Clinical Description
Together, isolated lissencephaly sequence (ILS) and subcortical band heterotopia (SBH) comprise the "agyria-pachygyria-band" spectrum of cortical malformations that are caused by deficient neuronal migration during embryogenesis. The term lissencephaly refers to a "smooth brain" with absent (agyria) or abnormally wide gyri (pachygyria).
To date, almost 200 individuals have been identified with an intragenic pathogenic variant in PAFAH1B1 [De Vita et al 2018, Di Donato et al 2018] causative of ILS and SBH. The following description of the phenotypic features associated with this condition is based on these reports.
Neurologic
Affected newborns may appear normal or may have mild-to-moderate hypotonia, feeding difficulties, and poor head control. During the first years, neurologic examination typically demonstrates poor visual tracking and response to sounds, axial hypotonia, and mild distal spasticity. Infants often demonstrate abnormal arching (opisthotonus). Later, distal spasticity becomes more prominent, although axial hypotonia remains. Affected individuals may develop moderate spastic quadriplegia and scoliosis.
ILS. Seizures occur in more than 90% of individuals with ILS, with onset usually in the first two years of life (mean onset age: 5 months) [Saillour et al 2009, Herbst et al 2016]. Approximately 80% of affected individuals have infantile spasms, although EEG does not always show the typical hypsarrhythmia pattern.
After the first months of life, most children have mixed seizure disorders including persisting infantile spasms, focal motor and generalized tonic seizures, and atypical absence, atonic, and myoclonic seizures [Herbst et al 2016].
Seizures are drug resistant in more than 65% of affected individuals. While polytherapy with lamotrigine and valproic acid can reduce the number of daily seizures effectively, two thirds of affected individuals continue to experience daily seizures [Herbst et al 2016].
SBH. Seizures typically occur in the first decade and are often drug resistant [Bahi-Buisson & Guerrini 2013, Herbst et al 2016].
Neurodevelopment
Prior to the onset of seizures, most infants have mild delay in development and mild hypotonia.
ILS. Onset of infantile spasms may be associated with a decline in function:
The developmental prognosis is poor for most individuals with ILS.
Even with good seizure control, the best developmental level achieved (excluding the few individuals with partial lissencephaly) is the equivalent of about age three to five months.
Affected individuals may achieve rolling over, limited creeping, and, very rarely, sitting. Few individuals have been reported to be able to walk with support.
Visual tracking is variable among affected individuals, with a subset achieving normal visual interactions while other do not achieve visual tracking.
With poor seizure control, children with ILS may function at or below the level of a newborn. Regression of acquired functioning due to poor seizure control has been reported [
Sicca et al 2003].
A few individuals with less severe (grade 4) lissencephaly (see
Neuroimaging), especially partial posterior lissencephaly or pachygyria, have a better developmental outcome [
Leventer et al 2001].
SBH. Developmental delay (ranging from mild to severe) has been seen in all of the limited number of individuals reported with SBH [Saillour et al 2009, Herbst et al 2016].
Growth
At birth, the occipitofrontal circumference is typically normal (between the mean and -2 SD). However, postnatal head growth is slow; most children develop microcephaly by age one year. The remainder of the growth parameters are often normal.
Feeding
Some have difficulty with feeding, and transient elevations in bilirubin in neonates are likely related to feeding difficulties. Feeding often improves during the first few months of life, but typically worsens again with seizure onset during the first year of life, and then again at several years of age for various reasons.
Poor feeding in newborns is usually managed by nasogastric tube feedings, as the feeding problems often improve during the first weeks of life (see Management,
Treatment of Manifestations).
However, feeding often worsens again with intercurrent illnesses and with advancing age and size. At least 50% of children with PAFAH1B1-related lissencephaly (but not PAFAH1B1-related SBH) eventually have a gastrostomy tube placed for feeding issues.
Individuals with low central tone frequently develop constipation; this is not specific to those with PAFAH1B1-related lissencephaly / subcortical band heterotopia.
Respiratory
Children with lissencephaly have poor control of their airway, which predisposes them to aspiration pneumonia, the most common terminal event. This is not typically seen in those with SBH.
Visual Impairment
Poor or absent visual interaction and limited visual-perceptual and eye-hand abilities have been reported in a subset of affected individuals, likely secondary to the severe cortical malformations [Leventer et al 2001, Mineyko et al 2010, de Wit et al 2011].
