Stimulation of group I mGluRs elicits ictal-like responses from normal quiescent hippocampal slices. Ictal-like responses once induced become persistent and show no fading even upon washout of the agonist. The conversion of normal neuronal activity to long-lasting epileptiform discharges resembles epileptogenesis and effects of group I mGluRs on hippocampal network provides a model for detailed studies of epileptogenesis. Experiments on the group I mGluR model reveal that epileptogenesis is sustained by specific long-lasting cellular responses (cellular plasticity). The latter include activation of a voltage-dependent cationic current and suppression of afterhyperpolarizations following action potentials. Induction of cellular plasticity (and epileptogenesis) requires synthesis of new proteins via local dendritic translation. The translation process is normally controlled by endogenous translation repressors including BC1 RNA and FMRP (fragile X mental retardation protein). Absence of FMRP causes fragile X syndrome (FXS) which is characterized by developmental mental retardation and enhanced susceptibility to epilepsy. Recent data indicate that group I mGluR-mediated translation is aberrantly enhanced in the absence of the repressor FMRP and that the enhanced translation constitutes a core co-morbidity mechanism of mental retardation and epilepsy observed in FXS. Thus, group I mGluRs represent a site of vulnerability and a potential therapeutic target against epilepsy.
In the early 1990’s the epileptogenic potential of metabotropic glutamate receptor (mGluR) activation in the hippocampus was first suggested by data using the then-newly-developed broad spectrum mGluR agonist (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD).1 These studies revealed that mGluR activation had the potent ability to recruit the hippocampal network to express robust synchronized discharges. These synchronized bursts had features suggestive of typical seizure discharges in that (1) their length was on the order of seconds, and (2) they were comprised of an intrinsic oscillatory series of discharges that began at a high frequency and gradually slowed. And indeed, work in other labs confirmed that ACPD application does elicit seizures in the intact organism.2 A hypothesis was developed proposing that the group I mGluRs, which are predominantly localized to the edges of synapses (“perisynaptic”),3 were likely to be activated at times of intense glutamate release, and this could result in the expression of acute seizures such as the impact seizure that occurs acutely in the setting of head trauma. However, subsequent studies using the selective group I mGluR agonist (S)-3,5-dihydroxyphenylglycine (DHPG) revealed a potential additional consequence of group I mGluR activation: long-lasting changes in network excitability.4
THE mGluR MODEL OF EPILEPTOGENESIS
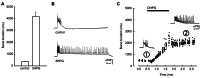
The impact of selective group I mGluR activation on the in vitro hippocampal network was profound. Not only did such activation result in the expression of ictal-like discharges, but removal of the mGluR agonist was insufficient to restore baseline network excitability; instead, ictal-like discharges continued to be expressed unabated for hours following agonist exposure and removal ().4 This network plasticity was strongly suggestive of the initiation of an epileptogenic process, one in which normal neuronal cortex is converted into a persistently hyperexcitable state with a lowered threshold for the production of seizure discharges. It was proposed that this form of epileptogenesis was likely to be clinically relevant because the instigating agent was acting at glutamate receptors, and glutamate was long recognized as the key excitatory transmitter in the CNS underlying the expression of seizure discharges.
Group I metabotropic glutamate receptor (mGluR) activation induces persistent ictal-like discharges in the hippocampus. A, Summary histogram of mean duration of synchronized bursts elicited by picrotoxin (control) and maximum duration of prolonged bursts (more...)
Numerous studies revealed that group I mGluR activation can induce long-lasting changes in cellular and synaptic properties (plasticity) in the CNS, including long-term potentiation and depression.5,6 As with all metabotropic receptors, a G-protein-coupled second-messenger signaling cascade is recruited upon stimulation, and the intracellular enzymes activated can have long-lasting consequences on cellular excitability. In the case of the group I mGluRs – receptors coupled to the Gq/11 protein and phospholipase C signaling pathway – two direct effects were well characterized early on: production of diacylglycerol, which would result in protein kinase C activation, and inositol triphosphate production, which would stimulate release of calcium from intracellular stores.7 Early studies attributed the synaptic long-lasting effects of group I mGluR activation to either or both of these intracellular pathways, and it was initially assumed that the network plasticity we observed was similarly mediated, but later studies revealed the additional involvement of a more complex set of proteins and enzymes, to be described later in this chapter.
Plasticity of any type has two critical components: the initiating factors necessary for induction of the modification, and the downstream effectors that underlie the sustained expression of the enhanced response. Experiments described below have delved into understanding the necessary contributors to each of these components.
