Both human and experimental evidence strongly supports the view of brain region-specific changes in phasic and tonic GABAA inhibition in typical absence seizures. In neocortex, a decrease in phasic GABAA inhibition characterizes this non-convulsive epileptic phenotype, though more studies are needed to disclose any neuronal type- and layer-specific alterations. The enhanced tonic GABAA inhibition, that is present in thalamocortical neurons of genetic and pharmacological models, is both necessary and sufficient for absence seizure generation, and in genetic models it results from a malfunction in the astrocytic GABA transporter GAT-1. Phasic GABAA inhibition is either unchanged or increased in thalamocortical neurons of absence models. Contradictory results from inbred and transgenic animals still do not allow us to draw firm conclusions on changes in phasic GABAA inhibition in the GABAergic neurons of the nucleus reticularis thalami. Thus, the long-standing view of a widespread GABAergic loss-of-function in typical absence seizures and childhood absence epilepsy is no longer tenable and model systems that use an indiscriminate block of GABAA receptors are of no value for understanding the cellular and network mechanisms operating in thalamocortical circuits during typical absence seizures.
A typical absence is a non-convulsive epileptic seizure that is characterized by a sudden and relatively brief impairment of consciousness, occurring concomitantly with a generalized, bilaterally synchronous ‘spike (or polyspike) and slow wave discharge’ (SWD) at 2.5–4 Hz in the EEG.1,2 Typical absence seizures are part of the multi-faceted clinical and EEG presentation of many idiopathic generalized epilepsies (IGEs), but in childhood absence epilepsy (CAE) these seizures are the only neurological symptom and are not accompanied by either metabolic, neuropathological or other neurological deficits.1,2 The human studies reviewed in this chapter, therefore, will be those that relate to CAE, since this is the only IGE where a putative causal link can be made between a typical absence seizure and the underlying genetic variants or pathophysiological mechanisms without the confounding effects of other epileptic and non-epileptic neurological phenotypes.
Typical absence seizures are genetically determined, and the classical 2.5–4 Hz SWD trait has been shown to be inherited in an autosomal dominant manner.1,2 Indeed, there is strong consensus in describing the occurrence of (typical) absence seizures as a familial disease with a complex multi-factorial genetic background. There is also a general consensus, based on some old invasive studies and modern non-invasive imaging investigations, that typical absence seizures originate from abnormal electrical activities in reciprocally connected thalamic and cortical territories - i.e. in what is generally referred to as thalamo-cortical networks, with little or no involvement of other brain areas, including hippocampus, cerebellum and limbic regions.3–7 Key cellular elements of thalamo-cortical networks include pyramidal cells and interneurons of different cortical layers, the thalamocortical (TC) neurons of sensory thalamic nuclei and their main inhibitory input, i.e. the GABAergic neurons of the nucleus reticularis thalami (NRT). Importantly, the notion that a typical absence seizure is ‘generalized’ from the very start of the SWD has been recently challenged by the observation that the onset of an absence seizure in humans is associated with paroxysmal activation of discrete, often frontal and parietal cortical regions, spreading then to other cortical regions and to the thalamus.3,5 The presence of a putative cortical ‘initiation site’ had previously been shown in genetic rat models of typical absence seizures where, differently from human absences, it is localized in the perioral region of the primary somatosensory cortex.8,9 Indeed, direct application of the anti-absence drug ethosuximide at this putative ‘initiation site’, but not 1 mm away from it, readily abolishes absence seizures in freely moving rats.10
It is now well established that GABAA receptor- (GABAAR)-mediated inhibition consists of a phasic component (i.e. the ‘classical’ IPSPs), that is generated by GABA interacting with synaptic GABAARs, and a tonic component (i.e. a persistent membrane hyperpolarization with increased conductance) that is due to GABA activation of perisynaptic or extrasynaptic GABAARs.11 Abnormalities both in synaptic and extrasynaptic GABAARs have undoubtedly been of primary significance among the various human molecular genetic alterations12–15 that have in recent years provided support for the idea that IGEs, and in particular absence seizures, are channelopathies.16 In this chapter, we first summarize recent evidence demonstrating that enhanced tonic GABAA inhibition is a common feature of both pharmacological and genetic models of absence epilepsy. In particular, enhanced tonic GABAA inhibition in TC neurons is both necessary and sufficient for the expression of typical absence seizures, and in genetic models it is not due to a neuronal abnormality but to a malfunction of the astrocytic GABA transporter GAT-1. The available data on phasic GABAA inhibition, on the other hand, suggest that the changes in this type of GABAergic function observed in animals and humans affected by typical absence seizures are both brain region- and neuronal type-specific. We will then discuss how all these findings on phasic and tonic GABAA inhibition fit within our current understanding of the pathophysiological mechanisms of typical absence seizures, and propose that the currently prevailing view of an overall GABAergic loss-of-function in typical absence seizures and CAE is no longer tenable.
TONIC GABAA INHIBITION
Enhanced Tonic GABAA Inhibition In Genetic Models Of Typical Absence Seizures
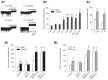
The tonic GABAA current measured in vitro in TC neurons of the somatosensory ventrobasal thalamus of different genetic models of absence seizures is enhanced compared to the current in their respective control animals.17 This is true for a polygenic rat model, i.e., the Genetic Absence Epilepsy Rat from Strasbourg (GAERS)18 () and for various mice models with known spontaneous monogenic mutations, including stargazer and lethargic mice19 (). In particular, there is a clear developmental profile of this increased GABAergic function since in GAERS up to postnatal day 16 the current is similar to that in the non-epileptic control (NEC) strain but almost doubles in amplitude at postnatal day 17 (), and remains elevated well past the time of seizure onset17 (around postnatal day 30 in this strain18). In contrast, no tonic GABAA current is detected in the GABAergic NRT neurons of GAERS and their respective non-epileptic control strain (unpublished observation), as it is indeed the case in normal Wistar rats.20
The enhanced tonic GABAA current in TC neurons of rat and mouse genetic models of typical absence seizures results from a malfunction in the GABA transporter GAT-1. A1. Current traces from TC neurons of postnatal day (P) 14 and 17 non-epileptic control (more...)
