nomenclature for DEB has changed four times in the last 15 years. The most recent classification system, referred to as the "onion skin" terminology, arose from an international consensus meeting, the recommendations of which were published in June 2014 [Fine et al 2014]. This classification system starts by dividing DEB into the inheritance pattern and follows with a histologic description of collagen VII staining, then the specific COL7A1 pathogenic variant that has been described in the affected individual (see Table 2).
For information on the newest nomenclature recommendations that pertain to epidermolysis bullosa simplex and junctional epidermolysis bullosa, see Table 3 (pdf).
Table 2.
Comparison of 2008 DEB Nomenclature with Proposed "Onion Skin" Terminology – Representative Examples
View in own window
| Old Name 1 | 2014 Nomenclature |
|---|
| RDEB, severe generalized | RDEB generalized severe, collagen VII absent, COL7A1 pathogenic variants (specify type) |
| RDEB, generalized other | RDEB generalized intermediate, collagen VII reduced staining, COL7A1 pathogenic variants (specify type) |
| DEB-BDN | DEB-BDN, granular intraepidermal collagen VII staining, COL7A1 AD or AR pathogenic variants (specify) |
| DDEB generalized | DDEB generalized, normal collagen VII staining, COL7A1 pathogenic variant (specify) |
BDN = bullous dermolysis of newborn; DDEB = dominant dystrophic epidermolysis bullosa; RDEB = recessive dystrophic epidermolysis bullosa
- 1.
Prevalence
According to the National EB Registry, the overall prevalence of EB is 11.07 per one million live births [Fine 2016]. The prevalence of DDEB and RDEB, respectively, is 1.49 and 1.35 per one million live births.
The carrier frequency of RDEB in the US population has been estimated at one in 370 [Pfendner et al 2001].
Differential Diagnosis
The four major types of epidermolysis bullosa (EB) syndrome, caused by pathogenic variants in 20 different genes, are EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB), and Kindler syndrome (see Table 4). While agreement exists as to diagnostic criteria for some types of epidermolysis bullosa, the validity of rarer subtypes and their diagnostic criteria are disputed. See Murrell [2010] for excellent clinical reviews and Fine et al [2014] (full text; note especially Tables I and VII) for the revised classification system.
The four major types of EB share fragility of the skin, manifested by blistering and/or erosions with little or no trauma. A positive Nikolsky sign (blistering of uninvolved skin after rubbing) is common to all types of EB. No clinical findings are specific to a given type; thus, establishing the EB type requires further laboratory evaluation. Molecular genetic testing may be used to establish a diagnosis (see Establishing the Diagnosis). Alternatively, a fresh skin biopsy from a newly induced blister that is stained by indirect immunofluorescence for critical basement membrane protein components can be performed. The diagnosis is established by determining the cleavage plane and the presence/absence and distribution of these protein components. Electron microscopy is also diagnostic and often more useful in milder forms of EB.
Clinical examination is useful in determining the extent of blistering, the presence of oral and other mucous membrane lesions, and the presence and extent of scarring.
The limitations of clinical findings in establishing the type of EB include the following:
In young children and neonates, the extent and severity of blistering and scarring may not be established or significant enough to allow identification of EB type.
Mucosal and nail involvement and the presence or absence of milia may not be helpful discriminators.
Post-inflammatory changes such as those seen in EBS, Dowling-Meara type (EBS-DM) are often mistaken for scarring or mottled pigmentation.
Scarring can occur in EB simplex and junctional EB as a result of infection of erosions or scratching, which further damages the exposed surface.
Congenital absence of the skin can be seen in any of the three major types of EB (i.e., EBS, JEB, DEB) and is not a discriminating diagnostic feature.
Table 4.
