

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Fitridge R, Thompson M, editors. Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists [Internet]. Adelaide (AU): University of Adelaide Press; 2011.

Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists [Internet].
Show detailsIntroduction
Atherosclerosis, the principal cause of heart attack, stroke, and peripheral vascular disease, remains a major contributor to morbidity and mortality in the Western World. Disease progression is slow, beginning in childhood and usually becoming clinically manifest in middle age or later. Although the aetiology of atherosclerosis is not fully understood, it is generally accepted that atherosclerosis is a multifactorial disease induced by the effects of various risk factors on appropriate genetic backgrounds1. Many risk factors, such as hypercholesterolemia, modified lipoproteins, hypertension, diabetes, infections and smoking have been identified in the development of atherosclerosis.
Atherosclerosis has been the focus of intense research for over 100 years. Since Anitschkow and Chalatow first reported that cholesterol can cause atherosclerosis, many investigators have intensively studied the role of blood cholesterol in the pathogenesis of atherosclerosis. Although formerly considered a bland lipid storage disease, new insights into the pathogenesis of atherosclerosis have emerged during the last decades, due to the progress of cellular and molecular approaches to the study of cell interactions in the arterial wall as well as alterations of lipid metabolism. These new insights were broadly summarized in three main theories, i.e. the ‘response to injury’,2 ‘oxidized low density lipoprotein (LDL)’, and ‘inflammation’1 hypotheses.
The response to injury hypothesis2 relies on the concept that the primary cause of atherosclerosis is an injury to the arterial endothelium induced by various factors, i.e. smoking, mechanical stress, oxidized-LDL, homocysteine, immunological events, toxins, viruses, etc. The oxidized LDL hypothesis postulates that LDL oxidized by various factors including endothelial cells, macrophages and smooth muscle cells of the arterial wall, plays a key role in the development of atherosclerosis. More recently, a widely accepted hypothesis is that atherosclerosis is an inflammatory disease, because recent advances in the basic science have established a fundamental role for inflammation in mediating all stages of this disease from initiation through progression and, ultimately, the thrombotic complications of atherosclerosis.1 The aim of the present chapter is to summarize the data from a vari ety of research areas providing an overview of atherosclerosis focusing on mechanistic studies.
Atherosclerotic Lesions
The intima of large and medium sized arteries is composed of a monolayer of endothelial cells and matrix proteins and occasional smooth muscle cells in the subendothelial space (Figure 3.1a). The media of the vessel contains smooth muscle cells and the elastic lamina built by matrix proteins, while the main component of adventitia is connective tissue. With increasing age, the diseased arterial wall slowly thickens and develops focal lesions of lipid accumulation in the intima. These early lesions are known as fatty streaks and are thought to be the sites of predisposition to advanced lesions called atherosclerotic plaques or atheroma, which may lead to clinical symptoms in certain circumstances.

FIGURE 3.1
Sections of rabbit arterial wall. Rabbits were fed with a standard chow-diet (a) or cholesterol-enriched diet (0.2%) for 3 weeks (b) or 16 weeks (c). Their aortas were harvested and sections prepared and stained with hematoxilin and eosin. Arrows indicate (more...)
Fatty streaks
Fatty streaks are generally the lesion types found in children, although they may also occur in adults. These lesions represent the early changes of atherosclerosis and are recognized as an increase in the number of intimal macrophages filled with lipid droplets (foam cells). A larger lesion which can be grossly visible is characterized by layers of macrophage foam cells and lipid droplets within intimal smooth muscle cells and minimal coarse grained particles and heterogeneous droplets of extracellular lipid (Figure 3.1b). With the progression of lesion development, intermediate lesions as described by pathologists, are the morphological and chemical bridge between fatty streaks and advanced lesions. These lesions appear in some adaptive intimal thickenings (progression-prone locations) in young adults and are characterized by pools of extracellular lipid in addition to other components of fatty streak lesions. The fatty streak is largely clinically benign, but is the precursor to later, clinically relevant lesions.
Plaque or atheroma
The advanced lesion, a dense accumulation of extracellular lipid, known as the lipid core, occupies an extensive but well-defined region of the intima.3 No increase in fibrous tissue and complications such as defects of the lesion surface and thrombosis are present at this stage of disease. This atherosclerotic plaque is also known as atheroma (Figure 3.1c). The characteristic core appears to develop from an expansion and confluence of the small isolated pools of extracellular lipid that characterize atheroma. Between the lipid core and the endothelial surface, the intima contains macrophages, smooth muscle cells, lymphocytes and mast cells. Capillaries surround the lipid core, particularly at the lateral margins and facing the lumen. Frequently macrophages, macrophage foam cells, and lymphocytes are more densely concentrated in the lesion periphery. Much of the tissue between the core and the surface endothelium corresponds to the proteoglycan-rich layer of the intima, although infiltrated with the cells just described. Advanced lesions may or may not narrow the arterial lumen, nor be visible by angiography, nor produce clinical manifestations. Such lesions may be clinically significant even though the arterial lumen is not narrowed, because complications may develop suddenly.3
In addition, two types of atherosclerotic plaques, i.e. ‘vulnerable’ and ‘stable’ plaques, have been recognized.4 Vulnerable plaques often have a well-preserved lumen, since plaques remodel outward initially. The vulnerable plaque typically has a substantial lipid core and a thin fibrous cap separating the thrombogenic macrophages bearing tissue factor (TF) from the blood. At sites of lesion disruption, smooth muscle cells are often activated, as detected by their expression of the transplantation antigen HLA-DR. In contrast, the stable plaque has a relatively thick fibrous cap protecting the lipid core from contact with the blood. Clinical data suggest that stable plaques more often show luminal narrowing detectable by angiography than do vulnerable plaques, but with much less chance of rupture.
Hypercholesterolemia and Oxidised-LDL
Accumulating evidence suggests a causal relationship between blood cholesterol and atherosclerosis. Blood cholesterol is carried by lipoproteins, including LDL, very low-density lipoprotein and high-density lipoprotein (HDL). LDL is believed to be ‘bad’ lipoprotein, while HDL is ‘good’ and plays a protective role in atherogenesis.5 It is established that familial hypercholesterolemia related to increased LDL levels causes premature atherosclerosis and heart disease,6 whereas non-genetic hypercholesterolemia is also associated with the development of atherosclerosis. The consensus of many trials using different cholesterol-lowering regimens indicate that for every 10% reduction in cholesterol level, the deaths of patients with coronary heart disease is reduced by at least 15%. It has been assumed that the reduction in adverse clinical events when plasma cholesterol levels are decreased is directly related to the magnitude of the cholesterol lowering.
