Glycosphingolipids (GSLs), a subclass of glycolipids found in the cell membranes of organisms from bacteria to humans, are the major glycolipids of animals. The emphasis of this chapter is on vertebrate glycosphingolipids. Information on glycolipids of fungi, plants, and invertebrates is covered elsewhere (Chapters 20 and 23–26), as are glycosylphosphatidylinositols (GPIs), glycolipids attached to proteins as membrane anchors (Chapter 12). This chapter describes the characteristic features of GSLs, pathways for their biosynthesis, and insights into their biological roles in membrane structure, host–pathogen interactions, cell–cell recognition, and modulation of membrane protein function.
DISCOVERY AND BACKGROUND
The first GSL to be characterized was galactosylceramide (GalCer). Among the simplest of glycolipids, it is also one of the most abundant molecules in the vertebrate brain. It consists of a single galactose residue in β-glycosidic linkage to the C-1 hydroxyl group of a lipid moiety called ceramide (Figure ). The structure of the lipid, a long-chain amino alcohol in amide linkage to a fatty acid, was so difficult to determine that the amino alcohol was dubbed “sphingosine” after the enigmatic Egyptian Sphinx. Animal cells synthesize various sphingosines and related long-chain amino alcohols, together referred to as sphingoid bases. Nearly all glycolipids in vertebrates are GSLs, which, in turn, are part of the larger family of sphingolipids (lipids built on sphingoid bases) that includes the major membrane phospholipid, sphingomyelin, and the second messenger sphingosine 1-phosphate that regulates angiogenesis and immune cell trafficking. Other GSLs were later identified because they accumulate to pathological levels in tissues of patients with lysosomal storage diseases, genetic disorders in which glycan degrading enzymes are faulty or missing (Chapter 44). For example, a sialic acid–containing GSL (GM2) was first isolated from the brain of a victim of Tay–Sachs disease, in which it accumulates, and was named “ganglioside” based on its location in nerve clusters or “ganglia” in the brain. Likewise, glucosylceramide (GlcCer) was first isolated from the spleen of a Gaucher disease patient, where it accumulates. As purification, separation, and analytical techniques improved, GSLs were found in all vertebrate tissues. Hundreds of unique GSL structures were found that vary in glycan structures alone, each of which are presented on several distinct ceramides.
Structures of representative glycosphingolipids (GSLs) and glycoglycerolipids. GSLs, such as GalCer, are built on a ceramide lipid moiety that consists of a long-chain amino alcohol (sphingosine) in amide linkage to a fatty acid. In comparison, glycoglycerolipids, (more...)
Glycoglycerolipids are distinguished from GSLs by their lipid, having glycans linked to the C-3 hydroxyl of diacylglycerol or alkyl(acyl)glycerol (Figure ). Very minor constituents of most animal tissues (other than the testes), glycoglycerolipids are widely distributed in microbes and plants. Glycolipids of fungi, plants, and invertebrates are covered in Chapters 20 and 23–26. Glycosylphosphatidylinositols (GPIs), a different family of glycolipids, are often attached to proteins as membrane, and may also exist as free glycolipids (discussed in Chapter 12). The lipopolysaccharides of Gram-negative bacteria are discussed in Chapter 21.
MAJOR CLASSES AND NOMENCLATURE
The ceramide lipid of GSLs consists of a sphingoid base with a fatty acid amide at the C-2 amine. Sphingosine is the most common sphingoid base in mammals, with hydroxyls at the C-1 and C-3 carbons and a trans double bond between C-4 and C-5 (Figure ). Sphinganine is the same structure without the double bond, and phytosphingosine lacks the double bond and has an additional hydroxyl on C-4. Ceramides containing sphinganine and phytosphingosine are less abundant in animals, whereas phytosphingosine is prominent in the glycosphingolipids of plants and fungi. The fatty acid components of ceramides vary widely, with lengths ranging from C14 to C30 or greater. Although often saturated, they may be variably unsaturated or have α-hydroxyl groups. Ceramide structures modulate membrane associations and functions of GSLs.
