The generation of interictal epileptiform discharges (IEDs) in partial epilepsies is commonly ascribed to enhanced excitatory interactions within glutamatergic neuronal networks. Recent evidence, however, supports the view that inhibitory networks do play a central role. Human and experimental EEG data indicate that IEDs (i) often present with superimposed high frequency activity; (ii) are followed by inhibition/depression of background activity; (iii) can generate delayed excitatory components; and (iv) can be sustained by either glutamatergic or GABAergic signalling. Pre-surgical intracranial EEG recordings performed in patients have confirmed that interictal spikes in the epileptogenic zone may be followed by either enhanced or depressed inhibition. In addition, recordings of neurons from post-surgical in vitro brain slices obtained from temporal lobe epilepsy patients have demonstrated that IEDs are abolished by GABAA receptor antagonists. Further evidence has emerged from studies of human brain slices with focal cortical dysplasia and several in vitro animal models of epileptiform synchronization showing GABAergic pre-ictal events occuring in neocortical and limbic structures. Together, these data indicate that diverse ligand-gated mechanisms activate IEDs and lead to network hyperexcitability in epileptic patients and in animal models of epilepsy.
Seizures (also termed ictal discharges) represent the critical events and the primary clinical burden of an active epileptic condition. Between seizures the brain of patients with epilepsy generates pathological patterns of activity, designated as interictal epileptiform discharges (IEDs), that are clearly distinguished from the activity observed during the seizure itself. The correlation between IEDs and ictal discharges in intractable partial epilepsies has been the subject of several studies (for review see 1–4), yet no conclusion regarding the reciprocal relationship and inter-dependence of IEDs and ictal discharges has been reached to date. Indeed, the existing data have led to two opposite views that assign to IEDs either a protective or a precipitating role in seizure occurrence.
Interest in the mechanisms underlying IEDs has been revived during the last decade by pre-surgical diagnostic studies that utilize prolonged video-EEG along with intracranial EEG monitoring over periods of several days. Analysis of IEDs recorded from the scalp and with intracranial electrodes in patients with partial epilepsy have focused on the relation between IED topographic distribution and seizure patterns, and on their occurrence during the pre-ictal state 5–10. The methods to localize the electric source(s) within the brain volume that generate IEDs recorded using scalp EEG electrodes have been extensively reviewed and will not be considered in the present chapter 11–13. Intracranial recordings either with cortical surface grids/strips or with intracerebral electrodes have been more useful to identify different IED patterns, since electrodes are positioned in closer proximity to the physiological IED generators. These studies, along with experimental evidence obtained from animal models of partial epilepsy, have demonstrated the existence of diverse IED electrical patterns. Focal IEDs show, a large pattern variability (including spikes, sharp waves, bursts of fast spikes, sequences of fast oscillation, etc.) even in the same patient/model (). Given this diversity, it is reasonable to assume that different types of IEDs may be mediated by distinct neurobiological mechanisms and play divergent functional roles with respect to ictogenesis. We will review in this chapter the clinical and experimental evidence that demonstrate the multiplicity of IED patterns, based on data obtained from humans and from experimental models of partial epilepsy and seizures. The neurobiological mechanisms responsible for the generation of different IEDs will also be considered.
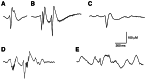
Interictal epileptic discharge (IED) patterns recorded in human partial epilepsies with intracranial electrodes. a Interictal spike; b group of interictal spikes from neocortical dysplasia, c sharp wave from a lesional partial epilepsy; d fast activity (more...)
Different IED Patterns in Epileptic Patients: Spikes, Spike Bursts, Sharp Waves
As mentioned above, interictal patterns are diverse and variable in partial epilepsies. Fast events defined as interictal spikes are characterized by a large-amplitude rapid component lasting 50–100 ms that is usually followed by a slow wave, 200–500 ms in duration 14;15 (). Highly reproducible interictal spikes are typical of cryptogenic and benign forms of epilepsy, such as epileptic disorders with Rolandic or occipital paroxysms 16;17. In these clinical conditions, interictal spikes show selective and specific regional distribution. In contrast, partial epilepsies secondary to brain lesions show more irregular interictal spikes, often associated with IED patterns that include sharp waves (characterized by a rapid component that lasts between 100 and 300 ms), bursts of spikes, fast oscillations, and repetitive, paroxysmal slow waves (; but see 18).
IEDs in partial pharmacoresistant epilepsies have been well characterized. In humans, simultaneous unit recordings and laminar field potential profiles obtained with intracortical multielectrodes during acute (surgical) corticography 19 or chronic pre-surgical monitoring 20;21, have revealed that interictal spikes are initiated by large postsynaptic depolarizations, consistent with a paroxysmal depolarization shift similar to those recorded experimentally 22–25. Moreover, the cortical layers where these depolarizations occur differed according to whether the spike was locally generated or remotely propagated from a distant area 19;21. Finally, characterization of unit firing during interictal spikes has demonstrated heterogeneity inside and outside the seizure onset zone, suggesting that IED’s are not a simple paroxysm of hypersynchronous excitatory activity, but rather represent the interplay of multiple distinct neuronal types within extended neuronal networks 26.
Certain forms of human ‘lesional’ partial epilepsies and cortical dysplasias show distinctive patterns that have specific diagnostic value 27–29. One such condition is the Taylor-type II focal cortical dysplasia that features IEDs characterized by high frequency spikes and polyspikes, defined as brushes
30–32 These brushes last 100–200 ms, recur with a periodicity of 1–2 s, and are enhanced during slow-wave sleep. Electrical stimulation has demonstrated that brushes in Taylor-type II focal dysplasias are followed by a desynchronization/depression of activity that lasts 0.5–1 s and are associated with a higher threshold for the generation of further IEDs. 33 IEDs in temporal lobe epilepsy (TLE) with hippocampal sclerosis are less frequent than in focal dysplasias, and consist of either spikes or sharp waves that are often undetectable on the scalp EEG 34;35. In patients with severe hippocampal atrophy, large amplitude spikes shorter than 100 ms with small post-spike slow activity have been reported. These IEDs increased in frequency and became rhythmic before and after ictal events 36.
Interictal Spikes in Acute and Chronic Animal Models In Vivo
The temporal correlation between IEDs and ictal discharges has been analyzed in vivo in animal models mimicking both acute seizures and chronic epilepsy. Pioneering intracellular recordings obtained from neurons located in the “epileptic focus” induced by application of convulsants (e.g., penicillin) have demonstrated that interictal spikes correlate with paroxysmal depolarizing shifts of the membrane potential leading to sustained action potential firing and at times followed by a robust hyperpolarization 22;23;37. These studies have also shown that the transition to seizure is characterized by IED acceleration along with a decrease or disappearance of the post-burst hyperpolarizing potential, a phenomenon that was proposed to result from the progressive accumulation of extracellular potassium 38;39.