KEY FEATURES RELEVANT TO INDUCTION OF GROUP I mGluR-DEPENDENT EPILEPTOGENESIS
Most studies examining the in vitro model of mGluR-induced epileptogenesis have been performed on the hippocampal slice preparation. In many instances, slices were initially exposed to picrotoxin, an antagonist of GABAA receptor-mediated inhibition, to elicit baseline interictal activity in the CA3 network. While there were data to indicate that this additional agent was not necessary for the induction of persistent ictal-length activity, the picrotoxin model was useful for the following reasons: (1) It helped to confirm the baseline health of the slice by establishing the initial responsiveness of the network. (2) It reduced the inter-slice variability of the mGluR-induced ictaform activity. (3) It made for easier interpretation of the site of action of the mGluR agonist: numerous studies had indicated that group I mGluRs are localized to principal neurons, interneurons, and glial cells,8 and mGluR activation could therefore potentially have direct suppressive effects on inhibitory responses. Nevertheless, elimination of GABAA receptor-mediated inhibition from the network activity in the mature hippocampal slice never elicited ictaform discharges; only interictal-length bursts (generally < 650 milliseconds in duration) were produced. Furthermore, additional suppression of GABAB receptor-mediated responses also failed to elicit ictal-length discharges.9
Under conditions of suppressed GABA-mediated inhibition, exposure to selective group I mGluR agonist invariably induced ictaform discharges.4 It was therefore suggested that group I mGluR activation participated in the interictal-to-ictal transition, eliciting ictaform discharges via a direct excitatory effect on the hippocampal network. But the persistence of the ictal discharges following transient agonist exposure was the finding of greatest interest due to its relevance to epileptogenesis. A series of experiments using co-applied antagonists indicated that the agonist was truly washing out of the chamber, suggesting that the persistence of the effect was due to a true network modification and not a pharmacological artifact.4 But was group I mGluR activation truly necessary to induce this modification?
Silent Induction
When hippocampal slices are perfused with the GABAA antagonist picrotoxin, interictal bursts recur spontaneously and rhythmically approximately every 10 seconds for as long as the slice is exposed to the agent. When group I mGluR agonist is subsequently introduced, the burst frequency promptly increases to 2–3 times its original rate, then returns close to baseline frequency as the length of the bursts gradually increase to ictal length.10 In light of this, it was hypothesized that perhaps the persistence of the effect was merely a kindling-like phenomenon resulting from the bombardment of recurrent bursting rather than a direct effect of the mGluR activation itself. To address this question, experiments were performed in which, after demonstration of normal interictal activity in the presence of picrotoxin, all bursting activity in the slice was suppressed via the perfusion of ionotropic glutamate receptor antagonists. Then, in the presence of this pharmacological blockade, the slice was transiently exposed to 20–40 min of group I mGluR agonist (DHPG), an experimental protocol referred to as “silent induction.”11 Upon removal of all agents except picrotoxin, the synchronized discharges that emerged were markedly potentiated to ictal length. The data demonstrated that (i) induction of ictaform events in this model is independent of ionotropic glutamatergic activation, and (ii) group I mGluR activation itself has a powerful direct long-lasting effect of enhancing hippocampal network responsiveness.
Group I mGluR subtypes: mGluR1 and mGluR5
Experiments in which the group I mGluR agonist was applied in the presence of antagonist confirmed that the epileptogenic effect was indeed driven by activation of group I mGluRs. The group I mGluR family is comprised of two receptor members, mGluR1 and mGluR5. Were both receptor subtypes necessary for induction of this epileptogenic effect? Utilizing selective antagonists against either mGluR1 or mGluR5, it was demonstrated that mGluR5 activation alone was often both necessary and sufficient to induce a persistent ictogenic effect.12 This was evident from two sets of data: (1) If group I mGluR agonist was transiently applied in the presence of mGluR1-selective antagonist, persistent ictal-length discharges often appeared following subsequent removal of the mGluR1 antagonist, uncovering the epileptogenic effect that was induced by activation of mGluR5 alone. (2) If group I mGluR agonist was applied in the presence of mGluR5-selective antagonist, persistent ictal-length discharges were generally not elicited, revealing that mGluR5 activation was a necessary component of the induction process.
NMDA receptors
NMDA receptor activation has frequently been associated with the induction of long-term potentiation and/or depression of synaptic responses.13 It was therefore natural to consider the possibility that NMDA receptor activation may be necessary for the induction of the mGluR-driven long-lasting network effect. Nevertheless, experiments performed in the presence of NMDA receptor antagonist demonstrated that the group I mGluR agonist could still elicit its full epileptogenic effect, in regards to both enhanced burst length and persistence following agonist removal.14
Protein synthesis
Protein synthesis has been shown to play a key role in various forms of long-lasting synaptic modifications.15 Experiments were performed to examine the protein synthesis dependence of the group I mGluR-induced epileptogenic effect. Utilizing anisomycin and cycloheximide, agents that suppress mRNA translation, it was shown that even prolonged group I mGluR agonist application failed to elicit burst prolongation in the presence of either protein synthesis inhibitor. By contrast, protein synthesis inhibitor had no effect on the expression of picrotoxin-induced interictal discharges.10 The dependence of group I mGluR-mediated induction of ictaform events on mRNA translation would turn out to be highly relevant to understanding the pathophysiology of seizures and other clinical features of fragile X syndrome, as will be discussed later in this chapter.