Although ceramide variations add significant diversity to GSL structures, major structural and functional classifications are based on the glycans. The first sugars linked to ceramide in vertebrates are typically β-linked galactose (GalCer) or glucose (GlcCer). GalCer and its analog sulfatide, with sulfate at the C-3 hydroxyl of galactose, are the major glycans in the kidney and the brain. In the brain they have essential roles in the structure and function of myelin, the insulator that allows for rapid nerve conduction. Interestingly, the related sulfogalactoglycerolipid, seminolipid (Figure ), is abundant only in the male reproductive tract, where it is essential for spermatogenesis. Sialylated GalCer (Neu5Acα2-3GalβCer; GM4) is also found in myelin. These galactolipids are seldom extended with larger saccharide chains; rather, most other members of the large and diverse family of GSLs in animals are built on GlcCer (Figure ). GlcCer itself is abundant in certain tissues. In skin, it is a precursor of special ceramides that are required for creating the essential surface water barrier when embedded into the stratum corneum of the epidermis (see below).
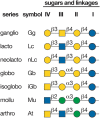
Glycosphingolipid (GSL) neutral cores and their designation based on IUPAC (International Union of Pure and Applied Chemistry) Nomenclature. In the official nomenclature, saccharide and other substituents that extend or branch from the neutral core structures (more...)
The vast majority of GSL structures are classified based on seven common tetrasaccharide neutral sugar core sequences (Figure ). GSLs sharing the same neutral core sequence are said to be of that “series”; the quantitatively major series in vertebrates are ganglio-, globo-, and neolacto-series, whereas in invertebrates the mollu- and arthro-series predominate.
GSL series are expressed in tissue-specific patterns. In mammals, ganglio-series GSLs, although broadly distributed, predominate in the brain, whereas neolacto-series glycolipids are common on certain hematopoietic cells including leukocytes. In contrast, lacto-series glycolipids are prominent in secretory organs and globo-series glycolipids are the most abundant in erythrocytes. This diversity presumably reflects important differences in GSL functions.
GSLs are further subclassified as neutral (no charged sugars or ionic groups), sialylated (having one or more sialic acid residues), or sulfated. Traditionally, all sialylated GSLs are known as “gangliosides” regardless of whether they are based on the ganglio-series neutral sugar core. In the official nomenclature, saccharide and other substituents that extend or branch from the neutral core structures are indicated by a roman numeral designating which of the neutral core sugars carries the substituent (counting the sugar closest to the ceramide as “I”), and a superscript designating which hydroxyl on that sugar is modified (Figure ). This nomenclature is too complex for daily use, so the most common GSLs are usually referred to by unofficial names. For example, in the widely used nomenclature of Svennerholm, the ganglioside Galβ1-3GalNAcβ1-4(Neu5Acα2-3)Galβ1-4GlcβCer is designated “GM1” (Figure ). In this nomenclature, G refers to ganglioside series, the second letter refers to the number of sialic acid residues (mono, di, tri, quattro [or tetra], penta, etc.), and the number (1, 2, 3, etc.) refers to the order of migration of the ganglioside on thin-layer chromatography (TLC) (e.g., GM3 > GM2 > GM1) with respect to the starting point. Therefore, the neutral core oligosaccharide shrinks in size (and moves farther by TLC) as the number increases.
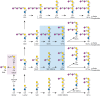
Glycosphingolipids (GSLs) are synthesized by the stepwise addition of sugars first to ceramide and then to the growing glycans. Shown as examples are major brain GSLs. Ceramide (Cer) is the acceptor for UDP-Gal:ceramide β-galactosyltransferase (more...)
ISOLATION, PURIFICATION, AND ANALYSIS
Organic solvents are used to extract glycolipids from tissues and cells, where they are found primarily in the external leaflet of plasma membranes. Extraction procedures, most often using defined chloroform–methanol–water mixtures, are optimized to precipitate and remove proteins and nucleic acids while maximizing solubilization of GSLs (along with other lipids). Because GSLs aggregate with one another and other lipids in aqueous solution, organic solvents are used throughout subsequent purification steps, which typically involve solvent partition, ion-exchange, and adsorption chromatographies.
