Historical Perspective
The excitatory potency of the acidic amino acids glutamate and aspartate in various regions of the central nervous system (CNS) has been recognized since the 1960s.1,2 Nevertheless, the earlier findings that these amino acids are (1) constituents of intermediary metabolism and are (2) located in the brain ubiquitously in high concentrations rendered them unlikely candidates as neurotransmitters. These findings fueled a sustained debate about their physiological role as neurotransmitters in the 1970s. Today, L-glutamate is accepted as the predominant fast excitatory neurotransmitter in the vertebrate brain.
In parallel to studies on the physiological role of these amino acids, it has been observed since the 1950s that glutamate can exert toxic effects on the nervous system under certain conditions. Following the systemic application of glutamate to mice, toxic effects on retinal neurons were described.3 Further studies in the 1970s corroborated these toxic effects and extended this view by showing neuronal cell death following oral intake of glutamate or aspartate in brain regions devoid of the blood-brain barrier in mice and nonhuman primates (Fig. 1).4–9
Thus, L-glutamate is the primary excitatory transmitter in the mammalian CNS but is cytotoxic under certain conditions. This relation between the physiological function as excitatory amino acid (EAA) and the pathological effect is reflected by the term “excitotoxicity” introduced in the 1970s by Olney et al.10
With the introduction of structural transmitter analogues, local injections of the glutamate agonist kainate were shown in the late 1970s and 1980s to induce cell death with a similar pattern of damage in different brain regions, thus confirming the neurotoxic effect.11–14 Today, it is well recognized, that exogenous or endogenous agonists of EAA receptors can induce cell death in CNS neurons.
Our knowledge about the role of glutamate as an excitatory neurotransmitter and its cytotoxic effects increased in parallel during the 1980s, and notably, the development of substances that antagonized the excitatory function15 also stimulated studies on mechanisms underlying the toxic effects.16–18 The discovery of different EAA receptor subtypes in conjunction with the introduction of selective receptor antagonists revealed that the glutamate-induced cell death was induced by excessive ionotropic glutamate receptor activation.19–26
The view that activation of different ionotropic EAA receptors can induce excitotoxic cell death was supported by subsequent studies on the ionic mechanisms underlying excitotoxicity. It was demonstrated that excessive calcium loading plays a pivotal role in neuronal cell death following the intense stimulation of ionotropic glutamate receptors,27,28 and since then the implication of ion homeostasis dysregulation and dysfunction in calcium signaling have been studied extensively (Fig. 2 ).29–33
Regarding the mode of cell death, glutamate-induced neuronal cell death has been judged originally as necrotic from the morphological appearance.6,34–36 However, the observation of a delayed neuronal cell death in the penumbra of ischemic lesions37 and after EAA exposure38,39 has stimulated studies in the 1990s focusing on the mode of cell death. Today, there is compelling evidence that failure in extracellular glutamate homeostasis can result in different modes of cell death with morphological and biochemical features of either apoptosis or necrosis depending on the severity of the insult, with more fulminant insults causing rapid energy failure because of lack of ionic homeostasis and thus necrosis.40–44
Clinical Relevance of Excitatory Amino Acid Neurotoxicity
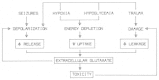
There is evidence that excitotoxicity is involved in acute brain damage under pathophysiological conditions following status epilepticus, mechanical trauma or ischemia (Fig. 3).45 Moreover, glutamate cytotoxicity seems to be partly involved in many neurodegenerative diseases.
Epilepsy
Histopathological studies on the brains of patients suffering from chronic epilepsy have revealed that certain brain regions show structural alterations with severe loss of neurons and reactive gliosis.46,47 Brain pathology in epilepsy is described best for human temporal lobe epilepsy, resulting in sclerosis of the hippocampus that extends into the amygdala and the parahippocampal gyrus and is termed “mesial temporal sclerosis”, “hippocampal sclerosis” or “Ammon's horn sclerosis”. Pronounced brain damage has been observed following sustained epileptiform activity with seizures lasting more than 30 min, called “status epilepticus” (SE).48–50 Importantly, neuronal cell loss after SE is not distributed equally across all hippocampal subfields and the extent of damage is in the order CA1 > CA4 > CA3 > CA2, thus indicating different susceptibility to SE-induced cell death.12,50
In animal models of epilepsy using chemoconvulsant-induced or electrical stimulation-induced SE, similar patterns of brain damage were observed.51–54 Experiments aimed at the observation of ultrastructural changes have demonstrated that certain features of cell damage, such as swelling of dendrites and soma, are independent of the mechanism used to induce SE.51–56
Evidence for the implication of EAA-mediated excitotoxicity in SE-induced neuronal cell death arises from several experiments. First, the morphological appearance of cell damage following SE is similar to damage following systemic or local application of the glutamate receptor agonists L-glutamate, NMDA or kainate.6,36,51,53,55,56 Second, the administration of ionotropic glutamate receptor antagonists that inhibit excitotoxic cell death in cultured neurons can prevent cell death induced by epileptiform activity.57–60
Apart from the above-mentioned evidence for excitotoxicity in epilepsy, there has been a considerable debate regarding the mode of cell death. There is now a large body of evidence suggesting the implication of both, necrotic and apoptotic cell death following pathologically relevant EAA receptor activation. Recently, several reports have added evidence for the implication of apoptotic pathways in epilepsy-associated neuronal cell death.61–66 In conclusion, the mode of cell death in SE-induced brain pathology may be the result of intensity and duration of glutamate receptor activation, with a shift from apoptotic death to necrotic death with increasing intensity and duration of receptor activation.40,43
Traumatic Brain Injury
Following head trauma, mechanical brain injury can be accompanied by secondary changes as ‘metabolic’ glutamate leaks uncontrollably from the neuronal cytoplasm67 causing subsequent excitotoxic damage of surrounding neurons. However, compared to research on mechanisms underlying the cell damage in epilepsy, studies on traumatic injury have been sparse and there is less evidence for the implicated modes of cell death. Some studies have suggested the sudden release of excitatory amino acids from the cytoplasm into the extracellular space and subsequent bioenergetic failure as well as ultrastructural damage that can be diminished by NMDA antagonists, all indicative of excitotoxicity,67–69 whereas others have demonstrated activation of caspase enzymes, internucleosomal DNA fragmentation and induction of immediate early genes, indicative of apoptosis.69–71 Most recently, in neonatal models of traumatic brain injury (TBI) (mortality and morbidity from head trauma is highest in children), the resulting excitotoxicity has been shown to elicit both apoptosis and necrosis. Necrosis occurs localized to the site of impact, and within 4 hr of the insult, whereas a secondary apoptotic damage occurs between 6–24 hr and is found in the areas surrounding the primary necrosis.72 In this model, the secondary damage was more severe than the primary damage, suggesting a preponderance of apoptosis over necrosis.
Hypoxia/Ischemia
Under conditions of local or global ischemia, neurons are deprived of glucose and oxygen, resulting in bioenergetic failure and subsequent decrease of ion gradients across the plasma membrane.73 The resulting plasma membrane depolarization leads to increased synaptic release of glutamate, and the diminished Na gradient is followed by attenuated Na-dependent glutamate uptake or even reversed glutamate transport in terms of transporter-mediated release of glutamate from neurons and astrocytes.74–76 Moreover, osmotic cell swelling following the influx of Na, Cl and H2O can result in plasma membrane rupture and further release of cytoplasmic glutamate into the extracellular space. In summary, hypoxia/ischemia results in a secondary net increase in the extracellular glutamate concentration.77–79 As in the other above-mentioned acute CNS insults, this increase in extracellular glutamate results in excitotoxic damage.80–83 The implication of excitotoxicity in hypoxia/ischemia is corroborated by the observation that NMDA-type glutamate receptor antagonists can reduce neuronal death in animal models of cerebral ischemia.84–87 However, the actual set of circumstances in humans appears to differ from the defined animal models.88 In addition to the well-accepted concept of excitotoxicity-associated acute necrotic cell death, there are several lines of evidence that neurons can undergo apoptosis following ischemia.41,89 Among various biochemical markers of apoptosis, DNA fragmentation90,91 and activation of caspase-3 have been observed63,92 in models for cerebral ischemia. Moreover, application of caspase-inhibitors results in reduced infarct size42,93,94 and decreases cell death in cultured neurons following ischemia.95
Neurodegenerative Diseases
In addition to acute neurological disorders, many chronic neurodegenerative diseases may exhibit a component of glutamate-dependent neuronal damage, including apoptosis or injury to dendrites and axons (Table 1). This arises when the primary disease causes neuronal injury which in turn may cause the leak or release of excessive glutamate. Additionally, elevated inflammatory responses in many of these diseases can also contribute to excessive glutamate release or decreased glutamate clearance from the synaptic cleft.96,97
Table 1
Neurodegenerative diseases thought to be mediated at least in part through stimulation of glutamate receptors.
Intoxication with Exogenous Excitatory Amino Acids
Several structural analogues of endogenous EAAs have been introduced to the neurosciences during the 1980s, which exhibit similar or even higher excitatory and neurotoxic potency compared to the endogenous EAAs. In 1987, an outbreak of domoic acid poisoning following ingestion of mussels occurred in Canada. Patients suffered from acute headache, seizures, sensory dysfunctions and showed motor signs. In four fatal cases, neuropathological studies revealed lesions predominantly in the hippocampus and amygdala, resembling the lesion pattern after application of the exogenous excitotoxin kainate.98 It was reconstructed that the mussels had accumulated domoic acid, synthesized by the phytoplankton Nitzchia pungens.98 Domoic acid is a structural analogue of kainic acid, which is also synthesized by seaweed, but compared to kainate, domoic acid has a higher excitatory potency.99,100 In parallel to its high excitatory potency, domoic acid exerts potent excitotoxic effects on CNS neurons (Table 2). Experimental administration of domoic acid to rodents and monkeys has resulted in brain damage with ultrastructural features resembling L-glutamate excitotoxicity.101–103
Table 2
Exogenous excitotoxins.
Ingestion of the chick pea Lathyrus sativus, which contains another EAA structural analogue results in acute spastic motor signs following the consumption. The clinical features of this motor disorder called “lathyrism” were known even by the ancient Greeks but the toxic component, the amino acid β-N-oxalyl-L-alanine (BOAA) was identified only some decades ago (Table 2).104 Experimental administration of BOAA is known to induce the features of lathyrism in nonhuman primates105 and application of BOAA to cell cultures results in cell death with structural features of excitotoxicity106 that can be attenuated by non-NMDA receptor antagonists.107,108 Importantly, the high excitotoxic potency of this glutamate receptor ligand is in accordance with its high excitatory potency as agonist at AMPA receptors.109
Implication of Distinct Glutamate Receptor Classes in Excitotoxicity
L-Glutamate, the most abundant excitatory transmitter in the brain, binds to different classes of receptors comprising different types of ionotropic receptors as well as metabotropic receptors. Ionotropic glutamate receptors are ligand-gated ion channels, which are named after agonists that bind preferentially to these receptor subtypes. They include α-amino-3-hydroxy-5-methyl-isoxalole-4-propionate (AMPA), kainate and N-methyl-D-aspartate (NMDA) receptors.
AMPA receptors are widely distributed in the CNS and are expressed on many different types of neurons. They control a cation channel that is permeable to Na and K ions with a single channel conductance <20 pS.110 Activation of AMPA receptors results in the fast onset of an excitatory postsynaptic current (EPSC) with rapid desensitization.111 This current shapes the fast component of glutamatergic EPSCs in CNS neurons.112 Importantly, certain AMPA receptor subtypes can exhibit substantial Ca2+ permeability and thereby contribute to the Ca2+-dependent form of excitotoxic cell damage.113–116 Ca2+-permeable channels are formed from the receptor subunits GluR1 or GluR3, whereas coassembly with GluR2 results in only poor Ca2+ permeability.117–119
Another type of ionotropic glutamate receptor termed kainate receptor is less well understood because of lack of sufficiently selective agonists and antagonists. As with AMPA receptors, kainate receptors control a cation channel that desensitizes rapidly. Since the agonist kainate activates kainate receptors but also AMPA receptors, their physiological and pathophysiological role remains elusive. Recently, results based on gene-targeted rodents lacking certain kainate receptor subtypes have extended our knowledge of localization and function of kainate receptors.120,121 From these studies one can conclude that kainate receptors are not only localized to postsynaptic sites but also appear to be localized presynaptically, suggesting a role for modulation of synaptic strength (for review see refs. 122,123).
The third type of ionotropic glutamate receptor, the NMDA receptor differs fundamentally from AMPA and kainate receptors in several ways. First, the pore is significantly permeable to Ca2+ ions,124,125 but also to K and Na ions.126 In contrast to AMPA receptors, NMDA receptors exhibit a high single channel conductance (50 pS) and desensitize much slower (for review see refs. 127,128). NMDA receptors are widely distributed in different types of CNS neurons and shape the late component of glutamatergic EPSCs. Second, the opening of the ligand-gated cation channel does not only depend on binding of agonist but is voltage-dependent, since the channel is blocked by Mg2+ions at resting membrane potentials and a depolarization of the plasma membrane is required to relieve the Mg2+-dependent block. Third, activation of NMDA receptors requires binding of a coagonist to the so-called glycine-binding site of the NMDA receptor. Very recently, the amino acid D-serine has been suggested to be an endogenous ligand for the glycine-binding site.129 This is supported by the findings that (1) D-serine has a high potency to potentiate NMDAR-mediated neurotransmission, (2) D-serine is colocalized with NMDARs in the forebrain and (3) enzymatic degradation of the amino acid attenuates NMDAR-mediated neurotransmission.
As discussed at the start of this chapter, experimental application of the endogenous nonselective agonist glutamate has been shown to induce excitotoxic cell damage.48 As with L-glutamate, administration of the agonists kainate11,12,14,20 or NMDA19 induced brain damage, thus confirming the implication of different glutamate receptor classes.
