See "Essentials of Glycobiology, 4th Edition"
See the updated version of this chapter
The term “glycome” describes the complete repertoire of glycans and glycoconjugates that cells produce under specified conditions of time, space, and environment. “Glycomics,” therefore, refers to studies that profile the glycome and is the topic of this chapter.
HISTORICAL PERSPECTIVE OF “OMICS” SCIENCE: GENOMICS, TRANSCRIPTOMICS, AND PROTEOMICS
The field of genomics arose from the availability of complete genome sequence data as well as computational methods for their analysis. One of the surprising findings from analysis of the human genome was the presence of fewer protein-encoding genes (a mere 25,000) than had been predicted earlier. Furthermore, the protein-encoding (i.e., “expressed”) genes comprise a small fraction, less than 2%, of the human genome. These genes are transcribed into mRNAs that are often referred to collectively as the “transcriptome.” The ability to analyze transcripts in a high-throughput parallel format using a DNA microarray, or “gene chip,” has enabled researchers to probe global differences in gene expression, for instance, between healthy and diseased cells, between neurons and muscle cells, and between drug-sensitive and drug-resistant cancer cells. Such “transcriptomic” comparisons have revealed networks of genes whose expression is linked to disease.
Although many scientific discoveries have emerged from genomic and transcriptomic approaches, this information still does not provide a complete picture of the physiology of a cell or organism. The proteins expressed by the cell, collectively termed the “proteome,” perform many of the cell’s functions. Most eukaryotic proteins are posttranslationally modified (e.g., by phosphorylation, oxidation, ubiquitination, lipidation, or glycosylation). These modifications, combined with alternative splicing in eukaryotes, render the proteome considerably more complex than the transcriptome. Although it is not known how many discrete proteins a particular human cell expresses, estimates between 50,000 and 120,000 have been suggested. Direct characterization of the proteome is required to understand both its complexity and its global functions. The global systems-level analysis of all proteins expressed by cells, tissues, or organisms is referred to as “proteomics.”
Unlike the genome, which is fixed for most cells, the proteome is dynamic. The repertoire of proteins expressed by a cell is highly dependent on its tissue type, microenvironment, and stage within its life cycle. As cells receive cues in the form of growth factors, hormones, metabolites, or other agents, various genes are turned on or off. Thus, proteomes vary during cell differentiation, activation, trafficking, and during malignant transformation. Also, many proteins are secreted from cells and circulate in the blood or lymphatic fluid or are excreted in the saliva, mucus, tear fluid, or urine. These bodily fluids also have distinct proteomes.
WHAT IS “GLYCOMICS”?
Glycomic analyses seek to understand how a collection of glycans relates to a particular biological event. As described throughout this book, glycans participate in almost every biological process, from intracellular signaling to organ development to tumor growth. Understanding how the totality of glycans governs these processes is a central goal of glycobiology.
The glycomes of life-forms include all of the glycan and glycoconjugate types that have been described in this book. For example, vertebrates possess protein-associated N- and O-glycans, glycosaminoglycans, and GPI anchors, as well as lipid-associated glycans and free glycans such as hyaluronan (see Chapters 8–18). Other organisms possess their own distinct glycomes, with those of plants (see Chapter 22) and prokaryotes (see Chapter 20) differing greatly in composition from the vertebrate and invertebrate glycomes (see Chapter 25). And as with the proteome, each cell type has its own distinct glycome that is governed by local cues and the cell’s internal state. The size of any particular glycome has not yet been established, but we know that glycomes can far exceed proteomes and transcriptomes with respect to complexity. For example, some estimates have placed the vertebrate glycome at more than one million discrete structures. Furthermore, it appears that the glycome is considerably more dynamic than the proteome or transcriptome.
The notion that glycans should be studied as a totality, as well as simply one at a time, is not a radical concept among glycobiologists. Indeed, researchers in the field have long known that glycans form patterns on cells that change during development (see Chapter 38) and cancer progression (see Chapter 44). Also, many glycan-binding proteins are oligomerized on cells and interact with multivalent arrays of glycans on opposing cells (see Chapter 27). In some cases, multiple discrete glycan epitopes work in concert to engage two cells or deliver a signal from one cell to the other. Thus, before “glycomics” was coined, scientists had already concluded that many aspects of glycobiology can be understood only with a systems-level analysis. Conversely, no systems-level analysis of a biological process is complete without interrogating the glycome in addition to the genome, transcriptome, and proteome.
