Clinical Description
Kindler syndrome (KS), a rare subtype of epidermolysis bullosa, is characterized by skin fragility and acral blister formation beginning at birth or in early infancy, diffuse cutaneous atrophy, photosensitivity (most prominent during childhood and usually decreasing after adolescence), poikiloderma, palmoplantar hyperkeratosis, and pseudosyndactyly. Mucosal manifestations are also common and include hemorrhagic mucositis and gingivitis, periodontal disease, premature loss of teeth, and labial leukokeratosis. Other mucosal findings include ectropion, urethral stenosis, and severe phimosis. Severe long-term complications of KS include periodontitis, mucosal strictures, and aggressive squamous cell carcinomas.
To date, about 400 individuals have been identified with biallelic pathogenic variants in FERMT1 [Guerrero-Aspizua et al 2019]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Kindler Syndrome: Frequency of Select Features
View in own window
| Feature | Frequency |
|---|
| Nearly all | Common | Infrequent |
|---|
| Blisters | ● | | |
| Skin atrophy | ● | | |
| Photosensitivity | | ● | |
| Poikiloderma | | ● | |
| Axillary freckling | | | ● |
| Hyperkeratosis of palms & soles | | ● | |
| Pseudosyndactyly | | ● | |
| Mucosal fragility | | ● | |
| Malignancy | | | ● |
The phenotypic spectrum ranges from mild to severe based on age of onset, organs involved, and severity of manifestations. The mild end of the spectrum is characterized by minimal skin involvement (such as that observed in adults with KS), with or without other mild manifestations. Some individuals with mild manifestations are not diagnosed until late in life; for example, two individuals were diagnosed by molecular genetic testing in their 60s and 70s after early-stage cutaneous precancerous lesions and epithelial skin cancer were identified and treated in their 50s [Has et al 2010]. In contrast, the severe end of the spectrum is characterized by such findings as severe mucosal involvement, severe esophageal stenosis, pseudoainhum, anemia, and/or malignancies.
Blisters are present at birth; those observed in childhood are mainly localized to extremities.
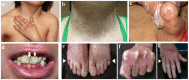
Skin atrophy, characterized by thin, wrinkled (cigarette-paper) skin is initially primarily localized to hands and feet; atrophy becomes generalized by adolescence and is often present on the abdomen, thighs, knees, and elbows (, , ) [Jobard et al 2003].
Characteristic clinical features of Kindler syndrome a. Child age nine years: atrophy of skin on the dorsum of the hands and poikiloderma on the neck and axillary area
Photosensitivity, characterized by erythema and burning after sun exposure, tends to improve with age; however, some degree of photosensitivity usually persists (e.g., facial erythema after minimal sun exposure) [Ashton et al 2004]. Note: Affected individuals can develop redness within minutes of sun exposure.
Poikiloderma, which is not present at birth, appears first on sun-exposed areas, progressing with age to non-sun-exposed areas (). This is characterized by reticular telangiectasia and mottled hypo- and hyperpigmentation of the skin, frequently appearing between ages two and three years. Generalized poikiloderma (in both sun-exposed and non-sun-exposed areas) eventually develops and persists throughout adult life in most affected individuals.
Axillary freckling may be observed in some () [Jobard et al 2003, Siegel et al 2003, Rayinda et al 2021].
Hyperkeratosis of the palms and soles has been reported as fissured or punctate [Penagos et al 2004] and is observed in about 65% of affected individuals. Palmar hyperkeratosis often has a waxy appearance, occasionally leading to the loss of the dermal ridges (i.e., fingerprints). Dermatoglyphics can be flattened or lost. Some individuals may have ridged, ribbed hyperkeratosis of the lateral and anterior ankles reminiscent of epidermolytic hyperkeratosis.
Pseudosyndactyly. Typically, interdigital webbing develops (), but without the scarring or milia noted in other forms of epidermolysis bullosa (EB) [Jobard et al 2003, Siegel et al 2003].
Constricting bands of the pseudoainhum type have also been reported (, ) [Penagos et al 2004].
Mucosal involvement can include the following [Jobard et al 2003, Penagos et al 2004]:
Mouth and periodontium. Severe periodontal disease (e.g., hemorrhagic mucositis, gingivitis, periodontitis, premature loss of teeth, and labial leukokeratosis), usually beginning in early adolescence [
Lai-Cheong & McGrath 2010]
Gastrointestinal tract. Esophageal stenosis, severe colitis, bloody diarrhea, constipation, and rectal mucosal fissures and stenosis. Affected children born with an imperforate anus that required surgical repair have also been reported [
Lai-Cheong & McGrath 2010].
Genitourinary tract
Malignancy. About half of individuals with KS will develop squamous cell carcinoma in their lifetime. An increased risk for malignancies has been reported:
Other findings that may be present:
Morbidity and mortality mostly result from mucosal strictures and associated complications, secondary infections or cutaneous bullae, and cancer.