Summary
Clinical characteristics.
Spondylocostal dysostosis (SCDO), defined radiographically as multiple segmentation defects of the vertebrae in combination with abnormalities of the ribs, is characterized clinically by a short trunk in proportion to height; short neck; and non-progressive mild scoliosis in most affected individuals – rarely, more significant scoliosis occurs. Respiratory function in neonates with severe disease may be compromised by reduced size of the thorax. By age two years lung growth may improve sufficiently to support relatively normal growth and development. In severely affected individuals with restricted pulmonary capacity, there is a possibility that pulmonary hypertension may eventually impact cardiac function. Males with SCDO appear to be at increased risk for inguinal hernia.
Diagnosis/testing.
The diagnosis of SCDO is based on radiographic features. Identification of biallelic pathogenic variants in DLL3, HES7, LFNG, MESP2, RIPPLY2, or TBX6 can confirm the diagnosis of autosomal recessive SCDO.
Management.
Treatment of manifestations: Surgical intervention may be necessary when scoliosis is significant; external bracing (e.g., by use of a vertical expandable prosthetic titanium rib) may be considered, as well as growing rods and other devices as appropriate. Respiratory support, including intensive care, is provided as needed for the small proportion of individuals with acute respiratory distress and chronic respiratory failure. Expert management is indicated for chronic respiratory failure, which can result in pulmonary hypertension and cardiac failure. Standard treatment of neurologic problems associated with LFNG-related SCDO. Inguinal hernias are treated per routine.
Surveillance: Growth, spinal curvature, respiratory function, neurologic and motor function, and development should be monitored. The parents / care providers of young males need to be alert for the signs of inguinal hernia and its potential complications.
Genetic counseling.
SCDO caused by biallelic pathogenic variants in DLL3, HES7, LFNG, MESP2, RIPPLY2, or TBX6 is inherited in an autosomal recessive manner. (Autosomal dominant inheritance of TBX6-related SCDO has been reported in a three-generation family.) If both parents are known to be heterozygous for an autosomal recessive SCDO-causing pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the autosomal recessive SCDO-related pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible. In experienced hands, detailed fetal ultrasound scanning is sensitive enough to detect multiple segmentation defects of the vertebrae as early as 13 weeks' gestation, especially when the malformation is anticipated and looked for specifically. However, molecular genetic testing of an at-risk pregnancy is considered the gold standard for accurate prenatal diagnosis.
Clinical Characteristics
Clinical Description
Spondylocostal dysostosis (SCDO), defined radiographically as multiple segmentation defects of the vertebrae that is usually generalized throughout the spine, is characterized clinically by a short trunk in proportion to height, short neck, and non-progressive mild scoliosis in most affected individuals. To date, nearly 100 individuals have been identified and/or reported with SCDO and biallelic pathogenic variants in one of the genes listed in Table 1. The following description of the phenotypic features associated with this condition is based on the cited reports.
Skeletal. Multiple segmentation defects of the vertebrae, which is usually generalized throughout the spine, results in:
A short trunk in proportion to height. The extent varies and the data is very limited, but based on leg length measurements, individuals with SCDO are 10% shorter than projected adult height. Some individuals have severe short stature, with height up to four standard deviations below the mean [
Sparrow et al 2006;
Schuhmann et al 2021; P Turnpenny, personal communication].
LNFG-related SCDO appears to be associated with shorter stature compared to other causes of SCDO (see ). The reason(s) for this variability is not well understood.
Short neck. The extent varies, and the data is limited, but – similar to the decrease in overall spine length – the neck is likely to be shortened by approximately 10%. The range of limitation in neck mobility has not been formally assessed.
Non-progressive, or mildly progressive but self-limiting, scoliosis occurs in most affected individuals, usually apparent radiographically in infancy. More significant scoliosis, with a greater degree of progression, especially at the thoracolumbar region, is apparent in individuals with
LFNG-related SCDO [
Sparrow et al 2006,
Takeda et al 2018,
Schuhmann et al 2021]. Severe scoliosis, including the need for scoliosis surgery, appears to be relatively rare [A Cornier, personal communication; P Turnpenny, personal communication].
