
© 2017 by Taylor & Francis Group, LLC.
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Kozak JA, Putney JW Jr., editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. doi: 10.1201/9781315152592-1
During the past two decades, great advances have been made in the electrophysiological and molecular identification of calcium entry pathways in non-excitable cells. The term “non-excitable” refers to a variety of cell types that are not capable of firing action potentials. Essentially, except for neurons, muscle cells, and some endocrine cells, all other cells in the body are non-excitable. For the most part, they lack the necessary levels of expression of voltage-gated Na+ channels (NaV family) and also voltage-gated Ca2+ channels (CaV family). Ca2+ influx in these cell types is therefore thought to rely on unrelated, voltage-independent channels, such as Orai (CRACM) and TRP family members. In this chapter, we will discuss direct electrophysiological methods used to record the electrical activity of these proteins, focusing on calcium-selective Orai/STIM and TRPV5/TRPV6 channels.
One of the first non-excitable cell types used to investigate the function of non-voltage-gated Ca2+ channels was cells of the immune system [1]. In T lymphocytes and mast cells, store-operated calcium entry is the main pathway providing cytoplasmic calcium elevations necessary for key cellular functions, such as antigenic activation, proliferation, and degranulation. Calcium stores inside the cell that were shown to be important for CRAC channel activation and functions are the endoplasmic reticulum (ER) and mitochondria [2–6].
Direct evidence for calcium entry following store depletion was demonstrated using the now classical calcium readdition protocol in cells loaded with calcium indicator dyes, such as Fura-2. Figure 1.1 shows a calcium imaging experiment in Jurkat T lymphocytes loaded with Fura-2 ratiometric calcium dye. Upon the removal of calcium and the simultaneous addition of sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) pump inhibitor cyclopiazonic acid (CPA) [7], a transient calcium elevation was observed. This is believed to represent the release of ionized calcium from the ER into the cytoplasm (Fura-2 indicator is in the cytoplasm). Calcium release in this case is mediated through a “Ca2+ leak” pathway operating in the ER membrane. Normally, SERCA acts to sequester cytoplasmic calcium into the ER, counteracting this outwardly directed calcium leak. When SERCA is blocked by CPA, however, the calcium flux into the cytoplasm due to the leak pathway is no longer counteracted and this is manifested as a calcium elevation. The transient nature of this elevation is likely due to plasma membrane Ca2+ ATPase (PMCA), which is not sensitive to CPA and can still expel calcium ions from the cytoplasm.
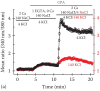
Fura-2 measurement of cytosolic calcium in intact human Jurkat T lymphocytes (a) and murine MIN6 β cells (b).
The basal ER “Ca2+ leak” pathway remains an enigma both in terms of its molecular identity and the factors that regulate it. It is thought to participate in determining the ER calcium content (also termed “calcium load”). Over the years several ion channels have been proposed to underlie the ER leak such as the translocon complex (translocation channel), pannexins, TRPP2 (polycystin), Bcl2 anti-apoptotic proteins, and even IP3R channels functioning in the absence of ligand binding [8–12]. It is presently unclear if Ca2+ leak channels are regulated by ER calcium content and cytoplasmic Ca2+ or if they are constitutively open. In whole-cell patch-clamp experiments, the leak pathway appears to function throughout the duration of the recording, enabling the prolonged continuous detection of CRAC channel activity with passive store depletion. Almost no information is available on their pharmacology. Basal leak pathways have also been described in the cardiac sarcoplasmic reticulum, and ryanodine receptors have been suggested to participate in basal leak under specific circumstances [10].
The reintroduction of calcium to the bathing solution (in the presence of CPA) results in a large calcium elevation, which represents store-operated calcium entry (SOCE). This process is a direct consequence of emptying the ER calcium store, as this pathway is not active without CPA or thapsigargin application. In lymphocytes, SOCE is usually larger than the ER release transient and also decays more slowly. When extracellular [K+] is increased from 4 to 140 mM (Figure 1.1b) calcium increases are drastically diminished. K+ elevation moves the potassium Nernst potential from -90 mV to approximately 0 mV, a 90 mV depolarization. This is expected to depolarize the membrane potential of the Jurkat T cell, which is set by the voltage-gated KV1.3 and calcium-activated KCa3.1 channels, as well as the two-pore voltage-independent K+ channels [13,14]. Depolarized potentials result in a reduced driving force for Ca2+, thereby decreasing Ca2+ influx. This in turn reduces KCa3.1 currents in a positive feedback loop, causing Ca2+ transients to decay faster [1]. Importantly, the shape (time course) of SOCE is set by PMCA activity [6,15–18]. In short, membrane depolarization reduces rather than increases SOCE, demonstrating that in lymphocytes, voltage-gated CaV channels are not the underlying calcium influx pathway. Accordingly, the blockade of KV1.3 in lymphocytes invariably inhibits SOCE and suppresses proliferation, which requires SOCE [19,20]. Similar SOCE-K+ channel systems exist in other cell types [21]. Note that the CPA-induced store calcium transient is not affected by the rise in the extracellular K+ concentration since it does not depend on the plasma membrane potential. SOCE is not entirely abolished in high K+, presumably because the calcium equilibrium potential is well above 0 mV [22]. By contrast, in excitable cells, such as pancreatic β cells, depolarizations caused by increasing [K+] result in a substantial Ca2+ influx through voltage-gated Ca2+ channels of the CaV family (Figure 1.1b). In β cells the major CaV subtype is the dihydropyridine-sensitive L-type channel, which opens at membrane potentials above -10 mV [23,24].