This transition pattern, however, has not been reproduced in chronic models of TLE. Thus in both kindling and drug-induced status epilepticus (SE) models, the IED frequency either did not change or it decreased before the onset of an ictal event 40–43 (for review see 2). Interictal spiking has also been analyzed in animals injected with kainic acid in one hippocampus; this represents a widely used chronic model that faithfully reproduces TLE. As in TLE patients, unilateral IEDs were reproducibly observed in this model in rats 44;45, mice 46 and guinea pigs (Carriero, Arcieri and de Curtis, unpublished observations). More recently, EEG-video monitoring of the epileptic activity recorded during the latent and chronic periods in rats undergoing pilocarpine-induced SE has revealed that following the appearance of seizures, IEDs diminish in duration in the CA3 region and occur at higher rates in the amygdala 47. Therefore, these findings suggest that IEDs undergo structure-specific changes following the appearance spontaneous seizure activity. Little information about IEDs is available for other chronic models of partial epilepsy, such as post-traumatic models or models of cortical dysplasia studied in vivo.
IEDs in Acute Animal Models In Vitro
IEDs can be studied in brain slices that are maintained in vitro following experimental procedures that favor epileptiform synchronization. Several reports have shown that IEDs are initiated by gradual enhancement and progressive recruitment of synaptic excitation that reaches the threshold for regenerative calcium currents 48–50. This process further sustains recurrent excitation and promotes the synchronous firing of a large number of neurons that contribute to the buildup of a population event recognizable as a population spike/sharp wave. Excitatory postsynaptic potentials associated with these IEDs 25;51;52 are mediated by glutamate receptors of the AMPA and NMDA subtypes 53–57 . Similar mechanisms of IED generation have also been identified in slices of the neocortex 58–60, piriform cortex 50;61, and entorhinal cortex 62 following a variety of pharmacological manipulations. Regenerative potentials sustained by high-voltage calcium spikes 61;63–66 and by a persistent fraction of the voltage-gated sodium current 67–69 also contribute to paroxysmal depolarizing shifts. Finally, interictal synchronization is further facilitated by non-synaptic interactions which can be mediated by extracellular electric fields (ephaptic interactions) or by intercellular gap junctions70;71 that exist between either principal neurons or interneurons. 72–74
IEDs and the associated glutamatergic paroxysmal shifts are typically observed during prolonged application of (i) drugs that interfere with GABAergic inhibition such as bicuculline, penicillin and picrotoxin (), (ii) agonists of glutamatergic transmission such as kainic acid, or (iii) solutions with a ionic composition that enhances neuronal excitability. However, epileptiform discharges can also be induced by drugs that boost both glutamatergic and GABAergic synaptic transmission such as the potassium blocked 4-aminopiridine (4AP). Early studies have shown that 4AP induces two types of IEDs within the hippocampal formation.. The first type is characterized by fast IEDs that are driven by the CA3 network, and are abolished by AMPA receptor antagonists. The second type consists of slow IEDs that were spared by glutamatergic receptor blockers but abolished by GABAergic antagonists 75. It was subsequently shown that these slow IEDs can be recorded from any limbic cortical area as well as from the neocortex in brain slices obtained from rats or mice (as well as in the human neocortex, see below). This evidence has recently been confirmed in several areas of the in vitro isolated guinea pig brain (). 76
IEDs analysed in in vitro models of epileptiform synchronization. A: Glutamatergic interictal spikes induced in hippocampal slices (left panel) by application of 20 μM bicuculline. Field potentials recorded in the entorhinal cortex (EC), in the (more...)
Intracellular recordings in brain slices have demonstrated that the slow IEDs induced by 4AP are coupled to a complex intracellular potential consisting of hyperpolarizing and depolarizing components. Recently, similar GABAergic IEDs - which correlated in entorhinal cortex (EC) neurons with inhibitory postsynaptic potentials - have been reported in the isolated guinea pig brain maintained in vitro during glutamatergic receptor blockade 76;77; under these pharmacological conditions, the slow IEDs continued to propagate within the hippocampal-entorhinal region and from one EC to the EC of the contralateral hemisphere () . These slow IEDs are abolished by GABAA receptor antagonists as well as by a mu-receptor agonist both in the brain slice and in the isolated guinea pig in vitro preparation, thus confirming that they reflect the synchronous activity of local GABAergic networks. It is unclear how the slow IEDs propagate during glutamatergic receptor blockade, but data obtained in brain slices suggest the involvement of non-synaptic mechanisms such as transient increases in extracellular potassium and subsequent redistribution of this ion (see below). However, the involvement of long-range GABAergic pathways or syncytia-like connections among interneurons cannot be ruled out78;79.
Experiments performed in the in vitro isolated guinea pig brain, in which inhibition was reduced circa 50% by short-lasting bicuculline systemic perfusions, have demonstrated the existence of IEDs sustained by bursting of inhibitory interneurons that precede (by about 1 minute) the generation of a seizure-like discharge 80. These IEDs were associated with inhibitory postsynaptic potentials in EC principal neurons of both superficial and deep layers. In addition, glutamatergic IEDs and GABAergic IEDs could be simultaneously induced by this short-lasting bicuculline application in the piriform cortex and in the EC, respectively. Hence, this evidence suggests that an epileptic brain can generate interictal activity that is sustained by both glutamatergic and GABAergic networks, and is in line with the occurrence of GABA-dependent IEDs as reported in human cortical slices obtained from post-surgical specimens 81–83. IEDs that correlate with synchronous inhibitory postsynaptic potentials in large groups of neurons are often associated with seizure onset 80;84. It is therefore tempting to speculate that GABA-mediated interictal events may contribute to enhance synchronization of local epileptic networks through a mechanisms of post-inhibition resetting of neuronal firing 85;86.
IEDs have recently been analyzed by coupling electrophysiological recordings with imaging of activity-depended intracellular changes of calcium concentration (for review see 87). Increases in calcium signals have been identified during IEDs and ictal discharges in neurons, presumably as a correlate of neuronal firing 88–90 In one of these studies, both IEDs and seizures appeared to be associated with calcium increases in astrocytes and these increases were independent of neuronal activation, thus suggesting that ictogenesis could be sustained exclusively by astrocyte activation and by the associated release of glutamate 90. However, even though the contribution of glutamatergic glio-transmission to ictogenesis has been confirmed by other authors, the unique role of astrocyte activity in seizure initiation has not been replicated 91. One recent report analyzed this issue in different experimental models of seizures and IEDs; these experiments found that astrocyte calcium signalling contributes to sustained ictal activity, but is not involved in the generation of IEDs 92.
High Frequency Oscillations as Interictal Events
High-frequency oscillations (HFOs) at >100 Hz have been recorded from cortical structures in humans and other animals, both under physiological conditions and in partial epilepsies (for review see 93). Cortical HFOs at 100–200 Hz occur under physiological conditions, during the interictal state in patients presenting with partial epilepsy, and in animal models 93–95. Intracranial EEG recordings obtained from pharmacoresistant patients suffering from mesial TLE have shown that HFOs are observed in coincidence with an interictal spike and in isolation. Further studies have confirmed these observations in TLE patients 97;98 and also in the epileptogenic region of patients with neocortical partial epilepsy 99–101. Physiological HFOs (also termed ripples) are implicated in the process of memory consolidation 102, and represent population inhibitory postsynaptic potentials generated by principal neurons entrained by synchronously active interneuron networks 103;104. Juxta-cellular recordings obtained with microelectrodes in the human hippocampus during physiological ripples have demonstrated that pyramidal cells fired preferentially at the highest amplitude of the ripple, while interneurons discharged earlier than pyramidal cells 105. Pathological fast ripples that express very high-rate oscillations (250–600 Hz) were recorded exclusively from epileptic tissue from epileptic patients and in animal models of TLE 96;106;107. Fast ripples can be observed in the interictal state, while components in the beta-gamma frequency range are usually associated with ictal discharges 108. Moreover, pathological fast ripples recorded in vivo, unlike ripples, are supported by synchronous burst firing of abnormally active principal neurons and are assumed to be independent of inhibitory neurotransmission 93.