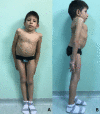
Individual with LFNG-related SCDO at age seven years nine months showing marked truncal shortening Courtesy of Prof Eleni Fryssira, Athens, Greece. LFNG analysis performed in Lausanne, Switzerland.
Respiratory. The most important consideration in neonates diagnosed with SCDO is impaired respiratory function, which may be compromised by reduced size of the thorax. In these infants, respiratory insufficiency may be the presenting clinical problem. Life-threatening respiratory insufficiency requiring neonatal intensive care appears to be rare but anecdotally has been known to occur. One individual died in infancy from respiratory insufficiency and at postmortem was found to have a membranous left hemidiaphragm [Turnpenny et al 1999]. This has not been reported in any other individuals with SCDO. The most significant potential secondary complication is chronic respiratory failure caused by reduced lung capacity in individuals with severe disease.
In children requiring early respiratory support, lung growth may improve sufficiently to support relatively normal growth and development by age two years. However, life-threatening complications can occur, especially pulmonary hypertension and cardiac failure in individuals with severely restricted lung capacity from birth. There is no systematic review concerning susceptibility to pulmonary infection and pneumonia or incidence of pulmonary hypertension.
Inguinal hernia. Males with SCDO appear to be at increased risk for inguinal hernia, which has been noted in the neonatal period [Turnpenny et al 1999, Turnpenny et al 2003, Otomo et al 2019].
Neurologic complications appear to be rare. Lumbosacral meningomyelocele was reported in one individual [Sparrow et al 2008] and a neural tube defect occurred in a second individual [Authors, personal communication] with HES7-related SCDO. Syringomyelia was identified at age seven years in one individual with LFNG-related SCDO on spine MRI performed due to new balance problems. By age ten years this individual had urinary incontinence; he was intellectually normal [E Fryssira and M Christodoulou, personal communication]. Distal arthrogryposis was reported in one individual with LFNG-related SCDO; it was not clear whether this was a primary abnormality or secondary to impingement of neural pathways in the cervical vertebrae [Sparrow et al 2006].
Other
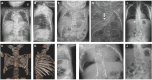
Cosegregation of dextrocardia was reported in a large consanguineous Middle Eastern kindred with
HES7-related SCDO (see ) [
Sparrow et al 2013a]; whether this was due to
HES7-related SCDO or a separate genetic cause has not been established.
Solitary pelvic kidney, uterine dysgenesis, absence epilepsy, and inner ear (presumed sensorineural) deafness were reported in one individual with
LFNG-related SCDO [
Schuhmann et al 2021].
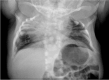
individuals with HES7-related SCDO compared to an unaffected individual. X-ray and MRI images showing vertebral and rib malformations and dextrocardia. Seven affected individuals from three families, all with the same homozygous pathogenic HES7 variant, (more...)
Prognosis. In the absence of restricted lung capacity, individuals with SCDO have normal life expectancy. The risk of pulmonary hypertension and associated complications is unknown.
Phenotype Correlations by Gene
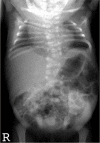
DLL3. Scoliosis is generally mild and non-progressive, and the need for surgical intervention to stabilize the spine is rare. However, more significant scoliosis has been observed in some individuals (see ) [A Cornier, personal communication].
Radiograph of infant with DLL3-related SCDO and unusually severe scoliosis
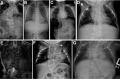
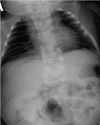
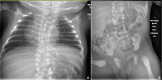
HES7. Radiographic features in the limited number of published individuals have ranged from resembling spondylothoracic dysostosis (STD) [Sparrow et al 2008] (see ) to those typical in DLL3-related SCDO; all vertebrae display abnormal segmentation.
Radiograph of child with HES7-related SCDO. Segmentation anomalies of all vertebrae are severe. The vertebral pedicles are relatively prominent ("tramline" sign) compared with those of DLL3-related SCDO. These radiographic findings resemble those of spondylothoracic (more...)
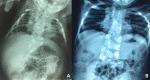
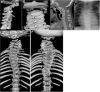
LFNG. Shortening of the spine and scoliosis appear to be more severe in individuals with LFNG-related SCDO compared to that seen in DLL3-, HES7-, and MESP2-related SCDO, because all vertebral bodies appear to show more severe segmentation defects (see , , and ) [Lefebvre et al 2018]. Rib anomalies are similar to those seen in DLL3- and MESP2-related SCDO.
