13.1. Introduction
The epithelial calcium (Ca2+) channels TRPV5 and TRPV6 are members of the transient receptor potential (TRP) channel family TRPV (“V” for vanilloid) subgroup. TRPV5 and TRPV6 play major roles in the maintenance of blood Ca2+ levels in higher organisms. Both channels exhibit similarities in many ways, as they share a high level (75%) of amino acid identity, comparable functional properties, and similar mechanisms of regulation. Also, they were discovered using similar cloning strategies [1,2]. Yet, their physiological contributions toward maintaining a systemic calcium balance are distinct. In addition, the following three key features distinguish TRPV5 and TRPV6 from other members of the TRP superfamily of cation channels: (1) high selectivity for Ca2+ over other cations, (2) apical membrane localization in Ca2+-transporting epithelial tissues, and (3) responsiveness to 1,25-dihydroxyvitamin D3 (1,25[OH]2D3) [3,4]. These features make TRPV5 and TRPV6 ideally suited to facilitate intestinal absorption and renal reabsorption of Ca2+, serving as apical Ca2+ entry channels in transepithelial Ca2+ transport [5,6]. A major difference between the properties of TRPV5 and TRPV6 lies in their tissue distribution: TRPV5 is predominantly expressed in the distal convoluted tubules (DCT) and connecting tubules (CNT) of the kidney, with limited expression in extrarenal tissues [1,7]. In contrast, TRPV6 exhibits a broader expression pattern, showing prominent expression in the intestine with additional expression in the kidney [8–10], placenta, epididymis, exocrine tissues (i.e., pancreas, prostate, salivary gland, sweat gland), and a few other tissues [11–13]. Thus, while TRPV5 plays a key role in determining the level of urinary Ca2+ excretion, the physiological roles of TRPV6 are not limited to intestinal Ca2+ absorption. Much progress has recently been made in understanding the roles of TRPV5 and TRPV6 channels in the kidney [14], intestine [15], placenta [16], and epididymis [17]. However, their roles in other organs have as yet not been fully investigated.
In this chapter, we review the current status of our knowledge of the physiological and pathological roles of TRPV5 and TRPV6 and discuss a variety of techniques that have led to a deeper understanding of these channels. We review the identification strategies of TRPV5 and TRPV6 in searches for Ca2+ absorption channels, as well as specific techniques used to reveal their key features. These include radiotracer Ca2+ uptake and electrophysiology procedures, structure-function studies, methods to identify regulatory interacting partners, genetically engineered animals, strategies to study the role of TRPV6 in cancers, procedures for the development of small-molecule modulators of TRPV6 and TRPV5, the evaluation of variations/mutations in humans, and 3D structural determination. For additional information about TRPV5 and TRPV6, we would like to refer the interested reader to other comprehensive review articles [3–6,18].
13.2. Ca2+ Transport across Epithelia
Epithelia form barriers to separate cells from the internal and external environment, allowing specific exchange of nutrients and other substances across epithelial cells. Among the many electrolytes, Ca2+ is an important intracellular messenger and a major component of the mineral phase of bones and teeth. Thus, at the cellular level, the cytosolic Ca2+ concentration is kept low (around 100 nM at rest), whereas its concentration in the extracellular fluids (ECF), such as blood, is maintained at a much higher level and within a relatively narrow limit (around 1 mM) [19]. The bone, which contains roughly 99% of the Ca2+ in the human body, is subject to constant Ca2+ exchange with the ECF. Any reduction in the extracellular Ca2+ concentration ([Ca2+]o) in the ECF is being monitored by the G-protein-coupled Ca2+-sensing receptor (CaSR) in the parathyroid gland, triggering secretion of the parathyroid hormone (PTH) [19]. The PTH increases bone resorption and renal reabsorption of Ca2+ and stimulates the hydroxylation of 25-hydroxyvitamin D3 [25(OH)2D3] in the proximal tubule of the kidney to form 1,25(OH)2D3 [20]. An important role of this activated form of vitamin D is the stimulation of intestinal Ca2+ absorption via the nuclear vitamin D receptor (VDR) [21]. Several organs and hormones work in concert to maintain stable levels of Ca2+ in the ECF. The intestinal and renal handling of Ca2+ is important as the intestine determines how much Ca2+ enters the body and the kidney determines how much Ca2+ is removed from the body.
Intestinal absorption and renal reabsorption of Ca2+ are similar processes, involving Ca2+ transport across the epithelia from the luminal side to the blood side [22,23]. In general, Ca2+ ions can cross the epithelia through the paracellular route, as well as the transcellular route (Figure 13.1). When [Ca2+]o is higher than that of the plasma, luminal Ca2+ predominantly enters the intestine via the paracellular route through tight junctions between the epithelial cells (Figure 13.1a). However, this route does not operate in the absence of favorable transepithelial [Ca2+] gradients. Thus, when luminal [Ca2+]o is lower than that in the plasma, Ca2+ will need to be actively absorbed across the epithelia via the transcellular route. For this to occur, Ca2+ first enters the cells across the apical membranes passively through TRPV6. This is followed by binding of Ca2+ to calbindin-D9K and transfer to the basolateral membrane. Basolateral exit is then mediated by the plasma membrane Ca2+ ATPase (PMCA), that is, by primary active transport, at the expense of stored ATP [22].
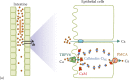
Figure 13.1
Mechanisms of Ca2+ transport in the intestine and kidney.
Similar transport mechanisms also take place in the kidney during Ca2+ reabsorption (Figure 13.1b). Based on our current knowledge from animal studies, up to 95% of filtered Ca2+ is being reabsorbed mainly via the paracellular route in renal proximal tubules and loops of Henle, while about 5%–10% of Ca2+ reabsorption is delayed till the distal tubules, where reabsorption occurs via the transcellular route and in a regulated manner in order to fine-tune the whole-body calcium homeostasis. At this point, it is worthy to note that TRPV6 in the human kidney likely plays an additional role in transcellular Ca2+ transport in specific parts of the tubular system (see Section 13.8 for further details). Similar to the intestine, the transcellular route in the kidney involves passive Ca2+ entry across the apical membranes through TRPV5, transfer to the basolateral membrane after binding to calbindin-D28K, and basolateral exit through the PMCA (primary active transport) or the Na+/Ca2+ exchanger NCX1 (at the expense of the inwardly directed Na+ gradient) [22,23].
Passive Ca2+ transport across the apical membrane via TRPV5 or TRPV6 is favored by the inwardly directed electrochemical gradient for Ca2+, which is warranted by the low cytosolic Ca2+ levels of approximately 100 nM. As already noted, once Ca2+ ions have entered the epithelial cell, they bind to calbindins during Ca2+ uptake (Figure 13.1). Two types of calbindins with different molecular weight and number of Ca2+-binding EF hands, calbindin-D9K and calbindin-D28K, are expressed in the Ca2+-transporting epithelial cells in the mammalian intestine and kidney, respectively [24]. They belong to a large family of high-affinity Ca2+-binding proteins (Kd = 10-8 - 10-6 M) featuring EF-hand structural motifs [25]. Calbindins and likely other Ca2+-binding proteins maintain the free cytosolic Ca2+ concentrations below toxic levels and help regulate TRPV5 and TRPV6 through their feedback mechanisms [26]. However, if apical Ca2+ entry exceeds the capacity of calbindins, Ca2+ will bind to calmodulin (CaM), resulting in a Ca2+ feedback inhibition of TRPV5 or TRPV6 channels (see Figure 13.1 on the right; also see Section 13.4.4).
In addition to the intestine and kidney, maternal-fetal transport of Ca2+ involves an active mechanism, as the serum [Ca2+] is higher in fetuses than in mothers [27]. In humans, Ca2+ is needed for the rapid growth of fetal skeleton, especially in the third trimester. The syncytiotrophoblast layer is a specialized epithelium responsible for the exchange of Ca2+ along with other nutrients and gases between mother and fetus [28]. Ca2+ transport in human syncytiotrophoblasts has been the subject of many studies [29]. These Ca2+ channels are insensitive to L-type voltage-gated Ca2+ channel modulators but sensitive to Mg2+ and ruthenium red [30], resembling the properties of TRPV6.
Active Ca2+ transport also takes place in specialized organs to create local environments where [Ca2+] needs to be controlled. For example, the luminal [Ca2+]o is lower in the distal portion of the epididymis [31]. Recent studies revealed that this lowered [Ca2+] is maintained by TRPV6-mediated Ca2+ transport into the epithelial cells of the epididymis and that this is important for preserving optimal motility of sperm and optimal male fertility [17]. Therefore, TRPV6 of the epididymis and the progesterone-regulated CatSper channel of sperm [32] both cooperate to ensure maximal motility of the sperm and fertility (see Section 13.5.4 for further details).
Another example is the observation that [Ca2+] in the vestibular endolymph is only one-fourth of that of the perilymph (∼1 mM). Indeed, [Ca2+]o is important in regulating the hair-bundle stiffness and gating-spring integrity in hair cells of the inner ear [33]. Active transcellular Ca2+ transport by TRPV5 and TRPV6 is present in the epithelial cells of the inner ear and maintains the low [Ca2+] of the vestibular endolymph [34,35].
The glandular tissue is also formed by epithelial cells. The release of content from secretory granules, which are loaded with Ca2+, is accompanied by the loss of Ca2+ [36]. Given that exocrine glands secrete large amounts of fluid, a Ca2+ reuptake mechanism may be useful to supply Ca2+ released in the apical pole of exocrine cells back into new secretory vesicles.
Lastly, carcinomas are cancer cells that originate from epithelial cells, and altered Ca2+ homeostasis is a characteristic feature of carcinomas. This may be related to cancer cell proliferation, survival, and metastasis formation [37–39]. The development of specific small-molecule inhibitors targeting Ca2+ channels overexpressed in carcinomas may offer novel strategies for cancer treatment [40,41] (see Section 13.7).
13.3. Identification of TRPV5 and TRPV6 by Expression Cloning
TRPV5 and TRPV6 were identified in 1999 by Bindels and colleagues and by our group from rabbit kidney cells and rat duodenum, respectively [1,2]. Both groups used expression cloning with radiotracer 45Ca2+ uptake assays, screening intestinal or renal cDNA libraries for the ability of clones to induce Ca2+ uptake in complementary RNA (cRNA)-injected Xenopus laevis oocytes. This approach has proven very useful in the identification of novel transporters, channels, and receptors [42]. Before the epithelial Ca2+ channels were identified, calbindins were known to facilitate the diffusion of Ca2+ from the apical to basolateral membranes, followed by cellular exit via Na+/Ca2+ exchangers and/or PMCAs. However, what mediated Ca2+ entry across the plasma membrane remained elusive for a long time [22,24]. To increase our chances for successfully cloning TRPV6, we fed rats a Ca2+-deficient diet for 2 weeks to boost the expression of Ca2+-reabsorptive gene products. Also, we isolated mRNA from the duodenum and cecum of the rat intestine, the sites where Ca2+ absorption primarily occurs. A twofold increase in radiotracer uptake was observed in oocytes injected with mRNA samples from the duodenum and cecum compared to control uninjected oocytes. We then fractionated the duodenal mRNA by preparative gel electrophoresis and tested the ability of the mRNA samples of different size fractions to stimulate Ca2+ uptake. An increase in Ca2+ uptake was observed in a 3 kb fraction. This fraction was used to make a cDNA library, and the library was divided into smaller pools of about 300 clones. Plasmid DNA was isolated from each pool and reverse-transcribed into capped cRNA. These cRNAs were then tested for their ability to stimulate Ca2+ uptake in injected oocytes. Once a positive pool was identified, it was divided into even smaller pools, and the earlier mentioned process was repeated in an iterative way, until a single positive clone was identified [2]. A similar approach was used to identify TRPV5 from rabbit kidney [1]. Because the distal tubule represents a very short portion of the nephron, cells from CT and cortical collecting ducts were isolated from rabbit kidney using a monoclonal antibody against these tubule segments to obtain enough mRNA for expression cloning of TRPV5 [1].
The identified rabbit TRPV5 and rat TRPV6 cDNAs encode proteins of 730 and 727 amino acid residues, respectively. In vivo evidence exists that the translation of TRPV6 protein may start from an upstream non-AUG codon. This does not change the TRPV6 function but may provide an additional scaffold for channel assembly [43]. TRPV5 was originally named epithelial Ca2+ channel (ECaC) [1] and TRPV6 Ca2+ transport protein subtype 1 (CaT1) [2]. The two proteins share 74% amino acid sequence identity. At the time of cloning, only two cloned transport proteins shared amino acid sequence similarity to TRPV5 and TRPV6: the capsaicin receptor TRPV1, a heat-activated cation channel that contributes to the sensations of pain and temperature [44], and OSM-9, a Caenorhabditis elegans protein involved in olfaction, mechanosensation, and olfactory adaptation [45]. The mammalian TRPV family now includes six members, whereby TRPV5 and TRPV6 share approximately 40%–45% amino acid sequence identity with the other four members [8]. While TRPV5 and TRPV6 represent specialized epithelial Ca2+ transport proteins, the other four members are nonselective cation channels that serve as sensors and can be activated by physical cues such as heat (TRPV1-TRPV3) or tonicity (TRPV4) [46].
13.4. Ca2+ Transport Properties Uncovered by Various Approaches
13.4.1. Functional Expression in Xenopus Oocytes
As both channels were identified by expression cloning using 45Ca2+ uptake assays in cRNA-injected oocytes to measure channel activity, the same assays were also utilized to uncover the basic transport properties of TRPV5 and TRPV6 [1,2]. In these assays, oocytes were first injected with cRNAs encoding the Ca2+ channel of interest, and 2–3 days later, when the channel protein expression reached stable levels, 45Ca2+ uptake assays and/or voltage-clamp experiments were performed. The uptake assay involved incubation of groups of oocytes (typically 7–9 oocytes/group) in an uptake solution containing 45Ca2+. Uptake measurements were performed during the initial uptake period, when the influx of Ca2+ was still in the linear range, typically within 30 min. The transport process was then terminated by removing the uptake solution and washing the oocytes with ice-cold solution 3–6 times, in order to remove 45Ca2+ nonspecifically absorbed to the oocyte surface. Individual oocytes were then lysed in 10% SDS solution, and the amount of 45Ca2+ in the oocytes was determined using scintillation counting. The amount of Ca2+ that entered the oocyte was calculated based on the level radiation in counts per minute (CPM) in oocyte lysates, following normalization against results from standard solutions with known 45Ca2+ values.