RELATIONSHIP OF THE GLYCOME TO THE GENOME AND PROTEOME
Clues regarding the composition and complexity of the glycome can be found in the cell’s genome, transcriptome, and proteome. As discussed in Chapter 7, genome “mining” using known sequences can identify many genes involved in glycan biosynthesis and processing. By such an analysis, more than 250 glycosyltransferases have been found encoded in the human genome as well as many nucleotide sugar biosynthetic enzymes and Golgi transporters (see Chapter 5). Some of the corresponding enzymes have been studied biochemically and their glycosyl donor and acceptor specificities have been defined, whereas others have been assigned predicted functions based on sequence relationships. Furthermore, expression patterns of many glycosyltransferases have been determined in human and mouse tissues using northern blots, quantitative PCR (polymerase chain reaction), and transcriptomic analyses.
In principle, one might use all of this information to construct “virtual glycomes.” However, this exercise is of limited value because the combinatorial action of glycosyltransferases in many competing biosynthetic pathways renders the complete glycome very difficult to predict with any accuracy. As an example, the reduced expression of a single glycosyltransferase can perturb the biosynthesis of dozens of glycan structures, some negatively and some positively. The direct glycan products of the glycosyltransferase will be reduced in expression, whereas glycans made by other enzymes that compete for common intermediates might increase in levels. Furthermore, unlike the genome and, to our knowledge, the proteome, the glycome can be sensitive to exogenous nutrient levels. Thus, variations in dietary monosaccharides, such as glucose, galactose, glucosamine, fucose, mannose (see Chapter 18), and N-glycolylneuraminic acid (see Chapter 14), can change the composition of the glycome. Because of these complexities, transcriptomic and proteomic data can at best guide hypotheses regarding the presence or absence of specific classes of structures. In contrast, the absence of particular genes (e.g., the sialic acid biosynthesis machinery in Caenorhabditis elegans; see Chapter 14) has been useful in assessing the relative compositions of various glycomes.
The numerous factors that influence the glycome (the genome, the proteome, and environmental nutrients, as well as the secretory machinery, pH, and many other determinants) create a system that is highly diverse and dynamic. Thus, the glycome can change dramatically in response to a subtle change in the cellular system. This feature makes glycomics research both exciting and also daunting. Since neither the proteome nor the transcriptome can accurately predict such a moving target, the glycome must be analyzed directly. Techniques that have been employed to characterize the glycome are summarized below.
TOOLS FOR CHARACTERIZING THE GLYCOME
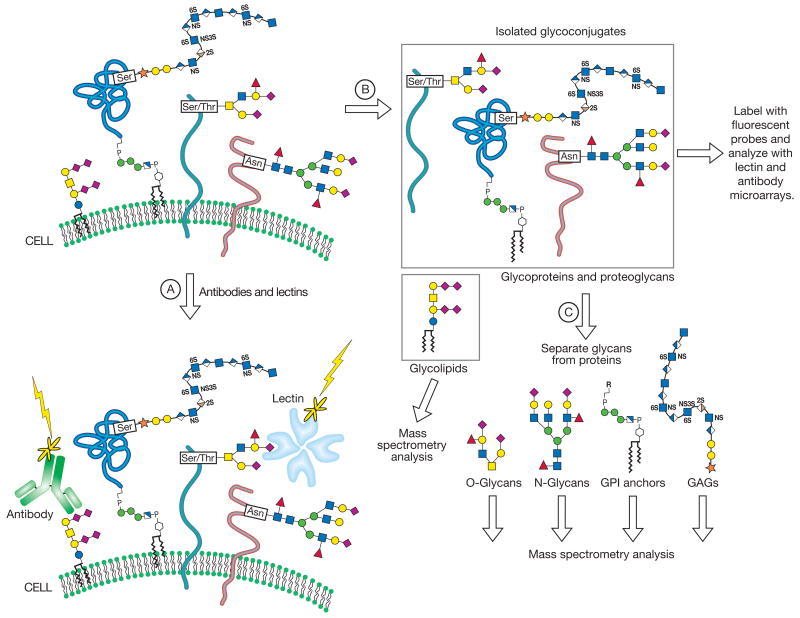
The glycome can be described at many hierarchical levels of complexity (Figure 48.1). First, the glycome can be deconstructed into an inventory of glycan structures separated from their protein or lipid scaffolds and independent of their location in the cell, organ, or organism. This first hierarchical level is essentially a catalog of structures. It is an important starting point for any comprehensive glycome analysis. But how the parts in the catalog assemble to form the intact system is also important for understanding function. Thus, a second hierarchical level of analysis involves defining which glycans