That assumption is supported by the fact that the benefit relates to the change in cholesterol level in much the same way whether the cholesterol lowering is achieved with diet or with drugs. These findings suggest that blood cholesterol exerts its role in the pathogenesis of atherosclerosis.
LDL can be modified by oxidation in vivo and in vitro and is detectable in the circulation as well as in atherosclerotic lesions. In vivo, the rate of production of oxidized-LDL in the arterial intima is a function of the concentration of native LDL present. The mechanism whereby hypercholesterolemia and oxidized-LDL trigger events leading to the generation of early atherosclerotic lesions i.e. fatty streak (Figure 3.2) remains uncertain. Although rabbits and pigs were often used in studying this issue, the apolipoprotein (apo) E–deficient mouse7 and the LDL receptor-deficient mouse have become preferred animal models. Deletion and overexpression of genes in animal models is now the gold standard for critically testing the relevance of candidate genes in atherogenesis. By using these models, it was observed that one of the earliest responses induced by hypercholesterolemia and oxidized-LDL is an increase in the expression of vascular cell adhesion molecule-1 (VCAM-1), a key adhesion molecule for monocytes and T cells, on the endothelial surface lining the major arteries.8 Oxidized-LDL is itself directly chemotactic for monocytes and T cells, and can also be cytotoxic for endothelial cells, mitogenic for macrophages and smooth muscle cells and stimulate the release of monocyte chemoattractant protein-1 (MCP-1) and monocyte colony stimulating factor (M-CSF) from endothelial cells. The oxidative modification hypothesis has been extensively reviewed.9
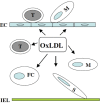
FIGURE 3.2
Schematic representation of the role of oxidized-LDL (oxLDL) in atherogenesis. Oxidized-LDL generated either locally or systemically stimulates endothelial cells (EC) expressing adhesion molecules, including ICAM-1, VCAM-1 and E-selectin, which are responsible (more...)
Oxidized-LDL can account for the loading of macrophages with cholesterol. Here, monocytes undergo phenotypic modification and take up oxidized-LDL to become foam cells, loaded with multiple cytoplasmic drop lets containing cholesterol esters.10 Recently there has been considerable progress in identifying the components of oxidized-LDL that make it a ligand for scavenger receptors. Extensive degradation of the polyunsaturated fatty acid (PUFA) in the sn-2 position of phospholipids by oxidation seems to be essential. Moreover, oxidized-LDL and apoptotic cells compete for binding to macrophage scavenger receptor, indicating that oxidized phospholipids in the membranes of apoptotic cells are involved in their binding to macrophage scavenger receptors. Therefore, oxidized-LDL promotes foam cell formation that forms the earliest lesions in the intima, which may progress to advanced lesions in the presence of other pro-atherogenic factors (Figure 3.2).
High-density lipoproteins role in atheroprotection
It has been known for many years that the plasma concentration of HDL-C correlates inversely with the incidence of cardiovascular disease. The Framingham Heart Study showed that people whose HDL-C was less than 35 mg/dL (0.91 mmol/L) at the beginning of the study had a future coronary risk more than eight times that in subjects whose HDL-C concentration was greater that 65 mg/dL (1.68 mmol/L).11 In the more recent Prospective Cardiovascular Munster (PROCAM) Study, men with an HDL-C concentration of less than 35 mg/dL (0.91 mmol/L) at baseline were shown to have a four times greater risk, at six years, than men whose HDL-C concentration was greater than 35 mg/dL (0.91 mmol/L).12 In both studies, the risk associated with lower plasma HDLC concentration was inde pendent of LDL-C concentration. HDLs have several properties that contribute to their ability to protect against the development of atherosclerosis.
The best known mechanism of atheroprotection relates to the ability of HDLs to promote efflux of cholesterol from foam cells. This process inhibits the progression of, and potentially promotes the regression of, atherosclerosis.13 High-density lipopro-teins can also inhibit the oxidative modification of LDLs and thus reduce their atherogenicity. The principle mechanism of this anti-oxidant function resides with the presence of paraoxonase enzyme residing in the HDL particle,14,15 although the main apolipoprotein, apolipoprotein AI (ApoAI), has also been demonstrated to have anti-oxidant capacity.16 Conceivably, the earliest observable cellular dysfunction in the normal blood vessel, leading to atherogenesis, is the expression of leukocyte adhesion molecules and chemokines. Many studies, both in vivo and in vitro have shown that HDLs can inhibit expression of endothelial cell adhesion molecules and MCP-1.17-20 Endothelial dysfunction, and subsequent platelet activation and aggregation are key elements of the progression of atherosclerotic plaque formation. The ability of HDLs to be anti thrombotic was demonstrated by the ability to induce prostacyclin (PGI2) synthesis, via induction of cyclooxygenase 2,21 and in addition to stimulate the generation of nitric oxide,22 thus reducing the endothelial dysfunction that may precede the development of atherosclerosis.
Hypertension and Biomechanical Stress
Hypertension is a well-established risk factor for atherosclerosis.23 Clinical trials have shown that, in the highest quintile for diastolic pressure, hypertension still contributes significantly to the risk of atherosclerosis, even with the added risks of high cholesterol and smoking. Induction of hypertension in the Watanabe heritable hyperlipidemic rabbit showed a synergistic effect, causing intensification of atherosclerosis. The fact that atherosclerotic lesions preferentially occur in the areas where hemodynamic or biomechanical stress is altered, e.g. bifurcation of the arteries, supports the idea that hypertension exerts its role in the pathogenesis of atherosclerosis via altered mechanical stress to the vessel wall.
In vivo, the vessel wall is exposed to two main hemodynamic forces or biomechanical stress: shear stress, the dragging frictional force created by blood flow, and mechanical stretch, a cyclic strain stress created by blood pressure.24 Shear stress stimulates endothelial cells to release nitric oxide and prostacyclin,25 resulting in vessel relaxation and protection of vascular cells, whereas smooth muscle cells are stimulated by cyclic strain stress. In humans, atherosclerotic lesions occur preferentially at bifurcations and curvatures where hemodynamic force is disturbed, i.e. lower shear stress and higher mechanical stretch. Although veins do not develop spontaneous atherosclerosis-like lesions, accelerated atherosclerosis occurs rapidly in venous bypass grafts, which bear increased biomechanical forces due to alterations in blood pressure, i.e. vein (0-30 mm Hg) vs. artery (120 mm Hg). This finding supports the hypothesis that mechanical stress could be a crucial factor in the pathogenesis of atherosclerosis.