13.4.2. Characterization of TRPV5 and TRPV6 at the Macroscopic Level
The 45Ca2+ uptake experiments revealed the properties of TRPV5 and TRPV6 at the macroscopic level. TRPV5- and TRPV6-mediated Ca2+ uptake exhibited saturation kinetics, with Km values for Ca2+ in the submillimolar range [1,2,11,47]. The Km values for Ca2+ were 0.2 mM for rabbit and 0.66 for rat TRPV5 [1,47]. Those for TRPV6 were 0.44 mM for rat and 0.25 mM for human TRPV6 [2,11]. [Ca2+]o that is normally present in the kidney DCT and in the intestinal lumen varies from submillimolar to millimolar ranges. Thus, given the Km values of renal TRPV5 and intestinal TRPV6 for Ca2+, these channels would be well suited to facilitate optimal Ca2+ uptake across the apical membranes.
TRPV5 and TRPV6 were found to be sensitive to protons and metal ions such as La3+, Gd3+, Cu2+, Pb2+, and Cd2+ [1,2]. The pH sensitivity of TRPV5 and TRPV6 may contribute to the elevated serum Ca2+ level in milk-alkali syndrome and the increased Ca2+ excretion in metabolic acidosis.
Subsequent voltage-clamp techniques allowed further measurements of the charge fluxes through these channels, controlling the voltage across the plasma membrane of Xenopus oocytes expressing TRPV5 or TRPV6. The Xenopus oocyte with a diameter of ∼1.2 mm is very suitable for a two-microelectrode voltage clamp, in order to determine transporter and channel characteristics, provided that the transport is accompanied by net electric charge movement. Two glass Ag-AgCl microelectrodes filled with 3 M KCl are inserted into an oocyte and connected to a commanding and recording system, allowing the voltage across the oocyte membrane to be controlled and the current flux to be measured accurately [48]. Employing this method, we demonstrated that Ca2+, Ba2+, and Sr2+, but not Mg2+, elicit measureable currents in oocytes expressing TRPV6 [2]. This approach also revealed that Na+ can permeate through TRPV6 in the absence of extracellular Ca2+ [2]. As a follow-up to this observation, Na+ currents were measured under divalent-free conditions with the patch-clamp technique. By measuring 45Ca2+ influx in parallel, using voltage-clamped TRPV6-expressing oocytes, the influx of each Ca2+ ion was shown to be accompanied by the movement of two positive charges across the membrane [2]. This confirmed the hypothesis that Ca2+-induced currents are solely due to Ca2+ passing through the channel, and not by movement of other ions.
13.4.3. Characterization of TRPV5 and TRPV6 by Patch Clamping
The properties of TRPV5 and TRPV6 at a microscopic level were investigated in great detail using the patch-clamp technique, which is discussed in more detail elsewhere in this book (see Chapter 1). Briefly, Na+ currents of TRPV5 were detected when divalent cations (e.g., Ca2+ and Mg2+) were removed from the extracellular solution [49]. Unitary Na+ conductance of TRPV5 and TRPV6 was subsequently determined [50,51]. Both TRPV5 and TRPV6 turned out to be highly selective for Ca2+, with Ca2+ to Na+ permeability ratios (PCa:PNa) of over 100 for both channels [50,51]. Together with site-directed mutagenesis studies, the key residue for Ca2+ permeation and Mg2+ blockade was identified as aspartate 542 in TRPV5 [52,53] (shown in Figure 13.2 as the site for Ca2+ permeation).

Figure 13.2
Model of the human TRPV5 channel based on the rat TRPV6 structure (PDB ID: 5IWK). (a) Tilted sideways view with one monomer of the tetrameric unit shaded in blue. Approximate position of the membrane is indicated. (b) Cutaway view of two oppositely positioned (more...)
13.4.4. Feedback Control Mechanisms
Both TRPV5 and TRPV6 exhibit Ca2+-dependent inactivation as feedback control mechanisms. However, differences exist between the two channels in this regard. The inactivation mechanism involves the intrinsic channel structure, interaction with Ca2+-binding proteins, and PI(4,5)P2 phospholipid. While the PI(4,5)P2-mediated inactivation mechanism is similar between TRPV5 and TRPV6, there are differences in the intrinsic inactivation kinetics and interactions with Ca2+-binding proteins. TRPV6 has a fast inactivation mechanism, compared to TRPV5 [54]. The first intracellular loop between transmembrane domains 2 and 3 is the molecular determinant of this mechanism [54]. The slower phase of Ca2+-dependent inactivation involves Ca2+/CaM binding (see Figure 13.2b). Flockerzi and colleagues showed that a CaM-binding site is present in the C-terminal of TRPV6 [55]. CaM binding mediates a slow phase of Ca2+-dependent inactivation of TRPV6 [55–57]. The interaction between Ca2+/CaM and TRPV6 increases dynamically as the intracellular Ca2+ level rises [56]. The CaM-binding site is not conserved between the two channels, and several CaM-binding sites were identified in TRPV5 [58,59]. Thus, the kinetics of the Ca2+/CaM interaction with TRPV5 is likely more complex. In addition, the vitamin D-regulated Ca2+-binding protein calbindin-D28K binds Ca2+ that enters through TRPV5 [26]. This may further prevent inactivation of TRPV5. TRPV5 activity is also regulated by a Ca2+ sensor, 80K-H [60] (Figure 13.2b).
The common mechanism for TRPV5 and TRPV6 inactivation involves PI(4,5)P2. Using the patch-clamp technique, Huang and colleagues demonstrated the regulation of TRPV5 by PI(4,5)P2 [61]. They found that PI(4,5)P2 activates TRPV5 and reduces Mg2+ blockade [61]. PI(4,5)P2 is necessary for maintaining TRPV5 and TRPV6 activity and appears to prevent Ca2+-dependent inactivation [61–64]. As the levels of PI(4,5)P2 decrease, the activities of TRPV5 and TRPV6 also decline [61,63]. Ca2+ influx as a result of channel activation stimulates phospholipase C (PLC), which in turn hydrolyzes PI(4,5)P2, leading to the reduction of channel activity [63]. As for the molecular site of PI(4,5)P2 binding, Rohacs and colleagues found that arginine 599 in mouse or rat TRPV5 (corresponding to R606 in rabbit and human TRPV5) in the “TRP domain” is likely the PI(4,5)P2-binding site [65] (Figure 13.2b). TRPV6 lacks a positively charged arginine residue between transmembrane domains 4 and 5 in TRPV1 that is important for high-affinity binding of PI(4,5)P2 in TRPV1 [66,67]. Introducing this arginine residue into TRPV6 produced high-affinity binding of PI(4,5)P2 in TRPV6 and abolished Ca2+-dependent inactivation of the channel [67]. Thus, the requirement of PI(4,5)P2 for function and low-affinity binding of PI(4,5)P2 are key features of TRPV6 (and likely of TRPV5 as well). Ca2+-dependent hydrolysis of PI(4,5)P2 by PLC underlies the Ca2+-dependent inactivation mechanism of these channels.
13.4.5. Ca2+ Imaging
Ca2+ imaging, an approach to study Ca2+ signaling, has been utilized for screening small-molecule inhibitors of TRPV6 [68]. Binding of Ca2+ ions to a fluorescent dye results in either an increase in fluorescence intensity or changes in emission/excitation wavelengths. When cells are loaded with a Ca2+-indicator dye, a rise in intracellular Ca2+, either by release from internal Ca2+ stores or by influx through Ca2+-permeant channels, can be captured with a camera. Bolanz and colleagues utilized Ca2+ imaging to study the inhibitory effect of tamoxifen on TRPV6 [69]. The same approach can also be utilized for high-throughput screening of inhibitors for TRPV6.
Employing the voltage-clamp approach, we found that Cd2+, which is a potent blocker of TRPV5-mediated 45Ca2+ uptake, is actually permeant through the TRPV5 and TRPV6 channels and generates current just like Ca2+ [47]. This forms the foundation for screening small-molecule modulators for TRPV5/6 using the FLIPR Tetra high-throughput cellular screening system (Molecular Devices), using the FLIPR Calcium 5 Assay Kit.
13.4.6. Additional Experimental Considerations
As shown in Figure 13.3, all of the aforementioned approaches have been utilized in evaluating channel function and regulation of TRPV5 and TRPV6. While radiotracer 45Ca2+ uptake is a sensitive approach that directly measures Ca2+ influx, it cannot reveal detailed kinetics of channel activity. Two-electrode voltage-clamp studies reveal charge movement and voltage-dependence of membrane transport processes. However, its application is mostly limited to Xenopus oocytes. The patch-clamp approach offers detailed channel activity measurements, and it is also well suited to uncover the mechanisms of channel regulation. However, most patch-clamp experiments of TRPV5 and TRPV6 were done in divalent cation-free bathing solutions. Such experimental conditions might miss channel regulatory mechanisms that are Ca2+ dependent, for example. Furthermore, single-channel activities using Ca2+ as charge carrier have not been demonstrated for TRPV5 or TRPV6 so far. Nevertheless, data from this array of techniques are complementary and provide a well-documented understanding of the biophysical properties and regulatory aspects of TRPV5 and TRPV6.
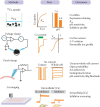
Figure 13.3
Major approaches to assess TRPV5 and TRPV6 function.
13.4.7. Crystal Structure of Rat TRPV6
The recently reported crystal structure of rat TRPV6 at 3.25 Å resolution [70] (discussed in detail in Chapter 14) allows us to view the functionally important regions of the protein in the context of its 3D structure. In Figure 13.2, we show a structural model of the tetrameric human TRPV5 channel that we built using the Rosetta modeling suite [71–73], with the rat TRPV6 structure as a template. Residues shaping pore selectivity, gating, and inactivation are highlighted, as well as interaction sites with CaM. While some residues cluster near the pore, the TRP helix also appears to be an important structural integrator of various processes modulating TRPV5 channel function.
13.5. Physiological Roles Revealed Using Genetically Engineered Animal Models
Genetically engineered mouse models were instrumental for exploring the physiological roles of TRPV5 and TRPV6. These include gene knockout (KO) models for TRPV5 [14] and TRPV6 [15], chemically induced gene mutations in TRPV5 [74], transgenic overexpression of TRPV6 in the small intestine [75], and a knock-in mouse model of TRPV6 [17]. These animal models and the findings from these models are summarized in the following sections.
13.5.1. TRPV5 and TRPV6 in Ca2+ Absorption and Reabsorption
Bindels and colleagues developed the TRPV5 gene KO mice with conditional potential. However, since TRPV5 expression is fairly restricted to the DCT and CNT, tissue-specific KO was unnecessary [14]. Our group developed a global TRPV6 KO mice [14]. The common features of the TRPV5 and TRPV6 KO mice include elevated 1,25(OH)2D3 serum levels, increased urinary Ca2+ excretion, and a certain degree of bone deficiency [14,15]. Urinary Ca2+ excretion increased sixfold in TRPV5 KO mice [14]. Intestinal Ca2+ absorption decreased in KO mice [15] but increased in TRPV5 KO mice as duodenal TRPV6 and calbindin-D9K mRNAs are upregulated due to elevated serum 1,25(OH)2D3 levels [14]. Interestingly, the serum Ca2+ levels of the TRPV5 and TRPV6 KO mice were largely normal, although the elevated PTH levels in TRPV6 KO mice [15] and in older TRPV5 KO mice [76] are an indication of a subtle reduction in serum Ca2+ levels that may have occurred in these mice. The elevated levels of 1,25(OH)2D3 are in further agreement with the overall deficiency in Ca2+ homeostasis in these mice. The increased urinary Ca2+ excretion in TRPV6 KO mice suggests that TRPV6 also plays a role in Ca2+ transport in the kidney. More recently, a chemical-induced S682P mutation of TRPV5 caused autosomal dominant hypercalciuria much like what was observed in the TRPV5 KO mice [74]. This confirms a key role of TRPV5 in regulating urinary Ca2+ excretion.
13.5.2. TRPV6 as a Central Component in Vitamin D-Regulated Active Ca2+ Absorption
While TRPV6 is considered the key apical channel for active Ca2+ absorption, a certain level of active Ca2+ absorption also occurs in the absence of TRPV6 [77]. Furthermore, the stimulating effect of 1,25(OH)2D3 on intestinal Ca2+ absorption was still partially present in TRPV6 KO mice [78]. This raised the question as to what extent vitamin D-regulated transcellular Ca2+ absorption depends on TRPV6 expression. To address this question, Fleet and colleagues developed a transgenic mouse model in which intestinal epithelium-specific expression of human TRPV6 is driven by the villin gene promoter [75]. This approach directly demonstrated that TRPV6 functions as an apical Ca2+ transporter and intestinal absorption was elevated 300% in mice with intestine-specific transgenic expression of TRPV6 (TRPV6TG). In this study, TRPV6TG and VDRKO mice were also crossed to generate TRPV6TG-VDRKO mice.
The TRPV6TG mice on chow diet (0.72% calcium) exhibited elevated serum and urinary Ca2+ levels, poor growth, and soft tissue calcification. Lowering the calcium level in the diet to 0.25% normalized the serum Ca2+ levels. Femur bone mineral density of TRPV6TG mice was increased by 28% compared to normal mice. The 1,25(OH)2D3 level was reduced by 56% likely due to the 98% lower CYP27B1 mRNA in the kidney of TRPV6TG mice. The reduced CYP27B1 mRNA level is likely a result of reduced PTH levels (46% lower). The phenotype of TRPV6TG mice is largely opposite to that of the TRPV6 KO mice, confirming the importance of TRPV6 in the intestine facilitating vitamin D-regulated transcellular Ca2+ absorption.
The key regulator of intestinal Ca2+ absorption is 1,25(OH)2D3. TRPV6 expression is strongly regulated by vitamin D as the TRPV6 gene promoter has multiple vitamin-responsive elements [79]. In VDRKO mice, duodenal TRPV6 mRNA is reduced down to 5%–10% compared to wild-type mice [21,80]. VDRKO mice fed a diet with slightly reduced calcium content (0.5% calcium, standard commercial chow diet contained 0.72% calcium) for 10 weeks were hypocalcemic, with drastically elevated serum 1,25(OH)2D3 and PTH levels, reduced bone mass, and decreased intestinal Ca2+ absorption [75].
