

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology. 2nd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2009.


Essentials of Glycobiology. 2nd edition.
Show details
This chapter covers the general characteristics of the enzymes involved in glycan biosynthesis and modification, including aspects of substrate specificity, primary sequence relationships, structures, and enzyme mechanisms.
GENERAL PROPERTIES
The biosynthesis of glycans is primarily determined by the glycosyltransferases that assemble monosaccharide moieties into linear and branched glycan chains. As might be expected from the complex array of glycan structures found in nature, the glycosyltransferases constitute a very large family of enzymes. However, they have in common the ability to catalyze a group-transfer reaction in which the monosaccharide moiety of a simple nucleotide sugar donor substrate (e.g., UDP-Gal, GDP-Fuc, or CMP-Sia; see Chapter 4) is transferred to the acceptor substrate (Figure 5.1). In some instances, the donor substrates are lipids, such as dolichol-phosphate linked to mannose or glucose or a dolichol-oligosaccharide precursor (see Chapter 8). Lipid-linked donor sugars (e.g., undecaprenyl-pyrophosphoryl-N-acetylmuramic acid-pentapeptide-N-acetylglucosamine) are also used by bacterial glycosyltransferases in the assembly of peptidoglycan, lipopolysaccharide, and capsules (see Chapter 20).

FIGURE 5.1
Glycosylation reactions. A glycosyltransferase uses a glycosyl donor and an acceptor substrate. In animals, glycosyl donors include nucleotide sugars and dolichol-phosphate-linked monosaccharides and oligosaccharides. Bacteria also use undecaprenyl-pyrophosphate (more...)
The glycosyltransferases that initiate the synthesis of glycoconjugates use acceptor substrates that include oligosaccharides, monosaccharides, polypeptides, lipids, small organic molecules, and even DNA. Only enzymes involved in the biosynthesis of glycoproteins, proteoglycans, and glycolipids are discussed in this chapter. The vast majority of these glycosyltransferases are responsible for elongating glycan chains. Generally speaking, these enzymes act sequentially, so that the product of one enzyme yields a preferred acceptor substrate for the subsequent action of another. The end result is a linear and/or branched polymer composed of monosaccharides linked to one another. In most cases, acceptor recognition does not involve the underlying polypeptide or glycolipid substrate, but several notable exceptions exist that are described below. Despite this finding, specific glycosylation sites of many glycoproteins carry predictable glycan structures, suggesting a complex mechanism for controlling composition.
A few of the enzymes involved in the biosynthesis of glycans are glycosidases that remove monosaccharides to form intermediates that are then acted on by glycosyltransferases. Processing of this type is especially relevant in the formation of N-glycans; in this case, the nascent glycoprotein glycan Glc3Man9GlcNAc2-Asn is sequentially trimmed by glucosidases and mannosidases before potential modification by glycosyltransferases that lead to complex and hybrid-type chains (see Chapter 8). In addition, glycans can be modified in many other ways, for example, by sulfotransferases, phosphotransferases, O-acetyl-transferases, O-methyltransferases, pyruvyltransferases, and ethanolamine phosphate transferases, to mention just a few (Figure 5.2).

FIGURE 5.2
Glycan-modifying enzymes. A variety of donors are used to modify glycans.
GLYCOSYLTRANSFERASE SPECIFICITY
The specificity of glycosyltransferases, with respect to nucleotide sugar donor and glycan acceptor, led early on to the concept that each glycosidic linkage is the product of a single enzyme. This so-called “one enzyme–one linkage” hypothesis was advanced by Saul Roseman and coworkers. The human B blood group α1–3 galactosyltransferase provides an excellent example of such specificity. This enzyme catalyzes a glycosylation reaction in which galactose is added in α linkage to the C-3 hydroxyl group of a galactose residue on the acceptor substrate (Figure 5.3). However, the enzyme only acts on galactose containing fucose in α1–2 linkage. Prior modification by other monosaccharides, such as α2–6-linked sialic acid, yields a glycan that is not a substrate (Figure 5.3).