Early electrophysiological evidence for the existence of a non-voltage-gated calcium channel was published in the 1990s (reviewed in [25]). Two groups demonstrated that the intracellular application of inositol trisphosphate (IP3) and high concentrations of Ca2+ buffer EGTA resulted in the gradual development of an inwardly rectifying current that did not exhibit any dependence of gating on voltage. This Ca2+ current was characterized in detail in Jurkat T and rat basophilic leukemia (RBL) cell lines. The channels responsible were named CRAC, for calcium release-activated Ca2+, even though they are in fact activated by store depletion and not calcium release per se [26,27]. It was debated for some time whether calcium release causes calcium influx [28] or if IP3 directly activates calcium influx channels in the plasma membrane [29–33].
In the past decade, the molecular identity of CRAC channels has been discovered to consist of two key components, STIM and Orai (CRACM). STIM1, stromal interaction molecule, is a single-pass transmembrane protein residing in the ER (but also in the plasma membrane) that via its EF hand domains senses Ca2+ concentration in the ER lumen [34]. Upon the emptying of the Ca2+ stores, STIM1 concentrates in junctional ER in close apposition to the plasma membrane, being able now to activate the pore subunit of the CRAC channel, Orai [35] (see Chapter 3). Orai1-3 form a three-member family of four transmembrane domain proteins, which can be activated by store depletion in overexpression systems [35–37]. STIM2, a homologue of STIM1, has been shown to have a role in setting the resting cytoplasmic calcium levels by virtue of its sensing smaller reductions of ER [Ca2+] [38] (see also Chapter 6).
In the following, we discuss in detail the steps required to record CRAC currents in whole-cell patch clamp. In order to deplete ER calcium stores, it is sufficient to simply include high concentrations of calcium chelators EGTA or BAPTA in internal recording solutions. Normally, CRAC channel activity is not detectable immediately after establishing the whole-cell configuration (i.e., break-in), because the calcium stores are full. As the chelator diffuses into the cell, cytoplasmic calcium is drastically lowered. The ER stores are then gradually emptied through ER leak channels [39,40], transporting calcium down its concentration gradient into the cytoplasm, where it is captured by the chelator and prevented from being pumped into ER by SERCA [41]. CRAC channel activation is proportional to the degree of store emptying, that is, inversely proportional to ER [Ca2+] [42,43].
Under physiological conditions, the ER calcium stores are emptied by the second messenger inositol trisphosphate (IP3) [2,44]. IP3 is generated from the hydrolysis of the plasma membrane phospholipid PI(4,5)P2 by phospholipase C enzymes [5,45]. Various types of PLC are stimulated by G proteins or tyrosine kinase-linked receptors, depending on the tissue and ligand [46]. IP3 can bind to its receptors (IP3R) in the ER membrane, which are calcium-permeable channels and provide a pathway for the diffusion of calcium into the cytoplasm. Compared to passive store depletion with chelators, the inclusion of IP3 in internal solutions results in a faster activation of CRAC channels due to rapid store depletion [47].
The most common protocol used to record CRAC channels is the application of command voltage ramps spanning -100 to +100 mV. Ramp durations can be anywhere between 50 and 500 ms in order to generate bona fide current-voltage (I-V) curves. Ramp durations shorter than 50 ms may result in deformed I-V curves. The advantage of a ramp protocol is that instantaneous I-V relations can be obtained every 1–2 s. This is important, particularly for monitoring the development of other, unrelated conductances, such as Mg2+-inhibited cation (MIC/TRPM7) channels discussed in the following section. The drawback of ramp protocols is lack of detailed kinetics information. Thus, CRAC channels inactivate when calcium influx is increased by raising the bathing Ca2+ concentration [48–51]. In cell types traditionally used for recording native CRAC channels (e.g., lymphocytes, mast cells), even when measured at -100 mV, a nonphysiological membrane potential, the current magnitude is usually quite small, in the vicinity of 5–10 pA (corresponding to current densities of 0.5–3 pA/pF). The time course of current development is monitored by plotting the inward current amplitude at negative potentials (usually at -100 or -80 mV) where current is maximal. Outward whole-cell current at a positive membrane potential can also be plotted to ascertain that the leak does not increase during the recording. Because of the steep inward rectification and calcium selectivity of CRAC channels, an outward ionic current at +50 mV or above should be minimal even when calcium stores are completely empty.