More recently, a different pattern of very low amplitude HFOs with high intrinsic rhythmicity (>250 Hz) has been identified in the seizure onset region in mesial TLE patients during the pre-ictal period 98;109;110. This activity - which is concealed in the intracranial recordings and can only be extracted by amplifying the appropriately filtered signal - occurs in coincidence with IEDs and sharp waves but also in the absence of any detectable IED 98;111. In summary, HFOs may be interpreted as typical IEDs in partial epilepsies, and their association with other types of IEDs is not the rule.
The cellular and network mechanisms responsible for interictal HFOs have been analyzed in detail in in vitro brain slices exposed to pro-epileptic drugs. Physiological fast oscillations, either pharmacologically induced 85;112–114 or occurring spontaneously during up-down states 115;116, were proposed to be supported by synchronization of inhibitory GABAergic networks via gap junctions with or without the contribution of glutamatergic networks. Faster oscillations, such as ripples, were also found to be supported by both excitatory and inhibitory transmission and gap junctions 117;118. It is not clear whether the HFOs seen during interictal discharges in epileptic tissue are the same as physiological fast activities. Dzhala and Staley proposed that epileptic HFOs are initiated and synchronized by excitatory interactions between pyramidal cells in the hippocampus 106;119. More recent reports proposed that HFOs generated at the onset of an ictal hippocampal discharge 120;121 correlate with transient GABAergic input 122, indicating GABAergic mechanisms as the source of epileptic HFO at least in this case.
The Slow Component After the Interictal Discharges
IEDs recorded from the epileptogenic zone are assumed to be generated by synchronous neuronal firing. In line with experimental evidence 23;24;123, presurgical intracranial studies in patients with partial epilepsy have demonstrated that the sharp component of a neocortical IED is followed by a depression of neuronal excitability. This phenomenon has been reported in mesial TLE patients analyzed with unit activity recordings 124–126 as well as by using the paired pulse stimulation paradigm. Thus, these studies provide evidence for the existence of inhibitory phenomena during the post-spike slow wave 127. Moreover, in Taylor type II focal dysplasia a refractory period of 0.5–1 sec has been identified after the interictal spike 33. In this study, enhanced threshold for spike generation was seen in brain regions that surround the epileptogenic zone during single-shock stimulations at 1 Hz that was performed to identify eloquent and symptomatogenic areas. Interestingly, no refractory period was observed after IEDs that were generated within the seizure-onset zone.
Early experimental studies 123, and more recent reports 128, have shown that glutamatergic IEDs are followed by a period of depression in excitability that may result from several mechanisms. Cortical surface IEDs are usually larger then 1 mV and they are associated with the simultaneous activation of large ensembles of neurons and possibly astrocytes. This synchronous activation generates massive neuronal firing that boosts recurrent synaptic activation and non-synaptic field and direct interactions. Since GABAergic inhibitory interneurons are presumably preserved in the area of IED generation 129–131, the activation of recurrent inhibitory networks during IED may be responsible for dampening neuronal excitability. Recurrent inhibition mediated by GABAA receptors lasts 100 ms and could be reinforced and prolonged up to circa 1 s by the activation of “slow” GABAB receptors. Since post-IED depression lasts longer than 1 s 123, other mechanisms should be implicated in its generation. In line with this view, it has been shown in the piriform cortex that intra/extracellular pH changes associated with IEDs contribute to this depression by decoupling gap junctions 132;133.
Intracranial studies in TLE patients have demonstrated that background activity and HFO are reduced in amplitude during the slow wave that follows an IED, suggesting post-IED depression 100. Preliminary findings obtained at the Claudio Munari Epilepsy Surgery Center in Milano suggest that fast activity is reduced after an IED generated in the “irritative zone” surrounding the area of seizure onset 134, whereas it is preserved and even enhanced after IEDs generated within the seizure-onset zone (). Thus, IEDs characterized by spikes or sharp waves generate a long-lasting period of inhibition/depression that dampens the fast and transient increase in excitability that occurs during the spike/sharp wave. Post-spike depression is typical of the tissue that borders the seizure-onset zone; hence, these findings may support that idea that some IEDs control brain hyperexcitability within the epileptic network and protect the region against seizure entrainment.
Intracranial recordings performed in a patient with cryptogenic partial epilepsy during pre-surgical evaluation of the epileptogenic region. The area of seizure onset is shaded in light grey in the central panels that illustrates 10 seconds of continuous (more...)
In Vitro Recordings of IEDs from Post-surgical Brain Tissue
The fundamental mechanisms of IEDs have also been analyzed in post-surgical cortical slices of human brain tissue incubated in vitro for electrophysiological analysis. Human cortical slices in vitro do not generate spontaneous ictal discharges in standard saline bath solutions unless excitability is enhanced with various experimental procedures 135. Yet, spontaneous IEDs can be recorded in post-surgical slices (that included the subiculum and the CA2 region) obtained from hippocampi of patients suffering from mesial TLE with Ammon horn sclerosis 82;136 as well as neocortical partial epilepsies 81. As detailed in the Chapter by Jefferys et al in this book, interictal spikes in these studies were blocked by antagonists of either glutamate or GABAA synaptic transmission 82;83. Hence, spontaneous IEDs in human cortical tissue in vitro are generated by both GABAAergic and glutamatergic synaptic conductances.
HFOs characterized by very fast frequencies at 80–400 Hz have been reported to occur in post-surgical neocortical slices obtained from patients with drug-resistant TLE 137. In this study, HFOs associated with interictal spikes did not require synaptic transmission as they continued to occur in the presence of glutamatergic and GABAergic receptor antagonists; they were however abolished by application of drugs that are known to decouple gap junctions, such as carbanoxolone. The effects induced by gap junction decouplers have also been documented in human neocortical slices obtained from patients with FCD as well as from TLE patients 138. It was shown in this study that spontaneous IEDs recorded in the presence of normal medium from FCD tissue were reduced, and even more importantly, were no longer synchronized during carbenoxolone application (); moreover, similar effects were seen when IEDs were elicited by 4AP in neocortical slices that had no obvious structural abnormality (). These IEDs, when recorded in the presence of glutamatergic receptor antagonists, correspond intracellularly to a complex sequence of potentials that are dominated by a long lasting depolarization () and are accompanied by transient increases in extracellular potassium 139.
Effects of gap-junction decouplers on the synchronous activity generated by human neocortical slices in vitro. A: Schematic drawing of the location of the two recording extracellular microelectrodes used in the experiments shown in B–D; inter-electrode (more...)
