The term genomics arose from the availability of complete genome sequence data as well as computational methods for their analysis. However, <2% of genes in the human genome encode proteins. These genes are transcribed into messenger RNAs (mRNAs) that make up the “transcriptome,” of which ∼30% are assigned to protein coding. The total complement of proteins expressed by the cell is collectively termed the “proteome.” Most eukaryotic proteins are post translationally modified (e.g., by phosphorylation, sulfation, oxidation, ubiquitination, acetylation, methylation, lipidation, or glycosylation). These modifications, combined with alternative RNA splicing in eukaryotes, render the proteome considerably more complex than the transcriptome. Although it has been estimated that approximately 120,000 different protein splice forms are expressed by human cells, the total number of modified proteoforms is likely to be at least an order of magnitude higher. The systems-level analysis of all proteins expressed by cells, tissues, or organisms is known as “proteomics.” The proteome, like the transcriptome, but unlike the DNA sequence of the genome, is fundamentally dynamic. The repertoire of proteins expressed by a cell is highly dependent on tissue type, microenvironment, and stage within the life cycle. As cells receive external and internal cues in the form of growth factors, hormones, metabolites, or cell–cell interactions, the expression of various genes is modulated and may be transcribed at levels ranging from silence to more than 104 mRNA copies per cell and 107 protein molecules per cell. Thus, proteomes and their modifications vary during cell differentiation, activation, trafficking, and malignant transformation.
However, no systems-level analysis of a biological process is complete without interrogating the glycome, defined as the entire complement of glycan structures (be it protein-/lipid-bound or -free) produced by cells, in addition to the genome, transcriptome, proteome, lipidome and metabolome.
THE GLYCOME
Vertebrates synthesize N-linked and O-linked glycoproteins, glycolipids (Chapters 11 and 12), proteoglycans, glycosaminoglycans (GAGs), and glycosylphosphatidylinositol (GPI) anchors covalently attached to proteins, as well as free oligosaccharides (Chapter 3). As with the proteome, each cell type has its own distinct glycome that is governed by local cues and the metabolic state of the cell. Other organisms have distinct glycomes; those of plants (Chapter 24) and prokaryotes (Chapter 21) are distinctly different from the vertebrate and invertebrate glycomes (Chapters 25–27).
The size of any particular cellular glycome has not yet been established, but the combinatorial possibilities that can occur with numerous glycan structures on multiple glycoconjugates means that determining a “complete” glycome is not straightforward. The notion that glycans should be studied as a totality (glycomics), as well as simply one glycan or glycoconjugate at a time, developed when it became apparent that glycans form patterns on cells that change during development (Chapter 41), cancer progression (Chapter 47), infection (Chapters 42 and 43), and many other diseases (Chapters 44–46). Many glycan-binding proteins, such as lectins, are oligomerized on the cell surface and interact with multivalent arrays of glycans on the same or opposing cells (Chapters 28–38). Sometimes, multiple discrete glycans and their matching glycan-binding proteins work together to engage two cells or to deliver signals between cells. Thus, the term “glycomics” was coined to describe the many aspects of glycobiology that can be understood only with a systems-level analysis of the glycome.
RELATIONSHIP OF THE GLYCOME TO THE GENOME AND PROTEOME
Clues regarding the composition and complexity of the glycome are found in the genome, transcriptome, and proteome of a cell. Thus, if a gene encoding a glycosyltransferase is not expressed (absent from the transcriptome), no glycans in that cell can carry the sugar transferred by that glycosyltransferase at that particular time. The action of many glycosyltransferases and glycoside hydrolases competing for the same substrates in the biosynthetic pathway renders the complete glycome impossible to predict with current tools and knowledge. As an example, the reduced expression of a single glycosyltransferase can perturb the biosynthesis of dozens of glycans. Furthermore, unlike the genome, the glycome is sensitive to exogenous nutrient levels and metabolic fluxes including salvage pathways. Thus, variations in dietary monosaccharides, such as glucose, galactose, glucosamine, fucose, mannose, and N-glycolylneuraminic acid (Chapter 15), may change the composition of the glycome. The numerous factors that influence the glycome (the transcriptome, the proteome, environmental nutrients, the secretory machinery, pH, and many other determinants) create a glycome that is highly diverse, adaptable, and dynamic. Thus, the glycome of a cell can change dramatically over time. It is this enormous structural plasticity in response to cellular and environmental states that underlies the essential roles of glycans in development, communication, and disease processes.
COMPARATIVE GLYCOMICS
Because the glycome is influenced by both genetic and environmental factors, the information contained therein sheds light on intra- and interspecies variations, including providing indicators of disease that can be used for diagnosis and for monitoring the efficacy of drugs. Comparative glycomics, the comparison of glycome profiles obtained from two or more individuals, tissues, or conditions of interest, is therefore an exciting frontier in biology and medicine.