All cell types have numerous ion channels in their membranes that contribute to the overall ionic conductance of the cells [52]. The particular channel complement is dependent on the cell type. In order to record CRAC channels in isolation, appropriate ion substitutions and membrane potentials are used. To minimize contribution from endogenous K+ channels, pipette K+ is substituted with Cs+, which permeates poorly through most K+-selective channels such as KV1.3 highly expressed in lymphocytes [13]. High concentrations of tetraethyl ammonium (TEA) or tetramethyl ammonium (TMA) can be included in recording solutions for this purpose. The addition of external Cs+ (1–10 mM) is often used to block inwardly rectifying K+ channels, such as Kir2.1 present in RBL cells and macrophages [53,54]. Alternatively, external K+ is entirely substituted with Cs+. The holding membrane potential between ramps is near 0 mV, which minimizes CRAC channel amplitude but is also depolarized enough to inactivate contaminating voltage-dependent channels, such as KV1.3. The frequency of voltage ramps is kept at 0.5–1 Hz, which favors KV channel inactivation.
Contributions from intermediate or small conductance calcium-activated K+ channels are reduced by Cs+ substitution and inclusion of large amounts of EGTA or BAPTA in the pipette solution. Most Ca2+-activated K+ channels require free calcium of 200 nM or higher to open [55]. Also, these channels can be blocked with relatively specific blockers such as charybdotoxin or TRAM-34 [56–58].
Chloride currents are reduced by substituting chloride both in the pipette and in the bathing solution with larger anions, such as aspartate, glutamate, gluconate, methanesulfonate, or isethionate. It should be kept in mind, however, that most organic anions bind Ca2+ and Mg2+ to some extent and can significantly reduce the concentrations of free divalent cations in the solution. This chelating effect is especially pronounced with citrate. In most cell types, large volume-sensitive chloride channels are expressed and an effort should be made to prevent cell swelling, which transiently activates these channels [59]. It needs to be noted that even the said large anions can pass through chloride channels to some degree [60]. Assuming that the background chloride conductance is constant throughout the recording, it can be subtracted out in most cases [61]. Maintaining correct and consistent osmolality of the internal and external recording solutions is also important to minimize volume-activated chloride channel activation. Osmolality can be adjusted with mannitol, which does not affect CRAC channels.
Magnesium-inhibited cation (MIC/TRPM7) channels are ubiquitously expressed and have in the past contaminated many recordings of CRAC channels. Like CRAC channels, MIC channels were first identified by patch-clamp electrophysiology in RBL and Jurkat cells [54,62,63]. All commonly used mammalian cell lines such as HEK293, CHO-K1, HeLa, COS-7, Jurkat, RBL1 and 2H3, Neuro-2A, and others express large Mg2+-inhibited cation currents encoded by TRPM7 (or possibly by TRPM6 which is also inhibited by Mg2+). TRPM7 channels are inhibited by micromolar to millimolar cytosolic Mg2+ concentrations in a non-voltage-dependent manner [54,64,65]. This current was initially confused with CRAC channels because the conditions for activation of both channels were similar; in both cases, high internal EGTA concentrations are used. While EGTA is used to passively deplete ER Ca2+ stores (see preceding text), it also chelates cellular Mg2+, albeit more weakly. In the absence of Mg2+, it can lead to the slow development of TRPM7 channels by virtue of gradual channel disinhibition. Moreover, the slow time course of current development is reminiscent of CRAC current development. However, major differences exist between these two conductances: the current voltage relation of TRPM7 is steeply outwardly rectifying although this is only apparent at membrane potentials above +40 mV. Upon removal of external divalent cations, TRPM7 I-V is drastically modified, becoming semilinear, as TRPM7 conducts Na+ and Cs+ equally well. By contrast, CRAC channels maintain their inward rectification both in the presence of Ca2+ and other divalent cations and in their absence. Na+ is permeant, whereas Cs+ is almost impermeant through CRAC channels [54,63,66,67]. TRPM7 inward unitary conductance is approximately 39 pS in the absence of divalent cations [64]. By contrast, CRAC channel unitary conductance is much smaller and cannot be measured directly. From noise analysis, it is inferred to be in the range of 100 fS (see succeeding text). CRAC channel inward rectification is not Mg2+ dependent [54,68] unlike TRPV6 or inwardly rectifying K+ channels (KIR) and appears to be intrinsic to the protein. It should be noted that the voltage-gated CaV open channel I-V plot is also inwardly rectifying (e.g., [69]). In addition to ion substitution, the pharmacological properties of CRAC channels can also be used for current separation and are discussed in some detail in Chapter 16.