Radiographs of a child with LFNG-related SCDO A. Spine radiograph as a neonate. The pattern of malsegmentation is not clearly distinguishable from typical findings in DLL3-related SCDO.
Segmentation defects of the vertebrae of the entire spine with angulated vertebral bodies (dotted lines) at birth in an individual with LNFG-related SCDO (patient 18 of Lefebvre et al [2018]) Reprinted with permission from Lefebvre et al [2018]
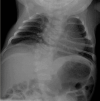
MESP2. Spine radiographs in individuals with MESP2-related SCDO show at least some disruption of all vertebral segments. However, lumbar vertebrae are relatively mildly affected compared to thoracic vertebrae (see ). In the limited reports thus far, the ribs tend to be straight and more regularly aligned than in other forms of SCDO (i.e., demonstrating fewer points of fusion).
Radiographs of a child with MESP2-related SCDO. The generalized segmentation defects of the vertebrae show more angular features than is typical of DLL3-related SCDO.
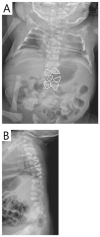
RIPPLY2. Two brothers with RIPPLY2-related SCDO had vertebral segmentation defects affecting the posterior elements of C1-C4 and hemivertebrae and butterfly vertebrae of T2-T7 (see ). Marked cervical kyphosis at C2-C3 was associated with cord compression, and mild thoracic scoliosis was present [McInerney-Leo et al 2015]. Three individuals from two families, with the same pathogenic variant [Serey-Gaut et al 2020], had agenesis of the posterior and lateral elements of most cervical vertebrae, with limited and variable involvement of some thoracic vertebrae. The radiologic pattern was distinct from other forms of SCDO, and RIPPLY2-related SCDO may be better categorized as a form of Klippel-Feil anomaly.
Imaging of individuals with RIPPLY-related SCDO A-C. 3D CT of male age 15 months with RIPPLY-related SCDO, showing failure of formation of the posterior elements of C1-C4 with descent of the occipital bone, resulting in canal stenosis and cord compression (more...)
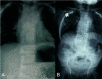
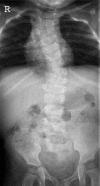
TBX6. Although the number of reported individuals with TBX6-related SCDO is limited, this phenotype resembles that of DLL3-related SCDO. Radiologically it is almost indistinguishable (see ) [C Shaw-Smith, personal communication].
Thoracic (A) and lumbar spine (B) radiographs of an infant with TBX6-related SCDO Courtesy of Charles Shaw-Smith, Exeter, UK
See for radiographic comparison of DLL3-, LFNG-, HES7-, and TBX6-related SCDO and MESP2-related STD.
Radiologic features for the different genes identified in a cohort of individuals with regional multiple segmentation defects of the vertebrae A-D. An individual with DLL3-related SCDO
Genotype-Phenotype Correlations
DLL3. The radiographic features of DLL3-related SCDO appear to be very consistent (see ). However, two individuals homozygous for DLL3 pathogenic missense variants in the region encoding the EGF domain had slightly milder phenotypes (see ). Some evidence suggests that these pathogenic missense variants would allow the EGF domains to adopt the correct fold in the DLL3 protein but perhaps be thermodynamically less stable than the wild type protein [Authors, unpublished data]. However, some of the pathogenic missense variants identified in affected individuals cause a phenotype that is indistinguishable from that caused by DLL3 pathogenic truncating variants. This probably results from the different effects conferred upon protein folding compared to those pathogenic missense variants associated with the slightly milder phenotype.
Radiograph of a child with a mild form of DLL3-related SCDO. All vertebrae show at least some relatively mild segmentation abnormality.
