Glycosyltransferases and glycosidases are responsible for the assembly, processing, and turnover of glycans. In addition, there are a number of transferases that modify glycans by the addition of acetyl, methyl, phosphate, sulfate, and other groups. This chapter covers the general characteristics of enzymes involved in glycan initiation, assembly, and processing, including aspects of substrate specificity, primary sequence relationships, structures, and enzyme mechanisms.
GENERAL PROPERTIES
The biosynthesis of glycans is primarily determined by glycosyltransferases that assemble monosaccharide moieties into linear and branched glycan chains. As might be expected from the complex array of glycan structures found in nature, glycosyltransferases constitute a very large family of enzymes. In many cases, they catalyze a group-transfer reaction in which the monosaccharide moiety of a simple nucleotide sugar donor (electrophile) substrate (e.g., UDP-Gal, GDP-Fuc, or CMP-Sia; Chapter 5) is transferred to the acceptor (nucleophile) substrate. In some instances, the donor substrates contain a lipid moiety, such as dolichol-phosphate, linked to mannose or glucose. For other glycosyltransferases, the donor substrate is dolichol-pyrophosphate, linked to an oligosaccharide, and in these cases the entire oligosaccharide is transferred en bloc to the acceptor substrate (Chapter 9). Similarly, other lipid-linked sugars serve as donor substrates for bacterial glycosyltransferases involved in the assembly of peptidoglycan (e.g., GlcNAc-MurNAc(pentapeptide)-undecaprenyl pyrophosphate), lipopolysaccharide, and capsules (Chapters 21 and 22).
Glycosyltransferases that use monosaccharides, oligosaccharides, proteins, lipids, small organic molecules, and DNA as acceptor substrates have been characterized (activity to RNA has also been suggested), but only glycosyltransferases involved in the biosynthesis of glycoproteins, proteoglycans, and glycolipids are discussed in this chapter. Among these enzymes, the vast majority is responsible for elongating the glycan moieties of these glycoconjugates; the remainder is responsible for the transfer of either a mono- or oligosaccharide directly to the polypeptide or lipid. Generally speaking, the enzymes that elongate glycans act sequentially so that the product of one enzyme yields a preferred acceptor substrate for the subsequent action of another. The end result is a linear and/or branched structure composed of monosaccharides linked to one another. Acceptor recognition by these glycan-elongating glycosyltransferases does not typically involve the polypeptide or lipid moiety of the acceptor substrate when it exists, although there are several notable exceptions as discussed below.
Glycosidases that remove monosaccharides to form intermediates that are then acted on by glycosyltransferases also play a role in the biosynthesis of some glycan types. These are to be contrasted with glycosidases that are involved in the degradation of glycans (e.g., in lysosomes [Chapter 44]). In addition, glycans can be modified by many other enzyme types, including sulfotransferases, phosphotransferases, O-acetyltransferases, O-methyltransferases, pyruvyltransferases, and phosphoethanolamine transferases.
GLYCOSYLTRANSFERASE SPECIFICITY
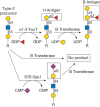
Most glycosyltransferases show a high degree of specificity for both their donor and acceptor substrates, and this led Saul Roseman and coworkers to advance the “one enzyme–one linkage” hypothesis. The human B blood group α1-3 galactosyltransferase exemplifies this concept. This enzyme catalyzes a glycosylation reaction in which galactose is added in α-linkage to the C-3 hydroxyl group of a galactose residue on the acceptor substrate (Figure ). However, the enzyme only acts on galactose modified by fucose in α1-2-linkage and prior modification by other monosaccharides, such as α2-6-linked sialic acid, yields a glycan that is not a substrate (Figure ).
The strict acceptor substrate specificity of glycosyltransferases is illustrated by the human B blood group α1-3 galactosyltransferase. The B transferase adds galactose in α1-3-linkage to the H antigen (top middle) to form the B antigen (more...)
