This title is an author manuscript version first made accessible on the NCBI Bookshelf website July 2, 2012.
Ionotropic glutamate receptors are ligand-gated integral transmembrane ion channel proteins that mediate fast synaptic transmission at the vast majority of excitatory synapses in the central nervous system. Glutamate receptor ion channels are tetrameric assemblies of subunits encoded by 18 genes. Tremendous advances in understanding how structure drives function of ionotropic glutamate receptors have been gained over the past decade by the combination of X-ray crystallography and site-directed mutagenesis. Likewise, some of the mechanisms (transcriptional, translational, and post-translational) underlying seizure-induced changes in expression of glutamate receptors have been elucidated. A wealth of new pharmacologic reagents, particularly allosteric receptor modulators, have been introduced that can facilitate study of the roles of specific glutamate receptors in epilepsy. The proposal that reactive astrocytes release glutamate, which then acts to synchronize neuron firing within local microdomains, has been developed within the last decade. The consequences of seizure-induced downregulation of glutamine synthase for both inhibitory and excitatory neurotransmission have received much attention. These and other topics are discussed.
The ionotropic glutamate receptors are ligand-gated ion channels that mediate the vast majority of excitatory neurotransmission in the brain. The past twenty years have been a golden age for glutamate receptor research. Even before that, in the early 1980’s the invention of the first selective antagonists for what would come to be known as NMDA receptors1 triggered a flood of investigations as the realization grew that NMDA receptors were critically involved in synaptic plasticity, learning, creation of the proper wiring diagram of the brain during development, excitotoxicity, and a host of neurological disorders involving aberrant circuitry organization including epilepsy (reviewed in 2,3). Cloning of the first glutamate receptor subunit was reported in December 1989,4 and within the next two years an additional 15 subunits were cloned.3,5 The subsequent application of molecular and gene ablation technologies have revealed a wealth of subtlety regarding control of synaptic transmission highlighted, perhaps, by a resurgence of interest in how excitatory input patterns to GABAergic interneurons regulate synchronous firing throughout the brain. Over the past decade our understanding of how these receptors work has been brought to the structural level by successful crystallization of numerous glutamate receptor subunits (see 6). Here I review the functional properties of glutamate receptors and discuss recent data pointing to their potential roles in epilepsy. But first, a word on:
NOMENCLATURE
The rapid growth of literature on glutamate receptors throughout the 1990s predictably spawned multiple names for the same subunit cloned in different species, or sometimes cloned in the same species but nearly simultaneously in different laboratories. The resulting confusion has abated slowly, but a strong effort has been mounted recently to replace the older names for glutamate receptor subunits by International Union of Basic and Clinical Pharmacology (IUPHAR) names7 (see http://www.iuphar-db.org/LGICNomenclature.jsp). The IUPHAR nomenclature, which was designed to harmonize with the gene names and will be used throughout this chapter, is presented in Table 1. Several excellent reviews have appeared in recent years that complement and extend the next three sections.6,8–13
Table 1
Glutamate receptor nomenclature.
GLUTAMATE RECEPTOR STRUCTURE
Ionotropic glutamate receptors are tetrameric protein complexes made up of four subunits surrounding an ion channel pore. Each protein complex draws subunits from the same receptor subfamily (AMPA, kainate, NMDA), with subunit assembly between subfamilies apparently strictly prohibited. Each subunit sports three transmembrane domains and a membrane reentrant loop (M2) that results in a cytoplasmically located carboxy terminus (Figure 1A). The cytoplasmic location of the C-terminus is important because residues near the C-terminus interact with numerous intracellular scaffolding and trafficking proteins that regulate both insertion of the receptors into the postsynaptic membrane and activity-triggered recycling.6

How structure drives molecular function of the ionotropic glutamate receptors is discussed in great detail in ref. 6. For all ionotropic glutamate receptors studied, the ligand-binding domain is in the form of a hinged clamshell consisting of two lobes that close around the agonist molecule (depicted as gold and blue sections in Figure 1A, B). Agonists bind deep within a gorge and make atomic contacts with both gold and blue lobes of the clamshell (Figure 1C, D). Agonist binding causes partial closure of the clamshell, trapping the agonist in the receptor and putting a torque on the transmembrane segments that apparently induces a twist in the transmembrane helices, thereby opening the channel. For AMPA receptors “full” agonists cause a more complete clamshell closure than partial agonists.14 Each subunit in a tetrameric structure typically binds the same agonist, however NMDA receptors are unique among all neurotransmitter receptors in requiring the binding of both glutamate (to GluN2 subunits) and glycine or D-serine (to GluN1 subunits) for channel opening.15
The amino-terminal domain of glutamate receptors also forms a clamshell,16–18 but in this case one that binds modulators of channel opening rather than agonists. The structure of the extracellular and transmembrane domains of one of the subunit dimers in a tetrameric glutamate receptor is shown in Figure 2 along with the location of binding sites for agonists and allosteric modulators. The pharmacology of ionotropic glutamate receptors has become quite rich in the past decade, with the creation or discovery of numerous antagonists and allosteric modulators that are selective for particular receptors or even particular subunit combinations.6,8 Many of the highly selective allosteric compounds target the amino-terminal domain.10,13 For example, cyclothiazide allosterically potentiates AMPA but not kainate receptors, whereas concanavalin A potentiates kainate but not AMPA receptors; in both cases potentiation occurs by relief from desensitization. Similarly, binding to the N-terminal domain of NMDA receptors, CP-101,606 selectively inhibits GluN2B-containing NMDA receptors, whereas zinc exerts high affinity (10–30 nM IC50) block only of GluN2A-containing NMDA receptors.

