Summary
Clinical characteristics.
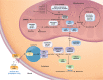
For this GeneReview, the term "isolated methylmalonic acidemia" refers to a group of inborn errors of metabolism associated with elevated methylmalonic acid (MMA) concentration in the blood and urine that result from the failure to isomerize (convert) methylmalonyl-coenzyme A (CoA) into succinyl-CoA during propionyl-CoA metabolism in the mitochondrial matrix, without hyperhomocysteinemia or homocystinuria, hypomethioninemia, or variations in other metabolites, such as malonic acid. Isolated MMA is caused by complete or partial deficiency of the enzyme methylmalonyl-CoA mutase (mut0 enzymatic subtype or mut– enzymatic subtype, respectively), a defect in the transport or synthesis of its cofactor, 5-deoxy-adenosyl-cobalamin (cblA, cblB, or cblD-MMA), or deficiency of the enzyme methylmalonyl-CoA epimerase. Prior to the advent of newborn screening, common phenotypes included:
Infantile/non-B12-responsive form (mut0 enzymatic subtype, cblB), the most common phenotype, associated with infantile-onset lethargy, tachypnea, hypothermia, vomiting, and dehydration on initiation of protein-containing feeds. Without appropriate treatment, the infantile/non-B12-responsive phenotype could rapidly progress to coma due to hyperammonemic encephalopathy.
Partially deficient or B12-responsive phenotypes (mut– enzymatic subtype, cblA, cblB [rare], cblD-MMA), in which symptoms occur in the first few months or years of life and are characterized by feeding problems, failure to thrive, hypotonia, and developmental delay marked by episodes of metabolic decompensation
Methylmalonyl-CoA epimerase deficiency, in which findings range from complete absence of symptoms to severe metabolic acidosis. Affected individuals can also develop ataxia, dysarthria, hypotonia, mild spastic paraparesis, and seizures.
In those individuals diagnosed by newborn screening and treated from an early age, there appears to be decreased early mortality, less severe symptoms at diagnosis, favorable short-term neurodevelopmental outcome, and lower incidence of movement disorders and irreversible cerebral damage. However, secondary complications may still occur and can include intellectual disability, tubulointerstitial nephritis with progressive impairment of renal function, "metabolic stroke" (bilateral lacunar infarction of the basal ganglia during acute metabolic decompensation), pancreatitis, growth failure, functional immune impairment, bone marrow failure, optic nerve atrophy, arrhythmias and/or cardiomyopathy (dilated or hypertrophic), liver steatosis/fibrosis/cancer, and renal cancer.
Diagnosis/testing.
The diagnosis of isolated MMA is established in a proband by identification of biallelic pathogenic variants in MCEE, MMAA, MMAB, MMADHC, or MMUT or (in some instances) by significantly reduced activity of one of the following enzymes: methylmalonyl-CoA mutase, methylmalonyl-CoA mutase enzyme cofactor 5'-deoxyadenosylcobalamin, or methylmalonyl-CoA epimerase. Because of its relatively high sensitivity, easier accessibility, and noninvasive nature, molecular genetic testing can obviate the need for enzymatic testing in most instances.
Management.
Treatment of manifestations / Prevention of primary manifestations: When isolated MMA is suspected during the diagnostic evaluation due to elevated propionylcarnitine (C3) on a newborn blood spot, metabolic treatment should be initiated immediately, while the suspected diagnosis is being confirmed. Development and evaluation of treatment plans, training and education of affected individuals and their families, and avoidance of side effects of dietary treatment (i.e., malnutrition, growth failure) require a multidisciplinary approach by experienced subspecialists from a specialized metabolic center. The main principles of treatment are to provide supplemental vitamin B12 to those who are known to be vitamin B12 responsive; restrict natural protein, particularly of propiogenic amino acid precursors, while maintaining a high-calorie diet; address feeding difficulties, recurrent vomiting, and growth failure; provide supplemental carnitine to those with carnitine deficiency; reduce propionate production from gut flora; and provide emergency treatment during episodes of acute decompensation with the goal of averting catabolism and minimizing central nervous system injury. In those with significant metabolic instability and/or renal failure, liver and/or renal transplantation may be considered.
Prevention of secondary complications: MedicAlert® bracelets and up-to-date, easily accessed, detailed emergency treatment and presurgical protocols to facilitate care.
Surveillance: Regular evaluations by a metabolic specialist and metabolic dietician; screening laboratory testing, including plasma amino acids, plasma and urine MMA levels, serum acylcarnitine profile and free and total carnitine levels, blood chemistries, and complete blood count at least every six months to one year, or more frequently in infants or in those who are unstable or require frequent changes in dietary management; measurement of renal function at least annually or as clinically indicated; assessment for liver disease at least annually or as clinically indicated; assessment of developmental progress and for signs of movement disorder at each visit; ophthalmology evaluation to monitor for optic atrophy at least annually or as clinically indicated; audiology evaluation at least annually in childhood and adolescence or as clinically indicated.
Agents/circumstances to avoid: Fasting, stress, increased dietary protein, supplementation with the individual propiogenic amino acids valine and isoleucine, nephrotoxic medications or agents, and agents that prolong QTc in the EKG.
Evaluation of relatives at risk: For at-risk newborn sibs when prenatal testing was not performed: in parallel with newborn screening, measure serum methylmalonic acid, urine organic acids, plasma acylcarnitine profile, plasma amino acids, and serum B12; test for the familial isolated methylmalonic acidemia-causing pathogenic variants if biochemistry is abnormal.
Pregnancy management for an affected mother: Monitor for complications including acute decompensation or hyperammonemia, deterioration of renal function, and obstetric complications including preeclampsia and preterm delivery.
Pregnancy management for an unaffected mother with an affected fetus: Oral and intramuscular vitamin B12 has been administered to women pregnant with a fetus with vitamin B12-responsive MMA, resulting in decreased maternal MMA urine output; however, further study of this treatment is needed.
Genetic counseling.
All forms of isolated MMA are inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an isolated MMA-causing pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of inheriting neither of the familial pathogenic variants. Once the isolated MMA-causing pathogenic variants have been identified in an affected family member, molecular genetic carrier testing and prenatal/preimplantation genetic testing are possible.
Clinical Characteristics
Clinical Description
The phenotypes of isolated methylmalonic acidemia (MMA) described below that are associated with the enzymatic subtypes mut0, mut–, cblA, cblB, and cblD-MMA share clinical presentations and a natural history characterized by periods of relative health and intermittent metabolic decompensation, usually associated with intercurrent infections and stress [Zwickler et al 2012]. Each such decompensation can be life threatening. Table 3 reviews the phenotypes, causative genes, enzymatic subtypes, and clinical correlations that will be discussed further in this section.
Table 3.
Phenotype Correlations by Gene and Enzymatic Subtype of Isolated Methylmalonic Acidemia
View in own window
| Methylmalonic Acidemia Phenotype | Gene | Enzymatic Subtype | Clinical Correlation |
|---|
| Infantile / non-B12-responsive 1 |
MMUT
|
mut
0
| Most common & severe form, typically presenting in infancy Higher rate of mortality & neurologic & other multisystem complications than in those w/mut– & cblA subtypes Renal disease may manifest in childhood in ~43%-60%, w/median age of onset 6-11 yrs. 2
|
|
MMUT
|
mut
–
| Onset may occur later, in 1st few mos or yrs of life. Symptoms often incl feeding problems, failure to thrive, hypotonia, & DD. Catastrophic decompensation can occur when diagnosis is delayed, incl injury in basal ganglia → movement disorder. Some persons have isolated renal tubular acidosis or chronic renal failure as primary finding.
|
|
MMAB
|
cblB
| Most affected persons have phenotype that resembles mut0, although certain pathogenic variants may be assoc w/milder phenotype. Higher rate of mortality & neurologic & other multisystem complications than in those w/mut– & cblA sybtypes Chronic renal failure occurs in ~66% & is less frequent than in those w/cblA subtype.
|
| B12-responsive 3 |
MMAA
|
cblA
| If diagnosed early & consistently treated w/injectable B12 4:
If not adherent to diet & injectable B12 therapy: at risk for significant neurologic & multiorgan complications 6 |
| MMADHC 7 | cblD-MMA | Metabolic acidosis, respiratory distress, hyperammonemia, & neurologic symptoms |
|
MMAB
|
cblB
|
|
|
MMUT
|
mut
–
| See Infantile/non-B12-responsive; this phenotype is rarely B12-responsive. |
| MCEE deficiency |
MCEE
|
MCEE
| In general, milder features ranging from no symptoms to severe metabolic acidosis. Not responsive to injectable B12 therapy A rare cause of persistent moderate MMA
|
DD = developmental delay; MCEE = methylmalonyl-coenzyme A epimerase
- 1.
The most common phenotype, which typically presents during infancy
- 2.
- 3.
Sometimes referred to as partial deficiency
- 4.
- 5.
- 6.
- 7.
Effect of Newborn Screening
Decreased early mortality, less severe symptoms at diagnosis, favorable short-term neurodevelopmental outcome, and lower incidence of movement disorders and irreversible cerebral damage were recorded in affected individuals identified through expanded NBS, though a number of infants with the mut0 enzymatic subtype present clinically before the NBS results become available [Leonard et al 2003, Dionisi-Vici et al 2006, Heringer et al 2016]. Limited observations in sibs with the cblA enzymatic subtype suggest that the IQs of the individuals treated from the newborn period were significantly higher than those of their older affected sibs who were diagnosed after the onset of symptoms [Hörster et al 2007].