Epidermolysis Bullosa Types
View in own window
| Clinical Features 1 | EB Type | EB Subtype | Gene | MOI | Skin Biopsy Findings
(Level of Cleavage) |
|---|
| Extent of Blistering | Presence of Oral/
Other Mucous
Membrane Lesions | Presence/Extent
of Scarring | Other (Associated Gene) 1 |
|---|
| Mild to severe depending on gene &/or variant | Mucous membrane involvement in severe forms |
| Dilated cardiomyopathy &/or woolly hair (DSP) Ectodermal dysplasia (PKP1) Right ventricular cardiomyopathy (JUP) Muscular dystrophy &/or pyloric atresia (PLEC) Hereditary sensory & autonomic neuropathy Type VI (DST) Alopecia & cardiomyopathy (KLHL24)
| EBS 3 | EBS suprabasal |
TGM5
| AR | In all EBS: a split above dermal-epidermal junction at ultrastructural level |
|
DSP
|
|
PKP1
|
|
JUP
|
| EBS basal |
KRT5
| AD
(AR 2) |
|
KRT14
|
|
CD151
|
|
EXPH5
| AR |
|
PLEC
|
|
DST
|
|
KLHL24
|
| Moderate to severe depending on type of pathogenic variant |
| Granulation tissue formed on skin around oral & nasal cavities, fingers, & toes, & internally around upper airway & nails suggests JEB-GS. | Hoarseness Prone to sepsis Alopecia (COL17A1 especially) Urogenital anomalies (ITGB4) Congenital interstitial lung disease, nephrotic syndrome (ITGA3)
| JEB | JEB severe generalized |
LAMA3
| AR | In all JEB: a split at level of lamina lucida, either:
W/in lamina lucida; or Occasionally, just above lamina lucida at hemidesmosomes (seen w/pathogenic variants in COL17A1, ITGB4, & ITGA6)
|
|
LAMB3
|
|
LAMC2
|
JEB
generalized
& localized |
LAMA3
|
|
LAMB3
|
|
LAMC2
|
|
COL17A1
|
|
ITGB4
|
| JEB late onset |
COL17A1
|
| JEB w/pyloric atresia |
ITGB4
|
|
ITGA6
|
| JEB w/respiratory & renal involvement |
ITGA3
|
| JEB-LOC syndrome 4 |
LAMA3A
|
| Extensive; predominantly on hands & feet | Esophageal strictures in RDEB | Pseudosyndactyly (mitten deformities) caused by scarring of hands & feet in older children & adults in RDEB | Corneal erosions Dystrophic or nail loss
| DEB | RDEB severe generalized |
COL7A1
| AR | Forms below the basement membrane (in superficial dermis) |
| RDEB generalized & localized |
| DDEB (all subtypes) | AD |
| Fragile thin skin w/acral blistering |
| Skin atrophy w/tissue paper appearance |
| Kindler syndrome | NA |
FERMT1
| AR | Multiple cleavage planes; specifically epidermal, lamina lucida, & sublamina densa (multiple cleavage planes are unique to mechanobullous disorders) |
AD = autosomal dominant; AR = autosomal recessive; DDEB = dominant dystrophic epidermolysis bullosa; EB = epidermolysis bullosa; EBS = epidermolysis bullosa simplex; JEB = junctional epidermolysis bullosa; JEB-GS = generalized severe junctional epidermolysis bullosa; MOI = mode of inheritance; RDEB = recessive dystrophic epidermolysis bullosa
- 1.
No clinical findings are specific to a given EB type; however, some clinical findings are more likely to be associated with a single type of EB.
- 2.
- 3.
EBS is divided into suprabasal and basal EBS defined by the level of the cleavage.
- 4.
Laryngoonychocutaneous (LOC) syndrome, or JEB-LOC (OMIM 245660), is described in Punjabi Indians. JEB-LOC has many phenotypic characteristics similar to non-Herlitz junctional epidermolysis bullosa (NH-JEB) [Figueira et al 2007, Pfendner et al 2007]. Skin fragility manifests as mild blistering and erosions of the hands and face that spread to other parts of the body and heal with crusted lesions. Neonates may have a hoarse cry and later laryngeal abnormalities and growths, conjunctival disease, abnormal nails, and hypoplastic dental enamel. Eventually, conjunctival disease may cause blindness and laryngeal disease may cause life-threatening airway obstruction requiring tracheotomy [Cohn & Murrell 2010].
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with dystrophic epidermolysis bullosa (DEB), the following evaluations are recommended if they have not already been completed.