GAT-1 Malfunction Underlies Increased Tonic GABAA Inhibition
The enhanced tonic GABAA current of TC neurons in genetic absence models is not due to increased vesicular GABA release, overexpression of extrasynaptic GABAARs, or mis-expression of synaptic GABAARs, but results from a malfunction of the GABA transporter GAT-1,17 despite it being far less abundant in the thalamus than GAT-3.21 As shown in in fact, i) block of GAT-1 (by the selective blocker NO711) in GAERS and stargazer has no effect on tonic current amplitude of TC neurons, ii) block of GAT-1 in the respective non-epileptic rats and mice increases tonic current amplitude to similar values to those seen in GAERS and stargazer mice, and iii) the compensatory increase in uptake by GAT-1 that is seen following block of GAT-3 (by the selective blocker SNAP5114) in non-epileptic animals is lost in both GAERS and stargazer mice.17 A malfunction in GAT-1 also underlies the increased tonic GABAA current in TC neurons of lethargic mice.17 These data support and enlarge previous observations that had shown a reduced GABA uptake by GAT-122 and an increased level of extracellular GABA23 in the ventrobasal thalamus of GAERS compared to NEC. Differently from GAERS and stargazer mice, however, GABA transport by GAT-1 is reversed in lethargic mice, with this transporter being a source of ambient GABA. Interestingly, NO711 increases tonic GABAA current by a similar amount in dentate gyrus granule cells of GAERS and NEC,17 indicating that GAT-1 activity is not compromised in a brain area that does not participate in the generation of typical absence seizures18 and where the distribution of this transporter is primarily neuronal.24 Indeed, the tonic current of dentate gyrus granule cells is not different between GAERS and NEC.17
In summary, therefore, genetic models of typical absence seizures (i.e., GAERS, stargazer, and lethargic mice) show a brain region-specific enhancement of tonic GABAA current, which in TC neurons is due to increased extracellular GABA level resulting from a malfunction in GABA uptake by astrocytic GAT-1.
Enhanced Tonic GABAA Inhibition In The GHB Model: Role of GABABRs
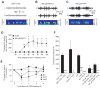
Systemic (or intrathalamic) injection of γ-hydroxybutyric acid (GHB) is undoubtedly the best-established pharmacological model of typical absence seizures,25,26 and systemic administration of 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol) (THIP), a selective agonist at δ subunit-containing extrasynaptic GABAARs,27 has been shown to elicit SWDs in normal animals.28 Therefore, whereas it is not surprising that THIP dose-dependently enhances the tonic GABAA current in TC neurons of normal Wistar rats,17 one would not expect GHB, which does not bind to GABAARs and is believed to elicit absence seizures by activation of GABABRs,26 to have an effect on the tonic GABAA current. However, GHB dose-dependently enhances the tonic GABAA current in TC neurons of normal Wistar rats17 () at concentrations that are similar to the brain concentrations that elicit absence seizures in vivo.29 This effect of GHB is not due to some aspecific action since it is abolished by the GABABR antagonist CGP55845 (). Interestingly, application of CGP55845 alone significantly reduces the tonic GABAA current amplitude in TC neurons of Wistar rats to 74% of control17 (), indicating that facilitation of extrasynaptic GABAARs by GABABRs contributes approximately one quarter of the tonic GABAA current in normal rats. Importantly, CGP55845 also reduces the tonic current in GAERS, stargazer and lethargic mice to about 55, 65 and 57%, respectively17 (), suggesting that facilitation of extrasynaptic GABAAR function by GABABR activation contributes up to half of the tonic current in these genetic models.
GABABRs involvement in the tonic GABAA current of TC neurons in genetic and pharmacological models of typical absence seizures. A. Current traces form TC neurons of normal Wistar rat showing the GHB-elicited increase in tonic GABAA current and its block (more...)
In summary, therefore, a GAT-1 malfunction in thalamic astrocytes of mouse and rat genetic models leads to an increase in ambient GABA in the sensory thalamus, which in turn elicits an enhancement in tonic GABAA inhibition through both direct activation of extrasynaptic GABAARs and indirect facilitation of extrasynaptic GABAARs via activation of GABABRs.