The mechanism whereby mechanical forces are sensed by cells and transmitted through intracellular signal transduction pathways to the nucleus resulting in quantitative and qualitative changes in gene expression in the vessel wall is not fully understood. However, recent evidence indicates that mechanical stretch initiates intracellular signal pathways, especially mitogen-activated protein kinase (MAPK) cascades26 which are thought to play a pivotal role in transmitting transmembrane signals required for cell proliferation, differentiation and apoptosis. MAPKs comprise a ubiquitous family of tyrosine/threonine kinases, and include extracellular signal-regulated kinases (ERKs), stress-activated protein kinases (SAPKs) or c-Jun NH2-terminal kinases (JNKs), and p38 MAPKs.27 They are highly activated or expressed in atherosclerotic lesions and vessel wall stimulated by acute hypertension.28
Biomechanical stress-induced cell death
While biomechanical force at physiological levels is essential to develop and maintain organic structure and function, at elevated levels mechanical stretch may result in cell death leading to pathological conditions. In recent years, however, it has become widely recognized that cell death, namely apoptosis, is not just a response to injury but a highly regulated and controlled process. Disturbances in the regulatory mechanisms of apoptosis often precede the development of atherosclerosis. Exploration of the molecular signalling mechanisms leading to mechanical stress induced apoptosis in cardiovascular disorders has revealed the crucial role of apoptosis in the pathogenesis of atherosclerosis.29 Recent data focussing on the molecular mechanisms of mechanical stress-induced apoptosis are summarised and the role of apoptosis in the development of atherosclerosis is highlighted.
Recently, the first mouse model of vein graft atherosclerosis was established by grafting autologous jugular vein or vena cava to carotid arteries in wild-type and apoE-deficient mice. In many respects, the morphological features of this murine vascular graft model resemble those of human graft atherosclerosis (Figure 3.3). Apoptosis occurred mainly in veins grafted to arteries, remaining unchanged in vein-to-vein grafts.30 The veins grafted to arteries were subjected to increased biomechanical forces in the form of stretch stress due to blood pressure. When mouse, rat and human arterial smooth muscle cells cultured on a flexible membrane were subjected to cyclic strain stress, apoptosis was observed in a time- and strength-dependent manner. Mechanical stretch resulted in p38 MAPK activation. Smooth muscle cell lines stably transfected with a dominant negative rac, an upstream signal transducer, or overexpressing MAPK phosphatase-1, a negative regulator for MAPKs, completely inhibited mechanical stress-stimulated p38 activation, and abolished mechanical stress-induced apoptosis.31 Interestingly, p53 deficient vein grafts had lower levels of apoptosis that correlated with increased atherosclerotic lesions.32 The sudden elevation in mechanical forces could be a strong stimulus to the grafted vessel wall and may result in activation of intra cellular signal pathways leading to gene expression and cell death. Thus, one of the earliest events in vein graft atherosclerosis is apoptosis, in which mechanical stress-induced p38MAPK-p53 activation is, at least in part, responsible for transducing signals leading to apoptosis.
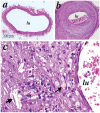
FIGURE 3.3
Hematoxilin and eosin-stained sections of arterialized mouse vein grafts. Under anesthesia, vena cava veins were removed and isografted into carotid arteries (of control mice)(a) of apoE-/- mice (b). Animals were sacrificed 8 weeks after surgery, and (more...)
Biomechanical stress and inflammation
Vein graft atherosclerosis has an inflammatory nature characterized by mononuclear cell infiltration followed by smooth muscle cell proliferation. It has been postulated that biomechanical stress plays a role in adhesion molecule expression via MAPK signal transduction pathways, leading to NF-κB activation. Supporting this concept is the fact that neointimal lesions of vein grafts in intercellular adhesion molecule (ICAM)-1-/mice were reduced from 50% to 30% compared to wildtype controls. ICAM-1 is critical in the development of venous graft atherosclerosis. It has been established that exposure of endothelial cells to shear (mechanical) stress results in increased expression of ICAM-1 and monocyte chemotactic protein-1 (MCP-1) via activation of transcription factor NF-κB and AP-1. These molecules are essential for leukocyte-endothelial cell interaction and subsequently cell infiltration, characteristic of the early lesions of vein grafts that undergo elevated blood pressure. Interestingly, mechanical stress also leads to smooth muscle cells expressing ICAM-1 via activation of NF-κB. In animal models, smooth muscle cells express ICAM-1 which is associated with monocyte/macrophage accumulation in vein grafts. Smooth muscle cells of ICAM-1 -/- mice do not express ICAM-1 which is correlated with reduced early lesions.33 Mechanical stress-induced adhesion molecules and chemokine expression in the vessel wall could be important for the inflammatory response.
Biomechanical stress-induced smooth muscle cell proliferation
It has been established that mechanical stress stimulates DNA synthesis and the proliferation of in vitro cultured smooth muscle cells. Hypertension increases mechanical force on the arterial wall up to 30%, resulting in marked alterations in signal transduction and gene expression in smooth muscle cells, which contribute to matrix protein synthesis, cell proliferation and differentiation.23 Recently, several reports demonstrated that angioplasty resulted in stretching of the arterial wall leading to rapid activation of the MAPKs in the regenerating carotids.34 The magnitude of Extracellular signal-regulated kinase p42 (ERK42) activation positively correlated with the degree of balloon injury to the arterial wall. Ex vivo stretching of the vessel wall also induced significant activation of ERK42 kinases. These findings suggested that the kinase activation in the early phase following injury may be due to mechanical stimulation of the vessel wall.
