

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Fitridge R, Thompson M, editors. Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists [Internet]. Adelaide (AU): University of Adelaide Press; 2011.

Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists [Internet].
Show detailsIntroduction
Ischaemia-Reperfusion injury (IRI) is defined as the paradoxical exacerbation of cellular dysfunction and death, following restoration of blood flow to previously ischaemic tissues. Reestablishment of blood flow is essential to salvage ischaemic tissues. However reperfusion itself paradoxically causes further damage, threatening function and viability of the organ. IRI occurs in a wide range of organs including the heart, lung, kidney, gut, skeletal muscle and brain and may involve not only the ischaemic organ itself but may also induce systemic damage to distant organs, potentially leading to multi-system organ failure. Reperfusion injury is a multi-factorial process resulting in extensive tissue destruction. The aim of this review is to summarise these molecular and cellular mechanisms and thus provide an insight into possible windows for effective therapeutic intervention.
Ischaemia
ATP and mitochondrial function
Ischaemia occurs when the blood supply is less than the demand required for normal function, resulting in deficiencies in oxygen, glucose and other substances required for metabolism. Derangements in metabolic function begin during this ischaemic phase. Initially, glycogen breakdown by mitochondrial anaerobic glycolysis produces two molecules of adenosine triphosphate (ATP) along with lactic acid, resulting in a decrease in tissue pH, which then acts by negative feedback to inhibit further ATP production. (Figure 18.1) ATP is then sequentially broken down into adenosine diphosphate (ADP), adenosine monophosphate (AMP) and inosine mono-phosphate (IMP) and then further into adenosine, inosine, hypoxanthine and xanthine. (Figure 18.2 upper panel)
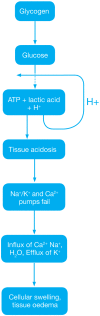
FIGURE 18.1
Dysregulation of metabolic pathways during ischaemia.
FIGURE 18.2
Generation of reactive oxygen species during reperfusion.
At the cellular level, a lack of ATP production causes ATP-dependent ionic pumps, including the Na+/K+ and Ca2+ pumps, to fail and the transmembrane ionic gradients are lost. Consequently, cytosolic sodium content rises, drawing with it, a volume of water to attempt to maintain the osmotic equilibrium and resulting in hydroponic swelling of the cells. To maintain the ionic balance, potassium ions escape from the cell into the interstitium (reviewed in1). Calcium is released from the mitochondria into the cytoplasm and into extracellular spaces, thereby activating mitochondrial calcium-dependent cytosolic proteases including calpain, which then converts the cellular enzyme xanthine dehydrogenase to xanthine oxidase (Figure 18.2 upper panel). Phospholipases are also activated during ischaemia, degrading membrane lipids and increasing the levels of circulating fatty acids.
Gene expression during ischaemia
As well as metabolic derangements, ischaemia induces expression of a large number of genes, which play a major role in the tissue’s response to ischaemic damage. An RNA expression microarray analysis, using mouse soleus muscle rendered ischaemic by femoral ligation, found that expression of 962 genes was induced and 327 genes were repressed.2 the activated genes were largely clustered into cytokine genes and mediators of inflammation and immune cell infiltration. The repressed genes were largely involved in energy production, including mitochondrial respiration and fatty acid oxidation.
Hypoxia itself also activates a number of genes, particularly transcription factors, including activating protein-1 (AP-1), hypoxia-inducible factor-1 (HIF-1) and nuclear factor-kappab (NF-kb). HIF-1 then activates transcription of other genes such as vascular endothelial growth factor (VEGF), erythropoietin and glucose transporter-1, which all play an important role in the cells’ adaptive responses to hypoxia (reviewed in3). Expression of both HIF-1 and cyclooxygenase-2 (COX-2) are also induced in the lungs of rats subjected to haemorrhagic shock. COX-2 may promote the inflammatory response through the rapid and exaggerated production of nitric oxide and prostaglandins, contributing to organ damage.4 Activation of NF-kb occurs during both the ischaemic and reperfusion phases and will therefore be discussed below.
Reperfusion
Reactive oxygen species
Table 18.1 illustrates the major reactive oxygen species (ROS), which play a role in tissue damage during IRI and the sources of generation of these species. Reactive oxygen species have a destructive role in mediating tissue damage during IRI. During ischaemia, the degradation of ATP produces hypoxanthine (Figure 18.2, upper panel). Once the ischaemic tissue is reperfused, an influx of molecular oxygen catalyses xanthine oxidase to degrade hypoxanthine to uric acid and thereby liberating the highly reactive superoxide anion (O2-) (Figure 18.2, lower panel). Superoxide is subsequently converted to hydrogen peroxide (H2O2) and the hydroxyl radical (OH•) (Figure 18.2, lower panel). The major consequence of hydroxyl radical production is peroxidation of the lipid structures of cell membranes resulting in the production and systemic release of proinflammatory eicosanoids, disruption of cell permeability and ultimately cell death. During IRI, ROS also activate endothelial cells, elevating the activity of the transcription factor, NF-κB. Once activated, the endothelial cell produces E-selectin, vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), endothelial-leukocyte adhesion molecule (EMLMl Am -1) plasminogen activator inhibitor-1 (PAi-1), tissue factor and interleukin-8 (il-8). These adhesion molecules contribute to important interactions between the neutrophil and the endothelium and will be discussed in more detail later.
TABLE 18.1
Reactive Oxygen species involved in IRI.
Superoxide anions can be detected within ischaemic muscle and also in the venous effluent of reperfused limbs,5 suggesting an additional role for superoxide in inducing damage to distant organs during skeletal muscle reperfusion injury. Xanthine oxidase is located within a spectrum of cell types and tissues to varying degrees, indicating widespread distribution and differing susceptibility to oxidant-mediated IRI. Inhibition of xanthine oxidase activity, by administration of allopurinol prior to ischaemia, reduces the production of superoxide and hence reduces the severity of reperfusion injury in animal models using a range of tissues including skeletal muscle, brain and gut. Results in humans are also promising. A systematic review6 provided evidence that allopurinol was effective in some studies in reducing the severity of post-operative cardiac dysfunction and arrhythmias after coronary artery bypass grafting, although larger trials are needed. Studies in other clinical settings of IRI remain limited.
Eicosanoids
As discussed above, ROS initiate lipid peroxidation of cellular membranes, releasing arachidonic acid, the main substrate for the production of prostaglandins, thromboxanes and leukotrienes (Figure 18.2, lower panel). These derivatives of arachidonic acid are collectively known as the eicosanoids and play a major role in the pathophysiology of IRI.
Prostaglandins, synthesised from arachidonic acid via the cyclo-oxygenase pathway, have a protective vasodilatory effect in IRI. However, since prostaglandins are short-lived molecules, their rapid depletion subsequently leads to uninhibited vasoconstriction, reduced local blood flow and exacerbation of ischaemia. The potential of prostaglandins to ameliorate the degree of metabolic and tissue derangement following IRI has been demonstrated in various tissues. In a placebo-controlled trial of human liver transplantation, administration of prostacyclin was shown to improve postoperative graft function.7 Patients who received prostacyclin demonstrated better post-operative myocardial oxygen consumption after coronary artery bypass surgery8 and improved muscle blood flow following skeletal muscle IRI.9
Plasma thromboxane A2, also synthesised from arachidonic acid, increases within minutes following skeletal muscle IRI, thus promoting vasoconstriction and platelet aggregation. These events coincide with a rapid rise in pulmonary artery pressure and a subsequent increase in pulmonary microvascular permeability,10 which correlates with sequestration of polymorphonuclear cells in the lungs. In animal models of lower limb IRI, thromboxane synthase inhibitors and synthetic thromboxane A2 receptor antagonists prevented pulmonary leukosequestration, thereby increasing blood flow to reperfused tissues and preserving tissue viability and function.11 Together these studies suggest that administration of thromboxane A2 antagonists may offer therapeutic potential to improve limb salvage rates after surgery for acute ischaemia.