MESP2. The 4-bp duplication c.500_503dup occurs after the basic helix-loop-helix (bHLH) domain and causes a frameshift resulting in a premature stop codon within the second (and final) MESP2 exon [Whittock et al 2004b]. Transcripts with this pathogenic variant would not be subject to nonsense-mediated decay. Individuals with this pathogenic variant are predicted to have a truncated protein containing an intact bHLH domain, which may retain some function. In contrast, the pathogenic nonsense variants identified in spondylothoracic dysostosis (STD) (see Genetically Related Disorders) are located within the first exon, and the resulting mutated mRNA transcripts are predicted to be susceptible to nonsense-mediated decay. Therefore, persons homozygous or compound heterozygous for these pathogenic nonsense variants are likely to have reduced or absent levels of MESP2 protein, which may account for the difference in severity between the MESP2-related SCDO and STD phenotypes.
TBX6. See Genetically Related Disorders for genotype-phenotype correlations observed in allelic disorders.
No genotype-phenotype correlations for HES7, LFNG, or RIPPLY2 have been identified.
Penetrance
To date, penetrance appears to be complete for the pathogenic variants implicated in autosomal recessive SCDO.
Nomenclature
The term Jarcho-Levin syndrome (JLS) [Jarcho & Levin 1938] has been used (confusingly) to refer to:
All radiologic phenotypes that include segmentation defects of the vertebrae (SDV) and abnormal rib alignment, including reports of phenotypes that are neither similar to the case description of
Jarcho & Levin [1938] nor consistent with spondylothoracic dysostosis (STD);
Use of the terms costovertebral dysplasia and spondylothoracic dysostosis/dysplasia for segmentation abnormalities of the spine and ribs has led to great confusion. Note: These disorders are dysostoses rather than dysplasias:
The wide range of radiologic phenotypes with multiple segmentation defects of the vertebrae (M-SDV) within SCDO has highlighted the need to rationalize nomenclature for these diverse and poorly understood disorders. The International Consortium for Vertebral Anomalies and Scoliosis (ICVAS), now subsumed into the International Consortium for Scoliosis Genetics Development and Disease (ICSGDD), proposed two algorithms:
The clinical algorithm, used for routine reporting of SDV, identifies seven broad categories (see ). For the purposes of clinical reporting, additional comments can describe SDV findings in more detail.
The research algorithm, used for more detailed documentation of SDV, employs ontology applicable to humans and animal models (see ).
ICVAS clinical classification algorithm All forms of SDV can be placed in one of seven broad categories. The classification combines a descriptive approach for the diverse radiologic phenotypes encountered in clinical practice with specific diagnoses (more...)
ICVAS research classification algorithm: a more detailed, systematic analysis of radiographic anatomic features. Documentation of phenotypes in a systematic ontology facilitates direct interspecies comparison and stratification of patient cohorts for (more...)
Note: In the classification system proposed by the ICVAS, SCDO is the preferred term for generalized segmentation defects of the vertebrae (G-SDV) with rib involvement [Turnpenny et al 2007, Offiah et al 2010].
Klippel-Feil anomaly (KFA) refers to cervical vertebral fusion anomalies. The term "KFA" is used broadly for a number of phenotypes.
Prevalence
DLL3-related SCDO. Seventy-five percent of individuals have been the offspring of consanguineous unions (Exeter Laboratory experience), mostly of Middle Eastern or Pakistani origin, and occasionally of European origin and elsewhere. A small number of individuals from northern Europe (England, Wales, the Netherlands, and Switzerland) have been shown to be compound heterozygotes [Bonafé et al 2003, Whittock et al 2004a]. Assuming a period of time during which approximately one million births occurred, the carrier frequency in the European population in the UK would be approximately 1:350.
HES7-, LFNG-, MESP2-, and RIPPLY2-related SCDO have been reported in only a small number of individuals [Whittock et al 2004b, Bonafé & Superti-Furga 2005, Sparrow et al 2006, Sparrow et al 2008, Sparrow et al 2010, Sparrow et al 2013a, Lefebvre et al 2017]. TBX6-related SCDO has been reported more often, suggesting it is the second most common form of SCDO after DLL3-related SCDO.
Differential Diagnosis
Rarely, spondylocostal dysostosis (SCDO) occurs in association with chromosome abnormalities; however, apart from trisomy 8 mosaicism, no consistent genomic region has been involved, and the significance of these associations is unknown.
Autosomal dominant SCDO. One family with autosomal dominant SCDO due to a heterozygous TBX6 pathogenic variant has been reported (OMIM 122600). Additional families with autosomal dominant SCDO without an identified gene have also been reported; in these families the extent of segmentation defects of the vertebrae is quite variable [Rimoin et al 1968, Kubryk & Borde 1981, Temple et al 1988, Lorenz & Rupprecht 1990].