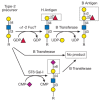
FIGURE 5.3
Strict acceptor substrate specificity of glycosyltransferases as exemplified by the human B blood group α1–3 galactosyltransferase. The B transferase adds galactose in α1–3 linkage to the H antigen (top). This enzyme requires (more...)
We now know that there are also instances in which more than one enzyme can use the same acceptor to make the same linkage. The human fucosyltransferases III–VII, for example, all attach fucose in α1–3 linkage to N-acetyllactosamine moieties on glycans (see Chapter 13). Other examples include α2–3 sialyltransferases that act on galactose, β1–4 galactosyltransferases that act on N-acetylglucosamine, and the family of GlcNAc 3-O-sulfotransferases that participate in heparin/heparan sulfate synthesis. In some rare cases, a single enzyme can catalyze more than one reaction. Human fucosyltransferase III can attach fucose in either α1–3 or α1–4 linkage, and an enzyme called EXTL2 can attach either N-acetylgalactosamine or N-acetylglucosamine in α linkage to glucuronic acid. The β1–4 galactosyltransferase involved in N-acetyllactosamine formation exhibits an unusual flexibility in specificity. When β1-4 galactosyltransferase binds α-lactalbumin (the complex is called lactose synthase), it switches its acceptor specificity from N-acetylglucosamine to glucose, which enables lactose synthesis during milk formation.
Finally, some glycosyltransferases (such as those that synthesize the backbones of glycosaminoglycans) have two separate active sites, for example, one that catalyzes the attachment of N-acetylglucosamine to glucuronic acid and another that attaches glucuronic acid to N-acetylglucosamine (see Chapters 15 and 16). However, the examples described above are all exceptions to the generally strict donor, acceptor, and linkage specificity exhibited by most glycosyltransferases, a property that serves to define and limit the number and type of glycan structures observed in a given organism.
In contrast to those glycosyltransferases that elongate glycan chains, initiation of the biosynthesis of glycoproteins and glycolipids requires glycosyltransferases that attach saccharides to either a polypeptide side chain or a sphingolipid base. As might be expected, these enzymes also show a high degree of specificity for their substrates. Among the polypeptide glycosyltransferases, those that initiate the biosynthesis of O-glycans typically transfer a specific monosaccharide to the side chain of serine or threonine (see Chapters 9 and 16). In contrast, N-linked glycans are initiated by the action of oligosaccharyltransferase, an enzyme that transfers the glycan Glc3Man9GlcNAc2 to the side chain of asparagine residues in the sequence motif Asn-X-Ser/Thr (where X can be any amino acid except proline). For amino acid–consensus sequences or glycosylation motifs used in the formation of glycopeptide bonds, see Table 5.1. The glycosyltransferases that initiate the synthesis of glycosphingolipids transfer a monosaccharide moiety to what was originally a serine residue in the ceramide lipid precursor of sphingolipids (see Chapter 10). Because different glycolipids have different ceramide moieties, it appears that some glycosyltransferases, such as the sialyltransferases, differentially recognize their substrates based on the nature of the ceramide moiety.
TABLE 5.1
Amino acid–consensus sequences or glycosylation motifs for the formation of glycopeptide bonds
PROTEIN/GLYCOPROTEIN ACCEPTORS AND GLYCOSYLTRANSFERASE ACTION
Among the glycosyltransferases that use proteins/glycoproteins as acceptor substrates, the underlying polypeptide chain of the acceptor is used in different ways to confer specificity on the glycosylation reaction. As mentioned above, all N-glycans are initiated by oligosaccharyltransferase, an enzyme that cotranslationally transfers the N-glycan precursor to asparagine residues in the sequence motif Asn-X-Ser/Thr. In contrast, many enzymes can glycosylate serine and threonine residues leading to O-glycans possessing O-GlcNAc, O-GalNAc, O-fucose, O-mannose, and O-xylose linkages. Among the enzymes responsible for these linkages, specificity for a particular serine or threonine residue is achieved in different ways. For example, O-xylosylation has an absolute requirement for a glycine residue located amino terminally to the serine with nearby acidic residues on one or both sides of the glycosylation site. Some polypeptide O-GalNAc transferases even possess a lectin domain that serves to direct the glycosyltransferase to regions of polypeptide already possessing glycan chains. In this way, regions of polypeptide that have a high degree of carbohydrate substitution, typical of mucin structures, can be synthesized.