For example, as discussed in detail in Chapter 47, numerous changes in the glycome have been associated with malignancy and metastasis, including altered N- and O- linked protein glycosylation, up-regulation of conjugated sialylated and fucosylated glycans, and altered GAGs. Regardless of functional consequence, a change in the glycome that is highly correlated with malignancy (or any disease) may serve as a diagnostic marker candidate. Notably, glycans altered in a disease may reflect downstream consequences of the disease on remote organs, changes in the patient's immune system, or other effects of the disease.
One major caveat is the currently unknown extent of natural variation among individual human glycomes. The glycome is known to respond to dietary and environmental changes and vary in elusive ways with age, gender, and acquired disease susceptibility (Chapter 46). Studies of evolutionary biology also have much to gain from comparative glycomics. Evolution of the vertebrate immune system, for example, was accompanied by the acquisition of new classes of glycan-binding proteins, including Siglecs (Chapter 35) and selectins (Chapter 34). Likewise, the glycomes and glycan-binding proteins of microbes and their vertebrate hosts appear to have coevolved in some instances (Chapter 42).
TOOLS FOR CHARACTERIZING THE GLYCOME
The glycome can be determined at different levels of granularity. First, “glycomics” constructs an inventory of glycans separated from their protein or lipid scaffolds from the cell, organ, or organism of interest. This is an important starting point for any comprehensive glycome analysis. The second level of analysis defines specific glycans associated with individual proteins or lipids. Analysis of the complete repertoire of a cell's glycoconjugates, including the microheterogeneity of glycans present at individual sites of attachment, lies, for example, at the intersection of glycomics and proteomics (“glycoproteomics”) and of glycomics and lipidomics (“glycolipidomics”). A third level of complexity involves determining which glycans and/or glycoconjugates are expressed in specific cells, tissues, or secretions at specific times or conditions. This level of analysis is essential if the goal is to reveal new functions in cell–cell communication or to correlate particular glycomes with specific diseases. Of course, none of these approaches recapitulates the actual arrangement of the complex glycan forest present on the surface of cells or in extracellular matrices. The spatial organization of the glycome on cell surfaces is currently only amenable to microscopy-, array- and flow cytometry–based imaging using various glycan-recognizing probes (lectins/GRPs; Chapter 48). The spatial organization of glycans in tissue sections can also be explored by recent matrix-assisted laser desorption/ionization (MALDI) MS imaging methods.
GLYCOMICS AND GLYCOPROTEOMICS
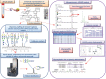
The term “glycomics” thus describes studies designed to define the complete repertoire of glycans that a cell, tissue, or organism produces under specified conditions of time, location, and environment. “Glycoproteomics” describes this glycome as it appears on the cellular proteome. Glycoproteomics determines which sites on each glycoprotein of a cell are glycosylated and ideally includes the identification and quantitation of the heterogeneous glycan structures at each site. The molecular complexity of the glycoproteome makes glycomics and glycoproteomics both exciting and daunting. Because neither the genome, transcriptome, nor the proteome can accurately predict the dynamically expressed protein-linked glycans, the glycome and glycoproteome must be analyzed directly. Techniques used to characterize a single glycoprotein after isolation (Figure ) or a complex mixture of glycoproteins (the glycoproteome) (Figure ) are described in this chapter.
Analysis of a purified single glycoprotein. Example of the characterization of an affinity purified serum glycoprotein, haptoglobin, including (left panel) a total glycomics profile of N-glycans released from the protein, fluorescently derivatized with (more...)
Glycomics-assisted glycoproteomics of a complex mixture of glycoproteins. In this workflow, the structures of the PNGase F released N-glycans from a total extract of rat brain membrane proteins are first analyzed in detail (1) and the sites of glycosylation (more...)
As described below, numerous techniques have been developed for interrogating the glycome and glycoproteome. Because no single technique can at present define all aspects of the glycome or glycoproteome, several approaches are typically used in parallel from assembling an individual cell type glycan repertoire to determining the global tissue expression for example. Different approaches and techniques are required to characterize, for example, the structures of glycoproteins versus glycolipids, N-glycans versus O-glycans, and sulfated GAGs versus neutral glycans (Chapter 50). In contrast, a single technique such as RNA sequencing (RNA-seq) can be used to identify and quantitate all mRNA transcripts at once, a much easier task.
GLYCOMIC ANALYSIS
Glycomic methods and analyses have been mostly directed toward protein glycosylation because of the current proteomics focus, although considerable effort is being directed toward methods that encompass all glycan conjugates. As an example of a typical experiment, a glycoprotein-enriched sample is prepared from a cell lysate and their released glycans analyzed by liquid chromatography (LC) and/or mass spectrometry (MS). In the case of glycoproteins, the N-glycans can be selectively released enzymatically or chemically, separated by high-performance liquid chromatography (HPLC) methods, and are often sequenced online by tandem mass spectrometry (MS/MS) with or without exoglycosidase treatments. Separately, the O-glycans may be released chemically and sequenced in a similar manner. In contrast, glycolipids can often be directly sequenced with or without release from the lipid component. GAGs are more problematic because of their large size and negative charge, but disaccharide fragments can be sequenced by LC or MS in conjunction with enzymatic digestion (Chapter 17).