Protein synthesis inhibitors had no effect on the expression of ictal-length discharges elicited with the broad spectrum mGluR agonist ACPD, even though these discharges were shown to be group I mGluR-dependent.16 Consistent with this finding, these ACPD-induced group I mGluR-driven but protein synthesis-independent ictal discharges were not persistent; rather, they readily disappeared upon removal of the broad spectrum agonist.4,16 Encouragingly, these data suggest that group I mGluR-mediated seizure discharges can be elicited without inducing an epileptogenic process, and further investigations clarifying the distinction between these two forms of mGluR activation may allow for the development of unique antiepileptogenic agents.
Phospholipase C
Group I mGluRs are coupled to intracellular signaling pathways via activation of phospholipase C (PLC);7 this PLC activation was shown to be a critical component of the epileptogenic process. Pharmacological inhibition of PLC prevented induction of ictal-like discharges by DHPG, and similarly, in slices prepared from PLCβ1 knockout mice (mice lacking the major isoform of PLC expressed in the hippocampus), group I mGluR activation failed to elicit ictal-like discharges.17 These data revealed that PLC activation is necessary for group I mGluR-induced epileptogenesis.
PLC activation stimulates two parallel signaling pathways: diacylglycerol-mediated protein kinase C activation, and inositol triphosphate-driven increases of intracellular Ca2+ via release from intracellular stores.7 Although protein kinase C (PKC) is a mediator of many group I mGluR-induced effects, experiments performed in the presence of chelerythrine, a PKC inhibitor, revealed no significant suppressive effect on the induction of persistent ictaform activity by group I mGluR agonist.18 On the other hand, agents that interfere with Ca2+ release from intracellular stores did prevent the expression of ictaform activity elicited by either DHPG19 or ACPD,20 suggesting that mGluR-induced epileptogenesis is dependent on intracellular Ca2+ mobilization.
Extracellular-signal-regulated kinase 1/2
Group I mGluR agonist elicits two phases of extracellular-signal-regulated kinase (ERK) 1/2 activation in hippocampal slices: an early response which is elicited directly by receptor activation and signaled via tyrosine kinase, and a later response driven by both neuronal firing and PKC activation. As indicated above, neither neuronal firing11 nor PKC activation18 is necessary for the mGluR-mediated induction of long-lasting ictaform activity. However, inhibition of either tyrosine kinase or of ERK 1/2 phosphorylation prevented DHPG from inducing ictal-like discharges, suggesting that the induction process is dependent on the early receptor-mediated ERK 1/2 response.21
Phospholipase D
Cysteine sulfinic acid (CSA) is an amino acid endogenous to the nervous system that is released in the hippocampus during periods of high-intensity activity22 and, among its many actions, activates PLD-coupled metabotropic receptors.23 CSA-mediated activation of PLD can be antagonized with the agent PCCG-13.24 Through the use of these two agents, it was shown that PLD activation impedes the epileptogenic process induced by selective group I mGluR activation, while it has no suppressive effect on baseline interictal bursting or on fully-induced persistent ictal activity.25 PLD activation also had no effect on reversible ictal activity elicited with the broad spectrum mGluR agonist ACPD.16 As the CSA-mediated suppression of epileptogenesis was blocked by chelerythrine,18 it was proposed that CSA’s antiepileptogenic effect may be mediated by PLD-driven PKC activation, which could then feedback to inhibit group I mGluR-mediated responses.
The role of PKC in the antiepileptogenic effect of CSA was tested with experiments in which phorbol esters were used to activate PKC. Phorbol ester failed to inhibit mGluR-driven epileptogenesis; in fact, the ictal activity induced in the presence of phorbol ester was more robust than that elicited by DHPG alone. Additional experiments revealed that phorbol esters elicited a powerful epileptogenic effect of their own, accelerating the epileptogenic process and potentiating the ictal discharge duration.26 It is possible that the PKC activation responsible for PLD-mediated suppression of epileptogenesis may need to be subtype-specific and/or localized to specific synapses on the activated neurons to evoke its suppressive effect.
WHAT SUSTAINS THE ONGOING EXPRESSION OF THE GROUP I mGluR-INDUCED ICTAL DISCHARGES?
As NMDA receptor mediated responses can been enhanced by group I mGluR activation,27 and potentiated NMDA responses can sustain enhanced synchronized activity in hippocampal networks,28 it was hypothesized that mGluR-induced NMDA potentiation may underlie the sustained expression of the ictal bursts in our system. However, experiments revealed that this was not the case; the ictal discharges continued unabated in the presence of NMDA antagonist.1,14 What, then, was responsible for the sustained expression of the prolonged ictal discharges?