The implication of different ionotropic glutamate receptor types in induction of brain damage has been further studied by the use of selective receptor antagonists. Experimental administration of competitive or uncompetitive NMDA antagonists21,22,85,86,130 or application of kainate antagonists attenuates cell death in cell cultures as well as brain damage.20 Unfortunately, the clinical use of these receptor antagonists has been hampered by the inhibition of physiological NMDAR-mediated neurotransmission, resulting in various adverse effects.131 However, the uncompetitive NMDA antagonist memantine (1-amino-3,5-dimethyladamantane hydrochloride), which has already been used for years for treatment of Parkinson's disease, is sufficiently tolerated. Memantine is an open-channel blocker, but has faster kinetics than MK-801. This results in substantial inhibitory drug action under conditions of prolonged exposure to glutamate but much less inhibition under millisecond exposure to glutamate.132 This raises the hypothesis, that memantine should be suited to treat disease conditions associated with elevated glutamate concentrations for prolonged periods. Indeed, several groups have demonstrated its neuroprotective effectiveness combined with only few adverse effects.133–135
Whereas the role of ionotropic glutamate receptors in excitotoxicity has been studied extensively, the significance of metabotropic glutamate receptors (mGluRs) in excitotoxicity is less well understood. Importantly, there is no indication, that activation or inhibition of mGluRs by itself exerts excitotoxicity.136 Instead, in view of the literature it appears more likely, that specific metabotropic receptor subtypes can be involved in modulation of ionotropic receptor-mediated excitotoxicity. This may be due to the coupling of different mGluR subtypes to different signal transduction pathways and effectors. Group I mGluRs comprise the subtypes mGluR1 and mGluR5 which in heterologous expression systems couple to phospholipid hydrolysis through phospholipase C. Among other effects, this results in second messenger-mediated activation of protein kinase C and Ca2+ release from IP3-sensitive Ca2+ stores.137–139 Application of recently developed selective receptor ligands suggests, that activation of group I, and particularly mGluR1 receptors amplifies NMDA-mediated excitotoxicity.136,140–144
Group II receptors comprise the subtypes mGluR2 and mGluR3 that are negatively coupled to the adenylate cyclase pathway. Recent pharmacological studies using selective agonists indicate that group II receptor activation results in protection against NMDA-mediated excitotoxicity.141,145–147 Likewise, group III receptors (mGluR4 and mGluR6–8) are negatively coupled to adenylyl cyclase, and activation of mGluR4 or mGluR8 group III receptors results in attenuation of NMDA-mediated neurotoxicity.147–149
Ionic Dependence of Excitotoxic Cell Damage
Activation of different ionotropic receptor types is linked to excitotoxic cell damage through the underlying ion currents. Depending on the predominance of either Na or Ca2+ influx, two different components of excitotoxicity have been suggested. Na ion influx mediated by activation of NMDA-type and non-NMDA-type glutamate receptors is followed by secondary influx of Cl and H2O and results in swelling of neurons.28,34,150,151 This acute form of cell damage depends on the transmembrane Na and Cl gradients28 and can be prevented by extracellular substitution of Na and Cl with impermeant ions.152,153 In contrast to the acute, primarily Nadependent osmotic damage, a more delayed mode of cell death has been attributed to Ca2+ influx.28,153 This delayed Ca2+−-dependent cell death can be largely attenuated by inhibition of NMDA receptors,19,29,84,154 removal of extracellular Ca2+ ions23,155,156 or buffering of cytoplasmic Ca2+ by membrane-permeable chelators.157,158 Therefore, the Ca2+-dependent component was traditionally believed to be induced exclusively by NMDA receptor activation.
The finding that the late component can be mimicked by calcium ionophores in presence of Ca2+ ions has corroborated the Ca2+ dependence of the delayed cell death.28 However, the discovery of Ca2+-permeable AMPA/kainate receptors, has lead to the view that neurons expressing these Ca2+-permeable nonNMDA receptors can also undergo Ca2+-dependent delayed cell death.32,159 In summary, different routes of Ca2+ influx such as through NMDA receptors, Ca2+-permeable AMPA/kainate receptors and voltage-dependent Ca2+ channels, which are all involved in physiological signaling, are implicated in excitotoxic cell death.
The suggested key role of Ca2+ in excitotoxicity has been subsequently confirmed by the finding, that the extent of excitotoxic cell death correlates with the total amount of Ca2+ uptake and is independent of the route of entry. In some cases Zn2+ can substitute for Ca2+ as the cation inducing excitotoxic damage.29,159,160
The Ca2+ overload observed following sustained stimulation of NMDA receptors results from the inability of cellular Ca2+ homeostasis, such as extrusion of Ca2+ across the plasma membrane by Na/Ca2+ antiporter161–163 and Ca2+ ATPase164 or Ca2+ sequestration by the endoplasmic reticulum and mitochondria165,166 to remove the large influx of Ca2+.
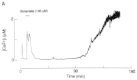
Interestingly, during sustained exposure to glutamate, the intracellular calcium concentration [Ca2+]c rises rapidly to a peak value and thereafter slowly recovers to an elevated plateau level.167 However, the [Ca2+]c can also manifest a second, delayed increase,30,168 immediately preceding cell death (Fig. 4 ). This delayed [Ca2+]c increase, termed “delayed calcium deregulation” (DCD) is irreversible and reflects irreversible loss of cellular Ca2+ homeostasis. Although the interval between the initial Ca2+ spike and the DCD varies within a cell population exposed to NMDA receptor agonists, once DCD occurs within a neuron, it invariably precedes cell death. The temporal relation between neuronal Ca2+ levels and delayed cell death suggests, that Ca2+-dependent effector mechanisms are involved, which do not need sustained high Ca2+ levels but are triggered by transient changes in [Ca2+]c.
Since many enzymes are activated by transient or sustained [Ca2+] elevation various effector mechanisms may be implicated in Ca2+-mediated excitotoxic cell death. Consequently, a variety of Ca2+-dependent hydrolytic enzymes, including lipases and proteases, have been suggested to be involved in excitotoxic neuronal damage. Activation of the Ca2+-dependent phospholipase A2 has been observed following NMDA receptor activation169 and the subsequent catabolism of released arachidonic acid by lipoxygenases and cyclooxygenases (COX), is also associated with concomitant production of reactive oxygen species (ROS).169–171 In addition, activation of phospholiase A2 and subsequent release of arachidonic acid may inhibit transporter-mediated glutamate uptake from the extracellular space.33,172,173
Among several Ca2+-activated proteases, the activity of the Ca2+-dependent cysteine protease calpain is increased following glutamate receptormediated Ca2+ loading. 174–177 Calpain activation results in proteolysis of structural proteins and degradation of the neuronal cytoskeleton.177 Furthermore, calpain may direct the mode of cell death to necrosis by preventing the cytochrome c-mediated activation of caspases (see following).174
Ca2+-mediated activation of nitric oxide synthase may be another pathway involved in excitotoxic cell death since neurons constitutively express the isoenzyme called “neuronal nitric oxide synthase” (nNOS). nNOS is activated by glutamate receptor-mediated [Ca2+]c increases178,179 and there is compelling evidence that nNOS activation is linked to excitotoxic damage31 since inhibition of NO formation results in protection of neurons from glutamate receptor-mediated cell death.179–182 Studies on nNOS-deficient neuronal cultures confirmed the role of NO in glutamate receptor-mediated neurotoxicity since in nNOS-deficient cortical cultures the toxic effects elicited by administration of NMDA are markedly attenuated.183 The role of NO in excitotoxicity is mediated at least partly through the reaction of NO with superoxide anions (O2 ) to form peroxynitrite (ONOO).184
Other classes of enzymes are also thought to be involved in the Ca2+-mediated cell death but in less direct ways. Calcineurin is a Ca2+/calmodulin-activated phosphatase which can dephosphorylate nNOS, thereby increasing its activity185 and potentially increasing excitotoxic damage. Calcineurin has been convincingly demonstrated to be involved in neuronal cell death since pharmacological experiments using the calcineurin inhibitors cyclosporin A and FK-506 have revealed attenuation of excitotoxic cell death.186,187 Accordingly, inhibition of calmodulin can also be shown to decrease excitotoxic cell death.182,188
Mitochondrial Dysfunction
Studies on the time course of glutamate receptor-induced [Ca2+]c increase have demonstrated that if the neuronal cell does not succumb to the insult, [Ca2+]c slowly recovers after the termination of agonist application.30,167 This recovery is due to several mechanisms that ensure the cellular Ca2+ homeostasis under physiological conditions (Fig. 5) such as extrusion across the plasma membrane by the Na/Ca2+ exchanger189 and the Ca2+-ATPase, or sequestration of Ca2+ into the endoplasmic reticulum189,190 and mitochondria.191,192

In theory, the driving force for Ca2+ ions to enter the mitochondrial matrix in energized mitochondria is formed by the strong electrochemical gradient for Ca2+ across the inner mitochondrial membrane, mainly due to the mitochondrial membrane potential Δψm(Δψm Å-180 mV). Therefore, normal energized mitochondria can take up substantial amounts of calcium.193 Net uptake of calcium occurs in isolated mitochondria whenever [Ca2+]c rises above a set point in the high nanomolar range (>0.5 μM), indicating the dynamic equilibrium of mitochondrial uptake and extrusion mechanisms.194 [Ca2+]c can be shown to increase above this set point195 during excessive cytoplasmic Ca2+ loading following ionotropic glutamate receptor activation and results in net mitochondrial Ca2+ uptake.191,192
It has been proposed that mitochondrial Ca2+ uptake can shape the time course of cytoplasmic [Ca2+]c.196 During excessive Ca2+ loading into neurons, mitochondrial Ca2+ uptake appears to blunt the [Ca2+]c increase since mitochondrial depolarization by protonophores prior to Ca2+ loading results in increased [Ca2+]c.163,196–199 After termination of the Ca2+ influx, [Ca2+]c slowly decreases and Ca2+ redistribution from mitochondria into the cytoplasm begins when [Ca2+]c decreases below the set point.192,196
Ca2+ uptake into mitochondria results in a decrease in Δψm that can be monitored in isolated energized mitochondria200–202 as well as in situ.203–207 This depolarization is due to the balancing of the charge transfer carried by Ca2+ ions by re-entry of protons into the mitochondrial matrix. Under normal conditions, Ca2+−induced depolarization is transient and serves to activate mitochondrial dehydrogenases208–214 and the mitochondrial ATP synthase,215 which in turn results in increased electron transport through the respiratory chain and consequently increased outward flux of protons.
However, studies on in situ mitochondrial membrane potential using cationic fluorochromes have demonstrated convincingly that the time course of Δψm depolarization can vary considerably, and depends on the time of Ca2+ loading. Short pulses of NMDA result in at least partial recovery of Δψm203 whereas prolonged NMDA exposure or higher agonist concentrations result in sustained depolarization with negligible recovery.40,203,206 This sustained depolarization indicates that excessive Ca2+ uptake triggers severe mitochondrial bioenergetic dysfunction. Sustained depolarization has been ascribed to various underlying mechanisms, and ROS seem to play a key role in several of the suggested hypotheses.
In neurons as in other tissues, mitochondria are a major source of ROS;216,217 nonetheless, mitochondria are themselves susceptible to oxidative damage. Excessive Ca2+ uptake into isolated mitochondria is known to result in increased formation of ROS (Fig. 6),218,219 which in turn inhibit pyruvate dehydrogenase220 and tricarboxylic acid cycle enzymes221,222 as well as complex I of the respiratory chain.223–225 In intact neurons, ROS formation has been monitored with redox-sensitive fluorochromes, which are oxidized by ROS to form fluorescent molecules.226–228 These studies indicate that ionotropic glutamate receptor-induced Ca2+ loading enhances the production of O2227,229–231
The main site of ROS formation within mitochondria is within the respiratory chain. Complex I232,233 as well as complex III234–236 are thought to participate in one electron reduction of molecular oxygen, resulting in the generation of O2 which in turn can lead to other ROS. Nevertheless, the exact biophysical link between mitochondrial Ca2+ uptake and increased mitochondrial ROS production remains unclear.
The Role of Reactive Oxygen Species in Excitotoxicity
Evidence for the implication of ROS in excitotoxic cell damage arises from experiments showing enhanced production of O2227,230,231,237 following ionotropic glutamate receptor overstimulation and from studies using radical scavengers or inhibitors of the formation of certain ROS.227 These studies unequivocally demonstrate that removal of ROS results in attenuation of glutamate receptor-induced cell death.238,239
Several Ca2+-dependent processes that increase the endogenous production of ROS have been described, which are all assumed to be implicated in excitotoxic cell death following NMDA receptor activation. As stated above, Ca2+ loading into mitochondria appears to be one important mechanism of ROS production under conditions associated with excitotoxicity.227,229–231,237,240 Apart from this mechanism, activation of Ca2+-dependent phospholipase A2 has been observed following NMDA receptor activation169 and catabolism of released arachidonic acid by lipoxygenases and cyclooxygenases has been implicated in excitotoxicity through the concomitant production of ROS.169–171
Some ROS exhibit a high reactivity with correspondingly short halflives and can undergo many different reactions. Generally speaking, ROS can exert multiple damaging reactions to proteins, lipids, carbohydrates and nucleic acids, thereby disrupting cellular functions. The increased production of ROS is therefore a potential threat to cellular homeostasis and neuronal survival if production is not balanced by the capacity of endogenous antioxidant mechanisms. Some basic characteristics of activated oxygen species are listed in Table 3.
Table 3
Characteristics of reactive oxygen species.