Depending on the level of detail desired, glycomic analyses may be divided into basic techniques: glycoprofiling, glycan class characterization, and full structural analysis of the glycans released from protein(s). The level of detail should ideally be tailored to the particular question at hand.
Glycoprofiling (fingerprinting, patterning) is the separation of a complex glycan mixture by a technique that provides a signature or fingerprint to give a simple overview or snapshot of the glycans in the sample. Technologies that provide different one-dimensional windows are
HPLC (separation by physiochemical parameters such as hydrophilicity, size or charge) with or without
MS, capillary electrophoresis (separation of labeled glycans by mass/charge), and MALDI and/or electrospray ionization (
ESI) MS (separation of unlabeled or labeled glycans by mass/charge).
Glycan class characterization uses technologies to separate glycan mixtures into types of glycans based on structural features. Examples include
MS separations of di-, mono-, and non-galactosylated IgG glycans or the weak anion-exchange (
WAX)
LC charge-based separations that separate glycans into neutral, mono-, di, tri-, and tetrasialylated structure types. This approach is a convenient way to highlight defined critical features and provide relative quantitation of the different glycan classes. LC-MS analysis can also be used to separate released N-glycan structures into paucimannose, oligomannose, and hybrid and complex sialylated and nonsialylated glycan types.
Detailed (full) structural analysis requires the determination of the sequence and any modifications to the monosaccharides, branch points, anomericity, and glycosidic linkages of the glycans in a glycome. In this detailed analysis, orthogonal technologies are usually required, first to assign preliminary structures and then to confirm the assignments. For example, an anion-exchange separation into differently charged glycan classes can be complemented by hydrophilic interaction liquid chromatography (
HILIC) separation of each class. Digestion by exoglycosidases can then be used to help determine the sequence, anomericity, and linkage of different glycans. On the other hand,
MS assigns compositions that are consistent with the mass data, and structural details can be resolved, for example, by (
LC)
ESI MS/MS or MS
n fragmentation. Separate release and analysis of glycans with sialic acid residues that have labile modifications such as O-acetylation or polysialylation may be needed, if preparation for MS is likely to destroy them. Full structural analysis also can include absolute or relative quantitation of the assigned glycan structures such as, for example, the level of core fucose on antibodies designed to initiate antibody-dependent cellular cytotoxicity (
ADCC), the levels of antigenic α-gal residues, sialyl-Lewis x epitopes that may be useful markers of inflammation and metastasis, or the specific structure binding to a bacterial protein.
Full structural analysis of glycans is challenging as different structures can have the same mass, often coelute on separation systems, and can require detailed manual annotation of MS/MS spectra. Usually glycans are given a preliminary structural assignment from one technology and are then confirmed by at least one orthogonal technology. Bioinformatic tools are being developed to try to mitigate this bottleneck (Chapter 52).
Release of Glycans from Proteins
The starting material in a glycomic analysis can be glycoproteins embedded in gels, protein extracts from whole cell lysates, homogenized tissues, enriched membrane fractions, or serum and other body fluids. For high-throughput analyses of the glycome, intact N-glycans are most often released from glycoproteins using an amidase (peptide N-glycosidase F [PNGase F]). PNGase F cleaves the linkage between the core GlcNAc and the asparagine residue in the NXT/S (X≠P) sequon, of all classes of N-glycans, with the exception of specific N-glycans found in plant and invertebrate glycoproteins that contain fucose α(1,3) linked to the core GlcNAc residue attached to the protein. PNGase A, an enzyme extracted from almond emulsion, may be used to release all core fucosylated N-glycans from protease-generated glycopeptides. Treatment with other endoglycosidase enzymes that cleave between the two GlcNAc residues within the chitobiose core (e.g., endoglycosidase D, which releases paucimannosidic N-linked glycans, endoglycosidase H, which selectively cleaves oligomannose- and hybrid-type structures and various types of endoglycosidase F) is also possible. Before treatment, denaturation both with and without trypsin digestion can be used to relax the three-dimensional (3D) structure of the protein and improve enzyme accessibility. N-glycans can also be conveniently released from glycoproteins purified in SDS-PAGE gel bands. After the N-glycans have been cleaved, the protein remaining in the gel can be identified by traditional proteomics.