Autopotentiation
The ictal-length persistent discharges that are expressed following the transient activation of group I mGluRs proved to be quite resistant to most of the agents described above that impede induction of the discharges: neither protein synthesis inhibitor10 nor cysteine sulfinic acid25 could suppress the ongoing ictal activity once it had been induced. However, there was one agent that could effectively suppress the sustained expression: group I mGluR antagonist.1,4,11 If introduced after induction had occurred and prolonged bursts were fully-expressed, the group I mGluR antagonist shortened the synchronized discharges to interictal length, but ictal bursts promptly reappeared upon removal of the antagonist. These data suggested that group I mGluRs undergo “autopotentiation,” i.e., transient agonist-mediated selective activation results in long-lasting enhanced responsiveness of group I mGluRs, allowing endogenous glutamate to now sustain the activation of the receptors. Experiments utilizing selective mGluR1 and mGluR5 antagonists revealed that both receptors participate in the expression of the prolonged bursts: either antagonist alone elicited significant reversible shortening of the sustained ictal-length bursts, although the mGluR1 antagonist was more effective, almost completely abbreviating the discharges back to their interictal length prior to agonist exposure.12
Although presynaptic enhancement of glutamate release remains a possible contributor to this observed potentiation of mGluR responses, it appears more likely that the primary site of modification is postsynaptic. In recent studies, postsynaptic long-lasting effects on intrinsic properties of CA3 pyramidal cells induced by group I mGluR activation have been identified.29,30 These cellular modifications may underlie epileptogenesis by way of inducing persistent changes in the cell firing pattern, thereby sustaining prolonged ictal-like discharges.
Persistent effects of group I mGluR activation
There are numerous reported cellular effects mediated by group I mGluR activation. Most have not been established as long-lasting, but even those that do persist (e.g. enhancement of NMDA responses27) do not necessarily contribute to the enduring nature of the epileptogenic effect.14 The following are two excitatory effects of group I mGluR activation that have been established as long-lasting and likely contribute to the ongoing expression of the persistent ictal discharges.
Induction of a voltage-dependent cationic current ImGluR(V)
A major response to group I mGluR stimulation in CA3 pyramidal cells is the appearance of a depolarization-activated, voltage-dependent ionic current (ImGluR(V)) that has an activation threshold around −60 mV, a reversal potential of about −10 mV, and shows no inactivation ().17,29,31
ImGluR(V) induces plateau potentials that sustain rhythmic periods of prolonged action potential firing of 2–7 seconds with intervals of similar durations.17
ImGluR(V) also promotes repetitive firing by eliciting spike afterdepolarizations (ADPs).32 This intrinsic pattern of neuronal firing could drive synchronized ictaform activity via the recurrent glutamatergic collateral connections in the CA3 cell population.33 Indeed, pharmacological blockade of ImGluR(V)17,29 or its abolition in transgenic preparations17,34 prevents the expression of group I mGluR-dependent ictal-like discharges, suggesting that ImGluR(V) underlies this mGluR-induced synchronized activity.
Group I mGluR activation induces a long-lasting voltage-dependent cationic current: ImGluR(V). Aa, Current response to a depolarizing ramp from −70 mV to −5 mV obtained from a CA3 pyramidal cell in a solution containing low Ca2+ (0.2 mM), (more...)
Suppression of action potential afterhyperpolarizations
DHPG-mediated activation of group I mGluRs elicits suppression of action potential afterhyperpolarizations (AHPs), an effect which enhances repetitive firing in CA3 pyramidal cells.32,35 This AHP suppression can be long-lasting,30 suggesting it may contribute to the prolonged neuronal firing during synchronized group I mGluR-mediated ictal activity.
Consistent with the persistent nature of these two cellular effects, DHPG cannot elicit ImGluR(V) or AHP suppression in the presence of protein synthesis inhibitors,29,30 lending support to the hypothesis that these cellular effects may underlie the sustained expression of the protein synthesis-dependent persistent ictal bursts.10
ENDOGENOUS REGULATION OF GROUP I mGluR-DEPENDENT EPILEPTOGENESIS
Studies on the endogenous control of group I mGluR-dependent epileptogenesis were prompted by the observation that persistent ictaform discharges are elicited by selective activation of group I mGluRS with DHPG4,10 but not by the broad-spectrum mGluR agonist ACPD4,16 nor by synaptic stimulation of group I mGluRs,29,36,37 suggesting that group I mGluR-mediated epileptogenesis is normally prevented. Concurrent activation of group II mGluRs does not account for the reversibility of the ACPD-induced ictogenesis.4,16 Active protein synthesis is critical to the induction process underlying the mGluR-driven epileptogenic effect,10,16,21 suggesting this to be a potential site of regulation.
