Summary
Clinical characteristics.
Disorders of intracellular cobalamin metabolism have a variable phenotype and age of onset that are influenced by the severity and location within the pathway of the defect. The prototype and best understood phenotype is cblC; it is also the most common of these disorders. The age of initial presentation of cblC spans a wide range:
In utero with fetal presentation of nonimmune hydrops, cardiomyopathy, and intrauterine growth restriction
Newborns, who can have microcephaly, poor feeding, and encephalopathy
Infants, who can have poor feeding and slow growth, neurologic abnormality, and, rarely, hemolytic uremic syndrome (HUS)
Toddlers, who can have poor growth, progressive microcephaly, cytopenias (including megaloblastic anemia), global developmental delay, encephalopathy, and neurologic signs such as hypotonia and seizures
Adolescents and adults, who can have neuropsychiatric symptoms, progressive cognitive decline, thromboembolic complications, and/or subacute combined degeneration of the spinal cord
Diagnosis/testing.
The diagnosis of a disorder of intracellular cobalamin metabolism in a symptomatic individual is based on clinical, biochemical, and molecular genetic data. Evaluation of the methylmalonic acid (MMA) level in urine and blood and plasma total homocysteine (tHcy) level are the mainstays of biochemical testing. Diagnosis is confirmed by identification of biallelic pathogenic variants in one of the following genes (associated complementation groups indicated in parentheses): MMACHC (cblC), MMADHC (cblD-combined and cblD-homocystinuria), MTRR (cblE), LMBRD1 (cblF), MTR (cblG), ABCD4 (cblJ), THAP11(cblX-like), ZNF143(cblX-like), or a hemizygous variant in HCFC1 (cblX, which can show a cblC complementation class).
Management.
Treatment of manifestations: Critically ill individuals must be stabilized, preferably in consultation with a metabolic specialist, by treating acidosis, reversing catabolism, and initiating parenteral hydroxocobalamin. Treatment of thromboembolic complications (e.g., HUS and thrombotic microangiopathy) includes initiation of hydroxocobalamin (OHCbl) and betaine or an increase in their doses. Long-term management focuses on improving the metabolic derangement by lowering plasma tHcy and MMA concentrations and maintaining plasma methionine concentrations within the normal range. Gastrostomy tube placement for feeding may be required; infantile spasms, seizures, congenital heart malformations, and hydrocephalus are treated using standard protocols.
Prevention of primary manifestations: Early institution of injectable hydroxocobalamin improves survival and may reduce but not completely prevent primary manifestations. To prevent metabolic decompensations, patients are advised to avoid situations that result in catabolism, such as prolonged fasting and dehydration, and always remain on a weight-appropriate dose of hydroxocobalamin.
Surveillance: During the first year of life, infants may need to be evaluated once or twice a month by a metabolic specialist to assess growth, nutritional status, feeding ability, and developmental and neurocognitive progress. Toddlers and school-age children should be evaluated at least twice a year to adjust medication dosing (hydroxocobalamin, betaine) during growth and evaluate nutritional status. Teens and adults may be seen on a yearly basis. Routine ophthalmologic, neurologic, and cardiac evaluations may also be appropriate.
Agents/circumstances to avoid: Prolonged fasting (longer than overnight without dextrose-containing intravenous fluids); dietary protein intake below the recommended dietary allowance for age or more than that prescribed by a metabolic specialist; methionine restriction including use of medical foods that do not contain methionine; and the anesthetic nitrous oxide.
Evaluation of relatives at risk: If the pathogenic variants in the family are known, at-risk sibs may be tested prenatally to allow initiation of treatment in utero or as soon as possible after birth.
If the newborn sib of an affected individual has not undergone prenatal testing, molecular genetic testing can be performed in the first week of life if the pathogenic variants in the family are known. Otherwise, evaluation of urine organic acids and plasma amino acids, measurement of total plasma homocysteine, serum methylmalonic acid analysis, and acylcarnitine profile analysis can be used for the purpose of early diagnosis and treatment.
Genetic counseling.
The majority of disorders of intracellular cobalamin metabolism are inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. The disorder of intracellular cobalamin metabolism caused by pathogenic variants in HCFC1 is inherited in an X-linked manner. The risk to sibs depends on the genetic status of the mother. If the mother of the proband has an HCFC1 pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected. Females who inherit the pathogenic variant will be heterozygous and will usually not be affected (no affected females have been described to date).
Once the pathogenic variant(s) have been identified in an affected family member, carrier testing for at-risk relatives, molecular genetic prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Diagnosis
The disorders of intracellular cobalamin metabolism result from deficient synthesis of the coenzymes derived from vitamin B12:
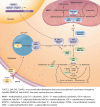
Intracellular metabolism of cobalamin. The intracellular cobalamin metabolism and related pathways – including the complementation groups and corresponding genes – are shown. Endocytosis of cobalamin bound to its blood carrier transcobalamin (more...)
