Clinical Description
Poikiloderma with neutropenia (PN) is characterized by post-inflammatory poikiloderma and chronic noncyclic neutropenia typically associated with recurrent sinopulmonary infections and often bronchiectasis. There is increased risk for myelodysplastic syndrome, which may evolve into acute myelogenous leukemia, and skin squamous cell carcinoma. Other ectodermal findings include thickened nails, nail dystrophy, and palmar/plantar hyperkeratosis. Most affected individuals also have reactive airway disease, and some have short stature, hypogonadotropic hypogonadism, midfacial retrusion, calcinosis cutis, and non-healing skin ulcers [Colombo et al 2012]. Intrafamilial clinical variability has been observed. The clinical information that follows is based on around 100 individuals with a clinical and molecular diagnosis of PN [Larizza 2024].
Table 2.
Poikiloderma with Neutropenia: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
|
Poikiloderma
| >95% | Post-inflammatory |
|
Palmar/plantar hyperkeratosis
| 50%-75% | |
|
Nail abnormalities
| >95% | Thick nails, dystrophic nails |
|
Hair abnormalities
| 50%-75% | |
|
Dental abnormalities
| 20%-40% | |
|
Neutropenia
| >95% | Chronic noncyclic, moderate to severe |
|
Recurrent infections
| 90% | Respiratory infections, otitis media, sinusitis, cellulitis |
|
Malignancies
| 5%-10% | Myelodysplasia, hematologic malignancies; rarely, skin cancer |
|
Facial dysmorphism
| 50%-75% | Frontal bossing, midface hypoplasia, depressed nasal bridge, hypertelorism |
Ectodermal Features
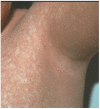
Skin. Typically, the skin is normal at birth, and at age six to 12 months, a nonpruritic acral eczematous-like rash develops that progresses to the trunk and face. Rarely, the rash can start on the face [Tadros et al 2021]. Over the next year or so the inflammatory rash resolves, the skin becomes dry, and poikiloderma becomes evident as areas of hyper- and hypopigmentation, atrophy, and telangiectasias develop (see ). Poikiloderma is not photodistributed, persists throughout life, and may be more noticeable in individuals who have constitutionally darker skin.
Palmar/plantar hyperkeratosis is common, can range from mild to severe, and can be debilitating [Concolino et al 2019, Akdogan et al 2020, Bilgic Eltan et al 2021].
Calcinosis cutis – small nodules that may be localized to the elbows, knees, and pinnae or can be more diffuse – may develop in childhood [Clericuzio et al 2011, Bishnoi et al 2021, Kreuter et al 2021, Tadros et al 2021, Parajuli et al 2024].
Children and adults are prone to cellulitis (a manifestation of neutropenia) that may progress to non-healing skin ulcers or even abscesses [Parperis et al 2020, Kreuter et al 2021, Roebke et al 2021, Parajuli et al 2024]. One individual has been described with pyoderma gangrenosum [Al Haddabi et al, unpublished data].
Photosensitivity and blistering have been reported in a few individuals [Kreuter et al 2021, Vahidnezhad et al 2021, Peterson et al 2022].
Squamous cell carcinoma of the skin has been reported in a handful of individuals at young ages (age 13 to 20 years) [Walne et al 2010, Rodgers et al 2013, Colombo et al 2018, Hertel et al 2018, Concolino et al 2019, Kreuter et al 2021].
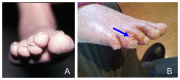
Nails. Thickened, hyperkeratotic toenails and/or fingernails (pachyonychia) are common; dystrophic nails, which can slough, may also be seen (see ) [Bilgic Eltan et al 2021, Bishnoi et al 2021, Kreuter et al 2021, Peterson et al 2022, Yan et al 2023]. Anonychia was observed in one individual [Colombo et al 2012].
A. Nail dystrophy in a girl age five years B. Dysplastic toenails and squamous cell carcinoma (blue arrow) in a girl age 14 years
Hair. Eyebrows and eyelashes are often sparse; hair can be dry and thin [Parperis et al 2020, Bilgic Eltan et al 2021, Kreuter et al 2021, Vahidnezhad et al 2021].
Teeth. Delayed dental eruption, abnormally shaped teeth, and dental abscesses have been observed. Gingivitis and dental caries leading to tooth loss are common [Concolino et al 2019, Bishnoi et al 2021, Kreuter et al 2021, Tadros et al 2021].
Hematologic Findings
Neutropenia/infections. Neutropenia is usually identified in early infancy. Chronic recurrent otitis media and sinusitis are common in childhood. After age five to ten years, the frequency of acute sinopulmonary infections decreases, but most individuals continue to have bronchiectasis, chronic non-productive cough, and reactive airway disease. Almost 90% of individuals with PN have recurrent pulmonary infections, including lung abscesses and lung granulomas.
Most individuals with PN who are not acutely ill have moderate neutropenia, although some have severe neutropenia [Bilgic Eltan et al 2021, Tadros et al 2021, Peterson et al 2022, Yan et al 2023, Parajuli et al 2024]. While the absolute neutrophil count (ANC) may rise to the low-normal range during acute infection, it is always inappropriately low, and with resolution of the infection reverts to baseline neutropenia.
Quantitative immunoglobulins and lymphocyte subset panels are normal.
Transient thrombocytopenia and variable anemia have been reported [Patiroglu & Akar 2015, Walne et al 2016, Colombo et al 2018, Bilgic Eltan et al 2021, Piccolo et al 2021].
Myelodysplasia and Hematologic Malignancies
Bone marrow studies have been described in a portion of reported individuals and have shown hypocellularity and premyelodysplastic changes (often defined as <10% abnormal cells), including increased number of immature cells and myeloid maturation defects. Some individuals with PN initially have normal bone marrow studies and subsequently develop bone marrow changes. Global impaired bone marrow production has been reported, affecting the development of all three major lineages [Concolino et al 2019, Bilgic Eltan et al 2021, Kreuter et al 2021]. Persons with PN can go on to develop myelodysplastic syndrome as well as acute myelogenous leukemia [Colombo et al 2018].
Other Findings
Additional abnormal laboratory findings. Virtually all individuals have elevated serum lactate dehydrogenase of unknown etiology; some have nonspecific mild elevation of aminotransferases, aspartate aminotransferase, ferritin, and creatine phosphokinase [Concolino et al 2019, Bilgic Eltan et al 2021, Kreuter et al 2021, Piccolo et al 2021, Peterson et al 2022].
Hepatosplenomegaly has been described in several individuals [Concolino et al 2019, Bilgic Eltan et al 2021, Piccolo et al 2021, Tadros et al 2021].
Development. Although early developmental delays (possibly related to chronic illness) have been reported, intellectual disability has not been reported.
Muscle weakness. Several individuals with muscle weakness had normal muscle biopsies.
Other rare features