Dermatologic and skin cancer risk
Thorough evaluation of total skin surface for blisters, erosions, and infections
Evaluation of crusted, nonhealing, or painful lesions in older individuals for squamous cell carcinoma (SCC)
Oral, gastrointestinal, and nutritional
Examination of the mouth including mucosal blistering and erosions, dental caries, and crowding
Barium swallow for esophageal strictures if there are symptoms of dysphagia
Measurement of height, weight, and BMI to evaluate nutritional status and need for gastrostomy feeding
Baseline laboratory examination for anemia and nutritional status (see
Surveillance)
Baseline DXA (dual-energy x-ray absorptiometry) scan for osteoporosis if diagnosis occurs in an older individual
Ocular. Ophthalmologic examination to evaluate for corneal abrasions and scars
Cardiac. Baseline echocardiogram
Urologic/renal. Baseline urinalysis to assess for hematuria and proteinuria
Orthopedic. Evaluation of hand function and mobility/dexterity status by a physical or occupational therapist
Gynecologic. Evaluation of pubertal status if individual is an adolescent at diagnosis
Psychosocial (psychology and social work)
Evaluation by professional to assess for anxiety, depression, substance dependency and abuse
Assistance with school-related issues
Assistance with access to needed services, insurance, and handicapped accommodations
Genetics. Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Dermatologic. Families must decide which of the many correct and effective methods of bandaging works for them: there is no "best" method. In general, however, blisters should be lanced and drained to prevent spread from fluid pressure [Denyer 2010, Pope et al 2012, El Hachem et al 2014].
In most cases, dressings for blisters and erosions involve three layers:
Primary (base) layer. A primary nonadherent dressing that does not stick to the skin. Tolerance to different primary layers varies. Primary layers may include any of the following:
Nonstick products with or without silicone surfaces that are nonadhesive
Dressings impregnated or covered with an emollient (such as petrolatum) or a topical antiseptic (such as silver, medical-grade honey, or topical antibiotics if there is infection)
Secondary layer. This layer provides stability for the primary layer and adds padding for protection. Rolls of soft gauze are commonly used.
Tertiary layer. This layer usually has some elastic properties that secure the integrity of the primary and secondary dressings.
Nonhealing wounds may require coverage with biologic skin substitutes or temporary porcine or human cadaver skin grafts. New products involving gene-corrected autologous cells are currently in clinical trials (see Therapies Under Investigation).
Pain and itch are major factors affecting quality of life in individuals with DEB [Goldschneider et al 2014, Danial et al 2015]. A variety of topical, oral, and psychological therapies have been advocated.
Skin infection is common in DEB. All open wounds in EB eventually become colonized with bacteria. Clinical judgment must determine when there is significant infection that requires treatment. Many affected individuals become infected with resistant bacteria, most often methicillin-resistant Staphylococcus
aureus (MRSA), Pseudomonas
aeruginosa, and Streptococcus. Both antibiotics and antiseptics need to be employed.
Squamous cell carcinoma (SCC). A variety of approaches to treatment of SCC are summarized in a 2016 publication [Mellerio et al 2016]. There is no well-accepted standard of care.
Oral, gastrointestinal, and nutritional. In infants and children with RDEB with more severe involvement, poor growth may be a problem, requiring additional nutritional support including a feeding gastrostomy when necessary to assure adequate caloric intake [Stehr et al 2008, Mortell & Azizkhan 2010, Hubbard 2016].
Esophageal strictures and webs can be dilated repeatedly to improve swallowing [Castillo et al 2002, Kay & Wyllie 2002, Azizkhan et al 2006].
Fluid and electrolyte problems, which can be significant and even life-threatening in the neonatal period and in infants with widespread disease, require careful management.
Anemia is a chronic problem with RDEB and can be treated with oral iron supplements, intravenous iron infusions, and/or red blood cell transfusions.
Treatment of other nutritional deficiencies includes:
Calcium and vitamin D supplementation and intravenous bisphosphonates for osteopenia and osteoporosis;
Selenium and carnitine replacement when levels are low to possibly help prevent dilated cardiomyopathy;
Zinc replacement when levels are low to enhance wound healing.
Good dental care is essential to insure the ability to eat and to allow for adequate caloric intake [Harris et al 2001]. Extractions for dental caries and crowding may be needed.
Ocular. Prophylactic use of eye lubricants and, in some cases, protective contact lenses, may prevent corneal abrasions.
