3.1. Introduction
3.1.1. ER Calcium Repository
Cells maintain an inward Ca2+ gradient from the extracellular fluid to the cytosol, entrenched by the action of plasma membrane pumps/transporters that actively “pump out” Ca2+ and organellar pumps/transporters that mold the Ca2+ transients by storing it away in intracellular reservoirs [1,2]. External stimuli trigger Ca2+ influx driven by the electrochemical gradient between a plenteous extracellular reservoir of Ca2+ (millimolar ranges) and a Ca2+-free cytosol. This Ca2+ surge is indispensible for its role as a second messenger, controlling a wide range of cellular functions like exocytosis, mast cell degranulation, and immune cell proliferation.
Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) family proteins drive excess Ca2+ from the cytosol into the ER lumen. Low-affinity, high-capacity buffers like calsequestrin and calreticulin soak up the incoming Ca2+, increasing the storage and Ca2+ buffering capacity of the ER [3,4]. Concurrently, Ca2+ overload and associated toxicity are prevented by the release channels inositol trisphosphate receptors (InsP3R) and ryanodine receptors. Maintaining ER Ca2+ concentration in the range of ∼100 μM is essential for its protein synthesis and folding activities.
The concept of the ER as the “capacitator” of Ca2+ entry was pioneered by James Putney and found traction in experiments where blocking ER Ca2+ uptake using thapsigargin, a noncompetitive inhibitor of SERCA pump, led to Ca2+ influx [5,6]. Exhaustibility of ER Ca2+ reserve is an essential feature that orchestrates cellular signaling initiated by Ca2+ entry. Plasma membrane receptor stimulation generates a second messenger, inositol 1,4,5-trisphosphate (IP3), which signals ER Ca2+ release by organellar channels [7]. Diminution of free Ca2+ from ER stores is sensed and compensated by a highly selective influx of the extracellular Ca2+, termed store-operated calcium entry (SOCE). Besides replenishment of the stores, initiation of specific local pathways that control downstream cellular responses is believed to be a driving force for SOCE [8].
3.1.2. CRAC Current and the Underlying Players
SOCE is measured as an inwardly rectifying Ca2+ current supporting a small but significant Ca2+ uptake (see Chapter 1). The current, ICRAC (CRAC: acronym for Ca2+ release-activated Ca2+), was first detected in Jurkat T cells and mast cells using a combination of whole-cell patch clamping and single-cell Ca2+ imaging [9,10]. Depletion of organellar stores, and not mere alteration in intracellular Ca2+, trigger ICRAC, substantiated by studies using IP3 or the Ca2+-specific ionophore ionomycin in mast cells and using thapsigargin or intracellular Ca2+ chelators like BAPTA in Jurkat T cells and the revocable ER Ca2+ chelator TPEN ([11–13]; reviewed in [14]). The biophysical features of ICRAC are indicative of a unique ion channel that conducts Ca2+ more precisely than any other characterized channel. In particular, it has a low conductance of <30 fS. This channel is ∼1000-fold selective for Ca2+ over Na+, blocked by low concentrations of Gd3+, and detectably permeable to other divalent cations (Ca2+, Ba2+, Sr2+). Both fast and slow inactivation of ICRAC, attributed to either a local increase in Ca2+ levels or a gradual refilling of store, are Ca2+-dependent feedback processes that regulate the channel activity [15,16]. CRAC channel can operate independent of the potential difference across the plasma membrane, necessitating the role of a ligand in gating [10,17].
CRAC channel identity remained elusive for years after ICRAC was first recorded, and a pair of ER Ca2+ sensor and plasma membrane Ca2+ channel controlling the Ca2+ influx was predicted. Two independent genome-wide RNAi screens identified the ER component that controls SOCE as STIM1, an ER membrane-located Ca2+-binding protein with the features of a signaling molecule [18,19]. Drosophila S2 cells that have mammal-like CRAC channel properties were instrumental in expounding the two molecular players responsible for ICRAC [20]. Another set of coincident genome-wide RNAi studies identified Drosophila Orai (dOrai) as the plasma membrane component responsible for store-operated Ca2+ influx in these cells [21–23]. Accompanying genetic analysis of human severe combined immunodeficiency (SCID) patients devoid of SOCE activity and a restoration of Ca2+ uptake function by ectopic expression of human ORAI1 confirmed its role as the CRAC channel [21].