Because of their amphipathic nature, glycolipids are well-suited for TLC analysis, which is useful for monitoring their purification, qualitative and quantitative determination of their expression in normal and diseased tissues, partial structural analysis, and detecting biological activities including immunoreactivity and binding activity toward toxins, viruses, bacteria, and eukaryotic cells. After separation by TLC, picomole to nanomole quantities of glycolipids can be chemically detected with orcinol reagent for hexoses and with resorcinol-HCl reagent for sialic acid. Normal phase TLC robustly resolves GSLs based on oligosaccharide diversity but is less effective at separating differences due to ceramides. To study glycolipid receptors, TLC-resolved glycolipids are overlaid with potential binding proteins or organisms (e.g., antibodies, lectins, toxins, viruses, bacteria, or cells). After washing, glycolipid species that bind can be identified by detection of the bound material at precise positions on the TLC plate. Two-dimensional TLC with exposure to ammonia vapor after the first dimension can be useful in identifying the presence of O-acetyl modifications on sialic acids, which are especially common in the brain.
Complete structural analyses of glycolipids require a combination of techniques to determine the composition, sequence, linkage positions, and anomeric configurations of the glycan moiety and the fatty acid and long-chain base of the ceramide moiety. Glycan composition is determined by hydrolysis and analysis of the released monosaccharides. Mass spectrometry (MS) of underivatized GSLs or of their permethylated derivatives is a powerful tool for sequence determination and ceramide identification and can sometimes be performed directly from the TLC plate. However, the methylation process causes destruction of labile substitutions like O-acetyl esters. Linkage determination is most rigorously performed by methylation analysis, and anomeric configurations can be obtained by nuclear magnetic resonance (NMR) spectroscopy. Information on glycan sequence, linkage, and anomeric configuration can also be obtained by combining enzymatic hydrolysis using specific glycosidases with TLC. Intact glycans are released from many GSLs enzymatically using ceramide glycanases or endoglycoceramidases. The resulting oligosaccharides can be analyzed using the methods described in Chapter 50. Additionally, there are many monoclonal antibodies that detect specific glycosphingolipid structures.
BIOSYNTHESIS, TRAFFICKING, AND DEGRADATION
Glycosphingolipid biosynthesis occurs in a stepwise fashion, with an individual sugar added first to ceramide and then subsequent sugars transferred by glycosyltransferases from nucleotide sugar donors. Ceramide is synthesized on the cytoplasmic face of the endoplasmic reticulum (ER), equilibrates to the luminal face, and then traffics to the Golgi apparatus. GlcCer is synthesized on the cytoplasmic face of the early Golgi apparatus. It is flipped to the luminal side of the Golgi (or after retrograde transport to the ER), and then is elongated by a series of Golgi glycosyltransferases. In contrast, GalCer is synthesized on the luminal face of the ER and then traffics through the Golgi, where it may be sulfated to form sulfatide. In both cases, the final orientation of GSLs during biosynthesis is consistent with their nearly exclusive appearance on the outer leaflet of the plasma membrane, facing the extracellular milieu. Specific exceptions occur, such as the high concentration of lactosylceramide in neutrophil granules. Although ceramide resides on intracellular organelles, such as mitochondria, GSLs beyond GlcCer are not typically found on the membrane leaflet facing the cytoplasm, although they appear in the nucleus.
The biosynthesis of GSLs in the brain provides an example of how competing biosynthetic pathways can lead to glycan structural diversity (Figure ). Stepwise biosynthesis of GalCer and sulfatide occurs in oligodendrocytes, the cells that elaborate myelin. Gangliosides, in contrast, are synthesized by all cells, with concentrations of the different forms varying according to cell type. Expression patterns of GSLs are determined by the expression and intracellular distribution of the enzymes required for their biosynthesis. In some cases, multiple glycosyltransferases compete for the same GSL precursor (Figure ). For example, the ganglioside GM3 may be acted on by N-acetylgalactosaminyltransferase, thereby forming GM2 and going down the “a-series” gangliosides, or by sialyltransferase, thereby forming GD3, the simplest of the “b-series” gangliosides. Each branch is a committed pathway, because sialyltransferases cannot directly convert a-series gangliosides (beyond GM3) to their corresponding b-series gangliosides. Because of this branch exclusivity, competition between two enzymes at a key branch point determines the relative expression levels of the final GSL products. The transfer of N-acetylgalactosamine to a-, b-, and c-series gangliosides, transforming GM3 into GM2, GD3 into GD2, or GT3 into GT2, is catalyzed by the same N-acetylgalactosaminyltransferase. Likewise, the transfer of galactose to GM2 to form GM1, to GD2 to form GD1b, or to GT2 to form GT1c is accomplished by a single galactosyltransferase. In addition, the levels of nucleotide sugar donors used by glycosyltransferases in the Golgi lumen (including UDP-Gal, UDP-Glc, UDP-GlcNAc, UDP-GalNAc, and CMP-Neu5Ac) ultimately affect the final structure of glycans and are regulated by synthetic enzymes in the cytoplasm or nucleus and by the activity of nucleotide sugar transporters in the Golgi membrane (Chapter 5). The sialic acids of human gangliosides are exclusively in the form of N-acetylneuraminic acid (Neu5Ac) and its O-acetylated derivatives, but those of many other mammals contain both Neu5Ac and N-glycolylneuraminic acid (Neu5Gc) (Chapter 15). Even in animals with predominantly Neu5Gc in the gangliosides of nonneural tissues, their brain gangliosides have nearly exclusively Neu5Ac. Sialic acids on gangliosides may be further modified by O-acetylation or removal of the N-acyl group, to generate a free amino group (Chapter 15).