Cardiac. Refer to a cardiologist if cardiomyopathy is detected on echocardiogram; medical treatment with beta blockers and/or ACE inhibitors may be able to control or reverse it.
Urologic/renal. Refer to a urologist for urethral erosions, strictures, or bladder dysfunction or to a nephrologist when hematuria or proteinuria is present, as glomerulonephritis and renal failure may occur [Almaani & Mellerio 2010].
Orthopedic
Gynecologic. Delayed puberty is common in girls with poor nutrition.
Psychosocial. Psychosocial support including social services and psychological counseling is essential for managing affected individuals and their families.
Prevention of Primary Manifestations
If a fetus is known to be affected with any form of DEB, cesarean delivery may reduce trauma to the skin during delivery.
Age-appropriate play involving activities that cause minimal trauma to the skin is encouraged.
Dressings and padding are needed to protect bony prominences from blister-inducing impact.
Surveillance
Table 5.
Recommended Surveillance for Individuals with DEB
View in own window
| System/Concern | Evaluation | Frequency |
|---|
|
Blood
|
| Every 6-12 mos to evaluate for anemia |
|
Kidney
|
| Every 6-12 mos to evaluate renal function & for cystitis |
|
Liver
| Liver function tests | Every 6-12 mos to evaluate for liver function 1 |
|
Skin
| Zinc | Every 6-12 mos for wound healing |
|
Bones
| 25-OH vitamin D3 | Every 6-12 mos due to risk of osteoporosis |
| Bone density scan | Annually for osteoporosis |
|
Heart
| Selenium & carnitine | Every 6-12 mos due to risk for cardiomyopathy w/low levels |
| Echocardiogram | Annually to monitor for cardiomyopathy |
|
Immune system
|
| Every 6-12 mos to evaluate for inflammation |
- 1.
Rarely, individuals with RDEB can develop fulminant and even fatal hepatic failure. Some of the medications used in severe RDEB for pain, itch, and/or infection may have adverse effects on liver function.
Because the lifetime risk of metastatic squamous cell carcinoma is greater than 90% in individuals with RDEB, surveillance at least yearly in the second decade of life for wounds that do not heal, have exuberant scar tissue, or otherwise look abnormal is essential. Frequent biopsies of suspicious lesions may be necessary followed by local excision. Affected individuals are often unwilling to undress completely in the clinic setting and home photographs during dressing changes may have to suffice.
Agents/Circumstances to Avoid
Poorly fitting or coarse-textured clothing and footwear should be avoided as they can cause trauma.
In general, activities that traumatize the skin (e.g., hiking, mountain biking, contact sports) should be avoided; affected individuals who are committed to participation in such activities should be encouraged to devise ways of protecting the skin.
Most persons with DEB cannot tolerate the use of ordinary medical tape or Band-Aids®.
Evaluation of Relatives at Risk
Evaluating an at-risk newborn for evidence of blistering is appropriate so that trauma to the skin can be avoided as much as possible.
Given the intrafamilial clinical variability, it is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual by molecular genetic testing of the COL7A1 pathogenic variant in the family in order to identify as early as possible those who would benefit from prompt initiation of treatment and preventive measures.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Data on pregnancy and EB are limited, but a recent survey did not detect increased risk for pregnancy-related complications in women with EB [Intong et al 2017].
Therapies Under Investigation
There are several promising therapies under current investigation including genetically corrected autologous epidermal grafts [Siprashvili et al 2016], various stem cell therapies including bone marrow transplant, mesenchymal stem cells, and IPS cells [Tamai & Uitto 2016], and gene-corrected fibroblasts [Jacków et al 2016].
A recent expedited clinical trial has shown the efficacy of autologous transgenic keratinocyte cultures used to regenerate an entire, fully functional epidermis on a child age seven years suffering from severe JEB [Hirsch et al 2017]. Similar studies are under way for DEB [Siprashvili et al 2016].