This GeneReview describes inborn errors of cobalamin metabolism, including disorders with combined methylmalonic acidemia and homocystinuria caused by AdoCbl and MeCbl deficiency (Table 1 – B) as well as disorders associated with homocystinuria (MeCbl deficiency) (Table 1 - C). For disorders associated with isolated methylmalonic acidemia (AdoCbl deficiency) (Table 1 – A) see Isolated Methylmalonic Acidemia.
Note: All the disorders of intracellular cobalamin metabolism are inherited in an autosomal recessive manner except for cblX (associated with pathogenic variants in HCFC1), which is inherited in an X-linked manner.
Table 1.
Disorders of Intracellular Cobalamin Metabolism by Biochemical Phenotype
View in own window
| Biochemical Phenotype | Complementation Group 1 | Gene |
|---|
| A. Methylmalonic acidemia (AdoCbl deficiency) 2 |
cblA
|
MMAA
|
|
cblB
|
MMAB
|
| cblD-methylmalonic aciduria | MMADHC 3 |
| B. Combined methylmalonic acidemia and homocystinuria 4 (AdoCbl and MeCbl deficiency) | cblC 5 | MMACHC 6
PRDX1 7
HCFC1 8 (cblX)
THAP11 7
ZNF143 9 |
| cblD-combined | MMADHC 3 |
|
cblF
|
LMBRD1
|
|
cblJ
| ABCD4 10 |
| C. Homocystinuria 4 (MeCbl deficiency) | cblD-homocystinuria | MMADHC 3 |
|
cblE
|
MTRR
|
|
cblG
|
MTR
|
Note: The terms methylmalonic acidemia and methylmalonic aciduria are synonymous, as are the terms hyperhomocysteinemia and homocystinuria.
- 1.
The nomenclature for inherited disorders of intracellular cobalamin metabolism is based on cellular complementation analysis that defines cobalamin groups A-J (cblA - cblJ). The name of each disorder is prefixed with "cbl" (for cobalamin) followed by a unique capital letter for its complementation group determined by somatic cell analysis (e.g., cblC represents complementation group C).
- 2.
- 3.
- 4.
The homocystinuria seen in disorders of intracellular cobalamin metabolism is associated with low/normal methionine in contrast to the homocystinuria seen in cystathionine beta-synthase deficiency, which is associated with high methionine (see ).
- 5.
Individuals with cblX can show a cblC complementation class.
- 6.
- 7.
- 8.
- 9.
- 10.
The diagnosis of a disorder of intracellular cobalamin metabolism in a symptomatic individual is based on clinical, biochemical, and molecular genetic data. With the availability of molecular genetic testing, complementation group analysis is no longer frequently used.
Suggestive Findings
A disorder of intracellular cobalamin metabolism should be suspected in individuals with the following physical and laboratory findings.
Physical findings
In utero. Nonimmune hydrops, cardiomyopathy, intrauterine growth restriction
Newborns. Microcephaly, poor feeding, encephalopathy
Infants. Poor feeding and slow growth, hypotonia, developmental delay, seizures including infantile spasms, infantile maculopathy. Rarely, hemolytic uremic syndrome and obtundation.
Toddlers. Poor growth, progressive microcephaly, cytopenias (including megaloblastic anemia), global developmental delay, encephalopathy, hypotonia, seizures
Adolescents and adults. Neuropsychiatric symptoms, progressive cognitive decline, thromboembolic complications, subacute combined degeneration of the spinal cord
Laboratory findings
Macrocytic anemia with normal B12 levels, thrombocytopenia, and/or neutropenia
Hyperammonemia and/or metabolic acidosis in infancy (rare)
Newborn with abnormal newborn screening based on elevated C3 propionylcarnitine or decreased methionine (See ACMG ACT Sheets for C3 and methionine.)
Establishing the Diagnosis
The diagnosis of a disorder of intracellular cobalamin metabolism is established in a proband with specific biochemical testing results (see Biochemical Testing and Table 2) and confirmed by identification of biallelic pathogenic variants in one of the genes listed in Table 3 – with the exception of HCFC1, in which a hemizygous pathogenic variant is confirmatory. For equivocal molecular genetic testing results, enzymatic testing on skin fibroblasts can be used.
Biochemical Testing
The identification of disorders of intracellular cobalamin metabolism relies on the following testing (Table 2):
Urine organic acid (UOA) analysis to screen for elevation of methylmalonic acid (MMA). Other secondary metabolites such as 3-hydroxypropionate, methylcitrate, and tiglylglycine may be seen transiently in symptomatic affected individuals.
Serum methylmalonic acid analysis is more quantitative than urine organic acid analysis.
Total plasma homocysteine (tHcy) analysis is the preferred method of detecting plasma homocysteine.
Note: Delays in separating serum from plasma after obtaining a blood sample can artificially increase total homocysteine by as much as 10% an hour [
Ubbink 2000,
Refsum et al 2004].