Common Phenotypes and Associated Features
As described prior to newborn screening (NBS) availability, the common phenotypes and associated features of isolated MMA included the following.
Infantile/non-B12-responsive phenotype (mut0 enzymatic subtype, cblB). The catastrophic neonatal presentation of isolated MMA can result in death despite aggressive intervention. Infants with the B12-responsive mut– enzymatic subtype or cblA can also present with an acute neonatal crisis.
The most common phenotype of isolated MMA presents during infancy. Infants are normal at birth but develop lethargy, tachypnea, hypothermia, vomiting, and dehydration on initiation of protein-containing feeds.
This can rapidly progress to coma due to hyperammonemic encephalopathy, if untreated.
Laboratory findings typically show a severe, high anion-gap metabolic acidosis, ketosis and ketonuria (highly abnormal in neonates and strongly suggestive of an organic aciduria), hyperammonemia, and hyperglycinemia [
Kölker et al 2015a].
Dialysis may be needed especially if hyperammonemia is significant and persistent.
Thrombocytopenia and neutropenia, suggestive of neonatal sepsis, can be seen.
Partially deficient or B12-responsive phenotypes (mut–, cblA, cblB [rare], cblD-MMA). This intermediate phenotype of isolated methylmalonic acidemia can occur in the first few months or years of life.
Affected infants can exhibit feeding problems (typically anorexia and vomiting), failure to thrive, hypotonia, and developmental delay.
Some have protein aversion and/or clinical symptoms of vomiting and lethargy after protein intake.
During such an episode of metabolic decompensation, the child may die despite intensive intervention if prompt treatment specific for MMA (see
Treatment of Manifestations) is not instituted and the symptoms are misdiagnosed (as, e.g., diabetic ketoacidosis) [
Ciani et al 2000].
Before the availability of NBS, or in cases that are false negative on NBS due to borderline C3 elevations, infants with the
cblA or
mut– subtypes would present with a devastating injury in the basal ganglia in the context of acute metabolic crisis / encephalopathy (more specifically lacunar infarcts in the globus pallidus) resulting in a debilitating movement disorder [
Korf et al 1986,
Heidenreich et al 1988].
Individuals with partial enzymatic deficiency (
mut–),
cblA, or
cblB can also present with isolated renal tubular acidosis or chronic renal failure [
Dudley et al 1998,
Coman et al 2006].
Methylmalonyl-coenzyme A epimerase (MCEE) deficiency. Findings in infants/children with biallelic pathogenic variants in MCEE have ranged from complete absence of symptoms to severe metabolic acidosis with increased MMA and 2-methylcitrate and ketones in the urine at initial presentation [Dobson et al 2006, Gradinger et al 2007, Heuberger et al 2019].
Screening of a large cohort of individuals with undefined MMA identified ten individuals with MCEE deficiency with symptoms including metabolic ketoacidosis, hypoglycemia, seizures, developmental delay, and spasticity. Cardiomyopathy was reported in a single individual, with a similarly affected sib with biochemical but no clinical findings [
Heuberger et al 2019].
Individuals with MCEE deficiency were not responsive to B
12 supplementation in vitro or in vivo and urine MMA concentrations ranged between 100 and 600 mmol/mol creatinine (normal: 0.3-1.1 mmol/mol) [
Heuberger et al 2019].
Secondary Complications
Secondary complications can be observed in any enzymatic subtype but may be dependent on the specific subtype and degree of metabolic control and adherence (see Table 3). Despite increased knowledge about isolated MMA and possibly earlier symptomatic diagnosis, isolated MMA continues to be associated with substantial morbidity and mortality [de Baulny et al 2005, Dionisi-Vici et al 2006, Kölker et al 2015b, Tuncel et al 2018] that correlates with the underlying defect [Hörster et al 2007, Hörster et al 2021]. Individuals with the mut0 and cblB subtypes have a higher rate of mortality and neurologic and other multisystem complications than those with the mut– and cblA subtypes. Multiorgan complications associated with secondary mitochondrial dysfunction accumulate with age and were shown to be associated with a higher total protein intake and imbalanced special metabolic food prescription [Manoli et al 2016b, Molema et al 2018, Haijes et al 2019a, Molema et al 2021a].
Therefore, primary and secondary biomarkers are important for monitoring affected individuals and supporting efficacy in therapeutic clinical trials [Longo et al 2022] (see Therapies Under Investigation and Molecular Genetics). As an example, plasma fibroblast growth factor 21 (FGF21) was shown to correlate with disease severity and long-term complications in different cohorts of affected individuals [Manoli et al 2018, Molema et al 2018, Manoli et al 2021].
The major secondary complications include the following.
Intellectual disability. Intellectual disability may or may not be present even in those with severe disease.
In one study about 50% of individuals with
mut0 subtype, 85% with
mut–, 48% with
cblA, and 70% with
cblB had an IQ above 90 [
Hörster et al 2007].
In a natural history study, the mean FSIQ of all individuals with isolated MMA (n=37) was 85.0 ± 20.68, which is in the low-average range (80≤IQ≤89) [
O'Shea et al 2012].
Individuals with cblA (n=7), cblB (n=6), and mut diagnosed prenatally or by NBS (n=3) had mean FSIQs in the average range (90≤IQ≤109).
The age of disease onset, the presence of severe hyperammonemia at diagnosis, and a history of seizures were associated with more severe impairments.
Tubulointerstitial nephritis with progressive impairment of renal function. All individuals with isolated MMA, even those who are mildly affected or who have received a liver allograft [Noone et al 2019], are at risk of developing renal insufficiency [Cosson et al 2009, Kruszka et al 2013, Manoli et al 2013, Morath et al 2013, Dao et al 2021, Hörster et al 2021], which can progress to end-stage renal disease requiring kidney transplantation (see Table 3).
Cystatin-C levels and age-appropriate equations to calculate estimated glomerular filtration rate (GFR) – or preferably, measurement of GFR by iohexol clearance or other methods – should be used for clinical monitoring, due to the fact that creatinine is a late marker of renal dysfunction in individuals with low muscle mass, as is seen in isolated MMA (see
Surveillance). This will allow for earlier referral to nephrology services and initiation of renoprotective measures – including, importantly, blood pressure control.
Renal tubular dysfunction presenting as a decrease in urine concentrating ability and acidification, hyporeninemic hypoaldosteronism, tubular acidosis type 4, and hyperkalemia have been reported in a number of affected individuals [
Dao et al 2021].
Comorbidities of renal disease including anemia, acidosis, hyperuricemia, secondary hyperparathyroidism, osteopenia/osteoporosis, hypertension, and short stature should be monitored regularly by a multidisciplinary care team (see
Surveillance).
Neurologic findings. Some individuals develop a "metabolic stroke" or bilateral lacunar infarction of the basal ganglia during acute metabolic decompensation, which can produce an incapacitating movement disorder.
Pancreatitis. Acute pancreatitis is a well-recognized complication of isolated MMA, with a reported incidence of 10%-27% in different cohorts [Marquard et al 2011, Forny et al 2018, Hwang et al 2021]. It can occur acutely or chronically. Several affected individuals have recurrent pancreatitis episodes. Pancreatitis may be underrecognized because it can manifest nonspecifically with vomiting and abdominal pain. It is therefore recommended that acutely ill individuals with MMA undergo testing for lipase and amylase (see Management).
Growth failure is frequent and multifactorial. It is the result of severe chronic illness and perhaps relative protein malnutrition that is complicated further by chronic renal failure. Many infants are more than three standard deviations below the mean for both length and weight [Manoli et al 2016b].
Functional immune impairment results in an increased susceptibility to severe infections, particularly by fungal and gram-negative organisms. Defects in both humoral and cellular immunity have been documented [Alizadeh Najjarbashi et al 2015, Harrington et al 2016, Altun et al 2022].
Bone marrow failure. During episodes of metabolic decompensation affected individuals can exhibit pancytopenia, with bone marrow hypoplasia and/or dysplasia that most frequently reverts to normal with supportive care [Bakshi et al 2018]. Anemia due to chronic disease or iron deficiency or secondary to progressive renal failure is common. Essential amino acid deficiencies can be a contributing factor in some individuals [Kölker et al 2015b].
Optic nerve atrophy. Late-onset optic atrophy associated with acute or subacute visual loss, resembling the presentation of the mitochondrial disorder Leber hereditary optic neuropathy (LHON), has been reported in 7%-11% of individuals with isolated MMA [Williams et al 2009, Pinar-Sueiro et al 2010, Traber et al 2011, Martinez Alvarez et al 2016] and up to 52% in a cohort of affected Middle Eastern individuals [Al-Owain et al 2019]. Response to antioxidant therapies (idebenone, coenzyme Q10, and vitamin E) has been variable.
Cardiac complications
Arrhythmias or cardiomyopathy (dilated or hypertrophic) have been reported in 10%-20% of individuals with isolated MMA, primarily
mut0 or
mut- (and
cblB subtypes, as well as in the B
12-responsive
cblA subtype) [
Prada et al 2011,
Chao et al 2012,
Hörster et al 2021].
Arterial hypertension associated with chronic kidney disease is common and necessitates monitoring and early intervention for renoprotection [
Kölker et al 2015b,
Park et al 2020].
Additional cardiometabolic risk factors, including obesity, insulin resistance, and hyperlipidemia, need to be monitored regularly to optimize cardiovascular health [
Gancheva et al 2020].
Liver steatosis, fibrosis, and cancer. Progressive liver toxicity associated with elevated transaminases (including GGT) and mild elevations of alpha-fetoprotein (AFP) has been observed in a number of individuals with isolated MMA. Liver ultrasound can show hepatomegaly and/or hyperechoic liver texture. Liver biopsies in three individuals showed steatosis, fibrosis, and (rarely) cirrhosis as early as age eight years [Imbard et al 2018].