The perforated-patch variety of the whole-cell patch clamp has been successfully used to record CRAC channels [70–73]. It was originally used to prevent a rundown of voltage-gated calcium channels [74]. In this method, the patch pipette is backfilled with internal solution supplemented with dissolved nystatin or amphotericin B. These are polyene antibiotic ionophores that form ion channels in mammalian cell membranes. Nystatin and amphotericin channels are permeable to monovalent cations and anions but not to divalent ions, such as Ca2+, Mg2+, or SO42- [75]. Such ion selectivity allows the maintenance of physiologically relevant buffering of these ions for the duration of the recording. The pipette tip is filled with an antibiotic-free internal solution. After seal formation, it takes roughly 10 min for the ionophore to diffuse to the pipette tip and the cell surface and begin forming channels. This is detectable by the increased slow membrane capacitance transients, a kind of whole-cell break-in “in slow motion.” Perforated-patch recording requires more skill than the simple whole-cell configuration, however, mostly because at high concentrations these ionophores prevent gigaohm seal formation. The shape of the pipette is crucial for facilitating the diffusion of pore-forming antibiotics to the tip. Both nystatin and amphotericin are light sensitive, particularly the former, and lose their activity in 4–5 h when diluted in the internal solution. In our experience, amphotericin B is easier to use because it forms pores in the membrane more readily, and access resistance falls to lower values (∼10 MΩ) than for nystatin [76]. If physiological anion concentrations are desired, then gramicidin can be used instead of nystatin or amphotericin, since gramicidin channels are believed not to conduct chloride, and the steady-state cellular anion concentrations will be undisturbed [24,75,77].
IP3, EGTA, and BAPTA cannot pass through the antibiotic-formed channels; therefore, CRAC channels can be elicited by the bath application of antigens, mitogens, hormones, or SERCA inhibitors thapsigargin and CPA, which are uncharged and membrane permeant [43,70,78]. The perforated patch should also prevent Mg2+ depletion and spontaneous TRPM7 activation [79]. In perforated patch, care must be taken to rule out the accidental rupture of the patch and formation of a conventional whole-cell configuration. This can be done by including high (millimolar) concentrations of Ca2+ in the internal solution expected to kill the cell after entering the cytoplasm. In order to minimize Ca2+-dependent inactivation [26], it is especially important to use a depolarized holding potential to avoid Ca2+ loading in the absence of EGTA or BAPTA.
As with CaV channels, in order to maximize the CRAC channel current, the external calcium concentration can be elevated to 50 or ∼110 mM [70,80]. (This concentration is close to the upper limit imposed by osmotic pressure.) This results in increased current amplitude, because the concentration of the conducting ion and driving force is increased. The increased calcium influx, however, also triggers calcium-dependent inactivation, both fast and slow, which eventually reduces the current amplitude [48,81]. Interestingly, the calcium-dependent inactivation appears to involve Orai and STIM proteins themselves but not calmodulin, unlike the case for TRPV5/6 channels (see succeeding text) [49–51]. A good trade-off between these two Ca2+ effects is a bathing concentration of 6–10 mM Ca2+, which is sufficiently low to prevent substantial inactivation [37,82]. Command voltage ramps can be preceded by a conditioning hyperpolarizing step to bring inactivation to a quasi-steady state and avoid the distortion of the instantaneous I-Vs due to fast Ca2+-dependent inactivation [78].
It is now firmly established that in the virtual absence of divalent cations in the external medium, Ca2+-selective channels become permeable to small monovalent cations. The amplitude of the monovalent cation current through Ca2+ channels in the absence of divalents is usually 6–10 times larger than when carried by Ca2+. This was originally demonstrated for L-type voltage-gated (CaV) Ca2+ channels [83–85]. Since then, the removal of divalent cations has been tested for other Ca2+ channels and shown to generally increase current amplitude. CRAC channels also belong to this group [26]. The removal of divalent cations results in a significantly larger current amplitude but with a twist: the increase is transient, unlike the Ca2+-mediated CRAC current recorded when internal solution contains high concentrations of chelators [86]. It is thought that this time-dependent reduction of monovalent CRAC current (termed inactivation) is a result of depotentiation caused by the removal of external Ca2+. Other divalent metal cations such as Ni2+ and Zn2+ could not substitute for the potentiating effect of Ca2+ in RBL cells [80], but Ni2+ was found to be effective in Jurkat T cells [87–89]. In an overexpression system, it was demonstrated that under certain conditions the monovalent Orai current can be stable [37]. It is now thought that this is a consequence of changes in Orai to STIM1 ratios during transfection [90]. The CRAC I-V shape in divalent-free (DVF) solutions is identical to its shape in Ca2+, preserving its inward rectification, unlike the case for TRPM7 [67].