A few glycosyltransferases are specific for a particular protein type, and in these cases recognition seems to be dependent on the final folded form of the protein acceptor. The O-fucosyltransferases that act on polypeptides selectively modify epidermal growth factor (EGF) or thrombospondin repeats (TSR), protein domains that must be properly folded and disulfide-bonded to serve as acceptor substrates. The glycoprotein hormone GalNAc transferase provides an interesting example in which modification of an N-glycan is dependent on the presence of the sequence motif Pro-X-Arg/Lys positioned several amino acids amino terminally to the N-glycan being modified. The motif is typically followed closely by additional positively charged residues. The X-ray crystal structure of the acceptor substrate, human chorionic gonadotropin, shows that the Pro-X-Arg/Lys motif is at the beginning of a short surface-exposed helix that also contains the additional positively charged residues (Figure 5.4). The N-acetylgalactosamine residues transferred by the glycoprotein hormone GalNAc transferase subsequently undergo a biologically important 4-O-sulfation reaction that, in the case of lutenizing hormone and follicle-stimulating hormone, generates a determinant recognized by specific liver clearance receptors (see Chapter 31). Additional examples are provided by the polysialyltransferases specific for neural cell adhesion molecule (N-CAM) (see Chapter 14) and by EXTL3, an N-acetylglucosaminyltransferase that adds GlcNAc in α1–4 linkage to glucuronic acid in the first committed step to heparan sulfate biosynthesis on proteoglycans (see Chapter 16).

FIGURE 5.4
Recognition site for glycoprotein hormone N-acetylgalactosaminyltransferase on human chorionic gonadotropin. Ribbon diagram of a fragment (residues 34–58) of human chorionic gonadotropin (PDB [protein data bank] ID 1hrp). The Pro40-Leu41-Arg42 (more...)
In a variation on this theme, GlcNAc-1-phosphotransferase is able to modify a family of enzymes that are specifically destined for lysosomes (see Chapter 30). In this case, lysine residues appropriately spaced and positioned relative to the N-glycan being modified have been shown to be important determinants of acceptor specificity.
The ER resident glucosyltransferase (GGT) has an acceptor substrate specificity unique among glycosyltransferases. It specifically reglucosylates N-glycans on misfolded glycoproteins, rendering them substrates for the chaperones calnexin and calreticulin (see Chapter 36). Its ability to differentiate between misfolded and properly folded glycoproteins is a critical component of the ER quality control machinery.
GLYCOSYLTRANSFERASE SEQUENCE FAMILIES AND FOLD TYPES
The cloning and sequencing of more than 500 genomes has now shown that glycosyltransferases are a very prevalent enzyme type, representing 1–2% of the genome. More than 30,000 glycosyltransferase sequences are known across all kingdoms, and they comprise approximately 90 glycosyltransferase families defined by primary sequence analysis. These are described in the carbohydrate-active enzymes (CAZy) database (see Chapter 7). Although the absence of significant sequence similarity between members of one family and another constitutes the basis for this classification, short sequence motifs common to the members of more than one family have been identified. These sequence elements are typically found among glycosyltransferases with a given donor substrate specificity; the sialyl motifs common to eukaryotic sialyltransferases are a good example (Figure 5.5). Sequence motifs common to galactosyltransferases, fucosyltransferases, and N-acetylglu-cosaminyltransferases have also been identified. In contrast, the so-called “DXD” motif (asp–any residue–asp) is not associated with any particular substrate specificity; rather, the motif is involved in metal ion binding and catalysis, and it is discussed in more detail below.

FIGURE 5.5
Sialyl motifs. Domain structure of a typical sialyltransferase, showing the sialyl motifs shared by this family of enzymes. The sialyl L motif of 48–49 amino acids shares significant similarity among members and may be up to 65% identical in amino (more...)