In cultured smooth muscle cells, mechanical forces evoked E RK activation followed by enhanced DNA-binding activity of transcription factor AP-1. Interestingly, physical forces rapidly result in phosphorylation of platelet-derived growth factor (PDGF) receptor,35 epithelial growth factor receptor and vascular endothelial growth factor receptor. Thus, mechanical stresses may directly perturb the cell surface or alter receptor conformation, thereby initiating signalling pathways normally used by growth factors. Suramin, a non-specific PDGF inhibitor, has been shown to be a growth factor receptor antagonist that inhibits cell proliferation. When vein isografts in mice were treated ex vivo and in vivo with suramin, intimal lesions were reduced up to 70% compared to untreated controls. The mechanism of suramin-inhibited neointimal hyperplasia mainly involves inhibition of smooth muscle cell migration and proliferation via blocking PDGF receptor-MAPK-AP-1 signal path-ways. Thus, research into biomechanical stress-regulated gene expression in atherosclerosis using these models could lead to a new therapeutic strategy in the treatment of this disease in humans.
Infections and Heat Shock Proteins
Risk factors, such as high blood cholesterol, hypertension and smoking only explain a proportion of the incident cases of all atherosclerosis. There is a body of evidence that microorganisms play a role in the pathogenesis of atherosclerosis and may be a primary risk factor in people who do not suffer from other established risk factors. Accumulating evidence suggests that infectious organisms reside in the wall of atherosclerotic vessels, including cyto–megalovirus (CMV) and Chlamydia (C) pneumoniae. Seroepidemiological studies demonstrate an association between the pathogen-specific IgG antibodies and atherosclerosis.26,27 However, the data are inconsistent, with other studies showing no increased risk for atherosclerosis.38,39 One possible explanation for this disparity is that infections contributing to atherosclerosis risk may depend, at least in part, on the host’s response to the pathogen, i.e. inflammatory and immune reactions.
Infections
Several papers reviewing the infections, ie C.pneumoniae, H.pylori and CMV and atherosclerosis have been published37 and these will be summarised. Saikku et al40 was first to show a link between C.pneumoniae infection, coronary artery disease and atherosclerosis. Since then, many studies have shown an association of C.pneumoniae with atherosclerosis. In vitro experiments have shown a preferential and specific attraction to and infection of macrophages, vascular endothelium and vascular smooth muscle, by C.Pneumoniae, thus resulting in their accumulation into atherosclerotic plaques. This is supported by studies of postmortem specimens of vascular tissue which found a high correlation between the distribution of atherosclerosis and C.pneumoniae41 and other organisms.
H.pylori, another gram negative bacteria which typically infects human gastric epithelial cells has been demonstrated in atherosclerotic plaques.38 Sero positivity to H.pylori was implicated as a risk factor in coronary heart disease (CHD) from the first report in 1994. However a meta-analysis 39 of 18 studies failed to show any correlation between sero-positivity against H.pylori and the presence or extent of CHD. Although the evidence supporting involvement of H.pylori in atherogenesis is not conclusive, it may be important to differentiate between virulent and avirulent strains of H.pylori to determine the effects on atherogenesis. Mayr et al42 conducted a population based study and investigated the effects of CagA (cytotoxin-associated gene A) positive and CagA negative strains of H.pylori. This study concluded that there was an increased risk of atherosclerosis in individuals who were infected with CagA positive strains of H.pylori. Another group has obtained similar results, indicating the role of this strain in the pathogenesis of atherosclerosis.
Heat shock proteins
The role of HSPs in disease with regard to their physiological functions and pathological involvement have been described in many reviews on the subject. The HSP family of proteins is subdivided into groups based on their molecular weight (e.g. HSP60 is a 60kDa protein) and are produced by almost all cells and play an important role in the organism’s general protective response to environmental and metabolic stresses (Table 3.1). They exist in all major cellular compartments. For example, HSP10, HSP60 and HSP75 are mainly located in mitochondria, while others are found in different compartments throughout the cell. They have important physiological functions, primarily as a molecular chaperone.7 HSPs also appear to be important in preventing cellular damage during repair processes following injury. Evidence indicates that HSPs may be autoantigens in some circumstances.43 HSP47, HSP60 and HSP70 have been identified as being involved in the pathogenesis of atherosclerosis.44
TABLE 3.1
Heat shock protein families.
Infections and HSP expression
In a recent study, increased HSP60 was demonstrated on the endothelium, smooth muscle cells, and mononuclear cells of all atherosclerotic carotid and aortic specimens, whereas vessels with normal intima showed no detectable expression of this HSP. The level of HSP60 expression positively correlated with atherosclerotic severity.45 Interestingly, chlamydial and human HSP60s have been shown to be co expressed in atherosclerotic lesions. These data support the concept that elevated HSP expression in lesions may be induced by the pathogen Chlamydiae species. During its normal cycle generating infectious progeny, Chlamydiae express basal levels of HSP. During the lytic phases of chlamydial infection, host cells release their own HSP60, and also chlamydial HSP60 that has been produced by these microorganisms. Soluble HSP60 (sHSP) levels were significantly elevated in subjects with prevalent/incident carotid atherosclerosis and correlated to intima-media thickness independent of age, sex and other risk factors. Interestingly, sHSP60 was also correlated with anti-LPS, anti-Chlamydia and anti-HSP60 antibodies, inflammation markers and chronic infections.
Infections, sHSP and innate immunity
Infectious agents contribute to atherogenesis in a variety of ways. One mechanism is a variety of ways. By triggering innate immune reactions leading to inflammatory responses. Innate immunity involves several different cell types, e.g. mononuclear phagocytes and endothelial cells. Both endothelial cells and macrophages express receptors that recognize molecular epitopes from a broad range of pathogens. These receptors include various scavenger and Toll-like receptors (TLRs). So far more than 10 human TLRs have been identified. A variety of bacterial and fungal components are known TLR ligands, including peptidoglycan for TLR2, LPS for TLR4, flagellin for TLR5, and unmethylated CpG (cytosine and guanine separated by a phosphate group, which links the two nucleotides together) motifs in bacterial DNA for TLR9. It is possible that TLRs may be collectively responsible for detecting a large range of microbial pathogens. TLRs are evolutionarily conserved innate immune receptors that are shared by IL-1 receptor signalling to activate the NF-KB pathway and release inflammatory cytokines. TLR ligation therefore induces expression of a wide variety of genes such as those encoding proteins involved in leukocyte recruitment, production of reactive oxygen species, and phagocytosis. Activation of TLRs will also elicit the production of cytokines that augment local inflammation. Finally, TLR ligation may directly induce apoptosis, probably of key importance in the first line of defence.46
Expression of TLR4 in atherosclerotic plaques has been found, preferentially in lipid-rich and macrophage-infiltrated areas of lesions. In vitro, basal expression of macrophage TLR4 was shown to be upregulated by oxidized-LDL. In addition, of the nine TLRs, expression of TLR1, TLR2, and TLR4 was shown to be markedly enhanced in human atherosclerotic plaques. A polymorphism or mutation of TLR4 was shown to be strongly correlated with the incidence and development of atherosclerosis in a large population study (Bruneck Study). Surprisingly, several groups reported that recombinant HSP60 and HSP70 from bacteria and humans specifically bind to TLR4 in macrophages, endothelial cells and smooth muscle cells. Recombinant HSP60 binding to the TLR4/CD14 complex of macro phages and endothelial cells led to activation of MyD88-NF-kB pathways. HSP70 and mycobacterial HSP65 have a similar binding activity to TLR4/CD14 that initiates MyD88-NF-κB signal pathways, suggesting that the TLR4/CD14 is a receptor for several HSPs that mediate the signal pathways leading to proinflammatory responses during infections.