In TRPV6TG-VDRKO mice, the serum Ca2+ remained elevated when kept on 0.5% calcium diet, serum 1,25(OH)2D3 and PTH levels were very close to that of the wild type, bone mass was close to that in the TRPV6TG mice, and intestinal Ca2+ reabsorption remained elevated [75]. Thus, overexpression of TRPV6 in the intestine is sufficient to overcome the deficiency caused by the reduction of intestinal Ca2+ absorption in the absence of VDR. The results of this study also show that, once TRPV6 is restored, other components relevant to transcellular Ca2+ transport such as levels of calbindin-D9K, 1,25(OH)2D3, and PTH are normalized in the absence of VDR [75]. Taken together, given the results from studies of TRPV6 KO, VDRKO, and TRPV6TG mice (Figure 13.4), TRPV6 is likely a key element in vitamin D-regulated Ca2+ transport. Given the importance of Ca2+ absorption, it is also unlikely that TRPV6 is the only Ca2+ channel for intestinal Ca2+ absorption. The presence of a vitamin D-regulated active Ca2+ transport mechanism in the intestine in mice lacking TRPV6 might be due to physiological adaptation, that is, upregulation of alternative, compensatory Ca2+ transport pathways. Also epigenetic and genetic alterations in the TRPV6 KO mice cannot be ruled out. The abovementioned studies suggest that an alternative transcellular Ca2+ entry pathway might exist in the intestine that is regulated by 1,25(OH)2D3. Further studies will be required to identify this putative additional transport mechanism in the intestinal brush border membrane.

Figure 13.4
Ca2+ homeostasis in mice with genetically engineered TRPV6 and VDR. Shown are changes in Ca2+ homeostasis-related phenotypes including serum levels of 1,25(OH)2D3 and PTH, intestinal Ca2+ absorption, renal Ca2+ reabsorption, and bone mineral density in (more...)
13.5.3. Role of TRPV6 in Maternal-Fetal Ca2+ Transport
The placenta transports Ca2+ from mother to fetus in order to maintain fetal bone mineralization [29,81]. It has been reported that blood [Ca2+] is higher in fetus compared to the mother during late pregnancy, when fetal bones are mineralized, suggesting that transcellular active Ca2+ transport is needed for fetal bone mineralization. Indeed, some Ca2+ deficiency symptoms in fetuses and neonates are thought to be derived from insufficient maternal-fetal Ca2+ transport in the placenta. However, the molecular identity of this process, as well as its mechanism of regulation, has, until recently, not been fully clarified. It was found that both TRPV5 and TRPV6 are expressed in the placenta [11,13,30,82,83]. However, TRPV6 mRNA levels are ∼1000 times higher than those of TRPV5 in human placenta [8]. Specifically, TRPV6 is expressed in trophoblasts and syncytiotrophoblasts of the human placenta [13]. TRPV6 in human syncytiotrophoblasts is associated with cyclophilin B, a member of the immunophilin family [84]. The physiological meaning of this association is unclear, but cyclophilin B increased TRPV6 activity in vitro, suggesting that the two proteins may act together in placental Ca2+ transport.
To elucidate the role of TRPV6 in placental Ca2+ transport, we examined the maternal-fetal Ca2+ transport pathway in TRPV6 KO mice [16]. The maternal-fetal 45Ca2+ transport activity was decreased by 40% in TRPV6 KO fetuses. TRPV6 KO mice exhibited a dramatic decrease in blood Ca2+ levels and ash weight in fetuses at 18 days post-fertilization (immediately before birth) [16]. These observations indicate that the fetal Ca2+ deficiency was caused by decreased maternal-fetal Ca2+ transport in the absence of TRPV6 [16].
A zebrafish loss-of-function TRPV6 mutation called “matt-und-schlapp” generated a 68% reduction of total body calcium content, with bone mineral defects, triggered by a marked reduction in Ca2+ uptake by the yolk sac and the gills [85]. It was found that the mutant possesses R304Stop before the transmembrane domain 1 (S1) of the TRPV6 gene. These results suggest that the role of TRPV6 in the Ca2+ transport in extraembryonic tissues involved in fetal bone formation is conserved among vertebrates.
13.5.4. Role of TRPV6 in Male Fertility
The reduced fertility in TRPV6 KO mice was another interesting observation [15]. The underlying mechanism was unclear until Weissgerber and colleagues observed that male fertility in mice depends on TRPV6-mediated Ca2+ absorption aimed at reducing the luminal Ca2+ levels in the epididymis [17]. This group generated a Trpv6D541A/D541A knock-in mutant mouse, whereby D541 corresponds to D542 in TRPV5, a key residue for Ca2+ permeation in TRPV6 [52]. The D541A mutation renders TRPV6 incapable of transporting Ca2+. While female Trpv6D541A/D541A mice were normal in fertility, male homozygous knock-in mice rarely impregnated females despite normal copulatory behavior. There was more than 50% reduction in the number of caudal sperm measured by the “swim out” method in Trpv6D541A/D541A mice. Most capacitated caudal sperm was immotile or had impaired motility. The ability of capacitated caudal sperm from knock-in mice to fertilize oocytes in vitro was reduced to 14.4%. A closer examination revealed that TRPV6 is expressed in the apical membrane of epididymal epithelia. [Ca2+]o determined by retrograde perfusion in the epididymal duct fluid of Trpv6D541A/D541A mice was 10 times that of wild-type littermates, and this matched the reduction of 45Ca2+ uptake in the epididymis of Trpv6D541A/D541A mice. Thus, experimental results from this study indicate that blocking TRPV6 function in the epididymis increased [Ca2+]o in the epididymal fluid, which is essential for sperm motility. This study shows that controlled [Ca2+] in the epididymal fluid by TRPV6 is critical in producing vital and healthy sperm cells. Raised [Ca2+] in the epididymal fluid in Trpv6D541A/D541A mice likely initiates premature motility of sperm cells and exhausts the energy that is needed for the motility after they exit the epididymis. The authors also examined this question in mice lacking Trpv6 and reached essentially the same conclusion [86].
Recent patch-clamp studies confirmed that the constitutive Ca2+ currents resemble that of TRPV6 in rat cauda epididymal principal cells [87]. The Ca2+-activated chloride channel transmembrane member 16A (TMEM16A) is coexpressed with TRPV6 in the apical membrane of principal cells, and functional coupling between TMEM16A and TRPV6 likely plays a role in Ca2+ absorption in the epididymis [87].
13.6. Proteins that Regulate TRPV5 and TRPV6
A number of proteins have been identified that interact and/or regulate TRPV5 and TRPV6 (see examples in Figure 13.2b). These proteins were identified because (1) they interact with TRPV5 or TRPV6 as detected by yeast two-hybrid screening, (2) they are expressed in the same tissues or cells, or (3) they are relevant to Ca2+ absorption or reabsorption in a physiological or pathological context [3]. Most of these proteins were identified for TRPV5. However, since TRPV6 and TRPV5 are structurally and functionally similar, they generally have analogous roles in regulating TRPV6, provided that they are adequately coexpressed.
13.6.1. Proteins That Interact with TRPV5 and TRPV6
Several TRPV5-interacting proteins were identified by Bindels and colleagues using the yeast two-hybrid approach, with the C-terminal region of TRPV5 as bait. A group of TRPV5-interacting protein, such as S100A10 [88], Rab11a [89], and NHERF2 [90–92], regulates the trafficking and plasma membrane abundance of TRPV5. Another group of TRPV5-interacting protein, including calbindin-D28K [26], CaM [58,59,93], 80H-K [60], BSPRY [94], and FKBP52 [95], also regulates TRPV5 activity. An interesting observation is that TRPV5 interacts with most of its binding partners through the region lying between 598 and 608 (human TRPV5 numbering) within the conserved “TRP domain” (Figure 13.2b). These include S100A10 [88], Rab11a [89], 80K-H [60], NHERF4 [96], and one CaM [59]. It is worth mentioning that the TRP domain that follows the sixth transmembrane domain also contains a PI(4,5)P2-binding site [65] and an intracellular pH sensor [97,98]. Histidine 711 (H712 in rabbit TRPV5), a critical residue involved in the constitutive internalization of TRPV5 [99], is also located in close proximity to the sites that interact with other proteins. It is very likely that TRPV5 function, membrane stability, and/or trafficking are affected when different binding partners interact with this channel. It is unclear whether all of the binding partners identified by the yeast two-hybrid approach or other in vitro approaches are physiologically relevant. Also, it is unlikely that all TRPV5-binding proteins regulate TRPV5 simultaneously. Some of the interactions may occur more often and some may occur at specific developmental or physiological stages. As already noted earlier, given that most of the interaction sites are conserved in TRPV5 and TRPV6 channels, it is possible that some of these interactions with TRPV5 are also relevant for TRPV6 in cells that express both TRPV6 and the interacting proteins.
13.6.2. Proteins That Are Physiologically Relevant to TRPV5 and TRPV6
Some proteins that regulate TRPV5 and TRPV6 were identified on the basis of physiological or pathological relevance. Because TRPV5 is fairly highly expressed in the DCT and CNT, proteins that are expressed in the DCT and associated with calcium homeostasis are possible candidates that regulate the TRPV5 channel.
An example is the study of the effect of the antiaging hormone Klotho on TRPV5 by Chang and colleagues [100]. These investigators showed that Klotho acts on the N-glycan moiety of the first extracellular loop of TRPV5 (see Figure 13.2b), thereby stimulating TRPV5-mediated Ca2+ uptake and stabilizing TRPV5 in the plasma membrane. This discovery revealed novel mechanisms of ion channel regulation [101–103]. This effect is dependent of the N-glycosylation state of TRPV5, since the N-glycosylation mutant TRPV5N358Q was not activated by Klotho. Subsequently, tissue transglutaminase was found to inhibit TRPV5 through a reduction of the pore diameter in a glycan-dependent manner [104]. Both, Klotho and tissue transglutaminase, act on TRPV5 through its N-glycan moiety, but in the opposite direction.
The novel regulation of TRPV5 by Klotho inspired a search for more relevant proteins that regulate TRPV5 from the extracellular side. As TRPV5 in the distal tubule faces proteins in the tubular fluid, some tubular proteins for which there was already evidence that they affect Ca2+ reabsorption were tested for their effects on TRPV5 function. Plasmin, a serine protease that is present in the nephrotic syndrome, suppresses TRPV5 through activating the protease-activated receptor-1 (PAR-1) [105]. The activation of the PAR-1/PLC/PKC pathway leads to altered CaM binding to TRPV5 and subsequent reduction in the pore size and open probability [105].
Wolf and colleagues found that the urinary protein uromodulin (Tamm-Horsfall glycoprotein/THP) and mucin-1 act extracellularly and decrease caveolin-mediated endocytosis of TRPV5 and thus enhance overall channel activity [106,107]. Because mutations of the uromodulin gene are associated with kidney stone disease, and mucin-1 protein is decreased in patients with hypercalciuric nephrolithiasis, it is tempting to speculate that uromodulin and mucin-1 prevent kidney stone formation in part through enhancing TRPV5 activity.
Other candidate proteins that may regulate TRPV5 function were identified because their gene mutations result in diseases associated with hypercalciuria. Studies of regulation of TRPV5 by the serine/threonine protein kinase WNK4, for example, were initiated because hypercalciuria was observed in pseudohypoaldosteronism type II (PHAII, also known as familial hyperkalemia and hypertension or Gordon’s syndrome) in patients with the WNK4Q565E mutation [108]. Jiang and colleagues found that WNK4 increases the activity of the TRPV5 channel by increasing its forward trafficking to the plasma membrane via the secretory pathway in Xenopus oocytes [109,110]. This regulation is enhanced and stabilized by the sodium-hydrogen exchange regulatory cofactor NHERF2 [92]. However, diseases causing mutations in WNK4 did not abolish the regulation of TRPV5 by WNK4. WNK3, another member of the WNK family, enhances both TRPV5 and TRPV6 [111].
Genetic mutations of the inositol polyphosphate-5-phosphatase gene OCRL are associated with Dent’s disease and Lowe syndrome (oculocerebrorenal syndrome), which often manifest hypercalciuria [112–114]. OCRL appears to inhibit TRPV6 by reducing the PI(4,5)P2 levels and affecting its trafficking [115]. OCRL mutations associated with hypercalciuria impaired the inhibitory effect of OCRL on TRPV6. This suggests that hyperabsorption of Ca2+ in the intestine due to elevated TRPV6 activity may contribute to hypercalciuria in patients with these OCRL mutations. The OCRL-mediated regulation of TRPV6 likely overrides other conditions (e.g., proteinuria) in Dent-2 disease in the kidney.
Additionally, as a follow-up to the hypercalciuria phenotype observed in mice lacking the serine protease called tissue kallikrein, it was found that tissue kallikrein stimulates Ca2+ reabsorption via PKC-dependent plasma membrane accumulation of TRPV5 and that its genetic defect delays retrieval of TRPV5 from the plasma membrane [116]. Furthermore, the activity of TRPV5 may be regulated by the CaSR. The activation of CaSR increases TRPV5-mediated currents and elevates [Ca2+]i in cells coexpressing TRPV5 and CaSR [117], providing an interesting type of regulation. Finally, TRPV5 and TRPV6 may be degraded in part through the ubiquitin E3 ligase Nedd4-2 and Nedd4, as demonstrated in vitro [118].
Thus, a variety of proteins have been found to regulate TRPV5 and TRPV6 using various approaches. It is unclear at this stage whether all of them are relevant in vivo. Therefore, the physiological significance of these regulatory pathways is awaiting further evaluation.
13.7. Potential Use of TRPV6 in Therapy and Development of Chemical Modulators
13.7.1. Evaluation of TRPV6 as a Therapeutic Target
Ca2+ regulates a wide array of physiological functions including cell differentiation, proliferation, muscle contraction, neurotransmission, and fertilization [37–39,119,120]. Several Ca2+ channels, pumps, Ca2+-binding proteins, and regulatory proteins work in concert to fine-tune cellular Ca2+ homeostasis. Thus, intracellular Ca2+ homeostasis plays a critical role in the regulation of growth, proliferation, survival, and apoptosis of normal and malignant cells. TRPV6 is a highly selective Ca2+channel that is, in general, thought to be constitutively active [51]. As already noted, TRPV6 mediates Ca2+ uptake in the duodenum where it is located within the apical membrane of intestinal enterocytes, and there is additional expression in epithelial tissues that bear tumors frequently [12]. Animal models indicate that alterations in TRPV6 expression and function lead to pathophysiological Ca2+ conditions, disruptions of renal transport, hypercalciuria, renal stone formation, preeclampsia, and osteoporosis [6]. Expression of TRPV6 is elevated in prostate, breast, colon, esophageal, and cervical tumor tissues, as well as in tumor cell lines, and increased expression stimulates the metastasis of cancer cells and confers chemotherapy resistance [4,12,121–129].