For O-linked glycan release from serine (S) and threonine (T) the method of choice is chemical reductive β-elimination. Drawbacks of the method include the fact that the resultant O-linked alditols cannot be further labeled at the reducing end, and that labile modifications can be destroyed during the release process. To date, there is no enzyme that can release all classes of O-linked glycans. The enzyme O-glycanase (in contrast to the PNGase enzymes) is restricted to releasing only simple core 1 (Galβ1-3GalNAcαS/T) O-glycans.
Analysis of Released Glycans
Derivatization of N- and O-Linked Glycans for LC, CE, and MS
Labeling of released glycans can optimize HPLC and capillary electrophoresis (CE) detectability and separability and may improve their MS properties. Many of these labeling approaches use reductive amination of the glycan at the reducing end or react the free amine at the reducing end that is left after PNGase F release. Fluorescent tags increase the sensitivity by lowering the limit of quantitation and detection. Permethylation and peracetylation, when all the mobile protons (present on hydroxyls, carboxyls, and amides) in a glycan are substituted by alkyls (e.g., -methyl) or esterified (e.g., -acetyl) convert the glycans from being hydrophilic to hydrophobic, making the glycans easier to purify and greatly improves the sensitivity and linkage determination by MS-based analyses.
Both N- and O-glycans can also be analyzed as alditols in which the reducing end monosaccharide ring is converted into a reduced linear alditol by reducing agents, such as sodium borohydride. This reduction removes the anomeric ambiguity of the carbohydrate, where the α- and β-isomers of the reducing end sugar may otherwise separate into two peaks chromatographically.
Tagging of the reducing ends of the released glycans (e.g., with 2-aminobenzamide [2-AB], aminobenzoic acid [2-AA], or aminopyridine [2-PA]) is often used for HILIC and reversed-phase LC separation of released glycans (Figure ). Labeled dextran oligomer ladders are commonly used in LC as external standards to help define composition and size based on comparable retention times, for which an “incremental value” can be calculated for each monosaccharide present in the structure (Figure ).
CE with laser-induced fluorescence (LIF) can also provide efficient, rapid, and quantitative separation of derivatized glycans. Glycans are mostly neutral structures so coupling a charged fluorescent label such as 1-aminopyrene-3,6,8-trisulfonic acid (APTS) is necessary to provide electrophoretic mobility and to enable sensitive fluorescence detection. Further details can be assigned after digestion of glycan mixtures by one or more exoglycosidases that specifically cleave glycosidic bonds of individual monosaccharide units from the terminal residue producing predictable shifts in the HPLC or CE (or MS) profiles of the digests.
Special derivatization protocols have also been developed to improve the quality of the MS fragmentation spectra of sialylated glycans—in particular, to target the charged carboxyl group of sialic acid residues. These can be converted into esters or amides to remove the acidic proton of the carboxylic acid that destabilizes the sialic acid in MALDI-MS and promotes undesirable in-source or post-source fragmentation in MS/MS. Specific derivatization of sialic acid residues can also help to determine by MS whether they are 2,3- or 2,6-linked to the glycan structure.
MS Profiling of N- and O-Linked Glycans
An advantage of mass spectrometric glycan profiling is that different glycans can be identified at once by their mass and diagnostic fragmentation ions, increasing the throughput of the glycomic analysis. However, mass spectrometry of glycans may miss potentially important labile modifications, such as sulfation and O-acetylation, depending on the sample preparation and MS techniques applied. MS has inherent challenges because of the isomeric and sometimes isobaric nature of the constituent monosaccharide units that form multiple glycan isomers exhibiting the same molecular mass.
Determining the molecular masses of glycans using MALDI- or ESI-MS gives a picture of the molecular distribution of glycans and allows a quantitative comparison of glycosylation between samples (Chapter 50). The limited number of masses of the monosaccharide units (Table ) makes combinatorial translation of molecular ion masses to monosaccharide composition possible albeit often with some remaining ambiguity. There are available search engines that can provide a suggested list of glycan compositions based on an experimentally determined mass (e.g., GlycoMod; Chapter 52). MS alone, however, cannot distinguish between isomeric monosaccharides, so a nomenclature for generic monosaccharide compositions has been adopted. For instance, all of the isomeric 6-carbon-containing monosaccharides, such as glucose, mannose, and galactose are given the unifying name of hexose (Hex) (Table ).
Families of common monosaccharides found in mammalian N- and O-linked glycans
Elucidation of variable linkage configurations, and glycan branch points is another analytical challenge.
Generic glycomic workflows using all the derivatization approaches described above have been developed for MS analysis. Both the neutral and sialylated glycans in a sample can be analyzed after permethylation and MALDI-MS, in which glycan masses are detected as their singly charged alkali ion adducts in positive ion mode (e.g., as [M+Na]+ ions or [M+K]+ ions). Negative ion ESI-MS is widely adopted for intact glycan profiling in their alditol form, in which both neutral and sialylated glycans are detected as deprotonated [M-nH]n− ions. The number of charges (n) will increase with the size of the glycan and is also dependent on the number of acidic moieties present (e.g., sialic acids, sulfates, and phosphates). Positive ion MALDI-MS without permethylation usually requires that sialic acid residues are derivatized as described above to prevent their loss in-source.