We now know that there are instances in which more than one glycosyltransferase can use the same acceptor to make the same linkage. The human fucosyltransferases III–VII, for example, all attach fucose in α1-3-linkage to N-acetyllactosamine moieties on glycans (Chapter 14). Examples of relaxed acceptor specificity are provided by the α2-3 sialyltransferases and β1-4 galactosyltransferases that act broadly on β-linked galactose and N-acetylglucosamine, respectively. In some rare cases, a single enzyme can catalyze more than one reaction. Human fucosyltransferase III can attach fucose in either α1-3- or α1-4-linkage, and an enzyme called EXTL2 can attach either N-acetylgalactosamine or N-acetylglucosamine in α-linkage to glucuronic acid (Chapter 17). The β1-4 galactosyltransferase involved in N-acetyllactosamine formation shows an unusual flexibility in specificity. When β1-4 galactosyltransferase binds α-lactalbumin (the complex is called lactose synthase), it switches its acceptor specificity from N-acetylglucosamine to glucose, which enables the synthesis of lactose and other oligosaccharides during milk production (Chapter 14). Finally, some glycosyltransferases have two separate active sites with different substrate specificities. For example, the enzymes that synthesize the backbones of heparan sulfate (EXT1) and hyaluronan (HAS) have one active site that catalyzes the attachment of N-acetylglucosamine to glucuronic acid and another that attaches glucuronic acid to N-acetylglucosamine (Chapters 16 and 17). However, the examples described above are all exceptions to the generally strict donor, acceptor, and linkage specificity shown by most glycosyltransferases, a property that serves to define and limit the number and type of glycan structures observed in a given cell type or organism.
The glycosyltransferases that transfer monosaccharides or oligosaccharides directly to polypeptide or lipid moieties also show a high degree of substrate specificity and this will be discussed in more detail below for those involving polypeptide. Glycosyltransferases that initiate the synthesis of glycosphingolipids transfer a monosaccharide moiety to what was originally a serine residue in the ceramide lipid precursor of sphingolipids (see Chapter 11). Because different glycolipids have different ceramide moieties, it appears that some glycosyltransferases, such as the sialyltransferases, differentially recognize their substrates based on the nature of the ceramide moiety.
GLYCOSYLTRANSFERASES THAT RECOGNIZE THE PROTEIN MOIETY OF THEIR ACCEPTOR SUBSTRATES
The glycosyltransferases that transfer sugar directly to the polypeptide chain of a protein or glycoprotein recognize their acceptor substrates in a number of different ways. All eukaryotic N-glycans are initiated by oligosaccharyltransferase (OST), generally an ER-resident multisubunit enzyme that transfers en bloc Glc3Man9GlcNAc2 to the side chain of asparagine residues in the sequence motif Asn-X-Ser/Thr (where X can be any amino acid except proline; Chapter 9). In contrast, the polypeptide GalNAc transferases, responsible for the initiation of mucin-type O-glycans, act after the protein has been folded and transported to the Golgi (Chapter 10). The polypeptide O-GalNAc transferases do not recognize a specific sequence motif and in general, they transfer N-acetyllgalactosamine to the side chain hydroxyl group of serine and threonine residues found in relatively unstructured regions of the folded protein. Some polypeptide O-GalNAc transferases possess a lectin domain that serves to direct the glycosyltransferase to regions of the polypeptide that already possess O-glycan chains. In this way, regions of polypeptide that have a high degree of O-glycan substitution, typical of mucin structures, can be synthesized.
In addition to the O-GalNAc linkage formed by the polypeptide O-GalNAc transferases, a number of other glycosyltransferases can glycosylate the side chain hydroxyl groups of serine and threonine to generate O-GlcNAc, O-Fuc, O-Glc, O-Man, and O-Xyl linkages (Chapters 13, 17, and 19). Specificity for a particular serine or threonine residue is achieved in different ways. The xylosyltransferase that O-xylosylates a serine residue in chondroitin and heparan sulfate proteoglycans, for example, has an absolute requirement for a glycine residue carboxy-terminal to the serine and/or more acidic residues in the vicinity of the glycosylation site. In contrast, the O-GlcNAc transferase (OGT), responsible for adding N-acetylglucosamine to serine and threonine residues on thousands of nuclear and cytoplasmic proteins (Chapter 19), lacks any obvious consensus sequence associated with acceptor substrate binding specificity. See Table for some amino acid–consensus sequences or glycosylation motifs used in the formation of glycopeptide bonds.