Leukotrienes are also synthesised from arachidonic acid through the activation of 5-lipoxygenase and participate in the inflammatory cascade of IRI. Leukotrienes lead to local and systemic injury by their direct proinflammatory action on endothelial and smooth muscle cells and indirectly by their effects on neutrophils. The leukotrienes C4, D4, and E4 modify the endothelial cytoskeleton, leading to increased vascular permeability and also enhance smooth muscle contraction, resulting in vasoconstriction. The lung produces leukotrienes following remote IRI. The direct effects of leukotrienes on pulmonary micro vessels lead to increased permeability, transient pulmonary hypertension and the activation of the endothelium to produce thromboxane, resulting in additional vaso-constriction. The leukotriene B4, released by activated neutrophils, leads to further pulmonary neutrophil accumulation.
The administration of 5-lipoxygenase synthesis inhibitors has been successfully used in animal studies to attenuate IRI. Such agents abolish the elevations in leukotrienes B4 and C4 and inhibit neutrophil infiltration normally induced by IRI, reducing mucosal permeability.12 However, there is currently very little up to date information on their use in a clinical situation.
Nitric oxide
Nitric oxide (NO) is a signalling molecule synthesised from L-arginine by the nitric oxide synthase enzyme (NOS) of which there are three types, constitutive (CNOS), inducible (INO S) and endothelial (ENO S). An initial surge in NO level in the first 15 minutes of the ischaemic phase is due to transient ENOS activation. This is followed during early reperfusion by a general decline in endothelial function and loss of functional ENOS, so that NO production falls, along with an increased production of reactive oxygen species. ENOS-derived NO is also necessary for the maintenance of vascular tone. The reduction in ENOS levels that occurs in IRI may therefore predispose to vasoconstriction, a common response seen in IRI. The second surge in NO production is largely due to cytokin-emediated up-regulation of INOS after about three hours of reperfusion.
The pathophysiological role of nitric oxide in reperfusion injury is variable, being dependent on the nature of its generation and appears to be tissue specific. In some instances, NO acts as an anti-oxidant and, in others, combines with the superoxide anion to form the peroxynitrite radical, a potent promoter of lipid peroxidation and hence cellular membrane disruption (Reviewed in13). Manipulation of nitric oxide production during IRI, using a range of techniques, has recently provided considerable evidence for a principal role for nitric oxide in the aetiology of IRI. Myocardial IRI has been well studied, with paradoxical results, where low doses of no were found to be protective and high doses harmful. The influence of NO in skeletal muscle IRI has been less well characterized, with some studies suggesting that NO may potentiate cytotoxicity and others suggesting a beneficial role for NO in extremity IRI. In skeletal muscle IRI, NO production may be deleterious and inhibition of NOS activity using a non-specific NOS inhibitor greatly reduced the severity of muscle damage.14
The assessment of experimental data derived from pharmacological NOS inhibition is difficult due to the non-specificity of NOS inhibitors; administration of these inhibitors at differing times during the injury merely adds to the complexity. In essence, augmentation of NO delivery may be beneficial with respect to protection, particularly in the ischaemic and early reperfusion phase. Inhibition of the INOS-induced surge in NO production at later times during reperfusion also mediates defense against IRI-induced tissue damage. However, in the clinical setting, systemic distortion of NO kinetics by administering NOS inhibitors would be likely to induce wide-ranging physiological disturbances. Further investigations will be needed to define a role for NOS inhibition in ameliorating the severity of IRI and local administration of these inhibitors may be required.
Endothelin
Endothelins are potent peptide vasoconstrictors produced by the vascular endothelium. Hypoxia, growth factors, angiotensin II and noradrenaline all stimulate their production resulting in Ca2+-mediated vasoconstriction. Endothelin-1 is elevated following skeletal muscle IRI during both the ischaemic and reperfusion phases and mediates capillary vasoconstriction, neutrophil aggregation and neutrophil-endothelial interactions. Endothelin-1 inhibitors, including bosentan and tezosentan, inhibit neutrophil infiltration, increase functional capillary density, microvascular perfusion and hence tissue viability and function Following IRI.15 However these inhibitors are not in widespread clinical use.
Cytokines
Hypoxia and IRI both induce the expression of numerous cytokines, including tumour necrosis factor-alpha (TNF- α,) interleukin-1 (IL-1), interleukin-6 (IL-6), interleukin-8 (IL-8) and platelet activating factor (PAF), in association with elevations in activity of the transcription factor, NF-kB (reviewed in 16) these cytokines are released systemically and are thus important in the development of systemic inflammatory response syndrome and ultimately multi-system organ failure.
TNF-α is a 17-kilodalton pro-inflammatory cytokine produced by activated macrophages, monocytes, T-lymphocytes, natural killer cells and fibroblasts. It is a potent chemoattractant and early response cytokine, which subsequently induces expression of IL-1, IL-6, IL-8 and PAF. Elevated serum levels of TNF-α have been detected during cerebral and skeletal muscle IRI and are known to increase neutrophil sequestration and permeability following pulmonary IRI. Serum TNF-α levels increased rapidly in an animal model of aortic clamping, thus inducing up-regulation of INOS, which increased NO production in the lungs, leading to more severe lung damage.17 in the same study, inhibition of TNF-α activity prior to limb ischaemia decreased pulmonary No production and reduced the severity of IRI. TNF-α can also induce the generation of ROS and enhance the susceptibility of the vascular endothelium to neutrophil mediated injury, by inducing the expression of ICAM-1, which mediates binding of neutrophils to the activated endothelium.
Numerous studies in animal models attest to the potential of TNF-α blockade as a therapeutic modality to reduce the severity of IRI. Anti-TNF-α antibody protected against IRI-induced pulmonary injury in a rat model by preventing microvascular damage. The introduction of humanised antibodies including etanercept and infliximab, has provided encouraging results in the treatment of other TNF-α-mediated inflammatory diseases, including a number of forms of arthritis and inflammatory bowel disease (reviewed in18). However, clinical trials to test the efficacy of TNF-α blockade in human IRI have not yet been reported.
The cytokines IL-1α and IL1 β are produced during IRI by tissue macrophages, neutrophils and the vascular endothelium. IL-1α is a potent chemotactic agent and stimulates neutrophil infiltration during hepatic IRI. Both IL-1α and TNF-α also increase levels of expression of ICAM-1 on the vascular endothelium. Exposure of endothelial cells in culture to IL-1α and TNF-α induces synthesis of E-selectin, which then interacts with L-selectin on the neutrophil surface leading to rolling on the endothelial surface. Permanent adhesion of the neutrophil to the endothelium is then mediated by expression of ICAM-1, IL-8 and PAF in the endothelial membranes (Figure 18.3).

FIGURE 18.3
Neutrophil rolling, adhesion to endothelium and extravasation.