CONTROL OF RECEPTOR PROPERTIES
Splice variants that control function or subcellular location have been identified for most of the subunits. The first example was the flip and flop variants of each of the four AMPA receptor subunits. This cassette of 38 amino acids, which is located in an extracellular loop shown in Figure 1A–C, influences desensitization rates of the receptors. C-terminal splice variants exist for most of the subunits and regulate coupling of the subunits to cytoplasmic proteins. For example alternative exon 5 incorporation into GluN1 strongly determines responsiveness to allosteric modulation by protons and polyamines.
A very interesting regulation is determined by editing of the primary RNA transcript for AMPA GluA2 and kainate GluK1-GluK2 subunits. This editing involves enzymatic conversion of a glutamine codon to an arginine codon in the pre-spliced RNA,19 which influences a wide range of functional properties including calcium permeability, single channel conductance, voltage-dependent block by cytoplasmic polyamines, and, in the case of AMPA receptors, assembly efficiency.
The particular subunits that each neuron chooses to express are strong determinants of synaptic phenotype, and subunit expression in turn is controlled both transcriptionally and translationally. The most intensively-studied ionotropic glutamate receptor in this respect is GluA2, which features a neuron-restrictive silencer element (NRSE) in its promoter that is responsible for downregulation of GluA2 after status epilepticus.20–22 Seizures cause rapid (within hours) induction of the transcriptional repressor, REST, which recruits histone deacetylases to the GluA2 promoter via the corepressor, Sin3A (Figure 3A). The mechanism of repression seems to involve deacetylation of histones that are physically bound to the Gria2 gene because a histone deacetylase inhibitor prevents both deacetylation of Gria2-bound histones and GluA2 downregulation after status epilepticus.22 Genes encoding GluN1 and GluN2C also have an NRSE but less is known about the conditions under which the REST repression system is brought into play for the NMDA receptors. Currently more than 1300 genes are known to have confirmed NRSE sequences, making the REST/NRSE system broadly seizure-responsive.