are associated with individual proteins or lipids. Analysis of the complete repertoire of a cell’s glycoproteins, including their glycan structures and sites of attachment, lies at the intersection of glycomics and proteomics and is often referred to with the term “glycoproteomics.” A third level of complexity involves determining which glycans or glycoconjugates are expressed on specific cells or tissues. This level of glycomic profiling is essential if the goal is to reveal new functions in cell–cell communication or to correlate particular glycomes with disease tissue. A final level that has yet to be investigated involves visualizing how glycoconjugates are actually organized relative to each other within the cell, at the cell surface, and in the extracellular matrix.
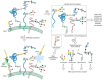
FIGURE 48.1
Multiple approaches for profiling a cell’s glycome at various hierarchical levels of complexity. (Step A) Cells can be directly probed for glycan expression using labeled lectins and glycan-specific antibodies. This top–down experiment (more...)
As described below, numerous techniques have been developed for interrogating the glycome at these various hierarchical levels. No single technique can define all aspects of the glycome. Thus, several approaches are typically employed in parallel, allowing one to assemble a picture of the glycome both from the “bottom up” (i.e., from the individual glycan repertoire) and from the “top down” (i.e., from a global tissue expression analysis). A significant challenge in analyzing the glycome derives from its enormous structural diversity. Different approaches and techniques are required to characterize the structures of glycoproteins versus glycolipids, N-glycans versus O-glycans, and sulfated glycosaminoglycans versus neutral glycans (see Chapter 47). By contrast, a single technique, the DNA microarray, can be used to interrogate all RNA transcripts at once. Thus, at present, the techniques for glycomic analysis remain relatively low-throughput and specific for a particular glycan type, although considerable effort is being directed toward methods that encompass all glycan classes.
Mass Spectrometry
High-resolution mass spectrometry (see Chapter 47) is the primary technique for characterizing the structures of individual glycans when only small quantities are available, as is the case in most glycomic studies. In a typical experiment, a glycoprotein- or glycolipid-enriched sample is prepared from cell lysates and analyzed by multiple rounds of mass spectrometry. In the case of glycoproteins, the N-glycans can be selectively released enzymatically or chemically, separated by HPLC (high-pressure liquid chromatography) methods, and actually sequenced. Separately, the O-glycans are released chemically and sequenced as well. Glycolipids can often be directly sequenced without separation of the lipid component. Glycosaminoglycans are more problematic because of their large size, but small fragments can be sequenced by mass spectrometry in conjunction with enzymatic digestion (see Chapter 16). An advantage of mass spectrometric glycan profiling is that multiple glycans of any given subtype can be profiled at once, increasing the throughput of the glycomic analysis. Still, there is no method at present by which highly complex samples possessing many glycan subtypes can be analyzed in one mass spectrometry experiment. Furthermore, many of the techniques routinely used tend to partially or completely destroy the sample or miss potentially important modifications such as sulfation and O-acetylation.