Numerous activating stimuli synthesised during IRI include H2O2), thrombin, leukotrienes C4, and D4. IL-lb, histamine, bradykinin and ATP; all of which induce the synthesis of PAF by monocytes, macrophages, neutrophils, eosinophils, basophils, platelets and endothelial cells. PAF functions as both an inter- and intra-cellular messenger, having three major effects, vasoconstriction, chemo-attraction and increased microvascular permeability. PAF is rapidly produced following skeletal muscle and renal IRI with peak levels after 15 minutes of reperfusion. PAF enhances the binding of neutrophils to endothelial cells since a PAF-receptor antagonist blocked adhesion to endothelial cells during IRI.19 Similarly pre-treatment with the PAF inhibitor lexipafant reduced the severity of intestinal barrier dysfunction and pulmonary and liver permeability in a rat model of intestinal IRI.20 However lexipafant is unlikely to be clinically useful as a pharmacotherapy for IRI since, alone, it failed to completely inhibit pulmonary endothelial damage after small bowel IRI.21
IL-6 is a proinflammatory 19-26kd a protein produced by monocytes, fibroblasts, keratinocytes and endothelial cells in response to IL-1 and TNF-α. IL-6 primes and stimulates the respiratory burst in neutrophils, stimulates endothelial cell expression of ICAM-1 and increases endothelial permeability. IL-6 is produced in hypoperfused skeletal muscle in patients with peripheral arterial disease and is released from the gut into the systemic circulation during reperfusion in aortic aneurysm surgery.22 in the setting of renal transplantation, IL-6 was released in large amounts from the reperfused transplanted kidney during the first 30 minutes of reperfusion.23
IL-8 is a potent neutrophil chemotactic and activating factor. It is produced by monocytes, T cells, NK cells, fibroblasts, endothelial cells, eosinophils and neutrophils in response to IL-1, TNF- α, endotoxin, histamine and hypoxia. The chemotactic activity of IL-8 induces diapedesis of activated neutrophils through the endothelium. (Figure 18.3) Elevated levels of serum IL-8 have been detected during early reperfusion following human lung transplantation and predict poor graft function.24 An anti IL-8 antibody prevented pulmonary neutrophil infiltration and tissue injury in a rabbit model of lung IRI.25
Neutrophil and endothelial interactions
Neutrophils play a major role in tissue damage incurred during IRI. Activated neutrophils are a major source of ROS, which are generated through the activity of the membrane-bound nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. Whilst oxidizing NADPH to NADP+,NADPH oxidase also reduces molecular oxygen to form the superoxide anion. Myeloperoxidase, stored in the azurophilic granules of neutrophils, converts hydrogen peroxide to toxic hypochlorous acid, which, in addition to its direct effects, is also capable of activating proteases. The activated neutrophils also secrete a number of proteases, including matrix metalloproteinases, which will degrade basement membrane and other tissue structures, contributing to the severity of tissue destruction.
Neutrophil infiltration is observed at sites of tissue damage26,27 and the depletion of neutrophils reduces the severity of organ damage in a mouse model of liver IRI.28 Depletion of neutrophils during cardiac surgery has been extensively investigated as a modality to reduce the severity of post-operative cardiac dysfunction with inconsistent results. Some studies have shown a reduction in markers of cardiac damage while others have been less successful in demonstrating a clinically relevant effect.
Selectins are a family of transmembrane molecules, expressed on the surface of leukocytes, activated endothelial cells and in platelets. Selectins mediate the initial phase of neutrophil–endothelial cell interactions, often termed rolling (Figure 18.3), which is essential for their subsequent adhesion and extravasation. L-selectin is expressed constitutively on the surface of neutrophils and initiates the reversible attachment of neutrophils to endothelial cells and platelets. Antibody-mediated blocking of L-selectin impairs the ability of neutrophils to roll on endothelial cells and reduces neutrophil infiltration following skeletal muscle and pulmonary IRI.29
P-selectin is stored in the α-granules of platelets and the Wiebel-Palade bodies of endothelial cells and is rapidly translocated to the cell surface along with PAF in response to thrombin, histamine, reactive oxygen species, complement and TNF-α. Typically, peak levels of endothelial P-selectin are detected 6 hours after reperfusion. Endothelial P-selectin plays a vital role in the rolling of neutrophils along the activated endothelium. Activation of the endothelium by pro-inflammatory mediators also results in de novo transcription and synthesis of E-selectin. Expression of endothelial E-selectin is induced during both renal and cerebral IRI. The focal expression of E-selectin at sites of endothelial activation promotes neutrophil adhesion and infiltration into adjacent tissues. In support of a vital role for E-selectin in mediating tissue damage during IRI, a study showed that antibodies against E-selectin reduced infarct size following cerebral IRI in mice.30 blocking the activity of selectins shows promise in ameliorating the severity of tissue damage in a number of animal models of IRI. Although some promising selectin inhibitors have been tested in animal models of IRI, this therapy has yet to be tested in a clinical situation (reviewed in31).
The integrin and immunoglobulin supergene families of adhesion molecules mediate the strong adhesion of activated neutrophils to the endothelium and hence allow their subsequent extravasation during IRI. The integrins form a large family of cell surface adhesion molecules that mediate intercellular recognition and cellular binding to the extracellular matrix. The neutrophil β2-integrin adhesion glycoprotein complex consists of a common polypeptide chain, CD18, which is non-covalently linked to three different α-polypeptide chains (CD11a, CD11b, CD11c). CD11a/CD18 is expressed on all leukocytes and mediates the attachment of stimulated neutrophils to the vascular endothelium through a specific interaction with ICAM-1 and ICAM-2. Chemotactic cytokines (IL-1, TNF- α) and ROS all induce neutrophil adherence to the endothelium by CD11/CD18-dependent mechanisms. The CD11b/18 complex on activated neutrophils interacts with ICAM-1 on the surface of the endothelial cell to mediate firm adhesion of neutrophils prior to their extravasation (reviewed in32). All of these molecules are required for the development of lung injury following skeletal muscle IRI. Using an anti-CD18 monoclonal antibody, inhibition of CD18-mediated leukocyte adhesion prevented vasoconstriction, inhibited vessel leakage and reduced vascular resistance in animal models of skeletal muscle IRI. However, despite encouraging animal studies, the clinical efficacy of blocking CD11/CD18-mediated interactions in IRI remains doubtful (reviewed in33). Clinical trials in humans failed to demonstrate any effect of CD11/CD18 in reducing infarct size following primary coronary angioplasty in the setting of acute myocardial infarction. A more recent review34 summarised the results from a number of clinical trials using antibodies to CD11/CD18, including for myocardial infarct and stroke, all of which failed to show any significant benefit to the patient.
The immunoglobulin supergene family (ligands for integrins) contains a large number of molecules with multiple immunoglobulin-G-like domains. Several members of this family are involved in leukocyte-endothelial cell interactions including ICAM-1, VCAM-1 and platelet-endothelial cell adhesion molecule-1 (PECAM-1). Levels of expression of ICAM-1 on endothelial cells are enhanced by exposure to circulating TNF-α that is generated in response to IRI. VCAM-1 was elevated during renal IRI in a mouse model but, unlike ICAM-1, was independent of TNF-α since renal IRI in TNF-α knockout mice also upregulated VCAM-1. PECAM-1 is expressed constitutively on platelets, leukocytes and endothelial cells. IRI induces elevated PECAM-1 levels thereby enhancing activation of neutrophil-endothelial interactions mediated by β-integrins and exacerbating neutrophil extravasation and tissue damage.