To obtain orthogonal separations prior to the mass analysis, ESI-MS is often connected to HPLC. HILIC columns can be used for separation of reducing end derivatized glycans and provides separation based on hydrophilicity (which correlates with the glycan size), with some isomeric structure resolution (Figures and 51.2). The retention of glycans on such columns is primarily based on hydrogen bonding to water surrounding the HILIC stationary phase (partitioning). Using an alternate stationary phase, porous graphitized carbon (PGC) has shown a unique ability to clearly separate isomers of released glycan alditols based on more complex retention mechanisms. If glycans are permethylated, the increased hydrophobicity allows separation using conventional C18 reversed-phase chromatography. Ion mobility has also been utilized to resolve glycan isomers entering the mass spectrometer. Data from two or more approaches can be used to give confidence in structural assignments and can be facilitated by software that allows automated data analysis (Chapter 52).
MS Fragmentation of N- and O-Linked Glycans
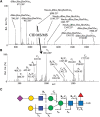
MS fragmentation has become the gold standard for glycan structural characterization. The goal is to generate information-rich fragment spectra that will allow unequivocal assignment of a glycan structure. However, in the current state of the art, fragments from colliding glycan molecular ions (collision-induced dissociation [CID]—either “beam-type” or “ion trap-type”) can only partly determine the structure of interest. We can distinguish three types of glycan fragment ions depending on the type and amount of information they carry (Figure ).
Collision-induced dissociation–tandem mass spectrometry (CID-MS/MS) of released N-glycans. (A) MS of released N-glycans: Masses are shown as doubly charged ions with corresponding monosaccharide compositions. (B) CID-MS/MS of m/z of [1098.25] (more...)
B- and C-type fragment ions comprise, by definition, the nonreducing end fragments that arise from a single glycosidic bond cleavage without or with the glycosidic oxygen, respectively. Reducing end fragments are assigned as Y- (with glycosidic oxygen) and Z-type (without glycosidic oxygen) fragment ions.
Cross-ring fragments can be assigned as nonreducing end fragments (A-type) and reducing end fragments (X-type) and require further annotation to specify which bonds in the carbon ring were dissociated to form the cross-ring fragment ion.
Internal fragments that occur from more than one fragmentation event from a combination of glycosidic and/or cross-ring fragmentation.
Ideally, a fragmentation spectrum containing all possible glycosidic fragments would allow the assignment of the primary sequence and branching of a glycan structure. In practice, however, most CID approaches provide glycosidic fragments, but several methods or a combination of methods (LC retention times, exoglycosidase digestions, ion mode, derivatization, multiple fragmentation steps, i.e., MSn) are usually used to fully define a particular structure of interest. Biosynthetic rules can be and often are applied to aid the glycan characterization.
Glycan Modifications
A further challenge is that many key glycan modifications such as O-acetylation, pyruvylation, etc., are labile to, and/or missed with, current analytical methods. This problem can result in populating databases with misleading or biased information. As just one example, although many databases assume that a sialic acid at the terminus of the vertebrate glycan chain is N-acetylneuraminic acid, there are in fact dozens of kinds of modified sialic acids in nature, and the differences can have profound effects on biological functions (Chapter 15). The same is true of N- and O-sulfate esters on HexNAc and hexosamine residues (Chapters 14 and 17). These analytical challenges are still being pursued. Meanwhile, in many instances, it would make sense to not make definitive positional assignments of modifications on the glycan chains.
THE FUTURE OF GLYCOMIC ANALYSES
Releasing the glycans from the protein(s) is currently a prerequisite for glycomics. The described techniques above are, however, complementary to support the assignment of glycan structures still attached to peptides, the techniques for which are less well-developed and present greater challenges (see below). N-linked and O-linked glycomics are still the only techniques that can detail the overall glycan structural landscape of the surface of a cell (if applied in combination with plasma membrane enrichment) so they will continue to be valid approaches in the foreseeable future to define cell–cell interactions and to discover disease biomarkers and new therapeutic targets.
However, there may not be a need for de novo analysis of every single glycan in the future. Matching of fragment spectra using fragmentation libraries containing many of the more ubiquitously expressed glycans (Chapter 52) is becoming a pathway to quickly assign a majority of structures fully or partially and allows researchers to focus on validation of only the structures that are important for addressing the biological question. Similar to proteomics, automated glycan identification should ideally be performed with stringent confidence thresholds by, for example, determining the false discovery rate of the reported identifications. Novel fragmentation techniques, the introduction of ion mobility MS, and in vivo or in vitro incorporation of heavy isotopic monosaccharides or tags are starting to provide other opportunities for the isomeric glycan structural characterization, quantitation, and visualization that are currently lacking in glycomic mass spectrometric techniques.