In summary, infections with proatherogenic organisms may be important in individuals lacking additional risk factors as well as acting synergistically with established risk factors. In this process, HSP may be a link between infections and the pathogenesis of atherosclerosis. Infectious agents may exert their role by producing their own HSPs and inducing host production which could be released into blood. The soluble form of HSPs contact endothelial cells and immune cells where innate immune responses are initiated. Innate immune reactions to HSPs result in proinflammatory responses in the vessel wall. Together, infections via HSPs contribute to the development of atherosclerosis (Figure 3.4).

FIGURE 3.4
Schematic representation of the likely mechanism of action of heat shock proteins (HSPs) in the development of atherosclerosis in response to risk factors (stressor), e.g. infections. TLR, Toll-like receptor; APC, antigen-presenting cells.
Immune Responses
The contribution of immune responses to the pathogenesis of atherosclerosis has been recognized and much progress in this research field has been achieved through the participation of many investigators.47 Involvement of the immune system in atherogenesis is supported by recent data, including the occurrence of granular deposits of immunoglobulins and co-distributed complement components, increased expression of C3b receptors (CR1) and C3b1 receptors (CR3) on macrophages within atherosclerotic lesions, but not in unaltered vessels. However, B cells are only found in very low numbers in various stages of atherosclerotic lesions, and the site of production for these immunoglobulins must, therefore, be sought elsewhere. Other than these humoral immune phenomena, it is now clear that T cells are among the first cells infiltrating the intima of arteries during the earliest stages of atherosclerosis, most probably before monocytes. A majority of these early T cells are CD4+, HLA-DR+ and interleukin-2 receptor+ (IL-2R+), i.e. activated. Others have shown that T cells in late atherosclerotic plaques express the low molecular variant of the leukocyte common antigen (CD45RO) and the integrin very late activation antigen-1 (VLA-1). Hansson and his group analyzing the rearrangement of T cell receptor (TCR) genes in these latter cells derived from advanced lesions, showed that they represent a polyclonal population rather than displaying restricted T cell receptor TCR usage. These findings support the role of the immune system in atherogenesis.
MHC class II antigen and T cells
Regardless of which antigen these lymphocytes may recognize, it seems improbable that endothelial cells (EC) which aberrantly express major histocompatibility complex (MHC) class II antigens act as primary antigen-presenting cells for T cell sensitization. MHC class II expression by EC occur concomitantly where T cells are found, and thus production of gamma-interferon (IFNγ), the major T-cell chemokine, is present in the intima directly beneath these areas. Therefore, it can be concluded that the expression of MHC class II molecules by endothelial cells represents a secondary rather than a primary phenomenon. The large majority of CD3+ cells in the mononuclear infiltrate in atherosclerotic lesions expresses TCRα/β, but an unexpectedly high proportion also express TCRγ/δ. While the latter type of cell only constitutes approximately 1 % of leukocytes in peripheral blood, enrichment to 10 % or more within early atherosclerotic lesions has been observed. The majority of these latter cells express the TCRγ2 chain, i.e. resemble the TCRγ/δ+ population found in the intestinal mucosa. On the other hand, TCR Vγ9δ2+ cells characteristic of circulating TCRγ/δ+ cells are not proportionally increased in the intima. Finally, endothelial cells and leukocytes express and synthesise a variety of immunological-inflammatory mediators, occurring in atherosclerotic lesions. Among others, these include interleukin-1 (IL-1), tumour necrosis factor α (TNFα), lymphotoxin, IL-2, IL-6, IL-8, monocytechemotactic peptide-1 and IFNγ1. Together, these molecules can modulate the local cellular immune response within emerging atherosclerotic lesions.
Oxidized-LDL as a candidate antigen
T cells isolated from human atherosclerotic plaques were shown to be specifically reactive to oxidized-LDL. One fourth of all CD4+ T cells cloned from human plaques recognized oxidized-LDL in an HLA-DR-dependent manner. Oxidized-LDL-specific T cells are present in lymph nodes of apoeE-KO mice, which have strong humoral as well as cellular immune responses to such modified lipoproteins. In humans, oxidized-LDL induces activation of a subset of peripheral T cells. In addition, antibodies to oxidized-LDL can be detected in atherosclerotic patients and are present in atherosclerotic lesions, suggesting that it is a quantitatively important antigen. The immune response to oxLDL plays a pathogenetic role in atherosclerosis because lesion progression can be inhibited by immunization or induction of neonatal tolerance to oxLDL. It seems paradoxical that both tolerization and hyperimmunization can reduce the extent of disease; this may be due to the different effector pathways activated by these two kinds of treatment.
HSP60 as a candidate antigen
As discussed above, HSPs have been implicated in activation of innate immune responses involved in the pathogenesis of atherosclerosis. Moreover, adaptive immune reactions to HSP60 have also been implicated in the development of atherosclerosis. In experimental models, rabbits immunised with HSP65/60 recorded induction of vascular inflammation, with endothelial activation and mononuclear cell adhesion demonstrated.48 The developing lesions also contained T cells, and cell lines derived from such infiltrates exhibited anti-HSP60 reactivity. Anti-HSP60 antibodies occurred in peripheral blood, and immunization with HSP60 was found to increase fatty streak development in hypercholesterolemic rabbits and mice. In humans, antibodies to HSP65/60 are elevated in early and late atherosclerosis and may predict progression of atherosclerotic disease. Since heat shock proteins of humans and microbes are structurally and antigenically similar, it is possible that molecular mimicry between immune responses to microbial HSP and homologues expressed by vascular cells could account for the association between infections and atherosclerosis.49 based on these findings, Maron and co-workers provided the evidence that atherosclerotic lesions were reduced by nasal immunization with HSP65 in apoE-deficient mice, suggesting that atherosclerosis might be inhibited by vaccination against HSP65.50
β2-glycoprotein Ib as a candidate antigen
A third autoantigen, β2-glycoprotein Ib (β2-GPI), is present on platelets but may also be expressed by endothelial cells. Autoantibodies to β2-GPI are produced in several inflammatory disorders, in addition to atherosclerosis. The immune response to β2-GPI appears to be proatherogenic, because hyperimmunization with β2-GPI116 or transfer of β2 GPI reactive T cells aggravates fatty streak formation in LDLR-/- mice. The pathogenic mechanism by which β2-GPI acts remains unclear, but it may be related to this protein’s capacity to bind phospholipids.