13.7.1.1. TRPV6 in Prostate and Breast Cancer
TRPV6 expression levels were shown to be upregulated in tissue samples originating from prostate, breast, thyroid, colon, ovarian, and pancreatic tumors [12,126]. Prostate and breast cancer are among the most lethal tumors in humans [130,131], yet the mechanisms underlying the development of aggressive tumor phenotypes are not well understood.
TRPV6 expression levels in prostate cancer are linked to tumor progression and have been proposed as a prognostic marker for tumor progression [132]. In the healthy prostate tissue and in benign prostate hyperplasia, TRPV6 is expressed at low levels, while high expression levels arise in prostate adenocarcinoma. TRPV6 mRNA levels were shown to significantly correlate with the Gleason grade score of prostate adenocarcinoma, and TRPV6 was found to be expressed in lymph node metastases of prostate origin [13,126,132]. Increased expression of TRPV6 mRNA is present in the human prostate cancer cell lines (LNCaP and PC-3) as compared to the normal and benign epithelial cells (PrEC and BPH-1) [126]. The androgen receptor (AR) agonist dihydrotestosterone was shown to inhibit TRPV6 expression in LNCaP cells while the AR antagonist bicalutamide (Casodex) increased TRPV6 expression [126,133,134]. TRPV6 expression has not been identified in the androgen-insensitive prostate cancer cell lines DU-145 and PC-3 [132].
Downregulation of TRPV6 in LNCaP cells using specific siRNA induces growth inhibition and S-phase cell cycle arrest and inhibits proliferating cell nuclear antigen expression [135]. The overexpression of TRPV6 in the LNCaP cell line led to increased resistance to apoptosis induced either by the SERCA Ca2+ pump inhibitor thapsigargin or DNA cross-linking by the chemotherapy drug cisplatin.
Recent studies by Raphael and colleagues indicate that TRPV6 is likely a tumor promoter in the prostate, functioning as a component of store-operated Ca2+ entry (SOCE) in these cells [127]. Being upregulated de novo, it significantly contributes to prostate cancer cell survival through proliferation and apoptosis resistance, promotes the formation of bone metastases, and potentiates tumorigenesis in vivo.
TRPV6 is also highly expressed in breast adenocarcinoma tissues and breast cancer cell lines (MDA-MB-231, MDA-MB-468, T47D, SKBR3, MCF-7) [69,136,137]. In vitro studies using the T47D cell line showed that TRPV6 can be regulated by estrogen, progesterone, tamoxifen, and 1α,25-dihydroxyvitamin D3 (vitamin D3), indicating that these agents affect breast cancer cell proliferation [136]. A recent study revealed that TRPV6 is mainly overexpressed in invasive breast cancer cells and that selective silencing of TRPV6 inhibited MDA-MB-231 migration and invasion, as well as MCF-7 migration [138].
TRPV1 agonist capsaicin has also been reported to upregulate TRPV6 protein expression levels and Ca2+ influx in AGS gastric cancer cell line and confers TRPV6-dependent proapoptotic activity [139]. A similar role of TRPV6 in capsaicin-induced apoptosis has been reported in small-cell lung carcinoma cell line [140].
Taken together, these data indicate that inhibition of TRPV6 may offer a promising therapeutic strategy for the treatment of prostate and breast cancers. As alluded to earlier mentioned text, selective TRPV6 blockers might also be useful for the treatment of hypercalciuria in kidney stone patients [6]. The development of specific TRPV6 inhibitors is therefore of great interest as it might open the door toward the development of more efficient therapeutics for cancer treatment.
Here we present an overview of the initiatives taken toward the discovery of TRPV6 modulators.
13.7.2. Development of Chemical Modulators of TRPV6
13.7.2.1. Development of Antiproliferative Compound TH-1177
Strategies to inhibit Ca2+ entry into electrically non-excitable cells by small-molecule compounds were addressed by Gray and colleagues in the early 2000s [40]. These investigators synthesized compounds such as TH-1177 and its stereoisomer TH-1211 that share similarities with known Ca2+ channel blockers derived from the molecules dihydropyridine, benzothiazepine, and phenylalkylamine (Figure 13.5a; compounds 1 and 2). In vitro testing revealed that TH-1177 was effective in blocking Ca2+ influx in prostate cancer cells. Efforts toward the identification of the molecular target of TH-1177 revealed that the 25B splice variant of the Cav3.2 T-type Ca2+ channel (also known as CACNA1H) is inhibited by TH-1177, whereas its stereoisomer TH-1211 had no effect. Moreover, these investigators postulated that increased expression levels and activity of T-type Ca2+ channels are likely responsible for Ca2+ influx and stimulation of proliferation in cancer cells. Indeed, TH-1177 reduced the proliferative rate of human prostate cancer cells LNCaP and PC-3 in vitro, with a potency that correlates with its inhibition of Ca2+ influx. Furthermore, in xenograft studies, administration of TH-1177 to nude mice inoculated with PC-3 human prostate cancer cells led to a significant dose-dependent increase in longevity. Paradoxically, T-type Ca2+ channels are actually known to open during membrane depolarization and thus are expected to mediate Ca2+ influx into cells after an action potential or in response to depolarizing signals, whereas the epithelial Ca2+ channel TRPV6 channels would seem more appropriate to facilitate Ca2+ entry into cancer cells, as they are open at the negative resting potentials of these cells. Thus, while TH-1177 is well established as an inhibitor of Ca2+ entry in prostate cancer cells, it is reasonable to speculate that it might inhibit TRPV6 in these cells as well.
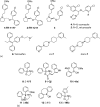
Figure 13.5
Chemical structures of known TRPV6 inhibitors: (a) heterocyclic compounds and (b) 2-APB analogs.
13.7.2.2. Development of TRPV6 Inhibitor “Compound #03”
To address the earlier question and to develop additional inhibitors of TRPV6 and also TRPV5, Landowski and colleagues synthesized compounds similar to TH-1177, (i.e., compounds #02, #03, #05, #06, and #09) [41]. Together with the antifungal agents econazole and miconazole that weakly inhibit TRPV6 [16,141,142] and TH-1177, these newly synthesized compounds were tested for their inhibitory effect on TRPV6 [41] in Xenopus oocytes expressing TRPV6 or TRPV5 and measuring 45Ca2+ influx. Figure 13.5a shows the chemical structure of “compound #03” (compound 3), as well as those of econazole and miconazole (compounds 4 and 5). The compounds were also tested using LNCaP prostate cancer and T47D breast cancer cells, studying their effects on proliferation. In LNCaP cells, TRPV6 is believed to be the most abundantly expressed Ca2+ entry channel, and it was shown that there is a negligible expression of TRPV5 in these cells [126]. Indeed, TH-1177 inhibited TRPV6 (and also TRPV5)-mediated Ca2+ uptake in oocytes, suggesting that the molecular target of TH-1177 in prostate cancer cells is TRPV6. However, since it is well established that TH-1177 mediates inhibition of Ca2+ entry via T-type Ca2+ channels and given that this compound also inhibits Ca2+ entry into cells that do not express TRPV6 [143], it is possible that, in prostate cancer cells, TH-1177 inhibits both T-type and TRPV6 Ca2+ channels.
Compound #03 of the study by Landowski and colleagues was found to be the most potent and selective inhibitor of TRPV6, significantly inhibiting prostate cancer cell proliferation in vitro, with an IC50 value of 0.44 μM [41]. Likewise, TRPV6-specific siRNAs were able to reduce proliferation. Indeed, it has been demonstrated that TRPV6 controls Ca2+ entry and proliferation in LNCaP cells (see Section 13.7.2.5 for further details).
13.7.2.3. Effects of Estrogen-Receptor Blocker Tamoxifen on TRPV6 Function
Looking for additional inhibitors, Bolanz et al. [69] demonstrated that the estrogen-receptor prodrug tamoxifen (Figure 13.5a, compound 6) that is currently used for treatment of breast cancer also inhibits TRPV6 in Xenopus oocytes, with an IC50 of 7.5 μM based on Ca2+ uptake studies. This finding is interesting as it may help explain why certain ER-negative breast tumors favorably respond toward tamoxifen treatment.
13.7.2.4. Ligand-Based Virtual Screening (LBVS) to Develop Improved TRPV6 Inhibitors
All of the abovementioned agents are still relatively weak TRPV6 inhibitors, and several of them, including econazole and miconazole, are rather unspecific. Thus, to push forward the development of more effective TRPV6 inhibitors, Reymond and colleagues employed the LBVS approach [68]. For this, the publicly available ZINC database was exploited (Department of Pharmaceutical Chemistry at the University of California, San Francisco), a curated collection of commercially available chemical compounds prepared especially for virtual screening, allowing commercially available compounds to be tested without having to synthesize them first. Specific LBVS tools that have been developed by Reymond and colleagues were used to enable efficient exploitation of the ZINC database [68]. The strategy was to initially search for analogs of the abovementioned TRPV6 inhibitors, that is, TH-1177, compound #03, econazole, miconazole, and tamoxifen. Starting with these inhibitors, a pharmacophore shape similarity search for analogs in the ZINC database was performed, followed by purchase of selected virtual hits, to form small, focused libraries of a few hundred compounds. These were then experimentally tested using HEK293 cells stably transfected with TRPV6, measuring Ca2+ or Cd2+ uptake using the fluorophore calcium-5 and the FLIPR Tetra microplate fluorescence reader, in order to identify positive hits. Hits were used for a second round of LBVS, and the resulting improved compounds hits were further optimized by chemical synthesis and structure-activity relationship studies.
The most effective inhibitor cis-22a (IC50 value of 0.12 μM) was further investigated (Figure 13.5a, compound cis-7). It showed high selectivity against other Ca2+ channels and related TRP targets. The antiproliferative activity of cis-22a on TRPV6 expressing T47D human breast cancer cells was determined and compared to that of SKOV3 ovarian carcinoma cells that do not express TRPV6. Treatment of T47D cells with cis-22a decreased cellular proliferation by 20% at 5 μM (IC50 = 25 μM), a concentration sufficient to block TRPV6-mediated Ca2+ influx, while SKOV3 cells were unaffected. By contrast, the less potent diastereomer, trans-22a (Figure 13.5a, compound trans-7), had no significant effect on proliferation in these cells lines. The selective but significantly smaller effect of cis-22a compared to siRNA knockdown (resulting in 50% reduction in cell growth) may suggest that TRPV6 expression affects cell growth by mechanisms other than reduced Ca2+ uptake. In addition, it is possible that the TRPV6 inhibitor only works if its expression in the plasma membrane is activated as part of SOCE, as recently described by Prevarskaya and colleagues [127]. Therefore, further studies are needed to evaluate the beneficial effects of this compound on tumor growth and metastasis formation in prostate and breast cancer.
13.7.2.5. Analogs of 2-Aminoethyl Diphenylborinate (2-APB) as Potential Modulators of TRPV6 Function
2-APB is known to influence SOCE via Orai Ca2+ channels with different effects in different cell lines. Recent studies revealed that 2-APB also affects TRP channels, including TRPV6.
As already alluded, Prevarskaya’s group demonstrated that TRPV6 translocates to the plasma membrane via an Orai1-mediated mechanism, controlling cancer cell survival [127]. According to their proposal, TRPV6 switches in prostate cancer cells from its constitutive activity to a store-operated mode. This switch is proposed to occur through STIM1, Orai1, and TRPC1-evoked translocation of TRPV6 to the plasma membrane, involving the Ca2+/Annexin I/S100A11 pathway. As TRPV6 is virtually absent in healthy prostate, it is upregulated de novo in prostate cancer cells, where it changes its constitutively active role, supplying intracellular Ca2+ in a store-operated mode that is used in cancer cells to increase cell survival.
To investigate whether 2-APB analogs modulate TRPV6 function, a SAR study was conducted by Lochner and colleagues [144] (see Figure 13.5b for the chemical structures of relevant analogs) in an attempt to develop synthetic analogs that are more potent and more specific for TRPV6, compared to SOCE. The synthesis of a small library of 2-APB analogs was made, and the effects of the compounds were tested in HEK293 cells. To test the effect on human TRPV6, transiently expressing HEK293 cells were used. To test the effect on SOCE, measurements were performed in HEK293 cells in the absence of TRPV6 and the compounds were administered together with 1 μM thapsigargin, in order to activate SOCE. Several 2-APB analogs were discovered that were more potent inhibitors of TRPV6 than 2-APB. However, in general, they inhibited SOCE as well. Analog 19b (Figure 13.5b; compound 13) was an exception as it exhibits an approximately 2.5-fold higher selectivity for TRPV6 compared to SOCE. It appears that if steric bulk around the central boron atom or rigidity is increased, for example, as in compounds 2j, 6a, 6b, and 11 (compounds 9–12 in Figure 13.5b), TRPV6 inhibitory activity is lost, while SOCE inhibitory activity is retained. The most beneficial effects were achieved when only one of the phenyl rings of 2-APB was replaced by a 2-methylthiophene (compound 19b).
Interestingly, 2-APB and its amino alcohol variants turned out to be rather unstable compounds since they rapidly hydrolyze and transesterify in solution, which complicates the analysis of their pharmacological effects and their use as therapeutic compounds.
Taken together, further studies are needed to evaluate the therapeutic potential of 2-APB analogs and to determine whether such compounds are suitable for developing therapeutic applications, targeting specifically the TRPV6 Ca2+ channel. An additional issue to be addressed may be cellular acidification caused by 2-APB at high concentrations [145].
13.8. TRPV5 and TRPV6 Mutations and Kidney Stone Diseases
Kidney stone disease is a common condition with an incidence of approximately 5%–10% in each population. Although it is known that the environmental factors such as Ca2+ intake are important for the stone formation, 40% of stone-forming patients have family history, suggesting that there should be genetic factors as well. Several lines of evidence including ones from a stone-forming rat (GHS rat) suggest that kidney stone disease is a polygenic disease. Hypercalciuria (higher urine Ca2+ level) is a common risk factor for stone formation [6].