3.1.3. SOCE Current as an Outcome of STIM-ORAI Coupling
Coexpression of STIM1 and ORAI1 proteins is sufficient for generating SOCE current of large amplitude across various cellular systems [24–26]. Light microscopy studies with fluorescent proteins show a strong correlation between Ca2+ influx and a major redistribution of homogeneous ER-localized STIM1 into puncta in ER compartments adjoining the plasma membrane [27,28]. Implication from this finding is that STIM1 physically interacts with ORAI1 across a narrow gap between the ER and the plasma membrane.
ORAI1 protein forms a Ca2+-permeable ion channel in the plasma membrane, and mutation of a conserved acidic residue in the pore can block Ca2+ flux through the channel [22,29] (Figure 3.1a). ER Ca2+ depletion is sensed by the Ca2+-binding luminal EF-hand domain of STIM1, a disruption of which leads to a constitutive activation of STIM1 (Figure 3.1b) [19,28]. Following activation, STIM1 can recruit ORAI channels into clusters, and both puncta and clusters spatially correspond with the sites of Ca2+ entry [30]. Store depletion is essential for STIM1 redistribution [27,28,31–-34], and interaction with STIM is sufficient for ORAI1 clustering and Ca2+ influx [30,31,35]. The obligatory role of STIM-ORAI interaction in generating SOCE current has been demonstrated through various biochemical studies, for the details of which, the reader is referred to other comprehensive literature reviews [36,37] (see Chapters 2 and 7).
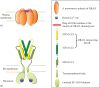
Figure 3.1
An illustration of ORAI1 and STIM1 proteins at rest under store replete conditions. (a) ORAI1 hexamer in the plasma membrane shown with Ca2+ bound to a ring of negatively charged E106 residues that mark the extracellular mouth of the channel. (b) Inactive (more...)
The entire process of STIM1- and ORAI1-mediated Ca2+ influx calls for both proteins to switch from a high-Ca2+ inactive state to a low-Ca2+ active state. In the case of STIM, Ca2+-binding status of the sensor itself can determine its conformation, while for ORAI1, STIM1 binding is essential for its gating. This conformational coupling can be regulated by several cellular factors, but STIM-ORAI interaction is adequate for SOCE. This chapter surveys the wealth of structural and biophysical studies to offer a mechanistic overview of STIM and ORAI functioning, focusing on the architectural changes these proteins undergo in response to store depletion.
3.2. ORAI Channel Structure
Mammalian ORAI1 (also known as CRACM1) is a ∼33 kDa four-transmembrane cell surface protein [21,22]. Mammals express three ORAI-family proteins, namely, ORAI1, ORAI2, and ORAI3, with high sequence similarity in the transmembrane (TM) regions. ORAI homologs are named after the three mythological gatekeepers of heaven. ORAI1 forms an ion-conducting pore in the plasma membrane, and its subunit topology, confirmed experimentally, shows both N and C termini to be cytoplasmic [29,38,39] (see Chapter 2). ORAI1 is the principal ORAI-family protein contributing to store-operated Ca2+ entry in most mammalian cells studied.
3.2.1. Calcium Binding Site
ORAI proteins distinctively lack sequence similarity to not just other ion channels but also any other known protein. However, like other Ca2+ channels, ORAI proteins utilize the negative charge on a glutamate residue to bind and select for Ca2+ (Figure 3.2a). TM1 residue E106 (E178 in dOrai) is central to the Ca2+ binding and conductance of ORAI1, and alanine or glutamine substitutions result in a drastic loss of SOCE current [22,29,38]. E106D, a smaller negatively charged substitution, restores channel function only partially, with a concomitant increase in permeability to monovalent cations, emphasizing the need for a glutamate to maintain ion selectivity [29]. Introduction of one copy of E106Q into a tandem tetramer of ORAI1 is sufficient to eliminate CRAC current [40]. E106C shows a high degree of oxidative cross-linking and coordinates Cd2+ (ionic size similar to Ca2+) efficiently to result in a current blockade, demonstrating that the sidechains at position 106 are positioned to directly coordinate Ca2+ ions and hence form the Ca2+ binding site(s) [41,42].
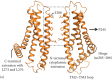
Figure 3.2
ORAI1 channel features highlighted in a homology model generated based on dOrai structure (PDB: 4HKR).