Three children had hepatoblastoma (diagnosed at 4 months, 19 months, and 11 years of age).
Two adults had hepatocellular carcinoma (diagnosed at age 22 years and 31 years).
Periodic screening (typically at least annually or as clinically indicated) including liver transaminases, serum AFP level, and liver ultrasound is recommended in individuals with severe MMA subtypes (see
Surveillance).
Renal cancer. A single case of a pediatric renal cell carcinoma has been reported in a female age six years with complete MMUT deficiency (mut0), complicated by renal tubular acidosis, Stage 4 chronic renal disease, and hypercalcemia with increasing parathyroid hormone-related protein. Inactivating somatic variants in TSC2 were identified in the tumor tissue [Potter et al 2017].
Survival in isolated methylmalonic acidemia has improved over time [Matsui et al 1983, van der Meer et al 1994, Baumgarter & Viardot 1995, Nicolaides et al 1998, Kölker et al 2015a]. Five-year survival improved from 33% in the 1970s to more than 80% in the 1990s.
Overall mortality was about 50% for those with the
mut0 enzymatic subtype (median age of death: 2 years) and for the
cblB enzymatic subtype (median age of death: 2.9 years) compared to 40% for the
mut– enzymatic subtype (median age of death: 4.5 years) and only about 5% for the
cblA enzymatic subtype (1 death at age 14 days) [
Hörster et al 2007].
More recent reports from the European Registry and Network for Intoxication Type Metabolic Diseases notes a 6% mortality for
mut MMA (combined
mut- and
mut0 populations) and a 100% survival for those with the B
12-responsive
cblA subtype of MMA [
Hörster et al 2021].
Improvements likely reflect changes in diagnosis and NBS, improved treatment guidelines for acute crises/hyperammonemia, optimized nutrition with gastrostomy tube feeding, access to intensive care, hemodialysis and N-carbamylglutamate for the management of hyperammonemia, as well as earlier referral and better morbidity and mortality associated with solid organ transplantation.
Genotype-Phenotype Correlations
Precise genotype-phenotype correlations are difficult to determine since most affected individuals are compound heterozygotes and many pathogenic variants are not recurrent in the population.
MMAB
MMADHC. Truncating pathogenic variants in the N-terminal region (exons 3, 4) cause isolated methylmalonic aciduria due to a defect in adenosylcobalamin synthesis; pathogenic variants elsewhere in this gene cause the other two biochemical phenotypes (see Genetically Related Disorders).
MMUT. The phenomenon of interallelic complementation makes prediction of genotype/phenotype/enzyme activity difficult because some individuals who have two pathogenic variants can have a mut– enzymatic subtype in the compound state but a mut0 enzymatic subtype in the homozygous state [Acquaviva et al 2005].
Persons with two truncating pathogenic variants usually have the mut0 enzymatic subtype.
Most of the pathogenic variants identified in the N-terminal domain have been associated with
mut0 enzymatic subtype of methylmalonic acidemia [
Acquaviva et al 2005,
Forny et al 2016]
The mut– enzymatic subtype is known to be associated mostly, but not exclusively, with pathogenic variants in the adenosylcobalamin-binding C-terminal domain of the MMUT protein.
The
mut– enzymatic subtype pathogenic variant usually plays a dominant role when in compound heterozygous state with a
mut0 enzymatic subtype pathogenic variant, given a OH-Cbl response in the in vitro assay [
Lempp et al 2007,
Forny et al 2016].
A linker domain spanning residues 482-585 separates the N-terminal, or substrate (methylmalonyl-CoA) binding domain from the C-terminal cobalamin-binding domain. This linker region is less conserved and has a lower frequency of pathogenic variants [
Forny et al 2016].
Table 4.
MMUT Pathogenic Missense Variants and Their Typical Enzymatic Subtype
View in own window
| Mut Enzymatic Subtype (when Homozygous) | DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
|
mut0
| c.19C>T | p.Gln7Ter |
NM_000255.4
NP_000246.2
|
|
mut0
| c.52C>T | p.Gln18Ter |
|
mut0
| c.91C>T | p.Arg31Ter |
|
mut0
| c.278G>A | p.Arg93His |
|
mut0
| c.284C>G | p.Pro95Arg |
|
mut0
| c.313T>C | p.Trp105Arg |
| mut0 1 | c.322C>T 2 | p.Arg108Cys |
|
mut0
| c.397G>A 3 | p.Gly133Arg |
|
mut0
| c.410C>T 4 | p.Ala137Val |
|
mut0
| c.415G>A 3 | p.Asp139Asn |
|
mut0
| c.521T>C | p.Phe174Ser |
|
mut0
| c.572C>A 5 | p.Ala191Glu |
|
mut0
| c.607G>A | p.Gly203Arg |
|
mut0
| c.643G>A 5 | p.Gly215Ser |
|
mut0
| c.655A>T 2, 3, 5 | p.Asn219Tyr |
|
mut0
| c.935G>T | p.Gly312Val |
|
mut0
| c.982C>T 3 | p.Leu328Phe |
|
mut0
| c.1105C>T | p.Arg369Cys |
|
mut0
| c.1106G>A 2, 3, 5, 6 | p.Arg369His |
|
mut0
| c.1280G>A | p.Gly427Asp |
|
mut0
| c.1553T>C | p.Leu518Pro |
|
mut0
| c.1843C>A 3 | p.Pro615Thr |
|
mut0
| c.1867G>A | p.Gly623Arg |
|
mut–
| c.299A>G 4, 6 | p.Tyr100Cys |
|
mut–
| c.566A>T 3, 7 | p.Asn189Ile |
|
mut–
| c.828G>C 3 | p.Glu276Asp |
|
mut–
| c.947A>G 2 | p.Tyr316Cys |
|
mut–
| c.970G>A 2 | p.Ala324Thr |
|
mut–
| c.1097A>G 4 | p.Asn366Ser |
|
mut–
| c.1160C>T 8 | p.Thr387Ile |
|
mut–
| c.1276G>A 2, 3 | p.Gly426Arg |
|
mut–
| c.1277G>A 3 | p.Gly426Glu |
|
mut–
| c.1663G>A 9 | p.Ala555Thr |
|
mut–
| c.1846C>T 2, 6 | p.Arg616Cys |
|
mut–
| c.1898T>G 10, 10 | p.Val633Gly |
|
mut–
| c.1924G>C 6 | p.Gly642Arg |
|
mut–
| c.2020C>T 8 | p.Leu674Phe |
|
mut–
| c.2054T>G 2, 10 | p.Leu685Arg |
|
mut–
| c.2080C>T 4, 6 | p.Arg694Trp |
|
mut–
| c.2099T>A 3, 5 | p.Met700Lys |
| mut– 11 | c.2150G>T 2, 3, 6 | p.Gly717Val |
|
mut–
| c.2206C>T 3 | p.Leu736Phe |
Data in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
mut0 = mut0 enzymatic subtype
mut– = mut– enzymatic subtype
- 1.
Observed in individuals of Mexican/Hispanic descent.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
Observed in individuals of African descent
Prevalence
Several studies have estimated the birth prevalence of isolated methylmalonic acidemia. Urine screening for isolated methylmalonic acidemia in Quebec identified "symptomatic methylmalonic aciduria" in approximately 1:80,000 newborns screened [Sniderman et al 1999].
The aggregate incidence from different newborn screening (NBS) programs in the US is reported as 1:159,614 [Therrell et al 2014, Chapman et al 2018]. A meta-analysis [Almási et al 2019] confirmed that the detection rate of MMA and isolated MMA in North America, Europe and Asia-Pacific regions was <1:100,000, while rates in the Middle East, North Africa, and Japan were higher [Shigematsu et al 2002].
Differential Diagnosis
Other genetic causes of elevated methylmalonic acidemia/aciduria are listed in Table 5. Biochemical findings typically allow differentiation of these disorders from isolated methylmalonic acidemia (MMA).
It is important to note that individuals with cblF or cblJ enzymatic subtypes can have decreased serum vitamin B12 levels (the finding of decreased serum vitamin B12 levels suggests a role for the lysosome in intestinal uptake of ingested cobalamin).
With the exception of cblX deficiency due to variants in HCFC1, which is inherited in an X-linked manner, the disorders summarized in Table 5 are inherited in an autosomal recessive manner.
Table 5.
Genetic Disorders with Methylmalonic Acidemia/Aciduria in the Differential Diagnosis of Isolated Methylmalonic Acidemia
View in own window
| Gene | Disorder | Biochemical Features | Clinical Features |
|---|
|
ABCD4
| cblJ deficiency (See Disorders of Intracellular Cobalamin Metabolism.) | Combined methylmalonic acidemia & hyperhomocysteinemia / homocystinuria; can present w/low serum B12 levels | 5 persons reported: 3 presented neonatally w/poor growth, feeding problems, hypotonia, respiratory distress, bone marrow suppression, & congenital heart defect. 2 presented in early childhood w/hyperpigmentation & premature graying, & transient ischemic attack (in 1 of 2 children). |
|
ACSF3
| Combined malonic & methylmalonic aciduria (OMIM 614265) | High MA & MMA levels in urine or plasma, w/MMA excretion typically higher than MA excretion (MMA/MA >5).