There are three types of hypercalciuria: (1) absorptive hypercalciuria caused by excess amount of intestinal Ca2+ absorption, (2) resorptive hypercalciuria that is derived from abnormal bone metabolism, and (3) renal leak hypercalciuria caused by a decreased Ca2+ reabsorption in the kidney. However, a hypercalciuria patient may have abnormal Ca2+ absorption and reabsorption and bone resorption at the same time. Since TRPV5 is suggested to be the molecular identity of the apical Ca2+ entry pathway for final renal Ca2+ reabsorption, and TRPV5 KO mice exhibited renal leak hypercalciuria-like phenotypes [14], it was reasonable to predict that TRPV5 mutations cause kidney stone disease with a renal Ca2+ leak. However, Muller et al. reported nine families with no association between TRPV5 gene and autosomal dominant hypercalciuria [146]. This is probably because the renal leak mutations are normally recessive mutations. However, TRPV5 cannot be excluded as a candidate gene for hypercalciuria. The same group also investigated 20 renal leak hypercalciuric patients, and 4 nonsynonymous polymorphisms were found. However, no association study was done in this report [147]. Later on, other groups investigated 365 kidney stone patients in Taiwan and found that the frequency of R154H (rs4236480) polymorphism in the TRPV5 gene was higher in the patients with multiple stones [148], indicating that the TRPV5 gene is somehow linked to renal stone formation.
Recently, the association between kidney stones and a novel TRPV5 gene mutation (L530R) was identified [149]. A genome-wide association study (GWAS) of 2636 Icelanders with kidney stones led to the identification of defects of the following genes that are responsible for kidney stone formation: (1) ALPL (tissue nonspecific alkali phosphatase for maintaining urine pyrophosphate that inhibits stone formation), (2) SLC34A1 (Na/Pi cotransporter for renal phosphate reabsorption), (3) claudin-14 (CLDN14), (4) CaSR, and (5) TRPV5. Claudin-14 and CaSR are thought to be important for renal Ca2+ handling to excrete excess Ca2+ via the paracellular pathway. With respect to the L530R mutation in the TRPV5 gene, it might represent a loss-of-function mutation, because it is localized in the pore loop of the TRPV5 channel. Thus, so far only one mutation of the TRPV5 gene has been identified that is associated with kidney stone disease, most likely resulting in renal leak hypercalciuria.
There are no further reports thus far showing an association of TRPV5 gene mutations with other human diseases. However, with respect to single nucleotide polymorphisms, 154H and 563T were more frequently distributed among African population compared to that of 154R or 563A. Renkema and colleagues showed that there were no functional differences in these polymorphisms with respect to Na+ currents, Ca2+ currents, and [Ca2+]i-dependent inactivation in patch-clamp recordings [147]. On the contrary, when using the in Xenopus oocyte expression system, an increased 45Ca2+ uptake in the 563T variant was reported [150]. Computational modeling based on the structure of TRPV1 revealed that the A563T variation induces structural, dynamical, and electrostatic changes in the TRPV5 pore, providing structural insights into the altered function of this variant [151]. The 154H variant exhibited the lowest Ca2+ uptake activity among five TRPV5 variants; however, the data did not reach statistical significance compared to the reference 154R variant [150]. In preliminary experiments, we found decreased 45Ca uptake in the 154H variant but not in A563T, without changing membrane surface expression of TRPV5 proteins (Y. Suzuki et al., unpublished data). Interestingly, there appears to be an association between R154H and bone density of the femoral neck and the tibial epiphysis in adults, most likely due to impaired osteoclastic bone resorption (A. Pasch, Y. Suzuki, and M.A. Hediger, unpublished data). These results and also data from TRPV5 KO mice [152] suggest that variants of the TRPV5 gene are risk factors for impaired bone quality. Genetic variants of TRPV5 might contribute to differences in bone quality and strength among different populations.
Given the numerous evidence, including the study of TRPV6 KO mice that lack TRPV6 function results in impaired intestinal Ca2+ absorption [15], TRPV6 is believed to be a key component of apical Ca2+ entry during intestinal absorption of Ca2+ from food and drinking water. Therefore, it is reasonable to speculate that genetic variations of the TRPV6 gene are associated with absorptive hypercalciuria, one of the major risk factors of kidney stone formation. Indeed, this is the case: a gain-of-function haplotype of TRPV6 gene has been identified from sequencing of 170 Ca2+-stone formers in Switzerland [153]. The frequency of this haplotype was statistically higher in the Ca2+-stone formers compared to normal individuals, suggesting that the haplotype might be a risk factor for kidney stone patients with absorptive hypercalciuria. Another line of evidence that stems from molecular evolution highlights the functional significance of this haplotype: it has been reported that this very same haplotype reflects a positive selection during human evolution [154,155] and that this “derived TRPV6 form” provided an evolutionary advantage for the survival of humans in response to certain past environmental changes, when compared to the ancestral form. An improvement of [Ca2+]i-dependent inactivation has been suggested in the derived TRPV6 form, compared to the ancestral form [154], which is consistent with the observed increase in 45Ca uptake in heterologous expression systems [155].
It is interesting to note that TRPV6 KO mice exhibit hypercalciuria [15]. This suggests that TRPV6 might also be involved in renal Ca2+ reabsorption. In fact, TRPV6 mRNA level appears to be higher than that of TRPV5 in the human kidney [8]. Also, prominent TRPV6 protein expression has been detected in both the proximal tubule and distal tubule in human kidney [10]. African-Americans exhibit lower urinary Ca2+ excretion and lower incidence of kidney stone disease [156]. The high prevalence of the ancestral haplotype of TRPV6 gene in African descendants suggest that TRPV6 may play a role in increased Ca2+ reabsorption in African populations.
13.9. Concluding Remarks
A variety of approaches have been utilized to achieve a better understanding of Ca2+ transport in epithelial cells, starting with expression cloning and moving all the way to in-depth cellular and biophysical analysis, generation of genetically engineered mouse models of TRPV5 and TRPV6, 3D structural studies, and chemical approaches. The remarkable Ca2+-selectivity and distinct Ca2+-dependent inactivation mechanisms involving PI(4,5)P2 are examples of what patch-clamp techniques could achieve in understanding these channels. Genetically engineered animal models not only provided the expected roles of TRPV5 and TRPV6 in intestinal absorption and renal reabsorption of Ca2+ but also revealed unique roles of the TRPV6 channel in maternal-fetal Ca2+ transport, male fertility, and auditory system. The growing evidence for the involvement TRPV6 in cancer proliferation and metastasis formation generates pharmaceutical interest. New approaches, such as ligand-based virtual screening, have been utilized to design inhibitors as a first step toward therapeutic developments. The novel crystal structure of TRPV6 will be instrumental to our understanding of the Ca2+ transport mechanisms and regulation and help refine the chemical design of TRPV6 modulators. As the involvement of genetic mutations/variations of TRPV5 and TRPV6 in human health is emerging, the integration of multidisciplinary approaches will further advance our knowledge and help develop new strategies for the treatment of kidney stone disease and cancer.
Acknowledgments
We thank our colleagues for their contributions to the research projects related to this book chapter. Our research was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK072154), the American Heart Association National Center (0430125N), the Greater Southeast Affiliate (09GRNT2160024), and the Swiss National Center for Competence in Research, NCCR TransCure (www.transcure.org). Gergely Gyimesi acknowledges the COFUND Marie Curie international fellowship program, IFP TransCure.
References
- 1.
- Hoenderop, J.G., van der Kemp, A.W., Hartog, A., van de Graaf, S.F., van Os, C.H., Willems, P.H., and Bindels, R.J. 1999. Molecular identification of the apical Ca2+ channel in 1,25-dihydroxyvitamin D3-responsive epithelia. J. Biol. Chem. 274:8375-8378. [PubMed: 10085067]
- 2.
- Peng, J.B., Chen, X.Z., Berger, U.V., Vassilev, P.M., Tsukaguchi, H., and Brown, E.M., and Hediger, M.A. 1999. Molecular cloning and characterization of a channel-like transporter mediating intestinal calcium absorption. J. Biol. Chem. 274:22739-22746. [PubMed: 10428857]
- 3.
- Na, T. and Peng, J.B. 2014. TRPV5: A Ca2+ channel for the fine-tuning of Ca2+ reabsorption. Handb. Exp. Pharmacol. 222:321-357. [PubMed: 24756712]
- 4.
- Fecher-Trost, C., Weissgerber, P., and Wissenbach, U. 2014. TRPV6 channels. Handb. Exp. Pharmacol. 222:359-384. [PubMed: 24756713]
- 5.
- Hoenderop, J.G., Nilius, B., and Bindels, R.J. 2005. Calcium absorption across epithelia. Physiol. Rev. 85:373-422. [PubMed: 15618484]
- 6.
- Suzuki, Y., Landowski, C.P., and Hediger, M.A. 2008. Mechanisms and regulation of epithelial Ca2+ absorption in health and disease. Annu. Rev. Physiol. 70:257-271. [PubMed: 17850211]
- 7.
- Bindels, R.J. 2000. Molecular pathophysiology of renal calcium handling. Kidney Blood Press. Res. 23:183-184. [PubMed: 11031716]
- 8.
- Peng, J.B., Brown, E.M., and Hediger, M.A. 2001. Structural conservation of the genes encoding CaT1, CaT2, and related cation channels. Genomics 76:99-109. [PubMed: 11549322]
- 9.
- Song, Y., Peng, X., Porta, A., Takanaga, H., Peng, J.B., Hediger, M.A., Fleet, J.C., and Christakos, S. 2003. Calcium transporter 1 and epithelial calcium channel messenger ribonucleic acid are differentially regulated by 1,25 dihydroxyvitamin D3 in the intestine and kidney of mice. Endocrinology 144:3885-3894. [PubMed: 12933662]
- 10.
- Wu, Y., Miyamoto, T., Li, K., Nakagomi, H., Sawada, N., Kira, S., Kobayashi, H., et al. 2011. Decreased expression of the epithelial Ca2+ channel TRPV5 and TRPV6 in human renal cell carcinoma associated with vitamin D receptor. J. Urol. 186:2419-2425. [PubMed: 22019165]
- 11.
- Peng, J.B., Chen, X.Z., Berger, U.V., Weremowicz, S., Morton, C.C., Vassilev, P.M., Brown, E.M., and Hediger, M.A. 2000. Human calcium transport protein CaT1. Biochem. Biophys. Res. Commun. 278:326-332. [PubMed: 11097838]
- 12.
- Zhuang, L., Peng, J.B., Tou, L., Takanaga, H., Adam, R.M., Hediger, M.A., and Freeman, M.R. 2002. Calcium-selective ion channel, CaT1, is apically localized in gastrointestinal tract epithelia and is aberrantly expressed in human malignancies. Lab. Invest. 82:1755-1764. [PubMed: 12480925]
- 13.
- Wissenbach, U., Niemeyer, B.A., Fixemer, T., Schneidewind, A., Trost, C., Cavalie, A., Reus, K., Meese, E., Bonkhoff, H., and Flockerzi, V. 2001. Expression of CaT-like, a novel calcium-selective channel, correlates with the malignancy of prostate cancer. J. Biol. Chem. 276:19461-19468. [PubMed: 11278579]
- 14.
- Hoenderop, J.G., van Leeuwen, J.P., van der Eerden, B.C., Kersten, F.F., van der Kemp, A.W., Merillat, A.M., Waarsing, J.H., et al. 2003. Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J. Clin. Invest. 112:1906-1914. [PMC free article: PMC297001] [PubMed: 14679186]
- 15.
- Bianco, S.D., Peng, J.B., Takanaga, H., Suzuki, Y., Crescenzi, A., Kos, C.H., Zhuang, L., et al. 2007. Marked disturbance of calcium homeostasis in mice with targeted disruption of the Trpv6 calcium channel gene. J. Bone Miner. Res. 22:274-285. [PMC free article: PMC4548943] [PubMed: 17129178]
- 16.
- Suzuki, Y., Kovacs, C.S., Takanaga, H., Peng, J.B., Landowski, C.P., and Hediger, M.A. 2008. Calcium channel TRPV6 is involved in murine maternal-fetal calcium transport. J. Bone Miner. Res. 23:1249-1256. [PMC free article: PMC2680174] [PubMed: 18348695]
- 17.
- Weissgerber, P., Kriebs, U., Tsvilovskyy, V., Olausson, J., Kretz, O., Stoerger, C., Vennekens, R., et al. 2011. Male fertility depends on Ca2+ absorption by TRPV6 in epididymal epithelia. Sci. Signal. 4:ra27. [PubMed: 21540454]
- 18.
- Stoerger, C. and Flockerzi, V. 2014. The transient receptor potential cation channel subfamily V member 6 (TRPV6): Genetics, biochemical properties, and functions of exceptional calcium channel proteins. Biochem. Cell Biol. 92:441-448. [PubMed: 25372600]
- 19.
- Brown, E.M. 1991. Extracellular Ca2+ sensing, regulation of parathyroid cell function, and role of Ca2+ and other ions as extracellular (first) messengers. Physiol. Rev. 71:371-411. [PubMed: 2006218]
- 20.
- Khundmiri, S.J., Murray, R.D., and Lederer, E. 2016. PTH and vitamin D. Compr. Physiol. 6:561-601. [PubMed: 27065162]
- 21.
- Van Cromphaut, S.J., Dewerchin, M., Hoenderop, J.G., Stockmans, I., Van, H.E., Kato, S., Bindels, R.J., et al. 2001. Duodenal calcium absorption in vitamin D receptor-knockout mice: Functional and molecular aspects. Proc. Natl. Acad. Sci. USA 98:13324-13329. [PMC free article: PMC60869] [PubMed: 11687634]
- 22.
- Bronner, F. 1998. Calcium absorption—A paradigm for mineral absorption. J. Nutr. 128:917-920. [PubMed: 9567004]
- 23.
- Friedman, P.A. 2000. Mechanisms of renal calcium transport. Exp. Nephrol. 8:343-350. [PubMed: 11014931]
- 24.
- Christakos, S., Gill, R., Lee, S., and Li, H. 1992. Molecular aspects of the calbindins. J. Nutr. 122:678-682. [PubMed: 1542030]
- 25.