3.2.2. Pore Features Mapped by Cd2+ Block and Disulfide Cross-Linking Experiments
Analysis of the packing of engineered cysteine substitutions against their counterparts from separate monomers of the ORAI1 channel has been instrumental in defining the ion permeation path. Cysteines replacing the TM1 residues along one helical face cross-link proficiently with oxidizing agents like copper phenanthroline and molecular iodine, demarcating the pore-facing amino acids [42]. The pore allows small probes like Cd2+ to penetrate and cause a current block due to coordination by the pore-lining cysteine substitutions while being impermeable to other cysteine-reactive probes with diameters >3 Å [41]. Both studies conclusively show a long narrow pore entirely composed of TM1 helices, with tightly packed E106 residues, a constricted nonpolar region spanning residues 99–104, and the TM1 apposition extending to at least residue 91 (Figure 3.2b). It is important to note that while oxidative cross-linking experiments report the architecture of resting channel pore, Cd2+ blocks the inward Ca2+ current through a conducting ORAI1 channel. Correspondence of the pore-facing residues in the two states implies that subtle changes in the pore can convert a closed channel into an open one.
3.2.3. Drosophila Orai Structure
The 3.35 Å crystal structure of the nonconducting dOrai channel (PDB: 4HKR) shows that TM1 helices indeed form a central pore independent of other TM helices, which are arranged concentrically around the pore [43]. The crystal structure, disuccinimidyl suberate cross-linking of the dOrai channel expressed in HEK293 cell membranes, and SEC-MALS measurements of the Orai channel mass indicate that the channel is hexameric. Although this conclusion differs from the tetramer model previously proposed on the basis of single-molecule photobleaching experiments and electrophysiology of concatenated ORAI1 constructs, a consensus has developed that this is the correct stoichiometry (discussed in Reference 44 and Chapters 2 and 14). The hexameric arrangement results in six negatively charged E178 residues (Drosophila equivalent of E106 discussed earlier) in close vicinity of each other, forming a Ca2+-binding ring at the extracellular mouth of the pore [43]. Electron density of a putative Ca2+ ion bound to the glutamate ring reiterates the role of E106 as a Ca2+ binding site. Since the pore is devoid of any other acidic residue forming an ancillary cation binding site, one hypothesis to be tested is whether backbone carbonyls of residues adjacent to E106 are ligands that assist the inward flux of Ca2+ ions.
3.2.4. Pore from Outside to Inside
The extracellular accessibility to E106 and the ORAI1 channel pore is through a loop connecting TM1 and TM2, a region featuring clusters of conserved aspartates. Structural information for the TM1-TM2 loop is absent in the dOrai structure, possibly due to the typical flexibility associated with protein loop regions [43]. Although Cd2+ block analysis indicates high accessibility and mobility of the loop, experiments with larger cysteine-modifying reagents ∼5–8 Å in diameter show an elevated block of D110C current, indicating a positioning of at least a part of the loop in line with the permeation pathway in the active state [41]. Alanine substitutions of all the loop aspartates result in altered ion selectivity and decrease substantially the luminescence intensity of Tb3+ bound in the vicinity of E106 in reconstituted ORAI1 channels [38,45,46a]. These findings and a molecular dynamics study simulating Ca2+ entry into ORAI1 pore indicate that TM1-TM2 loop might form a cation “focusing” vestibule that facilitates fast Ca2+ permeation [41,46a,47] (see Chapter 2).
The wide vestibule tapers at the mouth of the pore at the glutamate ring, juxtaposed to which is a constricted nonpolar section of the pore (Figure 3.2b). The hydrophobic residues of this region are well ordered in the engineered cysteine assays and show little variation in thermal motion in the Drosophila Orai structure [41–43]. As discussed in a later section on ORAI1 gating, this region evidently forms a barrier in the path of ion conductance and is presumably moved aside by STIM [46a,48].
A noteworthy feature of the pore captured in the crystallized state of dOrai is an anionic electron density trapped in the lysine-arginine-rich basic section that runs contiguous with the nonpolar region. The dOrai protein can cocrystallize with complex anions like iridium hexachloride, and mass spectrometric analysis of ions copurifying with dOrai detects considerable iron species, suggesting that binding an Fe-anion complex or other anions can overcome the strong repulsive forces and offer the constraint required for crystallization [43]. In a native closed channel, it is possible that common cytoplasmic anions remain bound in this region. However, cysteine cross-linking and Cd2+ block experiments find this basic region to be moderately accessible, suggesting flexibility in both states of the channel [41,42].