FROM GLYCOMIC TO GLYCOPROTEOMIC ANALYSIS
A key question that often follows the identification of important glycosylation features by glycomics is, which proteins carry the implicated glycan structures and at which sites? Given that protein glycosylation is dependent on the concerted action of multiple glycosyltransferases and glycoside hydrolases, one might expect all glycoproteins that traffic through the secretory pathway to be equally susceptible to similar modifications. However, protein- and site-specific glycosylation are well-recognized features of protein glycosylation. Although the underlying molecular basis for protein- and site-specific glycosylation is not entirely understood, contributing factors are likely to include common sequence motifs, protein structural conformation, unique physicochemical patches surrounding the glycosylation site, or some yet-unknown traits that collectively allow individual proteins to be sought out from among hundreds or thousands of other proteins to be acted on by the glycosylation machinery in a specific way.
MAPPING OF GLYCOSYLATION SITES
The mapping of N-glycosylation sites of formerly glycosylated (de-N-glycosylated) peptides of a complex protein sample is now routinely performed using conventional proteomic approaches. Notably, this approach does not give information on what glycan structure(s) are present on these defined glycosylation sites and as such does not fit well under glycoproteomics. By virtue of the action of PNGase F or Endo F/Endo H, the de-N-glycosylated peptides are mass-tagged by conversion of Asn to Asp (a +1 Da change in peptide mass) within the consensus sequon (Figure ) or by retention of a GlcNAc(±Fuc) at the Asn site, respectively. These previously glycosylated proteins can then be subjected to standard trypsin digestion and LC-MS/MS analysis, and the MS/MS data searched against proteomic databases for rapid identification of the N-glycosylation sites on the tryptic peptides. This approach can be coupled to initial affinity or chemical capture of the glycoproteome subset, by use of lectins at the glycoprotein and/or digested glycopeptide level, or by hydrazine capture of the oxidized glycans on the glycopeptide followed by PNGase F release of the captured peptide containing the deamidation of Asn to Asp. In this manner, thousands of N-glycosylation sites can now be routinely identified and their relative occupancy quantified in a single experiment. The use of sequential Endo H followed by PNGase F, given that Endo H only acts on oligomannosidic glycans and PNGase F cannot remove the remaining single GlcNAc attached to an Asn, can provide additional information about N-glycan classes at an individual site. Apart from not being able to inform the site-specific glycoform structures, the most commonly criticized aspect of this approach is the potential of false positives being introduced by spontaneous deamidation of Asn to Asp, which is independent of the action of the PNGase F. Enzymatic deglycosylation in heavy water and/or the use of Endo F/H are ways to reduce the number of false positive identifications arising from this spontaneous deamidation.
Innovative approaches using the zinc finger nuclease gene targeting technique, or by using CRISPR/Cas9 knockout of the C1GalT1 glycosyltransferase gene, which impairs O-glycosylation pathways by preventing extension of the O-GalNAc core, has led to the generation of so-called SimpleCells, in which all synthesized mucin-type O-glycoproteins carry only a single GalNAc or sialyl GalNAc (Chapter 56). The same approach applied to O-mannose initiated glycans resolved a long-standing question concerning the apparent abundance of O-mannose glycans in brain tissue although at the time only a few relatively low abundance proteins had been determined to possess this modification. The SimpleCell technology has greatly facilitated the experimental identification of hundreds and even thousands of O-glycosylation sites, although the actual sites and attached O-glycan structures under more natural and specific physiological states remain unknown.
GLYCOPROTEOMICS: DETERMINING THE HETEROGENEOUS SITE GLYCOSYLATION
Not every sequon (NXT/S) will carry an N-glycan, and in rare cases the T/S position of utilized sequons can be substituted with C. The actual glycosylated sites usually carry a heterogeneous collection of glycan structures. Glycoproteomics needs ideally to be able to identify all glycoproteins in a sample down to the level of which sites are occupied (macroheterogeneity) and to quantify and characterize their respective glycoforms at that site (microheterogeneity). The ultimate aim is to take snapshots of the distribution of disparate glycans in real-time on each glycoprotein in a cell to infer how site-specific glycosylation may promote or interfere with cellular interactions or signaling events. Ultimately, to understand the specific biological roles of protein glycoforms, we will need to define the population of each molecular species that arises from combinations of site-specific glycan diversity at multiple sites.