An additional level of regulation occurs via stable association of different GSL glycosyltransferases into “multiglycosyltransferase” complexes. The multiple enzymes are thought to act concertedly on the growing GSL without releasing intermediate structures, ensuring progression to the preferred end product.
Although the enzymes that catalyze the initial steps in GSL biosynthesis are specific and used only for GSL biosynthesis, outer sugars, such as the outermost sialic acid, fucose, or glucuronic acid residues, are sometimes added by glycosyltransferases that also act on glycoproteins, resulting in terminal structures being shared by GSL and glycoprotein glycans (Chapter 14). One example is the blood group ABO antigen system. The α1-3 N-acetylgalactosaminyltransferase encoded by the blood group A gene produces the blood group A determinant GalNAcα1-3(Fucα1-2)Galβ on glycoproteins and glycolipids. Correspondingly, the α1-3 galactosyltransferase encoded by the allelic blood group B gene transfers galactose to both glycoproteins and glycolipids.
Major brain ganglioside structures are highly conserved among all mammals and birds. In contrast, differences in blood cell glycolipids are well known even among humans, as in the case of the ABO, Lewis, and P blood group antigens (Chapter 14). Species differences among glycolipids also occur, one example being the expression of Forssman antigen, GalNAcα1-3GalNAcβ1-3Galα1-4Galβ1-4GlcβCer. This molecule is a good immunogen in Forssman antigen–negative species, such as rabbit, rat, and human, which have a mutated α1-3 N-acetylgalactosaminyltransferase that cannot transfer N-acetylgalactosamine to the precursor Gb4Cer. In contrast, guinea pig, mouse, sheep, and goat are Forssman antigen–positive.
The breakdown of GSLs occurs stepwise by the action of lysosomal hydrolases. GSLs on the outer surface of the plasma membrane are internalized, along with other membrane components, in invaginated vesicles that then fuse with endosomes, resulting in the GSL glycan facing the endosome lumen. GSL-enriched areas of the endosomal membrane may then invaginate once again to form intraluminal vesicles within the endosome. When endosomes fuse with primary lysosomes, GSLs become exposed to lysosomal hydrolases. Absence of any one of these glycosidases results in lysosomal storage diseases (Chapter 44).
As GSLs are successively cleaved to smaller structures, the remaining “core” monosaccharides become inaccessible to the water-soluble lysosomal hydrolases and require assistance from activator proteins that are referred to as “liftases.” These include GM2-activator protein and four structurally related saposins, all of which are derived from a single polypeptide precursor by proteolytic cleavage. Saposins are thought to bind to their glycolipid substrate, disrupt its interaction with the local membrane environment, and facilitate access of the glycans to the water-soluble hydrolytic enzymes. In certain lysosomal storage diseases (Chapter 44), mutations in activator proteins result in pathological accumulation of glycolipids, even though there is an abundance of the hydrolase responsible for degradation, thus demonstrating the essential role of activator proteins in GSL catabolism in vivo. The final breakdown products of GSL catabolism—monosaccharides, fatty acids, and free sphingoid bases—are then available for reuse by salvage pathways.