Electrophysiological analysis of slices of human FCD tissue has shown that during 4AP application, IEDs that are mainly dependent on GABA receptor-mediated conductances may be instrumental in eliciting NMDA receptor-mediated ictal discharges 140–142. As illustrated in , ictal discharges recorded from FCD slices were preceded by negative-going events resembling those seen in isolation during the interictal period; however, the field potentials leading to ictal discharge onset were always of larger amplitude and were followed by a secondary, slow negative field event from which ictal oscillations emerged. The ability of IEDs in initiating ictal synchronization in the human FCD tissue relies on the presence of a GABAA receptor mediated mechanism that leads to sizeable increases in extracellular potassium. As illustrated in , the transient elevations in extracellular potassium associated with the IEDs that shortly preceded the ictal discharge onset were characterized by rises in extracellular potassium larger than those seen in association with similar field potentials occurring during the interictal period. A similar association of large elevations in extracellular potassium and ictal discharge onset have been observed in the deep layers of the entorhinal cortex 143 as well as in isolated hippocampal slices obtained from young rats 144;145. Indeed, elevating extracellular potassium can disclose seizure activity both in vivo
146 and in vitro
147;148. It should be emphasized that a similar 4AP treatment in human neocortical tissue without obvious structural abnormality induces only periodic, synchronous, interictal-like GABA receptor-mediated potentials () 149. Therefore, these in vitro data support the view that epileptogenicity is a functional feature of FCD tissue.
Synchronous activity induced by bath application of 4AP in neocortical slices obtained from TLE (i.e., presenting with no architectural anomaly) and FCD patients. A: Isolated field potentials (arrow) occur spontaneously in a TLE slice analyzed with field (more...)
The role of GABA receptor-mediated synchronization in initiating ictal activity in FCD tissue is further supported by pharmacological manipulations aimed at decreasing or enhancing the function of GABAA receptors. GABAA receptor antagonists or activation of μ-opioid receptors (which blocks the release of GABA from interneuron terminals) made ictal discharges and GABA receptor-mediated interictal events disappear (). Under both conditions, FCD slices generated recurrent epileptiform activity that lacked the features of an electrographic ictal event. Conversely, potentiating GABAA receptor function with minimal concentrations of phenobarbital 150;151 caused a prolongation of the ictal discharges along with potentiation of the slow interictal events ().
GABAA receptor function modulates ictal discharges in brain FCD slices. A: Bath application of the μ-opioid receptor agonist DAGO (10 μM) reduces the amplitude of the isolated negative field events (arrows in a) and transforms ictal activity (more...)
Conclusions
Evidence reviewed in this chapter indicates that IEDs are heterogeneous in terms of both pattern and underlying mechanisms. It has been proposed that the core region in which focal seizures are generated is surrounded by an area that generates hypersynchronous activity (denominated the ‘irritative region’) interposed between the seizure-onset area and the surrounding normal tissue 134. IEDs are generated both in the epileptogenic zone and in the irritative region and can spread to (and thus be recorded from) adjacent ‘healthy’ brain structures. Therefore, it is reasonable to conclude that interictal events are sustained by cellular and pharmacological mechanisms that vary according to the site of generation. Indeed, these differences may result in a different functional role with respect to seizure generation. The existence of pre-ictal spikes and their recognition as GABA-mediated events in some experimental models suggest that different IEDs may have a different temporal correlation and possibly functional role with respect to seizure initiation. According to this view, post-IED depression could be a selective feature of IEDs that occur in brain regions (such as the irritative region) in which neuronal homeostasis and synaptic networks are not drastically altered by the epileptogenic process. In seizure-onset regions, tissue damage could be more intense and post-IED depression may not be present or insufficient to dampen excitability, allowing IED’s to effectively trigger seizures.
References
- 1.
Gotman J. Relationships between interictal spiking and seizures: human and experimental evidence.
Can J Neurol Sci. 1991;18:573–576. [
PubMed: 1777872]
- 2.
de Curtis M, Avanzini G. Interictal spikes in focal epileptogenesis.
Progr Neurobiol. 2001;63:541–567. [
PubMed: 11164621]
- 3.
Avoli M. Do interictal discharges promote or control seizures? Experimental evidence from an in vitro model of epileptiform discharge.
Epilepsia. 2001;42(Suppl 3):2–4. [
PubMed: 11520313]
- 4.
- 5.
Bast T, Oezkan O, Rona S, et al. EEG and MEG source analysis of single and averaged interictal spikes reveals intrinsic epileptogenicity in focal cortical dysplasia.
Epilepsia. 2004;45:621–31. [
PubMed: 15144427]
- 6.
Benar CG, Grova C, Kobayashi E, et al. EEG-fMRI of epileptic spikes: concordance with EEG source localization and intracranial EEG.
Neuroimage. 2006;30:1161–70. [
PubMed: 16413798]
- 7.
Baumgartner C, Pataraia E. Revisiting the role of magnetoencephalography in epilepsy.
Curr Opin Neurol. 2006;19:181–6. [
PubMed: 16538094]
- 8.
Plummer C, Harvey AS, Cook M. EEG source localization in focal epilepsy: where are we now.
Epilepsia. 2008;49:201–18. [
PubMed: 17941844]
- 9.
Rose S, Ebersole JS. Advances in spike localization with EEG dipole modeling.
Clin EEG Neurosci. 2009;40:281–7. [
PubMed: 19780349]
- 10.
Vulliemoz S, Lemieux L, Daunizeau J, Michel CM, Duncan JS. The combination of EEG source imaging and EEG-correlated functional MRI to map epileptic networks.
Epilepsia. 2010;51:491–505. [
PubMed: 19817805]
- 11.
Alarcon G, Guy CN, Binnie CD, Walker SR, Elwes RD, Polkey CE. Intracerebral propagation of interictal activity in partial epilepsy: implications for source localisation.
J Neurol Neurosurg Psych. 1994;57:435–49. [
PMC free article: PMC1072872] [
PubMed: 8163992]
- 12.
Bagshaw AP, Kobayashi E, Dubeau F, Pike GB, Gotman J. Correspondence between EEG-fMRI and EEG dipole localisation of interictal discharges in focal epilepsy.
Neuroimage. 2006;30:417–25. [
PubMed: 16269248]
- 13.
Rodin E, Constantino T, Rampp S, Wong PK. Spikes and epilepsy.
Clin EEG Neurosci. 2009;40:288–99. [
PubMed: 19780350]
- 14.
Kooi KA. Voltage-time characteristics of spikes and other rapid electroencephalographic transients: semantic and morphological considerations.
Neurology. 1966;16:59–66. [
PubMed: 5948007]
- 15.
Chatrian GE, Bergamini L, Dondey M, Klass DW, Lennox-Buchtal M. A glossary of terms most commonly used by clinical electroencephalographers.
Electroencheph Clin Neurol. 1974;37:538–548. [
PubMed: 4138729]
- 16.
Drury I, Beydoun A. Benign partial epilepsy of childhood with monomorphic sharp waves in centrotemporal and other locations.
Epilepsia. 1991;32:662–7. [
PubMed: 1915173]
- 17.
Frost JD Jr, Hrachovy RA, Glaze DG. Spike morphology in childhood focal epilepsy: relationship to syndromic classification.
Epilepsia. 1992;33:531–6. [
PubMed: 1592033]
- 18.
Jiruska P, Finnerty GT, Powell AD, Lofti N, Cmejla R, Jefferys JG. Epileptic high-frequency network activity in a model of non-lesional temporal lobe epilepsy.
Brain. 2010;133:1380–90. [
PMC free article: PMC2859153] [
PubMed: 20400525]
- 19.
Ulbert I, Halgren E, Heit G, Karmos G. Multiple microelectrode-recording system for human intracortical applications.