The therapeutic potential of blocking the activity of adhesion molecules has been tested in a number of animal models with encouraging results. Using monoclonal antibodies, inhibition of ICAM-1 activity attenuated neutrophil adhesion in the liver, reduced pulmonary sequestration and oedema following skeletal muscle IRI and also reduced intestinal dysfunction following IRI.35 Antisense oligonucleotides to ICAM-1 ameliorated renal IRI and prevented delayed graft dysfunction in a rat model of renal transplantation.36 However, results obtained in clinical trials have not been as positive. A recent clinical trial of anti-ICAM-1 antibody therapy in ischaemic stroke (Enlimomab Acute Stroke Trial) concluded that this was not an effective treatment and may significantly worsen stroke outcome, raising significant doubts regarding the efficacy of this therapeutic modality.37
Complement activation
Complement activation and deposition also contribute significantly to the pathogenesis of IRI. Rubin and colleagues have demonstrated that reperfusion of skeletal muscle is associated with systemic depletion of the complement protein, factor B, indicative of activation of the alternative complement pathway.38 the complex C5b-9 is also deposited into the endothelial cell membrane after IRI, leading to osmotic lysis.39 Pulmonary damage following bilateral hind limb ischemia was significantly reduced when the soluble complement receptor (SCR 1) was administered to rats, thus inhibiting complement activity.40 in the clinical setting, a relationship has been demonstrated between the severity of multi-system organ dysfunction and degree of complement activation after aortic cross clamping.41
Inhibition of the complement cascade has been demonstrated to improve outcomes following IRI in a number of different animal models. Complement depletion of circulating plasma improved the initial blood flow and decreased muscle necrosis and injury after ischaemia and prolonged reperfusion in dogs. Complement blockade also prevented leukocyte adhesion, leading to better capillary perfusion and muscle cell viability and attenuated the increase in permeability index in tissues.42 Unequivocal evidence for the importance of complement activation during skeletal muscle IRI has been provided from experiments where limb ischaemia was induced in C5-deficient mice. These mice had approximately 50% less tissue damage than the wild-type animals.39 An additive role of both complement and neutrophils in mediating skeletal muscle IRI has also been observed, with a greater reduction in histological damage in neutropenic C5-deficient animals than in neutropenic or C5-deficient mice alone.39 these data continue to demonstrate the multifactorial nature of tissue damage induced during IRI since complement blockade failed to completely ameliorate tissue damage.
Tissue Destruction
Proteases and metalloproteinases
The matrix metalloproteinases (MMPs) are a family of zinc dependent enzymes that have the ability to degrade components of the extracellular matrix. Together with their inhibitors, the tissue inhibitors of metalloproteinases (TIMPs), they are the major physiological regulators of the extracellular matrix. MMPs are intimately involved in all processes that necessitate degradation or synthesis of the extracellular matrix and important roles for these enzymes have been identified in wound healing, periodontal disease, cancer metastasis and, of particular relevance, vascular disease including the development of aneurysms, atherosclerotic plaques and reperfusion injury.
Elevations of MMP-2 and MMP-9 have been detected following pulmonary, hepatic and cardiac IRI. MMPs are also elevated following cerebral IRI, corresponding with opening of the blood-brain barrier, degradation of the basal lamina, increased capillary permeability and cerebral oedema.43 Definitive roles for MMP-9 in the pathophysiology of cerebral IRI have been demonstrated by using both selective MMP-9 inhibitors and MMP-9 knockout mice, which both significantly reduce cerebral infarct size.44 the role for MMPs in renal IRI is less clear. MMP-2 may have a late role in renal IRI with an elevation detected as late as 8 weeks after IRI.45 However the MMP inhibitor (Batimastat) did not alter the severity of IRI induced renal dysfunction.46
Barr and co-workers47 carried out a study examining acute ischaemic stroke patients by mri and correlated systemic plasma MMP-9 levels with a hyperintense acute reperfusion injury marker (HARM), measured by MRI 24 hours later. Plasma MMP-9 was a significant predictor of elevated HARM measures, supporting the hypothesis that elevated MMP-9 is associated with disruption of the blood brain barrier after ischaemic stroke. These results raise the possibility that inhibition of MMP-9 may be a useful modality to reduce the severity of cerebral damage.
Studies in our laboratory have demonstrated both a local and systemic role for MMP-2 and MMP-9 in the degradation of type IV collagen in pulmonary tissues and in skeletal muscle following lower limb IRI.27 Permanent ischaemia alone, without reperfusion, also results in elevation of MMP-2 and MMP-9, correlating with destruction of the basement membrane components, type IV collagen and laminin.
Apoptotic cell death during ischaemia-reperfusion injury
Tissue destruction resulting from IRI can be due to either necrotic or apoptotic cell death. Apoptosis or programmed cell death is an active process characterized by a series of gene-directed events leading to a characteristic cell morphology, controlled DNA fragmentation and eventually death of the cell. The role of apoptosis in IRI-induced tissue damage has been widely investigated in recent years. Oxidative stress and the production of ROS will induce apoptosis, the characteristics of which can readily be recognised following cerebral IRI. Similarly, renal and cardiac IRI all result in detectable levels of apoptosis in the damaged tissue. Apoptosis therefore appears to play a fundamental role in cellular damage occurring during IRI in a number of tissues. However the role of apoptosis in skeletal muscle IRI remains controversial. Studies conducted in our laboratory,26 in agreement with Knight and co-workers,48 have failed to detect any evidence of apoptosis in rat skeletal myocytes following IRI. This implicates a tissue-specific mechanism of cell death following IRI. Blocking the apoptotic cascade, using specific inhibitors directed against pro-apoptotic caspase enzymes, have been partially effective in animal models, reducing the severity and infarct size following hepatic and cardiac IRI.
No reflow phenomenon
No reflow is the failure of microvascular perfusion, following restoration of flow to previously ischaemic tissue. The cause of this phenomenon has not been fully elucidated (reviewed in49) but is certainly multifactorial. Cytokines and activated neutrophils act synergistically to produce microvascular barrier dysfunction. The resultant increase in permeability leads to the exudation of fluids and proteins, increasing the interstitial pressure and decreasing the net intravascular pressure. In addition, CD18dependent leukocyte plugging produces partial occlusion of post-capillary venules, further contributing to no-reflow. Neutrophil depletion virtually abolishes the phenomenon in the myocardium, brain and skeletal muscle, confirming a vital role for neutrophils in no-reflow.
Therapeutic Approaches to IRI
Ischaemic preconditioning
Ischaemic preconditioning consists of brief and repetitive episodes of IRI before the induction of sustained organ ischaemia and is effective in reducing the severity of tissue damage. The preconditioning effect can be delivered remotely instead of to the target organ. This treatment could be useful in a number of operative settings including transplantation, coronary bypass grafting and elective major vascular surgical procedures where the onset of ischaemia can be tightly controlled. In these settings, brief extremity IRI (10 minutes) administered by tourniquet before surgery has been widely investigated and shows promise as a therapy to reduce the severity of IRI.