Enhanced Tonic GABAA Inhibition Of TC Neurons Is Necessary And Sufficient For Typical Absence Seizure Generation
Assessing the impact of enhanced tonic GABAA current of TC neurons on the expression of absence seizures requires experiments in freely moving animals, where both the behavioural and EEG components of the seizures can be studied without bias introduced by the concomitant use of anaesthetics and/or analgesics. In fact, unrestrained and undrugged GAT-1 KO mice, which have an enhanced tonic GABAA current in TC neurons, express ethosuximide-sensitive typical absence seizures17 (). Similarly, direct injection of the selective GAT-1 blocker NO711 into the ventrobasal thalamus by reverse microdialysis in normal Wistar rats triggers ethosuximide-sensitive typical absence seizures17 (). Moreover, in δ GABAAR KO mice, which exhibit a markedly reduced GABAA inhibition in TC neurons, systemic administration of GHB fails to induce absence seizures17 (). Similarly, the intrathalamic injection of a δ subunit-specific antisense oligodeoxynucleotide in GAERS strongly decreases both the tonic GABAA current and spontaneous seizures 1–2 days after injection, whereas a missense oligodeoxynucleotide has no effect17 (). Finally, intrathalamic administration of the δ subunit-specific agonist THIP in normal Wistar rats elicits absence seizures in a concentration-dependent manner, an effect that is reversed by systemic administration of ethosuximide17 (). Taken together, these data show that enhanced tonic GABAA inhibition in TC neurons is both necessary and sufficient for the generation of typical absence seizures. In addition, they provide a mechanistic explanation for the aggravation of absence seizures that is observed in humans and experimental models following systemic and intrathalamic administration of drugs that increase GABA levels, including tiagabine, a GABA uptake blocker, and vigabatrine, a GABA transaminase blocker.18,30–32
The tonic GABAA inhibition in TC neurons is both necessary and sufficient for typical absence seizure generation. A. Bilateral (L = left, R = right hemispheres) EEG traces from a freely moving GAT-1 knockout (KO) mouse showing spontaneous SWDs (spectrogram (more...)
Significance Of Tonic GABAA Inhibition In Typical Absence Epilepsy
The discovery of an increased tonic GABAA current in TC neurons represents the first abnormality that i) is common to both well-established pharmacological and genetic models of typical absence seizures, irrespective of species and known up-stream mutations, ii) is manifested both before and after the developmental onset of seizures, iii) contributes to the exquisite sensitivity of experimental absence seizures to selective GABABR agonists and antagonists, and iv) explains the aggravating effects of GABAergic drugs in both human and experimental absence seizures. Importantly, these results, together with the presence of powerful GABAA IPSPs in the majority of TC neurons during absence seizures in vivo33,34 () (see below), lead to another significant conclusion, i.e., that studies aiming at reproducing typical absence seizures by indiscriminatly blocking GABAARs of TC neurons35–38 are inherently flawed.
Intracellular correlates of TC and NRT neuron activity during spontaneous SWDs in vivo. A1. Intracellular recording (bottom trace) from a TC neurons in GAERS in vivo during a spontaneous SWD (top trace) shows the presence of bursts of IPSPs occurring (more...)
The finding that the increased tonic GABAA inhibition in TC neurons of genetic models is due to a malfunction of GAT-1 shifts the emphasis from a neuronal to an astrocytic aetiology of absence epilepsy. The impaired GAT-1 activity in GAERS is not caused by a decreased expression of GAT-1 mRNA or protein levels in either thalamus or cortex.17 Also, GAT-1 cDNA from GAERS, stargazer and lethargic mice presents no genetic variants, whereas the mutations responsible for absence seizures in stargazer and lethargic mice are not present in GAERS.17 Potential abnormalities that may lead to a reduced GAT-1 function, but have not yet been tested, include the inability of this protein to reach the outer astrocytic membrane, an alteration in its subcellular location and/or abnormalities in its phosphorylation. Similalry, we are aware of no study that has investigated GAT-1 (or GAT-3) genetic variants in human CAE.
Experimental typical absence seizures can be elicited or aggravated by selective GABABR agonists and can be blocked by selective GABABR antagonists, applied either systemically or intrathalamically.39–41 Because about 50% of the tonic GABAA current observed in TC neurons of GAERS, stargazer and lethargic mice is abolished by a GABABR antagonist17 (), the behavioural and EEG effects of GABABR-selective drugs on typical absence seizures can no longer be simply explained by the ability of these drugs to affect GABAB-mediated IPSPs and/or presynaptic GABABRs, but should also take into account the positive modulation by GABABRs of the tonic GABAA inhibition in TC neurons.
PHASIC GABAA INHIBITION
The picture concerning phasic GABAA inhibition in typical absence seizures is much more complex than that of tonic GABAA inhibition, since contradictory results have emerged from studies in various experimental models. For sake of clarity, therefore, the analysis of available evidence on phasic GABAA inhibition will be presented separately for each main neuronal type of the thalamo-cortical network, i.e. cortical neurons, TC neurons and the GABAergic neurons of the NRT.
Phasic GABAA Inhibition In Cortex
Among the various mutations in GABAAR subunits, those that have been identified in related or unrelated CAE patients are: α1(S326fs328X), β3(P11S), 3(S15F), β3(G32R), γ2(R43Q), γ2(R139G) and γ2(IVS6+2T•G).12–15,42 Functional analysis of these mutant proteins expressed in heterelogous systems has shown that all of them bring about a marked decrease in GABA responses.12 Importantly, humans carrying the γ2(R43Q) mutation have been shown to have a decreased short-interval intracortical inhibition assessed with transcranial magnetic stimulation, leading to cortical facilitation.43 γ2(R43Q) mutant mice constructed by homologous recombination express spontaneous ethosuximide-sensitive absence seizures.44 Moreover, despite the fact that the γ2 subunit has an almost ubiquitous expression and is apparently required for functional synaptic GABAARs, these mice exhibit a brain region-specific alteration in phasic GABAA inhibition, since cortical layer 2/3 pyramidal cells but not TC or NRT neurons show a reduction in miniature IPSCs (mIPSCs) compared to age-matched wildtype littermates.44 Whether the region-specificity of this malfunction is also present in humans with the γ2(R43Q) mutation, or only occurs in mice because of some species-specific promoters or silencers, is not known at present. Nevertheless, together with showing the physiological effect of a human genetic variant in a “true” physiological system as opposed to hetereologous expression systems, the key significance of this finding is that it is the first clear evidence that the downstream abnormalities of a human genetic variant in CAE can be inhibition that brain region-specific. Importantly, the cortex-specific abnormality in phasic GABAA results from the human γ2(R43Q) mutation exquisitively complements previous experimental data showing that 1) in felines in vivo the localized block of GABAARs by cortical application of inhibition in the thalamus, does elicit SWDs,33 and bicuculline, which leaves intact phasic GABAA inhibition in TC neurons is either unaffected or 2) in rat and mouse genetic models phasic GABAAincreased compared to their respective non-epileptic controls17,44–47 (see below).