J Neurosci Methods. 2001;106:69–79. [
PubMed: 11248342]
- 20.
Fabo D, Magloczky Z, Wittner L, et al. Properties of in vivo interictal spike generation in the human subiculum.
Brain. 2008;131:485–99. [
PubMed: 18083752]
- 21.
Ulbert I, Heit G, Madsen J, Karmos G, Halgren E. Laminar analysis of human neocortical interictal spike generation and propagation: current source density and multiunit analysis in vivo.
Epilepsia. 2004;45(Suppl 4):48–56. [
PubMed: 15281959]
- 22.
Matsumoto H, Marsan CA. Cortical cellular phenomena in experimental epilepsy: interictal manifeststions.
Exp Neurol. 1964;80:286–304. [
PubMed: 14145629]
- 23.
Prince D. Inhibition in 'Epileptic' neurons.
Exp Neurol. 1968;21:307–321. [
PubMed: 5673646]
- 24.
Prince DA. Cortical cellular activities during cyclically occurring inter-ictal epileptiform discharges.
Electroencephalogr Clin Neurophysiol. 1971;31:469–84. [
PubMed: 4107801]
- 25.
Johnston D, Brown TH. Interpretation of voltage-clamp measurements in hippocampal neurons.
J Neurophysiol. 1983;50:464–486. [
PubMed: 6310063]
- 26.
- 27.
Gambardella A, Gotman J, Cendes F, Andermann F. The relation of spike foci and of clinical seizure characteristics to different patterns of mesial temporal atrophy.
Arch Neurol. 1995;52:287–93. [
PubMed: 7872883]
- 28.
Francione S, Kahane P, Tassi L, et al. Stereo-EEG of interictal and ictal electrical activity of a histologically proved heterotopic gray matter associated with partial epilepsy.
Electroencephalogr Clin Neurophysiol. 1994;90:284–90. [
PubMed: 7512909]
- 29.
Tyvaert L, Hawco C, Kobayashi E, LeVan P, Dubeau F, Gotman J. Different structures involved during ictal and interictal epileptic activity in malformations of cortical development: an EEG-fMRI study.
Brain. 2008;131:2042–60. [
PMC free article: PMC3792088] [
PubMed: 18669486]
- 30.
Tassi L, Colombo N, Garbelli R, et al. Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome.
Brain. 2002;125:1719–32. [
PubMed: 12135964]
- 31.
Francione S, Nobili L, Cardinale F, Citterio A, Galli C, Tassi L. Intra-lesional stereo-EEG activity in Taylor 's focal cortical dysplasia.
Epileptic Disord. 2003;5(Suppl 2):S105–14. [
PubMed: 14617429]
- 32.
Spreafico R, Blumcke I. Focal Cortical Dysplasias: clinical implication of neuropathological classification systems.
Acta Neuropathol. 2010;120:359–67. [
PubMed: 20607544]
- 33.
de Curtis M, Tassi L, Lo Russo G, Mai R, Cossu M, Francione S. Increased discharge threshold after an interictal spike in human focal epilepsy.
Eur J Neurosci. 2005;22:2971–6. [
PubMed: 16324132]
- 34.
Altafullah I, Halgren E, Stapleton JM, Crandall PH. Interictal spike-wave complexes in the human medial temporal lobe: typical topography and comparisons with cognitive potentials.
Electroencephalogr Clin Neurophysiol. 1986;63:503–16. [
PubMed: 2422000]
- 35.
Williamson PD, French JA, Thadani VM. characteristics of medial temporal lobe epilepsy: II. Interictal and ictal scalp EEG, neuropsychological testing, neuroimaging, surgical results, and pathology.
Ann Neurol. 1993;34:781–787. [
PubMed: 8250526]
- 36.
Wilson C, Nix J, Szostak J. Functional requirements for specific ligand recognition by a biotin-binding RNA pseudoknot.
Biochemistry. 1998;37:14410–9. [
PubMed: 9772167]
- 37.
Dichter M, Spencer WA. Penicillin-induced interictal discharges from the cat hippocampus. I. Characteristics and topographical features.
J Neurophysiol. 1969;32:649–62. [
PubMed: 4309021]
- 38.
Fertziger AP, Ranck JB Jr. Potassium accumulation in interstitial space during epileptiform seizures.
Exp Neurol. 1970;26:571–85. [
PubMed: 5435740]
- 39.
Dichter MA, Herman CJ, Selzer M. Silent cells during interictal discharges and seizures in hippocampal penicillin foci. Evidence for the role of extracellular K+ in the transition from the interictal state to seizures.
Brain Res. 1972;48:173–83. [
PubMed: 4645204]
- 40.
Ralston BL. The mechanism of transition of interictal spiking foci into ictal seizure discharges.
Electroencephalogr Clin Neurophysiol Suppl. 1958;10:217–32. [
PubMed: 13548068]
- 41.
Gotman J. Relationships between triggered seizures, spontaneous seizures, and interictal spiking in the kindling model of epilepsy.
Exp Neurol. 1984;84:259–73. [
PubMed: 6714340]
- 42.
Sherwin I. Ictal-interictal unit firing pattern differences in penicillin-induced primary and secondary epileptogenic foci.
Exp Neurol. 1984;84:463–77. [
PubMed: 6714353]
- 43.
Lange HH, Lieb JP, Engel J Jr, Crandall PH. Temporo-spatial patterns of pre-ictal spike activity in human temporal lobe epilepsy.
Electroencephalogr Clin Neurophysiol. 1983;56:543–555. [
PubMed: 6197273]
- 44.
Mathern GW, Cifuentes F, Leite JP, Pretorius JK, Babb TL. Hippocampal EEG excitability and chronic spontaneous seizures are associated with aberrant synaptic reorganization in the rat intrahippocampal kainate model.
Electroencephalogr Clin Neurophysiol. 1993;87:326–39. [
PubMed: 7693444]
- 45.
Bragin A, Engel J Jr, Wilson CL, Vizentin E, Mathern GW. Electrophysiologic analysis of a chronic seizure model after unilateral hippocampal KA injection.
Epilepsia. 1999;40:1210–21. [
PubMed: 10487183]
- 46.
Le Duigou C, Bouilleret V, Miles R. Epileptiform activities in slices of hippocampus from mice after intra-hippocampal injection of kainic acid.
J Physiol. 2008;586:4891–904. [
PMC free article: PMC2614071] [
PubMed: 18755752]
- 47.
Bortel A, Levesque M, Biagini G, Gotman J, Avoli M. Convulsive status epilepticus duration as determinant for epileptogenesis and interictal discharge generation in the rat limbic system.
Neurobiol Dis. 2010;40:478–89. [
PMC free article: PMC4878906] [
PubMed: 20682341]
- 48.
Ives AE, Jefferys JG. Synchronization of epileptiform bursts induced by 4-aminopyridine in the in vitro hippocampal slice preparation.
Neurosci Lett. 1990;112:239–45. [
PubMed: 2359522]
- 49.
Chamberlin NL, Traub RD, Dingledine R. Role of EPSPs in initiation of spontaneous synchronized burst firing in rat hippocampal neurons bathed in high potassium.
J Neurophysiol. 1990;64:1000–8. [
PubMed: 1977893]
- 50.
- 51.