Recent data reveal that there are multiple endogenous regulators of protein synthesis. Brain cytoplasmic 1 (BC1) RNA is a small non-protein-coding RNA expressed in neuronal dendrites,38 where it inhibits activity-dependent protein synthesis.39–41 Mice lacking BC1 RNA are susceptible to audiogenic seizures and their hippocampal CA3 neuronal network displays synaptic hyperexcitability leading to ictal discharges.37 These epileptogenic responses were shown to be protein synthesis dependent, and driven by enhanced activation of mGluR5 and ERK 1/2, as antagonism of either of these could suppress the epileptic effect.37 These data suggest that, in wild type mice, BC1 RNA is an endogenous translational repressor of group I mGluR-mediated epileptogenesis.37,42
The finding that group I mGluR-dependent LTD is abnormal in preparations lacking functional fragile X mental retardation protein (FMRP),43 an intracellular RNA-binding protein that represses mRNA translation,44 pointed to this protein as another candidate for control of protein synthesis-dependent DHPG-induced epileptogenesis. Indeed, in experiments carried out on hippocampal slices from transgenic mice lacking FMRP, synaptic stimulation of group I mGluRs was able to elicit the DHPG-like persistent cellular and network excitatory effects previously seen only with DHPG application: both ImGluR(V) and persistent ictaform discharges were readily elicited, and these effects could be blocked with protein synthesis inhibitors.29,36 These data are consistent with an inhibitory role of FMRP on group I mGluR-induced, protein synthesis-dependent plasticity in the normal condition, and with the hypothesis that absence of FMRP-mediated inhibition results in exaggerated group I mGluR-dependent protein synthesis-dependent responses responsible for the seizures seen in patients with fragile X syndrome ().45
Group I mGluR model of epileptogenesis. Activation of group I mGluRs induces local translation eliciting cellular plasticity. Cellular plasticity includes long-lasting ImGluR(V) and AHP suppression. Integrative expression of cellular plasticity causes (more...)
FRAGILE X SYNDROME: A CLINICAL CONDITION IN WHICH HYPEREXCITABLE GROUP I mGluRs UNDERLIE A PHENOTYPE THAT INCLUDES SEIZURES
Group I mGluRs appear to play a central role in the pathophysiology of fragile X syndrome (FXS) in humans.45 FXS is caused by a mutation that results in lack of expression of functional FMRP. The Fmr1 knockout mouse expresses the full phenotype associated with fragile X syndrome: memory deficits, learning disabilities, autism, epilepsy, altered body growth and macroorchidism are features common to both the knockout mouse and the human condition, and thus this mouse has become a useful model for studying the underlying pathophysiology of the disease.46 Studies have shown the efficacy of mGluR5 antagonist in suppressing audiogenic seizures in this model (),47 and clinical trials in patients with FXS have been encouraging as well.48 But even more remarkably, in Fmr1 knockout mice crossbred to express 50% fewer mGluR5 receptors, a startling observation was made - all phenotypic anatomic and behavioral abnormalities usually seen in the fragile X mouse were normalized except for the macroorchidism.46 The implications are profound: excessive mGluR5-driven protein synthesis is responsible for much more than just epileptogenesis; it has a broad impact on memory, behavior, and even prepubescent body growth in fragile X syndrome. These data lead us to believe that the clinical importance of the group I mGluR system may be even more widespread than is currently realized.
ADDITIONAL CLINICAL CONDITIONS IN WHICH GROUP I mGluR HYPEREXCITABILITY MAY PLAY A KEY ROLE
The involvement of group I mGluR activation in the epileptogenesis may well extend beyond FXS. The following are some examples of neurological conditions that are associated with enhanced susceptibility to seizures and for which recent data raise the issue of possible roles of group I mGluRs in their pathogenesis.
Alzheimer’s disease and Down syndrome
A common feature of Alzheimer’s disease (AD) and Down syndrome (DS) is overexpression of amyloid precursor protein (APP) and accumulation of β-amyloid (Aβ) in brain plaques, with associated dementia.49,50 AD and DS are both associated with an increased incidence of seizures,51 and recent data suggest that Aβ may be epileptogenic.52 Studies on AD and DS mouse models reveal increased susceptibility to audiogenic seizures53,54 and remarkably, mGluR5 antagonists reduce both the Aβ production and the expression of seizures.54,55 These data suggest that mGluR5 activation may underlie epileptogenesis in AD and DS, and one may speculate that mGluR5 hyperexcitability contributes to the cognitive deficits and growth abnormalities as well. Interestingly, it appears that FMRP normally inhibits mGluR5-stimulated translation of APP and that Aβ is overexpressed in FXS,56 indicating exaggerated mGluR5-mediated translation as a common pathogenic initiator in these distinct neurological diseases.
Post-traumatic and post-stroke epilepsy
In patient who have sustained head trauma or stroke, there is an increased risk of developing epilepsy over the ensuing years.57 Although the mechanisms for this epileptogenic process have not been fully elucidated and are likely multifactorial, the earliest inciting factor in both trauma and stroke is acute massive glutamate release, which may initiate an injurious process similar to kindling and culminating in the expression of recurrent unprovoked seizures. During the latent period, one can envision subclinical synchronized activity percolating in the network, contributing to a kindling-like phenomenon. The glutamate that instigates this cascade likely activates perisynaptic group I mGluRs, which may be key contributors to the epileptogenic process.