From a multitude of ROS-mediated disturbances following NMDA receptor activation, only some examples will be discussed. Since mitochondria appear to be the major source of Ca2+-induced increase in ROS formation and ROS are highly reactive, mitochondria are prone to damage by ROS. As discussed above, several mitochondrial enzymes, such as NADH:CoQ oxidoreductase, succinate dehydrogenase, ATP synthase, pyruvate dehydrogenase and the citric acid cycle enzyme aconitase, are inhibited by ROS including O2 , H2O2 or -OH.220,221,223,241,242 The delayed deregulation of cellular Ca2+ homeostasis (DCD) that has been observed after NMDA exposure could be caused by ROS-dependent mechanisms. This hypothesis is supported by the finding that experimentally induced production of O2 by menadione results in enhancement of DCD,238 whereas dismutation of O2 by the manganoporphyrin Mn-TBAP attenuates DCD.238 Importantly, the redox-sensitive mechanism underlying DCD is still unresolved. Although oxidative stress-induced dysfunction of the plasma membrane Ca2+−ATPase has been demonstrated243 and this dysfunction has been proposed as a mechanism underlying the NMDA receptor-induced DCD,238,244 the conclusion that mitochondrially generated ROS are involved in plasma membrane protein dysfunction should be judged cautiously because of the small reaction range.
Ca2+−-induced increase in ROS can result in mitochondrial dysfunction including dissipation of Δψm, bioenergetic failure and disturbance of cellular Ca2+ homeostasis. Apart from the inhibition of enzyme activity, the opening of a large nonselective pore in the inner mitochondrial membrane termed “mitochondrial permeability transition” (mPT) has been suggested as a key mechanism in neuronal excitotoxicity.203,206 The basic concept of a pore-mediated permeabilization of the inner mitochondrial membrane rather than membrane damage had been suggested as early as the 1970s. The early studies by Haworth and Hunter245 showed that various divalent cations could induce or prevent mitochondrial swelling and that the assumed pore is selective for the permeation of solutes below 1500 kDa. The idea of a large nonspecific channel was confirmed almost one decade later by electrophysiological experiments. Single-channel patch-clamp recordings from the inner mitochondrial membrane revealed a large conductance channel (1.3 nS),246 which opened in response to the addition of Ca2+ to the mitochondrial preparation. Notably, the activation and inhibition characteristics of this “mitochondrial megachannel” are similar to the proposed mPT. Application of Ca2+, ROS, inorganic phosphate and the adenine nucleotide translocator ligand atractylate induce permeability transition and opening of the megachannel, whereas Mg2+, antioxidants, ADP, cyclosporin A and the adenine nucleotide translocator ligand bongkrekic acid (BA) inhibit permeability transition and channel opening.219,247–256 This strikingly similar behavior has resulted in the conclusion that the mPT and the mitochondrial megachannel are virtually identical.257
Dissipation of Δψm, bioenergetic failure and disturbance of mitochondrial Ca2+ homeostasis following activation of NMDA receptors all could be explained by opening of the mPT.203,206 However, conclusions regarding the involvement of mPT in neuronal excitotoxicity until recently were based on pharmacological experiments using the mPT inhibitor cyclosporin A and should be treated cautiously since cyclosporin A also inhibits calcineurin and the multidrug resistance channel.258,259 Recently, however, our group succeeded in demonstrating that BA, a more specific inhibitor of mPT, could prevent NMDA-induced neuronal apoptosis, suggesting the involvement of mPT in this form of excitotoxic cell death.260
In summary, several ROS-dependent mechanisms have been suggested, mainly based on data from isolated mitochondria. In any event, the complex environment of in situ mitochondria, containing, for example, mPT enhancers as well as inhibitors, prevents a definitive interpretation of the physiological and pathophysiological relevance of the proposed mechanisms at this time.
Role of Nitric Oxide and other Reactive Nitrogen Species in Excitotoxicity
Nitric oxide (NO) is a well-recognized messenger molecule in the CNS that affects various cellular functions, e.g., neuronal transmitter release, synaptic plasticity and gene expression (see refs. 261–263). NO is produced by several types of cells expressing three distinct isoforms of the enzyme nitric oxide synthase (NOS, NADPHdiaphorase) that converts L-arginine into NO and citrulline: (1) neuronal NOS (nNOS), (2) inducible or immunologic NOS (iNOS) in microglia and astrocytes and (3) endothelial NOS (eNOS) predominantly in endothelial cells of brain blood vessels. Unlike classical neurotransmitters, NO is freely diffusible and therefore cannot be stored in synaptic vesicles. Once synthesized, NO diffuses across cell membranes and thus can reach (1) various compartments within the NO-producing cells and (2) different types of surrounding cells (Fig. 7). In contrast to conventional transmitters, the activity of the messenger molecule is not terminated by reuptake or enzymatic degradation but by chemical reactions with target molecules and its spontaneous formation of nitrite. The absence of control over its activity by release and reuptake mechanisms is functionally compensated by a tight regulation of its synthesis. However, the regulatory mechanisms involved in physiological signaling may also implicated under pathophysiological conditions. During the last decade, pharmacological experiments using NO donors, NOS inhibitors or exogenous substrates (reduced hemoglobin) for competitive reactions with NO suggested that NO is linked to neuronal cell death.31,43,182 Even more important, cell death under conditions assumed to induce excitotoxicity could be largely attenuated by inhibition of NO formation or blockade of NO effects.31,182,264 Subsequent studies on nNOS-deficient neuronal cultures confirmed the role of NO in glutamate receptor-mediated neurotoxicity since toxic effects elicited by administration of NMDA are markedly attenuated in nNOS-deficient cortical cultures compared with wild-type neurons.183 Since nNOS activity is known to increase with increasing calcium concentrations, the NMDAR-mediated calcium uptake provides the link between conditions associated with excitotoxicity and NO-mediated cell damage.178
Although NO is a free radical, it is not as reactive as most ROS265 and calculations based on its reactivity had estimated a reaction range of about 100 mm.261,263The well-accepted implication of NO in excitotoxicity has been suggested to depend on increased NO formation and the concomitant production of O2. Under this condition, O2 reacts with NO extremely fast to form peroxynitrite (ONOO).266–268 ONOO can undergo a multitude of different chemical reactions with various substrates, e.g., hydroxylation or nitration of tyrosine residues, lipid peroxidation or apparent decomposition into NO2 and -OH and subsequently, this toxic molecule has been implicated in NO-induced cell damage.43,182,184,269
So far, several pathways of ONOO toxicity have been suggested but with respect to its high reactivity, it appears unlikely that these are the only mechanisms involved in NO-mediated cell death.
Intense exposure to NO/O2 with resultant ONOO formation has been demonstrated convincingly to result in neuronal necrosis because of energy failure, while mild insults lead to apoptosis.43 This bioenergetic failure may be due to (1) increased ATP consumption under the condition of poly-(ADP ribose) polymerase (PARP) activation,270 which occurs following ONOO-mediated DNA damage and (2) decreased ATP synthesis through NO-mediated inhibition of the respiratory chain machinery.271–273
Very recently, p38 mitogen-activated protein (MAP) kinase has been suggested as another pathway implicated in NO-induced cell death. Application of a p38 MAP kinase inhibitor significantly attenuated NO-induced caspase activation, Bax translocation, and neuronal cell death.274
Excitotoxicity, Calcium Loading and Apoptosis
A great advance in excitotoxicity came with the understanding that neurons can die by apoptosis. In response to excitotoxicity the neuron activates apoptosis, i.e., dismantling of itself including DNA, cytoskeleton, and production of ATP. While in acute neurological situations such as stroke the cells in the ischemic core die rapidly by necrosis, at least in animal models, the cells in the penumbra also go on to die but show markers of apoptosis, such as oligonucleosomal DNA damage.275,276 Further evidence of apoptosis in excitotoxicity came with the examination of the activation of caspases in animal models of stroke.277 Inhibition of caspases in general by intracerebroventricular injections of N-benzyloxycarbonyl-Val-Ala-Aspfluoromethylketone (z-VAD.FMK), or selective inhibition of caspase-3 with N-benzyloxycarbonyl-Asp-Glu-Val-Aspfluoromethylketone (z-DEVD.FMK) reduced the ischemic infarct volume by as much as 60%.277 These caspase inhibitors remain effective even when applied hours after the insult, making them effective later than NMDA receptor blockade with MK801.94 While the morphology of dying ischemic cells does not precisely match that of “classical” apoptosis,278 and in vivo excitotoxic injury results in neurodegeneration along an apoptosis-necrosis continuum, studies with caspase inhibitors clearly indicate a role for apoptosis in ischemia.
Apoptosis is also a feature of neurons dying in neurodegenerative disorders. DNA fragmentation has been detected in human post mortem samples of Parkinson's disease,279,280 Alzheimer's disease,281,282 Huntington's disease,282,283 and amyotrophic lateral sclerosis.284 However, the presence of DNA fragmentation is not a guarantee that the cells are dying by apoptosis,285 and DNA fragmentation can be influenced by antemortem hypoxia.286 More recently other recognized apoptotic molecules such as caspases,287,288 and BclXL289 have been shown in Alzheimer's disease. It is now largely accepted that neuron loss in chronic neurodegeneration is mediated by apoptosis.
Key Signaling Players in Neuronal Apoptosis
Caspases are central to the induction of neuronal cell death.94,277,290 Their actions on key intracellular substrates, including other protease zymogens, make caspases the primary executioners of the cell death program. The 13 known caspases can be separated into two functional groups; those that initiate apoptosis by receiving the initiating signals, and those that effect the dismantling of the cell. The caspases that receive or integrate apoptotic signals (caspases-8, -10, -2, -9) cleave and activate the downstream effector caspases (caspases-3, -7, -6). Perhaps the last reversible step in the death of neurons is the activation of the caspase-3,291 although caspase-independent mechanisms almost certainly exist.174,292 As discussed above, peptide inhibitors of caspase-3 block the death of neurons in many different situations (for review see ref. 293). The central importance of caspase-3 in neurons was clearly shown in caspase-3-deficient mice which have a doubling of brain size, correlated with decreased apoptosis and premature death.294
Mitochondria appear to provide a link between the initiator caspases and the downstream effector caspases (Fig. 8). In nonneuronal cells, mitochondria have been shown to accelerate activation of effector caspases by releasing proapoptotic molecules, such as cytochrome c,295,296 the apoptosis-inducing factor,297 and SMAC/diabolo.298 Currently in neuronal systems, only the pathway utilized by cytochrome c has been fully elucidated. Cytochrome c triggers activation of caspase-9 which in turn cleaves caspases-3, -6, -7.299 Release of cytochrome c from mitochondria has received much attention as a commitment to apoptosis.295,296,300,301 Nevertheless, the release of cytochrome c from mitochondria has been shown to be not sufficient for neuronal apoptosis,260,302 and microinjection of cytochrome c into sympathetic neurons does not lead to death in the absence of additional stimuli.303 Therefore, cytochrome c requires other factors to initiate the caspase cascade. These factors, Apaf-1, dATP and procaspase-9 along with cytochrome c are known collectively as the apoptosome.304 The processed caspase-9 is then able to cleave and activate caspase-3.
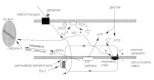
Considerable uncertainty surrounds the mechanism of release of proapoptotic factors from the mitochondria. Possibilities include (i) transport by poreforming Bcl-2 family proteins such as Bax,305 (ii) opening of the permeability transition pore (PTP) in the inner mitochondrial membrane leading to rupture of the outer mitochondrial membrane,306 or (iii) transport coupled to the voltage-dependent anion channel in the outer mitochondrial membrane.301,307 This remains an important question to answer, since cytochrome c release from the mitochondria will eventually bring about mitochondrial dysfunction and an energetic deficit. Studies with nonneuronal cells have shown that mitochondrial membrane potential is maintained by ATP hydrolysis following the exit of cytochrome c.308 The release of cytochrome c from mitochondria can be stimulated by some caspases and by the proapoptotic members of the Bcl-2 family Bid and Bax.300,305,306 On the contrary, survival factors can act to prevent release of cytochrome c, e.g., channel activity of Bax is inhibited by anti-apoptotic Bcl-2 family members such as Bcl-2 or Bcl-xL, and ordinarily Apaf-1 is sequestered at the membrane by Bcl-xL, and in the absence of cytochrome c cannot interact with procaspase-9.309 While the role of signaling molecules in neuronal apoptosis has been studied extensively in cell culture models, the in vivo data is sparse for many of the apoptotic pathways. However, several of the key signaling molecules in apoptosis have been detected following ischemia, e.g., the release of cytochrome c,310,311 Fas/TNFR,312 Bax,313 phospho-JNK,314 and caspase-9.315
The initiators of apoptosis in neurons are diverse including the withdrawal of trophic factors, and signaling through the transmembrane “death” receptors such as Fas/TNFR family as well as intrinsic signaling pathways that sense overload of ROS, Ca2+, and H (for review see ref. 290). In vitro, neuronal cell death can be induced by a variety of treatments: axotomy, excitotoxins, trophic deprivation, etc. These diverse stimuli have demonstrated that neurons indeed contain numerous apoptotic signaling pathways. Excitotoxicity has been achieved in cultures of primary neurons using either a direct insult such as activation of glutamate receptors, or with a secondary insult such as with Aβ (amyloid peptide), hypoxia, etc. These various paradigms which ultimately overstimulate glutamate receptors all result in the activation of caspases. Upstream of the caspases however, excitotoxic insults can simultaneously activate more than one pathway.
As discussed in earlier sections, overstimulation of NMDA receptors results in an influx of calcium. Calcium at high (>500 nM) concentrations inappropriately activates many enzymes, is accumulated within the nucleus, endoplasmic reticulum and mitochondria, and as a general disturbance to the cell's ionic homeostasis causes a large energy drain as the cell tries to extrude the excess calcium (reviewed in ref. 316). All of these occurrences can and do trigger the neuron to apoptosis. From an extensive and rapidly unfolding literature, the following points demonstrate some of the pathways that are concurrently activated by excitotoxic entry of calcium.