- Kawasaki, H., Nakayama, S., and Kretsinger, R.H. 1998. Classification and evolution of EF-hand proteins. Biometals 11:277-295. [PubMed: 10191494]
- 26.
- Lambers, T.T., Mahieu, F., Oancea, E., Hoofd, L., de Lange, F., Mensenkamp, A.R., Voets, T., et al. 2006. Calbindin-D28K dynamically controls TRPV5-mediated Ca2+ transport. EMBO J. 25:2978-2988. [PMC free article: PMC1500989] [PubMed: 16763551]
- 27.
- Schauberger, C.W. and Pitkin, R.M. 1979. Maternal-perinatal calcium relationships. Obstet. Gynecol. 53:74-76. [PubMed: 760023]
- 28.
- Smith, C.H., Moe, A.J., and Ganapathy, V. 1992. Nutrient transport pathways across the epithelium of the placenta. Annu. Rev. Nutr. 12:183-206. [PubMed: 1503803]
- 29.
- Stulc, J. 1997. Placental transfer of inorganic ions and water. Physiol. Rev. 77:805-836. [PubMed: 9234966]
- 30.
- Moreau, R., Daoud, G., Bernatchez, R., Simoneau, L., Masse, A., and Lafond, J. 2002. Calcium uptake and calcium transporter expression by trophoblast cells from human term placenta. Biochim. Biophys. Acta 1564:325-332. [PubMed: 12175914]
- 31.
- Jenkins, A.D., Lechene, C.P., and Howards, S.S. 1980. Concentrations of seven elements in the intraluminal fluids of the rat seminiferous tubules, rate testis, and epididymis. Biol. Reprod. 23:981-987. [PubMed: 7470534]
- 32.
- Carlson, A.E., Westenbroek, R.E., Quill, T., Ren, D., Clapham, D.E., Hille, B., Garbers, D.L., and Babcock, D.F. 2003. CatSper1 required for evoked Ca2+ entry and control of flagellar function in sperm. Proc. Natl. Acad. Sci. USA 100:14864-14868. [PMC free article: PMC299831] [PubMed: 14657352]
- 33.
- Marquis, R.E. and Hudspeth, A.J. 1997. Effects of extracellular Ca2+ concentration on hair-bundle stiffness and gating-spring integrity in hair cells. Proc. Natl. Acad. Sci. USA 94:11923-11928. [PMC free article: PMC23657] [PubMed: 9342338]
- 34.
- Yamauchi, D., Nakaya, K., Raveendran, N.N., Harbidge, D.G., Singh, R., Wangemann, P., and Marcus, D.C. 2010. Expression of epithelial calcium transport system in rat cochlea and vestibular labyrinth. BMC Physiol. 10:1. [PMC free article: PMC2825184] [PubMed: 20113508]
- 35.
- Nakaya, K., Harbidge, D.G., Wangemann, P., Schultz, B.D., Green, E.D., Wall, S.M., and Marcus, D.C. 2007. Lack of pendrin. Am. J. Physiol. Renal Physiol. 292:F1314-F1321. [PMC free article: PMC2515270] [PubMed: 17200157]
- 36.
- Nicaise, G., Maggio, K., Thirion, S., Horoyan, M., and Keicher, E. 1992. The calcium loading of secretory granules. A possible key event in stimulus-secretion coupling. Biol. Cell 75:89-99. [PubMed: 1393151]
- 37.
- Monteith, G.R., McAndrew, D., Faddy, H.M., and Roberts-Thomson, S.J. 2007. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 7:519-530. [PubMed: 17585332]
- 38.
- Prevarskaya, N., Skryma, R., and Shuba, Y. 2011. Calcium in tumour metastasis: New roles for known actors. Nat. Rev. Cancer 11:609-618. [PubMed: 21779011]
- 39.
- Roderick, H.L. and Cook, S.J. 2008. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 8:361-375. [PubMed: 18432251]
- 40.
- Gray, L.S., Perez-Reyes, E., Gomora, J.C., Haverstick, D.M., Shattock, M., McLatchie, L., Harper, J., Brooks, G., Heady, T., and Macdonald, T.L. 2004. The role of voltage gated T-type Ca2+ channel isoforms in mediating “capacitative” Ca2+ entry in cancer cells. Cell Calcium 36:489-497. [PubMed: 15488598]
- 41.
- Landowski, C.P., Bolanz, K.A., Suzuki, Y., and Hediger, M.A. 2011. Chemical inhibitors of the calcium entry channel TRPV6. Pharm. Res. 28:322-330. [PubMed: 21057859]
- 42.
- Romero, M.F., Kanai, Y., Gunshin, H., and Hediger, M.A. 1998. Expression cloning using Xenopus laevis oocytes. Methods Enzymol. 296:17-52. [PubMed: 9779438]
- 43.
- Fecher-Trost, C., Wissenbach, U., Beck, A., Schalkowsky, P., Stoerger, C., Doerr, J., Dembek, A., et al. 2013. The in vivo TRPV6 protein starts at a non-AUG triplet, decoded as methionine, upstream of canonical initiation at AUG. J. Biol. Chem. 288:16629-16644. [PMC free article: PMC3675598] [PubMed: 23612980]
- 44.
- Caterina, M.J., Schumacher, M.A., Tominaga, M., Rosen, T.A., Levine, J.D., and Julius, D. 1997. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 389:816-824. [PubMed: 9349813]
- 45.
- Colbert, H.A., Smith, T.L., and Bargmann, C.I. 1997. OSM-9, a novel protein with structural similarity to channels, is required for olfaction, mechanosensation, and olfactory adaptation in Caenorhabditis elegans. J. Neurosci. 17:8259-8269. [PMC free article: PMC6573730] [PubMed: 9334401]
- 46.
- Vennekens, R., Owsianik, G., and Nilius, B. 2008. Vanilloid transient receptor potential cation channels: An overview. Curr. Pharm. Des. 14:18-31. [PubMed: 18220815]
- 47.
- Peng, J.B., Chen, X.Z., Berger, U.V., Vassilev, P.M., Brown, E.M., and Hediger, M.A. 2000. A rat kidney-specific calcium transporter in the distal nephron. J. Biol. Chem. 275:28186-28194. [PubMed: 10875938]
- 48.
- Mackenzie, B. 1999. Selected techniques in membrane transport. In Biomembrane Transport. L.J.Van Winkle, ed. Academic Press, San Diego, CA, pp. 327-342.
- 49.
- Vennekens, R., Hoenderop, J.G., Prenen, J., Stuiver, M., Willems, P.H., Droogmans, G., Nilius, B., and Bindels, R.J. 2000. Permeation and gating properties of the novel epithelial Ca2+ channel. J. Biol. Chem. 275:3963-3969. [PubMed: 10660551]
- 50.
- Nilius, B., Vennekens, R., Prenen, J., Hoenderop, J.G., Bindels, R.J., and Droogmans, G. 2000. Whole-cell and single channel monovalent cation currents through the novel rabbit epithelial Ca2+ channel ECaC. J. Physiol. 527(Pt 2):239-248. [PMC free article: PMC2270079] [PubMed: 10970426]
- 51.
- Yue, L., Peng, J.B., Hediger, M.A., and Clapham, D.E. 2001. CaT1 manifests the pore properties of the calcium-release-activated calcium channel. Nature 410:705-709. [PubMed: 11287959]
- 52.
- Nilius, B., Vennekens, R., Prenen, J., Hoenderop, J.G., Droogmans, G., and Bindels, R.J. 2001. The single pore residue Asp542 determines Ca2+ permeation and Mg2+ block of the epithelial Ca2+ channel. J. Biol. Chem. 276:1020-1025. [PubMed: 11035011]
- 53.
- Dodier, Y., Banderali, U., Klein, H., Topalak, O., Dafi, O., Simoes, M., Bernatchez, G., Sauve, R., and Parent, L. 2004. Outer pore topology of the ECaC-TRPV5 channel by cysteine scan mutagenesis. J. Biol. Chem. 279:6853-6862. [PubMed: 14630907]
- 54.
- Nilius, B., Prenen, J., Hoenderop, J.G., Vennekens, R., Hoefs, S., Weidema, A.F., Droogmans, G., and Bindels, R.J. 2002. Fast and slow inactivation kinetics of the Ca2+ channels ECaC1 and ECaC2 (TRPV5 and TRPV6). Role of the intracellular loop located between transmembrane segments 2 and 3. J. Biol. Chem. 277:30852-30858. [PubMed: 12077127]
- 55.
- Niemeyer, B.A., Bergs, C., Wissenbach, U., Flockerzi, V., and Trost, C. 2001. Competitive regulation of CaT-like-mediated Ca2+ entry by protein kinase C and calmodulin. Proc. Natl. Acad. Sci. USA 98:3600-3605. [PMC free article: PMC30699] [PubMed: 11248124]
- 56.
- Derler, I., Hofbauer, M., Kahr, H., Fritsch, R., Muik, M., Kepplinger, K., Hack, M.E., et al. 2006. Dynamic but not constitutive association of calmodulin with rat TRPV6 channels enables fine tuning of Ca2+-dependent inactivation. J. Physiol. 577:31-44. [PMC free article: PMC2000671] [PubMed: 16959851]
- 57.
- Lambers, T.T., Weidema, A.F., Nilius, B., Hoenderop, J.G., and Bindels, R.J. 2004. Regulation of the mouse epithelial Ca2+ channel TRPV6 by the Ca2+-sensor calmodulin. J. Biol. Chem. 279:28855-28861. [PubMed: 15123711]
- 58.
- de Groot, T., Kovalevskaya, N.V., Verkaart, S., Schilderink, N., Felici, M., van der Hagen, E.A., Bindels, R.J., Vuister, G.W., and Hoenderop, J.G. 2011. Molecular mechanisms of calmodulin action on TRPV5 and modulation by parathyroid hormone. Mol. Cell Biol. 31:2845-2853. [PMC free article: PMC3133394] [PubMed: 21576356]
- 59.
- Kovalevskaya, N.V., Bokhovchuk, F.M., and Vuister, G.W. 2012. The TRPV5/6 calcium channels contain multiple calmodulin binding sites with differential binding properties. J. Struct. Funct. Genomics 13:91-100. [PMC free article: PMC3375010] [PubMed: 22354706]
- 60.
- Gkika, D., Mahieu, F., Nilius, B., Hoenderop, J.G., and Bindels, R.J. 2004. 80K-H as a new Ca2+ sensor regulating the activity of the epithelial Ca2+ channel transient receptor potential cation channel V5 (TRPV5). J. Biol. Chem. 279:26351-26357. [PubMed: 15100231]
- 61.
- Lee, J., Cha, S.K., Sun, T.J., and Huang, C.L. 2005. PIP2 activates TRPV5 and releases its inhibition by intracellular Mg2+. J. Gen. Physiol. 126:439-451. [PMC free article: PMC2266600] [PubMed: 16230466]
- 62.
- Thyagarajan, B., Benn, B.S., Christakos, S., and Rohacs, T. 2009. Phospholipase C-mediated regulation of transient receptor potential vanilloid 6 channels: Implications in active intestinal Ca2+ transport. Mol. Pharmacol. 75:608-616. [PMC free article: PMC2684912] [PubMed: 19073818]
- 63.
- Thyagarajan, B., Lukacs, V., and Rohacs, T. 2008. Hydrolysis of phosphatidylinositol 4,5-bisphosphate mediates calcium-induced inactivation of TRPV6 channels. J. Biol. Chem. 283:14980-14987. [PMC free article: PMC2397461] [PubMed: 18390907]
- 64.
- Rohacs, T. and Nilius, B. 2007. Regulation of transient receptor potential (TRP) channels by phosphoinositides. Pflugers Arch. 455:157-168. [PubMed: 17479281]
- 65.
- Rohacs, T., Lopes, C.M., Michailidis, I., and Logothetis, D.E. 2005. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 8:626-634. [PubMed: 15852009]
- 66.
- Poblete, H., Oyarzun, I., Olivero, P., Comer, J., Zuniga, M., Sepulveda, R.V., Baez-Nieto, D., Gonzalez, L.C., Gonzalez-Nilo, F., and Latorre, R. 2015. Molecular determinants of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) binding to transient receptor potential V1 (TRPV1) channels. J. Biol. Chem. 290:2086-2098. [PMC free article: PMC4303662] [PubMed: 25425643]
- 67.
- Velisetty, P., Borbiro, I., Kasimova, M.A., Liu, L., Badheka, D., Carnevale, V., and Rohacs, T. 2016. A molecular determinant of phosphoinositide affinity in mammalian TRPV channels. Sci. Rep. 6:27652. [PMC free article: PMC4904367] [PubMed: 27291418]
- 68.
- Simonin, C., Awale, M., Brand, M., van Deursen, R., Schwartz, J., Fine, M., Kovacs, G., et al. 2015. Optimization of TRPV6 calcium channel inhibitors using a 3D ligand-based virtual screening method. Angew. Chem. Int. Ed. Engl. 54:14748-14752. [PubMed: 26457814]
- 69.
- Bolanz, K.A., Kovacs, G.G., Landowski, C.P., and Hediger, M.A. 2009. Tamoxifen inhibits TRPV6 activity via estrogen receptor-independent pathways in TRPV6-expressing MCF-7 breast cancer cells. Mol. Cancer Res. 7:2000-2010. [PubMed: 19996302]
- 70.
- Saotome, K., Singh, A.K., Yelshanskaya, M.V., and Sobolevsky, A.I. 2016. Crystal structure of the epithelial calcium channel TRPV6. Nature 534:506-511. [PMC free article: PMC4919205] [PubMed: 27296226]
- 71.
- Alford, R.F., Koehler, L.J., Weitzner, B.D., Duran, A.M., Tilley, D.C., Elazar, A., and Gray, J.J. 2015. An integrated framework advancing membrane protein modeling and design. PLoS Comput. Biol. 11:e1004398. [PMC free article: PMC4556676] [PubMed: 26325167]
- 72.
- Mandell, D.J., Coutsias, E.A., and Kortemme, T. 2009. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 6:551-552. [PMC free article: PMC2847683] [PubMed: 19644455]
- 73.
- Kuhlman, B. and Baker, D. 2000. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. USA 97:10383-10388. [PMC free article: PMC27033] [PubMed: 10984534]
- 74.