Plasma amino acid (PAA) analysis. Hypomethioninemia, seen in disorders with defective MeCbl synthesis, helps differentiate disorders of intracellular cobalamin metabolism from other causes of homocystinuria, such as cystathionine beta-synthase deficiency, in which methionine level is elevated (see
Differential Diagnosis,
Cystathionine beta-synthase deficiency).
Other findings that can be seen on PAA analysis:
Serum vitamin B12 levels to exclude vitamin B12 deficiency
Note: cblF and cblJ disorders are the exceptions and have been reported to have low B12 levels at diagnosis.
Plasma acylcarnitine analysis to detect elevation of propionylcarnitine (C3) or confirm the elevated propionylcarnitine following newborn screening
Molecular Genetic Testing
Molecular genetic testing approaches can include a combination of gene-targeted testing (concurrent or serial single-gene testing, multigene panel) and comprehensive
genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
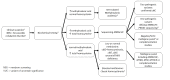
Individuals with the distinctive laboratory findings of a specific disorder of intracellular cobalamin metabolism described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas symptomatic individuals with nonspecific supportive clinical and laboratory findings in whom the diagnosis of a disorder of intracellular cobalamin metabolism has not been considered are more likely to be diagnosed using comprehensive genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of a disorder of intracellular cobalamin metabolism, molecular genetic testing approaches can include single-gene testing or use of a multigene panel.
Single-gene testing. Sequence analysis detects small intragenic deletions/insertions and
missense,
nonsense, and
splice site variants; typically,
exon or whole-gene deletions/duplications are not detected. Perform
sequence analysis of
MMACHC first in individuals with biochemical findings of combined AdoCbl and MeCbl deficiency as it is by far the most commonly associated gene (). If only one
pathogenic variant is found perform gene-targeted
deletion/duplication analysis to detect intragenic deletions or duplications. If single-gene testing is nondiagnostic, a
multigene panel is the next step.
A multigene panel for inherited disorders of intracellular cobalamin metabolism that includes the genes listed in
Table 3 and other genes of interest (see
Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Testing algorithm to confirm the diagnosis of a disorder of intracellular cobalamin metabolism in a proband Footnotes:
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by the wide of array of possible nonspecific clinical findings or an individual has atypical phenotypic features of a disorder of intracellular cobalamin metabolism, comprehensive
genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 3.
Molecular Genetic Testing Used in Disorders of Intracellular Cobalamin Metabolism
View in own window
| Gene 1 | Complementation Group / Disorder | Proportion of Disorders of Intracellular Cobalamin Metabolism Attributed to Pathogenic Variants in Gene | Proportion of Pathogenic Variants 2 Detected by Method |
|---|
| Sequence analysis 3 | Gene-targeted deletion/duplication analysis 4 |
|---|
|
MMACHC
|
cblC
| 80% | 96%-98% 5, 6, 7 | Unknown 8, 9 |
|
MMADHC
| cblD-combined; cblD-homocystinuria | <5% | 22/22 10 | Unknown 8 |
|
MTRR
|
cblE
| <5% | 21/22 11 | Unknown 8 |
|
LMBRD1
|
cblF
| <5% | 23/24 12 | One reported 13 |
|
MTR
|
cblG
| <5% | 64/74 11 | Unknown 8 |
|
ABCD4
|
cblJ
| <<1% | 12/12 14 | Unknown 8 |
|
HCFC1
|
cblX
| <1% | 14/17 15 | Unknown 8 |
|
THAP11
| Not yet defined-cblX-like | <1% | Single case report 16 | Unknown 8 |
|
ZNF143
| Not yet defined-cblX-like | <1% | Single case report 17 | Unknown 8 |
Genes are listed in order of complementation group number.
- 1.
- 2.
- 3.
- 4.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 5.
- 6.
- 7.
A rare complex variant of MMACHC and adjacent genes has been described [Guéant et al 2018].
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
Complementation Group Analysis
Complementation analysis is a method used historically to diagnose the specific defects of intracellular cobalamin metabolism using cultured skin fibroblasts [Watkins & Rosenblatt 1986]. Because of the availability of molecular genetic testing this method is now performed infrequently, but it may still be useful for individuals with equivocal molecular results.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with a disorder of intracellular cobalamin metabolism, the following evaluations are recommended.
In an unstable individual:
Serial metabolic evaluations of blood gases, electrolytes, glucose, ammonia, liver function, total and direct bilirubin, renal function, lactate dehydrogenase, plasma amino acids (methionine), plasma methylmalonic acid (MMA), and total plasma homocysteine (tHcy) to guide acute management until the individual stabilizes
Complete blood count (CBC) with differential to evaluate for megaloblastic anemia or cytopenias
Peripheral blood smear to evaluate for the presence of schistocytes, in the presence of other manifestations of hemolytic uremic syndrome (HUS)
Once the individual becomes stable:
Clinical assessment of growth parameters, head circumference, ability to feed, developmental status, and neurologic status
Laboratory assessment of nutritional status (electrolytes, albumin, prealbumin, plasma amino acids [with careful attention to methionine levels], vitamin levels [including thiamine and 25-hydroxyvitamin D], and trace minerals) and renal function; complete blood count to monitor for cytopenias
EEG and brain MRI in symptomatic individuals
Ophthalmologic examination
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
A set of guidelines for the diagnosis and management of cblC, cblD, cblE, cblF, cblG, cblJ, and MTHFR deficiency have been published [Huemer et al 2017].