Neuroimaging
In 2017, the imaging criteria for lissencephaly/SBH with a defined monogenic cause were revised. This classification system subdivides lissencephaly/SBH into 21 recurring patterns in order to aid clinicians in predicting the most likely genetic cause based on imaging features. Criteria to stratify lissencephaly/SBH are based on the anteroposterior gradient, grade of severity, cortical thickness and appearance, and associated non-cortical brain malformations [Di Donato et al 2017].
Imaging criteria for lissencephaly and subcortical band heterotopia in general
[Di Donato et al 2017]. Note: This is not specific to PAFAH1B1-related lissencephaly/SBH.
Gradient of gyral malformation:
Diffuse
Anterior more severe than posterior (a>p)
Posterior more severe than anterior (p>a)
Temporal more severe than posterior and p>a
Grade of gyral malformation:
SBH partial
SBH diffuse
LIS partial pachygyria
LIS diffuse pachygyria
LIS agyria-pachygyria
LIS diffuse agyria
Cortical thickness & appearance:
Non-cortical brain malformations:
Basal ganglia dysgenesis
Complete or partial agenesis (dysgenesis) of corpus callosum
Tectal hyperplasia
Brain stem hypoplasia & dysgenesis
Cerebellar hypoplasia (either diffuse or vermis predominant)
Applying the above imaging criteria allows correlation of major imaging features with the expected clinical outcome (although exceptions may exist, especially in those with severe epilepsy). The clinical severity of lissencephaly/SBH in general is characterized based on the criteria presented in Table 2 [Di Donato et al 2017]. Note: This is not specific to PAFAH1B1-related lissencephaly/SBH.
Table 2.
Clinical Severity of Lissencephaly and Subcortical Brain Heterotopia in General
View in own window
| Grade | Subtypes | Imaging Pattern | Typical Clinical Outcome |
|---|
|
1 (mild)
| 1-1 | Partial SBH a>p or p>a | Borderline-to-moderate ID, seizures of variable severity; survival into adulthood expected |
| 1-2 | Diffuse thin SBH (<10 mm) |
| 1-3 | Partial pachygyria a>p or p>a |
| 1-4 | Isolated "thin" or undulating LIS |
|
2 (moderate)
| 2-1 | Diffuse thick SBH (>10 mm) | Moderate-to-severe ID, severe language impairment, seizures often poorly controlled; life expectancy may be ↓, although many survive to adulthood. |
| 2-2 | Mixed pachygyria-SBH |
| 2-3 | Diffuse pachygyria a>p or p>a |
|
3 (severe)
| 3-1 | Mixed pachygyria-agyria | Profound ID, poorly controlled seizures, short survival typical: mortality rate ~50% by age 10 yrs w/normal cerebellum; much higher w/cerebellar hypoplasia |
| 3-2 | Diffuse agyria |
| 3-3 | Agyria w/cerebellar hypoplasia |
a = anterior; ID = intellectual disability; LIS = lissencephaly; p = posterior; SBH = subcortical band heterotopia
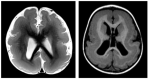
ILS. ILS consists of variable lissencephaly (grades 2-3, 3-1, or 3-2 in Table 2). Other associated brain MRI findings in the most common ("classic") form of lissencephaly:
Enlarged lateral ventricles, especially posteriorly
Mild hypoplasia of the corpus callosum (the anterior portion often appears flattened)
Cavum septi pellucidi et vergae
Normal brain stem and cerebellum in most individuals, mild cerebellar vermis hypoplasia in a few
SBH
Prognosis
In general, life expectancy in individuals with lissencephaly due to any cause is related to the severity of the malformation on neuroimaging [de Wit et al 2011].
ILS. In individuals with ILS, approximately 50% live to age ten years, and very few reach age 20 years. Cause of death is usually pneumonia or status epilepticus. The oldest known individual lived to age 30 years.
These estimates apply only to individuals with typical lissencephaly affecting the entire brain (the large majority of those with lissencephaly);
Shi et al [2019] reported a mildly affected individual with grade 4b lissencephaly who was still alive at age 49 years.
SBH. In general, affected individuals live into adult life. No reliable data regarding life span exist; it is likely to be shortened in those with severe intellectual disability, intractable epilepsy, or both.
Prevalence
Classic lissencephaly from any cause is rare. Birth prevalence is estimated to range from 11.7 to 40 per million births [personal communication with Metropolitan Atlanta Congenital Defects Program Personnel, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention, Atlanta, GA, 2002]. Even the latter is likely to be an underestimate, as the CDC program ascertains only hospitalized children in the first several years of life.