Despite the large number of sequence families that have been defined, structural analysis has shown that glycosyltransferases possess a limited number of fold types. To date, structures for members of 29 of the 90 families have been determined by X-ray crystallography, and of these all but a few possess what have been termed the GT-A or GT-B folds (Figure 5.6). The catalytic domain of the GT-A-fold enzymes can be viewed as a single domain. The first approximately 120 amino acids show similarity to a structural motif called the Rossmann fold, which is found in proteins that bind nucleotides and are responsible for binding the nucleotide sugar donor substrate. With only one exception, the GT-A enzymes have been found to possess a DXD motif and are metal-ion-dependent glycosyltransferases. The GT-B-fold enzymes possess two distinct domains separated by a cleft that binds the acceptor. The carboxy-terminal domain is primarily responsible for binding the nucleotide sugar donor substrate, but both domains possess elements similar to those of the Rossmann fold. Unlike enzymes that contain the GT-A fold, the GT-B glycosyltransferases are metal-ion independent and do not possess a DXD motif.

FIGURE 5.6
Ribbon diagrams of representative GT-A and GT-B folds. The GT-A and -B structures correspond to those of rabbit β1-2 N-acetylglucosaminyltransferase I (PDB ID 1foa) and T4 phage β-glucosyltransferase (PDB ID 1j39), respectively. In both cases, (more...)
CATALYTIC MECHANISMS
Glycosyltransferases catalyze their reactions with either inversion or retention of stereochemistry at the anomeric carbon atom of the donor substrate (Figure 5.7). For example, β1–4 galactosyltransferase, an inverting glycosyltransferase, transfers galactose from UDP-α-Gal (naturally occurring nucleotide sugars, except for CMP-Sia and CMP-KDO [3-deoxy-D-manno-octulosonic acid], are typically α linked) to generate a β1-4-linked galactose-containing product. Inversion of stereochemistry follows from the fact that the enzyme uses an SN2 (substitution nucleophilic bimolecular) reaction mechanism where an acceptor hydroxyl group attacks the anomeric carbon atom from one side and UDP leaves from the other (Figure 5.7a). Typically, enzymes of this type possess an aspartic acid or glutamic acid residue whose side chain serves to partially deprotonate the incoming acceptor hydroxyl group, rendering it a better nucleophile (as shown in Figure 5.8 for the β1–4 galactosyltransferase). In addition, these enzymes promote catalysis by features that help to promote leaving-group departure. In the GT-A enzymes, a metal ion, bound by the DXD motif, is typically positioned to interact with the diphosphate moiety. The positively charged metal ion serves to electrostatically stabilize the additional negative charge that develops on the UDP leaving group during bond breakage (Figure 5.8). In the one GT-A enzyme that is not metalion dependent, positively charged side chains stabilize the leaving group, a strategy also used by some of the GT-B-fold enzymes.
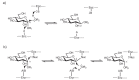
FIGURE 5.7
Schematic representation of (a) inverting and (b) retaining catalytic mechanisms. (a) SN2-like attack of the acceptor leads to inversion of stereochemistry at C1. For a glycosidase reaction, R2 would correspond to a proton and R1 would be the remainder (more...)

FIGURE 5.8
Catalytic site of bovine β1–4 galactosyltransferase. Composite figure shows selected residues/atoms of the superimposition of the donor complex (PDB ID 1tw1) on the acceptor complex (PDB ID 1tw5). O4 designates the C4 hydroxyl group of (more...)
Although the mechanism used by inverting glycosyltransferases is not well understood, insight into how they might work is provided by our knowledge of glycosidase mechanism. On the basis of much structural and enzyme kinetic analysis, it is well established that inverting glycosidases proceed via a single SN2 displacement mechanism (Figure 5.7a), whereas retaining glycosidases use a double displacement mechanism involving a covalent glycosyl-enzyme intermediate (Figure 5.7b). In the double displacement mechanism, an aspartic acid or glutamic acid side chain in the active site makes the first attack and inversion, followed by a second water-mediated attack (on the glycosyl-enzyme intermediate) and inversion, to give overall retention of stereochemistry. In fact, using mechanism-based inhibitors, the glycosyl-enzyme intermediate has been trapped and studied by X-ray crystallography for a number of glycosidases. However, similar attempts to trap and study glycosyltransferase reaction intermediates have not yet been successful. An alternate mechanism, also proposed for glycogen phosphorylase, is that of the so-called SNi mechanism. In this case, the incoming nucleophile attacks from the same side as the leaving group, leading to retention of configuration. Further work is required to establish whether glycosyltransferases use this mechanism.