Animal models of a number of settings of IRI have been used to investigate mechanisms of ischaemic preconditioning but the basic molecular mechanisms remain unclear, probably due to the multiple signal transduction pathways involved in this phenomenon. However it is generally recognised that brief ischaemic preconditioning induces a cascade of intracellular kinases, which subsequently modify mitochondrial function. A recent study in a rat model of lower limb IRI illustrated clearly that two brief 10 minute episodes of IRI before a full 60 minutes of ischaemia was effective in reducing pro-inflammatory neutrophil-endothelium interactions. This effect was noted in both the lower limb itself and in remote tissues, illustrating the systemic nature of this phenomenon.50 In a mouse model of hind limb IRI, preconditioning significantly reduced tissue damage in the limb itself and also in lung and small bowel. Preconditioned animals were also significantly protected against post-operative mortality.51
A large number of clinical trials have also been reported investigating the efficacy of ischaemic preconditioning but with varying degrees of success (reviewed in52). A small randomised clinical trial aimed to determine if remote lower limb ischaemic pre-conditioning before EVAR could reduce the severity of renal and cardiac damage.53 A significant reduction in urinary biomarkers of renal injury was detected in the preconditioning cohort but this small pilot trial was unable to detect any effect on clinical endpoints. However, in the setting of open AAA repair where operative ischemia is profound, promising results were obtained. Emote preconditioning significantly protected against post-operative myocardial injury, myocardial infarction, and renal impairment.54 An excellent ‘proof-of concept’ study of ischaemic preconditioning was recently reported in the setting of evolving ST-elevation acute myocardial infarction. Subjects were randomised while in the ambulance and received intermittent arm ischaemia during transport to hospital (four cycles of 5 minute inflation and 5 minute deflation of a bloodpressure cuff). The primary endpoint was the myocardial salvage index 30 days after primary percutaneous coronary intervention, measured by myocardial perfusion imaging. The data showed convincingly that remote ischaemic conditioning before hospital admission increased myocardial salvage.55 Further studies are needed to verify the effect of remote conditioning on clinical outcomes but this therapeutic modality currently appears very promising.
Ischaemic post-conditioning
Ischaemic post-conditioning is defined as rapid sequential intermittent interruption of blood flow applied during the early moments of reperfusion. This technique is particularly relevant where the initial ischaemic insult could not have been predicted, thus a preconditioning approach to limiting tissue damage could not have been applied. Experimental animal models have been used to successfully show attenuation of organ injury, including the heart, spinal cord, brain, kidney, liver, muscle, lung and intestines (reviewed in56). The mechanisms of post-conditioning are not yet entirely clear but appear to involve multiple signalling pathways and molecules, including protein kinases, ROS, pro-inflammatory cytokines and NO, as well as alterations in mitochondrial function (reviewed in57).
Animal models of particular relevance to vascular surgical procedures have been tested widely and results show promise for postconditioning as an effective therapy to reduce the severity of IRI. In a rat model of lower limb ischaemia induced by aortic clamping, rats underwent 180 minutes of ischaemia followed by post-conditioning consisting of six cycles of 10 seconds aortic occlusion followed by 10 seconds declamping at the beginning of reperfusion. Post-conditioning caused a significant reduction in both the severity of systemic inflammatory responses and degree of remote pulmonary and renal damage.58 In a similar study in the rat,59 60 minutes infrarenal aortic cross-clamping followed by intermittent 4 times 15 seconds reperfusion-15 seconds ischaemic episodes before reperfusion, was effective in reducing production of ROS, leukocyte-endothelial activation and cytokine production.
Based on the experimental models, ischaemic postconditioning thus appears to show promise as an effective therapy in vascular surgery to reduce reperfusion injuries after aortic surgery and revascularization procedures (reviewed in60). Some clinical studies have verified these findings, although this has been largely limited to cardiac IRI. However, the duration of the occlusion and reperfusion periods will be critical to the degree of protection and further studies are needed to calculate useful algorithms to plan therapeutic strategies after a significant ischaemic insult.
Conditioning effects of volatile anaesthetics
Anaesthetics have been widely demonstrated to reduce the severity of IRI-induced damage in the setting of myocardial ischaemia and reperfusion during cardiac surgery (reviewed in61). However, there is conflicting evidence regarding the relative contributions of preconditioning, conditioning during ischaemia and postconditioning to the significant cardioprotection provided by anaesthetics. The molecular mechanisms and signal transduction pathways involved in protection are an area of active investigation. A proteomic study demonstrated that volatile anaesthetics (isoflurane, sevoflurane or desflurane) induced long lasting changes in the expression of 106 proteins in the rat myocardium.62 Evidence also suggests that inhibition by anaesthetics of the opening of the mitochondrial permeability pore may be a key mechanism of anaesthetic-induced preconditioning. Anaesthetic-induced postconditioning mechanisms are also multi-factorial. Volatile anaesthetics are known to inhibit neutrophil adhesion in the coronary arteries during the reperfusion phase, thereby inhibiting the inflammatory action of activated neutrophils in post-ischaemic tissues (Figure 18.3).
There is good clinical evidence for the cardioprotective effects of volatile anaesthetics during cardiac surgery. A meta-analysis examined randomized trials comparing volatile with non-volatile anaesthesia in coronary bypass surgery. There was no sig-nificant difference in myocardial ischaemia, myocardial infarct, intensive care unit length of stay or in-hospital mortality. However, patients receiving volatile anaesthetics had significantly higher cardiac indices, lower troponin I serum concentrations and a lower requirement for inotropic support.63 A more recent large multicentre study provided excellent evidence that volatile anaesthesia significantly reduced mortality after coronary bypass grafting.64 Evidence for anaesthetic protection in vascular surgical settings other than in cardiac IRI is not currently available but is likely to be equally significant and should be actively investigated in the future.
Pharmacological treatments
As discussed in many of the sections above, a wide range of pharmacological therapies have been tested in both animal models and in the clinic. Although many of the animal models show considerable promise in reducing the severity of IRI, results from clinical trials have uniformly been disappointing. A recent Cochrane Review reported on treatments to reduce IRI during liver resection under vascular control.65 they identified 15 randomised trials, which examined 11 pharmacological interventions (methylprednisolone, multi-vitamin antioxidant infusion, vitamin E infusion, amrinone, prostaglandin E1, pentoxifylline, mannitol, trimetazidine, dextrose, allopurinol and a thromboxane A2 synthetase inhibitor). Although some therapies improved liver enzyme levels, there were no significant differences between the groups for mortality, liver failure, or perioperative morbidity. A second Cochrane review from the same authors66 examined the effects of prostaglandin E1, pentoxifylline, dopexamine, dopamine, ulinastatin, gantaile, sevoflurane, and propofol during liver IRI and reached the same conclusion that there were no significant differences.
Statin therapies have been widely accepted into clinical practice and there is also considerable evidence, both experimental and clinical, that statins will reduce the severity of IRI in a range of settings. Statins inhibit a range of cellular responses to IRI-induced inflammation, including inhibition of NFkb activity, which leads to decreased transcription OF MMPs, adhesion molecules and cytokine genes. Binding of adhesion molecules on activated neutrophils to endothelial cell surface receptors is also blocked. Secretion OF MMPs from activated neutrophils is also inhibited by statins. In the endothelium, levels of expression eNOS mRNA are increased and the eNOS protein is activated, while expression of endothelin-1 is inhibited. All of these effects will ameliorate the severity of tissue damage during IRI (reviewed in67).
Trials of lower limb IRI in the rat were carried out in our laboratory and illustrated convincingly that pre-treatment for a week with simvastatin before IRI markedly protected both skeletal muscle and remote organs including the lungs and kidneys.14,68 In the clinical setting, a recent review69 discussed the efficacy of statins in patients undergoing a range of vascular surgical procedures. Symptomatic patients with carotid artery stenosis and taking statins appear to have better outcomes after carotid endartarectomy than those not on statins, although the difference between the cohorts is not marked. In the setting of infrainguinal bypass for peripheral arterial disease, the indications that statins may protect against IRI during surgery are less definitive with some conflicting results although 1-year mortality was improved. Evidence for any effect of statin treatment on the severity of postoperative complications after AAA repair is lacking, although a retrospective observational study showed that all-cause mortality was reduced in those on long term statin therapy.70 However, since all vascular patients should be receiving statin treatment for secondary prevention of cardiovascular disease, prospective randomized trials to obtain definitive results can no longer ethically be performed.