Early treatment with hydroxocobalamin injections improves survival and biochemical, hematologic, and microangiopathic symptoms in individuals with cblC. Both newborn screening (NBS) and early treatment are recommended by current guidelines [Huemer et al 2015b, Huemer et al 2017].
Institution of therapy during acute illness results in rapid improvement of clinical, biochemical, and hematologic manifestations in individuals with early- and late-onset cblC [Bartholomew et al 1988, Rosenblatt et al 1997, Bodamer et al 2001, Tomaske et al 2001, Kind et al 2002, Van Hove et al 2002, Fowler et al 2008].
At the Time of Diagnosis
Goals of treatment are to reduce toxic metabolites and avoid low methionine levels.
Parenteral hydroxocobalamin (OHCbl) is the mainstay of therapy and should be instituted immediately when a disorder of intracellular cobalamin metabolism is suspected clinically or following positive NBS for propionylcarnitine.
Cyanocobalamin should not be used as it will not be effective in individuals with cblC.
Avoid treating individuals with a low-protein diet and medical foods designed for the treatment of individuals with
isolated MMA because they contain no methionine, which can further reduce methionine level.
Those with elevated total plasma homocysteine (tHcy) should also receive betaine (250 mg/kg/day) and folate or folinic acid. Betaine has a short effective half-life and should be given in divided doses (optimally divided into 3 or 4 doses per day). It can also be titrated to response while monitoring tHcy and plasma methionine.
Thromboembolic Complications
Thromboembolic complications as a cause of mortality in cblC are likely associated with increasing plasma concentrations of tHcy [Carrillo-Carrasco & Venditti 2012]. The proper management of thromboembolic complications, such as HUS and thrombotic microangiopathy, should include initiation (or dosage increase) of OHCbl and betaine [Van Hove et al 2002, Sharma et al 2007].
Long-Term Management
The goals of long-term management include improving the metabolic derangement by lowering plasma tHcy and methylmalonic acid (MMA) concentrations and maintaining plasma methionine concentrations within the normal range. These are accomplished by the following.
Parenteral hydroxocobalamin (OHCbl). The most experience derives from the treatment of individuals with cblC. Infants are usually started at a daily dose of 1.0 mg (~0.3 mg/kg/day) of OHCbl given intramuscularly or subcutaneously (SQ). Parenteral OHCbl (not the cyanocobalamin form or oral form) is the only effective preparation. Placement of a SQ catheter could be used to minimize cutaneous punctures [Maines et al 2017]; prefilled injections may increase compliance.
Weight-appropriate adjustment of OHCbl to 0.3 mg/kg/day to maintain the dosing in infancy is recommended and can be attained by the ability to concentrate OHCbl up to 30 mg/mL [Carrillo-Carrasco et al 2012]. Further titration of the dose may be empirically adjusted as needed for worsening clinical manifestations [Van Hove et al 2002] or for metabolic control of plasma tHcy, MMA, or methionine [Carrillo-Carrasco et al 2009]. Biochemical as well as clinical improvement has been demonstrated after increasing doses of OHCbl [Matos et al 2013], although clinical trials are needed to demonstrate long-term clinical outcomes [Dionisi-Vici et al 2013].
Betaine. Oral betaine (starting at ~250 mg/kg/day) is recommended in those with elevated plasma total homocysteine as it facilitates conversion of homocysteine to methionine through betaine homocysteine methyltransferase.
Folate and folinic acid. Folic acid and folinic acid can potentially augment remethylation and may help improve plasma tHcy and methionine concentrations. Folinic acid may be preferred as it crosses the blood-brain barrier more efficiently than folic acid. The adult dose of folate is 1.0 mg by mouth per day, titratable down to 0.5 mg for maintenance. Doses for children and infants are available in the Harriet Lane Handbook [Tschudy & Arcara 2012] and other common reference texts.
Dietary management. Individuals may be able to tolerate a normal diet.
Other Therapeutic Considerations
The following have not been fully validated:
Methionine supplementation. Hypomethioninemia is usually responsive to appropriate treatment with OHCbl, betaine, and optimal dietary management. The need for exogenous methionine supplementation may be minimized by these strategies, as the efficacy of this therapy is uncertain [
Smith et al 2006].
Pyridoxine. Vitamin B
6 is a cofactor for
cystathionine beta-synthase and therefore has been proposed as a means of maximizing the removal of homocysteine. Persons with disorders of intracellular cobalamin metabolism generally do not respond to pyridoxine unless they have a dietary deficiency.