BIOLOGICAL AND PATHOLOGICAL ROLES
Glycosphingolipid-Enriched Membrane Microdomains
GSLs comprise from <5% (erythrocytes) to >20% (myelin) of the total membrane lipids in the plasma membranes of vertebrate cells. However, they are not uniformly distributed in the plane of the membrane but cluster in “lipid rafts,” small lateral microdomains of self-associating membrane molecules. Although the precise structure and makeup of lipid rafts is a matter of ongoing debate, their outer leaflets are believed to be enriched in sphingolipids, including GSLs and sphingomyelin (the phosphocholine derivative of ceramide). The self-association of sphingolipids is driven through the unique biophysical properties imparted by their long saturated carbon chains (Figure ). Besides sphingolipids, lipid rafts are enriched in cholesterol and selected proteins, including GPI-anchored proteins and some transmembrane signaling proteins such as receptor tyrosine kinases. On the cytoplasmic side, acylated proteins, such as Src family protein tyrosine kinases and Gα subunits of G proteins, associate with lipid rafts.
Lipid rafts are apparently dynamic, short-lived (msec), and small (10–50 nm in diameter), each containing perhaps hundreds of lipid molecules along with a few protein molecules. It has been argued that external clustering of lipid rafts into larger structures might bring signaling molecules such as kinases and their substrates together to enhance intracellular signaling. Thus, GSLs may act as intermediaries in the flow of information from the outside to the inside of cells. This idea is supported by the observation that antibody-induced GSL clustering activates lipid-raft-associated signaling and has led to the concept of plasma membrane “glycosignaling domains” or “glycosynapse.” Interactions between glycans and glycan-binding proteins, as well as glycan–glycan interactions, are also very much influenced by the density of glycans in terms of binding affinity. Multiple glycans clustered in a limited area can increase the avidity of cognate binding proteins compared with a single molecule of a glycan (Chapter 29). Natural multivalency adds unique functional properties to plasma membrane glycolipids. Indeed, several growth factor receptors including the epidermal growth factor (EGF) receptor, insulin receptor, and the nerve growth factor receptor are localized in membrane microdomains, and structural studies identified binding sites occupied by specific GSLs that modulate receptor signaling functions.
Physiological Functions of Glycosphingolipids
When expressed in the outer leaflet of the plasma membrane of cells, with their glycans facing the external milieu, GSL functions fall into two major categories: mediating cell–cell interactions via binding to complementary molecules on apposing plasma membranes (trans recognition) and modulating activities of proteins in the same plasma membrane (cis regulation).
At the single-cell level, GSLs are not essential for life. Using specific chemical inhibitors and genetic ablation of biosynthetic genes, cells without GSLs survive, proliferate, and even differentiate. However, GSLs are required for development at the whole-animal level. Mice engineered to lack the gene for GlcCer synthesis fail to develop, with arrest occurring just past the gastrula stage because of extensive apoptosis in the embryo. These and other recent observations lead to a basic principle: GSLs mediate and modulate intercellular coordination in multicellular organisms. Sometimes this occurs in quite subtle ways, as exemplified by the role of GalCer and sulfatide in myelination.
GalCer and its 3-O-sulfated derivative, sulfatide, are predominant glycans in the brain, where they constitute >50% of the total glycoconjugates. In the brain, they are expressed by oligodendrocytes, which elaborate myelin, the multilayered membrane insulation that ensheathes nerve axons. GalCer and sulfatide constitute >20% of myelin lipids and were widely believed to be essential to myelin structure. This turned out to be true, but in a much subtler way than anticipated. Mice engineered to lack the enzyme responsible for GalCer synthesis (UDP-Gal:ceramide β-galactosyltransferase) do not make any GalCer or sulfatide. However, they myelinate axons, and the myelin appears grossly normal. Nevertheless, the mice show all the signs of failed myelination, including tremor, ataxia, slow nerve conduction, and early death. In both normal and mutant mice, myelination occurs in short stretches along axons, with intermittent gaps called “nodes of Ranvier” where concentrated ion channels pass nerve impulses along to the next gap. At the edge of the node, myelin membranes normally curve downward and attach to the axon to seal off the node. In animals lacking GalCer and sulfatide, these myelin “end feet” fail to attach to the axon, instead turning upward, away from the axon. The result is a faulty node of Ranvier, with ion channels and adhesion molecules in disarray. Without the proper structure at the node of Ranvier, rapid nerve conduction is disrupted. A similar phenotype is shared by mice lacking the enzyme that adds the sulfate group to GalCer to make sulfatide. The conclusion is that sulfatide is essential for myelin–axon interactions, and its absence results in severe neurological deficits.