Using murine models of RDEB and murine wound models, it has been demonstrated that cultured dermal fibroblasts (either from unaffected human subjects or from individuals with RDEB, engineered to express the collagen 7 protein [C7]) can be injected into murine skin or transplanted RDEB skin equivalents and that the injected cells then secrete C7 into the papillary dermis. There, C7 incorporates into the dermal-epidermal junction (DEJ), forms new anchoring fibrils (AFs), and reverses the RDEB phenotype of poor epidermal-dermal adherence. It has also been shown that the cells could be administered intravenously (IV) at home to open wounds in the skin, promoting healing. This suggests that such cells, injected IV into an individual with RDEB, could localize within healing wounds and continually secrete C7 that could then incorporate into the DEJ and form new AFs that promote healing [Woodley et al 2013].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
If a parent of the
proband is affected and/or is known to be
heterozygous for the
COL7A1 pathogenic variant, the risk to sibs of inheriting the variant is 50%. Intrafamilial clinical variability and reduced
penetrance have been observed.
Offspring of a proband. Each child of an individual with DDEB has a 50% chance of inheriting the COL7A1 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the COL7A1 pathogenic variant, the parent's family members may be at risk.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
The parents of a child with recessive dystrophic epidermolysis bullosa (RDEB) are obligate heterozygotes (i.e., carriers of one
COL7A1 pathogenic variant).
Heterozygous parents of a
proband with RDEB are typically asymptomatic.
Heterozygosity for a
COL7A1 glycine substitution variants in the Gly-X-Y
domain may or may not result in DEB; some parents
heterozygous for this type of
COL7A1 pathogenic variant are affected and some are asymptomatic. This variation can occur even within members of the same family [
Almaani et al 2011].
Sibs of a proband
At conception, each sib of an individual with RDEB whose parents are both carriers has a 25% chance of being affected, a 50% chance of being an asymptomatic
carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are typically asymptomatic (see
Parents of a proband).
Offspring of a proband. The offspring of an individual with RDEB are obligate heterozygotes (carriers) for a pathogenic variant in COL7A1.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a COL7A1 pathogenic variant.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the COL7A1 pathogenic variants in the family.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Dystrophic Epidermolysis Bullosa: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
View in own window
|
120120 | COLLAGEN, TYPE VII, ALPHA-1; COL7A1 |
|
131705 | TRANSIENT BULLOUS DERMOLYSIS OF THE NEWBORN; TBDN |
|
131750 | EPIDERMOLYSIS BULLOSA DYSTROPHICA, AUTOSOMAL DOMINANT; DDEB |
|
131850 | EPIDERMOLYSIS BULLOSA DYSTROPHICA, PRETIBIAL |
|
132000 | EPIDERMOLYSIS BULLOSA WITH CONGENITAL LOCALIZED ABSENCE OF SKIN AND DEFORMITY OF NAILS |
|
226600 | EPIDERMOLYSIS BULLOSA DYSTROPHICA, AUTOSOMAL RECESSIVE; RDEB |
|
226650 | EPIDERMOLYSIS BULLOSA, JUNCTIONAL 1A, INTERMEDIATE; JEB1A |
|
604129 | EPIDERMOLYSIS BULLOSA PRURIGINOSA |
Molecular Pathogenesis
COL7A1 is expressed in the keratinocytes including the basal keratinocytes of the epidermis where the protein products are assembled into homotrimeric molecules with a helical triple collagen domain. The homotrimers then associate via disulfide bonds into homodimeric structures in the extracellular matrix below the lamina densa and form the anchoring fibrils that anchor the basement membrane to the underlying dermis. The anchoring fibrils are linked to the basement membrane through attachment to laminin 5 and the keratinocyte hemidesmosomes directly above. The intracellular keratin intermediate filament network is linked directly to the hemidesmosomes that anchor the keratinocytes to the basal lamina and to the desmosomes that lead to strong attachment of the keratinocytes to one another. These associations along with the network itself supply stability and resistance to stress that enable the keratinocytes to maintain their structural integrity during minor trauma and remain anchored to the basement membrane and dermis [Bruckner-Tuderman 1999].
Pathogenic variants in COL7A1 can lead to reduced resistance to minor trauma and the resulting blistering that is the hallmark of DEB. The type of pathogenic variant, the biochemical properties of the substituted amino acid, and its location in the protein determine the severity of the blistering phenotype (see Genotype-Phenotype Correlations) and inheritance pattern. Intrafamilial phenotypic variability in dominant DEB suggests that other factors can affect the resistance of the cells to friction [Anton-Lamprecht & Gedde-Dahl 2002, Ortiz-Urda et al 2005].