In summary, adaptive immunity powerfully modulates initiation and progression of atherosclerosis. Atherogenesis involves intercommunication between shared pathways involved in adaptive and innate immunity. Various established and emerging risk factors for atherosclerosis modulate aspects of immune responses, including lipoproteins and their modified products, HSPs, and infectious agents. As the molecular details become understood, new potential targets for therapies will doubtless emerge.
Inflammation
It is generally accepted that atherosclerosis is an inflammatory disease,1 because recent findings have provided important links between all risk factors and the mechanisms of atherogenesis. Clinical studies have shown that this emerging biology of inflammation in atherosclerosis applies directly to human patients. Elevation of markers for inflammation predicts the outcome in patients with acute coronary syndromes.4 In addition, low-grade chronic inflammation, as indicated by levels of the inflammatory marker C reactive protein (CRP), prospectively defines the risk of atherosclerotic complications, thus adding to prognostic information provided by traditional risk factors. Certain treatments that reduce atherosclerosis risk also limit inflammation. For example, statins, used for lipid lowering,4 have anti inflammatory effects. Amongst the known inflammatory triggers on the vessel wall are known risk factors e.g. hypercholesterolemia, oxidized-LDL, hypertension, biomechanical stress and infection. These risk factors can directly or indirectly stimulate endothelial cells expressing adhesion molecules (VCAM-1, ICAM-1 and E-selectin) mediating subsequent mononuclear cell infiltration and foam cell formation in the subendothelial space.1 In this section, some recent findings that have not been described above will be summarised.
C-reactive protein
C reactive protein (CRP) is an acute phase protein that is involved in inflammatory processes. It is made up of 5 identical subunits which are arranged in a pentagonal shape. CRP is predominantly synthesized by hepatocytes, but in response to inflammation, CRP expression can be found in atherosclerotic plaque, aortic endothelial cells, monocytes and vascular smooth muscle cells. Inflammatory cytokines induce CRP gene expression and statins have been shown to reduce CRP levels.52
Biasucci et al53 studied a cohort of patients admitted with unstable angina and found that one half of this group had a persistently elevated CRP (>3mg/dL) at discharge. Those with elevated discharge CRP levels had a significantly elevated risk of recurrent unstable angina or MI during the subsequent 12 months. This group has also demonstrated that CRP elevation in individuals presenting with severe peripheral arterial disease was associated with an increased risk of MI, independent of other vascular risk factors.
CRP may be not only a marker of inflammation and atherosclerosis, it may also be an active component participating in atherogenesis. CRP can bind to lipoproteins and activate the complement system via the classical pathway. CRP deposits have been shown in the arterial wall early during lesion formation, which is colocalized with the terminal complement complex. This suggests that CRP may promote atherosclerotic lesion formation by activating the complement system and is involved in foam cell formation, which may be caused in part by the uptake of CRP-opsonized LDL.
CD40/CD40L
These antigens are ubiquitously expressed on the surface of endothelial cells, smooth muscle cells, macrophages, T lymphocytes, and platelets within human atheroma.51 The proatherogenic functions of CD40 ligation include augmented expression of matrix metalloproteinases, procoagulant tissue factor (TF), chemokines, and cytokines. Indeed, interruption of CD40 signalling not only reduced the initiation and progression of atherosclerotic lesions in hypercholesterolemic mice in vivo, but also modulated plaque architecture in ways that might lower the risk of causing thrombosis. In addition to the 39-kDa cell membrane–associated form, CD40L also exists as a soluble protein, termed sCD40L. Although lacking the cytoplasmic, the transmembrane region, and parts of the extracellular domains, this, the soluble form of CD40L, is considered to possess biological activity. Patients with unstable angina express higher sCD40L plasma levels than healthy individuals or patients with stable angina. Moreover, it was recently demonstrated that elevated plasma concentrations of sCD40L predict risk for future cardiovascular events. Although in vitro and in vivo studies established that CD40 signalling participates in atherosclerosis, the initial trigger for CD40/CD40L expression within atheroma may be regulated by oxidized-LDL. Thus CD40/CD40L may be a mediator in the inflammatory responses during the development of atherosclerosis.
Summary and Perspectives
Atherosclerosis is an inflammatory disease that is initiated by multiple risk factors, including hypercholesterolemia, oxidized-LDL, altered biomechanical stress, smoking and infections. Due to research achievements in recent decades, atherogenesis is no longer an inevitable consequence of aging—the statin revolution has left this in no doubt. Better control of hypercholesterolemia can clearly be achieved but many questions remain. For example, which factor is an initiator for the development of atherosclerotic lesions, and how do other factors participate in the disease process. Currently, atherosclerosis research is highly topical. The mystery of the molecular mechanisms in this disease will yield to the current multi disciplinary attack by academic institutions and the pharmaceutical industry using the powerful techniques of vascular biology and molecular approaches.
References
- 1.
- Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med 1999; 340: 115–126. [PubMed: 9887164]
- 2.
- Ross R. The pathogenesis of atherosclerosis – an update. N Engl J Med 1986; 314: 488–500 [PubMed: 3511384]
- 3.
- Stary HC, Chandler AB, Dinsmore RE, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb Vasc Biol 1995; 15: 1512–1531 [PubMed: 7670967]
- 4.
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 2002; 105: 1135–1143 [PubMed: 11877368]
- 5.
- GlassCK, Witztum JL. Atherosclerosis. The road ahead. Cell 2001; 104: 503–516 [PubMed: 11239408]
- 6.
- Goldstein JL, Kita T, Brown MS. Defective lipoprotein receptors and atherosclerosis. Lessons from an animal counterpart of familial hypercholesterolemia. N Engl J Med 1983; 309: 288–296 [PubMed: 6306464]
- 7.