A key function of GlcCer has been learned from studies on its catabolism in postnatal animals. Ceramide is a key component of the outer layer of the skin and is responsible for the epidermal permeability barrier—a key defense against dehydration. Infants with severe Gaucher disease, in which β-glucocerebrosidase activity is nearly absent and GlcCer is not catabolized, are prone to dehydration because of high skin permeability. The relationship among GlcCer, ceramide, and skin permeability was confirmed in mice engineered with the same mutation in β-glucocerebrosidase as found in these infants. The mice unable to catabolize GlcCer died within days of birth by dehydration through the skin. This established the role of GlcCer as the obligate precursor to the ceramide required to build the outermost protective layer (stratum corneum) of the skin. GlcCer is synthesized, transported to the stratum corneum, and then enzymatically hydrolyzed, resulting in ceramide deposition.
More complex GSLs function both in cell–cell recognition and in the regulation of signal transduction. As with sulfatide, these functions are sometimes subtle, as exemplified by the effects of blocking ganglioside biosynthesis on nervous system physiology. Given the complexity of complex ganglioside biosynthesis (Figure ), it was surprising to discover that major alterations in ganglioside expression resulted in only modest phenotypic changes in mice. When the N-acetylgalactosaminyltransferase responsible for ganglioside elongation (GM2/GD2 synthase) was inactivated in mice, none of the major complex gangliosides (GM1, GD1a, GD1b, or GT1b) were expressed, and a comparable concentration of the simple gangliosides GM3 and GD3 were found in the adult brain. The resulting mice were grossly normal, but as they aged, mice without the normal spectrum of brain gangliosides displayed signs of axon degeneration and demyelination, hallmarks of a problem in myelin–axon cell–cell communication. These deficits arise from loss of ganglioside binding to a protein on the myelin membrane, myelin-associated glycoprotein (MAG), a member of the Siglec family of sialic acid–dependent carbohydrate-binding proteins (Chapter 35). MAG is expressed on the innermost myelin wrap, directly across from the axon surface. Mice engineered to lack MAG have many of the same phenotypic changes as mice lacking GM2/GD2 synthase, and biochemical and cell-biological studies showed that the major brain gangliosides GD1a and GT1b are excellent ligands for MAG. These results support the conclusion that MAG on the innermost myelin membrane binds to GD1a and GT1b on the axon cell surface to stabilize myelin–axon interactions. Disruption of the same ganglioside biosynthetic gene in humans leads to a similar phenotype: hereditary spastic paraplegia.
A second trans recognition role for GSLs is evident in the interaction of leukocytes with the blood vessel wall during the process of inflammation, the body's protection against bacterial infection. As discussed in Chapter 34, the first step in inflammation is the binding of white blood cells (leukocytes) to the endothelial cells lining the blood vessel near sites of infection (activated endothelium). This cell–cell interaction is initiated when glycan-binding proteins of the selectin family, expressed on the activated endothelium, bind to complementary glycans on the surface of passing leukocytes. One of the selectins, E-selectin, binds to glycoconjugates found on human leukocytes. The glycan receptor(s) for E-selectin are resistant to protease treatment, indicating that they may be GSLs. A candidate class of GSLs, myeloglycans, has been identified in leukocytes. The candidate GSLs have long sugar chains consisting of a neutral core with Galβ1-4GlcNAcβ1-3 repeats (Chapter 14), substituted with a terminal sialic acid and fucose residues on one or more of the N-acetylglucosamine residues.
Another role for GSLs in mediating inflammatory responses involves lipid-specific antigen presentation. Natural killer T (NKT) cells, which carry both T- and NKT-cell receptors, are involved in the suppression of autoimmune reactions, cancer metastasis, and the graft rejection response. The MHC class I-related molecule (CD1d) of dendritic cells presents glycolipid antigens via T-cell receptor recognition to activate NKT cells. NKT cells can be activated by nonmammalian Galα-ceramide, expressed by some bacteria in the large intestine of mice, as well as the isoglobo-series GSL iGb3Cer (Galα1-3Galβ1-4GlcβCer), which has been proposed as an endogenous NKT activator.