Traumatic brain injury (TBI) has been reproduced with animal models to study the mechanisms of epileptogenesis.58 Chronic cellular modifications induced by TBI include loss of inhibitory interneurons, sprouting of excitatory connections, and increased excitability of pyramidal neurons.59,60 Interestingly, at least some of these changes are activity-dependent because they are prevented by tetrodotoxin.61 Contribution of group I mGluRs to post-traumatic neuronal injury is suggested by the findings in rat models that their activation exacerbates neuronal death in vitro while their pharmacological blockade decreases neuronal loss produced by traumatic brain injury in vivo.62,63 However, whether group I mGluR-dependent neuronal injury or activation of these receptors take any part in epileptogenic processes following TBI is unknown. To assess the therapeutic potential of group I mGluR antagonists in this context is of particular interest since most of the classical antiepileptic drugs tested so far are rather ineffective against post-traumatic epilepsy.64
It is clinically recognized that only a subpopulation of patients with strokes or head trauma develop epilepsy, and at present we do not have the means to predict accurately which patients will have this outcome. We can speculate, however, that any patient with hyperexcitable group I mGluRs, either due to direct mutations or lack of sufficient endogenous repression, will be more prone to the epileptogenic process and may benefit from treatments targeting this pathway during the latent period, before their first clinical seizure is experienced. Advances in genetic testing may someday allow us to identify these patients. It is interesting to note that animals deficient in the expression of group I mGluRs have been shown to be particularly resistant to developing epilepsy following an episode of status epilepticus.65 Although we are a long way from fully understanding the complex role of group I mGluRs in seizure expression and epileptogenesis, there are clear indications that such studies may have broad implications toward the development of new therapeutic strategies for a wide variety of neurological conditions associated with epilepsy.
REFERENCES
- 1.
Taylor GW, Merlin LR, Wong RKS. Synchronized oscillations in hippocampal CA3 neurons induced by metabotropic glutamate receptor activation.
J Neurosci. 1995;15:8039–8052. [
PMC free article: PMC6577924] [
PubMed: 8613741]
- 2.
McDonald JW, Fix AS, Tizzano JP, Schoepp DD. Seizures and brain injury in neonatal rats induced by 1S, 3R-ACPD, a metabotropic glutamate receptor agonist.
J Neurosci. 1993;13:4445–4455. [
PMC free article: PMC6576384] [
PubMed: 8410197]
- 3.
Baude A, Nusser Z, Roberts JDB, Mulvihill E, McIlhinney RAJ, Somogyi P. The metabotropic glutamate receptor (mGluR1α) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction.
Neuron. 1993;11:771–787. [
PubMed: 8104433]
- 4.
Merlin LR, Wong RKS. Role of group I metabotropic glutamate receptors in the patterning of epileptiform activities in vitro.
J Neurophysiol. 1997;78:539–544. [
PubMed: 9242303]
- 5.
Anwyl R. Metabotropic glutamate receptor-dependent long-term potentiation.
Neuropharmacology. 2009;56:735–740. [
PubMed: 19705571]
- 6.
- 7.
- 8.
Ferraguti F, Crepaldi L, Nicoletti F. Metabotropic glutamate 1 receptor: current concepts and perspectives.
Pharmacol Rev. 2008;60:536–581. [
PubMed: 19112153]
- 9.
Huszár P, Merlin LR. Contribution of GABA
B receptor-mediated inhibition to the expression and termination of group I mGluR-induced ictaform bursts.
Epilepsy Res. 2004;61:161–165. [
PubMed: 15451017]
- 10.
Merlin LR, Bergold PJ, Wong RKS. Requirement of protein synthesis for group I mGluR-mediated induction of epileptiform discharges.
J Neurophysiol. 1998;80:989–993. [
PubMed: 9705485]
- 11.
Merlin LR. Group I mGluR-mediated silent induction of long-lasting epileptiform discharges.
J Neurophysiol. 1999;82:1078–1081. [
PubMed: 10444701]
- 12.
Merlin LR. Differential roles for mGluR1 and mGluR5 in the persistent prolongation of epileptiform bursts.
J Neurophysiol. 2002;87:621–625. [
PubMed: 11784776]
- 13.
Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms.
Trends Neurosci. 1993;16:521–527. [
PubMed: 7509523]
- 14.
Galoyan S, Merlin LR. Long-lasting potentiation of epileptiform bursts by group I mGluRs is NMDA receptor independent.
J Neurophysiol. 2000;83:2463–2467. [
PubMed: 10758148]
- 15.
- 16.
- 17.
Chuang S-C, Bianchi R, Kim D, Shin H-S, Wong RKS. Group I metabotropic glutamate receptors elicit epileptiform discharges in the hippocampus through PLCβ1 signaling.