On the basis of indirect evidence, a decreased cortical GABAergic inhibition has also been suggested to occur in layer 2/3 regular spiking neurons48 as well as in layer 5 pyramidal neurons49 of adult Wistar Albino Glaxo/Rij (WAG) rats, another well established polygenic model of typical absence seizures.50 However, no change is observed in mIPSCs in pyramidal cell and interneurons of cortical layers 2–3 of young, pre-seizure GAERS,45 and cortical GABAergic inhibition is intact in the feline generalized penicillin epilepsy model.51
In summary, the evidence from a human mutation indicates that a decreased phasic GABAA inhibition in cortex characterizes typical absence seizures, though further studies are needed to clarify the extent of this decrease both in terms of different cortical regions, layers and neuronal types: this additional work might shed light on the existing discrepancies on changes in phasic GABAA inhibition among experimental models.
Phasic GABAA Inhibition In TC Neurons
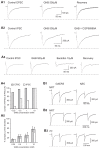
A reduced or absent phasic GABAAR function in the thalamus also characterizes the current view of the pathophysiology of typical absence seizures. While this may be partly true for the intra-NRT inhibition it certainly does not hold for TC neurons. Thus, both in felines that show spontaneous or cortical bicuculline-induced SWDs33 as well as in the well-established GAERS model34 the vast majority (60 and 94%, respectively) of TC neurons recorded in vivo during SWDs exhibit bursts of GABAA IPSPs, each tightly synchronized with the EEG spike and wave complex (). Indeed, the rise time, amplitude, frequency and decay time constant of mIPSCs and spontaneous IPSCs (sIPSCs) measured in TC neurons in vitro are not different between GAERS and NEC both prior to,45 and after spontaneous seizure manifestation17 (i.e. postnatal days 12–25, and postnatal day 30, respectively), and paired pulse depression of evoked IPSCs is similar between the two strains45 (). As mentioned earlier, no change in TC neuron IPSCs occurs in mice carrying the human γ2(R43Q) mutation,44 and no change in IPSC properties has been detected in TC neurons of β3 KO mice52 which show absence seizures as part of a much more complex neurological phenotype. Also, in lethargic, stargazer and tottering mice sIPSCs in TC neurons are similar to those in their respective littermates,17 and no difference has been found in evoked IPSCs in lethargic and tottering mice compared to control mice.47 Finally, an increase in mIPSCs frequency, potentially leading to enhanced phasic inhibition, is observed in TC neurons of the absence seizure-prone DAB/2J mouse strain.46
Phasic GABAA inhibition in TC and NRT neurons of genetic and pharmacological models of typical absence seizures. A1. GHB reversibly decreases the amplitude of a cortical EPSC recorded in TC neurons. A2. Example of an NRT-derived IPSC (recorded in a TC (more...)
In a similar manner to the genetic models, in the best-established pharmacological model of absence seizure, i.e. the GHB model, a net increase in phasic GABAA inhibition is observed in TC neurons of the ventrobasal thalamus.53 This is because whereas GHB reversibly and dose-dependently decreases the amplitude of all sensory54 and corticothalamic EPSCs53 (), the two main excitatory drives to TC neurons, at low concentrations (250 μM-1 mM) it only affects the GABAA IPSCs in some but not all TC neurons53 (). In particular, 250 μM GHB produces an effect in <10% of tested TC neurons, whereas at 0.5-1 mM it only decreases half of the recorded IPSCs (). Thus, at brain concentrations (250 μM - 1 mM) similar to those that elicit absence seizures in vivo GHB brings about not only the previously described GABABR- and K+-dependent hyperpolarization26,55 and an increase in tonic GABAA inhibition17 (see above) but also an imbalance between the excitatory and inhibitory drive to TC neurons, leading to a net increase in their phasic GABAA inhibition. This, in turn, will help to impose that periodic phasic inhibition that sculptures the electrical behaviour of TC neurons during SWDs as observed in vivo in feline56 and rat57 models ().
On the basis of all this evidence from both genetic and pharmacological models, as well as from mice expressing the human γ2(R43Q) mutation, it is therefore surprising that the data in support of the presence of an enhanced or unchanged phasic GABAergic function in TC neurons have been selectively dismissed over the last 15 years, such that textbooks and topical reviews58–62 still present the pathophysiological mechanisms of TC neuron activity during typical absence seizures as the one that is observed in vitro during application of a GABAAR antagonist.35,36,38 It is also surprising that the in vivo evidence which is often used to support this view is the ability of bicuculline (or other GABAAR antagonists) to generate, in ketamine-xylazine anaesthetized rats, strong field potentials at 3 Hz37, that are claimed to represent SWDs even if their sensitivity to ethosuximide or other anti-absence drugs was not tested. Indeed, it is highly unlikely that these field potentials represent SWDs, since the ketamine-xylazine combination, as other general anaesthetic, are known to abolish the SWDs of any well-established in vivo model of absence epilepsy.18,51,63
In summary, therefore, the presence of these serious issues of interpretation, together with the evidence showing an increased tonic and an unchanged/increased phasic GABAA inhibition in TC neurons of genetic and pharmacological models, strongly support the conclusion that the block of thalamic GABAARs that continues to be used in many in vitro and some in vivo studies does at best elict some form of thalamic hyperexcitability of as yet unknown pathophysiological relevance, but definitely not the activity that is present during typical absence seizures.