Because C3 (propionylcarnitine) is not ↑, affected infants are not detected by NBS based on a dried blood spot acylcarnitine analysis. 1 | Broad phenotypic spectrum ranging from completely asymptomatic to adults w/neurologic syndromes (seizures, memory problems, psychiatric disease, ±cognitive decline) to children w/a wide range of manifestations (e.g., coma, ketoacidosis, hypoglycemia, FTT, ↑ transaminases, microcephaly, dystonia, axial hypotonia, &/or DD). No biochemical or clinical response to B12 therapy. A largely benign clinical course was reported in an unselected cohort (children-young adult) ascertained through urine NBS in Quebec. 1 |
|
ALDH6A1
| Methylmalonate semialdehyde dehydrogenase deficiency (OMIM 614105) | Extremely variable biochemical phenotypes: may be assoc w/3-hydroxyisobutyric, 3-OH propionic aciduria, 3-aminoisobutyric, & β-alanine, &/or transient methylmalonic acidemia/aciduria 2 | Extremely variable clinical phenotypes incl severe ID, dysmorphic features; assoc w/significant brain myelination defects 2 |
AMN
CUBN
| Imerslund-Grasbeck syndrome (OMIM PS261100) | Low serum B12, combined methylmalonic acidemia & hyperhomocysteinemia / homocystinuria, proteinuria in ~50% of affected persons | Megaloblastic anemia, pallor, FTT, recurrent infections, mild proteinuria. |
|
CD320
| Transcobalamin receptor defect (TcblR) (OMIM 613646) | Identified on NBS w/an ↑ C3 & ↑ C3/C2 ratio, ↑ plasma & urine MMA, ± ↑ homocysteine & normal or mildly ↑ serum vitamin B12 levels 3 | Largely asymptomatic. Normal biochemistry w/parenteral hydroxocobalamin or oral B12 supplementation. Bilateral central retinal artery occlusion assoc w/hyperhomocysteinemia reported in 1 person. Most reported persons are homozygous for NM_016579.3:c.262_264del (p.Glu88del). 3, 4 |
HCFC1
THAP11
ZNF143
| cblX & cblX-like deficiency (See Disorders of Intracellular Cobalamin Metabolism.) | Combined methylmalonic acidemia & hyperhomocysteinemia | IUGR, congenital malformations, severe DD w/significant ID, early-onset intractable seizures; microcephaly, brain malformations, & dysmorphic features in some persons |
|
LMBRD1
| cblF deficiency (See Disorders of Intracellular Cobalamin Metabolism.) | Combined methylmalonic acidemia & hyperhomocysteinemia / homocystinuria; presents w/low serum B12 levels. | Often presents in infancy w/IUGR, poor postnatal growth, feeding difficulties, & DD; may also have stomatitis ± glossitis & congenital heart malformations |
MMACHC
PRDX1
| cblC deficiency (See Disorders of Intracellular Cobalamin Metabolism.) | ↑ plasma concentrations of homocysteine & methylmalonic acid, w/↓ levels of methionine | Frequently assoc w/DD, ID, progressive pigmentary retinopathy, "bull's eye" maculopathy, seizures; highly variable age of onset |
|
MLYCD
| Malonyl-CoA decarboxylase deficiency (OMIM 248360) | Combined methylmalonic & malonic aciduria w/significantly ↑ malonic vs methylmalonic acid levels; ↑ C3DC in acylcarnitine profile; ketotic dicarboxylic aciduria; hypoglycemia 5 | Hypoglycemia, metabolic acidosis, ketosis, cognitive impairment, seizures, microcephaly. Cardiomyopathy (left ventricular non-compaction, dilated or hypertrophic) is the leading cause of morbidity & mortality. 5 |
|
SUCLA2
| SUCLA2-related mtDNA depletion syndrome, encephalomyopathic form w/methylmalonic aciduria (succinyl-CoA ligase deficiency) | Methylmalonic aciduria ranges from 10 to 200 mmol/mol creatinine & is accompanied by ↑ plasma concentrations of lactate, methylcitrate, 3-hydroxyproprionic & 3-hydroxyisovaleric acid, proprionylcarnitine, & C4-dicarboxylic carnitine (C4DC). 6 | Hypotonia, muscle atrophy (presenting at age ~3-6 mos), hyperkinesia, seizures, severe hearing impairment, & growth failure. Leigh syndrome-like disorder, cortical & basal ganglia atrophy, & dystonia. ~30% of affected persons succumb during childhood. |
|
SUCLG1
| SUCLG1-related mtDNA depletion syndrome, encephalomyopathic form w/methylmalonic aciduria (succinyl-CoA ligase deficiency) | Methylmalonic aciduria ranges from 10 to 200 mmol/mol creatinine & is accompanied by ↑ plasma concentrations of lactate, methylcitrate, 3-hydroxyproprionic & 3-hydroxyisovaleric acid, proprionylcarnitine, & C4-dicarboxylic carnitine (C4DC) 6 | Hypotonia, muscle atrophy, feeding difficulties, & lactic acidosis. Affected infants commonly manifest DD/cognitive impairment, growth restriction/FTT, hepatopathy, hearing impairment, dystonia, & hypertonia. Life span is shortened (median survival: 20 mos). |
|
TCN2
| Transcobalamin II deficiency (OMIM 275350) | Combined methylmalonic acidemia & hyperhomocysteinemia. Mostly normal serum B12, but ↓ unsaturated B12 binding capacity & ↓TCII detected by immunoassay. 7 | Pallor, FTT, diarrhea, pancytopenia (can be misdiagnosed as leukemia), recurrent infections, megaloblastic anemia, immunodeficiency, neurologic abnormalities if delayed or inadequate treatment winjectable B12. 7 |
|
ZBTB11
| ZBTB11-related intellectual developmental disorder (OMIM 618383) | Biochemical phenotype similar to ACSF3 deficiency w/high MA & MMA levels in urine or plasma, w/MMA excretion typically higher than MA excretion (MMA/MA >5). Because C3 (propionylcarnitine) is not ↑, affected infants are not detected by NBS based on a dried blood spot acylcarnitine analysis. | DD, ID, FTT, microcephaly, cataracts, brain abnormalities; some persons can have isolated ID & no biochemical phenotype. 8 |
cbl = cobalamin; DD = developmental delay; FTT = failure to thrive; ID = intellectual disability; IUGR = intrauterine growth restriction; MA = malonic acid; MMA = methylmalonic acid; mtDNA = mitochondrial DNA; NBS = newborn screening
- 1.
- 2.
- 3.
- 4.
Polymorphisms in CD320 have been associated with increased risk for neural tube defects in an Irish cohort [Pangilinan et al 2010].
- 5.
- 6.
- 7.
- 8.
"Benign" MMA, "atypical"MMA, and MMA of unknown cause. Newborn screening (NBS) performed on urine rather than dried blood spots (a test method utilized in the province of Quebec and in the early years of the Massachusetts NBS program) identified infants with mild-to-moderate urinary methylmalonic acid excretion. Follow up of such infants revealed resolution in more than 50% of children, as well as an apparently benign, persistent, low-moderate methylmalonic acidemia in some [Giorgio et al 1976, Coulombe et al 1981, Ledley et al 1984, Sniderman et al 1999]. Relatively benign MMA with distal renal tubular acidosis (one sibship [Dudley et al 1998]) and isolated methylmalonic aciduria with normal plasma concentrations have also been reported [Sewell et al 1996, Martens et al 2002].
These older reports were published before the identification of ACSF3 pathogenic variants as a cause of CMAMMA (combined malonic and methylmalonic acidemia; OMIM 614265). Given the high minor allele frequency of known ACSF3 pathogenic variants (MAF ~ 0.005, with a predicted incidence of 1:37,000) and benign clinical phenotypes in some individuals [Levtova et al 2019], it is likely thaT many of these individuals harbor pathogenic variants in ACSF3. In a large cohort of individuals with MMA of unknown cause, 6% of individuals were found to have pathogenic variants in ACSF3, SUCLG1, or TCN2 [Pupavac et al 2016].
"Atypical" MMA has also been reported in an individual with mitochondrial depletion syndrome/complex IV deficiency and combined propionic and methylmalonic acidemia [Yano et al 2003]. The phenotype has similarities to the phenotypes in individuals with SUCLA2 or SUCLG1 deficiency.
Despite extensive genome and RNA sequencing, the genetic cause of isolated MMA and low propionate incorporation remains unknown in many individuals [Abdrabo et al 2020].
Vitamin B12 deficiency. Individuals with vitamin B12 deficiency can have elevated MMA and homocysteine and develop significant hematologic, neurologic, and psychiatric manifestations of B12 deficiency. Serum methylmalonic acid and plasma total homocysteine are more sensitive markers than B12 concentrations for detecting B12 deficiency [Stabler 2013].
Maternal B12 deficiency can produce an MMA syndrome in an infant that ranges from severe encephalopathy to elevated serum concentration of propionylcarnitine (C3) detected by NBS [Chace et al 2001, Campbell et al 2005, Hinton et al 2010, Scolamiero et al 2014]. This metabolic abnormality can also occur in a breastfed infant of a vegan mother, in an infant born to a mother with subclinical pernicious anemia [Marble et al 2008], and in infants born to mothers who have had gastric bypass surgery [Grange & Finlay 1994, Celiker & Chawla 2009, González et al 2016]. The mother does not necessarily have a very low serum concentration of vitamin B12. Intramuscular vitamin B12 replacement therapy to normalize vitamin B12 serum concentration reverses the metabolic abnormality.