The 5′-untranslated region (UTR) of many ionotropic glutamate receptor mRNAs is unusually long. These long 5′-UTRs often exhibit high GC content and contain out-of-frame AUG codons that could act as decoys for scanning ribosomes, reducing or preventing translation initiation at the true glutamate receptor AUG. At least two major transcriptional start sites exist for the Gria2 gene as depicted by the pair of bent arrows in Figure 3A. Interestingly, the longer transcripts contain an imperfect GU repeat that serves as a translational repressor, is conserved between rodents and man, and is polymorphic in length in man.23 Additionally, GluA2 transcripts have alternative 3′ untranslated regions that control the rate of initiation of protein synthesis. Translational suppression by the longer 3′UTR seems to be mediated in part by binding of CPEB324 and in the rat hippocampus is relieved by seizures.25,26 Thus both transcription and translation of GluA2 are regulated by seizure-induced signaling systems. The GluN1 and GluN2A subunits are also under translational control by the 5′UTR or 3′UTR, respectively.27,28
The combination of specific phosphoprotein antibodies, site-directed mutagenesis, chromophore-tagged receptors and, in some cases, fragmentation followed by mass spectrometry, has in the past decade led to the secure identification of phosphorylation sites on the C-terminal domains of ionotropic glutamate receptors, and in some cases to an understanding of the functional consequences of phosphorylation. The most intensively studied subunits are GluN1 and GluA1. The C-terminal domain of GluA1 exhibits four PKC targets, plus one PKA and one CAMKII site. Phosphorylation at each of these sites has been shown to regulate activity-dependent receptor trafficking, open probability or conductance of the channel.6 GluA2 has alternative C-terminal domains that are differentially regulated by phosphorylation as shown in Figure 3B. GluN1 has four C-terminal splice variants, only the longest of which appears to harbor phosphorylation sites. Status epilepticus causes rapid dephosphorylation of serines 890 and 897 on GluN1 then slowly-developing hyperphosphorylation of these serines by PKCγ and PKA, respectively.29,30 Phosphorylation of S890 disrupts surface clusters of NMDA receptors,31 whereas phosphorylation of S897 promotes insertion of receptors into the synaptic membrane.32
GLUTAMATERGIC MECHANISMS IN EPILEPSY
Currently available glutamate receptor antagonists, with the possible exception of those directed towards kainate receptors, are as a rule poor anticonvulsants due to meager selectivity towards rapidly firing neurons, yet activation of glutamate receptors on neurons and probably astrocytes contributes to the initiation and propagation of seizures. Given this conundrum, I will consider here several related topics that might be developed therapeutically.
Astrocytic Release of Glutamate
It is now clear that astrocytes in vitro can respond to chemicals (e.g., glutamate, prostaglandins, tumor necrosis factor alpha) released from surrounding cells during periods of repetitive firing or inflammatory stimuli. The astrocytic response in vitro involves generation of a cytoplasmic calcium signal and subsequent release of glutamate and/or D-serine.33 The consequence of astrocytic glutamate release has been the subject of numerous studies. One of the most convincing was that of Jourdain et al.34, who showed that repetitive depolarization of an astrocyte through a patch pipette in hippocampal slices increased the frequency of spontaneous miniature EPSCs recorded in nearby dentate granule cells. A number of internal controls ruled out neuronal depolarization as the culprit. Following such astrocytic stimulation, GluN2B-containing receptors on presynaptic terminals of the perforant path were activated, which in turn potentiated synaptic transmission by elevating the probability of transmitter release. GluN2B activation could be prevented by direct astrocytic infusion of the calcium chelator BAPTA or a tetanus toxin protease. The precise conditions under which such strong, prolonged depolarization of astrocytes would occur was not addressed by their study, but it can be supposed that status epilepticus could serve this purpose. Moreover, the key astrocytic event – exocytosis of glutamate or similar compound, or perhaps adjustment of the extracellular space to allow neuron-released glutamate to feed back onto presynaptic receptors – could not be definitively determined.
Although evidence is mounting for a specific role of astrocytic calcium waves in neurodegeneration during status epilepticus35 and ischemia,36 it is proving frustratingly difficult to determine exactly how and under what conditions the calcium wave contributes to neuropathology.33,37 One clue comes from the studies of Bezzi et al.,38 who showed that the TNFα produced by reactive astrocytes and activated microglia boosts glutamate release from astrocytes through a cyclooxygenase-mediated pathway. Thus it is possible that astrocytic glutamate release is minimal normally but enhanced in inflamed tissue (Figure 4).