Levocarnitine. Indicated for low plasma carnitine levels. Most individuals take 50-200 mg/kg/day to aid in the removal of excess propionyl-CoA groups.
Metanx. A supplement that contains L-methylfolate (active form of folate), methylcobalamin, and pyridoxal-5'-phosphate (active form of vitamin B6) is prescribed as a treatment for neuropathy. It may be used in individuals with cobalamin disorders (in particular teens and adults with clinical indication for neuropathy) and also considered in all individuals with cobalamin disorders to replace individual folate and B6 supplements.
Treatment of infantile spasms, seizures, congenital heart malformations, and hydrocephalus is done in a routine manner.
Prevention of Primary Manifestations
Early institution of injectable hydroxocobalamin improves survival and may reduce but not completely prevent primary manifestations.
To prevent metabolic decompensations, affected individuals should be advised to avoid situations that result in catabolism, such as prolonged fasting and dehydration, and always remain on a weight-appropriate dose of hydroxocobalamin. Of note, during an intercurrent illness individuals may be treated with glucose-containing IV fluid.
Flu prevention (i.e., immunization) should be a routine part of health maintenance.
Surveillance
The following evaluations are performed at different intervals depending on age and disease severity:
During the first year of life, infants may need to be evaluated once or twice a month by a metabolic specialist.
Toddlers and school-age children should be evaluated at least twice a year to adjust medication dosing (hydroxocobalamin, betaine) during growth and to evaluate nutritional status.
Teens and adults may be seen on a yearly basis.
Clinical evaluation should assess the following:
Growth including weight, linear growth, and head circumference
Nutritional status
Feeding ability
Developmental and neurocognitive progress, as age-appropriate
Laboratory evaluation should include the following:
Metabolic studies including urine organic acids, serum methylmalonic acid analysis, plasma amino acids (methionine), plasma tHcy concentration
CBC to monitor for cytopenias
Nutritional studies, if indicated: electrolytes, albumin, prealbumin, plasma amino acids, vitamin levels (including thiamine and 25-hydroxyvitamin D), essential fatty acids, and trace minerals
Routine evaluations should include the following:
Ophthalmologic evaluation for retinal and optic nerve changes in those with
cblC and
cblG and if visual symptoms are present. Recommendations for the ophthalmologic follow up of individuals with
cblC and related disorders have been published [
Weisfeld-Adams et al 2015].
Neurologic evaluations for early signs of developmental delays, behavioral disturbances, seizures, and myelopathy
Brain MRI and/or EEG as clinically indicated
Echocardiogram
Agents/Circumstances to Avoid
Potentially exacerbating circumstances:
Prolonged fasting (longer than overnight without dextrose-containing intravenous fluids)
Dietary protein intake below the recommended dietary allowance (RDA) for age
Dietary protein intake greater than that prescribed by a metabolic specialist especially in individuals with cblC, cblD-combined, cblF, or cblJ
Medical foods. Medical foods given to infants with
isolated methylmalonic acidemia do not contain methionine and should be avoided as the decreased methionine intake may worsen hypomethioninemia and long-term use may contribute to poor head and linear growth [
Manoli et al 2016,
Ahrens-Nicklas et al 2017], among other complications.
Evaluation of Relatives at Risk
If the pathogenic variant(s) in the family are known, at-risk sibs may be tested prenatally to allow initiation of treatment in utero or as soon as possible after birth.
If the newborn sib of an affected individual has not undergone prenatal testing, molecular genetic testing can be performed in the first week of life if the pathogenic variant(s) in the family are known. If the pathogneic variant(s) are not known, evaluation of urine organic acids and plasma amino acids, measurement of total plasma homocysteine, serum methylmalonic acid analysis, and acylcarnitine profile analysis can be used for the purpose of early diagnosis and treatment.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
A good pregnancy outcome was reported in two women with cblC [Brunel-Guitton et al 2010, Liu et al 2015]. In addition, an unaffected infant was born to an asymptomatic woman with cblC who was diagnosed following her child's positive NBS for low carnitine [Lin et al 2009].
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Disorders of intracellular cobalamin metabolism caused by pathogenic variants in ABCD4, LMBRD1, MMACHC, MMADHC, MTR, MTRR, THAP11, or ZNF143 are inherited in an autosomal recessive manner.
The disorder of intracellular cobalamin metabolism caused by pathogenic variants in HCFC1 is inherited in an X-linked manner.
A single report of a deceased child with a paternally inherited LMBRD1 pathogenic variant and a maternally inherited MTR pathogenic variant suggests that digenic inheritance may also be possible [Farwell Gonzalez et al 2015].
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
Sibs of a proband
Offspring of a proband. The offspring of an individual with a disorder of intracellular cobalamin metabolism are obligate heterozygotes (carriers) for an ABCD4, LMBRD1, MMACHC, MMADHC, MTR, MTRR, THAP11, or ZNF143 pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an ABCD4, LMBRD1, MMACHC, MMADHC, MTR, MTRR, THAP11, or ZNF143 pathogenic variant.