The overwhelming majority of CRAC channel recordings have been performed at room temperature. Increasing the bath temperature to 37°C significantly increased CRAC current amplitudes in RBL cells [71,78]. Additionally, in perforated-patch recording, CRAC channels could only be activated by antigen application at 37°C but not at room temperature [78], unlike the case for SERCA inhibitors.
One of the fundamental characteristics of an ion channel is its conductance. The first recordings of ICRAC in mast cells and lymphocytes suggested that the conductance of a single CRAC channel is well below the levels that can be resolved by patch clamping as single-channel currents [26,70,91]. The standard approach in cases when single-channel conductance is too small for patch-clamp recording is to use the so-called nonstationary noise analysis (analysis of variance) [92]. Due to the stochastic nature of single-channel opening, a steady-state macroscopic current mediated by a constant number of channels fluctuates with time around its mean value. The nonstationary noise analysis is based on the fact that the magnitude of these fluctuations depends on the amplitude of the single-channel current, the number of channels, and their open probability [92,93]. The relationship between current variance (σ2; averaged squared current fluctuations from its mean) and single-channel current is given by the following equation:
Although CRAC channels show some voltage dependence due to fast Ca2+-dependent inactivation at negative potentials [26,94], it is virtually impossible to record a large enough number of current sweeps in response to voltage steps below -100 mV in one cell due to an ICRAC drift and rundown. The first attempts of CRAC unitary conductance estimation used traces of ICRAC development in Jurkat T cells at a constant voltage of -80 mV in a bath solution containing 2 mM Ca2+, which was then switched to 110 mM Ca2+ [70]. This approach was based on the assumption that the changes in ICRAC amplitude during current development and its slow inactivation in a solution containing 110 mM Ca2+ were due to the changes in CRAC channel Po. Current variance was calculated from 200 ms episodes taken from every 2 s of continuous recording and plotted against corresponding mean current amplitudes [70]. In these plots, variance showed linear dependence on the current amplitude, which suggested a low Po of CRAC channels [70] (Figure 1.2). The calculated single-channel current amplitudes were -1.4 fA in 2 mM Ca2+ and -3.6 fA in 110 mM Ca2+ at -80 mV. The estimated single-channel conductances were 9 and 24 fS, respectively [70].
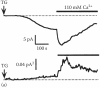
Fluctuation analysis of currents stimulated by thapsigargin in Jurkat T cells.
As discussed earlier, in the absence of Ca2+ and Mg2+, CRAC channels become permeable to monovalent cations. Replacing physiological (Ca2+-containing) bath solutions with divalent cation-free solutions causes a 7–8-fold increase in ICRAC amplitude followed by current deactivation to about 20%–30% of its peak value within 30–60 s, which suggests a significant change in CRAC channels Po in that time [86]. This property of ICRAC was utilized to estimate monovalent CRAC single-channel currents using an approach similar to that of Zweifach and Lewis [87]. In these experiments, the variance of Na+ current through ICRAC also exhibited linear dependence on the current amplitude, supporting the notion of low Po of CRAC channels [70]. However, the calculated unitary CRAC channel conductance of 2.6 pS in the absence of divalent cations was significantly higher than what would be expected from the estimates of CRAC unitary conductance in 110 mM Ca2+ [70], considering that the removal of divalent cations causes only about a 7–8-fold increase in ICRAC amplitude. The discrepancy was later explained by a possible contamination of these recordings with endogenous MIC/TRPM7 channels that were incompletely blocked by 3 mM MgCl2 in the pipette solution [63]. Using a higher internal concentration of Mg2+ (8 mM), Prakriya and Lewis reported a unitary monovalent cation (Na+) CRAC conductance of ∼0.2 pS, which agreed well with Ca2+ unitary conductance [63].