Ayala GF, Dichter M, Gumnit RJ. Genesis of epilectic interictal spikes. New knowledge of cortical feedback system suggest a neurophysiological explanation of brief paroxysms.
Brain Res. 1973;52:1–17. [
PubMed: 4573428]
- 52.
Traub RD, Wong RK. Cellular mechanism of neuronal synchronization in epilepsy.
Science. 1982;216:745–7. [
PubMed: 7079735]
- 53.
Hablitz JJ. Picrotoxin-induced epileptiform activity in hippocampus: role of endogenous versus synaptic factors.
J Neurophysiol. 1984;51:1011–27. [
PubMed: 6327932]
- 54.
Miles R, Traub RD, Wong RK. Spread of synchronous firing in longitudinal slices from the CA3 region of the hippocampus.
J Neurophysiol. 1988;60:1481–96. [
PubMed: 3193167]
- 55.
Traub RD, Miles R, Jefferys JG. Synaptic and intrinsic conductances shape picrotoxin-induced synchronized after-discharges in the guinea-pig hippocampal slice.
J Physiol. 1993;461:525–47. [
PMC free article: PMC1175271] [
PubMed: 8350274]
- 56.
Stanton PK, Jones RS, Mody I, Heinemann U. Epileptiform activity induced by lowering extracellular.
Epil Res. 1987;1:53–62. [
PubMed: 2904361]
- 57.
Bertram EH, Lothman EW. NMDA receptor antagonists and limbic status epilepticus: a comparison with standard anticonvulsant.
Epil Res. 1990;5:177–184. [
PubMed: 2166658]
- 58.
Lee WL, Hablitz JJ. Excitatory synaptic involvement in epileptiform bursting in the immature rat neocortex.
J Neurophysiol. 1991;66:1894–901. [
PubMed: 1687473]
- 59.
Chagnac-Amitai Y, Connors BW. Synchronized excitation and inhibition driven by intrinsically bursting neurons in neocortex.
J Neurophysiol. 1989;62:1149–62. [
PubMed: 2585046]
- 60.
Hwa GGC, Avoli M. Hyperpolarizing inward rectification in rat neocortical neurons located in the superficial layers.
Neurosci Lett. 1991;124:65–68. [
PubMed: 1857546]
- 61.
Forti M, Biella G, Caccia S, de Curtis M. Persistent excitability changes in the piriform cortex of the isolated guinea-pig brain after transient exposure to bicuculline.
Eur J Neurosci. 1997;9:435–51. [
PubMed: 9104586]
- 62.
Jones RSG, Heinemann V. Synaptic and intrinsic responses of medial entorhinal cortical cells in normal and magnesium-free medium “in vitro”
J Neurophysiol. 1988;59:1476–1496. [
PubMed: 2898511]
- 63.
Schwartzkroin PA, Slawsky M. Probable calcium spikes in hippocampal neurons.
Brain Res. 1977;135:157–61. [
PubMed: 912429]
- 64.
- 65.
- 66.
de Curtis M, Radici C, Forti M. Cellular mechanisms underlying spontaneous interictal spikes in an acute model of focal cortical epileptogenesis.
Neuroscience. 1999;88:107–17. [
PubMed: 10051193]
- 67.
Auerbach JM, Segal M. A novel cholinergic induction of long-term potentiation in rat hippocampus.
J Neurophysiol. 1994:2034–2040. [
PubMed: 7823117]
- 68.
Franceschetti S, Guatteo E, Panzica F, Sancini G, Wanke E, Avanzini G. Ionic mechanisms underlying burst firing in pyramidal neurons: intracellular study in rat sensorimotor cortex.
Brain Res. 1995;696:127–39. [
PubMed: 8574660]
- 69.
Azouz R, Jensen MS, Yaari Y. Ionic base of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells.
Jo Physiol. 1996;492:211–223. [
PMC free article: PMC1158874] [
PubMed: 8730596]
- 70.
Traub RD, Dudek FE, Snow RW, Knowles WD. Computer simulations indicate that electrical field effects contribute to the shape of the epileptiform field potential.
Neuroscience. 1985;15:947–58. [
PubMed: 4047402]
- 71.
Jefferys JGR. Nonsynaptic modulation of neuronal activity in the brain: electric currents and extracellular ions.
Physiol Rev. 1995;75:689–723. [
PubMed: 7480159]
- 72.
Draguhn A, Traub RD, Schmitz D, Jefferys JG. Electrical coupling underlies high-frequency oscillations in the hippocampus in vitro [see comments]
Nature. 1998;394:189–92. [
PubMed: 9671303]
- 73.
Galarreta M, Hestrin S. A network of fast-spiking cells in the neocortex connected by electrical synapses.
Nature. 1999;402:72–75. [
PubMed: 10573418]
- 74.
Skinner FK, Zhang L, Velazquez JL, Carlen PL. Bursting in inhibitory interneuronal networks: A role for gap-junctional coupling.
J Neurophysiol. 1999;81:1274–83. [
PubMed: 10085354]
- 75.
Perreault P, Avoli M. 4-aminopyridine-induced epileptiform activity and a GABA-mediated long-lasting depolarization in the rat hippocampus.
J Neurosci. 1992;12:104–15. [
PMC free article: PMC6575697] [
PubMed: 1309571]
- 76.
Uva L, Avoli M, de Curtis M. Synchronous GABA-receptor-dependent potentials in limbic areas of the in-vitro isolated adult guinea pig brain.
Eur J Neurosci. 2009;29:911–20. [
PMC free article: PMC4873282] [
PubMed: 19291222]
- 77.
Carriero G, Uva L, Gnatkovsky V, de Curtis M. Distribution of the olfactory fiber input into the olfactory tubercle of the in vitro isolated guinea pig brain.
J Neurophysiol. 2009;101:1613–9. [
PubMed: 18922946]
- 78.
Aram JA, Michelson HB, Wong RKS. Synchronized GABAergic IPSPs recorded in the neocortex after blockade of synaptic transmission mediated by excitatory amino acids.
J Neurophysiol. 1991;65:1034–1041. [
PubMed: 1678421]
- 79.
- 80.
Gnatkovsky V, Librizzi L, Trombin F, de Curtis M. Fast activity at seizure onset is mediated by inhibitory circuits in the entorhinal cortex in vitro.
Ann Neurol. 2008;64:674–86. [
PubMed: 19107991]
- 81.
Kohling R, Lucke A, Straub H, et al. Spontaneous sharp waves in human neocortical slices excised from epileptic patients.
Brain. 1998;121(Pt 6):1073–87. [
PubMed: 9648543]
- 82.
Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro.
Science. 2002;298:1418–21. [
PubMed: 12434059]
- 83.
- 84.
Lopantsev V, Avoli M. Participation of GABAA-mediated inhibition in ictallike discharges in the rat entorhinal cortex.
J Neurophysiol. 1998;79:352–60. [
PubMed: 9425204]
- 85.
Whittington M, Traub RD, Jefferys JGR. Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation.
Nature. 1995;373:612–615. [
PubMed: 7854418]
- 86.
Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons.
Nature. 1995;378:75–8. [
PubMed: 7477292]
- 87.
Seifert G, Carmignoto G, Steinhauser C. Astrocyte dysfunction in epilepsy.
Brain Res Rev. 2010;63:212–21. [
PubMed: 19883685]
- 88.