It is important to screen pregnant mothers by testing maternal serum B12, as well as serum methylmalonic acid and plasma total homocysteine, especially in all infants with positive NBS for elevated propionylcarnitine (C3) [Hinton et al 2010, Held et al 2022]. The addition of second-tier strategies of measuring methylmalonic/3-OH-propionic/methylcitric and homocysteine in dried blood spots can greatly improve detection of acquired vitamin B12 deficiency during NBS and allow treatment to prevent serious neurologic manifestations that can result from prolonged B12 deficiency in both infant and mother [Gramer et al 2020, Pajares et al 2021].
Reye-like syndrome. A Reye-like syndrome of hepatomegaly and obtundation in the face of a mild intercurrent infection can be seen as an unrecognized presentation of a number of inborn errors of metabolism, including isolated MMA [Chang et al 2000].
Management
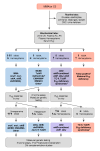
Consensus guidelines on the diagnosis, management, and follow-up for individuals with methylmalonic acidemia were published in 2014 [Baumgartner et al 2014] (full text) and revised in 2021 [Forny et al 2021] (full text). Several additional expert reviews and publications detail management in acute crises and chronic monitoring, treatment of hyperammonemia, dietary practices, and other aspects of clinical care: Ktena et al [2015b], Fraser & Venditti [2016], Manoli et al [2016b], Valayannopoulos et al [2016], Aldubayan et al [2017], Evans et al [2017], Molema et al [2019], Pinto et al [2020], and Molema et al [2021b], among others.
When isolated MMA is suspected during the diagnostic evaluation due to elevated propionylcarnitine (C3) on a newborn blood spot, metabolic treatment should be initiated immediately, while the suspected diagnosis is being confirmed.
Once confirmed, development and evaluation of treatment plans, training and education of affected individuals and their families, and careful monitoring of dietary treatment (to avoid malnutrition, growth failure) require a multidisciplinary approach including multiple subspecialists, with oversight and expertise from a specialized metabolic center.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with isolated MMA, the evaluations summarized Table 6 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 6.
Recommended Evaluations Following Initial Diagnosis of Isolated Methylmalonic Acidemia
View in own window
| Evaluation | Comment |
|---|
| Consultation w/metabolic physician / biochemical geneticist & specialist metabolic dietitian 1 | Transfer to specialist center w/experience in mgmt of inherited metabolic diseases is strongly recommended. Consider short hospitalization at a center of expertise for inherited metabolic conditions to provide caregivers w/detailed education (natural history, maintenance & emergency treatment, prognosis, & risks for acute encephalopathic crises). Review diet/food records w/metabolic dietitian. Provide patient/family w/sick-day diet instructions & emergency treatment letter detailing mgmt plan & specialist contact information (see Table 12).
|
| Assessment of vitamin B12 responsiveness | Generally, 1.0-mg injections (preferably of OHCbl) daily for 3-5 days Obtain >1 baseline & follow-up measures over 10 days to assess for a ↓ in serum & urine methylmalonic acid (>50% ↓ is considered a positive B12 response).
|
Consider screening laboratory testing, which may incl:
Serum vitamin B12 concentration (in newborns; see above for vitamin B12 responsiveness.) Serum chemistry panel incl renal function, liver enzymes 2 CBC w/differential, iron status, folate Arterial or venous blood gas Plasma ammonium & lactic acid concentration Urinalysis & urine ketone measurement Quantitative plasma amino acids Urine organic acids 3 Serum methylmalonic acid & (if available) methylcitrate levels Measurement of free & total carnitine levels Pancreatic enzymes (amylase, lipase) Serum albumin, total protein, & prealbumin to assess for nutritional status
| The choice of screening labs depends on the patient’s current age & clinical status. |
| Cardiac eval, which may incl:
| To assess for hypertension, abnormal QT interval, or other cardiac issues |
| Measure growth parameters (weight, length/height, head circumference). | To assess for failure to thrive, poor growth, &/or short stature |
| Baseline bone age & bone density (DXA) | Assess for evidence of growth failure, need for gastrostomy tube to meet caloric needs, growth hormone treatment. Prevent & treat osteopenia due to low-protein diet, renal osteodystrophy, delayed puberty.
|
| Developmental assessment | Consider referral to developmental pediatrician after newborn period. |
| Consultation w/neurologist | To assess for signs & symptoms of mvmt disorder, seizures, neuropathy Brain imaging (MRI, MRS) in case of abnormal neurologic exam findings
|
| Ophthalmology eval | To assess for optic nerve atrophy, which typically develops in older persons |
| Audiology eval | To assess for hearing loss 4 |
| Consultation w/psychologist &/or social worker | To ensure understanding of diagnosis & assess parental / affected person's coping skills & resources |
| Genetic counseling by genetics professionals 5 | To inform affected persons & families re nature, MOI, & implications of isolated MMA in order to facilitate medical & personal decision making |
CBC = complete blood count; OHCbl = hydroxocobalamin (as opposed to cyanocobalamin); MOI = mode of inheritance
- 1.
After a new diagnosis of isolated methylmalonic acidemia in a child, the closest hospital and local pediatrician should also be informed. The family needs to have an updated emergency treatment letter and plan.
- 2.
Na+, K+, CI–, glucose, urea, creatinine, bicarbonate, AST, ALT, alkaline phosphatase, bilirubin [T/U], lipid panel, and cystatin-C.
- 3.
By gas chromatography and mass spectrometry (GC-MS)
- 4.
Hearing loss may occur in those who have experienced episodes of metabolic decompensation. The risk of hearing loss likely increases with age and can be seen along with optic nerve atrophy.
- 5.
Medical/biochemical geneticist, certified genetic counselor, certified advanced genetic nurse
Treatment of Manifestations
Guidelines developed by professionals across 12 European countries and the US based on rigorous literature evaluation and expert group meetings outline the current management recommendations and areas for further research. See Baumgartner et al [2014] (full text) and Forny et al [2021] (full text).
Table 7.
Routine Daily Treatment in Individuals with Isolated Methylmalonic Acidemia
View in own window
| Principle/Manifestation | Treatment | Considerations/Other |
|---|
|
Vitamin B12 supplementation in those known to be vitamin B12 responsive (See
Table 6
.)
| 1 mg hydroxocobalamin administered by intramuscular injections, 1-3x/wk to daily, depending on metabolic response | Treatment w/cyanocobalamin is contraindicated in persons w/cobalamin C deficiency. |
|
Restriction of natural protein, particularly of propiogenic amino acid precursors 1, while maintaining a high-calorie diet 2
| Safe levels of natural protein per age group should be the aim (see 2007 FAO/WHO/UNU report). The individual protein amount prescribed depends on growth parameters, metabolic stability, & stage of renal failure. A propiogenic amino acid-deficient formula 3, 4 & a protein-free formula 5 (medical foods) are often used to provide addl calories Use medical foods in moderation, w/relative intake of natural protein to propiogenic amino-acid-deficient formula not exceeding a ratio of 1:1
| Natural protein must be carefully titrated to allow for normal growth. 6 As infants grow, total protein load is slowly ↓, based on growth, plasma amino acid concentrations, & plasma & urine methylmalonic acid concentrations. Adjustment of dietary whole (complete)-protein intake (based on lab findings) is required lifelong (see Surveillance). Isolated valine & isoleucine deficiencies may be caused in part by overuse of propiogenic amino-acid deficient formula; individual amino acid supplementation should be avoided (see Agents/Circumstances to Avoid). A ratio of complete protein to medical formula of 60%/40% to 70%/30% of total protein prescription is usually not assoc w/deficiency of valine or isoleucine [Authors, personal observation]. Attn to protein:energy ratio is important; when available, accurate assessment of resting energy expenditure can guide dietary & caloric prescriptions & avoid overfeeding. 7 Plasma amino acids should be drawn ~4 hrs after food intake. Continue protein restriction & dietary monitoring after liver transplantation to avoid extrahepatic disease complications.
|
|
Addressing feeding difficulties, recurrent vomiting, growth failure
| Fundoplication, gastrostomy, or jejunostomy | Adequate provision of dietary information & education to parents, affected persons, & caregivers |
|
Secondary carnitine deficiency
| Oral dosage of 50-100 mg/kg/day, up to ~300 mg/kg/day, of L-carnitine divided into 3-4 doses is common. Dose is adjusted on an individual basis to maintain plasma free carnitine concentration w/in normal age-appropriate reference range.
| Lifelong carnitine supplementation is generally recommended. 8 |
|
Reduction in propionate production from gut flora
| Metronidazole at a dose of 10-15 mg/kg/day typically given 1 wk to 10 days every 1-3 mos | Rotating antibiotic regimens may be considered in some persons. Responsiveness to antibiotic should be determined by a ↓ in serum methylmalonic acid concentration compared to patient's baseline value, or a ↓ in whole-body output of methylmalonic acid on antibiotic therapy by a timed urine collection compared to patient's baseline value. Chronic cyclic antibiotic therapy is not innocuous; it introduces the risk of repopulation w/resistant flora & has been assoc w/peripheral neuropathy. 9
|
- 1.
Propiogenic amino acid precursors include isoleucine, valine, methionine and threonine
- 2.
- 3.
For example, Propimex®-1/2, XMTVI-1/2, or OA-1/2
- 4.
- 5.
For example, Pro-Phree® or Duocal®
- 6.
In patients with low protein tolerance, severe restriction of propiogenic amino acid precursors (isoleucine, valine, methionine, and threonine) can produce a nutritional deficiency state.
- 7.
- 8.
Carnitine may replace the free carnitine pool and enhance the conjugation and excretion of propionylcarnitine. The contribution of propionylcarnitine excretion to the total propionate load is, however, small. The relief of intracellular CoA accretion may be the mechanism by which carnitine supplementation benefits some individuals.
- 9.