Carrier Detection
Molecular genetic carrier testing for at-risk relatives requires prior identification of the ABCD4, LMBRD1, MMACHC, MMADHC, MTR, MTRR, THAP11, or ZNF143 pathogenic variants in the family.
Biochemical testing is not reliable for carrier detection.
X-Linked Inheritance – Risk to Family Members
Parents of a male proband
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother.
If the mother of the
proband has an
HCFC1 pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected. Females who inherit the pathogenic variant will be
heterozygous and will usually not be affected (no affected females have been described to date).
If the
proband represents a
simplex case (i.e., a single occurrence in a family) and if the
HCFC1 pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is slightly greater than that of the general population (though still <1%) because of the theoretic possibility of maternal
germline mosaicism.
Offspring of a proband. Affected males transmit the HCFC1 variant to:
Other family members. The proband's maternal aunts may be at risk of being heterozygotes (carriers) for the HCFC1 pathogenic variant, and the aunts' offspring, depending on their sex, may be at risk of being heterozygotes for the pathogenic variant (carrier females) or of being affected (males).
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the pathogenic variant(s) have been identified in an affected family member, molecular genetic prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Biochemical testing. If the pathogenic variant(s) have not been identified in an affected family member, prenatal testing for pregnancies at risk for a disorder of intracellular cobalamin metabolism may be possible using these methods:
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Disorders of Intracellular Cobalamin Metabolism: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
OMIM Entries for Disorders of Intracellular Cobalamin Metabolism (View All in OMIM)
View in own window
|
156570 | 5-@METHYLTETRAHYDROFOLATE-HOMOCYSTEINE S-METHYLTRANSFERASE; MTR |
|
236270 | HOMOCYSTINURIA-MEGALOBLASTIC ANEMIA, cblE COMPLEMENTATION TYPE; HMAE |
|
250940 | HOMOCYSTINURIA-MEGALOBLASTIC ANEMIA, cblG COMPLEMENTATION TYPE; HMAG |
|
277380 | METHYLMALONIC ACIDURIA AND HOMOCYSTINURIA, cblF TYPE; MAHCF |
|
277400 | METHYLMALONIC ACIDURIA AND HOMOCYSTINURIA, cblC TYPE; MAHCC |
|
277410 | METHYLMALONIC ACIDURIA AND HOMOCYSTINURIA, cblD TYPE; MAHCD |
|
300019 | HOST CELL FACTOR C1; HCFC1 |
|
309541 | METHYLMALONIC ACIDURIA AND HOMOCYSTINURIA, cblX TYPE; MAHCX |
|
602568 | METHIONINE SYNTHASE REDUCTASE; MTRR |
|
603214 | ATP-BINDING CASSETTE, SUBFAMILY D, MEMBER 4; ABCD4 |
|
603433 | ZINC FINGER PROTEIN 143; ZNF143 |
|
609119 | THAP DOMAIN-CONTAINING PROTEIN 11; THAP11 |
|
609831 | METABOLISM OF COBALAMIN ASSOCIATED C; MMACHC |
|
611935 | METABOLISM OF COBALAMIN ASSOCIATED D; MMADHC |
|
612625 | LMBR1 DOMAIN-CONTAINING PROTEIN 1: LMBRD1 |
See . For detailed summaries of gene and protein information for the genes discussed in this section, see Table A, Gene.
HCFC1
Gene structure.
HCFC1 is a gene of about 24 kb; it comprises 26 exons.
Pathogenic variants. Five missense variants were identified in 14 individuals with cblX: c.202C>G, c.217G>A, c.218C>T, c.343G>A, and c.344C>T [Yu et al 2013]. These variants were located in the first two Kelch domains, which are conserved ~50-amino-acid sequences that interact to form a β-propeller structure and are involved in protein-protein interactions. An additional variant, c.307T>C, was reported in another family [Gérard et al 2015].
Other HCFC1 variants have been reported in individuals with intellectual disability with or without congenital malformations, in the absence of known biochemical abnormalities (although biochemical testing has not always been performed) [Huang et al 2012, Jolly et al 2015, Koufaris et al 2016].
Variants listed in Table 5 are only those HCFC1 variants reported to cause a cobalamin disorder phenotype.
Table 5.
HCFC1 Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.202C>G | p.Gln68Glu |
NM_005334.2
NP_005325.2
|
| c.217G>A | p.Ala73Thr |
| c.218C>T | p.Ala73Val |
| c.307T>C | p.Tyr103His |
| c.343G>A | p.Ala115Thr |
| c.344C>T | p.Ala115Val |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
HCFC1 encodes host cell factor C-1 (aka HCF-1), a transcriptional coregulator. The protein is 2,035 amino acids long and is known to interact with transcription factors including those encoded by THAP11 and ZNF143. The HCFC1 complex has been reported to regulate hundreds of genes [Dejosez et al 2010].