Despite similarities in the mechanisms and transition states used by glycosidases and glycosyltransferases, small-molecule inhibitors of many glycosidases exist (see Chapter 50), whereas there are relatively few glycosyltransferase inhibitors. This probably reflects the fact that many good enzyme inhibitors mimic the transition state, a relatively simple task in the case of the glycosidase reaction where the acceptor substrate is a simple water molecule. Simple high-affinity competitive inhibitors of substrate binding provide another means of inhibiting an enzyme reaction (see Chapter 50).
KINETIC MECHANISMS
In addition to understanding the structure and catalytic mechanisms of glycosyltransferases, there is much interest in understanding the kinetic mechanism. Many glycosyltransferases have been shown to possess a Bi Bi sequential kinetic mechanism in which the donor substrate binds before the acceptor substrate, and the glycosylated acceptor is released before the nucleoside monosphosphate or disphosphate, depending on the reaction. Such kinetics are easily explained by a structural model in which the active site represents a deep pocket, with the nucleotide sugar substrate at the bottom and the acceptor substrate stacked on top. If the acceptor substrate were to bind first, it would sterically preclude donor substrate binding. Necessarily, release of the glycosylated product must precede release of the nucleoside phosphate. Although largely consistent with such a model, the X-ray crystal structures of glycosyltransferase-substrate complexes also shows that substrate-dependent ordering of flexible loops is a feature common to glycosyltransferases. Typically, donor substrate binding orders a loop(s) that in turn facilitates acceptor substrate binding.
SULFATION AND OTHER MODIFICATIONS
Glycans can undergo a number of modifications, depending on the major subclass and species of origin. The sulfotransferases are a large family of enzymes found in both the cytoplasm and the Golgi. Enzymes from the cytoplasm sulfate low-molecular-weight substrates such as steroids and exogenous compounds, whereas those from the Golgi sulfate a large number of cell-surface and secreted glycans. Sulfotransferases have a particularly important role in the production of glycosaminylglycans, molecules involved in embryological development and physiology (see Chapters 16 and 24). They are also involved in the formation of L-selectin ligands, glycans required for the trafficking of lymphocytes across high endothelial venules in lymph nodes (see Chapter 31). All sulfotransferases use 3′-phospho-adenosine-5′-phosphosulfate (PAPS) as the sulfate donor (see Chapter 4). Although the sequence similarity among sulfotransferases can be very low, they all possess conserved sequence motifs responsible for binding the 5′ and 3′ phosphate groups of PAPS. Moreover, structural analysis has shown that all of the sulfotransferases solved to date possess the same basic structure. Mechanistically, the enzyme proceeds via an SN2-like reaction, with the hydroxyl group of the acceptor making an in-line attack on the sulfate group. In conjunction with structural analysis, mutagenesis has provided insight into the catalytic role played by a number of residues highly conserved among the sulfotransferases. Of these, a histidine residue serves to activate the hydroxyl nucleophile and a lysine assists in stabilizing the PAP leaving group. Interestingly, a conserved serine seems to be involved in modulating the activity of these enzymes to prevent PAPS hydrolysis in the absence of acceptor substrate.
Phosphorylation of sugar residues also occurs (e.g., at C-2 of xylose in proteoglycans), but little is known about the nature of this reaction. Phosphoglycosylation, a process in which a sugar phosphate is transferred from a nucleotide sugar donor directly to a serine residue on a protein (e.g., GlcNAc-P-serine), occurs in Dictyostelium. Mannose-6-phosphate on the N-glycans of lysosomal enzymes serves as a targeting signal. However, the formation of this phosphate ester is not mediated by a typical ATP-using kinase, but rather by a two-step reaction involving the donor UDP-GlcNAc (see Chapter 30). Detailed studies of reaction mechanisms have not yet been undertaken.