Phasic GABAA Inhibition In NRT Neurons
The existance of contradictory data makes it difficult at present to draw a unifying picture of the changes in the phasic GABAA inhibition of NRT neurons that are associated with an absence seizure phenotype. Starting from inbred genetic models, mIPSCs recorded from NRT neurons of pre-seizure GAERS show a 67% higher frequency, a 25% larger amplitude and a 40% faster decay than those in age-matched NEC.45 Moreover, paired-pulse depression of evoked GABAA IPSCs is significantly smaller (46%) in NRT neurons of GAERS than NEC45 (). This smaller paired-pulse depression of IPSCs in GAERS would undoubtedly ensure a consistent hyperpolarizing drive to help promote the expression of the strong, low threshold Ca2+ potential-mediated bursts of action potentials that are the hallmark of NRT neuron firing at each spike and wave complex in vivo56,57 (). Moreover, an increase in mIPSC frequency, potentially leading to enhanced phasic GABAA inhibition, is observed in NRT neurons of the absence seizure-prone DAB/2J mouse strain.46 Finally, an almost complete disappearance of the α3 subunit, that in the thalamus is selectively expressed in NRT neurons64, has been reported in these GABAergic neurons from WAG rats,65 suggesting a potential loss of function.
Surprisingly, however, α3 KO mice do not show spontaneous absence seizures and indeed exibit a small reduction in GHB-elicited seizures compared to wildtype littermates, a result that has been interpreted as resulting from a powerful compensatory gain in phasic GABAA inhibition in NRT neurons.66 On the other hand, since unknown mechanisms lead to this unexpected compensatory increase in NRT GABAA inhibition following deletion of the α3 subunit, the lack of spontaneous absence seizures and the relative resistance to pharmacologically induced absence seizures may also be due to additional compensatory changes in phasic (or tonic?) GABAA inhibition in cortex, or tonic inhibition in thalamus, all of which were not investigated in that study.66 Further evidence for a pro-absence role of a decreased phasic GABAAR function in NRT neurons has also come from the observation of an increase in high frequency discharges at 3 Hz in β3 subunit KO mice, which show a massive reduction in sIPSP frequency and amplitude in NRT, but no change in TC neurons.52 Since these β3 KO mice exhibit a large variety of neurological deficits and are considered a model of Angelman’s syndrome, it is difficult to unequivocally assign a causative role for this decreased intra-NRT inhibition in typical absence seizures.
In summary, whereas in inbred models and models with spontaneous mutations there is either an increase or no change in intra-NRT phasic GABAA inhibition, data from two transgenic mice suggest that a decrease in this NRT synaptic function has a pro-absence effect. Thus, it may be tempting to conclude that abnormalities in intra-NRT phasic GABAA inhibition are not a necessary condition for the expression of typical absence seizures, a view supported by the lack of changes in NRT IPSCs of mice carrying the human γ 2(R43Q) mutation44. In this respect, it is worth noting that the anti-absence effect of clonazepam in humans and experimental models67,68 may not be solely due to its ability to increase phasic GABAA inhibition in NRT,69 since α3 subunit-containing GABAARs that are the target of this benzodiazepine are also present in neocortical neurons.70
ROLE FOR PHASIC AND TONIC GABAA INHIBITION IN THE GENESIS OF ABSENCE SEIZURES
The evidence reviewed in this chaper suggests the following scenario concerning the cellular contribution of phasic and tonic GABAA inhibition to typical absence seizures. A decreased phasic GABAA inhibition in the neocortex leads or contributes to a paroxysmal development of normal 5–9 Hz oscillations in a discrete somatosensory cortical “initiation site”. This strong and highly synchronous cortical output powerfully excites the GABAergic neurons of the NRT, which respond by generating low-threshold Ca2+ potential-mediated bursts of action potentials at every spike and wave complex of the SWD. This rhythmic burst firing, in turn, results in bursts of IPSPs in TC neurons which override cortical excitation. Concomitantly, ambient GABA levels around TC neurons abnormally increase due to reduced GABA uptake by GAT-1, enhancing extrasynaptic GABAAR function directly, and indirectly by a GABABR-dependent facilitation. Enhanced tonic inhibition persistently hyperpolarises the TC neurons and increases their membrane conductance, reducing their action potential output, with low threshold Ca2+ potential-mediated bursts of action potentials rarely occurring. Furthermore, the responsiveness of TC neurons to excitatory, sensory synaptic inputs is reduced, ‘gating’ information flow through the thalamus, abolishing responsiveness to external stimuli and causing behavioural arrest. Importantly, the rhythmic IPSP bursts entrain TC neuron output to each cycle of a SWD, providing a sparse but synchronised input to the cortex and maintaining paroxysmal activity in thalamo-cortical networks.
CONCLUDING REMARKS
Despite the long-standing notion of an overall decrease in GABAAR function in typical absence seizures, a critical appraisal of the human and experimental evidence strongly supports the view of different, brain region-specific changes of both phasic and tonic GABAAR-mediated inhibition in this non-convulsive epileptic phenotype. In particular, a decreased phasic GABAA inhibition characterizes cortical activity, though more studies are needed to disclose any neuronal type- and layer-specific abnormalities. An enhanced tonic inhibition, which results from a malfunction of the astrocytic GAT-1 transporters, together with an unchanged or increased phasic inhibition is present in TC neurons of both genetic and pharmacological models of typical absence seizures. Contradictory results in different species and transgenic animals and the reliance on unsuitable in vitro model systems still do not allow a clear view of the changes in phasic GABAA inhibition that accompany typical absence seizure in NRT neurons. Thus, thalamic and cortical model systems where GABAARs are indiscriminately blocked do not reproduce the complex pattern of alterations in these neurotransmitter receptors that are observed in human and experimental absence seizures, and are therefore of no value for understanding the cellular mechanisms operating in thalamo-cortical networks during typical absence seizures.