Although promising glycoproteomics methods have been developed in recent years, the unambiguous identification of intact glycopeptides in complex mixtures using LC-MS/MS remains challenging. The workflows are comparably less mature than the corresponding proteomics methods for the analysis of unmodified peptides or peptides carrying simple modifications. Appropriate false discovery rate (FDR)-based glycopeptide identification strategies are emerging but are still not sufficiently integrated into the workflows such that confident identifications are still dependent on manual interrogation of data by experienced users. Most, if not all, glycoproteomics methods rely on accurate mass analysis of the monoisotopic glycopeptide precursor ions (low ppm) to accurately specify the molecular mass of the intact glycopeptides. The fragment ions also benefit from detection at high resolution to aid the spectral assignment.
For N-glycopeptides, CID, either on an ion trap or quadrupole time-of-flight (QTOF) platform, or increasingly by higher-energy collision dissociation (HCD), available on a range of Orbitrap mass analyzers, induces mostly glycosidic cleavages, giving rise to abundant glycan oxonium ions in the low mass region, complemented by successive neutral losses of glycosyl residues from the glycopeptide precursors down to a single GlcNAc on the Asn (Figure ). The abundant diagnostic oxonium ions enable quick and accurate broad classification of all glycopeptide-containing spectra, with details of the monosaccharide composition and topology, and usually is sufficient to allow for confident glycopeptide identification. In favorable cases, the Y1 ion (peptide backbone+GlcNAc) can be assigned and its m/z can be used to define the molecular mass of the peptide carrying the glycan Thereafter, the precursor ion mass gives the composition of the attached glycan after the peptide mass has been subtracted. More recently the use of step-HCD (generating multiple fragmentation spectra of the same species with different collision energies) has been shown to generate rich spectra for assigning both the glycan and the peptide.
Complementary tandem mass spectrometry (MS/MS) fragmentation of N-glycopeptides. (A) Both collision-induced dissociation (CID)-MS/MS and higher-energy collision dissociation (HCD)-MS/MS of glycopeptides produce cleavages of the glycan to give diagnostic (more...)
When submitting the more labile O-glycopeptides to CID/HCD fragmentation the glycosyl residues are usually detached from the peptide carrier and therefore produce b- and y-ions without the conjugated glycan. The limitation is also that the modification site(s) cannot be specified.
Electron transfer dissociation (ETD) performed with or without additional CID- or HCD-based activation, termed ETciD and EThcD, respectively, represents an alternative fragmentation strategy to achieve more information-rich glycopeptide spectra. In principle, ETD yields c- and z-type ions arising from peptide bond cleavages along the peptide backbone without inducing glycosidic cleavages (Figure ). The resulting c- and z-ions that still carry the intact glycan are therefore useful to identify both the peptide carrier and the modification site, a feature particularly useful for O-glycopeptides in which site allocation cannot reliably be predicted based on the peptide sequence. The practical problem is that the ETD efficiency is dependent on the charge density of the glycopeptide. Relative low dissociation efficiency is typically observed with doubly and triply charged N-glycopeptides, which constitutes a substantial portion of all tryptic peptides. One way to address this issue is to increase the charge state of the glycopeptides by either chemical derivatization, such as using a tandem mass tag (TMT), by using supercharging agents in the LC solvents and/or by using a different proteolytic enzyme such as LysC or GluC instead of trypsin to generate larger glycopeptides. Using EThcD (supplemental activation) and/or longer ETD activation times may also produce more information rich spectra by promoting dissociation of already charge-reduced nondissociated precursors. ETD works reasonably well with O-glycopeptides, particularly those decorated with only one or two glycosyl residues including O-GlcNAc, O-GalNAc (Tn), O-GalNAc-Gal (T), O-Fuc, and O-Man. By retaining these glycosyl substituents, ETD ideally allows identification of their distribution over several closely placed Ser/Thr residues which is not possible with CID or HCD.
Innovative LC-MS/MS acquisition strategies are increasingly being used to enhance the performance of glycoproteomics experiments. For example, abundant HexNAc oxonium ions at m/z 204 arising from HCD-MS/MS fragmentation are useful diagnostic ions for any type of N- and O-linked glycopeptides. Product ion-dependent ETD, or EThcD triggering of precursors giving rise to such HexNAc (or other) oxonium ions, are elegant strategies to tailor the instrument to acquire more glycopeptide specific informative data enabling a higher glycoproteome coverage without the need of prior glycopeptide enrichment.
Similar to the quantitation of peptides in proteomics, the quantitation of intact glycopeptides can be performed using either label-free or label-assisted (e.g., TMT) strategies. The type of quantitation depends on the question at hand but may involve relative quantitation of all glycans at each glycosylation site (glycoproteome mapping), comparison of the glycoprofiles of individual sites between conditions (comparative glycoproteomics), or, more simply, determination of the relative abundance of a single glycopeptide form between two or more conditions (biomarker discovery). Multiple quantitative approaches as well as information of the protein level and degree of site occupancy are often required to extract biologically relevant information from glycoproteomics data.