In addition to their action as trans recognition molecules, GSLs also interact laterally with proteins in the same membrane to modulate their activities (cis regulation). Notable among these cis-regulatory interactions are those between gangliosides and members of the receptor tyrosine kinase family. Gangliosides regulate the activity of the EGF receptor, platelet-derived growth factor receptor, fibroblast growth factor receptor, TrkA neurotrophin receptor, and insulin receptor. Ganglioside GM3, for example, down-regulates insulin receptor responsiveness to insulin. Mice that lack the enzyme responsible for the biosynthesis of GM3 display increased insulin receptor phosphorylation, enhanced glucose tolerance, and enhanced insulin sensitivity and are less susceptible to induced insulin resistance. Cis-regulatory interactions such as GM3-mediated insulin sensitivity may arise from direct interactions with signaling receptors or from the contribution of GSLs to the biophysical characteristics of membrane microdomains.
Glycosphingolipids in Human Pathology
Mutations in glycosphingolipid biosynthetic genes are exceedingly rare in humans, perhaps because of their devastating effects (Chapter 45). A single case of UDP-glucose ceramide glucosyltransferase (Ugcg) deficiency has been described, resulting in a lethal form of ichthyosis. This mutation evidently left some residual activity, because complete loss is associated with embryonic lethality in mice. Mutations in a gene required for biosynthesis of ganglioside GM3, ST3GAL5, result in severe infantile seizures accompanied by profound motor and intellectual deficits as well as blindness and deafness. Mutations in another ganglioside-specific biosynthetic gene, B4GALNT1, responsible for biosynthesis of GM2 and GD2, are less severe, resulting in hereditary spastic paraplegia accompanied by intellectual disability. Loss of globo-series GSLs does not cause obvious disease, but increases spontaneous abortions associated with the rare p blood group resulting from α4-galactosyltransferase deficiency. Mutations in GSL degradation genes, which are also rare, cause GSL storage diseases that lead to the accumulation of GSLs in lysosomes. They typically result from mutations in glycosidases, and less frequently from mutations in activator proteins (Chapter 44). The symptoms depend on the tissues in which the unhydrolyzed GSL accumulates and on the extent of loss of enzyme activity. The most common GSL storage disease is Gaucher disease, which is caused by mutations in the enzyme β-glucocerebrosidase, resulting primarily in accumulation of GlcCer in the liver and spleen (and other tissues in more severe cases). Enzyme replacement therapy has been successful in treating Gaucher disease, and drugs to block GlcCer synthesis (“substrate reduction therapy”) are in clinical use (Chapter 55). Another example, Tay–Sachs disease, is caused by mutations in a β-hexosaminidase and results in the buildup of GM2, culminating in irreversible fatal deterioration of brain function. Unfortunately, enzyme replacement delivery to the brain has not yet been successfully developed, which is also a problem for long-term treatment of Gaucher disease. Glycosphingolipid storage and related diseases are considered more extensively in Chapter 44.
Anti-GSL antibodies are involved in certain autoimmune diseases (Chapter 46). Some forms of Guillain–Barré syndrome, the most common form of paralytic disease worldwide, clearly involve autoantibodies against gangliosides. One form of Guillain–Barré syndrome occurs subsequent to infection with particular strains of the common diarrheal bacterial agent Campylobacter jejuni. These bacteria produce near-exact replicas of brain ganglioside glycans (such as GD1a) attached to their lipooligosaccharide cores. Following infection and immune clearance of the bacteria, the antiglycan antibodies produced to fight the bacteria go on to attack the patient's own nerves, causing paralysis. In some patients with multiple myeloma (a malignancy of antibody-producing plasma cells), the tumor cells secrete monoclonal antibodies against glycolipids, such as the rare sulfoglucuronyl epitope of nervous system GSLs termed HNK-1 (IV3GlcA[3-sulfate]-nLc4Cer). These patients suffer severe peripheral neuropathy.