Isolated brain mitochondria when exposed to high calcium release cytochrome c.317 Accordingly, prolonged activation of NMDA receptors in cultured neurons and subsequent calcium loading results in release of cytochrome c from mitochondria followed by the activation of caspase-3.260,318 Thus in excitotoxicity, cytochrome c release from mitochondria, in the presence of the other members of the apoptosome, is a direct signal to apoptosis. Apart from the mitochondrion as a key player in the apoptosis of neurons, the endoplasmic reticulum (ER) seems to participate in apoptotic signaling. The ER regulates cellular responses to stress and intracellular calcium levels. Treatment of cultured neurons with glutamate or thapsigargin induces the expression of caspase-12 on the ER outer membrane.319,320 This results in cleavage and activation of cytosolic caspase-3.319
As discussed in an earlier section, glutamate receptor activation also activates the Ca2+-dependent cysteine protease calpain.174–177 Calpain inhibitors appear to be at least as effective as caspase inhibitors in preventing neuronal death and DNA fragmentation,293 and have been demonstrated to reduce neuronal cell death caused by hypoxia.321
Other concomitantly activated pathways include the p38 MAP kinase and c-jun N-terminal kinase or stress-activated kinase (JNK/SAPK) which promote cell death.322 SAPK/JNK is activated by tyrosine kinase and elevated intracellular calcium, and in primary striatal cultures, treatment with glutamate causes both increased phosphorylation of its substrate cjun and increased activity of JNK.323 Glutamate has been shown to activate p38 MAP kinase in rat cerebellar granule neurons,324 and inhibition of p38 MAP kinase has been demonstrated to protect in vitro retinal ganglion neurons from NMDA-induced apoptosis.325
DNA damage is a common trigger for apoptosis in many different tissues, and in the mature nervous system, p53 is essential for neuronal death in response to DNA damage, ischemia and excitotoxicity.326 Lately, signaling pathways that are responsible for triggering p53-dependent neuronal apoptosis are starting to be elucidated and involve cell cycle deregulation and also activation of the JNK pathway.
Conclusions
A large body of evidence has accumulated showing that excessive activation of EAA receptors by the primary excitatory amino acid L-glutamate under pathophysiological conditions or intoxication with exogenous agonists results in excitotoxic cell death. Studies on the ion currents occurring under conditions of excitotoxicity have revealed that imbalanced Na and Cl gradients as well as Ca2+ overloading are implicated in cell death. Calcium ions (and in some cases zinc ions) seem to play a pivotal role in EAA neurotoxicity, since this ubiquitous intracellular messenger can activate a multitude of enzymatic and nonenzymatic signaling and execution pathways implicated in neuronal cell death. There is compelling evidence that the mode of cell death that neurons undergo in excitotoxicity varies considerably along an apoptosis-necrosis continuum, depending on the severity of the insult. To date, numerous mechanisms involved in necrosis as well as various signaling and execution pathways that are implicated in apoptosis have been shown convincingly. However, while the role of signaling molecules in neuronal apoptosis has been studied extensively in in vitro models, the in vivo data is sparse for many of the pathways.
References
- 1.
- Curtis DR, Watkins JC. The excitation and depression of spinal neurons by structurally related amino acids. J Neurochem. 1960;6:117–141. [PubMed: 13718948]
- 2.
- Krnjevic K, Phillis JW. Actions of certain amines on cerebral cortical neurones. Br Pharm Chemother. 1963;20:471–490. [PMC free article: PMC1703804] [PubMed: 14035890]
- 3.
- Lucas DR, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. Ama Arch Opthalmol. 1957;58:193–201. [PubMed: 13443577]
- 4.
- Olney JW, Sharpe LG. Brain lesions in an infant rhesus monkey treated with monsodium glutamate. Science. 1969;166(903):386–388. [PubMed: 5812037]
- 5.
- Olney JW, Ho OL. Brain damage in infant mice following oral intake of glutamate, aspartate or cysteine. Nature. 1970;227(258):609–611. [PubMed: 5464249]
- 6.
- Olney JW. Glutamate-induced neuronal necrosis in the infant mouse hypothalamus. An electron microscopic study. J Neuropathol Exp Neurol. 1971;30(1):75–90. [PubMed: 5542543]
- 7.
- Olney JW, Sharpe LG, Feigin RD. Glutamate-induced brain damage in infant primates. J Neuropathol Exp Neurol. 1972;31(3):464–488. [PubMed: 4626680]
- 8.
- Olney JW, Rhee V, Gubareff TD. Neurotoxic effects of glutamate on mouse area postrema. Brain Res. 1977;120(1):151–157. [PubMed: 832113]
- 9.
- Olney JW. Neurotoxicity of excitatory amino acids In: McGeer EG, Olney JW, McGeer PL, eds. Kainic acid as a tool in neurobiology New York: Raven, 197895–121.
- 10.
- Olney JW, Misra CH, de Gubareff T. Cysteine S-sulfate: brain damaging metabolite in sulfite oxidase deficiency. J Neuropathol Exp Neurol. 1975;34(2):167–177. [PubMed: 1123650]
- 11.
- Coyle JT, Molliver ME, Kuhar MJ. In situ injection of kainic acid: a new method for selectively lesioning neural cell bodies while sparing axons of passage. J Comp Neurol. 1978;180(2):301–323. [PubMed: 659663]
- 12.
- Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271(5646):676–677. [PubMed: 625338]
- 13.
- Schwarcz R, Coyle JT. Neurochemical sequelae of kainate injections in corpus striatum and substantia nigra of the rat. Life Sci. 1977;20(3):431–436. [PubMed: 14287]
- 14.
- Schwarcz R, Coyle JT. Kainic acid: neurotoxic effects after intraocular injection. Invest Ophthalmol Vis Sci. 1977;16(2):141–148. [PubMed: 832974]
- 15.
- Watkins JC, Krogsgaard LP, Honore T. Structure-activity relationships in the development of excitatory amino acid receptor agonists and competitive antagonists. Trends Pharmacol Sci. 1990;11(1):25–33. [PubMed: 2155495]
- 16.
- Greenamyre JT. The role of glutamate in neurotransmission and in neurologic disease. Arch Neurol. 1986;43(10):1058–1063. [PubMed: 2428340]
- 17.
- Olney JW, Price MT, Fuller TA. et al. The antiexcitotoxic effects of certain anesthetics, analgesics and sedative hypnotics. Neurosci Lett. 1986;68(1):29–34. [PubMed: 3523314]
- 18.
- Schwarcz R, Meldrum B. Excitatory amino acid antagonists provide a therapeutic approach to neurological disorders. Lancet. 1985;2(8447):140–143. [PubMed: 2862329]
- 19.
- Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8(1):185–196. [PMC free article: PMC6569373] [PubMed: 2892896]
- 20.
- Koh JY, Goldberg MP, Hartley DM. et al. Non-NMDA receptor-mediated neurotoxicity in cortical culture. J Neurosci. 1990:10693–705. [PMC free article: PMC6570171] [PubMed: 2406381]
- 21.
- Michaels RL, Rothman SM. Glutamate neurotoxicity in vitro: Antagonist pharmacology and intracellular calcium concentrations. J Neurosci. 1990;10(1):283–292. [PMC free article: PMC6570346] [PubMed: 1967639]
- 22.
- Frandsen A, Drejer J, Schousboe A. Direct evidence that excitotoxicity in cultured neurons is mediated via N-methyl-D-aspartate (NMDA) as well as non-NMDA receptors. J Neurochem. 1989;53(1):297–299. [PubMed: 2566655]
- 23.
- Rothman SM, Thurston JH, Hauhart RE. Delayed neurotoxicity of excitatory amino acids in vitro. Neuroscience. 1987;22(2):471–480. [PubMed: 3670595]
- 24.
- Garthwaite G, Garthwaite J. Amino acid neurotoxicity: intracellular sites of calcium accumulation associated with the onset of irreversible damage to rat cerebellar neurones in vitro. Neurosci Lett. 1986;71(1):53–58. [PubMed: 3537848]
- 25.
- Garthwaite G, Hajos F, Garthwaite J. Ionic requirements for neurotoxic effects of excitatory amino acid analogues in rat cerebellar slices. Neuroscience. 1986;18(2):437–447. [PubMed: 3526174]
- 26.
- Garthwaite G, Garthwaite J. Differential dependence on Ca2+ of N-methyl-D-aspartate and quisqualate neurotoxicity in young rat hippocampal slices. Neurosci Lett. 1989;97(3):316–322. [PubMed: 2469997]
- 27.
- Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58(3):293–297. [PubMed: 2413399]
- 28.
- Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7(2):369–379. [PMC free article: PMC6568907] [PubMed: 2880938]
- 29.
- Hartley DM, Kurth MC, Bjerkness L. et al. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13(5):1993–2000. [PMC free article: PMC6576563] [PubMed: 7683048]
- 30.
- Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12(5):1882–1895. [PMC free article: PMC6575874] [PubMed: 1349638]
- 31.
- Dawson VL, Dawson TM, London ED. et al. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A. 1991;88(14):6368–6371. [PMC free article: PMC52084] [PubMed: 1648740]
- 32.
- Brorson JR, Manzolillo PA, Miller RJ. Ca2+ entry via AMPA/KA receptors and excitotoxicity in cultured cerebellar Purkinje cells. J Neurosci. 1994;14(1):187–197. [PMC free article: PMC6576870] [PubMed: 7904304]
- 33.
- Volterra A, Trotti D, Cassutti P. et al. High sensitivity of glutamate uptake to extracellular free arachidonic acid levels in rat cortical synaptosomes and astrocytes. J Neurochem. 1992;59(2):600–606. [PubMed: 1629731]
- 34.
- Olney JW. Inciting excitotoxic cytocide among central neurons. Adv Exp Med Biol. 1986;203:631–645. [PubMed: 3024464]
- 35.
- Ingvar M, Morgan PF, Auer RN. The nature and timing of excitotoxic neuronal necrosis in the cerebral cortex, hippocampus and thalamus due to flurothyl-induced status epilepticus. Acta Neuropathol Berl. 1988;75(4):362–369. [PubMed: 3364160]
- 36.
- Olney JW. Glutamate-induced retinal degeneration in neonatal mice. Electron microscopy of the acutely evolving lesion. J Neuropathol Exp Neurol. 1969;28(3):455–474. [PubMed: 5788942]
- 37.
- Siesjo BK. Pathophysiology and treatment of focal cerebral ischemia. Part I: Pathophysiology. J Neurosurg. 1992;77(2):169–184. [PubMed: 1625004]
- 38.
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. [PubMed: 1970230]
- 39.
- Hahn JS, Aizenman E, Lipton SA. Central mammalian neurons normally resistant to glutamate toxicity are made sensitive by elevated extracellular Ca2+: Toxicity is blocked by the N-methyl-D-aspartate antagonist MK801. Proc Natl Acad Sci USA. 1988;85(17):6556–6560. [PMC free article: PMC282012] [PubMed: 2901101]
- 40.
- Ankarcrona M, Dypbukt JM, Bonfoco E. et al. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15(4):961–973. [PubMed: 7576644]
- 41.
- Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6(5):667–672. [PubMed: 8937832]
- 42.
- Gottron FJ, Ying HS, Choi DW. Caspase inhibition selectively reduces the apoptotic component of oxygen-glucose deprivation-induced cortical neuronal cell death. Mol Cell Neurosci. 1997;9(3):159–169. [PubMed: 9245499]
- 43.
- Bonfoco E, Krainc D, Ankarcrona M. et al. Apoptosis and necrosis: Two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A. 1995;92(16):7162–7166. [PMC free article: PMC41299] [PubMed: 7638161]
- 44.
- Kure S, Tominaga T, Yoshimoto T. et al. Glutamate triggers internucleosomal DNA cleavage in neuronal cells. Biochem Biophys Res Commun. 1991;179(1):39–45. [PubMed: 1679329]
- 45.
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1(8):623–634. [PubMed: 2908446]
- 46.
- Margerison JH, Corsellis JA. Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain. 1966;89(3):499–530. [PubMed: 5922048]
- 47.
- Sagar HJ, Oxbury JM. Hippocampal neuron loss in temporal lobe epilepsy: correlation with early childhood convulsions. Ann Neurol. 1987;22(3):334–340. [PubMed: 3674798]
- 48.
- Corsellis JA, Bruton CJ. Neuropathology of status epilepticus in humans. Adv Neurol. 1983;34:129–139. [PubMed: 6829328]
- 49.
- Hauser WA. Status epilepticus: frequency, etiology, and neurological sequelae. Adv Neurol. 1983;34:3–14. [PubMed: 6829339]
- 50.
- DeGiorgio CM, Tomiyasu U, Gott PS. et al. Hippocampal pyramidal cell loss in human status epilepticus. Epilepsia. 1992;33(1):23–27. [PubMed: 1733757]
- 51.
- Olney JW, deGubareff T, Sloviter RS. “Epileptic” brain damage in rats induced by sustained electrical stimulation of the perforant path. II. Ultrastructural analysis of acute hippocampal pathology. Brain Res Bull. 1983;10(5):699–712. [PubMed: 6871738]
- 52.
- Turski WA, Cavalheiro EA, Bortolotto ZA. et al. Seizures produced by pilocarpine in mice: a behavioral, electroencephalographic and morphological analysis. Brain Res. 1984;321(2):237–253. [PubMed: 6498517]
- 53.
- Clifford DB, Olney JW, Maniotis A. et al. The functional anatomy and pathology of lithium pilocarpine and high-dose pilocarpine seizures. Neuroscience. 1987;23(3):953–968. [PubMed: 3437996]
- 54.