Amino acid–consensus sequences or glycosylation motifs for the formation of glycopeptide bonds
The endoplasmic reticulum (ER)-resident O-fucosyltransferases, POFUT1 and POFUT2, that specifically fucosylate epidermal growth factor (EGF)-like domains and thrombospondin type 1 repeats (TSRs), respectively (Chapter 13), differ fundamentally from most other glycosyltransferases. In addition to recognizing a specific sequence motif containing the target serine or threonine residue (Table ), these enzymes only act on EGF-like domains and TSRs that are properly folded and disulfide-bonded. Glycosyltransferases that add O-Glc and O-GlcNAc to other serine and threonine residues in the EGF-like domain also exist and these enzymes also recognize a specific sequence motif and require a folded EGF-like domain.
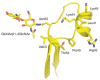
A number of the glycan-elongating glycosyltransferases that act on glycoproteins also recognize the polypeptide moiety of their acceptor substrates. The glycoprotein hormone GalNAc transferase (a member of the family of β1-4GalNAcTs) provides an interesting example in which modification of an N-glycan is dependent on the presence of the protein sequence motif Pro-X-Arg/Lys positioned several amino acids amino-terminally to the N-glycan being modified. The motif is typically followed closely by additional positively charged residues. The X-ray crystal structure of its acceptor substrate, human chorionic gonadotropin, shows that the Pro-X-Arg/Lys motif is at the beginning of a short surface-exposed helix that also contains the additional positively charged residues (Figure ). The N-acetylgalactosamine residue transferred by this enzyme subsequently undergoes a biologically important 4-O-sulfation reaction that, in the case of luteinizing hormone and follicle-stimulating hormone, generates a determinant recognized by specific liver clearance receptors that remove them from the blood (Chapter 34). Glycosyltransferases that only elongate the fucose moiety added by POFUT1 or POFUT2 also exist (Chapter 13). The specificity shown by these enzymes stems from their ability to recognize both the fucose moiety and the EGF-like and TSR components of their acceptor substrates. Additional examples of enzymes that elongate glycans on specific glycoprotein substrates include the polysialyltransferases that act on neural cell adhesion molecule (NCAM) and neuropilin-2 (Chapter 15), and the N-acetylglucosaminyltransferase, EXTL3, that adds N-acetylglucosamine in α1-4-linkage to glucuronic acid in the first committed step to heparan sulfate biosynthesis on proteoglycans (Chapter 17).
Human chorionic gonadotropin showing the determinants of recognition used by glycoprotein hormone N-acetylgalactosaminyl (GalNAc) transferase. Ribbon diagram of a fragment (residues 34–58) of human chorionic gonadotropin (PDB [protein data bank] (more...)
In a variation on this theme, GlcNAc-1-phosphotransferase selectively modifies the N-glycans found on a large family of lysosomal enzymes that differ in three-dimensional structure and that lack any obvious and common protein sequence motif. In this case, modification by this enzyme has been shown to be dependent on lysine residues appropriately spaced and positioned relative to the N-glycan site (Chapter 33). The ER-resident glucosyltransferase, UGGT, is also able to transfer sugar to the N-glycans on a large but unique subset of glycoprotein substrates (Chapter 39). In this case, the enzyme adds a glucose moiety to the N-glycans of misfolded glycoproteins rendering them substrates for the ER-resident lectins calnexin and calreticulin. These lectins in turn recruit folding catalysts that promote correct disulfide bond formation and the cis–trans isomerization of proline residues.
GLYCOSYLTRANSFERASE SEQUENCE FAMILIES AND FOLD TYPES
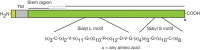
Approximately 1% of genes in the mammalian genome are involved in the production or modification of glycans. More than 750,000 glycosyltransferase sequences are known across all kingdoms, and the numbers are growing rapidly. Based on sequence analysis, they have been categorized into more than 110 glycosyltransferase families as described in the Carbohydrate-Active Enzymes (CAZy) database (Chapter 8). Although the absence of significant sequence similarity between members of one family and another constitutes the basis for this classification, short sequence motifs common to the members of more than one family have been identified. These sequence elements are typically found among glycosyltransferases with a given donor substrate specificity; the sialyl motifs common to eukaryotic sialyltransferases are a good example (Figure ). Sequence motifs common to galactosyltransferases, fucosyltransferases, and N-acetylglucosaminyltransferases have also been identified. In contrast, the so-called “DXD” motif (Asp–any residue–Asp) is not associated with any particular substrate specificity; this motif is involved in metal ion binding and catalysis as discussed in more detail below.