Spondylothoracic dysostosis (STD), despite similarities to autosomal recessive SCDO, has distinctive phenotypic features that warrant this separate designation. Infants with STD are at the highest risk for respiratory insufficiency and have a nearly 50% mortality rate by the end of infancy [Cornier et al 2004]. To date, most individuals reported with STD have had pathogenic nonsense variants in exon 1 of MESP2 (see Genetically Related Disorders). The differences in the radiographic findings in STD that distinguish it from SCDO include the following (see ):
More severe shortening of the spine (all vertebral segments affected), especially the thoracic spine, leading to impaired respiratory function in infancy
Rib fusions typically occurring posteriorly at the costovertebral origins, where the spinal shortening is most severe. The ribs usually appear straight and neatly aligned without points of fusion along their length. On anteroposterior x-ray the ribs characteristically "fan out" from their costovertebral origins in a "crab-like" fashion.
A distinctive radiographic appearance called the "tramline sign" that results from early radiographic prominence of the vertebral pedicles, in contrast to the vertebral bodies, which have no regular form or layout [
Turnpenny et al 2007].
Segmentation defects of the vertebrae (SDV) are estimated to occur in 0.5-1.0 in 1,000 live births, but in clinical practice the radiologic phenotypes and syndromic associations are extremely diverse. Syndromic forms of multiple segmentation defects of the vertebrae (M-SDV) should be considered if the diagnostic criteria for SCDO or STD are not met. For most individuals the underlying cause is not known, but an increasing number of genes are being identified. Some of the M-SDV syndromes to consider are listed in Tables 2a and 2b.
Table 2a.
Selected Genes Associated With M-SDV (SCDO and STD excluded)
View in own window
AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance; M-SDV = multiple segmentation defects of the vertebrae; RAPADILINO = radial ray defect, patellae hypoplasia or aplasia and cleft or highly arched palate, diarrhea and dislocated joints, little size and limb malformation, nose slender and normal intelligence; SCDO = spondylocostal dysostosis; STD = spondylothoracic dysostosis; XL = X-linked
- 1.
KBG syndrome is caused by either a heterozygous pathogenic variant in ANKRD11 or deletion of 16q24.3 that includes ANKRD11. Recurrence risk for sibs of a proband with KBG syndrome depends on the genetic alteration.
- 2.
Table 2b.
Other Syndromes/Conditions That Include M-SDV (SCDO and STD excluded)
View in own window
| Syndrome/Condition |
|---|
| 22q11.2 deletion syndrome (DiGeorge syndrome / velocardiofacial syndrome) |
| Chromosome abnormalities |
| Maternal diabetes mellitus |
| Syndromes of unknown genetic cause | Casamassima-Morton-Nance syndrome (OMIM 271520) (SDV & urogenital anomalies) |
| Cleft-limb-heart malformation syndrome (OMIM 215850) |
| Dyssegmental dysplasia, Rolland-Desbuquois type (OMIM 224400) |
| Facial dysmorphism with multiple malformations (OMIM 227255) |
| Femoral hypoplasia-unusual facies syndrome (OMIM 134780) |
| Lower mesodermal agenesis |
| OEIS complex (OMIM 258040) |
| Phaver syndrome (OMIM 261575) |
| Spinal dysplasia, Anhalt type (OMIM 601344) |
| Limb deficiency-vertebral hypersegmentation-absent thymus 1 |
| VATER/VACTERL (OMIM 192350) |
| Wildervanck syndrome (OMIM 314600) |
M-SDV = multiple segmentation defects of the vertebrae; SCDO = spondylocostal dysostosis; STD = spondylothoracic dysostosis
- 1.
Note: A single individual with an SCDO-like phenotype with multiple regional segmentation defects of the vertebrae, multiple intervertebral fusions of laminae, dysmorphic features, and cleft palate has been reported in association with homozygosity for a start-loss variant in DMRT2 [Bouman et al 2018]. With severe left-sided rib cage deficiency, the infant died at age nine days. It is not yet known if DMRT2-related SCDO is a distinct entity.