Several bacterial toxins take advantage of GSLs to gain access to cells (Chapter 37). Cholera toxin and the structurally related Escherichia coli heat-labile enterotoxins are produced in the intestinal tract of infected individuals, bind to intestinal epithelial cell surfaces, and insert their toxic polypeptide “payload” through the cell membrane, where it disrupts ion fluxes, causing severe diarrhea. The toxins behave as docking modules with gangliosides acting as the site of attachment. Five identical polypeptide B subunits in a ring each bind to ganglioside GM1 on the cell surface, and a sixth A subunit (the “payload”) is then inserted through the membrane. A similar mechanism is used by Shiga toxin (also called verotoxin), which binds to the glycolipid Gb3Cer (globotriaosylceramide, Galα1-4Galβ1-4GlcβCer) via five subunits in a ring, each with three GSL-binding sites. In contrast, tetanus and related botulinum toxins are multidomain single polypeptides. One domain binds b-series gangliosides on nerve cells, whereas the other domains translocate the toxin into cells and disrupt proteins essential for synaptic transmission. Custom-designed multivalent glycans and glycoconjugates are being evaluated as high-affinity blockers of certain bacterial toxins. In addition to soluble toxins, certain intact bacteria also bind to specific GSLs via bacterial surface proteins called adhesins. This adherence is essential for successful colonization and symbiosis. Microbial adhesins are addressed in more detail in Chapter 37.
Malignant transformation in cancer progression is often associated with changes in the glycan structures of glycoproteins and glycolipids. The changes result mainly from altered levels of glycosyltransferase activities involved in glycolipid biosynthesis. The increase of GD3 or GM2 in melanoma, and of sialyl-Lewis a antigen (Neu5Acα2-3Galβ1-3[Fucα1-4]GlcNAcβ1-3Galβ1-4GlcβCer) in gastrointestinal cancers, and of GD2 in neuroblastoma are typical examples (Chapter 47). Certain cancers also produce and shed gangliosides that have immunosuppressive effects.
ACKNOWLEDGMENTS
The authors acknowledge contributions to previous versions of this chapter by Akemi Suzuki and appreciate helpful comments and suggestions from Anabel Gonzalez-Gil, Tetsuya Okajima, and Ryan N. Porell.
FURTHER READING
Hakomori S. 1981. Glycosphingolipids in cellular interaction, differentiation, and oncogenesis. Ann Rev Biochem
50: 733–764. doi:10.1146/annurev.bi.50.070181.003505 [
PubMed: 7023369] [
CrossRef]
Todeschini AR, Hakomori S-I. 2008. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim Biophys Acta
1780: 421–433. doi:10.1016/j.bbagen.2007.10.008 [
PMC free article: PMC2312458] [
PubMed: 17991443] [
CrossRef]
Simons K, Gerl MJ. 2010. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol
11: 688–699. doi:10.1038/nrm2977 [
PubMed: 20861879] [
CrossRef]
D'Angelo G, Capasso S, Sticco L, Russo D. 2013. Glycosphingolipids: synthesis and functions. FEBS J
280: 6338–6353. doi:10.1111/febs.12559 [
PubMed: 24165035] [
CrossRef]
Jennemann R, Grone HJ. 2013. Cell-specific in vivo functions of glycosphingolipids: lessons from genetic deletions of enzymes involved in glycosphingolipid synthesis. Prog Lipid Res
52: 231–248. doi:10.1016/j.plipres.2013.02.001 [
PubMed: 23473748] [
CrossRef]
Feingold KR, Elias PM. 2014. Role of lipids in the formation and maintenance of the cutaneous permeability barrier. Biochim Biophys Acta
1841: 280–294. doi:10.1016/j.bbalip.2013.11.007 [
PubMed: 24262790] [
CrossRef]
Platt FM. 2014. Sphingolipid lysosomal storage disorders. Nature
510: 68–75. doi:10.1038/nature13476 [
PubMed: 24899306] [
CrossRef]
Schnaar RL, Gerardy-Schahn R, Hildebrandt H. 2014. Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease and regeneration. Physiol Rev
94: 461–518. doi:10.1152/physrev.00033.2013 [
PMC free article: PMC4044301] [
PubMed: 24692354] [
CrossRef]
Sandhoff R, Schulze H, Sandhoff K. 2018. Ganglioside metabolism in health and disease. Prog Mol Biol Transl Sci
156: 1–62. doi:10.1016/bs.pmbts.2018.01.002 [
PubMed: 29747811] [
CrossRef]
Yu J, Hung J-T, Wang S-H, Cheng J-Y, Yu AL. 2020. Targeting glycosphingolipids for cancer immunotherapy. FEBS Lett
594: 3602–3618. doi:10.1002/1873-3468.13917 [
PubMed: 32860713] [
CrossRef]