Abnormal gene product. Fibroblasts from individuals with p.Ala73Val and p.Ala115Val showed decreased MMACHC RNA and protein expression, suggesting the failure of mutated HCFC1 protein to regulate MMACHC expression – potentially the result of disrupted protein-protein interactions via the Kelch domains.
LMBRD1
Gene structure.
LMBRD1 is a gene of 121 kb; it comprises 16 exons [Rutsch et al 2009].
Pathogenic variants.
Rutsch et al [2009] studied 12 unrelated individuals with cblF confirmed by complementation analysis. A common variant, c.1056delG, was seen in 18/24 independent alleles; this variant causes a frameshift yielding a premature stop codon in exon 11 [Rutsch et al 2009]. Altogether five different DNA variants accounted for 22 of 24 observed pathogenic variants. Eight additional variants have been reported including small deletions, splice site variants, and a large (6-kb) deletion, the latter described by Miousse et al [2011].
Table 6.
LMBRD1 Pathogenic Variants Discussed in This GeneReview
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. The probable lysosomal cobalamin transporter LMBR1 domain-containing one protein (LMBD1) is 540 amino acids and has a predicted molecular weight of 61.3 kd (the longest isoform). The predicted protein structure includes nine transmembrane regions and multiple potential glycosylation sites. The protein has been shown by immunocytofluorescence to colocalize with the lysosomal marker LAMP1. The protein is predicted to be a lysosomal membrane transporter [Rutsch et al 2009] and interacts in a complex with ABCD4 (cblJ) [Fettelschoss et al 2017].
Abnormal gene product. Pathogenic variants cause the defective release of cobalamin from lysosomes.
MMACHC
Gene structure.
MMACHC is 11 kb and comprises four exons [Lerner-Ellis et al 2006].
Pathogenic variants. More than 90 variants have been identified in persons with cblC. Table 7 includes variants common in certain populations, such as c.609G>A, which is common in individuals of Chinese ancestry. The most common variant, present in an estimated 30%-50% of alleles, is c.271dupA, historically associated with infantile-onset disease.
Table 7.
MMACHC Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change 1 | Predicted Protein Change | Reference Sequences |
|---|
| c.271dupA 2 | p.Arg91LysfsTer140 |
NM_015506.2
NP_056321.2
|
| c.331C>T 3 | p.Arg111Ter |
| c.347T>C | p.Leu116Pro |
| c.394C>T 4 | p.Arg132Ter |
| c.440G>C | p.Gly147Ala |
| c.482G>A 5 | p.Arg161Gln |
| c.609G>A 6 | p.Trp203Ter |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
- 2.
- 3.
- 4.
- 5.
Associated with milder disease, and may be more common in individuals of Hispanic descent [Almannai et al 2017]
- 6.
Recently, three individuals who are double heterozygous for pathogenic variants in MMACHC and PRDX1 have been identified. PRDX1 is a neighboring gene on chromosome 1 transcribed from the reverse strand. Variants identified in PRDX1 located at the intron 5 splice acceptor site caused skipping of exon 6, transcription of antisense MMACHC, and hypermethylation of the MMACHC promoter/exon 1, resulting in no gene expression from that allele [Guéant et al 2018].
Normal gene product. The methylmalonic aciduria and homocystinuria type C (MMACHC) protein has 282 amino acids and a predicted molecular weight of 31.7 kd. The MMACHC protein has been crystallized and in vitro studies have shown that MMACHC removes the upper B-axial ligand of cobalamin derivatives (e.g., CN, OH, Ado, Me) through dealkylation or decyanation using glutathione; it is also thought to be involved in intracellular trafficking of cobalamins [Koutmos et al 2011]. MMACHC resides in a complex with MMADHC and other proteins in the pathway [Gherasim et al 2013, Bassila et al 2017].
Abnormal gene product. Defects in MMACHC disrupt its ability to process newly internalized cobalamins in the cytosol [Hannibal et al 2009].
MMADHC
Gene structure.
MMADHC, previously known as C2orf25, is 18 kb and comprises eight exons (NM_015702.2) [Coelho et al 2008].
Pathogenic variants. There are no clearly identified common disease-causing variants; 13 variants have been reported to date. Pathogenic variants in MMADHC can result in three different phenotypes – cblD-MMA (AdoCbl deficiency), cblD-Hcy (MeCbl deficiency), and cblD-combined (AdoCbl and MeCbl deficiency). The pathogenic variants described to date are either missense or truncating (nonsense, splice, or frameshift) variants.
Normal gene product. The MMADHC product is predicted to have 296 amino acids with a calculated molecular mass of 32.8 kd (NP_056517.1) [Stucki et al 2012]. There is an N-terminal mitochondrial leader sequence [Coelho et al 2008]. The C-terminus is thought to guide vitamin B12 to methionine synthase [Plesa et al 2011]. In vitro studies suggest that MMADHC does not directly bind cobalamin but may act as a chaperone given that it binds to MMACHC [Deme et al 2012, Gherasim et al 2013]. The crystal structure showed that MMADHC is a modified nitroreductase fold with structural similarity to MMACHC [Froese et al 2015, Yamada et al 2015].