Neural tube defects are also frequently associated with adjacent severe segmentation anomalies of one or more vertebrae. However, current consensus is that the diagnosis of SCDO should be reserved for individuals with abnormal segmentation of at least ten contiguous vertebrae.
Management
No clinical practice guidelines for autosomal recessive spondylocostal dysostosis (SCDO) have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with autosomal recessive SCDO, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Autosomal Recessive Spondylocostal Dysostosis: Recommended Evaluations Following Initial Diagnosis
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Skeletal
| Full spine x-rays, AP & lateral x-rays, & chest x-rays | |
|
Respiratory
| Assessment of respiratory function per pulmonologist, esp if tachypnea &/or feeding difficulties suggest possibility of respiratory insufficiency | |
|
Gastrointestinal
|
| |
Kidneys /
Urinary tract
| Ultrasound eval of kidneys & urinary tract | In persons w/LFNG-related SCDO |
|
Neurologic
| MRI of full spinal cord to assess for assoc neurologic complications |
|
Genetic counseling
| By genetics professionals 1 | To inform affected persons & their families re nature, MOI, & implications of AR SCDO to facilitate medical & personal decision making |
Family support
& resources
| Assess need for:
| |
AR = autosomal recessive; MOI = mode of inheritance; SCDO = spondylocostal dysostosis
- 1.
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Treatment of Manifestations
In the majority of individuals, treatment is conservative because the clinical manifestations of the vertebral and rib malformations do not increase with age.
Table 4.
Autosomal Recessive Spondylocostal Dysostosis: Treatment of Manifestations
View in own window
| Manifestation/Concern | Treatment | Considerations/Other |
|---|
|
Growth
| No specific nutritional needs to be considered other than maintaining appropriate weight for height | Persons w/SCDO all have variable short-trunk short stature. |
|
Scoliosis
| Surgical intervention as needed if scoliosis is significant; severe scoliosis is unusual. | External bracing (e.g., using vertical expandable prosthetic titanium rib) 1 may be considered, as well as growing rods & other devices as appropriate. |
|
Respiratory distress/failure
| Respiratory support, depending on extent of pulmonary compromise (usually only necessary in severe disease) Assessment for complications of respiratory disease, incl pulmonary hypertension & cardiac failure as indicated
| |
|
Neurologic
| Standard treatment of neurologic problems assoc w/LFNG-related SCDO according to findings & symptoms | |
|
Inguinal hernia
|
| |
SCDO = spondylocostal dysostosis
- 1.
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 5 are recommended.
Table 5.
Autosomal Recessive Spondylocostal Dysostosis: Recommended Surveillance
View in own window
| System/Concern | Evaluation | Frequency |
|---|
|
Growth/Nutrition
| Assess growth. | At each visit throughout childhood |
|
Skeletal
| Assessment for spinal curvature | Annually or as needed |
|
Respiratory
| Assessment of respiratory function |
|
Neurologic
| Assessment of neurologic & motor function | Annually or as needed in those w/LFNG-related SCDO |
|
Development
| Developmental assessment | Annually or as needed |
|
Gastrointestinal
| The parents /care providers of young males need to be alert for signs of inguinal hernia & its potential complications. | Annually or as needed |
SCDO = spondylocostal dysostosis
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Virtually all individuals with SCDO have relative truncal shortening, and some have generalized short stature. For affected women, pregnancy may give rise to exaggerated intra-abdominal pressure problems, though there is no published research on this issue. As the spine is distorted, there are likely to be concerns with offering spinal and/or epidural anesthesia. However, spinal anesthesia has been successfully administered [Dolak & Tartt 2009].
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Spondylocostal dysostosis (SCDO) caused by biallelic pathogenic variants in DLL3, HES7, LFNG, MESP2, RIPPLY2, or TBX6 is inherited in an autosomal recessive manner.
Note: Autosomal dominant inheritance of TBX6-related SCDO has been reported in a three-generation family (all affected family members were male) [Sparrow et al 2013b]. Autosomal dominant inheritance is not discussed further in this section.