Sialyl motifs. Domain structure of a typical sialyltransferase, showing the sialyl motifs shared by this family of enzymes. The sialyl L motif of 48–49 amino acids shares significant similarity among members and may be up to 65% identical in amino (more...)
Despite the large number of sequence families that have been defined, structural analysis has shown that glycosyltransferases possess a limited number of fold types. To date, structures for 262 members representing 59 of the CAZy families have been determined by X-ray crystallography or cryo-electron microscopy (cryo-EM); of these, all but a few possess what have been termed the GT-A, GT-B, GT-C, and lysozyme-type folds (Figure ). The GT-A and GT-B enzymes use nucleotide sugar donor substrates, whereas the GT-C and lysozyme-type enzymes use lipid-linked sugar donors. The GT-A fold glycosyltransferases appear to have evolved from a common ancestor harboring a ∼231 amino acid core structure and a conserved set of structural features. The core contains elements of the Rossmann fold, which mediates the interaction with the nucleotide sugar, as well as the generally conserved DXD motif responsible for divalent cation binding (usually Mn++ or Mg++) and catalysis. Recent analysis has shown how the insertion of loops into the core structure has facilitated the acquisition, over evolutionary time, of the diverse acceptor specificities shown by the GT-A enzymes. The GT-B enzymes possess two distinct domains, and although the carboxy-terminal domain is primarily responsible for binding the nucleotide sugar donor substrate, both domains possess elements of the Rossmann fold. Acceptor substrates are typically bound in the cleft between the two domains, and unlike the GT-A enzymes, the GT-B glycosyltransferases are metal-ion-independent and do not possess a DXD motif. The GT-C fold enzymes are multi-spanning integral membrane proteins characterized by their use of lipid-linked sugar donors. Recent X-ray and cryo-EM structures are now providing important insights into how these enzymes mediate substrate binding and catalysis (Figure ). Notable among them are the structures of both prokaryotic (PglB and AglB) and eukaryotic (yeast and human) oligosaccharyltransferases that have provided a model for N-glycosylation, a modification conserved across all three domains of life. Structural comparison among the GT-C glycosyltransferases has identified a conserved core of transmembrane helices involved in binding their polyisoprenoid(pyro)phosphate-sugar donors and a variable number of transmembrane- and nonmembrane-associated segments involved in acceptor interactions. The lysozyme-type glycosyltransferases (CAZy family 51) are involved in the biosynthesis of bacterial peptidoglycan and use lipid II (GlcNAc-MurNAc(pentapeptide)-undecaprenyl pyrophosphate) as donor substrate (Chapter 21). In addition to the lysozyme-type domain, these glycosyltransferases possess the jaw subdomain, which embeds in the extracellular face of the cytoplasmic membrane and provides access to the lipid II substrate. In a very recent advance, it has been shown that the dual activity glycosyltransferase-phosphorylases, important for the turnover of the storage polysaccharide mannogen and virulence in Leishmania parasites, possess a β-propeller fold catalytic domain not previously observed for a glycosyltransferase.
Ribbon diagrams of representative GT-A, GT-B, GT-C, and lysozyme-type fold glycosyltransferases. The GT-A and GT-B structures correspond to those of rabbit β1-2 N-acetylglucosaminyltransferase I (PDB ID 1FOA) and T4 phage β-glucosyltransferase (more...)