3.2.5. Cytoplasmic Extensions of ORAI1
A protracted cytoplasmic region was inferred from experiments where forced narrowing of ER-plasma membrane distance to ≤9 nm causes an exclusion of ORAI1 channels [49]. According to the dOrai structure, TM1 helix extends deeper into the cytoplasm than what is observed in chemical cross-linking studies and forms an N-terminal extension, contiguous with the pore (Figure 3.2a). The C-terminal extension is comprised of a helix that originates from the TM4 helix through a hinge formed by residues 263–266 of ORAI1 (aa306-309 in dOrai), and an intramembrane bend at TM4 residue P245 (P288 in dOrai) causes the C-terminal half of TM4 to run more or less parallel to the membrane with the extension angled away from the body of the channel (Figure 3.2a). Neighboring C-terminal extensions homodimerize through a hydrophobic patch containing L273 and L276 (I316 and L319 in dOrai) (Figure 3.2a). C-terminal dimerization confers upon ORAI the threefold down-up symmetry, as viewed from the cytoplasm, and forms the primary interaction site for STIM binding [32,43,50].
3.3. Overview of STIM1 Structure
STIM1 is a multidomain protein anchored in the ER membrane through a single-pass transmembrane helix (Figure 3.1b). STIM1, an abbreviation for Stromal Interaction Molecule 1, was initially believed to be either a protein secreted by bone marrow stromal cells or a leukemia cell surface protein [51,52], but it has since been shown to act as an ER-localized regulator of SOCE [19]. STIM1 is largely localized in the ER, with a smaller fraction in the plasma membrane [52–54]. In addition to its well-studied role in SOCE, it has additional functions in both locations [55–57]. Mammals express two STIM proteins with complementary, and nonredundant, functions in Ca2+ signaling [58]. STIM1 and STIM2 are likely to share common mechanisms of sensing and communicating store depletion, given the high degree of sequence conservation between the proteins in their core functional domains.
The function of STIM proteins in Ca2+ sensing is manifest in their structure and orientation in the ER membrane, with a Ca2+-sensing domain facing the ER lumen, and an extensible region and ORAI1-binding domain in the cytoplasm, enabling STIM to relay a Ca2+ depletion signal from the ER to the plasma membrane (reviewed in References 59 and 60 and Chapter 2).
3.3.1. Ca2+ Sensing by STIM1 Luminal Domain
The luminal “sensor” domain of STIM1 was predicted, from its sequence, to contain an EF-hand motif, a common helix-loop-helix structural element that binds Ca2+. A solution nuclear magnetic resonance (NMR) structure of the luminal region of human STIM1 (residues 58–201; PDB: 2K60) in the presence of millimolar Ca2+, which is likely to represent the conformation of STIM1 in the ER lumen in resting cells, revealed the presence of two EF-hands in tandem: a canonical EF-hand followed in the linear sequence by a noncanonical “hidden” one [61]. Although the hidden EF-hand retains a recognizable EF-hand fold, the expected Ca2+-binding residues at positions 1 and 3 in the loop are replaced by Phe and Gly, respectively, consistent with the finding that only one Ca2+ is bound per luminal domain [34]. This leads to a model in which Ca2+ is bound in the canonical EF-hand site, and the second EF-hand provides structural stabilization, both through the β-strand interaction typical of paired EF-hands and through its participation in a hydrophobic pocket that partially envelops the STIM1 sterile alpha motif (SAM) domain that follows immediately in the linear sequence. Ca2+ binds to recombinant STIM(58–201) with a relatively low affinity, whether binding is monitored with 45Ca2+ or by following changes in intrinsic protein fluorescence or far-UV circular dichroism, the Kd ranging between 200 and 600 μM [34].
3.3.2. Cytoplasmic Domain of STIM1
The cytoplasmic “effector” domain of STIM1, expressed as a soluble protein fragment, can activate native ORAI1 channels in Jurkat T cells [62] and recombinant ORAI1 channels expressed in other cells [32,63,64]. The Ca2+ influx through ORAI1 channels under these conditions is constitutive and uncoupled from regulation by ER Ca2+ stores. “Effector” function of the STIM cytoplasmic domain is further verified by the ability of purified recombinant STIM1(233–685) to trigger Ca2+ efflux from lipid vesicles or artificial liposomes containing ORAI1 channels [46a,65]. The region sufficient to activate ORAI1 channels is STIMSOAR (residues 344–442; STIM1 Orai Activating Region) or a similar fragment containing SOAR [32,66,67].