Acknowledgments
Work in our laboratories is supported by Wellcome Trust, MRC, EU (Framework 7), Fondation NRJ-Institut de France, and CNRS LEA 528 (Thalamic Function in Health and Disease States). DWC is a Research Fellow of Epilepsy Research UK.
REFERENCES
- 1.
Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks.
Nat Rev Neurosci. 2002;3:371–382. [
PubMed: 11988776]
- 2.
Blumenfeld H. Cellular and network mechanisms of spike-wave seizures.
Epilepsia. 2005;46:21–33. [
PubMed: 16302873]
- 3.
Holmes MD, Brown M, Tucker DM. Are “generalized” seizures truly generalized? Evidence of localized mesial frontal and frontopolar discharges in absence.
Epilepsia. 2004;45:1568–1579. [
PubMed: 15571515]
- 4.
Hamandi K, Salek-Haddadi A, Laufs H, Liston A, Friston K, Fish DR, Duncan JS, Lemieux L. EEG-fMRI of idiopathic an secondarily generalized epilepsies.
Neuroimage. 2006;31:1700–1710. [
PubMed: 16624589]
- 5.
Westmijse I, Ossenblok P, Gunning B, van Luijtelaar G. Onset and propagation of spike and slow wave discharges in human absence epilepsy: A MEG study.
Epilepsia. 2009;50:2538–2548. [
PubMed: 19519798]
- 6.
Bai X, Vestal M, Berman R, Negishi M, Spann M, Vega C, Desalvo M, Novotny EJ, Constable RT, Blumenfeld H. Dynamic time course of typical childhood absence seizures: EEG behaviour, and functional magnetic resonance imaging.
J Neurosci. 2010;30:5884–5893. [
PMC free article: PMC2946206] [
PubMed: 20427649]
- 7.
Szaflarski JP, Difrancesco M, Hirschauer T, Banks C, Privitera MD, Gotman J, Holland SK. Cortical and subcortical contributions to absence seizure onset examined with EEG/fMRI.
Epilepsy Behav. 2010. [Epub ahead of print] [
PMC free article: PMC2922486] [
PubMed: 20580319]
- 8.
Meeren HKM, Pijn JPM, Van Liujtelaar ELJM, Coenen AML, da Silva FHL. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats.
J Neurosci. 2002;22:1480–1495. [
PMC free article: PMC6757554] [
PubMed: 11850474]
- 9.
Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somotosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures.
J Neurosci. 2007;27:6590–6599. [
PMC free article: PMC6672429] [
PubMed: 17567820]
- 10.
Manning JP, Richards DA, Leresche N, Crunelli V, Bowery NG. Cortical-area specific block of genetically determined absence seizures by ethosuximide.
Neuroscience. 2004;123:5–9. [
PubMed: 14667436]
- 11.
Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA
A receptors.
Nat Rev Neurosci. 2005;6:215–229. [
PubMed: 15738957]
- 12.
- 13.
Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABA (A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures.
Nat Genet. 2001;28:49–52. [
PubMed: 11326275]
- 14.
Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K, Rebstock J, Heils A, Steinlein OK. A splice-site mutation in GABRG3 associated with childhood absence epilepsy and febrile convulsions.
Arch Neurol. 2002;59:1137–1141. [
PubMed: 12117362]
- 15.
Tanaka M, Olsen RW, Medina MT, Schwarz E, Alonso ME, Duron RM, Castro-Ortega R, Martinez-Juarez IE, Pascual-Castroviejo I, Machado-Salas J, Silva R, Bailey JN, Bai D, Ochoa A, Jara-Prado A, Pineda G, MacDonald RL, Delgado-Escueta AV. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy.
Am J Hum Gen. 2008;82:1249–1261. [
PMC free article: PMC2427288] [
PubMed: 18514161]
- 16.
Noebels JL. The biology of epilepsy genes.
Ann Rev Neurosci. 2003;26:599–625. [
PubMed: 14527270]
- 17.
Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lörincz ML, Gould TM, Carter DA, Crunelli V. Enhanced tonic GABA
A inhibition in typical absence epilepsy.
Nat Med. 2009;15:1392–1398. [
PMC free article: PMC2824149] [
PubMed: 19966779]
- 18.
Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. Pathophysiological mechanisms of genetic absence epilepsy in the rat.
Prog Neurobiol. 1998;55:27–57. [
PubMed: 9602499]
- 19.
Fletcher CF, Frankel WN. Ataxic mouse mutants and molecular mechanisms of absence epilepsy.
Hum Mol Genet. 1999;8:1907–1912. [
PubMed: 10469844]
- 20.
- 21.
Pow DV, Sullivan RK, Williams SM, Scott HL, Dodd PR, Finkelstein D. Differential expression of the GABA transporters GAT-1 and GAT-3 in brains of rats, cats, monkeys and humans.
Cell Tissue Res. 2005;320:379–392. [
PubMed: 15821932]
- 22.
Sutch RJ, Davies CC, Bowery NG. GABA release and uptake measured in crude synaptosomes from Genetic Absence Epilepsy Rats from Strasbourg (GAERS).
Neurochem Int. 1999;3:15–425. [
PubMed: 10397370]
- 23.
Richards DA, Lemos T, Whitton PS, Bowery NG. Extracellular GABA in the ventrolateral thalamus of rats exhibiting spontaneous absence epilepsy: a microdialysis study.