GLYCOSIDASES
Glycosidases are a very large group of enzymes with more than 870,000 members falling into more than 170 CAZy database families (Chapter 8). Unlike the glycosyltransferases, members of this family have evolved independently many times, a fact reflected in the diverse array of three-dimensional structures observed for these enzymes. Glycosidases play important roles in the degradation of glycan structures for the uptake and metabolism of sugars and for the turnover of glycoconjugates in various cellular processes. Glycosidases are also involved in the formation of intermediates that are used as substrates for glycosyltransferases in the biosynthesis of glycans. The use of glycosidases in this way is particularly important in the biosynthesis of N-glycan-containing glycoproteins in more evolutionarily advanced eukaryotes and is thought to be associated with the acquisition of complex N-glycans during the evolution of multicellular organisms. In this case, the nascent glycoprotein glycan, Glc3Man9GlcNAc2-Asn, is trimmed by glucosidases and mannosidases in the ER and Golgi to generate substrates for the glycosyltransferases that lead to elaborated complex and hybrid-type N-glycans (Chapter 9). Glucosidase II, one of the two ER-resident glucosidases involved, also works in conjunction with UGGT during glycoprotein folding to allow repeated deglucosylation/reglucosylation during what has been termed the calnexin/calreticulin quality control cycle (Chapter 39). An interplay between glycosylation and deglycosylation is also found to occur with the O-GlcNAc moiety that appears in many nuclear and cytoplasmic proteins (transferred by O-GlcNAc transferase; Chapter 19). In this case, removal of the N-acetylglucosamine moiety by the glycosidase, O-GlcNAcase, provides a means of dynamically regulating the extent of O-GlcNAcylation and the diverse processes that it mediates.
CATALYTIC AND KINETIC MECHANISMS
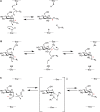
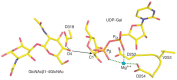
Glycosyltransferases catalyze their reactions with either inversion or retention of stereochemistry at the anomeric carbon atom of the donor substrate (Figure ). For example, β1-4 galactosyltransferase, an inverting glycosyltransferase, transfers galactose from UDP-α-Gal to generate a β1-4-linked galactose-containing product. Inversion of stereochemistry follows from the fact that the enzyme uses an SN2 (substitution nucleophilic bimolecular) reaction mechanism in which an acceptor hydroxyl group attacks the anomeric carbon atom of UDP-Gal from one side and UDP leaves from the other (Figure ). Typically, enzymes of this type possess an aspartate, glutamate, or histidine residue whose side chain serves to partially deprotonate the incoming acceptor hydroxyl group (acting as a general base), rendering it a better nucleophile (as shown in Figure for the β1-4 galactosyltransferase). Additional structures for GT-B fold inverting glycosyltransferases have been identified in which such a base appears to be lacking. In these cases, a water-mediated proton-shuttle mechanism has been proposed to achieve the required acceptor deprotonation. In addition, these enzymes possess features that help to promote leaving-group departure. In the GT-A enzymes, a metal ion, bound by the DXD motif, is typically positioned to interact with the diphosphate moiety. The positively charged metal ion serves to electrostatically stabilize the additional negative charge that develops on the terminal phosphate moiety of the UDP leaving group during breakage of the sugar-phosphate bond of the donor substrate (Figure ). In the few GT-A enzymes that are not metal-ion-dependent, positively charged side chains stabilize the leaving group, a strategy also used by some of the GT-B-fold enzymes.
Schematic representation of inverting and retaining catalytic mechanisms. (A) SN2-like attack of the acceptor leads to inversion of stereochemistry at C1 of the donor sugar (the anomeric carbon). For a glycosidase reaction, R2 would correspond to a proton (more...)
Catalytic site of bovine β1-4 galactosyltransferase. Composite figure shows selected residues/atoms of the superimposition of the donor complex (PDB ID 1TW1) on the acceptor complex (PDB ID 1TW5). O4 designates the C4 hydroxyl group of the GlcNAcβ1-4GlcNAc (more...)
Although the mechanisms used by retaining glycosyltransferases remain open, initial catalytic models based on glycosidase mechanisms (see below) proposed a double-displacement mechanism (Figure ). In this case, a nucleophile (e.g., an aspartic acid or glutamic acid side chain) in the enzyme active site makes the first attack with inversion of anomeric configuration, followed by a second attack (on a glycosyl-enzyme intermediate) and inversion, to give overall retention of stereochemistry. However, no enzyme-associated catalytic nucleophile has yet been identified for a retaining glycosyltransferase and attempts to trap and study glycosyltransferase covalent reaction intermediates have not yet clearly supported this mechanism. Thus, while a “double-displacement” mechanism cannot yet be discounted and/or may be limited to specific families of glycosyltransferases, other mechanisms have been proposed. In several cases, retaining glycosyltransferases appear to position the hydroxyl nucleophile of the acceptor adjacent to the β-phosphate group of the donor nucleotide. Substrate-assisted acceptor deprotonation by the donor phosphate has been proposed with a dissociative, same/front-side mechanism in which the leaving group “guides” the nucleophilic acceptor to the same face of the anomeric carbon with resulting retention of anomeric configuration for the resulting glycosidic bond (Figure ). This SNi or SNi-like mechanism is supported by computational modeling and, in specific cases, by kinetic analyses and X-ray crystal structures of transition-state analog inhibitor complexes.