- BenAri Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14(2):375–403. [PubMed: 2859548]
- 55.
- Meldrum BS. Cell damage in epilepsy and the role of calcium in cytotoxicity. Adv Neurol. 1986;44:849–855. [PubMed: 3706026]
- 56.
- Olney JW, Collins RC, Sloviter RS. Excitotoxic mechanisms of epileptic brain damage In: Delgado-Escueta AV, Ward AA, eds. Basic mechanisms of the epilepsies New York: Raven Press 1986857–877. [PubMed: 3706027]
- 57.
- Clifford DB, Olney JW, Benz AM. et al. Ketamine, phencyclidine, and MK801 protect against kainic acid-induced, seizure-related brain damage. Epilepsia. 1990;31(4):382–390. [PubMed: 2196169]
- 58.
- Clifford DB, Zorumski CF, Olney JW. Ketamine and MK801 prevent degeneration of thalamic neurons induced by focal cortical seizures. Exp Neurol. 1989;105(3):272–279. [PubMed: 2670599]
- 59.
- Fariello RG, Golden GT, Smith GG. et al. Potentiation of kainic acid epileptogenicity and sparing from neuronal damage by an NMDA receptor antagonist. Epilepsy Res. 1989;3(3):206–213. [PubMed: 2543557]
- 60.
- Sparenborg S, Brennecke LH, Jaax NK. et al. Dizocilpine (MK801) arrests status epilepticus and prevents brain damage induced by soman [published erratum appears in Neuropharmacology 1993 Mar;32(3):313] Neuropharmacology. 1992;31(4):357–368. [PubMed: 1522953]
- 61.
- Becker AJ, Gillardon F, Blumcke I. et al. Differential regulation of apoptosisrelated genes in resistant and vulnerable subfields of the rat epileptic hippocampus. Brain Res Mol Brain Res. 1999;67(1):172–176. [PubMed: 10101244]
- 62.
- Charriaut MC, Aggoun ZD, Represa A. et al. Apoptotic features of selective neuronal death in ischemia, epilepsy and gp-120 toxicity. Trends Neurosci. 1996;19(3):109–114. [PubMed: 9054057]
- 63.
- Gillardon F, Bottiger B, Schmitz B. et al. Activation of CPP32 protease in hippocampal neurons following ischemia and epilepsy. Brain Res Mol Brain Res. 1997;50(12):16–22. [PubMed: 9406913]
- 64.
- Henshall DC, Chen J, Simon RP. Involvement of caspase-3-like protease in the mechanism of cell death following focally evoked limbic seizures. J Neurochem. 2000;74(3):1215–1223. [PubMed: 10693954]
- 65.
- Henshall DC, Clark RS, Adelson PD. et al. Alterations in Bcl-2 and caspase gene family protein expression in human temporal lobe epilepsy. Neurology. 2000;55(2):250–257. [PubMed: 10908900]
- 66.
- Pollard H, Charriaut-Marlangue C, Cantagrel S. et al. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63(1):7–18. [PubMed: 7898662]
- 67.
- Faden AI, Demediuk P, Panter SS. et al. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244(4906):798–800. [PubMed: 2567056]
- 68.
- Faden AI, Simon RP. A potential role for excitotoxins in the pathophysiology of spinal cord injury. Ann Neurol. 1988;23(6):623–626. [PubMed: 2841902]
- 69.
- Bittigau P, Pohl D, Sifringer M. et al. Modeling pediatric head trauma: Mechanisms of degeneration and potential strategies for neuroprotection. Restor Neurol Neurosci. 1998;13(12):11–23. [PubMed: 12671284]
- 70.
- Shah PT, Yoon KW, Xu XM. et al. Apoptosis mediates cell death following traumatic injury in rat hippocampal neurons. Neuroscience. 1997;79(4):999–1004. [PubMed: 9219962]
- 71.
- Yakovlev AG, Knoblach SM, Fan L. et al. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17(19):7415–7424. [PMC free article: PMC6573442] [PubMed: 9295387]
- 72.
- Pohl D, Bittigau P, Ishimaru MJ. et al. N-Methyl-D-aspartate antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc Natl Acad Sci USA. 1999;96(5):2508–2513. [PMC free article: PMC26815] [PubMed: 10051673]
- 73.
- Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiol Rev. 1985;65(1):101–148. [PubMed: 3880896]
- 74.
- Hirsch JA, Gibson GE. Selective alteration of neurotransmitter release by low oxygen in vitro. Neurochem Res. 1984;9(8):1039–1049. [PubMed: 6149480]
- 75.
- Nicholls D, Attwell D. The release and uptake of excitatory amino acids [see comments] Trends Pharmacol Sci. 1990;11(11):462–468. [PubMed: 1980041]
- 76.
- Szatkowski M, Attwell D. Triggering and execution of neuronal death in brain ischaemia: two phases of glutamate release by different mechanisms. Trends Neurosci. 1994;17(9):359–365. [PubMed: 7529438]
- 77.
- Benveniste H, Drejer J, Schousboe A. et al. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;43(5):1369–1374. [PubMed: 6149259]
- 78.
- Drejer J, Benveniste H, Diemer NH. et al. Cellular origin of ischemia-induced glutamate release from brain tissue in vivo and in vitro. J Neurochem. 1985;45(1):145–151. [PubMed: 2860206]
- 79.
- Obrenovitch TP, Richards DA. Extracellular neurotransmitter changes in cerebral ischaemia. Cerebrovasc Brain Metab Rev. 1995;7(1):1–54. [PubMed: 7742171]
- 80.
- Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11(10):465–469. [PubMed: 2469166]
- 81.
- Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann Neurol. 1986;19(2):105–111. [PubMed: 2421636]
- 82.
- Rothman S. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J Neurosci. 1984;4(7):1884–1891. [PMC free article: PMC6564878] [PubMed: 6737044]
- 83.
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399(6738 Suppl):A7–14. [PubMed: 10392575]
- 84.
- Albers GW, Goldberg MP, Choi DW. Do NMDA antagonists prevent neuronal injury? Yes. Arch Neurol. 1992;49(4):418–420. [PubMed: 1558524]
- 85.
- Simon RP, Swan JH, Griffiths T. et al. Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science. 1984;226(4676):850–852. [PubMed: 6093256]
- 86.
- Wieloch T. Hypoglycemia-induced neuronal damage prevented by an N-methyl-D-aspartate antagonist. Science. 1985;230(4726):681–683. [PubMed: 2996146]
- 87.
- Benveniste H. The excitotoxin hypothesis in relation to cerebral ischemia. Cerebrovasc Brain Metab Rev. 1991;3(3):213–245. [PubMed: 1931486]
- 88.
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22(9):391–397. [PubMed: 10441299]
- 89.
- Martin LJ, Al Abdulla NA, Brambrink AM. et al. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46(4):281–309. [PubMed: 9671259]
- 90.
- Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24(12):2002–2008. [PubMed: 8248983]
- 91.
- MacManus JP, Buchan AM, Hill IE. et al. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett. 1993;164(12):89–92. [PubMed: 8152622]
- 92.
- Namura S, Zhu J, Fink K. et al. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18(10):3659–3668. [PMC free article: PMC6793169] [PubMed: 9570797]
- 93.
- Fink K, Zhu J, Namura S. et al. Prolonged therapeutic window for ischemic brain damage caused by delayed caspase activation. J Cereb Blood Flow Metab. 1998;18(10):1071–1076. [PubMed: 9778183]
- 94.
- Endres M, Namura S, Shimizu SM. et al. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18(3):238–247. [PubMed: 9498840]
- 95.
- Chen J, Nagayama T, Jin K. et al. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18(13):4914–4928. [PMC free article: PMC6792571] [PubMed: 9634557]
- 96.
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410(6831):988–994. [PubMed: 11309629]
- 97.
- Bezzi P, Domercq M, Brambilla L. et al. CXCR4-activated astrocyte glutamate release via TNF-alpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4(7):702–710. [PubMed: 11426226]
- 98.
- Teitelbaum JS, Zatorre RJ, Carpenter S. et al. Neurologic sequelae of domoic acid intoxication due to the ingestion of contaminated mussels [see comments] N Engl J Med. 1990;322(25):1781–1787. [PubMed: 1971710]
- 99.
- Biscoe TJ, Evans RH, Headley PM. et al. Domoic and quisqualic acids as potent amino acid excitants of frog and rat spinal neurones. Nature. 1975;255(5504):166–167. [PubMed: 1128682]
- 100.
- Biscoe TJ, Evans RH, Headley PM. et al. Structure-activity relations of excitatory amino acids on frog and rat spinal neurones. Br J Pharmacol. 1976;58(3):373–382. [PMC free article: PMC1667529] [PubMed: 990592]
- 101.
- Tryphonas L, Iverson F. Neuropathology of excitatory neurotoxins: the domoic acid model. Toxicol Pathol. 1990;18(1 Pt 2):165–169. [PubMed: 2195636]
- 102.
- Tryphonas L, Truelove J, Nera E. et al. Acute neurotoxicity of domoic acid in the rat. Toxicol Pathol. 1990;18(1 Pt 1):1–9. [PubMed: 2362984]
- 103.
- Tryphonas L, Truelove J, Iverson F. Acute parenteral neurotoxicity of domoic acid in cynomolgus monkeys (M. fascicularis) [published erratum appears in Toxicol Pathol 1990;18(3):431]. Toxicol Pathol 199018(2):297–303. [PubMed: 2268382]
- 104.
- Rao S L N, Adiga PR, Sarma PS. Biochemistry. 1964. pp. 1452185–220. [PubMed: 14155110]
- 105.
- Spencer PS, Roy DN, Ludolph A. et al. Lathyrism: evidence for role of the neuroexcitatory amino acid BOAA. Lancet. 1986;2(8515):1066–1067. [PubMed: 2877226]
- 106.
- Nunn PB, Seelig M, Zagoren JC. et al. Stereo-specific acute neuronotoxicity of “uncommon” plant amino acids linked to human motor-system diseases. Brain Res. 1987;410(2):375–379. [PubMed: 3109690]
- 107.
- Ross SM, Seelig M, Spencer PS. Specific antagonism of excitotoxic action of “uncommon” amino acids assayed in organotypic mouse cortical cultures. Brain Res. 1987;425(1):120–127. [PubMed: 3123008]
- 108.
- Ross SM, Spencer PS. Specific antagonism of behavioral action of “uncommon” amino acids linked to motor-system diseases. Synapse. 1987;1(3):248–253. [PubMed: 3145580]
- 109.
- Bridges RJ, Stevens DR, Kahle JS. et al. Structure-function studies on N-oxalyldiaminodicarboxylic acids and excitatory amino acid receptors: evidence that beta-LODAP is a selective non-NMDA agonist. J Neurosci. 1989;9(6):2073–2079. [PMC free article: PMC6569742] [PubMed: 2542485]
- 110.
- Ascher P, Nowak L. Quisqualate- and kainate-activated channels in mouse central neurones in culture. J Physiol. 1988;399:227–245. [PMC free article: PMC1191661] [PubMed: 2457088]
- 111.
- Mosbacher J, Schoepfer R, Monyer H. et al. A molecular determinant for submillisecond desensitization in glutamate receptors. Science. 1994;266(5187):1059–1062. [PubMed: 7973663]
- 112.
- Hestrin S, Nicoll RA, Perkel DJ. et al. Analysis of excitatory synaptic action in pyramidal cells using whole-cell recording from rat hippocampal slices. J Physiol. 1990;422:203–225. [PMC free article: PMC1190128] [PubMed: 1972190]
- 113.
- Murphy SN, Miller RJ. Regulation of Ca influx into striatal neurons by kainic acid. J Pharmacol Exp Ther. 1989;249(1):184–193. [PubMed: 2565388]
- 114.
- Gilbertson TA, Scobey R, Wilson M. Permeation of calcium ions through non-NMDA glutamate channels in retinal bipolar cells. Science. 1991;251(5001):1613–1615. [PubMed: 1849316]
- 115.
- Ogura A, Akita K, Kudo Y. Non-NMDA receptor mediates cytoplasmic Ca2+ elevation in cultured hippocampal neurones. Neurosci Res N Y. 1990;9(2):103–113. [PubMed: 2177531]
- 116.
- Pruss RM, Akeson RL, Racke MM. et al. Agonist-activated cobalt uptake identifies divalent cation-permeable kainate receptors on neurons and glial cells. Neuron. 1991;7(3):509–518. [PubMed: 1716930]
- 117.
- Jonas P, Racca C, Sakmann B. et al. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluRB subunit expression. Neuron. 1994;12(6):1281–1289. [PubMed: 8011338]
- 118.
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KAAMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252(5007):851–853. [PubMed: 1709304]
- 119.
- Nakanishi N, Shneider NA, Axel R. A family of glutamate receptor genes: evidence for the formation of heteromultimeric receptors with distinct channel properties. Neuron. 1990;5(5):569–581. [PMC free article: PMC4481242] [PubMed: 1699567]
- 120.
- Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short and longterm plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29(1):209–216. [PubMed: 11182092]
- 121.
- Contractor A, Swanson GT, Sailer A. et al. Identification of the kainate receptor subunits underlying modulation of excitatory synaptic transmission in the CA3 region of the hippocampus. J Neurosci. 2000;20(22):8269–8278. [PMC free article: PMC6773182] [PubMed: 11069933]
- 122.
- Ben Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23(11):580–587. [PubMed: 11074268]
- 123.
- Frerking M, Nicoll RA. Synaptic kainate receptors. Curr Opin Neurobiol. 2000;10(3):342–351. [PubMed: 10851174]
- 124.
- MacDermott AB, Mayer ML, Westbrook GL. et al. NMDA receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones [published erratum appears in Nature 1986 Jun 26Jul 2;321(6073):888] Nature. 1986;321(6069):519–522. [PubMed: 3012362]
- 125.