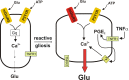
Impaired Glutamine Cycle in Sclerotic Tissue
Much of the glutamate released from excitatory nerve terminals is transported by active membrane pumps from the extrasynaptic space into surrounding astrocytes. Glutamate is then largely converted to glutamine by an enzyme, glutamine synthase which is expressed by astrocytes and oligodendrocytes but not by neurons. Glutamine, in turn, is transported to extracellular fluid and is taken up by both inhibitory and excitatory neurons, where it is converted back to glutamate by phosphate-activated glutaminase. The movement of glutamate and glutamine between neurons and astrocytes is called the ‘glutamine cycle’ and is an important feature of the close metabolic relationship that exists between neurons and glia in the brain. The glutamine cycle supplies this glutamate precursor to both glutamatergic and GABAergic neurons. The role of the glutamine cycle in epilepsy is somewhat controversial. Inhibition of glutamine transport or glutamine synthase reduces the amplitude of GABAergic evoked or spontaneous IPSCs,39,40 resulting in disinhibition and suggesting a major function of the glutamine cycle in the dynamic regulation of inhibitory synaptic strength. The notion that disruption of the glutamine cycle should promote seizures is reinforced by the observations that astrocytic glutamine synthase is downregulated in sclerotic tissue resected from epilepsy patients,41 that selective induction of astrogliosis causes reduced GABAergic inhibition in the hippocampus that could be reversed by exogenous glutamine,42 and that recurrent seizures develop in rats after local inhibition of glutamine synthase in the hippocampus.43 However, when GABAergic inhibition is blocked, the resulting epileptiform activity in hippocampal44 and cortical45 slices maintained in vitro is dramatically attenuated by inhibition of glutamine transport or glutamine synthase. This finding suggests that a continual supply of glutamine is needed to fuel glutamate synthesis for excitatory synaptic transmission in rapidly firing neurons. Taken together, these studies indicate that inhibition of the glutamine cycle can reduce both GABAergic inhibition and synaptic excitation mediated by glutamatergic synapses. To develop this field further it will be important to explore the anticonvulsant potential of potentiating residual glutamine synthase in reactive astrocytes under the more realistic conditions of chronic epilepsy. For additional discussion of the potential roles for disruption of the glutamine cycle in epilepsy see the chapter by J. V. Nadler in this volume.
Special Role for Kainate Receptors in Epilepsy
The year 1974 brought the first demonstration that a newly-developed antihelminthic, kainic acid, caused convulsions in mice.46 Much has been learned about the pharmacology and biology of kainate receptors in the last decade (reviewed in refs. 8 and 47). Because kainate is a powerful agonist at AMPA receptors as well as kainate receptors, consideration of the roles of kainate receptors in epileptiform activity awaited the development of selective drugs and genetically-modified mice. Genetically engineered mice lacking GluK2 showed an interesting phenotype.48,49 First, the potency with which kainate induced an inward current in CA3 pyramidal cells from these mice was reduced about 6-fold. Kainate-induced inward currents on CA3 interneurons, and kainate-induced increase in frequency of spontaneous IPSCs on pyramidal cells, were also absent in mice lacking GluK2. Second, kainate was unable to induce gamma oscillations (38 Hz) in the CA3 region of mice lacking GluK2. Finally, mice injected with a moderate (20 mg/kg ip) but not a higher (30 mg/kg ip) dose of kainate were protected from seizure development. These results point to an important role for GluK2 in mediating epileptiform activity produced by kainic acid.
The role of GluK1 in seizure development, on the other hand, is more controversial with superficially opposing results of pharmacologic and genetic experiments. Genetic ablation of GluK1 increased the potency with which kainate induced gamma oscillations and epileptiform activity in CA3,49 and yet a first generation GluK1-selective antagonist could prevent pilocarpine-induced seizures in rats as well as seizure activity produced by 6 Hz corneal stimulation.50 This antagonist, LY377770, was 10 to 100-fold selective for GluK1 vs the four AMPA receptors in ligand binding assays, and >40-fold selective for GluK2 or GluK2/GluK5 receptors. Kainate was still able to elicit seizure activity in GluK1 knockouts (unpublished data reported in ref. 47), but the sensitivity to kainate in the GluK1 nulls was not reported. It would be simple to dismiss the pharmacologic results based on insufficient selectivity, but one must also consider the consequence of homeostatic adjustments in circuitry or receptor expression in the GluK2 knockouts (as shown by Christensen et al.51).
CHALLENGES AND OPPORTUNITIES
In the past decade we have seen the first fruits of understanding the molecular function of ionotropic glutamate receptors based on their protein structure and post-translational processing. In addition, roles for reactive astrocytes in the buildup of extracellular glutamate during seizures have been proposed. A major challenge for using this information to develop new anticonvulsants is the near ubiquitous distribution of ionotropic glutamate receptors, which mediate the vast majority of excitatory neurotransmission in virtually every brain region. However, the tremendous strides made in developing subunit-selective antagonists of kainate receptors8 represent a real opportunity to explore the potential for use of kainate receptor antagonists in epilepsy. A related opportunity relies on the finding that activation of Gαq-coupled receptors such as M1 muscarinic receptors (by, e.g., pilocarpine) can potentiate the activation of heteromeric kainate receptors.52 It might therefore be possible to use selective antagonists of group 1 metabotropic glutamate receptors, muscarinic receptors, or Gαq-coupled prostaglandin receptors to allosterically dampen kainate receptor activity. More gentle allosteric inhibition could be preferable to frank antagonism of the ionotropic glutamate receptors to minimize adverse effects.