Then why does STIM1 not activate ORAI1 channels in resting cells? The short answer is that SOAR is retained near the ER under these conditions. The first evidence that the cytoplasmic domain folds back on itself came from the determination of the distance in recombinant STIM1CT between a label placed adjacent to residue 233 and a label at the C terminus [68]. The measurement by luminescence resonance energy transfer (LRET) showed that the two sites are in close proximity, only 3–4 nm distant, implying that STIM1CT has a preferred conformation in which the polybasic tail would be near the ER rather than near the plasma membrane (see Chapter 7). Furthermore, the purified CC1 fragment (STIM1(233–343)) interacts with purified SOAR in vitro [68], and membrane-anchored STIMEFSAM-TM-CC1 expressed in cells recruits cytosolic SOAR to the ER as evidenced by colocalization and by Förster resonance energy transfer (FRET) between the labeled proteins [69]. In the latter experiment, even a truncated EFSAM-TM-CC1(233–261) construct is effective, showing that SOAR is held very close to the ER membrane. These findings indicate that CC1 is not simply a tether linking SOAR to the ER but is intimately involved in maintaining the inactive conformation of STIM1, a point that will be taken up in the next section.
3.3.3. ORAI-Interacting Machinery
Critical features of the STIM1CT structure remain to be filled in, with definition of the structural interactions that retain SOAR near the ER being a prime goal. One piece of the puzzle is a crystal structure of human SOAR (PDB: 3TEQ) showing intramonomer antiparallel coiled coils that supercoil to form a V-shaped dimer [70]. There is a plausible argument that this structure might reflect the inactive conformation of the STIM1SOAR [50], but even if so, there is very limited information about how this subdomain is packed with the remainder of the STIM1CT. An elegant analysis of peptide-peptide interactions using short fragments of STIM1CT [71] is consistent with available data from mutational studies and points to intramolecular contacts that might stabilize the inactive form of STIM1CT but has not so far provided sufficient constraints to predict a three-dimensional structure. A crystal structure of Caenorhabditis elegans SOAR (PDB: 3TER) captures in addition a short piece of the CC1 region, equivalent to residues 318–337 in human STIM1, making contact with the SOAR domain [70]. A caveat in extending this structural finding to the human protein is that the neighboring CC1 residues 310–317 and residues 275–300 of human STIM1 have no counterparts in the C. elegans STIM sequence, suggesting that the mode of CC1-SOAR packing in the human protein may be distinct. Another cautionary note is that in an NMR structure of human STIM1 residues 312–387 (PDB: 2MAJ), representing a part of CC1 and a part of SOAR, the positioning of the 313–340 helix differs appreciably from that of the 257–279 helix in the C. elegansSOAR structure [50]. On the other hand, the STIM1(312–387) structure also displays an altered arrangement of helices when compared to the human SOAR structure, and the difference could be satisfactorily explained if the STIM1(312–387) structure represents an activated conformation of STIM1 [50], as discussed below.
3.4. STIM Active State
Experimental depletion of the resting ER Ca2+ stores triggers a development of CRAC current that correlates inversely to the level of Ca2+ remaining in the ER. Current is highly cooperative with the Ca2+ concentration and reaches its half-maximal value at ∼170 μM Ca2+ [72]. Store depletion acts as a switch for STIM1 activation, a process that can be visualized by confocal, total internal reflection fluorescence (TIRF), and electron microscopy as a redistribution of appropriately tagged STIM1 [19,27,28]. The newly redistributed STIM1 is localized in “puncta” or ∼100–300 nm wide regions where ER is closely apposed to the plasma membrane (reviewed in Reference 73). ICRAC requires STIM1 puncta formation and shows a similar Ca2+ concentration dependence and cooperativity [72,74]. The studies discussed here have utilized STIM1 fragments or mutant proteins to gain valuable insights into the conformational changes in STIM1 that enable communication between the ER lumen and the plasma membrane. However, the precise sequence of events in STIM1 activation following Ca2+ store depletion and the detailed architecture of activated STIM1 are still active areas of investigation.