Summary
In summary, IRI is a highly complex series of interwoven pro-inflammatory and pathological events. The production, release and activation of cytokines, ROS, proteases and complement if left unchecked, leads to both local and systemic injury with potentially fatal consequences. The failure of therapeutic interventions to translate into clinical practice is a reflection of this complexity and redundancy within the system. New therapeutic agents directed towards multiple areas within this cascade may be required to overcome this difficult clinical challenge.
References
- 1.
- Allen DG, Xiao XH. Activity of the NA+/H+ exchanger contributes to cardiac damage following ischaemia and reperfusion. Clin Exp Pharmacol Physiol 2000; 27: 727–33. [PubMed: 10972541]
- 2.
- Paoni NF, Peale F, Wang F, Errett-baroncini C, Steinmetz H, Toy K,Bai W, Williams PM, Bunting S, Gerritsen ME, Powell-braxton L. Time course of skeletal muscle repair and gene expression following acute hind limb ischemia in mice. Physiol Genomics 2002; 11: 263–72. [PubMed: 12399448]
- 3.
- Safronova O,Morita I. Transcriptome remodeling in hypoxic inflammation. J Dent Res 2010; 89: 430–44. [PubMed: 20348484]
- 4.
- Hierholzer C, Harbrecht BG, Billiar TR, Tweardy DJ. Hypoxia-inducible factor-1 activation and cyclo-oxygenase-2 induction are early reperfusion-independent inflammatory events in hemorrhagic shock. Arch Orthop Trauma Surg 2001; 121: 219–22. [PubMed: 11317684]
- 5.
- Yokoyama K, Kimura M, Nakamura K, Itoman M. Time course of post-ischemic superoxide generation in venous effluent from reperfused rabbit hindlimbs. J Reconstr Microsurg 1999; 15: 215–21. [PubMed: 10226957]
- 6.
- Pacher P, Nivorozhkin A, Szabo C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev 2006; 58: 87–114. [PMC free article: PMC2233605] [PubMed: 16507884]
- 7.
- Neumann UP, Kaisers U, Langrehr JM, Glanemann M, Muller AR, Lang M, Jorres A, Settmacher U, Bechstein WO, Neuhaus P. Administration of prostacyclin after liver transplantation: a placebo controlled randomized trial. Clin Transplant 2000; 14: 70–4. [PubMed: 10693639]
- 8.
- Katircioglu SF, Kucukaksu DS, Bozdayi M, Tasdemir O, Bayazit K. Beneficial effects of prostacyclin treatment on reperfusion of the myocardium. Cardiovasc Surg 1995; 3: 405–8. [PubMed: 7582995]
- 9.
- Rowlands TE, Gough MJ, Homer-Vanniasinkam S. DO prostaglandins have a salutary role in skeletal muscle ischaemia-reperfusion injury? Eur J Vasc Endovasc Surg 1999; 18: 439–44. [PubMed: 10610833]
- 10.
- Slupski M, Szadujkis-Szadurska K, Szadujkis-Szadurski R, Szadujkis-Szadurski L, Wlodarczyk Z, Andruszkiewicz J, Sinjab AT. Nitric oxide and thromboxane A2 modulate pulmonary pressure after ischemia and intestinal reperfusion. Transplant Proc 2006; 38: 334–7. [PubMed: 16504740]
- 11.
- Mazolewski PJ, Roth AC, Suchy H, Stephenson LL, Zamboni WA. Role of the thromboxane A2 receptor in the vasoactive response to ischemia-reperfusion injury. Plast Reconstr Surg 1999; 104(5): 1393–6. [PubMed: 10513923]
- 12.
- Mangino MJ, Murphy MK, Anderson CB. Effects of the arachidonate 5–lipoxygenase synthesis inhibitor A-64077 in intestinal ischemia-reperfusion injury. J Pharmacol Exp Ther 1994; 269: 75–81. [PubMed: 8169854]
- 13.
- Khanna A, Cowled PA, Fitridge RA. Nitric oxide and skeletal muscle reperfusion injury: current controversies (research review). J Surg Res 2005; 128: 98–107. [PubMed: 15961106]
- 14.
- Cowled PA, Khanna A, Laws PE, Field JB, Varelias A, Fitridge RA. Statins inhibit neutrophil infiltration in skeletal muscle reperfusion injury. J Surg Res 2007; 141: 267–76. [PubMed: 17559881]
- 15.
- Kiris I, Narin C, Gulmen S, Yilmaz N, Sutcu R, Kapucuoglu N. Endothelin receptor antagonism by tezosentan attenuates lung injury induced by aortic ischemia-reperfusion. Ann Vasc Surg 2009; 23: 382–91. [PubMed: 19135850]
- 16.
- Lutz J, Thurmel K, Heemann U. Anti-inflammatory treatment strategies for ischemia/reperfusion injury in transplantation. J Inflamm 2010; 7: 27. [PMC free article: PMC2894818] [PubMed: 20509932]
- 17.
- Tassiopoulos AK, Carlin RE, Gao Y, Pedoto A, Finck CM, Landas SK, Tice DG, Marx W, Hakim TS, McGraw DJ. Role of nitric oxide and tumor necrosis factor on lung injury caused by ischemia/reperfusion of the lower extremities. J Vasc Surg 1997; 26: 647–56. [PubMed: 9357467]
- 18.
- Esposito E, Cuzzocrea S. TNF-alpha as a therapeutic target in inflammatory diseases, ischemia-reperfusion injury and trauma. Curr Med Chem 2009; 16: 3152–67. [PubMed: 19689289]
- 19.
- Duran WN, Milazzo VJ, Sabido F, Hobson RW, 2nd. Platelet-activating factor modulates leukocyte adhesion to endothelium in ischemia-reperfusion. Microvasc Res 1996; 51: 108–15. [PubMed: 8812764]
- 20.
- Sun Z, Wang X, Deng X, Lasson A, Soltesz V, Borjesson A, Andersson R. Beneficial effects of lexipafant, a PAF antagonist on gut barrier dysfunction caused by intestinal ischemia and reperfusion in rats. Dig Surg 2000; 17: 57–65. [PubMed: 10720833]
- 21.
- Borjesson A, Wang X, Sun Z, Inghammar M, Truedsson L, Andersson R. Early treatment with lexipafant, a platelet-activating factor-receptor antagonist, is not sufficient to prevent pulmonary endothelial damage after intestinal ischaemia and reperfusion in rats. Dig Liver Dis 2002; 34: 190–6. [PubMed: 11990391]
- 22.
- Norwood MG, Bown MJ, Sutton AJ, Nicholson ML, Sayers RD. Interleukin 6 production during abdominal aortic aneurysm repair arises from the gastrointestinal tract and not the legs. Br J Surg 2004; 91: 1153–6. [PubMed: 15449266]
- 23.
- de Vries DK, Lindeman JH, Tsikas D, de Heer E, Roos A, de Fijter JWW, Baranski AG, van Pelt J, Schaapherder AF. Early renal ischemia-reperfusion injury in humans is dominated by IL-6 release from the allograft. Am J Transplant Jul; 9: 1574–84. [PubMed: 19459788]
- 24.