Pseudodominant inheritance. Although rare, there have been reports of SCDO appearing to be inherited in an autosomal dominant manner, although the extent of segmentation defects of the vertebrae (SDV) is variable [Temple et al 1988, Gucev et al 2010, Sparrow et al 2012]. In one such family [Floor et al 1989] the inheritance pattern was shown to be an example of pseudodominant inheritance (i.e., an autosomal recessive condition present in individuals in two or more generations of a family, thereby appearing to follow a dominant inheritance pattern) of DLL3-related SCDO in a highly consanguineous family [Turnpenny et al 1999, Whittock et al 2004a].
Risk to Family Members (Autosomal Recessive Inheritance)
Parents of a proband
The parents of an affected child are presumed to be heterozygous for an autosomal recessive SCDO-causing pathogenic variant.
If a molecular diagnosis has been established in the proband, molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an autosomal recessive SCDO-causing pathogenic variant and to allow reliable recurrence risk assessment.
If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the proband occurred as a
de novo event in the proband or as a postzygotic
de novo event in a mosaic parent [
Jónsson et al 2017]. If the proband appears to have homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
A single- or multiexon deletion in the proband that was not detected by sequence analysis and that resulted in the artifactual appearance of homozygosity;
Uniparental isodisomy for the parental chromosome with the pathogenic variant that resulted in homozygosity for the pathogenic variant in the proband.
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
If both parents are known to be heterozygous for an autosomal recessive SCDO-causing pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. Unless an affected individual's reproductive partner* has heterozygous or biallelic pathogenic variants in the same autosomal recessive SCDO-related gene as that involved in the proband, offspring will be obligate heterozygotes (carriers) for an autosomal recessive SCDO-causing pathogenic variant.
* Molecular genetic testing for reproductive partners is appropriate, particularly if consanguinity is likely. Approximately 75% of individuals with autosomal recessive SCDO are from consanguineous families, usually from communities in which cousin partnerships are common.
Other family members. Each sib of the proband's parents is at 50% risk of being a carrier of an autosomal recessive SCDO-causing pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the autosomal recessive SCDO-causing pathogenic variants in the family. See Related Genetic Counseling Issues, Family planning.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the autosomal recessive SCDO-related pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Fetal ultrasound examination. In experienced hands, detailed fetal ultrasound scanning is sensitive enough to detect multiple segmentation defects of the vertebrae (M-SDV) as early as 13 weeks' gestation, especially when the malformation is anticipated and looked for specifically. However, molecular genetic testing of an at-risk pregnancy is considered the gold standard for accurate prenatal diagnosis. Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Chapter Notes
Acknowledgments
Research on spondylocostal dysostosis (SCDO) in Exeter has been funded by Action Medical Research, British Scoliosis Research Foundation, and the Skeletal Dysplasia Group, to whom the authors are indebted. In the Exeter Molecular Genetics Laboratory the work was undertaken by Mike Bulman, June Duncan (deceased), Neil Whittock, and recently Melissa Sloman, all under the supervision of Sian Ellard. The work greatly benefited from collaboration with Kenro Kusumi, Sally Dunwoodie, and, more recently, Olivier Pourquié, Philip Giampietro, Alberto Cornier, Amaka Offiah, and Ben Alman, through the ICVAS consortium. Many clinicians have sent images of individuals with segmentation defects of the vertebrae (SDV), but for this review particular thanks are due to Dr Oivind Braaten, Oslo, Norway, and Drs Karin van Spaendonck-Zwarts and Mirjam M de Jong, Groningen, the Netherlands, Professor Eleni Fryysira and Dr Michael Christadoulou, Athens, Greece, and Dr Charles Shaw-Smith, Exeter, UK. Sally Dunwoodie received SCDO research funds from the National Health and Medical Research Council (ID142006, 404804,1044543, 1042002, 1135886).
Author History
Sally Dunwoodie, BSc PhD (2017-present)
Melissa Sloman, BSc, DipRCPath (2017-present)
Peter D Turnpenny, BSc, MB, ChB, FRCP, FRCPCH, FRCPath (2009-present)
Elizabeth Young, PhD; Royal Devon & Exeter NHS Foundation Trust (2009-2017)
Revision History
17 August 2023 (sw) Comprehensive update posted live
21 December 2017 (sw) Comprehensive update posted live
17 January 2013 (me) Comprehensive update posted live
25 August 2009 (et) Review posted live
6 February 2009 (pdt) Original submission