Tashiro A, Goldberg J, Yuste R. Calcium oscillations in neocortical astrocytes under epileptiform conditions.
J Neurobiol. 2002;50:45–55. [
PubMed: 11748632]
- 89.
Kang N, Xu J, Xu Q, Nedergaard M, Kang J. Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons.
J Neurophysiol. 2005;94:4121–30. [
PubMed: 16162834]
- 90.
- 91.
Fellin T, Gomez-Gonzalo M, Gobbo S, Carmignoto G, Haydon PG. Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices.
J Neurosci. 2006;26:9312–22. [
PMC free article: PMC6674496] [
PubMed: 16957087]
- 92.
- 93.
Engel J Jr, Bragin A, Staba R, Mody I. High-frequency oscillations: what is normal and what is not.
Epilepsia. 2009;50:598–604. [
PubMed: 19055491]
- 94.
Allen PJ, Fish DR, Smith SJ. Very high-frequency rhythmic activity during SEEG suppression in frontal lobe epilepsy.
Electroencephalogr Clin Neurophysiol. 1992;82:155–9. [
PubMed: 1370786]
- 95.
Bragin A, Engel J Jr, Wilson CL, Fried I, Buzsaki G. High-frequency oscillations in human brain.
Hippocampus. 1999;9:137–42. [
PubMed: 10226774]
- 96.
Bragin A, Engel J Jr, Wilson CL, Fried I, Mathern GW. Hippocampal and entorhinal cortex high-frequency oscillations (100--500 Hz) in human epileptic brain and in kainic acid--treated rats with chronic seizures.
Epilepsia. 1999;40:127–37. [
PubMed: 9952257]
- 97.
Staba RJ, Wilson CL, Bragin A, Fried I, Engel J Jr. Quantitative analysis of high-frequency oscillations (80–500 Hz) recorded in human epileptic hippocampus and entorhinal cortex.
J Neurophysiol. 2002;88:1743–52. [
PubMed: 12364503]
- 98.
Jacobs J, LeVan P, Chander R, Hall J, Dubeau F, Gotman J. Interictal high-frequency oscillations (80–500 Hz) are an indicator of seizure onset areas independent of spikes in the human epileptic brain.
Epilepsia. 2008;49:1893–907. [
PMC free article: PMC3792077] [
PubMed: 18479382]
- 99.
Worrell GA, Parish L, Cranstoun SD, Jonas R, Baltuch G, Litt B. High-frequency oscillations and seizure generation in neocortical epilepsy.
Brain. 2004;127:1496–506. [
PubMed: 15155522]
- 100.
Urrestarazu E, Jirsch JD, LeVan P, Hall J. High-frequency intracerebral EEG activity (100–500 Hz) following interictal spikes.
Epilepsia. 2006;47:1465–76. [
PubMed: 16981862]
- 101.
Schevon CA, Trevelyan AJ, Schroeder CE, Goodman RR, McKhann G Jr, Emerson RG. Spatial characterization of interictal high frequency oscillations in epileptic neocortex.
Brain. 2009;132:3047–59. [
PMC free article: PMC2768661] [
PubMed: 19745024]
- 102.
Buzsáki G. The hippocampo-neocortical dialogue.
Cereb Cort. 1996;6:81–92. [
PubMed: 8670641]
- 103.
Buzsáki G, Horváth Z, Urioste R, Hetke J, Wise K. High-frequency network oscillation in the hippocampus.
Science. 1992;256:1025–7. [
PubMed: 1589772]
- 104.
Ylinen A, Bragin A, Nadasdy Z, et al. Sharp wave-associated high-frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms.
J Neurosci. 1995;15:30–46. [
PMC free article: PMC6578299] [
PubMed: 7823136]
- 105.
Le Van Quyen M, Bragin A, Staba R, Crepon B, Wilson CL, Engel J Jr. Cell type-specific firing during ripple oscillations in the hippocampal formation of humans.
J Neurosci. 2008;28:6104–10. [
PMC free article: PMC2693199] [
PubMed: 18550752]
- 106.
- 107.
Jiruska P, Csicsvari J, Powell AD, et al. High-frequency network activity, global increase in neuronal activity, and synchrony expansion precede epileptic seizures in vitro.
J Neurosci. 2010;30:5690–701. [
PMC free article: PMC6632330] [
PubMed: 20410121]
- 108.
de Curtis M, Gnatkovsky V. Reevaluating the mechanisms of focal ictogenesis: The role of low-voltage fast activity.
Epilepsia. 2009 [
PubMed: 19674056]
- 109.
Jacobs J, Zelmann R, Jirsch J, Chander R, Dubeau CE, Gotman J. High frequency oscillations (80–500 Hz) in the preictal period in patients with focal seizures.
Epilepsia. 2009;50:1780–92. [
PMC free article: PMC3764053] [
PubMed: 19400871]
- 110.
Brazdil M, Halamek J, Jurak P, et al. Interictal high-frequency oscillations indicate seizure onset zone in patients with focal cortical dysplasia.
Epilepsy Res. 2010;90:28–32. [
PubMed: 20362416]
- 111.
- 112.
Whittington MA, Traub RD. Interneuron diversity series: inhibitory interneurons and network oscillations in vitro.
Trends Neurosci. 2003;26:676–82. [
PubMed: 14624852]
- 113.
Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks.
Nat Rev Neurosci. 2007;8:45–56. [
PubMed: 17180162]
- 114.
Mann EO, Paulsen O. Role of GABAergic inhibition in hippocampal network oscillations.
Trends Neurosci. 2007;30:343–9. [
PubMed: 17532059]
- 115.
Gnatkovsky V, Wendling F, de Curtis M. Cellular correlates of spontaneous periodic events in the medial entorhinal cortex of the in vitro isolated guinea pig brain.
Eur J Neurosci. 2007;26:302–11. [
PubMed: 17650108]
- 116.
Compte A, Reig R, Descalzo VF, Harvey MA, Puccini GD, Sanchez-Vives MV. Spontaneous high-frequency (10–80 Hz) oscillations during up states in the cerebral cortex in vitro.
J Neurosci. 2008;28:13828–44. [
PMC free article: PMC6671899] [
PubMed: 19091973]
- 117.
Maier N, Nimmrich V, Draguhn A. Cellular and network mechanisms underlying spontaneous sharp wave-ripple complexes in mouse hippocampal slices.
J Physiol. 2003;550:873–87. [
PMC free article: PMC2343079] [
PubMed: 12807984]
- 118.
Nimmrich V, Maier N, Schmitz D, Draguhn A. Induced sharp wave-ripple complexes in the absence of synaptic inhibition in mouse hippocampal slices.
J Physiol. 2005;563:663–70. [
PMC free article: PMC1665611] [
PubMed: 15661820]
- 119.
Behrens CJ, van den Boom LP, Heinemann U. Effects of the GABA(A) receptor antagonists bicuculline and gabazine on stimulus-induced sharp wave-ripple complexes in adult rat hippocampus in vitro.
Eur J Neurosci. 2007;25:2170–81. [
PubMed: 17419756]
- 120.
Khosravani H, Pinnegar CR, Mitchell JR, Bardakjian BL, Federico P, Carlen PL. Increased high-frequency oscillations precede in vitro low-Mg seizures.
Epilepsia. 2005;46:1188–97. [
PubMed: 16060927]
- 121.