3.4.1. Activation of the Luminal Domain
The first response to a decline in store Ca2+ levels comes from the luminal EF-hand. Mutating one of the consensus Ca2+-coordinating positions, D76, to alanine causes STIM1 to localize into puncta independent of store depletion [19]. Constitutive STIM1 activation is likewise observed with the E87Q mutant of the EF-hand, corroborating that an inability to bind Ca2+, and by extrapolation a loss of bound Ca2+, is responsible for STIM1 redistribution [28]. The high micromolar Kds estimated for Ca2+ binding to isolated STIM1EFSAM or to the STIM1 EF-hand grafted into a scaffolding protein domain are arguably suited to sensing physiological changes in ER-luminal Ca2+ [34,75,76]. Direct evidence that Ca2+ depletion is coupled to a conformational change comes from far-UV CD spectra and NMR spectra of isolated recombinant STIMEFSAM, which indicate a striking loss of α-helical structure in the absence of Ca2+. This change in secondary structure is accompanied by a transition of STIMEFSAM from monomer to dimer and, in one study, the formation of higher aggregates [34,76]. In cells, an increase in intramolecular FRET efficiency between fluorophores attached to the luminal domain of STIM1 occurs upon store depletion and, notably, precedes the relocalization of STIM1 to puncta, reinforcing the idea that a STIM1 conformational change might drive subsequent relocalization [33,63]. The FRET change in cells could represent both the dimerization that has been observed with isolated STIM1 luminal domains and a further STIM1 oligomerization step as discussed in the following section.
3.4.2. Activation Involves Extension of STIM1 Cytoplasmic Domain
A key insight into the active conformation of the STIM cytoplasmic domain came from the constitutively activated L251S and L416S/L423S mutants of STIM1 [77]. When expressed in cells together with ORAI1, the mutant proteins are constitutively colocalized with ORAI1 channels in puncta and trigger a constitutive La3+-blockable current. The mutations result in an extended conformation of the recombinant ORAI1-activating small fragment (OASF) (STIM1(233–474)) as assessed by intramolecular FRET. LRET analysis of the purified recombinant STIM1 cytoplasmic domain carrying the L251S mutation shows a physical extension of the molecule by at least several nanometers compared to wild-type STIM1CT [68]. LRET is not suitable for measuring distances greater than 10 nm, but it is arguable that STIM1 has been directly visualized in EM and cryo-electron tomography studies of cells overexpressing STIM1, as filaments spanning the 10–20 nm distance between cortical ER and plasma membrane [78,79]. These data support a model in which the ORAI-interacting SOAR/CAD domain of resting STIM1 is retained near the ER, whereas it is freed to interact with plasma membrane ORAI1 in the active form of the protein (Figure 3.3).
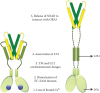
Figure 3.3
Proposed series of events in the process of STIM1 activation, based on the current research findings. Left panel shows a resting state STIM1 that undergoes at least the five steps listed in the figure, resulting in an active form of the protein that is (more...)
3.4.3. CC1 and the Release of ORAI1-Interacting Machinery
A series of experiments with engineered proteins dissected the mechanism underlying STIM1 activation. As noted earlier, recombinant STIM1CC1 interacts with immobilized MBP-tagged STIM1SOARin vitro [68]. The individual replacements L251S, L258G, and L261G in STIM1CC1 reduce this binding [68,69]. Parallel experiments in cells have shown that the mutations L258G and, to a lesser extent, L261G in full-length STIM1 resemble L251S in causing constitutive Ca2+ entry, suggesting that they also free SOAR to interact with plasma membrane ORAI [69]. It is unlikely that these mutations are disrupting a folded structure of the protein. Despite its designation as a predicted coiled coil, the isolated CC1 fragment is partially α-helical and partially unstructured, and monomeric [68]. Moreover the thermal melting curve for CC1 obtained by monitoring its CD spectrum offers no suggestion that intramolecular interactions stabilize parts of the α-helical secondary structure. Thus, it is most likely that L251, L258, and L261 engage SOAR directly.