O-Acetylation occurs in bacteria and in the modification of sialic acids, but little information is available about the chemistry of these reactions. N-Deacetylation of N-acetylglu-cosamine residues occurs during heparin/heparan sulfate formation (see Chapter 16), lipopolysaccharide assembly (see Chapter 20), and GPI-anchor synthesis (see Chapter 11). The bacterial enzyme is zinc dependent, but in-depth studies of the vertebrate N-deacetylases have not been performed. N-Deacetylation of N-acetylneuraminic acid (the most common sialic acid) has also been reported (see Chapter 14).
Finally, glycans can be modified in many other ways, some of which are listed in Figure 5.2. These include pyruvylation (e.g., in the formation of N-acetylmuramic acid; see Chapter 20), the addition of ethanolamine phosphate (e.g., during GPI-anchor synthesis; see Chapter 11), and alkylation, deoxygenation, and halogenation in microbial glycans. All of these reactions are catalyzed by unique transferases or oxidoreductases and represent areas of active research.
FURTHER READING
- Manzella SM, Hooper LV, Baenziger JU. Oligosaccharides containing β1,4-linked N-acetyl-galactosamine, a paradigm for protein-specific glycosylation. J. Biol. Chem. 1996;271:12117–12120. [PubMed: 8647799]
- Negishi M, Pedersen LG, Petrotchenko E, Shevtsov S, Gorokhov A, Kakuta Y, Pedersen LC. Structure and function of sulfotransferases. Arch. Biochem. Biophys. 2001;390:149–157. [PubMed: 11396917]
- Spiro RG. Protein glycosylation: Nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. 2002;12:43R–56R. [PubMed: 12042244]
- Coutinho PM, Deleury E, Davies GJ, Henrissat B. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 2003;328:307–317. [PubMed: 12691742]
- Negishi M, Dong J, Darden TA, Pedersen LG, Pedersen LC. Glucosaminylglycan biosynthesis: What we can learn from the X-ray crystal structures of glycosyltransferases GlcAT1 and EXTL2. Biochem. Biophys. Res. Commun. 2003;303:393–398. [PubMed: 12659829]
- Ten Hagen KG, Fritz TA, Tabak LA. All in the family: The UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology. 2003;13:1R–16R. [PubMed: 12634319]
- Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu. Rev. Biochem. 2004;73:491–537. [PubMed: 15189151]
- Lairson LL, Withers SG. Mechanistic analogies amongst carbohydrate modifying enzymes. Chem. Commun. 2004;20:2243–2248. [PubMed: 15489968]
- Davies GJ, Gloster TM, Henrissat B. Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr. Opin. Struct. Biol. 2005;15:637–645. [PubMed: 16263268]
- Jones J, Krag SS, Betenbaugh MJ. Controlling N-linked glycan site occupancy. Biochim Biophys Acta. 2005;1726:121–137. [PubMed: 16126345]
- Qasba PK, Ramakrishnan B, Boeggeman E. Substrate-induced conformational changes in glycosyltransferases. Trends Biochem. Sci. 2005;30:53–62. [PubMed: 15653326]
- Blanchard S, Thorson JS. Enzymatic tools for engineering natural product glycosylation. Curr. Opin. Chem. Biol. 2006;10:263–271. [PubMed: 16675288]
- Letts JA, Rose NL, Fang YR, Barry CH, Borisova SN, Seto NOL, Palcic MM, Evans SV. Differential recognition of the Type I and II H antigen acceptors by the human ABO(H) blood group A and B glycosyltransferases. J. Biol. Chem. 2006;281:3625–3632. [PubMed: 16326711]
- Luo Y, Nita-Lazar A, Haltiwanger RS. Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J. Biol. Chem. 2006;281:9385–9392. [PubMed: 16464858]
- Narimatsu H. Human glycogene cloning: Focus on β3-glycosyltransferase and β4-glycosyltrans-ferase families. Curr. Opin. Struct. Biol. 2006;16:567–575. [PubMed: 16979334]
- Yu H, Chen X. Carbohydrate post-glycosylational modifications. Org. Biomol. Chem. 2007;5:865–872. [PMC free article: PMC2769254] [PubMed: 17340000]
- Glycosyltransferases and Glycan-processing Enzymes - Essentials of GlycobiologyGlycosyltransferases and Glycan-processing Enzymes - Essentials of Glycobiology
Your browsing activity is empty.
Activity recording is turned off.
See more...