- Loh, N.Y., Bentley, L., Dimke, H., Verkaart, S., Tammaro, P., Gorvin, C.M., Stechman, M.J., et al. 2013. Autosomal dominant hypercalciuria in a mouse model due to a mutation of the epithelial calcium channel, TRPV5. PLoS One 8:e55412. [PMC free article: PMC3559602] [PubMed: 23383183]
- 75.
- Cui, M., Li, Q., Johnson, R., and Fleet, J.C. 2012. Villin promoter-mediated transgenic expression of transient receptor potential cation channel, subfamily V, member 6 (TRPV6) increases intestinal calcium absorption in wild-type and vitamin D receptor knockout mice. J. Bone Miner. Res. 27:2097-2107. [PMC free article: PMC3430830] [PubMed: 22589201]
- 76.
- van Abel, M., Huybers, S., Hoenderop, J.G., van der Kemp, A.W., van Leeuwen, J.P., and Bindels, R.J. 2006. Age-dependent alterations in Ca2+ homeostasis: Role of TRPV5 and TRPV6. Am. J. Physiol. Renal Physiol. 291:F1177-F1183. [PubMed: 16705151]
- 77.
- Benn, B.S., Ajibade, D., Porta, A., Dhawan, P., Hediger, M., Peng, J.B., Jiang, Y., et al. 2008. Active intestinal calcium transport in the absence of transient receptor potential vanilloid type 6 and calbindin-D9k. Endocrinology 149:3196-3205. [PMC free article: PMC2408805] [PubMed: 18325990]
- 78.
- Kutuzova, G.D., Sundersingh, F., Vaughan, J., Tadi, B.P., Ansay, S.E., Christakos, S., and Deluca, H.F. 2008. TRPV6 is not required for 1alpha, 25-dihydroxyvitamin D3-induced intestinal calcium absorption in vivo. Proc. Natl. Acad. Sci. USA 105:19655-19659. [PMC free article: PMC2605002] [PubMed: 19073913]
- 79.
- Meyer, M.B., Watanuki, M., Kim, S., Shevde, N.K., and Pike, J.W. 2006. The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25-dihydroxyvitamin D3 in intestinal cells. Mol. Endocrinol. 20:1447-1461. [PubMed: 16574738]
- 80.
- Song, Y., Kato, S., and Fleet, J.C. 2003. Vitamin D receptor (VDR) knockout mice reveal VDR-independent regulation of intestinal calcium absorption and ECaC2 and calbindin D9k mRNA. J. Nutr. 133:374-380. [PubMed: 12566470]
- 81.
- Pitkin, R.M. 1985. Calcium metabolism in pregnancy and the perinatal period: A review. Am. J. Obstet. Gynecol. 151:99-109. [PubMed: 3881031]
- 82.
- Bernucci, L., Henriquez, M., Diaz, P., and Riquelme, G. 2006. Diverse calcium channel types are present in the human placental syncytiotrophoblast basal membrane. Placenta 27:1082-1095. [PubMed: 16564089]
- 83.
- Moreau, R., Hamel, A., Daoud, G., Simoneau, L., and Lafond, J. 2002. Expression of calcium channels along the differentiation of cultured trophoblast cells from human term placenta. Biol. Reprod. 67:1473-1479. [PubMed: 12390878]
- 84.
- Stumpf, T., Zhang, Q., Hirnet, D., Lewandrowski, U., Sickmann, A., Wissenbach, U., Dorr, J., Lohr, C., Deitmer, J.W., and Fecher-Trost, C. 2008. The human TRPV6 channel protein is associated with cyclophilin B in human placenta. J. Biol. Chem. 283:18086-18098. [PubMed: 18445599]
- 85.
- Vanoevelen, J., Janssens, A., Huitema, L.F., Hammond, C.L., Metz, J.R., Flik, G., Voets, T., and Schulte-Merker, S. 2011. Trpv5/6 is vital for epithelial calcium uptake and bone formation. FASEB J. 25:3197-3207. [PubMed: 21670068]
- 86.
- Weissgerber, P., Kriebs, U., Tsvilovskyy, V., Olausson, J., Kretz, O., Stoerger, C., Mannebach, S., et al. 2012. Excision of Trpv6 gene leads to severe defects in epididymal Ca2+ absorption and male fertility much like single D541A pore mutation. J. Biol. Chem. 287:17930-17941. [PMC free article: PMC3365704] [PubMed: 22427671]
- 87.
- Gao, D.Y., Zhang, B.L., Leung, M.C., Au, S.C., Wong, P.Y., and Shum, W.W. 2016. Coupling of TRPV6 and TMEM16A in epithelial principal cells of the rat epididymis. J. Gen. Physiol. 148:161-182. [PMC free article: PMC4969799] [PubMed: 27481714]
- 88.
- Lewit-Bentley, A., Rety, S., Sopkova-de Oliveira, S.J., and Gerke, V. 2000. S100-annexin complexes: Some insights from structural studies. Cell Biol. Int. 24:799-802. [PubMed: 11067764]
- 89.
- van de Graaf, S.F., Chang, Q., Mensenkamp, A.R., Hoenderop, J.G., and Bindels, R.J. 2006. Direct interaction with Rab11a targets the epithelial Ca2+ channels TRPV5 and TRPV6 to the plasma membrane. Mol. Cell Biol. 26:303-312. [PMC free article: PMC1317621] [PubMed: 16354700]
- 90.
- Embark, H.M., Setiawan, I., Poppendieck, S., van de Graaf, S.F., Boehmer, C., Palmada, M., Wieder, T., et al. 2004. Regulation of the epithelial Ca2+ channel TRPV5 by the NHE regulating factor NHERF2 and the serum and glucocorticoid inducible kinase isoforms SGK1 and SGK3 expressed in Xenopus oocytes. Cell Physiol. Biochem. 14:203-212. [PubMed: 15319523]
- 91.
- Palmada, M., Poppendieck, S., Embark, H.M., van de Graaf, S.F., Boehmer, C., Bindels, R.J., and Lang, F. 2005. Requirement of PDZ domains for the stimulation of the epithelial Ca2+ channel TRPV5 by the NHE regulating factor NHERF2 and the serum and glucocorticoid inducible kinase SGK1. Cell Physiol. Biochem. 15:175-182. [PubMed: 15665527]
- 92.
- Jing, H., Na, T., Zhang, W., Wu, G., Liu, C., and Peng, J.B. 2011. Concerted actions of NHERF2 and WNK4 in regulating TRPV5. Biochem. Biophys. Res. Commun. 404:979-984. [PMC free article: PMC3031669] [PubMed: 21187068]
- 93.
- Holakovska, B., Grycova, L., Bily, J., and Teisinger, J. 2011. Characterization of calmodulin binding domains in TRPV2 and TRPV5 C-tails. Amino Acids 40:741-748. [PubMed: 20686800]
- 94.
- van de Graaf, S.F., van der Kemp, A.W., van den Berg, D., van Oorschot, M., Hoenderop, J.G., and Bindels, R.J. 2006. Identification of BSPRY as a novel auxiliary protein inhibiting TRPV5 activity. J. Am. Soc. Nephrol. 17:26-30. [PubMed: 16380433]
- 95.
- Gkika, D., Topala, C.N., Hoenderop, J.G., and Bindels, R.J. 2006. The immunophilin FKBP52 inhibits the activity of the epithelial Ca2+ channel TRPV5. Am. J. Physiol. Renal Physiol. 290:F1253-F1259. [PubMed: 16352746]
- 96.
- van de Graaf, S.F., Hoenderop, J.G., van der Kemp, A.W., Gisler, S.M., and Bindels, R.J. 2006. Interaction of the epithelial Ca2+ channels TRPV5 and TRPV6 with the intestine- and kidney-enriched PDZ protein NHERF4. Pflugers Arch. 452:407-417. [PubMed: 16565876]
- 97.
- Yeh, B.I., Kim, Y.K., Jabbar, W., and Huang, C.L. 2005. Conformational changes of pore helix coupled to gating of TRPV5 by protons. EMBO J. 24:3224-3234. [PMC free article: PMC1224685] [PubMed: 16121193]
- 98.
- Cha, S.K., Jabbar, W., Xie, J., and Huang, C.L. 2007. Regulation of TRPV5 single-channel activity by intracellular pH. J. Membr. Biol. 220:79-85. [PubMed: 18004496]
- 99.
- de Groot, T., Verkaart, S., Xi, Q., Bindels, R.J., and Hoenderop, J.G. 2010. The identification of Histidine 712 as a critical residue for constitutive TRPV5 internalization. J. Biol. Chem. 285:28481-28487. [PMC free article: PMC2937873] [PubMed: 20628046]
- 100.
- Chang, Q., Hoefs, S., van der Kemp, A.W., Topala, C.N., Bindels, R.J., and Hoenderop, J.G. 2005. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310:490-493. [PubMed: 16239475]
- 101.
- Lu, P., Boros, S., Chang, Q., Bindels, R.J., and Hoenderop, J.G. 2008. The beta-glucuronidase klotho exclusively activates the epithelial Ca2+ channels TRPV5 and TRPV6. Nephrol. Dial. Transplant. 23:3397-3402. [PubMed: 18495742]
- 102.
- Leunissen, E.H., Nair, A.V., Bull, C., Lefeber, D.J., van Delft, F.L., Bindels, R.J., and Hoenderop, J.G. 2013. The epithelial calcium channel TRPV5 is regulated differentially by klotho and sialidase. J. Biol. Chem. 288:29238-29246. [PMC free article: PMC3795225] [PubMed: 23970553]
- 103.
- Wolf, M.T., An, S.W., Nie, M., Bal, M.S., and Huang, C.L. 2014. Klotho up-regulates renal calcium channel transient receptor potential vanilloid 5 (TRPV5) by intra- and extracellular N-glycosylation-dependent mechanisms. J. Biol. Chem. 289:35849-35857. [PMC free article: PMC4276853] [PubMed: 25378396]
- 104.
- Boros, S., Xi, Q., Dimke, H., van der Kemp, A.W., Tudpor, K., Verkaart, S., Lee, K.P., Bindels, R.J., and Hoenderop, J.G. 2012. Tissue transglutaminase inhibits the TRPV5-dependent calcium transport in an N-glycosylation-dependent manner. Cell Mol. Life Sci. 69:981-992. [PubMed: 21952826]
- 105.
- Tudpor, K., Lainez, S., Kwakernaak, A.J., Kovalevskaya, N.V., Verkaart, S., van Genesen, S., van der Kemp, A., Navis, G., Bindels, R.J., and Hoenderop, J.G. 2012. Urinary plasmin inhibits TRPV5 in nephrotic-range proteinuria. J. Am. Soc. Nephrol. 23:1824-1834. [PMC free article: PMC3482726] [PubMed: 23024298]
- 106.
- Wolf, M.T., Wu, X.R., and Huang, C.L. 2013. Uromodulin upregulates TRPV5 by impairing caveolin-mediated endocytosis. Kidney Int. 84:130-137. [PMC free article: PMC3700562] [PubMed: 23466996]
- 107.
- Nie, M., Bal, M.S., Yang, Z., Liu, J., Rivera, C., Wenzel, A., Beck, B.B., Sakhaee, K., Marciano, D.K., and Wolf, M.T. 2016. Mucin-1 increases renal TRPV5 activity in vitro, and urinary level associates with calcium nephrolithiasis in patients. J. Am. Soc. Nephrol. 27:3447-3458. [PMC free article: PMC5084893] [PubMed: 27036738]
- 108.
- Mayan, H., Munter, G., Shaharabany, M., Mouallem, M., Pauzner, R., Holtzman, E.J., and Farfel, Z. 2004. Hypercalciuria in familial hyperkalemia and hypertension accompanies hyperkalemia and precedes hypertension: Description of a large family with the Q565E WNK4 mutation. J. Clin. Endocrinol. Metab. 89:4025-4030. [PubMed: 15292344]
- 109.
- Jiang, Y., Ferguson, W.B., and Peng, J.B. 2007. WNK4 enhances TRPV5-mediated calcium transport: Potential role in hypercalciuria of familial hyperkalemic hypertension caused by gene mutation of WNK4. Am. J. Physiol. Renal Physiol. 292:F545-F554. [PubMed: 17018846]
- 110.
- Jiang, Y., Cong, P., Williams, S.R., Zhang, W., Na, T., Ma, H.P., and Peng, J.B. 2008. WNK4 regulates the secretory pathway via which TRPV5 is targeted to the plasma membrane. Biochem. Biophys. Res. Commun. 375:225-229. [PMC free article: PMC2570258] [PubMed: 18703016]
- 111.
- Zhang, W., Na, T., and Peng, J.B. 2008. WNK3 positively regulates epithelial calcium channels TRPV5 and TRPV6 via a kinase-dependent pathway. Am. J. Physiol. Renal Physiol. 295:F1472-F1484. [PMC free article: PMC2584897] [PubMed: 18768590]
- 112.
- Hoopes, R.R., Jr., Shrimpton, A.E., Knohl, S.J., Hueber, P., Hoppe, B., Matyus, J., Simckes, A., et al. 2005. Dent disease with mutations in OCRL1. Am. J. Hum. Genet. 76:260-267. [PMC free article: PMC1196371] [PubMed: 15627218]
- 113.
- Shrimpton, A.E., Hoopes, R.R., Jr., Knohl, S.J., Hueber, P., Reed, A.A., Christie, P.T., Igarashi, T., et al. 2009. OCRL1 mutations in Dent 2 patients suggest a mechanism for phenotypic variability. Nephron Physiol. 112:27-36. [PubMed: 19390221]
- 114.
- Sliman, G.A., Winters, W.D., Shaw, D.W., and Avner, E.D. 1995. Hypercalciuria and nephrocalcinosis in the oculocerebrorenal syndrome. J. Urol. 153:1244-1246. [PubMed: 7869519]
- 115.
- Wu, G., Zhang, W., Na, T., Jing, H., Wu, H., and Peng, J.B. 2012. Suppression of intestinal calcium entry channel TRPV6 by OCRL, a lipid phosphatase associated with Lowe syndrome and Dent disease. Am. J. Physiol. Cell Physiol. 302:C1479-C1491. [PMC free article: PMC3361998] [PubMed: 22378746]
- 116.
- Gkika, D., Topala, C.N., Chang, Q., Picard, N., Thebault, S., Houillier, P., Hoenderop, J.G., and Bindels, R.J. 2006. Tissue kallikrein stimulates Ca2+ reabsorption via PKC-dependent plasma membrane accumulation of TRPV5. EMBO J. 25:4707-4716. [PMC free article: PMC1618098] [PubMed: 17006539]
- 117.