The predicted propensity of CC1 to form a coiled coil comes into play in a further series of experiments. When STIMCC1 is forced to form dimers by oxidation of an N-terminally appended cysteine, there is an increase in α-helical content, along with a partial stabilization against thermal denaturation suggesting intradimer helix packing [68]. Dimerized STIM1CC1 binds less well to SOAR, and similar cross-linking of CC1 within the dimeric STIM1CT fragment triggers an extended conformation of the protein as measured in LRET experiments [68]. It has not been rigorously proven that the conformational change detected upon dimerization of CC1 involves the formation of a partial coiled coil. However, this is the most straightforward explanation, in light of the facts that the residues L251, L258, and L261 are predicted to be buried in a coiled coil interface, that an L251S replacement in CC1 prevents the increase in α-helical content and a stabilization against thermal denaturation [68], and that each of the leucine replacements is effective in releasing SOAR [68,69].
The final link connecting depletion of ER Ca2+ stores and the STIM1CT conformational change that releases SOAR comes from the FRET-based assay in cells in which STIMEFSAM-TM-CC1 recruits cytosolic SOAR to the ER. The assay detects a considerable loss of FRET between ER-localized STIMEFSAM-TM-CC1 and cytosolic SOAR after Ca2+ store depletion [69]. The current model for STIM1 activation [68] is that inactive STIM1 is a dimer because of its dimeric SOAR domains, that association of the ER-luminal domains within a STIM dimer brings together the CC1 segments, and that, by burying the critical CC1 residues that contact SOAR in the inactive conformation, this structural rearrangement of CC1 triggers the activating conformational change in STIM1CT. Note that the model does not require that CC1 associate into a stable coiled coil. Temporary coiled coil formation in the segment containing residues 248–261 may trigger release of SOAR, and thereafter either an α-helical or partially α-helical extended CC1 chain would be of sufficient length to position SOAR in the vicinity of ORAI1 in the plasma membrane.
The demonstrated CC1-SOAR interaction implies that CC1 must have a partner surface in SOAR. A candidate region in SOAR was suggested by the early finding that L416S/L423S is an activating mutation [77]. Recent evidence that L416G, V419G, and L423G mutations in STIM1 all result in constitutive Ca2+ entry [69] gives additional support to this possibility. Indeed, studies using CC1 and SOAR fragments find an interaction between the CC1α1 helix (residues 233–276) and the SOAR helix CC3430 (residues 388–430), and this interaction is abolished by the L251S substitution [71]. Special emphasis has been given to the role of CC1α1 in maintaining the inactive conformation, based on the finding that STIM1 with a deletion of residues 278–337 is still fully regulated by Ca2+, and an L251S mutation in this protein results in constitutive current [71]. The emphasis is undoubtedly justified, but other parts of CC1 help to stabilize the inactive conformation of full-length STIM1. For example, the 318EEELE322 > AAALA substitutions constitutively activate STIM1 and lead to physical extension of OASF and STIM1CT [68,77,80], and the single Y316A replacement causes appreciable though incomplete constitutive activation [81]. A full understanding of STIM1 activation will require the determination of how intramolecular packing changes throughout the CC1-SOAR region.
3.4.4. Higher-Order STIM1 Oligomerization
Because of the very steep Ca2+ concentration dependence of STIM relocalization to ER-plasma membrane junctions [72,74], it has been thought that STIM assembles into higher-order oligomers at some point during the activation process. Any oligomerization is likely to be through the SOAR/CAD domain [82,83], and a possible structural basis for oligomerization has been proposed in a documented CC3-CC3 interaction [71]. Persuasive as the data are, it should be recognized that all the studies thus far have been carried out with STIM1 fragments and with overexpressed STIM1. It remains to be tested whether the strength of STIM-STIM interactions is sufficient to drive the formation of higher-order oligomers of STIM1 in cells, at native levels of STIM1 and prior to the relocalization that concentrates STIM1 in ER-plasma membrane junctions.
3.5. Activation of ORAI1 Channels by STIM1
SOCE is linked with the redistribution of ORAI1 to plasma membrane sites that correspond precisely to STIM puncta [30,31,35]. STIM1 activation and redistribution are independent of ORAI1 expression [27,28,31–33], whereas ORAI1 is recruited to ER-plasma membrane junctions only in the presence of activated STIM1 [30,31,35]. Close proximity between STIM1 and ORAI1 is evident in FRET measurements of appropriately labeled proteins, indicating that activated STIM1 at ER-plasma membrane junctions couples to ORAI1 itself or to a closely associated protein [63,84,85] (see Chapter 2).