- Zhang SH, Reddick RL, Piedrahita JA, et al. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein e. Science 1992; 258: 468–471 [PubMed: 1411543]
- 8.
- Li H, Cybulsky MI, Gimbrone MA, Jr., et al. An atherogenic diet rapidly induces VCAM-1, a cytokine regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb 1993; 13: 197–204. [PubMed: 7678986]
- 9.
- Navab M, Berliner JA, Watson AD, et al. The Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arterioscler Thromb Vase Biol 1996; 16: 831–842. [PubMed: 8673557]
- 10.
- Witztum JL, Steinberg D. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends Cardiovasc Med 2001; 11: 93–102. [PubMed: 11686009]
- 11.
- Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977; 62: 707–714. [PubMed: 193398]
- 12.
- Assman G and Schulte H. Relation of high-density lipoprotein cholesterol and triglycerides to incidence of atherosclerotic coronary artery disease (the PROCAM experience). Prospective Cardiovascular Minister Study. Am J Cardiol. 1992; 70: 733–737. [PubMed: 1519522]
- 13.
- Assman G, Nofer J-R. Atheroprotective effects of high-density lipoproteins. Ann Rev Med. 2003; 54: 321–341. [PubMed: 12414916]
- 14.
- Mackness B, Hine D, Lui Y, Mastorikou M, Mackness M. Paraoxonase-1 inhibits oxidised LDL-induced MCP-1 production by endothelial cells. Biochem Biophys Res Comm 2004; 318: 680–683. [PubMed: 15144891]
- 15.
- Navab M, Hama SY, Anantharamaiah GM, Hassan K, Hough GP, WatsonAD et al., Normal high density lipoprotein inhibits three steps in the formation of mildly oxidised loe density lipoprotein; steps 2 and 3. J Lipid Res 2000; 41: 1495–1508. [PubMed: 10974057]
- 16.
- Stocker R, Keaney JF Jr., Role of oxidative modification in atherosclerosis. Physiol Rev 2004; 84: 1381–1478. [PubMed: 15383655]
- 17.
- Cockerill GW, K-A Rye, JR Gamble, MA Vadas and P Barter. High density lipoproteins inhibit cytokine-induced expression of adhesion molecules on endothelial cells. Arterioscler Thromb Vasc Biol 1995; 15: 1987–1994. [PubMed: 7583580]
- 18.
- Cockerill GW, T Huehns, C Stocker, NE Miller and DO Haskard. Elevation of plasma HDL inhibits cytokine-induced induction of E-selectin in a porcine model of acute inflammation. Circulation 2001; 103: 108–112. [PubMed: 11136694]
- 19.
- Cockerill GW, M McDonald, S Cruzzocrea, C Thiemermann. High density lipoproteins reduce organ injury and dysfunction following hemorrhagic shock. FASEB J 2001; 15: 1945–1951. [PubMed: 11532974]
- 20.
- Nicholls SJ, Dusting GJ, Cutri B, Bao S, Drummond GR, Rye KA, Barter PJ. Reconstituted high-density lipoproteins inhibit the acute prooxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolaemic rabbits. Circulation 2005; 111; 1543–1550. [PubMed: 15781735]
- 21.
- Cockerill GW, J Saklatvala, SH Ridley, H Yarwood, NE Miller, B Oral, S Nithyanathan, G Taylor and DO Haskard. High density lipoproteins differentially modulate cytokine induced expression of E-selectin and cyclooxygenase-2. Arterioscler Thromb Vasc Biol 1999; 19: 910–917. [PubMed: 10195917]
- 22.
- Drew BG, Fidge NH, Gallon-Beaumier G, Kemp BE, Kingwell BA. High density lipoprotein and apolipoprotein AI increase endothelial NO synthase activation by protein association and multisite phosphorylation. Proc Natl Acad Sci (USA). 2004\; 101; 6999–7004. [PMC free article: PMC406455] [PubMed: 15107497]
- 23.
- Alexander RW. Theodore Cooper Memorial Lecture. Hypertension and the pathogenesis of atherosclerosis. Oxidative stress and the mediation of arterial inflammatory response: a new perspective. Hypertension 1995; 25: 155–161. [PubMed: 7843763]
- 24.
- Xu Q. Biomechanical stress induced signaling and gene expression in the development of arteriosclerosis. Trends Cardiovasc Med 2000; 10: 35–41. [PubMed: 11150727]
- 25.
- Bhagyalakshmi A, Frangos JA. Mechanism of shear-induced prostacyclin production in endothelial cells. Biochem Biophys Res Commun 1989; 158: 31–37. [PubMed: 2643433]
- 26.
- Li C, Xu Q. Mechanical stress-initiated signal transductions in vascular smooth muscle cells. Cell Signal 2000; 12: 435–445. [PubMed: 10989277]
- 27.
- Hu Y, Dietrich H, Metzler B, et al. Hyperexpression and activation of extracellular signal-regulated kinases (eRk1/2) in atherosclerotic lesions of cholesterol-fed rabbits. Arterioscler Thromb Vasc Biol 2000; 20: 18–26. [PubMed: 10634796]
- 28.
- Xu Q, Liu Y, Gorospe M, et al. Acute hypertension activates mitogen-activated protein kinases in arterial wall. J Clin Invest 1996; 97: 508–514. [PMC free article: PMC507044] [PubMed: 8567974]
- 29.
- Wernig F, Xu Q. Mechanical stress induced apoptosis in the cardiovascular system. Prog Biophys Mol Biol 2002; 78: 105–137. [PubMed: 12429110]
- 30.
- Mayr M, Li C, Zou Y, et al. Biomechanical stress induced apoptosis in vein grafts involves p38 mitogen-activated protein kinases. FASEB J 2000; 14: 261–270. [PubMed: 10660448]
- 31.
- Mayr M, Hu Y, Hainaut H, et al. Mechanical stress-induced DNA damage and racp38MAPK signal pathways mediate p53-dependent apoptosis in vascular smooth muscle cells. FASEB J 2002; 16: 1423–1425. [PubMed: 12205035]
- 32.
- Mayr U, Mayr M, Li C, et al. Loss of p53 accelerates neointimal lesions of vein bypass grafts in mice. Circ Res 2002; 90: 197–204. [PubMed: 11834713]
- 33.