Gene structure. The normal cDNA comprises 9.2 kb with an open reading frame of 8,833 nucleotides encoding 2,944 amino acids in 118 exons spanning 32 kb. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Glycine substitution variants in the triple helical domain (Gly-X-Y; especially in exons 73, 74, and 75) predominate (>75%) in DDEB. p.Gly2034Arg and p.Gly2043Arg are the most common DDEB-causing pathogenic variants, making up 50% of the dominant pathogenic variants reported in the largest US cohort [Varki et al 2007]. Glycine substitutions as well as other amino acid substitutions and splice junction variants outside of this region may also be found in dominant DEB; often, however, inheritance pattern cannot be predicted without determination of parental phenotype and corresponding genotype.
More than 700 COL7A1 recessive DEB-causing variants spanning the entire gene have been described for all forms of DEB [Ashton et al 1999, Mellerio et al 1999b, Whittock et al 1999, Gardella et al 2002a, Murata et al 2004, Sawamura et al 2005, Varki et al 2007]. Common pathogenic variants have been described in certain ethnic backgrounds – including c.497dupA [Ashton et al 1999, Gardella et al 2002a], c.2470dupG [Mellerio et al 1999b], p.Arg578Ter [Whittock et al 1999], c.3840delC [Whittock et al 1999], and c.4919delG [Whittock et al 1999] – and are recurrent in the US population. Each, however, accounts for no more than 1%-2% of the total number of pathogenic variants described. Null variants predominate in RDEB, though glycine substitutions and other amino acid substitutions have been described. Milder forms of RDEB are often caused by splice junction variants or other missense variants.
Table 6.
COL7A1 Variants Discussed in This GeneReview
View in own window
DNA Nucleotide Change
(Alias 1) | Predicted Protein Change | Reference Sequences |
|---|
c.497dupA
(c.497insA) | p.Val168GlyfsTer12 |
NM_000094.3
NP_000085.1
|
| c.1732C>T | p.Arg578Ter |
c.2470dupG
(c.2470insG) | p.Asn825LysfsTer41 |
| c.3840delC | p.Gly1281ValfsTer44 |
| c.4919delG | p.Gly1640ValfsTer70 |
| c.6100G>A | p.Gly2034Arg |
| c.6127G>A | p.Gly2043Arg |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
Normal gene product. Collagen VII is a monomer of 2,944 amino acids that associates into a homotrimer with a triple helical collagenous domain. The homotrimers then associate via disulfide bonds into homodimeric structures that form the anchoring fibrils.
Abnormal gene product. Recessive DEB usually results from the absence of the COL7A1 gene product as a result of premature termination codon variants on both alleles, or splicing variants leading to large deletions or out-of-frame gene products – although some glycine substitution variants have been demonstrated to have autosomal recessive as well as autosomal dominant inheritance and may lead presumably to an absent gene product (recessive) or defective gene product (dominant).
In dominant DEB, collagen VII with a glycine substitution in the collagenous domain (Gly-X-Y) may result in abnormal triple helical coiling and a partially nonfunctional protein product. These proteins may exhibit altered morphology on electron microscopy while immunofluorescent staining may be normal or slightly reduced in intensity, making diagnosis by immunofluorescent staining of a skin biopsy difficult unless a cleavage plane is present. In addition, in-frame exon skipping may serve to modulate disease severity in recessive disease and generate a partially functional gene product [McGrath et al 1999, Varki et al 2007].
Individuals with recessive DEB severe generalized (RDEB-sev gen) have a greater-than-90% lifetime risk of aggressive metastasizing squamous cell carcinoma. The reason for the elevated risk was not clear until the study of Ortiz-Urda et al [2005], which examined Ras-driven tumorigenesis in RDEB keratinocytes and found that cells lacking collagen VII did not form tumors in mice, whereas those retaining a specific collagen VII fragment (the amino-terminal noncollagenous domain NC1) were tumorigenic. Restoring NC1 expression restored tumorigenicity in collagen VII-deficient cells. They concluded that tumor-stroma interactions mediated by collagen VII promote neoplasia, and retention of NC1 sequences in a subset of individuals with RDEB may be a factor in their increased susceptibility to squamous cell carcinoma.