- Mayer ML, MacDermott AB, Westbrook GL. et al. Agonist and voltage-gated calcium entry in cultured mouse spinal cord neurons under voltage clamp measured using arsenazo III. J Neurosci. 1987;7(10):3230–3244. [PMC free article: PMC6569186] [PubMed: 2444678]
- 126.
- Mayer ML, Westbrook GL. Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol. 1987;394:501–527. [PMC free article: PMC1191974] [PubMed: 2451020]
- 127.
- Ascher P, Nowak L. Electrophysiological studies of NMDA receptors. Trends Neurosci. 1987;10(7):284–288.
- 128.
- Mayer ML, Westbrook GL. The physiology of excitatory amino acids in the vertebrate central nervous system. Prog Neurobiol. 1987;28(3):197–276. [PubMed: 2883706]
- 129.
- Mothet JP, Parent AT, Wolosker H. et al. D-Serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 2000;97(9):4926–4931. [PMC free article: PMC18334] [PubMed: 10781100]
- 130.
- Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor. Trends Neurosci. 1987;10(7):299–302. [PubMed: 7537407]
- 131.
- Lees KR. Cerestat and other NMDA antagonists in ischemic stroke. Neurology. 1997;49(5 Suppl 4):S66–S69. [PubMed: 9371155]
- 132.
- Chen HS, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol. 1997;499(Pt 1):27–46. [PMC free article: PMC1159335] [PubMed: 9061638]
- 133.
- Chen HS, Pellegrini JW, Aggarwal SK. et al. Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci. 1992;12(11):4427–4436. [PMC free article: PMC6576016] [PubMed: 1432103]
- 134.
- Chen HS, Wang YF, Rayudu PV. et al. Neuroprotective concentrations of the N-methyl-D-aspartate openchannel blocker memantine are effective without cytoplasmic vacuolation following postischemic administration and do not block maze learning or long-term potentiation. Neuroscience. 1998;86(4):1121–1132. [PubMed: 9697119]
- 135.
- Seif eN, Peruche B, Rossberg C. et al. Neuroprotective effect of memantine demonstrated in vivo and in vitro. Eur J Pharmacol. 1990;185(1):19–24. [PubMed: 2226632]
- 136.
- Bruno V, Copani A, Knopfel T. et al. Activation of metabotropic glutamate receptors coupled to inositol phospholipid hydrolysis amplifies NMDA-induced neuronal degeneration in cultured cortical cells. Neuropharmacology. 1995;34(8):1089–1098. [PubMed: 8532158]
- 137.
- Sharp AH, McPherson PS, Dawson TM. et al. Differential immunohistochemical localization of inositol 1,4,5-trisphosphate and ryanodine-sensitive Ca2+ release channels in rat brain. J Neurosci. 1993;13(7):3051–3063. [PMC free article: PMC6576698] [PubMed: 8392539]
- 138.
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361(6410):315–325. [PubMed: 8381210]
- 139.
- Henzi V, MacDermott AB. Characteristics and function of Ca(2+) and inositol 1,4,5-trisphosphate-releasable stores of Ca2+ in neurons. Neuroscience. 1992;46(2):251–273. [PubMed: 1311812]
- 140.
- Bruno V, Battaglia G, Kingston A. et al. Neuroprotective activity of the potent and selective mGlu1a metabotropic glutamate receptor antagonist, (+)2-methyl-4-carboxyphenylglycine (LY367385): comparison with LY357366, a broader spectrum antagonist with equal affinity for mGlu1a and mGlu5 receptors. Neuropharmacology. 1999;38(2):199–207. [PubMed: 10218860]
- 141.
- Kingston AE, O'Neill MJ, Bond A. et al. Neuroprotective actions of novel and potent ligands of group I and group II metabotropic glutamate receptors. Ann NY Acad Sci. 1999;890:438–449. [PubMed: 10668448]
- 142.
- Strasser U, Lobner D, Behrens MM. et al. Antagonists for group I mGluRs attenuate excitotoxic neuronal death in cortical cultures. Eur J Neurosci. 1998;10(9):2848–2855. [PubMed: 9758154]
- 143.
- Sagara Y, Schubert D. The activation of metabotropic glutamate receptors protects nerve cells from oxidative stress. J Neurosci. 1998;18(17):6662–6671. [PMC free article: PMC6792973] [PubMed: 9712638]
- 144.
- Mukhin A, Fan L, Faden AI. Activation of metabotropic glutamate receptor subtype mGluR1 contributes to posttraumatic neuronal injury. J Neurosci. 1996;16(19):6012–6020. [PMC free article: PMC6579189] [PubMed: 8815884]
- 145.
- Ambrosini A, Bresciani L, Fracchia S. et al. Metabotropic glutamate receptors negatively coupled to adenylate cyclase inhibit N-methyl-D-aspartate receptor activity and prevent neurotoxicity in mesencephalic neurons in vitro. Mol Pharmacol. 1995;47(5):1057–1064. [PubMed: 7746273]
- 146.
- Battaglia G, Bruno V, Ngomba RT. et al. Selective activation of groupII metabotropic glutamate receptors is protective against excitotoxic neuronal death. Eur J Pharmacol. 1998;356(23):271–274. [PubMed: 9774259]
- 147.
- Bruno V, Battaglia G, Copani A. et al. Activation of class II or III metabotropic glutamate receptors protects cultured cortical neurons against excitotoxic degeneration. Eur J Neurosci. 1995;7(9):1906–1913. [PubMed: 8528465]
- 148.
- Bruno V, Copani A, Bonanno L. et al. Activation of group III metabotropic glutamate receptors is neuroprotective in cortical cultures. Eur J Pharmacol. 1996;310(1):61–66. [PubMed: 8880068]
- 149.
- Bruno V, Battaglia G, Ksiazek I. et al. Selective activation of mGlu4 metabotropic glutamate receptors is protective against excitotoxic neuronal death. J Neurosci. 2000;20(17):6413–6420. [PMC free article: PMC6772963] [PubMed: 10964947]
- 150.
- Churchwell KB, Wright SH, Emma F. et al. NMDA receptor activation inhibits neuronal volume regulation after swelling induced by veratridine-stimulated Na influx in rat cortical cultures. J Neurosci. 1996;16(23):7447–7457. [PMC free article: PMC6579111] [PubMed: 8922400]
- 151.
- Inglefield JR, SchwartzBloom RD. Activation of excitatory amino acid receptors in the rat hippocampal slice increases intracellular Cl and cell volume. J Neurochem. 1998;71(4):1396–1404. [PubMed: 9751170]
- 152.
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13(8):3510–3524. [PMC free article: PMC6576549] [PubMed: 8101871]
- 153.
- Dessi F, Charriaut MC, Ben Ari Y. Glutamate-induced neuronal death in cerebellar culture is mediated by two distinct components: a sodium chloride component and a calcium component. Brain Res. 1994;650(1):49–55. [PubMed: 7953676]
- 154.
- Hartley DM, Choi DW. Delayed rescue of N-methyl-D-aspartate receptor-mediated neuronal injury in cortical culture. J Pharmacol Exp Ther. 1989;250(2):752–758. [PubMed: 2569534]
- 155.
- Murphy TH, Malouf AT, Sastre A. et al. Calcium-dependent glutamate cytotoxicity in a neuronal cell line. Brain Res. 1988;444(2):325–332. [PubMed: 2896063]
- 156.
- Garthwaite G, Garthwaite J. Neurotoxicity of excitatory amino acid receptor agonists in rat cerebellar slices: dependence on calcium concentration. Neurosci Lett. 1986;66(2):193–198. [PubMed: 3014386]
- 157.
- Tymianski M, Wallace MC, Spigelman I. et al. Cell-permeant Ca(2+) chelators reduce early excitotoxic and ischemic neuronal injury in vitro and in vivo. Neuron. 1993;11(2):221–235. [PubMed: 8102532]
- 158.
- Malcolm CS, Ritchie L, Grieve A. et al. A prototypic intracellular calcium antagonist, TMB8, protects cultured cerebellar granule cells against the delayed, calcium-dependent component of glutamate neurotoxicity. J Neurochem. 1996;66(6):2350–2360. [PubMed: 8632157]
- 159.
- Lu YM, Yin HZ, Chiang J. et al. Ca(2+)-permeable AMPA/kainate and NMDA channels: high rate of Ca2+ influx underlies potent induction of injury. J Neurosci. 1996;16(17):5457–5465. [PMC free article: PMC6578887] [PubMed: 8757258]
- 160.
- Eimerl S, Schramm M. The quantity of calcium that appears to induce neuronal death. J Neurochem. 1994;62(3):1223–1226. [PubMed: 8113805]
- 161.
- Andreeva N, Khodorov B, Stelmashook E. et al. Inhibition of Na/Ca2+ exchange enhances delayed neuronal death elicited by glutamate in cerebellar granule cell cultures. Brain Res. 1991;548(12):322–325. [PubMed: 1678303]
- 162.
- Mattson MP, Guthrie PB, Kater SB. A role for Nadependent Ca2+ extrusion in protection against neuronal excitotoxicity. FASEB J. 1989;3(13):2519–2526. [PubMed: 2572500]
- 163.
- White RJ, Reynolds IJ. Mitochondria and Na/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15(2):1318–1328. [PMC free article: PMC6577803] [PubMed: 7869100]
- 164.
- Carafoli E. Calcium pump of the plasma membrane. Physiol Rev. 1991;71(1):129–153. [PubMed: 1986387]
- 165.
- Gunter TE, Gunter KK, Sheu SS. et al. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267(2 Pt 1):C313–C339. [PubMed: 8074170]
- 166.
- Pozzan T, Rizzuto R, Volpe P. et al. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev. 1994;74(3):595–636. [PubMed: 8036248]
- 167.
- Dubinsky JM. Intracellular calcium levels during the period of delayed excitotoxicity. J Neurosci. 1993;13(2):623–631. [PMC free article: PMC6576661] [PubMed: 8093901]
- 168.
- Tymianski M, Charlton MP, Carlen PL. et al. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13(5):2085–2104. [PMC free article: PMC6576557] [PubMed: 8097530]
- 169.
- Dumuis A, Sebben M, Haynes L. et al. NMDA receptors activate the arachidonic acid cascade system in striatal neurons. Nature. 1988;336(6194):68–70. [PubMed: 2847054]
- 170.
- Chan PH, Fishman RA. Transient formation of superoxide radicals in polyunsaturated fatty acidinduced brain swelling. J Neurochem. 1980;35(4):1004–1007. [PubMed: 6256498]
- 171.
- Lazarewicz JW, Wroblewski JT, Palmer ME. et al. Activation of N-methyl-D-aspartate-sensitive glutamate receptors stimulates arachidonic acid release in primary cultures of cerebellar granule cells. Neuropharmacology. 1988;27(7):765–769. [PubMed: 2843786]
- 172.
- Williams JH, Errington ML, Lynch MA. et al. Arachidonic acid induces a long-term activity-dependent enhancement of synaptic transmission in the hippocampus. Nature. 1989;341(6244):739–742. [PubMed: 2571939]
- 173.
- Pellegrini-Giampietro DE, Cherici G, Alesiani M. et al. Excitatory amino acid release from rat hippocampal slices as a consequence of free-radical formation. J Neurochem. 1988;51(6):1960–1963. [PubMed: 2903225]
- 174.
- Lankiewicz S, Marc LC, Truc BN. et al. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem. 2000;275(22):17064–17071. [PubMed: 10828077]
- 175.
- Lee KS, Frank S, Vanderklish P. et al. Inhibition of proteolysis protects hippocampal neurons from ischemia. Proc Natl Acad Sci U S A. 1991;88(16):7233–7237. [PMC free article: PMC52268] [PubMed: 1871130]
- 176.
- Siman R, Noszek JC, Kegerise C. Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci. 1989;9(5):1579–1590. [PMC free article: PMC6569848] [PubMed: 2542478]
- 177.
- Siman R, Noszek JC. Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron. 1988;1(4):279–287. [PubMed: 2856162]
- 178.
- Garthwaite J, Garthwaite G, Palmer RM. et al. NMDA receptor activation induces nitric oxide synthesis from arginine in rat brain slices. Eur J Pharmacol. 1989;172(45):413–416. [PubMed: 2555211]
- 179.
- Gunasekar PG, Kanthasamy AG, Borowitz JL. et al. NMDA receptor activation produces concurrent generation of nitric oxide and reactive oxygen species: implication for cell death. J Neurochem. 1995;65(5):2016–2021. [PubMed: 7595485]
- 180.
- Dawson TM, Hung K, Dawson VL. et al. Neuroprotective effects of gangliosides may involve inhibition of nitric oxide synthase. Ann Neurol. 1995;37(1):115–118. [PubMed: 7529474]
- 181.
- Cazevieille C, Muller A, Meynier F. et al. Superoxide and nitric oxide cooperation in hypoxia/reoxygenation-induced neuron injury. Free Radic Biol Med. 1993;14(4):389–395. [PubMed: 8096826]
- 182.
- Dawson VL, Dawson TM, Bartley DA. et al. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13(6):2651–2661. [PMC free article: PMC6576487] [PubMed: 7684776]
- 183.
- Dawson VL, Kizushi VM, Huang PL. et al. Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase-deficient mice. J Neurosci. 1996;16(8):2479–2487. [PMC free article: PMC6578778] [PubMed: 8786424]
- 184.
- Lipton SA, Choi YB, Pan ZH. et al. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitrosocompounds [see comments] Nature. 1993;364(6438):626–632. [PubMed: 8394509]
- 185.