This could pose a serious infectious threat and could be especially dangerous to individuals with isolated methylmalonic acidemia, since most deaths are related to metabolic decompensation, often precipitated by infection [Diodato et al 2018, Forny et al 2021].
Table 8.
Treatment of Secondary Complications in Individuals with Isolated Methylmalonic Acidemia
View in own window
| Manifestation | Treatment | Consideration/Other |
|---|
Developmental delay /
Intellectual
disability
| Supportive developmental therapies (may incl PT, OT, speech & cognitive therapies) Coordination of individualized educational plan in school
| Specialists in physiatry, PT, & OT & developmental pediatrician can help address the complex challenges faced by patients & families, maximize functionality, & improve quality of life. 1 |
|
Tubulointerstitial nephritis
| Standard therapy per nephrologist incl mgmt of chronic acidosis (bicitra or sodium bicarbonate), hypertension, anemia, hyperuricemia, & renal osteodystrophy/osteopenia | See Table 13 for recommended surveillance of renal function.
|
End-stage renal
disease
| Standard therapy, which may incl renal replacement therapy such as dialysis | Renal transplantation should be considered ideally before the need for hemodialysis, as those w/MMA are at risk for exacerbation of complications (e.g., hospitalizations, optic nerve disease) |
Anemia /
Bone marrow
suppression 2
| Iron supplementation & erythropoietin may be considered; per nephrologiat | This is a typical complication of chronic renal failure & may resolve after renal transplantation. |
|
Pancreatitis
| Standard therapy incl bowel rest, analgesia, institution of IVF hydration & calories, & careful enteral alimentation w/low-fat preparations | Providing TPN w/intralipids can exacerbate pancreatitis. |
|
Liver disease
| Liver transplantation may be considered. | See also Prevention of Primary Manifestations. |
Optic nerve
atrophy
| No specific treatment is available. | Community vision services |
|
Hearing loss
| Hearing aids may be helpful; per otolaryngologist. | Community hearing services through early intervention or school district |
Growth hormone
deficiency
| Growth hormone therapy | Dose & diet must be carefully adjusted. 3 |
Movement
disorders /
Dystonia
| Standard therapy per neurologist | Antispasmodic medications (trihexyphenidil, baclofen pump) or deep brain stimulation have been used in persons w/severe basal ganglia strokes. |
|
Spasticity
| Orthopedics / physical medicine & rehab / PT/OT incl stretching to help avoid contractures & falls | Consider need for positioning & mobility devices, disability parking placard. |
IVF = intravenous fluids; OT = occupational therapy; PT = physical therapy; TPN = total parenteral nutrition
- 1.
- 2.
- 3.
Table 9.
Emergency Outpatient Treatment in Individuals with Isolated Methylmalonic Acidemia
View in own window
| Manifestation | Treatment | Consideration/Other |
|---|
|
Vomiting, mildly increased catabolism 1
| Carbohydrate supplementation orally or via tube feed 2 Reduce natural protein intake 3 Increase carnitine supplementation 4
| Trial of outpatient treatment at home for up to 12 hours Initiation of sick-day dietary plan Reassessment (~every 2 hours) for clinical changes 5
|
|
Fever
| Administration of antipyretics (acetaminophen) if temperature rises >38.5°C |
|
|
Occasional vomiting
| Antiemetics 6 | Avoid repeat doses of ondansetron as it can prolong the QTc interval on EKG. |
- 1.
Fever <38.5°C (101°F); enteral or gastrostomy tube feeding is tolerated without recurrent vomiting or diarrhea; absence of neurologic symptoms (altered consciousness, irritability)
- 2.
Stringent guidelines to quantify carbohydrate/caloric requirements are available to guide nutritional arrangements in the outpatient setting, with some centers recommending frequent provision of carbohydrate-rich, protein-free beverages every two hours, with frequent reassessment.
- 3.
Some centers advocate additional steps such as reducing natural protein intake to zero or to 50% of the normal prescribed regimen for short periods (<24 hours) in the outpatient setting during intercurrent illness. Protein restriction more than 24-48 hours could lead to catabolism and should be avoided.
- 4.
Temporarily increasing L-carnitine doses (e.g., to 200 mg/kg/day in infants) may be considered.
- 5.
Alterations in mentation/alertness, fever, and enteral feeding tolerance, with any new or evolving clinical features should be discussed with the designated center of expertise for inherited metabolic diseases.
- 6.
Some classes of antiemetics can be used safely on an occasional basis to temporarily improve enteral tolerance of food and beverages at home or during transfer to hospital.
Acute manifestations (e.g., lethargy, encephalopathy, seizures, or progressive coma), often occurring in the setting of intercurrent illness and/or inadequate caloric intake, should be managed symptomatically and with generous caloric support in a hospital setting, with aggressive treatment and supportive care. Immediate consultation with a metabolic/biochemical geneticist is essential. Individuals with MMA can deteriorate rapidly and consultations with neurology, nephrology, and ICU teams are often required during crises (see Table 10).
Table 10.
Acute Inpatient Treatment in Individuals with Methylmalonic Acidemia
View in own window
| Manifestation | Treatment 1 | Consideration/Other |
|---|
|
↑ catabolism (due to fever, perioperative/peri-interventional fasting periods, repeated vomiting/diarrhea)
| Administration of high-energy IV fluids (D10/0.45 or 0.9 saline) at 1.5x maintenance rate to achieve age-appropriate glucose infusion rate (GIR), &, if needed insulin 2, 3 Lipid emulsion is often necessary to provide sufficient calories at a dose of 1- 2 g/kg/day. Address electrolytes & pH imbalances w/bicarbonate bolus, expect need for potassium replacement, as needed. 4 ↓ or omit total protein for ≤24-48 hours. 5 L-carnitine IV supplementation at 50-100 mg/kg/day either BID or QID
| Blood glucose, electrolyte concentrations (particularly sodium, potassium & bicarbonate concentrations), blood gases (w/monitoring of the anion gap), complete blood count & differential, serum lactate, urine ketones & urine output should be followed serially. Central or peripheral TPN, which typically contains glucose & amino acids, & in some instances lipids, may be required. Thiamine may be added, esp in the presence of lactic acidosis. Lipid infusions must be used w/caution due to risk of pancreatitis. Dietary protein should be reintroduced enterally as soon as is feasible given the clinical scenario & may need to be further augmented w/TPN. Nasograstric or orogastric feeding should be strongly considered so that enteral feedings can be reintroduced w/o delay.
|
|
Hyperammonemia
| N-carbamylglutamate (NCG, Carbaglu®) 4, 6 Administer IV sodium benzoate 4, 7; if hyperammonemia persists consider sodium phenylbutyrate/acetate. Hemodialysis or hemofiltration in consultation w/nephrologist may be required in the event of treatment failure (uncontrollable acidosis &/or hyperammonemia).
| A STAT plasma ammonia level should be obtained in the ED or on admission. NCG activates the first step in the urea cycle (CPS1 enzyme) & is effective in lowering ammonia concentration during acute crises in patients w/MMA. Chronic or periodic use has been attempted in cases w/frequent decompensations, but has not obtained regulatory approval. 6 Use of phenylacetate may accentuate low glutamine levels by generating phenylacetylglutamine & deplete 2-ketoglutarate in the TCA cycle.
|
|
New or evolving neurologic symptoms (↓ consciousness, seizures, dystonic/ choreoathetotic movements of face/extremities, changes in visual acuity)
|
| Symptoms of mvmt disorder can evolve gradually & periodic neurologic exam during crises is important for early initiation of PT to preserve function. |
|
Bone marrow failure 8
| Granulocyte-colony stimulating factor may be considered. | Supportive care of the metabolic disease typically results in resolution of this finding. |
BID = twice a day; ED = emergency department; PT = physical therapy; QID = four times a day; TPN = total parenteral nutrition
- 1.
Inpatient emergency treatment should:
(1) take place at the closest medical facility,
(2) be started without delay, and
(3) be supervised by physicians and specialist dieticians at the responsible metabolic center, who should be contacted without delay.
- 2.
Intravenous glucose solutions should preferably consist of D10 or D12.5 (10 - 12.5% dextrose).
- 3.
Use of insulin if hyperglycemia emerges; intravenous insulin given at a starting dose of 0.01-0.02 IU/kg/hour in the event of persistent hyperglycemia (>150-180 mg/dL in plasma, or glucosuria)
- 4.
- 5.
Total protein can be gradually reintroduced depending on the patient's acid-base balance and remaining laboratory values, including ammonia, lactic acid, and plasma amino acids, among others.
- 6.
- 7.
The dose of sodium benzoate is 250 mg/kg as a bolus given over 90-120 min, followed by 250 mg/kg/day for maintenance, administered in 10% dextrose IV (intravenously). The same dose regimen is used for sodium phenylbutyrate (PBA). The maximum dose of sodium benzoate or sodium PBA is 5.5 g/m2 or 12 g/d.
- 8.
May include both bone marrow hypoplasia and/or dysplasia
Transition from pediatric to adult-centered multidisciplinary care settings. As MMA is a lifelong disorder with varying implications according to age, smooth transition of care from the pediatric setting is essential for long-term management and should be organized as a well-planned, continuous, multidisciplinary process integrating resources of all relevant subspecialties. Standardized procedures for transitional care do not exist for isolated MMA due to the absence of multidisciplinary outpatient departments.
Transitional care concepts have been developed in which adult internal medicine specialists initially see individuals with isolated methylmalonic acidemia together with pediatric or adult metabolic experts, dietitians, psychologists, and social workers.