All measurements described earlier were based on the assumption that the observed changes in ICRAC amplitude resulted from changes in Po and not the number of functional CRAC channels on the plasma membrane. The linear dependence of variance on current amplitude was explained by low Po of CRAC channels [70]. The linear variance-current plot, however, could result from the changes in the number of active CRAC channels during ICRAC development and depotentiation in divalent cation-free solutions, with constant and high Po [89]. To investigate this possibility, Prakriya and Lewis used nonstationary noise analysis of Na+ current through CRAC channels in the presence of a range of micromolar Ca2+ concentrations in the bath solution [89]. An application of up to 20 μM of Ca2+ to the bath solution increased the Na+ current noise, although the amplitude of the current declined [89], which suggested that, indeed, the Po of CRAC channels was above 0.5 and the previous assumption of low Po of CRAC channels was incorrect. The variance-current plot of the data obtained using a Ca2+ block of Na+ CRAC conductance followed a parabolic function (Equation 1.1), giving a CRAC Na+ conductance of ∼0.7 pS and Po of about 0.8 [89]. Similar results were later obtained for overexpressed Orai1/STIM1 and Orai3/STIM1 channels [95,96]. In conclusion, a higher than previously thought single-channel conductance and high Po of CRAC channels opens a possibility of direct single-channel current measurements. The single-channel conductance of 0.7 pS corresponds to a 70 fA unitary current per 100 mV of driving force. Assuming that the equilibrium potential for Na+ is about +50 mV, the amplitude of the monovalent cation unitary CRAC current at a membrane potential of -100 mV should be in the vicinity of 0.1 pA. A single-channel current of such amplitude can be recorded using low-noise patch-clamp amplifiers and appropriate filtering, provided that the right conditions for the current activation are met [97]. It remains to be determined, however, if this can be achieved in practice for CRAC channels.
The molecular components of CRAC channels were discovered more than a decade ago and include Orai1-3 (CRACM1-3) pore subunits and STIM1-2, ER resident luminal calcium sensors (reviewed in [33,35] and Chapters 2 and 3). Thus, mammalian cells express three different pore subunits, which was not predicted before they were cloned. Orai1 and Orai2 amino acid sequences are ∼60% homologous. Orai3 also has a high percentage of homology with the remaining members: ∼63% with Orai1 and ∼66% with Orai2. In the transmembrane domains the homology percentages are even higher, reaching 90% [98,99]. All three isoforms are expressed in human T lymphocytes [82]; however, a familial defect in Orai1 results in severe combined immunodeficiency even in the presence of normal Orai2 and Orai3 expressed in these cells. This strongly suggests that Orai2 and 3 cannot substitute for Orai1 function, at least in that case [100]. It should be noted that in cells where Orai1 expression has been knocked down with siRNA, small SOCE signals mediated by Orai2 or Orai3 are still evident [101,102].
When coexpressed with STIM1, all three Orai proteins give rise to robust inwardly rectifying currents (Figure 1.3). Orai1-3 isoforms are permeable to Ca2+ and some other divalent cations (Ba2+, Sr2+) [90,103]. For Orai2 and Orai3, inward Ba2+ currents were larger than for Orai1, although they could only be detected in the presence of Na+ but not TEA, suggesting that Na+ may carry part of the current [103]. In the complete absence of divalent cations, Orai1-3 readily conduct monovalents, such as Na+, Li+, and Rb+ [103,104]. Interestingly, Orai isoforms can be distinguished in heterologous expression systems by their sensitivity to 2-aminoethyl diphenyl borinate (2-APB), a compound originally identified as an IP3 receptor blocker [105] (see Chapter 16). Orai1 and 2 are potentiated by low (5–10 μM) but inhibited by high 2-APB (above 50 μM) concentrations [106]. By contrast, 2-APB activates Orai3/STIM1, altering its I-V relation, which is normally inwardly rectifying (Figures 1.3 and 1.4). 2-APB-mediated activation results in an outwardly rectifying current, which can be conducted by both Cs+ and Na+. Similar to native CRAC channels, overexpressed Orai1 and Orai2 conduct Cs+ poorly (∼10% of Na+ conductance) in the absence of divalent cations. Consequently, it has been suggested that 2-APB widens the Orai3 pore and allows it to conduct monovalent cations even in the presence of Ca2+ [107]. In contrast to Orai3, 2-APB does not change Orai1 and Orai2 selectivity [106]. Whether this divergence of 2-APB effects for overexpressed Orai1-3 can be used to distinguish endogenous Orai isoform activity is still unclear. It remains to be discovered if the Orai3 nonselective channel state can be achieved under physiological conditions without 2-APB and what that means in terms of cellular calcium metabolism.

Instantaneous I-V plots of Orai1-3/STIM1-mediated ICRAC. Traces were recorded in response to 100 ms voltage ramps ranging from -120 to 120 mV in HEK293T cells transfected with STIM1 and Orai1, Orai2, or Orai3 cDNA as previously described [90]. External (more...)