J Neurochem. 1995;65:1674–1680. [
PubMed: 7561864]
- 24.
De Biasi S, Vitellaro-Zuccarello L, Brecha NC. Immunoreactivity for the GABA transporter-1 and GABA transporter-3 is restricted to astrocytes in the rat thalamus. A light and electron-microscopic immunolocalization.
Neuroscience. 1998;83:815–828. [
PubMed: 9483565]
- 25.
Banerjee PK, Hirsch E, Snead OC III. γ-hydroxybutyric acid induced spike and wave discharges in rats: relation to high-affinity [
3H]γ-hydroxybutyric acid binding sites in the thalamus and cortex.
Neuroscience. 1993;56:11–21. [
PubMed: 8232911]
- 26.
- 27.
Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA. Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors.
Br J Pharm. 2002;136:965–974. [
PMC free article: PMC1573424] [
PubMed: 12145096]
- 28.
Fariello RG, Golden GT. The THIP-induced model of bilateral synchronous spike and wave in rodents.
Neuropharmacology. 1987;26:161–165. [
PubMed: 3587530]
- 29.
Snead OC. The gamma-hydroxybutyrate model of absence seizures: correlation of regional brain levels of gamma-hydroxybutyric acid and gamma-butyrolactone with spike wave discharges.
Neuropharmacology. 1991;30:161–167. [
PubMed: 2030821]
- 30.
Hosford DA, Wang Y. Utility of the lethargic (
lh/
lh) mouse model of absence seizures in predicting the effects of lamotrigine, vigabatrin, tiagabine, gabapentin, and topiramate against human absence seizures.
Epilepsia. 1997;38:408–414. [
PubMed: 9118845]
- 31.
Perucca E, Gram L, Avanzini G, Dulac O. Antiepileptic drugs as a cause of worsening seizures.
Epilepsia. 1998;39:5–17. [
PubMed: 9578007]
- 32.
Ettinger AB, Bernal OG, Andriola MR, Bagchi S, Flores P, Just C, Pitocco C, Rooney T, Tuominen J, Devinsky O. Two cases of nonconvulsive status epilepticus in association with tiagabine therapy.
Epilepsia. 1999;40:1159–1162. [
PubMed: 10448832]
- 33.
Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity.
J Neurosci. 1995;15:623–642. [
PMC free article: PMC6578306] [
PubMed: 7823168]
- 34.
Pinault D, Leresche N, Charpier S, Deniau J-M, Marescaux C, Vergnes M, Crunelli V. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy.
J Physiol. 1998;509:449–456. [
PMC free article: PMC2230966] [
PubMed: 9575294]
- 35.
von Krosigk M, Bal T, McCormick DA. Cellular mechanisms of a synchronized oscillation in the thalamus.
Science. 1993;261:361–364. [
PubMed: 8392750]
- 36.
Bal T, von Krosigk M, McCormick DA. Synaptic and membrane mechanisms underlying synchronized oscillations in the ferret lateral geniculate nucleus in vitro.
J Physiol. 1995;483:641–663. [
PMC free article: PMC1157808] [
PubMed: 7776249]
- 37.
- 38.
Kleiman-Weiner M, Beenhakker MP, Segal WA, Huguenard JR. Synergistic roles of GABA
A receptors and SK channels in regulating thalamocortical oscillations.
J Neurophysiol. 2009;102:203–213. [
PMC free article: PMC2712277] [
PubMed: 19386752]
- 39.
Lui Z, Vergnes M, Depaulis A, Marescaux C. Involvement of intrathalamic GABA
B neurotransmission in the control of absence seizures in the rat.
Neuroscience. 1992;48:87–93. [
PubMed: 1316571]
- 40.
Hosford DA, Lin FH, Kraemer DL, Cao Z, Wang Y, Wilson JT Jr. Neural network of structures in which GABA
B receptors regulate absence seizures in the lethargic (lh/lh) mouse model.
J Neurosci. 1995;15:7367–7376. [
PMC free article: PMC6578045] [
PubMed: 7472490]
- 41.
Snead OC 3rd. Antiabsence seizure activity of specific GABA
B and gamma-Hydroxybutyric acid receptor antagonist.
Pharmacol Biochem Behav. 1996;53:73–79. [
PubMed: 8848463]
- 42.
Maljevic KK, Cobilanschi J, Tilgen N, Beyer S, Weber YG, Schlesinger F, Ursu D, Melzer W, Cossette P, Bufler J, Lerche H, Heils A. A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy.
Ann Neurol. 2006;59:983–987. [
PubMed: 16718694]
- 43.
Fedi M, Berkovic SF, MacDonell RA, Curatolo JM, Marini C, Reutens DC. Intracortical hyperexcitability in humans with a GABA
A receptor mutation.
Cereb Cortex. 2008;18:664–669. [
PubMed: 17615250]
- 44.
Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S, Clarke AL, Dibbens L, Krestel H, Mulley JC, Jones MV, Seeburg PH, Sakmann B, Berkovic SF, Sprengel R, Petrou S. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy.
Proc Natl Acad Sci USA. 2007;104:17536–17541. [
PMC free article: PMC2077291] [
PubMed: 17947380]
- 45.
Bessaïh T, Bourgeais L, Badiu CI, Carter DA, Toth TI, Ruano D, Lambolez B, Crunelli V, Leresche N. Nucleus-specific abnormalities of GABAergic synaptic transmission in a genetic model of absence seizures.
J Neurophysiol. 2006;96:3074–3081. [
PubMed: 16971676]
- 46.
- 47.
Caddick SJ, Wang C, Fletcher CF, Jenkins NA, Copeland NG, Hosford DA. Excitatory but not inhibitory synaptic transmission is reduced in lethargic (
Cacnb4lh) and tottering (
Cacna1atg) mouse thalami.