As the long-term course of pediatric metabolic diseases in this age group is not yet fully characterized and there is limited availability of clinics for adults with IEMs, continuous supervision by a center with expertise in metabolic diseases with sufficient resources is essential.
Prevention of Primary Manifestations
See also Table 7, which outlines dietary therapies that can help to prevent a metabolic crisis.
Large case series of affected individuals undergoing elective liver or combined liver/kidney transplantation (as opposed to isolated kidney transplantation) have detailed the indications, peri-operative complications, surgical and anesthesia approaches, anti-rejection regimens, and long-term outcomes in people with MMA undergoing these procedures. Inclusion of enzymatic and genotype information in case series of transplanted individuals allows for better comparisons of the outcomes and genotype-phenotype associations that could inform decisions about the indication and timing of transplantation in individual cases.
Liver transplantation is increasingly offered to younger affected individuals with significant metabolic instability, often in infancy, as a measure to prevent neurologic damage from recurrent metabolic crises associated with hyperammonemia. Referral to centers with experience in managing people with organic acidemias and continued monitoring and dietary therapy are essential for all MMA transplant recipients.
Table 11.
Prevention of Primary Manifestations in Individuals with Isolated Methylmalonic Acidemia
View in own window
| Principle | Prevention | Considerations/Other |
|---|
|
Protection against metabolic instability 1
| Liver transplantation 2 | The underlying biochemical parameters & frequency of metabolic decompensation improve significantly in persons undergoing liver transplantation despite persistent metabolic abnormalities. Liver transplantation is not curative. Patients remain at risk for long-term complications incl renal disease, basal ganglia injury & neurologic complications, & optic nerve atrophy. 3 High CSF concentrations of methylmalonic acid have been reported, especially when protein intake is liberalized. Neurotoxicity due to calcineurin inhibitors has been described in transplanted patients. 4
|
| Kidney transplantation 5 | More mildly affected persons w/mut- or cblA MMA subtypes who have primarily renal failure may undergo isolated renal transplantation. Elective kidney transplantation, before the onset of renal disease, cannot stabilize persons w/mut0 MMA and is not recommended. Double liver kidney transplant offers a higher amount of enzyme activity and allows for better control of kidney rejection. 6
|
- 1.
Most of the metabolic conversion of propionate occurs in the liver, so liver transplantation has the potential to provide enough enzymatic activity to avert severe metabolic crises for the most significantly affected individuals (MMA mut0 subtype) and is performed electively in younger people to avoid recurrent hospitalizations.
- 2.
More than 100 individuals with MMA have undergone living-donor [Kasahara et al 2006, Morioka et al 2007, Kasahara et al 2014, Sakamoto et al 2016, Jang et al 2021] or cadaveric, orthotopic, or partial liver transplantation, or combined liver-kidney transplantation [van 't Hoff et al 1998, van't Hoff et al 1999, Kayler et al 2002, Nyhan et al 2002, Hsui et al 2003, McGuire et al 2011, Niemi et al 2015, Sloan et al 2015, Spada et al 2015, Critelli et al 2018, Jiang & Sun 2019, Chu et al 2019, Pillai et al 2019, Brassier et al 2020, Yap et al 2020, Molema et al 2021b]. Living-related donor transplants from heterozygote (carrier) parents may be associated with higher incidence of steatosis in the graft liver [Irie et al 2020].
- 3.
- 4.
Neurotoxicity from calcineurin inhibitors, including posterior reversible encephalopathy syndrome (PRES), has been reported [Molema et al 2021b].
- 5.
- 6.
Brassier et al [2013], Brassier et al [2020]. One patient died after developing hepatoblastoma, neurologic deterioration accompanied by CSF lactic acidosis, and multiorgan failure; a second patient developed progressive neurologic symptoms; and two others developed metabolic decompensations post-transplant.
Antioxidants. One individual with isolated MMA who was documented to be glutathione deficient after a severe metabolic crisis responded to ascorbate therapy [Treacy et al 1996]. Several studies document increased oxidative stress, glutathione depletion, and specific respiratory chain complex deficiencies in persons with the mut0 enzymatic subtype of MMA [Atkuri et al 2009, Chandler et al 2009, de Keyzer et al 2009, Manoli et al 2013], suggesting a potential benefit of treatment with antioxidants or other mitochondria-targeted therapies in these individuals.
A regimen of coenzyme Q10 and vitamin E has been shown to prevent progression of acute optic nerve involvement in a patient with MMA [Pinar-Sueiro et al 2010]. Thiamine can help with severe lactic acidosis by overcoming pyruvate dehydrogenase inhibition during the treatment of acute metabolic crises (see Table 10). Whether chronic administration of CoQ10, vitamin E, or N-acetylcysteine could prevent long-term complications requires further study [Haijes et al 2019b].
Base replacement. Individuals with MMUT methylmalonic academia (subtype mut0 or mut-) have renal tubular dysfunction and low-grade chronic acidosis that can accelerate the progression of their chronic kidney disease. Sodium bicarbonate or citrate (Bicitra®) replacement aiming for a serum bicarbonate concentration of 22-24 µmol/L, is recommended per standard guidelines for management of chronic kidney disease in children [KDOQI Work Group 2009, Brown et al 2020]. Bicitra has the additional benefit of offering citrate for TCA cycle anaplerosis and was studied in propionic acidemia [Longo et al 2017]. Polycitra contains potassium, which should be monitored closely due to the risk of developing hyperkalemia in individuals with kidney disease.
Prevention of Secondary Complications
One of the most important components of management (as it relates to prevention of secondary complications) is education of parents and caregivers such that diligent observation and management can be administered expediently in the setting of intercurrent illness or other catabolic stressors (see also Tables 9 and 10). Adherence to a low-protein diet and frequent monitoring by the primary metabolic clinic (see Table 13), as well as continued care by other specialists (nephrologist, neurologist, gastroenterologist, cardiologist, and others), is necessary throughout life.
Table 12.
Prevention of Secondary Manifestations in Individuals with Isolated Methylmalonic Acidemia
View in own window
Manifestation/
Situation | Prevention | Considerations/Other |
|---|
Acute
encephalopathic
crisis
| Intense & ongoing education of affected persons & caregivers re natural history, maintenance & emergency treatment, prognosis, & risks of acute encephalopathic crises Treatment protocols & provision of emergency letters or cards to incl guidance for care in the event of illness while on holiday/vacation MediAlert® bracelets/pendants, or car seat stickers Adequate supplies of specialized dietary products (protein-free or propiogenic amino acid deficient formulas); medication required for maintenance & emergency treatment (vitamin B12, carnitine, antipyretics, base replacement, in some cases Carbaglu®,& other medications, as well as gastrostomy or tube feeding supplies) should always be maintained at home.
| Written protocols for maintenance & emergency treatment should be provided to parents & primary care providers/pediatricians, & to teachers & school staff. 1, 2 Emergency letters/cards should be provided summarizing key information & principles of emergency treatment for MMA & containing contact info for the primary treating metabolic center. For any planned travel or vacations, consider contacting a center of expertise near the destination prior to travel dates.
|
Surgery or
procedure (incl
dental) 3
| Notify designated metabolic center in advance of the procedure to discuss perioperative management w/surgeons & anesthesiologists. 4 Emergency surgeries/procedures require planning input from physicians w/expertise in inherited metabolic diseases (w/respect to perioperative fluid & nutritional management).
| Consider placing a "flag" in the affected person's medical record so that all care providers are aware of the diagnosis & the need to solicit opinions & guidance from designated metabolic specialists in the setting of certain procedures. |
- 1.
Essential information including written treatment protocols should be in place in anticipation of possible future need for inpatient emergency treatment.
- 2.
Parents or local hospitals should immediately inform the designated metabolic center if: (1) temperature rises >38.5°C; (2) vomiting/diarrhea or other symptoms of intercurrent illness develop; or (3) new neurologic symptoms occur.
- 3.
- 4.
Perioperative/perianesthetic management precautions may include visitations at specialist anesthetic clinics for affected persons deemed to be at high risk for perioperative complications.
Surveillance
During the first year of life, infants may need to be evaluated as frequently as every week and continued at intervals determined by the frequency of metabolic crises/admissions, growth patterns, and dietary needs. Attention to transition periods (e.g., after the first two years, in adolescence) with other stressors in the family are necessary for modification of dietary prescription.
In addition to regular evaluations by a metabolic specialist and metabolic dietician, the following are recommended. See Table 13.
Table 13.
Recommended Surveillance for Individuals with Isolated Methylmalonic Acidemia
View in own window
| Manifestation | Evaluation | Frequency/Comment |
|---|
|
Poor growth
| Measurement of growth & head circumference | At each visit |
Metabolic
abnormalities
| Screening lab testing, incl:
| At least every 6-12 mos; more frequently in infants or in those who are unstable or require frequent changes in mgmt |
Renal
insufficiency 3
| Measurement of creatinine, cystatin-C, & (if available) GFR (e.g., iohexol plasma decay) 4, 5, 6 Renal imaging Bone mineral density (DXA) 7 Early referral to nephrologist is critical for consideration of renoprotective measures. Monitoring of renal comorbidities by multidisciplinary team
| At least annually, or as clinically indicated |
|
Liver disease
|
| Annually, or as clinically indicated 9 |
Delayed
acquisition of developmental
milestones
| Monitor developmental milestones. 10 | At each visit |
| Neuropsychological testing using age-appropriate standardized assessment batteries, development of an individualized education plan. | As clinically indicated |
| Standardized quality-of-life assessment tools for affected persons & parents/caregivers | As needed |
Movement
disorder
| Assessment for clinical symptoms & signs of mvmt disorders, severity, & responses to treatment, PT, & pharmacologic interventions | At each visit |
Optic nerve
atrophy
| Ophthalmology eval 11 | At least annually, or as clinically indicated |
|
Hearing loss 12
| Audiology eval | At least annually in childhood & adolescence, or as clinically indicated |
CBC = complete blood count; GFR = glomerular filtration rate; PT = physical therapy
- 1.