Mass spectrometry can also be employed to define sites of attachment of glycans to the underlying protein scaffold (i.e., for glycoproteomic analysis). Typically, the glycoprotein-associated glycans are first trimmed to remove peripheral epitopes such as sialic acid and fucose residues. This procedure simplifies the diversity of the pool and therefore sacrifices some of the information in the glycome. Then, the glycoprotein is subjected to tryptic digest and the peptides are analyzed by mass spectrometry using a technique that leaves the glycans attached to the peptide during the analysis (termed electron transfer dissociation [ETD] mass spectrometry). In parallel, the protein can be stripped of its glycans before the tryptic digest and the masses of those naked peptides can be compared to those of the glycopeptides. The differential allows prediction of the attached glycan structure. Furthermore, if an endoglycosidase such as PNGase F is used to release N-glycans (see Chapter 47), the resulting change from asparagine to aspartic acid can mark the site of the original glycosylation. Similarly, β-elimination of O-glycans changes the amino acid at the site of elimination, and can also be followed by Michael addition of nucleophiles such as dithiothreitol to mark the original O-linked sites.
A common problem encountered in proteomic studies is a lack of sensitivity for low-abundance species. The range of protein levels in cells and bodily fluids is thought to span more than eight orders of magnitude. Mass spectrometry detection often suffers from saturation by the most abundant species, leaving those with lower abundance impossible to detect. For glycoproteomic analyses, therefore, it is often beneficial to enrich the sample in glycoproteins and to discard those proteins that do not bear glycans. Lectins have been artfully employed for this purpose. As discussed in Chapter 45, a variety of glycan-binding proteins (e.g., the plant lectins) are commercially available. These proteins can be immobilized on agarose beads and used for affinity purification of glycoproteins from cell lysates or body fluids. Once enriched, the glycoproteins can be analyzed in the absence of abundant unglycosylated protein contaminants.
Lectin and Antibody Arrays
A major benefit of mass spectrometry is the detailed information it provides regarding the structure of a glycan. A drawback, however, is its relatively low throughput and the need for different experimental protocols for each glycan subtype. Lectin and antibody arrays can be employed to interrogate the glycome with much higher throughput, although in considerably less structural detail. As described in Chapter 45, nature has provided a considerable collection of lectins and many have been biochemically characterized. These lectins possess a range of specificities; some recognize a particular monosaccharide in virtually any context, whereas others are very specific for higher-order glycan epitopes or single residues within a defined context. For those epitopes that lack a naturally occurring lectin, one can generate monoclonal antibodies (see Chapter 45). This can be accomplished by immunization of a rodent with a synthetic or isolated version of the glycan attached to an antigenic carrier protein such as keyhole limpet hemocyanin. The monoclonal antibodies generated in this fashion can serve as “artificial lectins.” Single-chain-fragment antibodies generated in bacteria have also proven useful for analysis of glycosaminoglycans. However, recent studies have noted that some antibodies once thought to be highly specific for certain glycans can cross-react with others.
The lectin array employs the same architecture as the DNA microarray or the glycan microarray described in Chapter 27. Lectins (or glycan-specific antibodies) are spatially arrayed on a glass chip by covalent attachment (Figure 48.2). Glycoproteins from the cell lysate or fluid sample of interest are nonspecifically labeled with a fluorescent dye. The sample is then incubated with the array and the fluorescence associated with each pixel is quantified. The pattern of bright spots reflects the glycome of the particular sample. For comparative purposes, two samples can be analyzed in parallel, one labeled with a green dye and one with a red dye. Combining the two samples onto one array allows direct analysis of changes in the glycome. In principle, samples that are even more complex than glycoproteins can be probed using lectin arrays, and indeed intact bacterial cells have been probed using this technique.

FIGURE 48.2
Analysis of cellular glycomes using lectin and antibody arrays. The arrays are generated by immobilization of lectins and glycan-specific antibodies on a chip. Glycoproteins from cell or tissue samples are labeled with a fluorescent dye and then incubated (more...)
The lectin array provides global information about the types of glycan epitopes that are present in the sample but does not give any detailed structural information, nor does the experiment provide information regarding which proteins the glycans are attached to. However, the high-throughput platform allows for rapid comparison of many glycomes in search of global changes that might motivate further mass spectrometry studies.
Cell and Tissue Analysis Using Lectins and Antibodies
Glycobiologists routinely use lectins and glycan-specific antibodies as histological probes of glycan expression. This approach still holds an important place in any comprehensive glycomic analysis, as the cellular and tissue distribution of glycans is an important element of the glycome. In the modern era, tissue-expression patterns observed using lectins and antibodies can be correlated with lectin array data and mass spectrometry profiling data as well as genomic and proteomic data to create a more complete picture of the glycome. In the future, such in situ labeling followed by laser-capture microdissection of specific regions from tissue sections could potentially allow all the techniques to come together.