- Dawson TM, Steiner JP, Dawson VL. et al. Immunosuppressant FK506 enhances phosphorylation of nitric oxide synthase and protects against glutamate neurotoxicity [see comments] Proc Natl Acad Sci U S A. 1993;90(21):9808–9812. [PMC free article: PMC47661] [PubMed: 7694293]
- 186.
- Ankarcrona M, Dypbukt JM, Orrenius S. et al. Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Lett. 1996;394(3):321–324. [PubMed: 8830666]
- 187.
- Marcaida G, Kosenko E, Minana MD. et al. Glutamate induces a calcineurin-mediated dephosphorylation of Na,K()ATPase that results in its activation in cerebellar neurons in culture. J Neurochem. 1996;66(1):99–104. [PubMed: 8522995]
- 188.
- Marcaida G, Minana MD, Grisolia S. et al. Lack of correlation between glutamate-induced depletion of ATP and neuronal death in primary cultures of cerebellum. Brain Res. 1995;695(2):146–150. [PubMed: 8556324]
- 189.
- Blaustein MP. Calcium transport and buffering in neurons. Trends Neurosci. 1988;11(10):438–443. [PubMed: 2469161]
- 190.
- Berridge MJ. Elementary and global aspects of calcium signalling. J Physiol Lond. 1997;499(Pt 2):291–306. [PMC free article: PMC1159305] [PubMed: 9080360]
- 191.
- Peng TI, Greenamyre JT. Privileged access to mitochondria of calcium influx through N-methyl-D-aspartate receptors. Mol Pharmacol. 1998;53(6):974–980. [PubMed: 9614198]
- 192.
- Peng TI, Jou MJ, Sheu SS. et al. Visualization of NMDA receptorinduced mitochondrial calcium accumulation in striatal neurons. Exp Neurol. 1998;149(1):1–12. [PubMed: 9454610]
- 193.
- Scarpa A, Azzone GF. The mechanism of ion translocation in mitochondria. 4. Coupling of K efflux with Ca2+ uptake. Eur J Biochem. 1970;12(2):328–335. [PubMed: 5459571]
- 194.
- Nicholls DG, Scott ID. The regulation of brain mitochondrial calciumion transport. The role of ATP in the discrimination between kinetic and membranepotentialdependent calciumion efflux mechanisms. Biochem J. 1980;186(3):833–839. [PMC free article: PMC1161720] [PubMed: 7396840]
- 195.
- Nicholls DG. The regulation of extramitochondrial free calcium ion concentration by rat liver mitochondria. Biochem J. 1978;176(2):463–474. [PMC free article: PMC1186255] [PubMed: 33670]
- 196.
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14(1):348–356. [PMC free article: PMC6576848] [PubMed: 8283242]
- 197.
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem. 1996;66(1):403–411. [PubMed: 8522981]
- 198.
- Wang GJ, Thayer SA. Sequestration of glutamate-induced Ca2+ loads by mitochondria in cultured rat hippocampal neurons. J Neurophysiol. 1996;76(3):1611–1621. [PubMed: 8890280]
- 199.
- Kiedrowski L, Costa E. Glutamate-induced destabilization of intracellular calcium concentration homeostasis in cultured cerebellar granule cells: role of mitochondria in calcium buffering. Mol Pharmacol. 1995;47(1):140–147. [PubMed: 7838122]
- 200.
- Akerman KE. Changes in membrane potential during calcium ion influx and efflux across the mitochondrial membrane. Biochim Biophys Acta. 1978;502(2):359–366. [PubMed: 418807]
- 201.
- Nicholls DG. Calcium transport and porton electrochemical potential gradient in mitochondria from guinea pig cerebral cortex and rat heart. Biochem J. 1978;170(3):511–522. [PMC free article: PMC1183926] [PubMed: 348200]
- 202.
- Heaton GM, Nicholls DG. The calcium conductance of the inner membrane of rat liver mitochondria and the determination of the calcium electrochemical gradient. Biochem J. 1976;156(3):635–646. [PMC free article: PMC1163798] [PubMed: 949345]
- 203.
- White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16(18):5688–5697. [PMC free article: PMC6578963] [PubMed: 8795624]
- 204.
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67(6):2282–2291. [PubMed: 8931459]
- 205.
- Khodorov B, Pinelis V, Vergun O. et al. Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Lett. 1996;397(23):230–234. [PubMed: 8955353]
- 206.
- Schinder AF, Olson EC, Spitzer NC. et al. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16(19):6125–6133. [PMC free article: PMC6579180] [PubMed: 8815895]
- 207.
- Vergun O, Keelan J, Khodorov BI. et al. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol. 1999;519 Pt:2451–466. [PMC free article: PMC2269520] [PubMed: 10457062]
- 208.
- Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J. 1978;176(3):899–906. [PMC free article: PMC1186314] [PubMed: 218557]
- 209.
- Hansford RG. Relation between cytosolic free Ca2+ concentration and the control of pyruvate dehydrogenase in isolated cardiac myocytes. Biochem J. 1987;241:145–151. [PMC free article: PMC1147536] [PubMed: 2436608]
- 210.
- Hansford RG, Zorov D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol Cell Biochem. 1998;184:359–369. [PubMed: 9746330]
- 211.
- McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J. 1979;180(3):533–544. [PMC free article: PMC1161091] [PubMed: 39549]
- 212.
- McCormack JG, England PJ. Ruthenium Red inhibits the activation of pyruvate dehydrogenase caused by positive inotropic agents in the perfused rat heart. Biochem J. 1983;214:581–585. [PMC free article: PMC1152284] [PubMed: 6193784]
- 213.
- McCormack JG, Denton RM. Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Dev Neurosci. 1993;15(35):165–173. [PubMed: 7805568]
- 214.
- Robb GL, Burnett P, Rutter GA. et al. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998;17(17):4987–5000. [PMC free article: PMC1170827] [PubMed: 9724635]
- 215.
- Moreno SR. Contribution of the translocator of adenine nucleotides and the ATP synthase to the control of oxidative phosphorylation and arsenylation in liver mitochondria. J Biol Chem. 1985;260(23):12554–12560. [PubMed: 2864340]
- 216.
- Skulachev VP. Role of uncoupled and noncoupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys. 1996;29(2):169–202. [PubMed: 8870073]
- 217.
- Dykens JA. Mitochondrial Free Radical Production and Oxidative Pathophysiology: Implications for Neurodegenerative DiseaseIn: Beal MF, Howell N, BodisWollner J, eds. Mitochondria and Free Radicals in Neurodegenerative Diseases NewYork: Wiley-Liss, 199729–55.
- 218.
- Dykens JA. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated CA2+ and Na: implications for neurodegeneration. J Neurochem. 1994;63(2):584–591. [PubMed: 8035183]
- 219.
- Kowaltowski AJ, Castilho RF, Grijalba MT. et al. Effect of inorganic phosphate concentration on the nature of inner mitochondrial membrane alterations mediated by Ca2+ ions. A proposed model for phosphate-stimulated lipid peroxidation. J Biol Chem. 1996;271(6):2929–2934. [PubMed: 8621682]
- 220.
- Tabatabaie T, Potts JD, Floyd RA. Reactive oxygen species-mediated inactivation of pyruvate dehydrogenase. Arch Biochem Biophys. 1996;336:290–296. [PubMed: 8954577]
- 221.
- Patel M, Day BJ, Crapo JD. et al. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16(2):345–355. [PubMed: 8789949]
- 222.
- Sheu KF, Blass JP. The alpha-ketoglutarate dehydrogenase complex. Ann N Y Acad Sci. 1999;893:61–78. [PubMed: 10672230]
- 223.
- Zhang Y, Marcillat O, Giulivi C. et al. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J Biol Chem. 1990;265(27):16330–16336. [PubMed: 2168888]
- 224.
- Hillered L, Ernster L. Respiratory activity of isolated rat brain mitochondria following in vitro exposure to oxygen radicals. J Cereb Blood Flow Metab. 1983;3(2):207–214. [PubMed: 6841468]
- 225.
- Malis CD, Bonventre JV. Mechanism of calcium potentiation of oxygen free radical injury to renal mitochondria. A model for postischemic and toxic mitochondrial damage. J Biol Chem. 1986;261(30):14201–14208. [PubMed: 2876985]
- 226.
- Bindokas VP, Jordan J, Lee CC. et al. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16(4):1324–1336. [PMC free article: PMC6578569] [PubMed: 8778284]
- 227.
- Budd SL, Castilho RF, Nicholls DG. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997;415(1):21–24. [PubMed: 9326361]
- 228.
- Rothe G, Valet G. Flow cytometric analysis of respiratory burst activity in phagocytes with hydroethidine and 2',7'-dichlorofluorescin. J Leukoc Biol. 1990;47(5):440–448. [PubMed: 2159514]
- 229.
- Lafon CM, Pietri S, Culcasi M. et al. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364(6437):535–537. [PubMed: 7687749]
- 230.
- Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15(5 Pt 1):3318–3327. [PMC free article: PMC6578215] [PubMed: 7751912]
- 231.
- Dugan LL, Sensi SL, Canzoniero LM. et al. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J Neurosci. 1995;15(10):6377–6388. [PMC free article: PMC6578019] [PubMed: 7472402]
- 232.
- Herrero A, Barja G. ADP-regulation of mitochondrial free radical production is different with complex I or complex IIlinked substrates: implications for the exercise paradox and brain hypermetabolism. J Bioenerg Biomembr. 1997;29(3):241–249. [PubMed: 9298709]
- 233.
- Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191(2):421–427. [PMC free article: PMC1162232] [PubMed: 6263247]
- 234.
- Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237(2):408–414. [PubMed: 2983613]
- 235.
- Demin OV, Kholodenko BN, Skulachev VP. A model of O2 generation in the complex III of the electron transport chain. Mol Cell Biochem. 1998;184(12):21–33. [PubMed: 9746310]
- 236.
- Boveris A, Cadenas E, Stoppani AO. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem J. 1976;156(2):435–444. [PMC free article: PMC1163765] [PubMed: 182149]
- 237.
- Prehn JH. Mitochondrial transmembrane potential and free radical production in excitotoxic neurodegeneration. Naunyn Schmiedebergs Arch Pharmacol. 1998;357(3):316–322. [PubMed: 9550304]
- 238.
- Castilho RF, Ward MW, Nicholls DG. Oxidative stress, mitochondrial function, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1999;72(4):1394–1401. [PubMed: 10098841]
- 239.
- Lafon CM, Culcasi M, Gaven F. et al. Nitric oxide, superoxide and peroxynitrite: putative mediators of NMDA-induced cell death in cerebellar granule cells. Neuropharmacology. 1993;32(11):1259–1266. [PubMed: 7509050]
- 240.
- Bondy SC, Lee DK. Oxidative stress induced by glutamate receptor agonists. Brain Res. 1993;610(2):229–233. [PubMed: 8319085]
- 241.
- Huschenbett J, Zaidi A, Michaelis ML. Sensitivity of the synaptic membrane Na/Ca2+ exchanger and the expressed NCX1 isoform to reactive oxygen species. Biochim Biophys Acta. 1998;1374(12):34–46. [PubMed: 9814850]
- 242.
- Yan LJ, Levine RL, Sohal RS. Oxidative damage during aging targets mitochondrial aconitase [published erratum appears in Proc Natl Acad Sci U S A 1998 Feb 17;95(4):1968] Proc Natl Acad Sci U S A. 1997;94(21):11168–11172. [PMC free article: PMC23404] [PubMed: 9326580]
- 243.
- Hoyal CR, Thomas AP, Forman HJ. Hydroperoxide-induced increases in intracellular calcium due to annexin VI translocation and inactivation of plasma membrane Ca2+ ATPase. J Biol Chem. 1996;271(46):29205–29210. [PubMed: 8910578]
- 244.
- Castilho RF, Hansson O, Ward MW. et al. Mitochondrial control of acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 1998;18(24):10277–10286. [PMC free article: PMC6793348] [PubMed: 9852565]
- 245.
- Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys. 1979;195(2):460–467. [PubMed: 38751]
- 246.
- Petronilli V, Szabo I, Zoratti M. The inner mitochondrial membrane contains ionconducting channels similar to those found in bacteria. FEBS Lett. 1989;259(1):137–143. [PubMed: 2480918]
- 247.
- Szabo I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem. 1991;266(6):3376–3379. [PubMed: 1847371]
- 248.
- Bernardi P, Vassanelli S, Veronese P. et al. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem. 1992;267(5):2934–2939. [PubMed: 1737749]
- 249.
- Szabo I, Bernardi P, Zoratti M. Modulation of the mitochondrial megachannel by divalent cations and protons. J Biol Chem. 1992;267(5):2940–2946. [PubMed: 1371109]
- 250.
- Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem. 1989;264(14):7826–7830. [PubMed: 2470734]
- 251.
- Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ Biochemistry. 1996;35(26):8483–8488. [PubMed: 8679608]
- 252.
- Chernyak BV, Bernardi P. The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur J Biochem. 1996;238(3):623–630. [PubMed: 8706660]
- 253.
- Kowaltowski AJ, Castilho RF, Vercesi AE. Ca(2+)induced mitochondrial membrane permeabilization: role of coenzyme Q redox state. Am J Physiol. 1995;269(1 Pt 1):C141–C147. [PubMed: 7631741]
- 254.
- Kowaltowski AJ, Castilho RF, Vercesi AE. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett. 1996;378(2):150–152. [PubMed: 8549822]
- 255.
- Kowaltowski AJ, Netto LE, Vercesi AE. The thiol-specific antioxidant enzyme prevents mitochondrial permeability transition. Evidence for the participation of reactive oxygen species in this mechanism. J Biol Chem. 1998;273(21):12766–12769. [PubMed: 9582302]
- 256.
- Vercesi AE, Kowaltowski AJ, Grijalba MT. et al. The role of reactive oxygen species in mitochondrial permeability transition. Biosci Rep. 1997;17(1):43–52. [PubMed: 9171920]
- 257.