- Topala, C.N., Schoeber, J.P., Searchfield, L.E., Riccardi, D., Hoenderop, J.G., and Bindels, R.J. 2009. Activation of the Ca2+-sensing receptor stimulates the activity of the epithelial Ca2+ channel TRPV5. Cell Calcium 45:331-339. [PubMed: 19157541]
- 118.
- Zhang, W., Na, T., Wu, G., Jing, H., and Peng, J.B. 2010. Down-regulation of intestinal apical calcium entry channel TRPV6 by ubiquitin E3 ligase Nedd4-2. J. Biol. Chem. 285:36586-36596. [PMC free article: PMC2978587] [PubMed: 20843805]
- 119.
- Berridge, M.J., Lipp, P., and Bootman, M.D. 2000. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1:11-21. [PubMed: 11413485]
- 120.
- Berridge, M.J., Bootman, M.D., and Roderick, H.L. 2003. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4:517-529. [PubMed: 12838335]
- 121.
- Fan, H., Shen, Y.X., and Yuan, Y.F. 2014. Expression and prognostic roles of TRPV5 and TRPV6 in non-small cell lung cancer after curative resection. Asian Pac. J. Cancer Prev. 15:2559-2563. [PubMed: 24761864]
- 122.
- Giusti, L., Cetani, F., Da, V.Y., Pardi, E., Ciregia, F., Donadio, E., Gargini, C., et al. 2014. First evidence of TRPV5 and TRPV6 channels in human parathyroid glands: Possible involvement in neoplastic transformation. J. Cell Mol. Med. 18:1944-1952. [PMC free article: PMC4244010] [PubMed: 25164318]
- 123.
- Huhn, S., Bevier, M., Pardini, B., Naccarati, A., Vodickova, L., Novotny, J., Vodicka, P., Hemminki, K., and Forsti, A. 2014. Colorectal cancer risk and patients’ survival: Influence of polymorphisms in genes somatically mutated in colorectal tumors. Cancer Causes Control 25:759-769. [PubMed: 24706189]
- 124.
- Jiang, Y., Gou, H., Zhu, J., Tian, S., and Yu, L. 2016. Lidocaine inhibits the invasion and migration of TRPV6-expressing cancer cells by TRPV6 downregulation. Oncol. Lett. 12:1164-1170. [PMC free article: PMC4950910] [PubMed: 27446413]
- 125.
- Kim, S.Y., Hong, C., Wie, J., Kim, E., Kim, B.J., Ha, K., Cho, N.H., Kim, I.G., Jeon, J.H., and So, I. 2014. Reciprocal positive regulation between TRPV6 and NUMB in PTEN-deficient prostate cancer cells. Biochem. Biophys. Res. Commun. 447:192-196. [PubMed: 24704446]
- 126.
- Peng, J.B., Zhuang, L., Berger, U.V., Adam, R.M., Williams, B.J., Brown, E.M., Hediger, M.A., and Freeman, M.R. 2001. CaT1 expression correlates with tumor grade in prostate cancer. Biochem. Biophys. Res. Commun. 282:729-734. [PubMed: 11401523]
- 127.
- Raphael, M., Lehen’kyi, V., Vandenberghe, M., Beck, B., Khalimonchyk, S., Vanden Abeele, F., Farsetti, L., et al. 2014. TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc. Natl. Acad. Sci. USA 111:E3870-E3879. [PMC free article: PMC4169956] [PubMed: 25172921]
- 128.
- Skrzypski, M., Khajavi, N., Mergler, S., Szczepankiewicz, D., Kolodziejski, P.A., Metzke, D., Wojciechowicz, T., Billert, M., Nowak, K.W., and Strowski, M.Z. 2015. TRPV6 channel modulates proliferation of insulin secreting INS-1E beta cell line. Biochim. Biophys. Acta 1853:3202-3210. [PubMed: 26384871]
- 129.
- Zhang, S.S., Xie, X., Wen, J., Luo, K.J., Liu, Q.W., Yang, H., Hu, Y., and Fu, J.H. 2016. TRPV6 plays a new role in predicting survival of patients with esophageal squamous cell carcinoma. Diagn. Pathol. 11:14. [PMC free article: PMC4730645] [PubMed: 26818094]
- 130.
- Cooperberg, M.R., Moul, J.W., and Carroll, P.R. 2005. The changing face of prostate cancer. J. Clin. Oncol. 23:8146-8151. [PubMed: 16278465]
- 131.
- Schroder, F.H. 2010. Prostate cancer around the world. An overview. Urol. Oncol. 28:663-667. [PubMed: 21062647]
- 132.
- Fixemer, T., Wissenbach, U., Flockerzi, V., and Bonkhoff, H. 2003. Expression of the Ca2+-selective cation channel TRPV6 in human prostate cancer: A novel prognostic marker for tumor progression. Oncogene 22:7858-7861. [PubMed: 14586412]
- 133.
- Bodding, M., Fecher-Trost, C., and Flockerzi, V. 2003. Store-operated Ca2+ current and TRPV6 channels in lymph node prostate cancer cells. J. Biol. Chem. 278:50872-50879. [PubMed: 14534305]
- 134.
- Vanden Abeele, F., Roudbaraki, M., Shuba, Y., Skryma, R., and Prevarskaya, N. 2003. Store-operated Ca2+ current in prostate cancer epithelial cells. Role of endogenous Ca2+ transporter type 1. J. Biol. Chem. 278:15381-15389. [PubMed: 12584203]
- 135.
- Lehen’kyi, V., Flourakis, M., Skryma, R., and Prevarskaya, N. 2007. TRPV6 channel controls prostate cancer cell proliferation via Ca2+/NFAT-dependent pathways. Oncogene 26:7380-7385. [PubMed: 17533368]
- 136.
- Bolanz, K.A., Hediger, M.A., and Landowski, C.P. 2008. The role of TRPV6 in breast carcinogenesis. Mol. Cancer Ther. 7:271-279. [PubMed: 18245667]
- 137.
- Peters, A.A., Simpson, P.T., Bassett, J.J., Lee, J.M., Da, S.L., Reid, L.E., Song, S., et al. 2012. Calcium channel TRPV6 as a potential therapeutic target in estrogen receptor-negative breast cancer. Mol. Cancer Ther. 11:2158-2168. [PubMed: 22807578]
- 138.
- Dhennin-Duthille, I., Gautier, M., Faouzi, M., Guilbert, A., Brevet, M., Vaudry, D., Ahidouch, A., Sevestre, H., and Ouadid-Ahidouch, H. 2011. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cell Physiol. Biochem. 28:813-822. [PubMed: 22178934]
- 139.
- Chow, J., Norng, M., Zhang, J., and Chai, J. 2007. TRPV6 mediates capsaicin-induced apoptosis in gastric cancer cells—Mechanisms behind a possible new “hot” cancer treatment. Biochim. Biophys. Acta 1773:565-576. [PubMed: 17292493]
- 140.
- Lau, J.K., Brown, K.C., Dom, A.M., Witte, T.R., Thornhill, B.A., Crabtree, C.M., Perry, H.E., et al. 2014. Capsaicin induces apoptosis in human small cell lung cancer via the TRPV6 receptor and the calpain pathway. Apoptosis 19:1190-1201. [PMC free article: PMC4072851] [PubMed: 24878626]
- 141.
- Nilius, B., Prenen, J., Vennekens, R., Hoenderop, J.G., Bindels, R.J., and Droogmans, G. 2001. Pharmacological modulation of monovalent cation currents through the epithelial Ca2+ channel ECaC1. Br. J. Pharmacol. 134:453-462. [PMC free article: PMC1572972] [PubMed: 11588099]
- 142.
- Schwarz, E.C., Wissenbach, U., Niemeyer, B.A., Strauss, B., Philipp, S.E., Flockerzi, V., and Hoth, M. 2006. TRPV6 potentiates calcium-dependent cell proliferation. Cell Calcium 39:163-173. [PubMed: 16356545]
- 143.
- Cove-Smith, A., Mulgrew, C.J., Rudyk, O., Dutt, N., McLatchie, L.M., Shattock, M.J., and Hendry, B.M. 2013. Anti-proliferative actions of T-type calcium channel inhibition in Thy1 nephritis. Am. J. Pathol. 183:391-401. [PMC free article: PMC3730775] [PubMed: 23746655]
- 144.
- Hofer, A., Kovacs, G., Zappatini, A., Leuenberger, M., Hediger, M.A., and Lochner, M. 2013. Design, synthesis and pharmacological characterization of analogs of 2-aminoethyl diphenylborinate (2-APB), a known store-operated calcium channel blocker, for inhibition of TRPV6-mediated calcium transport. Bioorg. Med. Chem. 21:3202-3213. [PubMed: 23602525]
- 145.
- Chokshi, R., Fruasaha, P., and Kozak, J.A. 2012. 2-Aminoethyl diphenyl borinate (2-APB) inhibits TRPM7 channels through an intracellular acidification mechanism. Channels (Austin) 6:362-369. [PMC free article: PMC3508775] [PubMed: 22922232]
- 146.
- Muller, D., Hoenderop, J.G., Vennekens, R., Eggert, P., Harangi, F., Mehes, K., Garcia-Nieto, V., et al. 2002. Epithelial Ca2+ channel (ECAC1) in autosomal dominant idiopathic hypercalciuria. Nephrol. Dial. Transplant. 17:1614-1620. [PubMed: 12198212]
- 147.
- Renkema, K.Y., Lee, K., Topala, C.N., Goossens, M., Houillier, P., Bindels, R.J., and Hoenderop, J.G. 2009. TRPV5 gene polymorphisms in renal hypercalciuria. Nephrol. Dial. Transplant. 24:1919-1924. [PubMed: 19131347]
- 148.
- Khaleel, A., Wu, M.S., Wong, H.S., Hsu, Y.W., Chou, Y.H., and Chen, H.Y. 2015. A single nucleotide polymorphism (rs4236480) in TRPV5 calcium channel gene is associated with stone multiplicity in calcium nephrolithiasis patients. Mediators Inflamm. 2015:375427. [PMC free article: PMC4452106] [PubMed: 26089600]
- 149.
- Oddsson, A., Sulem, P., Helgason, H., Edvardsson, V.O., Thorleifsson, G., Sveinbjornsson, G., Haraldsdottir, E., et al. 2015. Common and rare variants associated with kidney stones and biochemical traits. Nat. Commun. 6:7975. [PMC free article: PMC4557269] [PubMed: 26272126]
- 150.
- Na, T., Zhang, W., Jiang, Y., Liang, Y., Ma, H.P., Warnock, D.G., and Peng, J.B. 2009. The A563T variation of the renal epithelial calcium channel TRPV5 among African Americans enhances calcium influx. Am. J. Physiol. Renal Physiol. 296:F1042-F1051. [PMC free article: PMC2681372] [PubMed: 19261737]
- 151.
- Wang, L., Holmes, R.P., and Peng, J.B. 2016. Molecular modeling of the structural and dynamical changes in calcium channel TRPV5 induced by the African-specific A563T variation. Biochemistry 55:1254-1264. [PMC free article: PMC5304435] [PubMed: 26837804]
- 152.
- van der Eerden, B.C., Hoenderop, J.G., de Vries, T.J., Schoenmaker, T., Buurman, C.J., Uitterlinden, A.G., Pols, H.A., Bindels, R.J., and van Leeuwen, J.P. 2005. The epithelial Ca2+ channel TRPV5 is essential for proper osteoclastic bone resorption. Proc. Natl. Acad. Sci. USA 102:17507-17512. [PMC free article: PMC1297662] [PubMed: 16291808]
- 153.
- Suzuki, Y., Pasch, A., Bonny, O., Mohaupt, M.G., Hediger, M.A., and Frey, F.J. 2008. Gain-of-function haplotype in the epithelial calcium channel TRPV6 is a risk factor for renal calcium stone formation. Hum. Mol. Genet. 17:1613-1618. [PubMed: 18276610]
- 154.
- Akey, J.M., Swanson, W.J., Madeoy, J., Eberle, M., and Shriver, M.D. 2006. TRPV6 exhibits unusual patterns of polymorphism and divergence in worldwide populations. Hum. Mol. Genet. 15:2106-2113. [PubMed: 16717058]
- 155.
- Hughes, D.A., Tang, K., Strotmann, R., Schoneberg, T., Prenen, J., Nilius, B., and Stoneking, M. 2008. Parallel selection on TRPV6 in human populations. PLoS One 3:e1686. [PMC free article: PMC2246018] [PubMed: 18301763]
- 156.
- Sarmina, I., Spirnak, J.P., and Resnick, M.I. 1987. Urinary lithiasis in the black population: An epidemiological study and review of the literature. J. Urol. 138:14-17. [PubMed: 3599198]
Ji-Bin Peng
Division of Nephrology
Nephrology Research and Training Center
and
Department of Urology
University of Alabama at Birmingham
Birmingham, Alabama
Yoshiro Suzuki
Division of Cell Signaling
National Institutes of Natural Sciences
and
Department of Physiological Sciences
SOKENDAI (The Graduate University for Advanced Studies)
Okazaki, Japan
Gergely Gyimesi
Institute of Biochemistry and Molecular Medicine
and
Swiss National Center of Competence in Research
TransCure
University of Bern
Bern, Switzerland
Matthias A. Hediger
Institute of Biochemistry and Molecular Medicine
and
Swiss National Center of Competence in Research
TransCure
University of Bern
Bern, Switzerland
Publication Details
Author Information and Affiliations
Authors
Ji-Bin Peng, Yoshiro Suzuki, Gergely Gyimesi, and Matthias A. Hediger.Copyright
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
Publisher
CRC Press/Taylor & Francis, Boca Raton (FL)
NLM Citation
Peng JB, Suzuki Y, Gyimesi G, et al. TRPV5 and TRPV6 Calcium-Selective Channels. In: Kozak JA, Putney JW Jr., editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. Chapter 13. doi: 10.1201/9781315152592-13


.&p=BOOKS&id=531440_fig13_2.jpg "Click on image to zoom")


%20heterocyclic%20compounds%20and%20(b)%202-APB%20analogs.&p=BOOKS&id=531440_fig13_5.jpg "Click on image to zoom")