Imaging the Glycome by Metabolic and Covalent Labeling
A recent addition to the arsenal of tools for glycome analysis is the use of metabolic labels that allow covalent tagging of glycans with imaging probes. As shown in Figure 48.3, cells or organisms can be treated with monosaccharide substrates bearing azido groups, and the downstream metabolic products are incorporated into cellular glycans. The azido groups can be covalently reacted with azide-specific imaging reagents. Once labeled, the glycans can be visualized on cells or tissues by fluorescence microscopy. This procedure has been used to image changes in the glycome during zebrafish embryonic development. The technique provides little structural information regarding the elements of the glycome that are labeled but has the advantage that global changes in the glycome can be monitored in vivo and in real time. By contrast, lectin and antibody reagents are largely restricted to ex vivo analysis of glycomes in cultured cells or tissues.
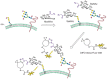
FIGURE 48.3
Metabolic and covalent labeling of glycans for in vivo imaging of the glycome. Cells metabolize azido sugars such as N-azidoacetylmannosamine (ManNAz), which is converted into the corresponding azido sialic acid (SiaNAz) and incorporated into cellular (more...)
COMPARATIVE GLYCOMICS
Because the glycome is influenced by both genetic and environmental factors, the information contained therein might shed light on intraspecies and interspecies variations as well as on changes that have occurred over evolutionary history. From the immediate clinical perspective, the glycome might provide indicators of disease that can be used for diagnosis and for monitoring the efficacy of drugs. Comparative glycomics is therefore an exciting frontier in biology and medicine.
As discussed in detail in Chapter 44, numerous changes in the glycome have been associated with malignancy and metastasis, including altered N- and O-glycosylation, up-regulation of sialylated and fucosylated antigens, and altered heparan sulfates. In some cases, the tumor-associated glycans have been shown to be functionally relevant, whereas in other cases, the association of the glycan with cancer remains correlative. No matter its functional consequence, however, a change in the glycome that is highly correlated with malignancy (or any disease) can serve as a diagnostic biomarker. Given the dearth of such biomarkers for cancer screening, it is not surprising that considerable effort is being directed toward analysis of cancer-associated glycomes.
Already, mass spectrometry has been artfully employed in several glycomic studies of serum samples from healthy and cancer patients. In these studies, O-glycans were released from serum glycoproteins and analyzed by mass spectrometry. Although many structures were similar in the samples from healthy donors and cancer patients, a handful were markedly elevated in the latter. This observation suggests that changes in the serum glycome accompany cancer and that such changes might be used for diagnostic purposes. Notably, we do not need to understand how the disease of interest, in this case cancer, causes changes in the serum glycome in order to use that information for clinical benefit. Indeed, glycans that are altered in the disease sample might not be directly related to the disease at all. Rather, they may reflect downstream consequences of the disease on remote organs or they may reflect changes in the patient’s immune system.
A fundamental question that remains unanswered is what is the extent of natural variation among individual human glycomes? Since the glycome can respond, in principle, to dietary and environmental changes, an equally interesting question is how glycomes around the world vary as a function of local dietary habits and/or medicine use, and further, how do glycome variations relate to acquired disease susceptibility (see Chapter 43)?
Studies of evolutionary biology have also much to gain from comparative glycomics. Evolution of the vertebrate immune system, for example, was accompanied by the acquisition of new glycan-binding proteins including the Siglec (see Chapter 32) and selectin (see Chapter 31) family members. The process by which the glycome evolved to accommodate these developments is of considerable interest. Likewise, the glycomes of microbes and their vertebrate hosts may have coevolved in some instances. The human blood group antigens, for example, are thought to reflect selective pressure induced by bacterial pathogens bearing similar glycan epitopes (see Chapter 39). This dramatic observation may be one of many examples of glycome coevolution across species. Comparative glycomic analysis of humans and their resident microbes, both pathogenic and symbiotic, may be highly revealing.