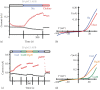
2-APB modified Orai3 conducts both Na+ and Ca2+. Whole-cell patch-clamp recordings of HEK293 cells transfected with Orai3 and STIM1. (a, c) Time courses of inward (black) and outward (red) current development in the presence of 50 μM 2-APB. (b, (more...)
Orai1 to STIM1 transfection ratios significantly influence the electrophysiological properties of these channels. Specifically, the kinetics of the channel activation and inactivation were changed in addition to selectivity to divalent metals (Ba2+ and Ca2+). Increasing STIM1 to Orai1 expression ratios results in an inactivating current whereas Orai1 excess to STIM1 leads to non-inactivating currents [90]. Orai currents can be evoked by coexpressing full length STIM1 but also by overexpressing only the SOAR region [37] (discussed in Chapter 2).
Orai/STIM channels have primarily been studied in human embryonic kidney (HEK) cells transfected with their various isoforms. The recording conditions are essentially the same as for recording the native CRAC channels. The internal solution should contain calcium chelators alone or with IP3 for depleting ER Ca2+ stores [104,106]. However, in most cases, overexpressed Orai/STIM channels are to some extent constitutively active, unlike CRAC channels in Jurkat or RBL cells [37]. The overexpression of either Orai or STIM proteins in isolation does not usually result in large CRAC currents.
The murine and human Orai2 exist in two forms, long and short [108–110]. The Orai2 expression pattern is somewhat different from that of Orai1, although in many cases they are coexpressed [109]. For example, neurons and chondrocytes show high expression of Orai2, even though its function in these cells is unknown [111]. When coexpressed with STIM1, human Orai2 form inwardly rectifying channels (Figure 1.3) sharing many biophysical properties with Orai1 [103]. In an early detailed study of overexpressed murine Orai2, Gross and colleagues recorded currents from HEK cells overexpressing both long and short splice variants [108]. Orai2 and STIM1 were transfected at a 2:1 ratio. Infusion of cells with high EGTA and IP3-containing buffers evoked currents reminiscent of native CRAC channels. Interestingly, the levels of expression depended on the cell type chosen for transfection, HEK vs. RBL. One striking difference of Orai2 from Orai1 was the apparently increased (slow) calcium-dependent inactivation. It was concluded that calcium influx through Orai2 channels causes the inactivation that can be relieved by the fast removal of calcium from the vicinity of the channels. Inactivation was proportional to the peak current amplitude. The dependence of channel expression on the type of the background cell line was explained by the fact that HEK cells express very little Orai1 and Orai2 protein detected in Western blots, whereas RBL cells express both channels at high levels. Accordingly, the overexpression of STIM1 alone in HEK cells did not increase CRAC current density but doubled it in RBL cells. When coexpressed with Orai1, Orai2 was shown to reduce current expression, suggesting that it can form heteromers with other Orai isoforms [108]. Similar conclusions were reached by Inayama and colleagues by coexpressing Orai1 E106Q point mutant with the wild-type Orai2 in chondrocyte cell lines [111].
TRPV5 and TRPV6 channels are present in kidney and gastrointestinal epithelial cells at high levels [112,113]. TRPV5 (ECaC1) is thought to be expressed mostly in the kidney, whereas TRPV6 (CaT1, ECaC2) expression is higher in the gastrointestinal tract, placenta, and epididymal epithelia [114–118] (see Chapter 13). Both are absent in lymphocytes [119]. TRPV5/6 channels were first identified by patch-clamp electrophysiology only after their cDNA sequence was discovered. In an overexpression system (primarily plasmid-transfected HEK or CHO cells but also mRNA-injected Xenopus oocytes; Chapter 13), they are either constitutively active or activated (i.e., revealed) by calcium removal from the cytosol when they become permeable to monovalent cations [120–126]. Increasing bathing calcium concentration potentiates the currents as expected for a Ca2+-selective channel. This appears to be a simple increase in ionic current due to increased driving force for calcium. The extracellular calcium-dependent potentiation effect described for CRAC channels has not been reported for these channels. Notably, the holding potential essentially determines the current magnitude in a given cell [121,127]. Current increase resulting from increased [Ca2+]o is usually short-lived and depends on the holding membrane potential [120]. This is thought to be the consequence of calcium loading into the cell (in the vicinity of the channels) via constitutively open TRPV5/6 channels [122]. Accordingly, the dependence on the holding potential disappears when external DVF solutions are used. Internal Ca2+ inhibits TRPV5/6 channels with an IC50 of ∼130 nM, explaining the current inactivation seen when Ca2+ is present in the bath solution [128]. As for Orai/STIM channels, internal recording solutions should contain high concentrations of EGTA or BAPTA, in this case to avoid Ca2+-dependent inactivation.