- Cheng GC, Libby P, Grodzinsky AJ, et al. Induction of DNA synthesis by a single transient mechanical stimulus of human vascular smooth muscle cells. Role of fibroblast growth factor-2. Circulation 1996; 93: 99–105. [PubMed: 8616949]
- 34.
- Yamazaki T, Komuro I, Kudoh S, et al. Mechanical stress activates protein kinase cascade of phosphorylation in neonatal rat cardiac myocytes. J Clin Invest 1995; 96: 438–446. [PMC free article: PMC185217] [PubMed: 7615816]
- 35.
- Hu Y, Bock G, Wick G, et al. Activation of PDGF receptor alpha in vascular smooth muscle cells by mechanical stress. FASEB J 1998; 12: 1135–1142. [PubMed: 9737716]
- 36.
- Epstein SE, Zhu J, Burnett MS, et al. Infection and atherosclerosis: potential roles of pathogen burden and molecular mimicry. Arterioscler Thromb Vasc Biol 2000; 20: 1417–1420. [PubMed: 10845851]
- 37.
- Epstein SE. The multiple mechanisms by which infection may contribute to atherosclerosis development and course. Circ Res 2002; 90: 2–4. [PubMed: 11786508]
- 38.
- Folsom AR, Nieto FJ, Sorlie P, et al. Helicobacter pylori seropositivity and coronary heart disease incidence. Atherosclerosis Risk In Communities (ARIC) Study Investigators. Circulation 1998; 98: 845–850. [PubMed: 9738638]
- 39.
- Danesh J, Peto R. Risk factors for coronary heart disease and infection with Helicobacter pylori: metaanalysis of 18 studies. BMJ 1998; 316: 1130–1132. [PMC free article: PMC28515] [PubMed: 9552950]
- 40.
- Saikku P, Leinonen M, Mattila K, et al. Serological evidence of an association of a novel Chlamydia, TWAR, with chronic coronary heart disease and acute myocardial infarction. Lancet 1988; 2: 983–986. [PubMed: 2902492]
- 41.
- Fuzio G, Giovino M, Gullot A, Bacarella D, Novo G, Novo S. Atherosclerosis, inflammation and Chlamydia pneumonia. World J Cardiol 2010. 1.31–40.
- 42.
- Mayr M, Kiechl S, Willeit J, et al. Increased Risk of Atherosclerosis is Confined to CagA Positive H. pylori Strains: Prospective Results from the Bruneck Study. Stroke 2002. [PubMed: 12624280]
- 43.
- Mollenhauer J, Schulmeister A. The humoral immune response to heat shock proteins. Experientia 1992; 48: 644–649. [PubMed: 1639172]
- 44.
- Pockley AG. Heat shock proteins, inflammation, and cardiovascular disease. Circulation 2002; 105: 1012–1017. [PubMed: 11864934]
- 45.
- Xu Q. Role of heat shock proteins in atherosclerosis. Arterioscler Thromb Vasc Biol 2002; 22: 1547–1559. [PubMed: 12377729]
- 46.
- GK Hansson, Libby P, Schonbeck U, et al. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res 2002; 91: 281–291. [PubMed: 12193460]
- 47.
- GK Hansson. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol 2001; 21: 1876–1890. [PubMed: 11742859]
- 48.
- Xu Q, Dietrich H, Steiner HJ, et al. Induction of arteriosclerosis in normocholesterolemic rabbits by immunization with heat shock protein 65. Arterioscler Thromb 1992; 12: 789–799. [PubMed: 1616904]
- 49.
- Mayr M, Kiechl S, Willeit J, et al. Infections, immunity, and atherosclerosis: associations of antibodies to Chlamydia pneumoniae, Helicobacter pylori, and cytomegalovirus with immune reactions to heat-shock protein 60 and carotid or femoral atherosclerosis. Circulation 2000; 102: 833–839. [PubMed: 10952949]
- 50.
- Maron R, Sukhova G,Faria AM et al., Mucosal administration of heat shock protein-65 decreases atherosclerosis and inflammation in aortic arch of low-density lipoprotein receptor-deficient mice. Circulation 2002; 106: 1708–1715. [PubMed: 12270867]
- 51.
- Mach F, Schonbeck U, Sukhova GK, et al. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci USA 1997; 94: 1931–1936. [PMC free article: PMC20020] [PubMed: 9050882]
- 52.
- Singh SK, Suresh MV, Voletti B and Agrawal A. The connection between C-reactive protein and atherosclerosis. Annals of Medicine. 2008; 40: 110–120. [PMC free article: PMC3364506] [PubMed: 18293141]
- 53.
- Biasucci LM, Liuzzo G, Grillo RL, Caliguri G, Rebuzzi AG, Buffon A, Summaria F, Ginnetti F, Fadda G, Maseri A. Elevated levels of C-Reactive protein at discharge in patients with unstable angina predict recurrent instability. Circulation 1999; 99: 855–860. [PubMed: 10027805]
- [Progress in researches on ancient history of atherosclerosis].[Zhongguo Zhong Xi Yi Jie He Za...][Progress in researches on ancient history of atherosclerosis].Chen KJ, Fu CG. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2013 Oct; 33(10):1301-4.
- Review Immune mechanisms in atherosclerosis and potential for an atherosclerosis vaccine.[Discov Med. 2011]Review Immune mechanisms in atherosclerosis and potential for an atherosclerosis vaccine.Chyu KY, Nilsson J, Shah PK. Discov Med. 2011 May; 11(60):403-12.
- Review [Atherosclerosis (hypertension)].[Nihon Rinsho. 2012]Review [Atherosclerosis (hypertension)].Ai M. Nihon Rinsho. 2012 May; 70(5):840-5.
- Study on atherosclerosis treated with theory of detoxification.[J Tradit Chin Med. 2006]Study on atherosclerosis treated with theory of detoxification.Xu Y, Wang H, Ding J. J Tradit Chin Med. 2006 Jun; 26(2):115-9.
- [Transdisciplinary Approach for Sarcopenia. Sarcopenia and atherosclerosis].[Clin Calcium. 2014][Transdisciplinary Approach for Sarcopenia. Sarcopenia and atherosclerosis].Kohara K. Clin Calcium. 2014 Oct; 24(10):1487-92.
- Atherosclerosis - Mechanisms of Vascular DiseaseAtherosclerosis - Mechanisms of Vascular Disease
Your browsing activity is empty.
Activity recording is turned off.
See more...