- De Perrot M, Sekine Y, Fischer S, Waddell TK, McRae K, Liu M, Igle A, Keshavjee S. Interleukin-8 release during early reperfusion predicts graft function in human lung transplantation. Am J Respir Crit Care Med 2002; 165: 211–5. [PubMed: 11790657]
- 25.
- Sekido N, Mukaida N, Harada A, Akanishi I, Watanabe Y, Matsushima K. Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nature 1993; 365: 654–7. [PubMed: 8413628]
- 26.
- Cowled PA, Leonardos L, Millard SH, Fitridge RA. Apoptotic Cell Death Reperfusion Injury in Skeletal muscle in the Rat. Eur J Vasc Endovasc Surg 2001; 21: 28–34. [PubMed: 11170874]
- 27.
- Roach DM, Fitridge RA, Laws PE, Millard SH, Varelias A, Cowled PA. UP-regulation of MMP-2 and MMP-9 leads to degradation of type IV collagen during skeletal muscle reperfusion injury; protection by the MMP inhibitor, doxycycline. Eur J Vasc Endovasc Surg 2002; 23: 260–9. [PubMed: 11914015]
- 28.
- Martinez-Mier G, Toledo-Pereyra LH, McDuffie JE, Warner RL, Ward PA. Neutrophil depletion and chemokine response after liver ischemia and reperfusion. J Invest Surg 2001; 14: 99–107. [PubMed: 11396626]
- 29.
- Levine AJ, Parkes K, Rooney SJ, Bonser RS. The effect of adhesion molecule blockade on pulmonary reperfusion injury. Ann Thorac Surg 2002; 73: 1101–6. [PubMed: 11996249]
- 30.
- Huang J, Choudhri TF, Winfree CJ, McTaggart RA, Kiss S, Mocco J, Kim LJ, Protopsaltis TS, Zhang Y, Pinsky DJ, Connolly ES, Jr. Postischemic cerebrovascular E-selectin expression mediates tissue injury in murine stroke. Stroke 2000; 31: 3047–53. [PubMed: 11108771]
- 31.
- Calvey CR, Toledo-Pereyra LH. Selectin inhibitors and their proposed role in ischemia and reperfusion. J Invest Surg 2007; 20: 71–85. [PubMed: 17454392]
- 32.
- Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res 2008; 30: 783–93. [PMC free article: PMC2748428] [PubMed: 18826804]
- 33.
- McKenzie ME, Gurbel PA. The potential of monoclonal antibodies to reduce reperfusion injury in myocardial infarction. BioDrugs 2001; 15: 395–404. [PubMed: 11520250]
- 34.
- Yonekawa K, Harlan JM. Targeting leukocyte integrins in human diseases. J Leukoc Biol 2005; 77: 129–40. [PubMed: 15548573]
- 35.
- Sun Z, Wang X, Lasson A, Bojesson A, Annborn M,Andersson R. Effects of inhibition of PAF, ICAM-1 and PECAM-1 on gut barrier failure caused by intestinal ischemia and reperfusion. Scand J Gastroenterol 2001; 36: 55–65. [PubMed: 11218240]
- 36.
- Dragun D, Tullius SG, Park JK, Maasch C, Lukitsch I, Lippoldt A, Gross V, Luft FC, Haller H. ICAM-1 antisense oligodesoxynucleotides prevent reperfusion injury and enhance immediate graft function in renal transplantation. Kidney Int 1998; 54: 590–602. [PubMed: 9690227]
- 37.
- Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology 2001; 57: 1428–34. [PubMed: 11673584]
- 38.
- Rubin BB, Smith A, Liauw S, Isenman D, Romaschin AD, Walker PM. Complement activation and white cell sequestration in postischemic skeletal muscle. Am J Physiol 1990; 259: H525–31. [PubMed: 2167024]
- 39.
- Kyriakides C, Austen W, Wang Y, Favuzza J, Kobzik L, Moore FD, Hechtman HB. Skeletal muscle reperfusion injury is mediated by neutrophils and the complement membrane attack complex. Am J Physiol 1999; 277: C1263–8. [PubMed: 10600778]
- 40.
- Lindsay TF, Hill J, Ortiz F, Rudolph A, Valeri CR, Hechtman HB, Moore FD, Jr. Blockade of complement activation prevents local and pulmonary albumin leak after lower torso ischemia-reperfusion. Ann Surg 1992; 216: 677–83. [PMC free article: PMC1242715] [PubMed: 1334645]
- 41.
- Harkin DW, Marron CD, Rother RP, Romaschin A, Rubin BB, Lindsay TF. C5 complement inhibition attenuates shock and acute lung injury in an experimental model of ruptured abdominal aortic aneurysm. Br J Surg 2005; 92: 1227–34. [PubMed: 16078298]
- 42.
- Kyriakides C, Wang Y, Austen WG, Jr., Favuzza J, Kobzik L, Moore FD, Jr., Hechtman HB. Moderation of skeletal muscle reperfusion injury by a sLe(x)-glycosylated complement inhibitory protein. Am J Physiol Cell Physiol 2001; 281: C224–30. [PubMed: 11401845]
- 43.
- Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res 1999; 842: 92–100. [PubMed: 10526099]
- 44.
- Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, LOEH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with bb-94. J Cereb Blood Flow Metab 2000; 20: 1681–9. [PubMed: 11129784]
- 45.
- Jain S, Bicknell GR, Nicholson ML. Molecular changes in extracellular matrix turnover after renal ischaemia-reperfusion injury. Br J Surg 2000; 87: 1188–92. [PubMed: 10971426]
- 46.
- Ziswiler R, Daniel C, Franz E, Marti HP. Renal Matrix Metalloproteinase Activity Is Unaffected by Experimental Ischemia Reperfusion Injury and Matrix Metalloproteinase Inhibition Does Not Alter Outcome of Renal Function. Exp Nephrol 2001; 9: 118–24. [PubMed: 11150860]
- 47.
- Barr TL, Latour LL, Lee KY, Schaewe TJ, Luby M, Chang GS, El-Zammar Z, Alam S, Hallenbeck JM, Kidwell CS, Warach S. Bloodbrain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. Stroke 2010; 41: e123–8. [PMC free article: PMC2827673] [PubMed: 20035078]
- 48.
- Knight KR, Messina A, Hurley JV, Zhang B, Morrison WA, Stewart AG. Muscle cells become necrotic rather than apoptotic during reperfusion of ischaemic skeletal muscle. Int J Exp Pathol 1999; 80: 169–75. [PMC free article: PMC2517767] [PubMed: 10469272]
- 49.
- Niccoli G, Marino M, Spaziani C, Crea F. Prevention and treatment of no-reflow. Acute Card Care 2010; 12: 81–91. [PubMed: 20698732]
- 50.
- Szabo A, Varga R, Keresztes M, Vizler C, Nemeth I, Razga Z, Boros M. Ischemic limb preconditioning downregulates systemic inflammatory activation. J Orthop Res 2009; 27: 897–902. [PubMed: 19105227]
- 51.
- Eberlin KR, McCormack MC, Nguyen JT, Tatlidede HS, Randolph MA, Austen WG, Jr. Ischemic preconditioning of skeletal muscle mitigates remote injury and mortality. J Surg Res 2008; 148: 24–30. [PubMed: 18570927]
- 52.
- Kharbanda RK, Nielsen TT, Redington AN. Translation of remote ischaemic preconditioning into clinical practice. Lancet 2009; 374: 1557–65. [PubMed: 19880021]
- 53.