J Neurosci. 2001;21:6387–6394. [
PMC free article: PMC6763182] [
PubMed: 11487662]
- 18.
Cuellar JC, Griffith EL, Merlin LR. Contrasting roles of protein kinase C in induction versus suppression of group I mGluR-mediated epileptogenesis in vitro.
J Neurophysiol. 2005;94:3643–3647. [
PubMed: 16049142]
- 19.
Zhao W, Bianchi R, Wong RKS. Ca2+ store-dependent and -independent population activity induced by the activation of group I mGluRs in the hippocampus Program #55914. 2001 Neuroscience Meeting Planner; San Diego, CA: Society for Neuroscience; (Online abstract)
- 20.
McDonald JW, Fix AS, Tizzano JP, Schoepp DD. Seizures and brain injury in neonatal rats induced by 1S,3R-ACPD, a metabotropic glutamate receptor agonist.
J Neurosci. 1993;13:4445–4455. [
PMC free article: PMC6576384] [
PubMed: 8410197]
- 21.
Zhao W, Bianchi R, Wang M, Wong RKS. Extracellular signal-regulated kinase 1/2 is required for the induction of group I metabotropic glutamate receptor-mediated epileptiform discharges.
J Neurosci. 2004;24:76–84. [
PMC free article: PMC6729577] [
PubMed: 14715940]
- 22.
Klancnik JM, Cuénod M, Gähwiler BH, Jiang ZP, Do KQ. Release of endogenous amino acids, including homocysteic acid and cysteine sulphinic acid, from rat hippocampal slices evoked by electrical stimulation of Schaffer collateral-commissural fibres.
Neuroscience. 1992;49:557–570. [
PubMed: 1354337]
- 23.
Boss V, Nutt KM, Conn PJ. L-cysteine sulfinic acid as an endogenous agonist of a novel metabotropic receptor coupled to stimulation of phospholipase D activity.
Mol Pharmacol. 1994;45:1177–1182. [
PubMed: 8022410]
- 24.
Albani-Torregrossa S, Attucci S, Marinozzi M, Pellicciari R, Moroni F, Pellegrini-Giampietro DE. Antagonist pharmacology of metabotropic glutamate receptors coupled to phospholipase D activation in adult rat hippocampus: focus on (2R,1′S,2′R,3′S)-2-(2′-carboxy-3′-phenylcyclopropyl)glycine versus 3,5-dihydroxyphenylglycine.
Mol Pharmacol. 1999;55:699–707. [
PubMed: 10101028]
- 25.
Rico MJ, Merlin LR. Evidence that phospholipase D activation prevents group I mGluR-induced persistent prolongation of epileptiform bursts.
J Neurophysiol. 2004;91:2385–2388. [
PubMed: 14695353]
- 26.
- 27.
O’Connor JJ, Rowan MJ, Anwyl R. Long-lasting enhancement of NMDA receptor-mediated synaptic transmission by metabotropic glutamate receptor activation.
Nature. 1994;367:557–559. [
PubMed: 7906392]
- 28.
Traub RD, Jefferys JGR, Whittington MA. Enhanced NMDA conductance can account for epileptiform activity induced by low Mg
2+ in the rat hippocampal slice.
J Physiol (Lond). 1994;478:379–393. [
PMC free article: PMC1155660] [
PubMed: 7965853]
- 29.
- 30.
Young SR, Bianchi R, Wong RKS. Signaling mechanisms underlying group I mGluR-induced persistent AHP suppression in CA3 hippocampal neurons.
J Neurophysiol. 2008;99:1105–1118. [
PubMed: 18184892]
- 31.
Chuang S-C, Bianchi R, Wong RKS. Group I mGluR activation turns on a voltage-gated inward current in hippocampal pyramidal cells.
J Neurophysiol. 2000;83:2844–2853. [
PubMed: 10805682]
- 32.
Young SR, Chuang S-C, Wong RKS. Modulation of afterpotentials and firing pattern in guinea pig CA3 neurones by group I metabotropic glutamate receptors.
J Physiol. 2004;554:371–385. [
PMC free article: PMC1664775] [
PubMed: 14578486]
- 33.
Wong RKS, Chuang S-C, Bianchi R. Plasticity mechanisms underlying mGluR-induced epileptogenesis.
Adv Exp Med Biol. 2004;548:69–75. [
PubMed: 15250586]
- 34.
Chuang S-C, Zhao W, Young SR, Conquet F, Bianchi R, Wong RKS. Activation of group I mGluRs elicits different responses in murine CA1 and CA3 pyramidal cells.
J Physiol. 2002;541:113–121. [
PMC free article: PMC2290298] [
PubMed: 12015424]
- 35.
Charpak S, Gähwiler BH, Do KQ, Knöpfel T. Potassium conductances in hippocampal neurons blocked by excitatory amino-acid transmitters.