On the basis of much structural and enzyme kinetic analysis, it is well established that inverting glycosidases proceed via a single SN2 displacement mechanism (Figure ), whereas retaining glycosidases use a double-displacement mechanism involving a covalent glycosyl-enzyme intermediate (Figure ). In the double-displacement mechanism, an aspartic acid or glutamic acid side chain in the enzyme active site makes the first attack and inversion, followed by a second water-mediated attack (on the glycosyl-enzyme intermediate) and inversion, to give overall retention of stereochemistry. Using mechanism-based inhibitors, the glycosyl-enzyme intermediate has been trapped and studied by X-ray crystallography for a number of glycosidases.
Many glycosyltransferases have been shown to possess a Bi-Bi sequential kinetic mechanism in which the donor substrate binds before the acceptor substrate, and the glycosylated acceptor is released before the nucleoside monophosphate or diphosphate, depending on the reaction. Such kinetics are readily explained by a structural model in which the active site represents a deep pocket, with the nucleotide sugar substrate at the bottom and the acceptor substrate stacked on top. If the acceptor substrate was to bind first, it would sterically preclude donor substrate binding and, as such, catalysis would not occur. Because of the stacked arrangement, it also follows that release of the glycosylated product must precede release of the nucleotide leaving group. Although largely consistent with such a model, the X-ray crystal structures of glycosyltransferase-substrate complexes also show that substrate-dependent ordering of flexible loops is a feature common to glycosyltransferases. Typically, donor substrate binding orders a loop(s) that in turn facilitates acceptor substrate binding. Moreover, loop ordering may serve to exclude bulk water from the active site, a strategy proposed to be used by most enzymes to create an active site environment that reduces the energy of the transition state and promotes catalysis. For glycosyltransferases, this would also serve to remove waters as a competing hydrolytic nucleophile, thereby favoring the acceptor substrate.
SULFATION AND OTHER MODIFICATIONS
The sulfotransferases are a large family of enzymes found in both the cytoplasm and the Golgi. Sulfotransferases play a particularly important role in the production of glycosaminoglycans (Chapter 17) and the formation of L-selectin ligands, glycans, and glycoconjugates required for the trafficking of lymphocytes across high-endothelial venules in lymph nodes (Chapter 34). All sulfotransferases use 3′-phospho-adenosine-5′-phosphosulfate (PAPS) as the sulfate donor (Chapter 5). Although the sequence similarity among sulfotransferases can be very low, they all possess conserved sequence motifs responsible for binding the 5′ and 3′ phosphate groups of PAPS. Moreover, structural analysis has shown that all of the sulfotransferases solved, to date, possess the same basic structure. Mechanistically, the enzyme proceeds via an SN2-like reaction, with the hydroxyl group of the acceptor making an in-line attack on the sulfate group, although it should be understood that substitution at sulfur may be mechanistically very different to that at carbon. Structural studies and mutagenesis have shown that a histidine residue serves to activate the hydroxyl nucleophile and that a lysine residue assists in stabilizing the PAP leaving group. Interestingly, a conserved serine residue seems to be involved in modulating the activity of these enzymes to prevent PAPS hydrolysis in the absence of acceptor substrate, another example of the need for enzymes to control the role of the competing water.