- Walsh SR, Boyle JR, Tang TY, Sadat U, Cooper DG, Lapsley M, Norden AG, Varty K, Hayes PD, Gaunt ME. Remote ischemic preconditioning for renal and cardiac protection during endovascular aneurysm repair: a randomized controlled trial. J Endovasc Ther 2009; 16: 680–9. [PubMed: 19995115]
- 54.
- Ali ZA, Callaghan CJ, Lim E, Ali AA, Nouraei SA, Akthar AM, Boyle JR, Varty K, Kharbanda RK, Dutka DP, Gaunt ME. Remote ischemic preconditioning reduces myocardial and renal injury after elective abdominal aortic aneurysm repair: a randomized controlled trial. Circulation 2007; 116: 198–105. [PubMed: 17846333]
- 55.
- Botker HE, Kharbanda R, Schmidt MR, Bottcher M, Kaltoft AK, Terkelsen CJ, Munk K, Andersen NH, Hansen TM, Trautner S, Lassen JF, Christiansen EH, Krusell LR, Kristensen SD, Thuesen L, Nielsen SS, Rehling M, Sorensen HT, Redington AN, Nielsen TT. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet 2010 Feb 27; 375: 727–34. [PubMed: 20189026]
- 56.
- Zhao ZQ. Postconditioning in Reperfusion Injury: A Status Report. Cardiovasc Drugs Ther 2010; 24: 265–79. [PubMed: 20532970]
- 57.
- Kaur S, Jaggi AS, Singh N. Molecular aspects of ischaemic postconditioning. Fundam Clin Pharmacol 2009; 23: 521–36. [PubMed: 19674116]
- 58.
- Gyurkovics E, Aranyi P, Stangl R, Onody P, Ferreira G, Lotz G, Kupcsulik P, Szijarto A. Postconditioning of the Lower Limb-Protection Against the Reperfusion Syndrome. J Surg Res 2010; In Press (ePub doi: 10.1016/j.jss.2009.10.014). [PubMed: 20085841] [CrossRef]
- 59.
- Sinay L, Kurthy M, Horvath S, Arato E, Shafiei M, Lantos J, Ferencz S, Bator A, Balatonyi B, Verzar Z, Suto B, Kollar L, Weber G, Roth E, Jancso G. Ischaemic postconditioning reduces peroxide formation, cytokine expression and leukocyte activation in reperfusion injury after abdominal aortic surgery in rat model. Clin Hemorheol Microcirc 2008; 40: 133–42. [PubMed: 19029638]
- 60.
- Mockford KA, Girn HR, Homer-Vanniasinkam S. Postconditioning: current controversies and clinical implications. Eur J Vasc Endovasc Surg. 2009; 37: 437–42. [PubMed: 19217807]
- 61.
- Frassdorf, J De Hert S, Schlack W. Anaesthesia and myocardial ischaemia/reperfusion injury. Br J Anaesth 2009; 103: 89–98. [PubMed: 19502287]
- 62.
- Kalenka A, Maurer MH, Feldmann RE, Kuschinsky W, Waschke KF. Volatile anesthetics evoke prolonged changes in the proteome of the left ventricule myocardium: defining a molecular basis of cardioprotection? Acta Anaesthesiol Scand 2006; 50: 414–27. [PubMed: 16548853]
- 63.
- Symons JA, Myles PS. Myocardial protection with volatile anaesthetic agents during coronary artery bypass surgery: a meta-analysis. Br J Anaesth 2006; 97: 127–36. [PubMed: 16793778]
- 64.
- Bignami E, Biondi-Zoccai G, Landoni G, Fochi O, Testa V, Sheiban I, Giunta F, Zangrillo A. Volatile anesthetics reduce mortality in cardiac surgery. J Cardiothorac Vasc Anesth 2009; 23: 594–9. [PubMed: 19303327]
- 65.
- Abu-Amara M, Gurusamy KS, Hori S, Glantzounis G, Fuller B, Davidson BR. Pharmacological interventions versus no pharmacological intervention for ischaemia reperfusion injury in liver resection surgery performed under vascular control. Cochrane Database Syst Rev 2009; CD007472. [PubMed: 19821421]
- 66.
- Abu-Amara M, Gurusamy KS, Glantzounis G, Fuller B, Davidson BR. Pharmacological interventions for ischaemia reperfusion injury in liver resection surgery performed under vascular control. Cochrane Database Syst Rev 2009; CD008154. [PMC free article: PMC7182152] [PubMed: 19821445]
- 67.
- Laws PE, Spark JI, Cowled PA, Fitridge RA. The role of statins in vascular disease. Eur J Vasc Endovasc Surg 2004; 27: 6–16. [PubMed: 14652831]
- 68.
- Cowled PA, Khanna A, Laws PE, Field JB, Fitridge RA. Simvastatin plus nitric oxide synthase inhibition modulates remote organ damage following skeletal muscle ischemia-reperfusion injury. J Invest Surg 2008; 21(3): 119–26. [PubMed: 18569431]
- 69.
- Stalenhoef AF. The benefit of statins in non-cardiac vascular surgery patients. J Vasc Surg 2009; 49: 260–5. [PubMed: 19174265]
- 70.
- Kertai MD, Boersma E, Westerhout CM, van Domburg R, Klein J, Bax JJ, van Urk H, Poldermans D. Association between long-term statin use and mortality after successful abdominal aortic aneurysm surgery. Am J Med 2004; 116: 96–103. [PubMed: 14715323]
- Review [Pathophysiology of ischaemia-reperfusion injury].[Lijec Vjesn. 2006]Review [Pathophysiology of ischaemia-reperfusion injury].Sirotković-Skerlev M, Plestina S, Bilić I, Kovac Z. Lijec Vjesn. 2006 Mar-Apr; 128(3-4):87-95.
- Ischemia-Reperfusion Injury : Pathophysiology and Clinical Implications.[Eur J Trauma Emerg Surg. 2007]Ischemia-Reperfusion Injury : Pathophysiology and Clinical Implications.Dorweiler B, Pruefer D, Andrasi TB, Maksan SM, Schmiedt W, Neufang A, Vahl CF. Eur J Trauma Emerg Surg. 2007 Dec; 33(6):600-12. Epub 2007 Nov 20.
- Review [Pathophysiology of myocardial reperfusion injury].[Dtsch Med Wochenschr. 2008]Review [Pathophysiology of myocardial reperfusion injury].Piper HM, Kasseckert SA, Schlüter KD, Abdallah Y. Dtsch Med Wochenschr. 2008 Mar; 133(12):586-90.
- Review [Pathophysiology of ischemia-reperfusion injury in renal transplantation].[Postepy Hig Med Dosw (Online)....]Review [Pathophysiology of ischemia-reperfusion injury in renal transplantation].Boratyńska M, Kamińska D, Mazanowska O. Postepy Hig Med Dosw (Online). 2004 Feb 5; 58:1-8. Epub 2004 Feb 5.
- Review Myocardial reperfusion injury: the critical challenge.[Crit Care Nurs Clin North Am. ...]Review Myocardial reperfusion injury: the critical challenge.Coombs VJ, Black L, Townsend SN. Crit Care Nurs Clin North Am. 1992 Jun; 4(2):339-46.
- Pathophysiology of Reperfusion Injury - Mechanisms of Vascular DiseasePathophysiology of Reperfusion Injury - Mechanisms of Vascular Disease
Your browsing activity is empty.
Activity recording is turned off.
See more...