In addition to Ca2+, TRPV5 and 6 also conduct Sr2+, Ba2+, and Mn2+ [128]. When Ba2+ is used as the charge carrier instead of Ca2+, inactivation is drastically reduced for both TRPV5 and 6 [128]. Mn2+ permeation through TRPV5/6 becomes relevant when Mn2+ quenching of Fura-2 signals is used to evaluate calcium entry [129,130].
Like Orai channels, TRPV5 and TRPV6 are steeply inwardly rectifying. And as with Orai channels, the removal of divalent cations from the bathing medium results in vastly magnified TRPV5 and TRPV6 currents. This simple maneuver has been used to record channel activity without the fast inactivation caused by Ca2+ influx through these channels (see preceding text) [127,131]. Primarily Na+ has been used, but K+ and especially Li+ are also highly permeant [128]. Cs+ is less permeant (∼80% of Na+), as is the case for voltage-gated Ca2+ channels. It appears that channel sensitivity to blockers, ruthenium red or econazole, is not affected by the permeating ion [131].
It was discovered early on that TRPV5 and 6 channels exhibit a rundown of activity during prolonged electrophysiological recordings [120]. Intracellular MgATP is required to maintain TRPV5 and TRPV6 channel activity and prevent rundown, whereas NaATP cannot substitute for this effect of MgATP [128]. Most likely the MgATP effect reflects maintaining PI(4,5)P2 levels through lipid kinases, although other mechanisms have also been proposed [128,132,133]. Cytosolic Mg2+ alone induces slow inhibition which can be relieved by PI(4,5)P2 binding [134]. The strong inward rectification of TRPV6 also depends on intracellular Mg2+ and is mediated by an aspartate residue [135].
Endogenous TRPV5/6 channel activity has been reported mainly in two studies: TRPV5 channel currents were measured in rat renal cells [136] and TRPV6 in a human colonic cell line [137] using whole-cell patch clamp. In epithelial cells, native TRPV5/6 channels are not normally active/detectable in the presence of physiological concentrations of Ca2+ (1–2 mM). In order to detect measurable currents, bathing calcium was increased to 20 or 100 mM combined with EGTA-containing internal solutions [136,137]. The addition of the steroid hormone 17β-estradiol markedly increased TRPV5 and TRPV6 currents carried by Ca2+. It remains to be discovered under what metabolic conditions native TRPV5/6 channels conduct measurable current at more physiological calcium and intracellular buffering. To date, however, most patch-clamp studies of TRPV5/6 have been performed in heterologous expression systems. It should be kept in mind that in the conventional whole-cell recording configuration used for TRPV5/6 recording, the ER stores will also most likely be depleted due to low calcium and the presence of chelators in the internal solution. A way around this would be to use a perforated patch, which however will prevent the depletion of cytoplasmic calcium and reduce current amplitudes by inactivation [121,122].
For (rabbit) TRPV5, a range of single-channel conductances has been determined in DVF Na+-based solutions, 59 pS [138], 64 pS [139], and 77.5 pS [140], and has been shown to depend on pH [138]. Inside-out patch recording configuration was used in these studies. For TRPV6 the unitary conductance in DVF Na+-based solutions was found to be 51 pS [127]. The single-channel conductance for calcium or other permeant divalent cations has not been reported for TRPV5 or TRPV6 but is expected to be much smaller than their Na+ conductance. Unlike CRAC channels, TRPV5/6 channels do not exhibit depotentiation after the removal of Ca2+, enabling prolonged recordings of Na+ currents.
In summary, Orai/STIM and TRPV5/6 exhibit many common properties, such as inward rectification and calcium selectivity but also significant differences, such as single-channel properties and mechanism of inward rectification. Chapters 9, 11, and 15 provide useful details on the more technical aspects of electrophysiological recordings (amplifiers, vibration isolation tables, perfusion systems, micromanipulators, etc.). Chapter 9 discusses the patch-clamp recording of overexpressed Orai/STIM and site-directed mutagenesis. Chapter 13 describes the expression of TRPV5/6 channels in Xenopus oocytes. Detailed compositions of solutions used for electrophysiological recordings of Orai/STIM channels can be found in several recent publications: [47,141] describe the recording of native CRAC channels in the whole-cell mode.
We thank Pavani Beesetty, Wright State University, for performing the single-cell calcium imaging experiments depicted in Figure 1.1.
J. Ashot Kozak
Department of Neuroscience, Cell Biology and Physiology
Boonshoft School of Medicine
Wright State University
Dayton, Ohio
Grigori Rychkov
School of Medicine
University of Adelaide
and
South Australian Health and Medical Research Institute
Adelaide, South Australia, Australia
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
Your browsing activity is empty.
Activity recording is turned off.
See more...