Glutamine synthase represents another intriguing molecular target, even though this astrocytic enzyme supplies glutamate to both inhibitory and excitatory neurons. A potentiator of residual glutamine synthase expressed by reactive astrocytes might restore inhibitory balance. No chemical potentiator exists yet but the application of high throughput screening of small molecules followed by medicinal chemistry would be the preferred route to developing an allosteric potentiator.
REFERENCES
- 1.
- Davies J, Francis AA, Jones AW, Watkins JC. 2-Amino-5-phosphonovalerate (2APV), a potent and selective antagonist of amino acid-induced and synaptic excitation. Neurosci Lett. 1981;21:77–81. [PubMed: 6111052]
- 2.
- Dingledine R, McBain CJ, McNamara JO. Excitatory amino acid receptors in epilepsy. Trends in Pharmacol. 1990;11:334–338. [PubMed: 2168104]
- 3.
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacological Reviews. 1999;51:7–62. [PubMed: 10049997]
- 4.
- Hollmann M, O’Shea-Greenfield A, Rogers SW, Heinemann S. Cloning by functional expression of a member of the glutamate receptor family. Nature. 1989;342:643–648. [PubMed: 2480522]
- 5.
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. [PubMed: 8210177]
- 6.
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological Rev. 2010;62:405–496. [PMC free article: PMC2964903] [PubMed: 20716669]
- 7.
- Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. [PMC free article: PMC2847504] [PubMed: 18655795]
- 8.
- Jane DE, Lodge D, Collingridge GL. Kainate receptors: pharmacology, function and therapeutic potential. Neuropharmacology. 2009;56:90–113. [PubMed: 18793656]
- 9.
- Penn AC, Greger IH. Sculpting AMPA receptor formation and function by alternative RNA processing. RNA Biol. 2009;6:517–21. [PubMed: 19717983]
- 10.
- Mony L, Kew JN, Gunthorpe MJ, Paoletti P. Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol. 2009;157:1301–1317. [PMC free article: PMC2765303] [PubMed: 19594762]
- 11.
- Swanson GT. Targeting AMPA and kainate receptors in neurological disease: therapies on the horizon? Neuropsychopharmacology. 2009;34:249–250. [PMC free article: PMC2715329] [PubMed: 19079074]
- 12.
- Bassani S, Valnegri P, Beretta F, Passafaro M. The GLUR2 subunit of AMPA receptors: synaptic role. Neuroscience. 2009;158:55–61. [PubMed: 18977416]
- 13.
- Hansen KB, Furukawa H, Traynelis SF. Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol Pharmacol. 2010;78:535–549. [PMC free article: PMC2981397] [PubMed: 20660085]
- 14.
- Jin R, Banke TG, Mayer ML, Traynelis SF, Gouaux E. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci. 2003;6:803–810. [PubMed: 12872125]
- 15.
- Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA receptors expressed in Xenopus oocytes. Science. 1988;241:835–837. [PubMed: 2841759]
- 16.
- Kumar J, Schuck P, Jin R, Mayer ML. The N-terminal domain of GluR6-subtype glutamate receptor ion channels. Nat Struct Mol Biol. 2009;16:631–638. [PMC free article: PMC2729365] [PubMed: 19465914]
- 17.
- Jin R, Singh SK, Gu S, Furukawa H, Sobolevsky AI, Zhou J, Jin Y, Gouaux E. Crystal structure and association behaviour of the GluR2 amino-terminal domain. EMBO J. 2009;28:1812–1823. [PMC free article: PMC2699365] [PubMed: 19461580]
- 18.
- Karakas E, Simorowski N, Furukawa H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009;28:3910–3920. [PMC free article: PMC2797058] [PubMed: 19910922]
- 19.
- Higuchi M, Single FN, Köhler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell. 1993;75:1361–1370. [PubMed: 8269514]
- 20.
- Myers SJ, Peters J, Comer M, Barthel F, Dingledine R. Transcriptional regulation of the GluR2 gene: neural-specific expression, multiple promoters, and regulatory elements. J Neuroscience. 1998;18:6723–6739. [PMC free article: PMC6792970] [PubMed: 9712644]
- 21.