Phosphorylation of sugar residues by ATP-dependent kinases occurs at C-2 of O-xylose in proteoglycans (Chapter 17) and at the C-6 position of O-mannose in α-dystroglycan after the mannose has been further glycosylated (Chapter 13). Phosphoglycosylation (or glycophosphorylation), a process in which a sugar phosphoryl is transferred from a nucleotide sugar donor directly to a serine residue on a protein (e.g., GlcNAc-P-serine and Man-P-serine), occurs in Dictyostelium and Leishmania, respectively. In eukaryotic cells, mannose-6-phosphate on the N-glycans of lysosomal enzymes occurs by a two-step process involving UDP-GlcNAc as the phosphate donor. In the first step, GlcNAc-1-phosphotransferase uses UDP-GlcNAc to generate GlcNAcα1-P-6-Manα1-(N-glycan), and in the second step the N-acetylglucosamine moiety is removed by a second enzyme to give P-6-Manα1-(N-glycan) (Chapter 33).
Glycan O-acetylation occurs in bacteria and plants and the O-acetylation of sialic acids is found in bacteria, parasites, and vertebrates. The structure of a plant acetyltransferase suggests the use of a catalytic triad and a double-displacement mechanism for acetyl group transfer analogous to esterases and serine proteases. CASD1, the sialate O-acetyltransferase (SOAT) in humans is a multi-spanning membrane protein with a globular extramembranous catalytic domain. The enzyme transfers the acetyl group from acetyl-CoA to CMP-Neu5Ac. As such, it is the sialytransferase-mediated delivery of 9-O-acetylated sialic to acceptor substrates that leads to the 9-O-acetylated glycoconjugates observed. 9-O-acetylated sialic acid plays a role in CD22 signaling, a process regulated by the opposing action of a sialate 9-O-acetylesterase (SIAE). It also serves as receptor or coreceptor for influenza C and a number of coronaviruses. N-Deacetylation of N-acetylglucosamine residues to glucosamine occurs during heparin/heparan sulfate formation (Chapter 17), lipopolysaccharide assembly (Chapters 21 and 22), and GPI-anchor synthesis (Chapter 12). The bacterial enzyme is zinc-dependent, but in-depth studies of the vertebrate N-deacetylases have not been performed. N-Deacetylation of N-acetylneuraminic acid (the most common sialic acid) has also been reported (Chapter 15).
Finally, glycans can be modified in many other ways, including pyruvylation (e.g., in the formation of N-acetylmuramic acid; Chapters 21 and 22), the addition of ethanolamine phosphate (e.g., during GPI-anchor synthesis; Chapter 12), and alkylation, deoxygenation, and halogenation in microbial glycans. All of these reactions are catalyzed by unique transferases or oxidoreductases and represent areas of active research.
ACKNOWLEDGMENTS
The authors appreciate helpful comments and suggestions from David Vocadlo, Mike Ferguson, Susan Bellis, and Tetsuya Okajima.
FURTHER READING
Roseman S. 2001. Reflections on glycobiology. J Biol Chem
276: 41527–41542. doi:10.1074/jbc.R100053200 [
PubMed: 11553646] [
CrossRef]
Lairson LL, Henrissat B, Davies GJ, Withers SG. 2008. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem
77: 521–555. doi:10.1146/annurev.biochem.76.061005.092322 [
PubMed: 18518825] [
CrossRef]
Schwarz F, Aebi M. 2011. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol
21: 576–582. doi:10.1016/j.sbi.2011.08.005 [
PubMed: 21978957] [
CrossRef]
Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA. 2012. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology
22: 736–756. doi:10.1093/glycob/cwr182 [
PMC free article: PMC3409716] [
PubMed: 22183981] [
CrossRef]
Hurtado-Guerrero R, Davies GJ. 2012. Recent structural and mechanistic insights into post-translational enzymatic glycosylation. Curr Opin Chem Biol
16: 479–487. doi:10.1016/j.cbpa.2012.10.013 [
PubMed: 23142486] [
CrossRef]
Albesa-Jové D, Giganti D, Jackson M, Alzari PM, Guerin ME. 2014. Structure–function relationships of membrane-associated GT-B glycosyltransferases. Glycobiology
24: 108–124. doi:10.1093/glycob/cwt101 [
PMC free article: PMC3907083] [
PubMed: 24253765] [
CrossRef]
Taujale R, Venkat A, Huang L-C, Yeung W, Rasheed K, Edison AS, Moremen KW, Kannan N. 2020. Deep evolutionary analysis reveals the design principles of fold A glycosyltransferases. eLife
9: e54532. doi:10.7554/eLife.54532 [
PMC free article: PMC7185993] [
PubMed: 32234211] [
CrossRef]