LIMITATIONS AND PROSPECTS OF GLYCOPROTEOMICS
A limitation of current glycoproteomics data is that the glycopeptide fragment data do not afford detailed glycan topology, linkage, and stereochemistry information of the attached glycans. Diagnostic fragment ions can sometimes be found confirming terminal epitopes such as a fucosylated Hex-HexNAc or sialylated fucosylated Hex-HexNAc (Lewis x and sialyl-Lewis x, respectively) but these cannot be distinguished from Lewis a and sialyl-Lewis a. Detailed glycan profiling achieved with site resolution (structure-focused glycoproteomics) is an important frontier that should be a goal of the next-generation of glycoproteomics. At this time, continued development of selective enrichment and/or preseparation of specific subsets of glycopeptides needs to be pursued, because no single LC-MSn method or instrument is sufficient to handle the full dynamic range of the entire glycopeptide pool derived from a complete glycoproteome.
To limit the search space and enhance the detailed structural knowledge of the glycans encountered in a given glycoproteome, parallel profiling of the glycome can be performed. Such a “glycomics-assisted glycoproteomics” approach (Figure ) can also be complemented with quantitative proteome and de-glycoproteome profiling (mapping of glycosylation sites), which reduce the search space even further and provide supporting evidence to pinpoint the mechanism(s) driving the observed glycoproteome alterations.
Although glycoproteomics is rapidly maturing and continues to reach a larger group of scientists, significant analytical challenges remain and the technology is still best performed in “specialist” laboratories because of the demand for manual expert interrogation and interpretation of data. Current glycoproteomics methods cannot identify the full complement of glycans on every site of a mixture of glycoproteins and cannot easily tackle multiply glycosylated peptides especially with both N- and O-linked glycan structures. Defining the relative proportions of different intact glycoform populations is important for understanding biological function. This has only been achieved in a few cases (e.g., by MS analysis of the intact IgG glycoprotein).
Several promising informatics initiatives aimed at automating the glycopeptide identification process, aiding the quantitation, and supporting the interpretation of the data output have appeared over recent years. A recent interlaboratory study conducted by the Human Glycoproteomics Initiative of HUPO highlights the importance of different software search parameters for the successful identification and characterization of glycopeptides in order to try to deal with false positives and the reporting of thousands of nonverified glycopeptides. At this stage, for most of the available glycopeptide identification software tools it is strongly advised that manual validation of the glycopeptide assignments still be performed to generate sufficient confidence in the reported identifications. It is therefore clear that the computational solutions require further improvements to advance the field of glycoproteomics and ultimately achieve full integration among other disciplines within the glycosciences (Chapter 52).
ACKNOWLEDGMENTS
The authors acknowledge contributions to previous versions of this chapter by Carolyn R. Bertozzi and Ram Sasisekharan and appreciate helpful comments and suggestions from Daron Freedberg and Chad Slawson.
FURTHER READING
Berger EG, Buddecke E, Kamerling JP, Kobata A, Paulson JC, Vliegenthart JF. 1982. Structure, biosynthesis and functions of glycoprotein glycans. Experientia
38:
1129–1162. doi:10.1007/bf01959725 [
PubMed: 6754417] [
CrossRef]
Domann PJ, Pardos AC, Fernandes DL, Spencer DI, Radcliffe CM, Royle L, Dwek RA, Rudd PM. 2007. Separation-based glycoprofiling approaches using fluorescent labels. Proteomics
1:
70–76. doi:10.1002/pmic.200700640 [
PubMed: 17893855] [
CrossRef]
Tissot B, North SJ, Ceroni A, Pang PC, Panico M, Rosati F, Capone A, Haslam SM, Dell A, Morris H. 2009. Glycoproteomics: past, present and future. FEBS Letters
583:
1728–1735. doi:10.1016/j.febslet.2009.03.049 [
PMC free article: PMC2753369] [
PubMed: 19328791] [
CrossRef]
Jensen PH, Karlsson NG, Kolarich D, Packer NH. 2012. Structural analysis of N- and O-glycans released from glycoproteins. Nat Protocols
7:
1299–1310. doi:10.1038/nprot.2012.063 [
PubMed: 22678433] [
CrossRef]
Kolarich D, Jensen PH, Altmann F, Packer NH. 2012. Determination of site-specific glycan heterogeneity on glycoproteins. Nat Protocols
7:
1285–1298. doi:10.1038/nprot.2012.062 [
PubMed: 22678432] [
CrossRef]
Harvey DJ. 2015. Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: an update for 2011–2012. Mass Spectrom Rev
34:
268–422. doi:10.1002/mas.21411 [
PMC free article: PMC7168572] [
PubMed: 24863367] [
CrossRef]
Thaysen-Andersen M, Kolarich D, Packer NH. 2021. Glycomics & glycoproteomics: from analytics to function. Mol Omics
17:
8–10. doi:10.1039/d0mo90019b [
PubMed: 33295916] [
CrossRef]