FUNCTIONAL GLYCOMICS USING GLYCAN MICROARRAYS
Taking inventory of the glycome provides the basis for hypotheses regarding biological function. Studying the functions of glycans is, of course, the central goal of the field of glycobiology. When this goal is pursued using high-throughput techniques, the term “functional glycomics” is often applied. As an example, the relative binding activity of glycans to glycan-binding proteins can be probed in high-throughput parallel fashion using glycan microarrays. These arrays seek to represent a fraction of the glycome and are often generated using glycans isolated from cells or tissue sources. The glycans are typically immobilized on a chip and then exposed to the protein of interest labeled with a fluorescent dye. Binding of the protein to the various glycans is detected by fluorescence imaging.
Arrays have been used for the analysis of a number of glycan-binding proteins, such as plant and microbial lectins, glycan-binding proteins involved in the innate and adaptive immune system, glycan-specific antibodies, viral glycan-binding proteins, and whole cells (see Chapter 27). Two examples of such applications are briefly described below.
DC-SIGN and DC-SIGNR
DC-SIGN and DC-SIGNR belong to the type II transmembrane receptor subfamily of C-type lectins (see Chapter 31). DC-SIGN is abundantly expressed on dendritic cells and plays a key role in adhesion of T cells as well as in the recognition of pathogens such as HIV. Indeed, binding of HIV to DC-SIGN on dendritic cells enhances T-cell infection. The related protein DC-SIGNR shares 77% sequence identity with DC-SIGN, but screening of these two similar proteins using glycan arrays revealed distinct ligand specificities. In addition to the high-mannose structures bound by both receptors, DC-SIGN recognized certain fucosylated ligands that were not bound by DC-SIGNR. This finding may shed light on the functional distinction between the two related proteins.
Influenza Virus Hemagglutinin
Influenza A virus subtypes are avian viruses that are named according to their surface antigens: hemagglutinin (HA) and neuraminidase (NA) (see Chapter 39). These viruses bind to sialylated glycans on host epithelial cells to initiate infection. The HA glycoprotein mediates host-cell recognition and is therefore an important determinant of species tropism. Human viral HA preferentially recognizes glycans terminated by NeuAcα2-6Gal, whereas avian HA preferentially recognizes glycans containing NeuAcα2-3Gal. Likewise, the upper airway epithelial cells in humans contain mainly NeuAcα2-6Gal, whereas in birds both the airways and intestine contain mainly NeuAcα2-3Gal linkages (see Chapters 13 and 14, and the cover figure).
In view of the rapid geographic spread of avian influenza A subtypes such as H5N1 and the increasing numbers of confirmed human cases, it is critical to survey potential influenza outbreaks and monitor human adaptation, which is a key step in the emergence of a pandemic virus. Glycan array technologies have proven to be powerful tools for this purpose. Arrays patterned with sialylated glycans of various linkages and topologies have been generated and screened with viral HA proteins as well as intact viruses. The microarray studies revealed striking glycan binding preferences that were governed not only by the sialic acid linkage and glycan modifications such as fucosylation or sulfation but also by underlying glycan structures. Once integrated into a portable format, glycan arrays may be employed in the field for influenza surveillance, which is a very practical application of functional glycomics techniques.
THE INFORMATICS CHALLENGES OF DIVERSE GLYCOMIC DATA
As mentioned above, a complete picture of the glycome can only be assembled using both top–down and bottom–up approaches. The structures of the individual glycans, their assembly on proteins and lipids, their distribution on cells and tissues, and their relation to each other on cells and within the extracellular matrix all warrant interrogation at the systems level. However, each corresponding experimental platform produces very different types of data. Mass spectral data, which can guide the assignment of a glycan’s primary sequence, have a different form than lectin microarray data. The integration of disparate forms of data to generate a comprehensive picture of the glycome is a major frontier in informatics associated with glycomics research.