- Huang Y-F, Myers SJ, Dingledine R. Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nature Neuroscience. 1999;2:867–872. [PubMed: 10491605]
- 22.
- Huang Y, Doherty JJ, Dingledine R. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci. 2002;22:8422–8428. [PMC free article: PMC6757766] [PubMed: 12351716]
- 23.
- Myers SJ, Huang Y, Genetta T, Dingledine R. Inhibition of glutamate receptor 2 translation by a polymorphic repeat sequence in the 5′-untranslated leaders. J Neurosci. 2004;24:3489–3499. [PMC free article: PMC6729757] [PubMed: 15071096]
- 24.
- Huang YS, Kan MC, Lin CL, Richter JD. CPEB3 and CPEB4 in neurons: analysis of RNA-binding specificity and translational control of AMPA receptor GluR2 mRNA. EMBO J. 2006;25:4865–4876. [PMC free article: PMC1618119] [PubMed: 17024188]
- 25.
- Irier HA, Quan Y, Yoo J, Dingledine R. Control of glutamate receptor 2 (GluR2) translational initiation by its alternative 3′ untranslated regions. Mol Pharmacol. 2009;76:1145–1149. [PMC free article: PMC2784723] [PubMed: 19794129]
- 26.
- Irier HA, Shaw R, Lau A, Feng Y, Dingledine R. Translational regulation of GluR2 mRNAs in rat hippocampus by alternative 3′ untranslated regions. J Neurochem. 2009;109:584–594. [PMC free article: PMC2727682] [PubMed: 19222700]
- 27.
- Wood MW, VanDongen HMA, VanDongen AMJ. The 59-untranslated region of the N-methly-D-aspartate receptor NR2A subunit controls efficiency of translation. J Biol Chem. 1996;271:8115–8120. [PubMed: 8626498]
- 28.
- Wells DG, Dong X, Quinlan EM, Huang YS, Bear MF, Richter JD, Fallon JR. A role for the cytoplasmic polyadenylation element in NMDA receptor-regulated mRNA translation in neurons. J Neurosci. 2001;21:9541–9548. [PMC free article: PMC6763028] [PubMed: 11739565]
- 29.
- Sanchez-Perez AM, Felipo V. Serines 890 and 896 of the NMDA receptor subunit NR1 are differentially phosphorylated by protein kinase C isoforms. Neuroche Int. 2005;47:84–91. [PubMed: 15936117]
- 30.
- Niimura M, Moussa R, Bissoon N, Ikeda-Douglas C, Milgram NW, Gurd JW. Changes in phosphorylation of the NMDA receptor in the rat hippocampus induced by status epilepticus. J Neurochem. 2005;92:1377–1385. [PubMed: 15748156]
- 31.
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. [PubMed: 9030583]
- 32.
- Scott DB, Blanpied TA, Ehlers MD. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology. 2003;45:755–767. [PubMed: 14529714]
- 33.
- Wetherington J, Serrano G, Dingledine R. Astrocytes in the epileptic brain. Neuron. 2008;58:168–178. [PMC free article: PMC4124883] [PubMed: 18439402]
- 34.
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 2007;10:331–339. [PubMed: 17310248]
- 35.
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27:10674–10684. [PMC free article: PMC2917229] [PubMed: 17913901]
- 36.
- Ding S, Wang T, Cui W, Haydon PG. Photothrombosis ischemia stimulates a sustained astrocytic Ca2+ signaling in vivo. Glia. 2009;57:767–776. [PMC free article: PMC2697167] [PubMed: 18985731]
- 37.
- Hamilton NB, Attwell D. Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci. 2010;11:227–238. [PubMed: 20300101]
- 38.
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4- activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001;4:702–710. [PubMed: 11426226]
- 39.
- Liang SL, Carlson GC, Coulter DA. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J Neurosci. 2006;26:8537–8548. [PMC free article: PMC2471868] [PubMed: 16914680]
- 40.
- Fricke MN, Jones-Davis DM, Mathews GC. Glutamine uptake by System A transporters maintains neurotransmitter GABA synthesis and inhibitory synaptic transmission. J Neurochem. 2007;102:1895–1904. [PubMed: 17504265]
- 41.
- Eid T, Thomas MJ, Spencer DD, Rundén-Pran E, Lai JC, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, de Lanerolle NC. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. 2004;363:28–37. [PubMed: 14723991]
- 42.