- Szabo I, Zoratti M. The mitochondrial megachannel is the permeability transition pore. J Bioenerg Biomembr. 1992;24(1):111–117. [PubMed: 1380498]
- 258.
- Friche E, Jensen PB, Nissen NI. Comparison of cyclosporin A and SDZ PSC833 as multidrug-resistance modulators in a daunorubicin-resistant Ehrlich ascites tumor. Cancer Chemother Pharmacol. 1992;30(3):235–237. [PubMed: 1628375]
- 259.
- Tiberghien F, Loor F. Ranking of P-glycoprotein substrates and inhibitors by a calcein-AM fluorometry screening assay. Anticancer Drugs. 1996;7(5):568–578. [PubMed: 8862725]
- 260.
- Budd SL, Tenneti L, Lishnak T. et al. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci U S A. 2000;97(11):6161–6166. [PMC free article: PMC18575] [PubMed: 10811898]
- 261.
- Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. [PubMed: 7539993]
- 262.
- Zhang J, Snyder SH. Nitric oxide in the nervous system. Annu Rev Pharmacol Toxicol. 1995;35:2133–333. [PubMed: 7598492]
- 263.
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271(5 Pt 1):C1424–C1437. [PubMed: 8944624]
- 264.
- Schulz JB, Matthews RT, Jenkins BG. et al. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J Neurosci. 1995;15(12):8419–8429. [PMC free article: PMC6577929] [PubMed: 8613773]
- 265.
- Butler AR, Flitney FW, Williams DL. NO, nitrosonium ions, nitroxide ions, nitrosothiols and iron-nitrosyls in biology: a chemist's perspective. Trends Pharmacol Sci. 1995;16(1):18–22. [PubMed: 7732599]
- 266.
- Beckman JS, Beckman TW, Chen J. et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87(4):1620–1624. [PMC free article: PMC53527] [PubMed: 2154753]
- 267.
- Radi R, Beckman JS, Bush KM. et al. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266(7):4244–4250. [PubMed: 1847917]
- 268.
- Radi R, Beckman JS, Bush KM. et al. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991;288(2):481–487. [PubMed: 1654835]
- 269.
- Leist M, Fava E, Montecucco C. et al. Peroxynitrite and nitric oxide donors induce neuronal apoptosis by eliciting autocrine excitotoxicity. Eur J Neurosci. 1997;9(7):1488–1498. [PubMed: 9240406]
- 270.
- Zhang J, Dawson VL, Dawson TM. et al. Nitric oxide activation of poly(ADPribose) synthetase in neurotoxicity. Science. 1994;263(5147):687–689. [PubMed: 8080500]
- 271.
- Bolanos JP, Almeida A, Stewart V. et al. Nitric oxide-mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. J Neurochem. 1997;68(6):2227–2240. [PubMed: 9166714]
- 272.
- Bolanos JP, Heales SJ, Land JM. et al. Effect of peroxynitrite on the mitochondrial respiratory chain: differential susceptibility of neurones and astrocytes in primary culture. J Neurochem. 1995;64(5):1965–1972. [PubMed: 7722484]
- 273.
- Brorson JR, Schumacker PT, Zhang H. Nitric oxide acutely inhibits neuronal energy production. J Neurosci. 1999;19(1):147–158. [PMC free article: PMC6782368] [PubMed: 9870946]
- 274.
- Ghatan S, Larner S, Kinoshita Y. et al. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150(2):335–347. [PMC free article: PMC2180235] [PubMed: 10908576]
- 275.
- Li Y, Chopp M, Jiang N. et al. Induction of DNA fragmentation after 10 to 120 minutes of focal cerebral ischemia in rats. Stroke. 1995;26(7):1252–1257. [PubMed: 7541573]
- 276.
- Linnik MD, Miller JA, SprinkleCavallo J. et al. Apoptotic DNA fragmentation in the rat cerebral cortex induced by permanent middle cerebral artery occlusion. Brain Res Mol Brain Res. 1995;32(1):116–124. [PubMed: 7494449]
- 277.
- Hara H, Friedlander RM, Gagliardini V. et al. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci U S A. 1997;94(5):2007–2012. [PMC free article: PMC20033] [PubMed: 9050895]
- 278.
- LookerenCampagne M, Gill R. Ultrastructural morphological changes are not characteristic of apoptotic cell death following focal cerebral ischaemia in the rat. Neurosci Lett. 1996;213(2):111–114. [PubMed: 8858621]
- 279.
- Mochizuki H, Goto K, Mori H. et al. Histochemical detection of apoptosis in Parkinson's disease. J Neurol Sci. 1996;137(2):120–123. [PubMed: 8782165]
- 280.
- Tompkins MM, Basgall EJ, Zamrini E. et al. Apoptotic-like changes in Lewy body-associated disorders and normal aging in substantia nigral neurons. Am J Pathol. 1997;150(1):119–131. [PMC free article: PMC1858540] [PubMed: 9006329]
- 281.
- Su JH, Anderson AJ, Cummings BJ. et al. Immunohistochemical evidence for apoptosis in Alzheimer's disease. Neuroreport. 1994;5(18):2529–2533. [PubMed: 7696596]
- 282.
- Dragunow M, Faull RL, Lawlor P. et al. In situ evidence for DNA fragmentation in Huntington's disease striatum and Alzheimer's disease temporal lobes. Neuroreport. 1995;6(7):1053–1057. [PubMed: 7632894]
- 283.
- Portera-Cailliau C, Hedreen JC, Price DL. et al. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15(5 Pt 2):3775–3787. [PMC free article: PMC6578226] [PubMed: 7751945]
- 284.
- Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58(5):459–471. [PubMed: 10331434]
- 285.
- Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H. et al. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21(5):1465–1468. [PubMed: 7737654]
- 286.
- Kingsbury AE, Mardsen CD, Foster OJ. DNA fragmentation in human substantia nigra: apoptosis or perimortem effect? Mov Disord. 1998;13(6):877–884. [PubMed: 9827610]
- 287.
- Uetsuki T, Takemoto K, Nishimura I. et al. Activation of neuronal caspase-3 by intracellular accumulation of wild-type Alzheimer amyloid precursor protein. J Neurosci. 1999;19(16):6955–6964. [PMC free article: PMC6782871] [PubMed: 10436052]
- 288.
- Selznick LA, Holtzman DM, Han BH. et al. In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58(9):1020–1026. [PubMed: 10499444]
- 289.
- Drache B, Diehl GE, Beyreuther K. et al. BclxL-specific antibody labels activated microglia associated with Alzheimer's disease and other pathological states. J Neurosci Res. 1997;47(1):98–108. [PubMed: 8981243]
- 290.
- Pettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20(4):633–647. [PubMed: 9581757]
- 291.
- Villa P, Kaufmann SH, Earnshaw WC. Caspases and caspase inhibitors. Trends Biochem Sci. 1997;22(10):388–393. [PubMed: 9357314]
- 292.
- Oppenheim RW, Flavell RA, Vinsant S. et al. Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J Neurosci. 2001;21(13):4752–4760. [PMC free article: PMC6762357] [PubMed: 11425902]
- 293.
- Villa PG, Henzel WJ, Sensenbrenner M. et al. Calpain inhibitors, but not caspase inhibitors, prevent actin proteolysis and DNA fragmentation during apoptosis. J Cell Sci. 1998;111(Pt 6):713–722. [PubMed: 9472000]
- 294.
- Kuida K, Zheng TS, Na S. et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384(6607):368–372. [PubMed: 8934524]
- 295.
- Kluck RM, BossyWetzel E, Green DR. et al. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275(5303):1132–1136. [PubMed: 9027315]
- 296.
- Yang J, Liu X, Bhalla K. et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275(5303):1129–1132. [PubMed: 9027314]
- 297.
- Susin SA, Zamzami N, Castedo M. et al. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184(4):1331–1341. [PMC free article: PMC2192812] [PubMed: 8879205]
- 298.
- Du C, Fang M, Li Y. et al. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42. [PubMed: 10929711]
- 299.
- Li P, Nijhawan D, Budihardjo I. et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–489. [PubMed: 9390557]
- 300.
- BossyWetzel E, Green DR. Caspases induce cytochrome c release from mitochondria by activating cytosolic factors. J Biol Chem. 1999;274(25):17484–17490. [PubMed: 10364179]
- 301.
- Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC [see comments] Nature. 1999;399(6735):483–487. [PubMed: 10365962]
- 302.
- Rosse T, Olivier R, Monney L. et al. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c [see comments] Nature. 1998;391(6666):496–499. [PubMed: 9461218]
- 303.
- Deshmukh M, Johnson EM. Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21(4):695–705. [PubMed: 9808457]
- 304.
- Zou H, Li Y, Liu X. et al. An Apaf-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274(17):11549–11556. [PubMed: 10206961]
- 305.
- Marzo I, Brenner C, Zamzami N. et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. [PubMed: 9748162]
- 306.
- Zamzami N, Susin SA, Marchetti P. et al. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183(4):1533–1544. [PMC free article: PMC2192517] [PubMed: 8666911]
- 307.
- Van der Heiden MG, Li XX, Gottleib E. et al. BclxL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem. 2001;276(22):19414–19419. [PubMed: 11259441]
- 308.
- Rego AC, Vesce S, Nicholls DG. The mechanism of mitochondrial membrane potential retention following release of cytochrome c in apoptotic GT17 neural cells. Cell Death Differ. 2001;8(10):995–1003. [PubMed: 11598797]
- 309.
- Hu Y, Benedict MA, Wu D. et al. BclXL interacts with Apaf-1 and inhibits Apaf-1dependent caspase-9 activation. Proc Natl Acad Sci U S A. 1998;95(8):4386–4391. [PMC free article: PMC22498] [PubMed: 9539746]
- 310.
- Fujimura M, Morita FY, Murakami K. et al. Cytosolic redistribution of cytochrome c after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998;18(11):1239–1247. [PubMed: 9809513]
- 311.
- Ouyang YB, Tan Y, Comb M. et al. Survival and death-promoting events after transient cerebral ischemia: phosphorylation of Akt, release of cytochrome c and activation of caspase-like proteases. J Cereb Blood Flow Metab. 1999;19(10):1126–1135. [PubMed: 10532637]
- 312.
- Matsuyama T, Hata R, Yamamoto Y. et al. Localization of Fas antigen mRNA induced in postischemic murine forebrain by in situ hybridization. Brain Res Mol Brain Res. 1995;34(1):166–172. [PubMed: 8750874]
- 313.
- Cao G, Minami M, Pei W. et al. Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab. 2001;21(4):321–333. [PubMed: 11323518]
- 314.
- Hayashi T, Sakai K, Sasaki C. et al. c-Jun N-terminal kinase (JNK) and JNK interacting protein response in rat brain after transient middle cerebral artery occlusion. Neurosci Lett. 2000;284(3):195–199. [PubMed: 10773432]
- 315.
- Krajewski S, Krajewska M, Ellerby LM. et al. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci U S A. 1999;96(10):5752–5757. [PMC free article: PMC21932] [PubMed: 10318956]
- 316.
- Budd SL. Mechanisms of neuronal damage in brain hypoxia/ischemia: focus on the role of mitochondrial calcium accumulation. Pharmacol Ther. 1998;80(2):203–229. [PubMed: 9839772]
- 317.
- Andreyev A, Fiskum G. Calcium induced release of mitochondrial cytochrome c by different mechanisms selective for brain versus liver. Cell Death Differ. 1999;6(9):825–832. [PubMed: 10510464]
- 318.
- Luetjens CM, Bui NT, Sengpiel B. et al. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. 2000;20(15):5715–5723. [PMC free article: PMC6772544] [PubMed: 10908611]
- 319.
- Nakagawa T, Zhu H, Morishima N. et al. Caspase-12 mediates endoplasmic reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98–103. [PubMed: 10638761]
- 320.
- Rao RV, Hermel E, Castro-Obregon S. et al. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem. 2001;276(36):33869–33874. [PubMed: 11448953]
- 321.
- Wang KK, Nath R, Posner A. et al. An alpha-mercaptoacrylic acid derivative is a selective nonpeptide cell-permeable calpain inhibitor and is neuroprotective. Proc Natl Acad Sci U S A. 1996;93(13):6687–6692. [PMC free article: PMC39087] [PubMed: 8692879]
- 322.
- Harper SJ, LoGrasso P. Signalling for survival and death in neurones: the role of stress-activated kinases, JNK and p38. Cell Signal. 2001;13(5):299–310. [PubMed: 11369511]
- 323.
- Schwarzschild MA, Cole RL, Hyman SE. Glutamate, but not dopamine, stimulates stress-activated protein kinase and AP1-mediated transcription in striatal neurons. J Neurosci. 1997;17(10):3455–3466. [PMC free article: PMC6573695] [PubMed: 9133371]
- 324.
- Kawasaki H, Morooka T, Shimohama S. et al. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem. 1997;272(30):18518–18521. [PubMed: 9228012]
- 325.
- Kikuchi M, Tenneti L, Lipton SA. Role of p38 mitogen-activated protein kinase in axotomy-induced apoptosis of rat retinal ganglion cells. J Neurosci. 2000;20(13):5037–5044. [PMC free article: PMC6772303] [PubMed: 10864961]
- 326.
- Miller FD, Pozniak CD, Walsh GS. Neuronal life and death: an essential role for the p53 family. Cell Death Differ. 2000;7(10):880–888. [PubMed: 11279533]
Publication Details
Author Information and Affiliations
Authors
Thomas Gillessen, Samantha L. Budd, and Stuart A. Lipton.Copyright
Publisher
Landes Bioscience, Austin (TX)
NLM Citation
Gillessen T, Budd SL, Lipton SA. Excitatory Amino Acid Neurotoxicity. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.