Abnormal gene product. The type and location of pathogenic variants within the protein is thought to determine whether the synthesis of AdoCbl, MeCbl, or both is affected. Variants in the region encoding the mitochondrial targeting sequence (amino acid position 1-61) affect AdoCbl synthesis.
MTR
Gene structure.
MTR is 105.24 kb and comprises 33 exons [Brody et al 1999].
Pathogenic variants. About 40 pathogenic variants have been identified. The most common disease-causing allele in persons with cblG is a missense variant, c.3518C>T, accounting for 40% of alleles [Watkins et al 2002].
A subset of severe pathogenic variants (including frameshifting deletions and nonsense variants) [Watkins et al 2002] and a variant resulting in a cryptic splice site are thought to result in premature translation termination and mRNA instability [Wilson et al 1998, Watkins et al 2002].
Leclerc et al [1996] identified two pathogenic variants specifically near the cobalamin-binding domain.
Table 8.
MTR Pathogenic Variants Discussed in This GeneReview
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. The MTR enzyme has 1,265 amino acids and weighs 140.5 kd. There are at least three functional domains:
The 38-kd C-terminal
domain binds AdoMet.
A
domain comprising amino acids 650 to 896 includes the binding domain for the required cofactor methylcobalamin.
The 70-kd N-terminal
domain binds homocysteine and methyltetrahydrofolate.
The latter two activities may be on separate domains within this region [Goulding et al 1997].
Abnormal gene product. Pathogenic variants are expected to decrease enzymatic activity.
MTRR
Gene structure. The longest transcript variant of MTRR (NM_002454.2) comprises 15 exons and encodes the shorter protein isoform of 698 amino acids (NP_002445.2). In comparison, the transcript variant NM_024010.2 uses an alternate splice site in the 5' region and initiates translation at an alternate start codon. The encoded protein isoform NP_076915.2 has 725 amino acids with a longer N-terminus.
Pathogenic variants. More than 25 variants in MTRR have been reported.
The c.1361C>T variant is common in persons of Iberian ancestry. Reports suggest a milder
phenotype with no neurologic involvement [
Zavadáková et al 2005].
Other variants have been described [Leclerc et al 1999, Zavadáková et al 2002]. Most reported variants have been nonconservative missense variants.
Table 9.
MTRR Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change (Alias 1) | Predicted Protein Change | Reference Sequences |
|---|
c.903+469T>C
(903_904ins140) | -- |
NM_002454.2
NP_002445.2
|
| c.1361C>T | p.Ser545Leu |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
Normal gene product. The MTRR protein isoform NP_002445.2 has 698 amino acids. MTRR shares homology with human cytochrome P450 reductase [Leclerc et al 1999]. MTRR has some chaperone-like activity with regard to MTR [Yamada et al 2006]. The longer N-terminus isoform NP_076915.2 was initially predicted to have a mitochondrial leader sequence [Leclerc et al 1999], but in vitro experiments suggest that the protein is primarily cytosolic [Froese et al 2008].
Abnormal gene product. Pathogenic variants are expected to decrease enzymatic activity.
THAP11
Gene structure. The THAP11 transcript NM_020457.2 is ~2 kb and contains a single exon.
Pathogenic variants. A single variant, NM_020457.2:c.240C>G (p.Phe80Leu), was identified in the homozygous state in a single individual with a cblX-like clinical and biochemical phenotype [Quintana et al 2017].
Table 10.
THAP11 Variants Discussed in This GeneReview
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org).See Quick Reference for an explanation of nomenclature.
Normal gene product. The transcript (NM_020457) encodes a 314-amino-acid protein (NP_065190.2). THAP11 (THAP containing protein 11) is a transcription factor located in a complex with HCFC1, ZNF143, and other proteins that regulate the expression of hundreds of genes including MMACHC.
Abnormal gene product. The variant reported may interfere with DNA binding or disrupt the interactions with HCFC1, ZNF143, and other proteins in the complex, resulting in decreased expression of MMACHC and other genes.
ZNF143
Gene structure. The gene is 67 kb and is composed of 16 exons.
Pathogenic variants. The two variants shown in Table 11 have been reported in a single case [Pupavac et al 2016].
Table 11.
ZNF143 Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | ReferenceSequences |
|---|
| c.851T>G | p.Leu284Ter |
NM_003442.5
NP_003433.3
|
| c.1019C>T | p.Thr340Ile |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org).See Quick Reference for an explanation of nomenclature.
Normal gene product. The longest transcript is 2.9 kb (NM_003442) encoding a protein of 638 amino acids. ZNF143 is a transcription factor located in a complex with HCFC1, THAP11, and other proteins that regulate the expression of hundreds of genes.
Abnormal gene product. The variants reported may interfere with DNA binding or disrupt the interactions with HCFC1, ZNF143, and other proteins in the complex, resulting in decreased expression of MMACHC and other genes.