A comprehensive systems-level analysis would correlate data that define the glycome at various hierarchical levels with data derived from transcriptomic and proteomic experiments. One would like to know, for example, how the relative expression levels of genes that encode glycan biosynthetic enzymes compare to the glycome observed in a cell or tissue type. The effects of pharmacological agents or gene knockdowns on the glycome as compared to the transcriptome and proteome are also of interest. The functional consequences of perturbing a cell’s glycome on its interactions with other cells might also be cataloged and correlated with additional systems-level data.
At present, we do not have a clear picture of how expression levels of glycan biosynthesis and processing genes relate, at the systems level, to the composition of the glycome. Efforts to correlate large data sets obtained from glycomic, transcriptomic, genomic, and proteomic studies have met with several challenges. Representation of glycan chemical structures is difficult because of their complexity and branching patterns. The use of single alphabet codes, as employed to describe nucleic acid and amino acid sequences, is not applicable to glycans. Rather than the conventional character-based codes used for sequence information in transcriptomic and proteomic data sets, numerical or object-based codes are better suited to link the complex glycan structure information in various glycomic data sets to each other and, ultimately, to the transcriptomic and proteomic data sets. The field is in need of a comprehensive bioinformatics platform that stores, integrates, and processes data from glycomic and other “omic” studies and disseminates them in a meaningful fashion via the Internet to the scientific community.
In recent years, academic and commercial organizations have made a significant effort toward building new databases and bioinformatics platforms that fulfill this goal (GlycoSuiteDB, Sweet, KEGG GLYCAN). International organizations have formed to develop community resources. The Consortium for Functional Glycomics (CFG), EuroCarb, and the Japanese Glycomics Consortia are collaborating to develop technologies for advancing glycomics. These collaborative efforts have resulted in the development of novel experimental resources as well as online searchable databases.
With growing effort directed toward new technology and bioinformatics platforms, the future of comprehensive “omics” science is bright and potentially exciting. The reformatting of existing genomic and proteomic data sets for compatibility with emerging glycomic data sets is under way and stand-alone glycan structure databases are already in place. Once achieved, the integration of large data sets that link the glycome, genome, transcriptome, and proteome will generate a wealth of hypotheses for pursuit by future generations of scientists.
FURTHER READING
- Hirabayashi J. Lectin-based structural glycomics: Glycoproteomics and glycan profiling. Glycoconj J. 2004;21:35–40. [PubMed: 15467396]
- Campbell CT, Yarema KJ. Large-scale approaches for glycobiology. Genome Biol. 2005;6:236. [PMC free article: PMC1297640] [PubMed: 16277754]
- Paulson JC, Blixt O, Collins BE. Sweet spots in functional glycomics. Nat Chem Biol. 2006;2:238–248. [PubMed: 16619023]
- Prescher JA, Bertozzi CR. Chemical technologies for probing glycans. Cell. 2006;126:851–854. [PubMed: 16959565]
- Raman R, Venkataraman M, Ramakrishnan S, Lang W, Raguram S, Sasisekharan R. Advancing glycomics: Implementation strategies at the consortium for functional glycomics. Glycobiology. 2006;16:82R–90R. [PubMed: 16478800]
- Sasisekharan R, Raman R, Prabhakar V. Glycomics approach to structure–function relationships of glycosaminoglycans. Annu Rev Biomed Eng. 2006;8:181–231. [PubMed: 16834555]
- Pilobello KT, Mahal LK. Deciphering the glycocode: The complexity and analytical challenge of glycomics. Curr Opin Chem Biol. 2007;11:300–305. [PubMed: 17500024]
- Timmer MS, Stocker BL, Seeberger PH. Probing glycomics. Curr Opin Chem Biol. 2007;11:59–65. [PubMed: 17208037]
- Turnbull JE, Field RA. Emerging glycomics technologies. Nat Chem Biol. 2007;3:74–77. [PubMed: 17235338]
- Mahal LK. Glycomics: Towards bioinformatic approaches to understanding glycosylation. Anticancer Agents Med Chem. 2008;8:37–51. [PubMed: 18220504]
Publication Details
Author Information and Affiliations
Authors
Carolyn R Bertozzi and Ram Sasisekharan.Copyright
Publisher
Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY)
NLM Citation
Bertozzi CR, Sasisekharan R. Glycomics. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology. 2nd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2009. Chapter 48.