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–591. [PMC free article: PMC3225960] [PubMed: 20418874]
- 43.
- Eid T, Ghosh A, Wang Y, Beckström H, Zaveri HP, Lee TS, Lai JC, Malthankar-Phatak GH, de Lanerolle NC. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain. 2008;131:2061–70. [PMC free article: PMC2724901] [PubMed: 18669513]
- 44.
- Bacci A, Sancini G, Verderio C, Armano S, Pravettoni E, Fesce R, Franceschetti S, Matteoli M. Block of glutamate-glutamine cycle between astrocytes and neurons inhibits epileptiform activity in hippocampus. J Neurophysiol. 2002;88:2302–2310. [PubMed: 12424271]
- 45.
- Tani H, Dulla CG, Huguenard JR, Reimer RJ. Glutamine is required for persistent epileptiform activity in the disinhibited neocortical brain slice. J Neurosci. 2010;30:1288–1300. [PMC free article: PMC2821093] [PubMed: 20107056]
- 46.
- Olney JW, Rhee V, Ho OL. Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 1974;77:507–512. [PubMed: 4152936]
- 47.
- Vincent P, Mulle C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience. 2009;158:309–323. [PubMed: 18400404]
- 48.
- Mulle C, Sailer A, Pérez-Otaño I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392:601–5. [PubMed: 9580260]
- 49.
- Fisahn A, Contractor A, Traub RD, Buhl EH, Heinemann SF, McBain CJ. Distinct roles for the kainate receptor subunits GluR5 and GluR6 in kainate-induced hippocampal gamma oscillations. J Neurosci. 2004;24:9658–68. [PMC free article: PMC6730151] [PubMed: 15509753]
- 50.
- Smolders I, Bortolotto ZA, Clarke VR, Warre R, Khan GM, O’Neill MJ, Ornstein PL, Bleakman D, Ogden A, Weiss B, Stables JP, Ho KH, Ebinger G, Collingridge GL, Lodge D, Michotte Y. Antagonists of GLU(K5)-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nat Neurosci. 2002;5:796–804. [PubMed: 12080343]
- 51.
- Christensen JK, Paternain AV, Selak S, Ahring PK, Lerma J. A mosaic of functional kainate receptors in hippocampal interneurons. J Neurosci. 2004;24:8986–93. [PMC free article: PMC6730072] [PubMed: 15483117]
- 52.
- Benveniste M, Wilhelm J, Dingledine R, Mott DD. Subunit-Dependent Modulation of Kainate Receptors by Muscarinic Acetylcholine Receptors. Brain Res. 2010;1352:61–69. [PMC free article: PMC2926221] [PubMed: 20655886]
Publication Details
Author Information and Affiliations
Authors
Raymond Dingledine1.
Affiliations
Ray Dingledine, PhD, Professor and Chair, Department of Pharmacology, Executive Associate Dean, Research, Emory University School of Medicine, Atlanta, GA 30322,
Email: ude.yrome.mrahp@enidelgnidrCopyright
All Jasper's Basic Mechanisms of the Epilepsies content, except where otherwise noted, is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported license, which permits copying, distribution and transmission of the work, provided the original work is properly cited, not used for commercial purposes, nor is altered or transformed.
Publisher
National Center for Biotechnology Information (US), Bethesda (MD)
NLM Citation
Dingledine R. Glutamatergic Mechanisms Related to Epilepsy: Ionotropic Receptors. In: Noebels JL, Avoli M, Rogawski MA, et al., editors. Jasper's Basic Mechanisms of the Epilepsies [Internet]. 4th edition. Bethesda (MD): National Center for Biotechnology Information (US); 2012.
%20and%20crystal%20structure%20of%20the%20agonist-binding%20domain%20(B%02013D)%20of%20the%20GluA2%20subunit%20protein.&p=BOOKS&id=98189_dingledinef1.jpg "Click on image to zoom")
%2C%20amino%20terminal%20domain%20(ATD)%2C%20and%20transmembrane%20domain%20(TMD).&p=BOOKS&id=98189_dingledinef2.jpg "Click on image to zoom")
%2C%20translational%20repression%20mediated%20by%20alternative%205%02032UTRs%20(A)%2C%20and%20post-translational%20processing%20on%20alternative%20C-terminal%20domains%20(B).&p=BOOKS&id=98189_dingledinef3.jpg "Click on image to zoom")