Frequent monitoring of plasma amino acids is necessary to avoid deficiencies of essential amino acids (particularly isoleucine, valine, and methionine) as a result of excessive protein restriction and the development of acrodermatitis-enteropathica-like cutaneous lesions in methylmalonic aciduria, as in other organic acidurias (glutaric aciduria-I) and amino acid disorders (maple syrup urine disease) [De Raeve et al 1994].
- 2.
Including Na+, K+, CI–, glucose, urea, creatinine, bicarbonate, AST, ALT, alkaline phosphatase, bilirubin (T/U), triglycerides, and cholesterol
- 3.
Comorbidities of renal disease may include anemia, acidosis, hyperuricemia, secondary hyperparathyroidism, osteopenia/osteoporosis, hypertension, and short stature. In addition to cystatin-C, biochemical markers of bone health (Ca, P, alkaline phosphatase, parathyroid hormone, 1.25 dihydroxy-vit D (D3), and uric acid should be assessed periodically.
- 4.
Combined equations based on creatinine and cystatin-C and measured GFR by iohexol clearance or other methods are expected to reflect more accurately the kidney function in people with MMA [Dao et al 2021]. Age-appropriate formulas to estimate GFR are available for both pediatric patients and adult patients.
- 5.
- 6.
- 7.
DXA scan is typically done in older individuals, starting in adolescence, unless there is evidence for renal disease earlier.
- 8.
- 9.
Particularly in individuals with severe MMA subtypes
- 10.
Enrollment in early intervention programs for physical, occupational, and speech therapy is recommended.
- 11.
To assess for optic nerve thinning/pallor
- 12.
Hearing loss can occur in isolated MMA and may be a result of episodes of metabolic decompensation.
Agents/Circumstances to Avoid
The following should be avoided:
Fasting. During acute illness, intake of adequate calories is necessary to arrest/prevent decompensation.
Stress
Increased dietary protein
Nephrotoxic medications or agents (e.g. ibuprofen)
Agents that prolong QTc in the EKG
Evaluation of Relatives at Risk
Evaluation of all at-risk sibs of any age is warranted to allow for early diagnosis and treatment of isolated methylmalonic acidemia.
For at-risk newborn sibs when prenatal testing was not performed: in parallel with newborn screening, measure serum methylmalonic acid, urine organic acids, plasma acylcarnitine profile, plasma amino acids, and serum B12; and test for the familial isolated methylmalonic acidemia-causing pathogenic variants if biochemistry is abnormal.
Prenatal diagnosis of at-risk sibs may allow for prompt treatment of affected newborns at the time of delivery or prenatal administration of vitamin B12 in responsive subtypes, especially cblA.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Affected mother
In pregnancies of affected women with MMA, complications observed included acute decompensation or hyperammonemia, deterioration of renal function, and obstetric complications including preeclampsia, preterm delivery, and cæsarean section [
Raval et al 2015].
Despite high maternal MMA levels, fetal growth and development have been reported to be normal, suggesting negligible teratogenic effects to the fetus from exposure to high methylmalonic acid levels in utero, though long-term follow up with age-appropriate neurocognitive testing is limited [
Wasserstein et al 1999,
Deodato et al 2002].
Pregnancies in transplant recipients are rare and further studies on the health of the offspring are needed [
Marcellino et al 2021].
Unaffected mother with an affected fetus. Oral and intramuscular vitamin B12 has been administered to women pregnant with a fetus with vitamin B12-responsive MMA, resulting in decreased maternal MMA urine output [Ampola et al 1975, van der Meer et al 1990]. These observations notwithstanding, maternal vitamin B12 supplementation for isolated MMA needs further study.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
13-C-propionate breath test. A stable isotope 13-C-propionate breath test has been developed as a surrogate biomarker of disease severity and was shown to correlate with in vitro 14-C-propionate incorporation, isolated MMA subtype, and several disease-related manifestations (rate of progression of chronic renal disease, growth parameters, and cognitive outcomes). Moreover, it showed a response to B12 supplementation or solid organ transplantation [Manoli et al 2021]. It can be used in specialized centers to help prognosticate disease severity and select affected individuals with very low oxidation rates for referral to transplantation or clinical trials testing novel genomic therapies.
Increased understanding of the underlying pathophysiology and the generation of disease-specific animal or cellular models has allowed the development of several novel therapies for isolated MMA [Chandler & Venditti 2019, Luciani et al 2020, Dimitrov et al 2021, Head et al 2022]. The effect of each of these therapeutic approaches on the long-term clinical outcomes of MMA remains to be elucidated.
Review the following for more information on current clinical trials on isolated MMA:
clinicaltrials.gov/ct2/show/NCT04581785
clinicaltrials.gov/ct2/show/NCT04732429
clinicaltrials.gov/ct2/show/NCT04899310
clinicaltrials.gov/ct2/show/NCT04836494
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
All forms of isolated methylmalonic acidemia (MMA) – including complete or partial deficiency of the enzyme methylmalonyl-CoA mutase; defect in transport or synthesis of the methylmalonyl-CoA mutase cofactor, 5'deoxyadenosyl-cobalamin; and deficiency of the enzyme methylmalonyl-CoA epimerase – are inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
The parents of an affected child are presumed to be heterozygous for an MMUT, MMAA, MMAB, MCEE, or MMADHC pathogenic variant.
If a molecular diagnosis has been established in the proband, molecular genetic testing is recommended for the parents of the proband to confirm that both parents are heterozygous for an isolated MMA-causing pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
One of the pathogenic variants identified in the proband occurred as a
de novo event in the proband or as a postzygotic
de novo event in a mosaic parent [
Jónsson et al 2017].
Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband. Uniparental isodisomy has been reported (
MMUT [id(6)pat] and
MMAA [segmental upd(4)mat] [
Abramowicz et al 1994,
Chen et al 2020].)
Heterozygotes (carriers) of a pathogenic variant in an isolated MMA-related gene (i.e., MMUT, MMAA, MMAB, MCEE, or MMADHC) have normal metabolite concentrations.
Sibs of a proband
If both parents are known to be heterozygous for an isolated MMA-causing pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of inheriting neither of the familial pathogenic variants.
Heterozygotes (carriers) of a pathogenic variant in an isolated MMA-causing gene (i.e., MMUT, MMAA, MMAB, MCEE, or MMADHC) have normal metabolite concentrations.
Offspring of a proband. Unless an affected individual's reproductive partner also has isolated MMA or is a carrier, offspring will be obligate heterozygotes (carriers) for a pathogenic variant in an isolated MMA-related gene.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an isolated MMA-causing pathogenic variant.
Carrier Detection
Methods other than molecular genetic testing are not reliable for carrier testing.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the isolated MMA-causing pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Biochemical testing. Both amniotic fluid measurements for methylmalonic acid and cellular biochemical assays (14C propionate incorporation and complementation assays) on cultured fetal cells obtained by amniocentesis or chorionic villus sampling have been used for prenatal testing [Morel et al 2005]. However, due to the limited availability and longer turnaround time for cellular biochemical assays, the preferred method for prenatal diagnosis is molecular genetic testing.
Chapter Notes
Author Notes
Dr Manoli is a pediatrician and clinical and biochemical geneticist. She is a clinician associate investigator and senior staff clinician in the Organic Acid Research Section of the National Human Genome Research Institute, and an attending physician at the National Institutes of Health Clinical Center.
Dr Sloan is a genetic counselor, molecular geneticist, and cytogeneticist. She is a staff scientist in the Organic Acid Research Section of the National Human Genome Research Institute.
Dr Venditti is a pediatrician and clinical and biochemical geneticist. He is a senior investigator and the director of the Organic Acid Research Section at the National Human Genome Research Institute and an attending physician at the National Institutes of Health Clinical Center.
Websites:
www.genome.gov/staff/Charles-P-Venditti-MD-PhD
www.genome.gov/about-nhgri/Division-of-Intramural-Research/Metabolic-Medicine-Branch
Acknowledgments
The authors are supported by the Intramural Research Program of the National Human Genome Research Institute, Bethesda, MD. They have a longitudinal natural history protocol on methylmalonic acidemias and cobalamin disorders at the NIH (Study URL: clinicaltrials.gov/ct2/show/NCT00078078) and a focused interest on translational research in these disorders. Further details and contact information are provided at the group's website: www.genome.gov/Current-NHGRI-Clinical-Studies/Methylmalonic-Acidemia-MMA.
The authors wish to acknowledge the non-profit organizations "Angels for Alyssa," "A Cure for Clark," and the Organic Acidemia Association (OAA), all founded by families and friends of patients with MMA, for their ongoing dedication and support of MMA research.
Revision History
8 September 2022 (ma) Comprehensive update posted live
1 December 2016 (cpv) Revision: Molecular Genetics: MMAB and MMUT
7 January 2016 (me) Comprehensive update posted live
28 September 2010 (me) Comprehensive update posted live
18 January 2007 (cd) Revision: testing for mutations in MMAA and MMAB clinically available
16 August 2005 (me) Review posted live
11 May 2004 (cpv) Original submission
Note: Pursuant to 17 USC Section 105 of the United States Copyright Act, the GeneReview "Isolated Methylmalonic Acidemia" is in the public domain in the United States of America.