J Neurophysiol. 1999;81:2066–2074. [
PubMed: 10322048]
- 48.
Luhmann HJ, Mittmann T, van Luijtelaar G, Heinemann U. Impairment of introcortical GABAergic inhibition in a rat model of absence epilepsy.
Epilepsy Res. 1995;22:43–51. [
PubMed: 8565966]
- 49.
D’Antuono M, Inaba Y, Biagini G, D’Arcangelo G, Trancredi V, Avoli M. Synaptic hyperexcitability of deep layer neocortical cells in a genetic model of absence seizures.
Genes Brain Behav. 2006;5:73–84. [
PubMed: 16436191]
- 50.
Coenen AM, Drinkenburg WH, Inoue M, van Luijtelaar EL. Genetics models of absence epilepsy, with emphasis on the WAG/Rij strain of rats.
Epilepsy Res. 1992;12:75–86. [
PubMed: 1396543]
- 51.
Giaretta D, Kostopoulos G, Gloor P, Avoli M. Intracortical inhibitory mechanisms are preserved in feline generalized penicillin epilepsy.
Neurosci Letts. 1985;59:203–208. [
PubMed: 4058793]
- 52.
Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus.
Science. 1999;283:541–543. [
PubMed: 9915702]
- 53.
Gervasi N, Monnier Z, Vincent P, Paupardin-Tritsch D, Hughes SW, Crunelli V, Leresche N. Pathway-specific action of gamma-hydroxybutyric acid in sensory thalamus and its relevance to absence seizures.
J Neurosci. 2003;23:11469–11478. [
PMC free article: PMC6740512] [
PubMed: 14673012]
- 54.
Emri Z, Antal K, Crunelli V. Gamma-hydroxybutyric acid decreases thalamic sensory excitatory postsynaptic potentials by an action on presynaptic GABA
B receptors.
Neurosci Letts. 1996;216:121–124. [
PubMed: 8904798]
- 55.
Williams SR, Turner JP, Crunelli V. Gamma-hydroxybutyrate promotes oscillatory activity of rat and cat thalamocortical neurons by a tonic GABA
B receptor-mediated hyperpolarization.
Neuroscience. 1995;66:133–144. [
PubMed: 7637863]
- 56.
Steriade M, Contreras D. Spike-wave complexes and fast components of cortically generated seizures. I. Role of neocortex and thalamus.
J Neurophysiol. 1998;80:1439–1455. [
PubMed: 9744951]
- 57.
Slaght SJ, Leresche N, Deniau J-M, Crunelli V, Charpier S. Activity of thalamic reticular neurons during spontaneous genetically determined spike and wave discharges.
J Neurosci. 2002;22:2323–2334. [
PMC free article: PMC6758255] [
PubMed: 11896171]
- 58.
McCormick DA, Contreras D. On the cellular and network bases of epileptic seizures.
Annu Rev Physiol. 2001;63:815–846. [
PubMed: 11181977]
- 59.
- 60.
Budde T, Pape C-H, Kumar SS, Huguenard JR. Thalamic, thalamocortical and corticocortical models of epilepsy with an emphasis on absence seizures. In: Pitkanen A, Schwartzkroin PA, Moshe SL, editors. Models of Seizures and Epilepsy. Amsterdam: Elsevier; 2006. pp. 73–88.
- 61.
Huguenard JR, McCormick DA. Thalamic Synchrony and dynamic regulation of global forebrain oscillations.
Trends Neurosci. 2007;30:350–357. [
PubMed: 17544519]
- 62.
- 63.
Gloor P. Generalized cortico-reticular epilepsies. Some considerations on the pathophysiology of generalized bilaterally synchronous spike and wave discharges.
Epilepsia. 1968;9:249–263. [
PubMed: 4975028]
- 64.
Browne SH, Kang J, Akk G, Chiang LW, Schulman H, Huguenard JR, Prince DA. Kinetic and pharmacological properties of GABA
A receptors in single thalamic neurons and GABA
A receptor subunit expression.
J Neurophysiol. 2001;86:2312–2322. [
PubMed: 11698521]
- 65.
Liu XB, Coble J, van Luijtelaar G, Jones EG. Reticular nucleus-specific changes in alpha3 subunit protein at GABA synapses in genetically epilepsy-prone rats.
Proc Natl Acad Sci USA. 2007;104:12512–12517. [
PMC free article: PMC1916487] [
PubMed: 17630284]
- 66.
Schofield CM, Kleiman-einer M, Rudolph U, Huguenard JR. A gain in GABA
A receptor synaptic strength in thalamus reduces oscillatory activity and absence seizures.
Proc Natl Acad Sci USA. 2009;106:7630–7635. [
PMC free article: PMC2678654] [
PubMed: 19380748]
- 67.
Mattson RH. General Principles: Selection of Antiepileptic Drug Therapy. In: Levy R, editor. Antiepileptic Drugs. 4th ed. New York: Raven Press; 1995. pp. 123–135.
- 68.
Hosford DA, Wang Y, Cao Z. Differential effects mediated by GABA
A receptors in thalamic nuclei in lh/lh model of absence seizures.
Epilepsy Res. 1997;27:55–65. [
PubMed: 9169291]
- 69.
Sohal VS, Keist R, Rudolph U, Huguenard JR. Dynamic GABA
A receptor subtype-specific modulation of the synchrony and duration of thalamic oscillations.
J Neurosci. 2003;23:3649–3657. [
PMC free article: PMC6742195] [
PubMed: 12736336]
- 70.
Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult.
Neuroscience. 2000;101:815–850. [
PubMed: 11113332]