Lasztoczi B, Nyitrai G, Heja L, Kardos J. Synchronization of GABAergic inputs to CA3 pyramidal cells precedes seizure-like event onset in juvenile rat hippocampal slices.
J Neurophysiol. 2009;102:2538–53. [
PubMed: 19675286]
- 122.
Lasztoczi B, Antal K, Nyikos L, Emri Z, Kardos J. High-frequency synaptic input contributes to seizure initiation in the low-[Mg2+] model of epilepsy.
Eur J Neurosci. 2004;19:1361–72. [
PubMed: 15016094]
- 123.
Lebovitz LB. Autorhythmicity of spontaneous interictal spike discharge at hippocampal penicillin focus.
Brain Research. 1979;172:35–55. [
PubMed: 466466]
- 124.
Wyler AR, Ojemann GA, Ward AA Jr. Neurons in human epileptic cortex: correlation between unit and EEG activity.
Ann Neurol. 1982;11:301–8. [
PubMed: 7092182]
- 125.
Babb TL, Wilson CL, Isokawa-Akesson M. Firing patterns of human limbic neurons during stereoencephalography (SEEG) and clinical temporal lobe seizures.
Electroencephalogr Clin Neurophysiol. 1987;66:467–82. [
PubMed: 2438112]
- 126.
Isokawa-Akesson M, Wilson CL, Babb TL. Inhibition in synchronously firing human hippocampal neurons.
Epil Res. 1989;3:236–47. [
PubMed: 2731521]
- 127.
Wilson CL, Khan SU, Engel J Jr, Isokawa M, Babb TL, Behnke EJ. Paired pulse suppression and facilitation in human epileptogenic hippocampal formation.
Epil Res. 1998;31:211–30. [
PubMed: 9722031]
- 128.
de Curtis M, Librizzi L, Biella G. Discharge threshold is enhanced for several seconds after a spontaneous interictal spike in a model of focal epileptogenesis.
Eur J Neurosci. 2001;14:1–6. [
PubMed: 11488962]
- 129.
Davenport CJ, Brown WJ, Babb TL. GABAergic neurons are spared after intrahippocampal kainate in the rat.
Epil Res. 1990;5:28–42. [
PubMed: 2303020]
- 130.
Esclapez M, Hirsch JC, Khazipov R, Ben-Ari, Bernard C. Operative GABAergic inhibition in hippocampal CA1 pyramidal neurons in experimental epilepsy.
Proc Natl Acad Sci U S A. 1997;94:12151–6. [
PMC free article: PMC23733] [
PubMed: 9342378]
- 131.
Prince DA, Jacobs K. Inhibitory function in two models of chronic epileptogenesis.
Epil Res. 1998;32:83–92. [
PubMed: 9761311]
- 132.
de Curtis M, Manfridi A, Biella G. Activity-dependent pH shifts and periodic recurrence of spontaneous interictal spikes in a model of focal epileptogenesis.
Jo Neurosci. 1998;18:7543–51. [
PMC free article: PMC6793250] [
PubMed: 9736672]
- 133.
Spray D, Harris A, Bennet M. Gap junctional conductance is a simple and sensitive function of intracellular pH.
Science. 1981;211:712–715. [
PubMed: 6779379]
- 134.
Talairach J, Bancaud J. Lesion, “irritative” zone and epileptogenic focus.
Confin Neurol. 1966;27:91–4. [
PubMed: 5334025]
- 135.
Avoli M, Louvel J, Pumain R, Kohling R. Cellular and molecular mechanisms of epilepsy in the human brain.
Prog Neurobiol. 2005;77:166–200. [
PubMed: 16307840]
- 136.
Wittner L, Huberfeld G, Clemenceau S, et al. The epileptic human hippocampal cornu ammonis 2 region generates spontaneous interictal-like activity in vitro.
Brain. 2009;132:3032–46. [
PubMed: 19767413]
- 137.
Roopun AK, Simonotto JD, Pierce ML, et al. A nonsynaptic mechanism underlying interictal discharges in human epileptic neocortex.
Proc Natl Acad Sci U S A. 2010;107:338–43. [
PMC free article: PMC2806783] [
PubMed: 19966298]
- 138.
Gigout S, Louvel J, Kawasaki H, et al. Effects of gap junction blockers on human neocortical synchronization.
Neurobiol Dis. 2006;22:496–508. [
PubMed: 16478664]
- 139.
Louvel J, Papatheodoropoulos C, Siniscalchi A, et al. GABA-mediated synchronization in the human neocortex: elevations in extracellular potassium and presynaptic mechanisms.
Neuroscience. 2001;105:803–13. [
PubMed: 11530219]
- 140.
Mattia D, Olivier A, Avoli M. Seizure-like discharges recorded in human dysplastic neocortex maintained in vitro.
Neurology. 1995;45:1391–5. [
PubMed: 7617202]
- 141.
Avoli M, Bernasconi A, Mattia D, Olivier A, Hwa GG. Epileptiform discharges in the human dysplastic neocortex: in vitro physiology and pharmacology.
Ann Neurol. 1999;46:816–26. [
PubMed: 10589533]
- 142.
D'Antuono M, Louvel J, Kohling R, et al. GABAA receptor-dependent synchronization leads to ictogenesis in the human dysplastic cortex.
Brain. 2004;127:1626–40. [
PubMed: 15175227]
- 143.
Barbarosie M, Louvel J, D'Antuono M, Kurcewicz I, Avoli M. Masking synchronous GABA- mediated potentials controls limbic seizures.
Epilepsia. 2002;43:1469–79. [
PubMed: 12460247]
- 144.
Avoli M, Louvel J, Kurcewicz I, Pumain R, Barbarosie M. Extracellular free potassium and calcium during synchronous activity induced by 4-aminopyridine in the juvenile rat hippocampus.
J Physiol. 1996;493(Pt 3):707–17. [
PMC free article: PMC1159019] [
PubMed: 8799893]
- 145.
Borck C, Jefferys JG. Seizure-like events in disinhibited ventral slices of adult rat hippocampus.
J Neurophysiol. 1999;82:2130–42. [
PubMed: 10561393]
- 146.
Zuckermann EC, Glaser GH. Hippocampal epileptic activity induced by localized ventricular perfusion with high-potassium cerebrospinal fluid.
Exp Neurol. 1968;20:87–110. [
PubMed: 5637118]
- 147.
Traub RD, Dingledine R. Model of synchronized epileptiform bursts induced by high potassium in CA3 region of rat hippocampal slice. Role of spontaneous EPSPs in initiation.
J Neurophysiol. 1990;64:1009–18. [
PubMed: 2230914]
- 148.
Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice.
J Neurophysiol. 1988;59:259–76. [
PubMed: 3343603]
- 149.
Avoli M, Mattia D, Siniscalchi A, Perreault P, Tomaiuolo F. Pharmacology and electrophysiology of a synchronous GABA-mediated potential in the human neocortex.
Neuroscience. 1994;62:655–66. [
PubMed: 7870297]
- 150.
- 151.
Barker JL, McBurney RN. Phenobarbitone modulation of postsynaptic GABA receptor function on cultured mammalian neurons.
Proc R Soc Lond B Biol Sci. 1979;206:319–27. [
PubMed: 43977]
The authors declare no conflicts of interest.