Nature. 1990;347:765–767. [
PubMed: 2172830]
- 36.
Chuang S-C, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RKS. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model.
J Neurosci. 2005;25:8048–8055. [
PMC free article: PMC6725444] [
PubMed: 16135762]
- 37.
Zhong J, Chuang S-C, Bianchi R, Zhao W, Lee H, Fenton AA, Wong RKS, Tiedge H. BC1 regulation of metabotropic glutamate receptor-mediated neuronal excitability.
J Neurosci. 2009;29:9977–9986. [
PMC free article: PMC2866649] [
PubMed: 19675232]
- 38.
- 39.
- 40.
- 41.
Wang H, Iacoangeli A, Popp S, Muslimov IA, Imataka H, Sonenberg N, Lomakin IB, Tiedge H. Dendritic BC1 RNA: functional role in regulation of translation initiation.
J Neurosci. 2002;22:10232–10241. [
PMC free article: PMC1828542] [
PubMed: 12451124]
- 42.
- 43.
- 44.
Aschrafi A, Cunningham BA, Edelman GM, Vanderklish PW. The fragile X mental retardation protein and group I metabotropic glutamate receptors regulate levels of mRNA granules in brain.
Proc Natl Acad Sci USA. 2005;102:2180–2185. [
PMC free article: PMC548595] [
PubMed: 15684045]
- 45.
Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation.
Trends Neurosci. 2004;27:370–377. [
PubMed: 15219735]
- 46.
- 47.
Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP.
Neuropharmacology. 2005;49:1053–1066. [
PubMed: 16054174]
- 48.
Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchison J, Snape M, Tranfaglia M, Nguyen DV, Hagerman R. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome.
J Med Genet. 2009;46:266–271. [
PMC free article: PMC2658751] [
PubMed: 19126569]
- 49.
Isacson O, Seo H, Lin L, Albeck D, Granholm AC. Alzheimer’s disease and Down’s syndrome: roles of APP, trophic factors and ACh.
Trends Neurosci. 2002;25:79–84. [
PubMed: 11814559]
- 50.
Lott IT, Head E, Doran E, Busciglio J. Beta-amyloid, oxidative stress and Down syndrome.
Curr Alzheimer Res. 2006;3:521–528. [
PubMed: 17168651]
- 51.
Menéndez M. Down syndrome, Alzheimer’s disease and seizures.
Brain Dev. 2005;27:246–252. [
PubMed: 15862185]
- 52.
Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy.
J Neurosci. 2009;29:3453–3462. [
PMC free article: PMC6665248] [
PubMed: 19295151]
- 53.
Westmark CJ, Westmark PR, Beard AM, Hildebrandt SM, Malter JS. Seizure susceptibility and mortality in mice that over-express amyloid precursor protein.
Int J Clin Exp Pathol. 2008;1:157–168. [
PMC free article: PMC2480559] [
PubMed: 18784809]
- 54.
- 55.
- 56.
- 57.
Hauser WA, Annegers JF, Kurland LT. Prevalence of epilepsy in Rochester, Minnesota: 1940–1980.
Epilepsia. 1991;32:429–445. [
PubMed: 1868801]
- 58.
Pitkänen A, McIntosh TK. Animal models of post-traumatic epilepsy.
J Neurotrauma. 2006;23:241–261. [
PubMed: 16503807]
- 59.
- 60.
Prince DA, Parada I, Scalise K, Graber K, Jin X, Shen F. Epilepsy following cortical injury: cellular and molecular mechanisms as targets for potential prophylaxis.
Epilepsia. 2009;50(Suppl 2):30–40. [
PMC free article: PMC2710960] [
PubMed: 19187292]
- 61.
Graber KD, Prince DA. Tetrodotoxin prevents posttraumatic epileptogenesis in rats.
Ann Neurol. 1999;46:234–242. [
PubMed: 10443889]
- 62.
- 63.
Movsesyan VA, O’Leary DM, Fan L, Bao W, Mullins PGM, Knoblach SM, Faden AI. mGluR5 antagonists 2-methyl-6-(phenylethynyl)-pyridine and (E)-2-methyl-6-(2-phenylethenyl)-pyridine reduce traumatic neuronal injury in vitro and in vivo by antagonizing N-methyl-D-aspartate receptors.
J Pharmacol Exp Ther. 2001;296:41–47. [
PubMed: 11123360]
- 64.
Temkin NR. Preventing and treating posttraumatic seizures: the human experience.
Epilepsia. 2009;50(Suppl 2):10–13. [
PubMed: 19187289]
- 65.
Chen J, Larionov S, Pitsch J, Hoerold N, Ullmann C, Elger CE, Schramm J, Becker AJ. Expression analysis of metabotropic glutamate receptors I and III in mouse strains with different susceptibility to experimental temporal lobe epilepsy.
Neurosci Lett. 2005;375:192–197. [
PubMed: 15694259]