SOAR/CAD decorates the plasma membrane of cells when ORAI1 is coexpressed, and SOAR/CAD coimmunoprecipitates with ORAI1, indicating a close interaction between the two proteins [32,66]. Purified STIM1CT protein triggers Ca2+ efflux from PC/PS liposomes into which purified ORAI1 has been reconstituted and from membrane vesicles isolated from yeast expressing human ORAI1, conclusively proving that direct STIM1-ORAI1 interactions are sufficient to gate the channel [46a,65]. Considerable work has been directed toward defining these interactions. The current model favors multiple sites of contact between STIM and the ORAI1 channel [32,63,86–88]. This section considers the proposed sites of interaction and the role(s) attributed to each site in activation of ORAI1.
3.5.1. Interaction of Activated STIM1 with ORAI1
Truncated ORAI1 channels missing either aa267-301 at the C terminus or aa74-90 at the N terminus fail to generate CRAC currents, although only the C-terminal deletion shows a lack of ORAI1 redistribution [35,66]. The single replacement L273S in the C-terminal region of ORAI1 blocks both STIM-ORAI interaction and CRAC channel activation [63,66]. Purified STIM1CT can interact specifically with a GST-tagged C-terminal peptide of ORAI1 [65,66]. ORAI1ΔC and other C-terminal mutants fail to show ORAI1 clustering and STIM-ORAI FRET after store depletion [63,84,89,90]. ORAI1 mutants L273D, L276D, and truncations of the C-terminal helix that remove aa272-283 indicate that the hydrophobic face of this helix is important for STIM interaction [32,84,91]. Although earlier studies focused on the role of STIM1 interaction with the ORAI1 C terminus in recruiting ORAI1 to ER-plasma membrane junctions, McNally et al. [86] have given persuasive evidence that mutations and deletions in the C-terminal region of ORAI1 affect channel gating as well as STIM-ORAI clustering.
The C-terminal ORAI1 segment implicated in STIM1 binding maps within a juxtamembrane cytoplasmic helix in the dOrai structure [43]. These juxtamembrane helices are connected to their TM4 helices by a short flexible segment that adopts two configurations in the crystals, permitting the cytoplasmic helices of adjacent ORAI1 monomers to pack in a short antiparallel coiled coil stabilized by L273-L276′ and L276-L273′ contacts [43]. STIM1 binding appears to displace the cytoplasmic C-terminal helices, since STIM1-ORAI1 interaction and CRAC current are disrupted when cysteine side chains of engineered L273C ORAI1 monomers are chemically cross-linked [87]. This cross-linking has been interpreted as locking the ORAI1 C-terminal helices in a STIM-inaccessible conformation resembling that in the Drosophila Orai crystal structure.
There are two distinct proposals on the mode of STIM-ORAI interaction. In one, based on the solution NMR structure of the complex of ORAI1(272–292) peptide with a STIM1(312–387) fragment, an active STIM1 dimer binds a pair of ORAI1 helices, supplanting the ORAI-ORAI interaction by providing two hydrophobic grooves on the surface of STIM1 to accommodate the ORAI1 helices [50]. The antiparallel orientation of the ORAI1 helices is maintained in this complex, but there is a shift in the relative positions of the ORAI1 helices compared to the dOrai crystal structure. An alternative proposal [43] is that the individual C-terminal ORAI1 helices extend away from the plasma membrane, unpaired, to interact with STIM. This possibility gains some support from the recent finding that SOAR dimers with an F394H substitution in only one subunit are fully able to bind and activate ORAI1 channels, while those with an F394H substitution in both subunits are severely impaired in interacting with ORAI [92] (see Chapter 7). No specific structural model for monomeric ORAI1 peptide binding to STIM has been put forward. Both proposals are consistent with the experimental finding that ORAI1 C-terminal helices locked in the crystal configuration cannot interact productively with STIM [87].
In the ORAI1 N-terminal region, the segment spanning residues 74–90, corresponding to a cytoplasmic extension of the TM1 helix in the dOrai structure [43], is implicated in channel gating. SOAR/CAD can elicit normal currents with ORAI1Δ1-73 but no current with ORAI1Δ1-88 [35,93,94], even though STIM1 still recruits the truncated proteins to ER-plasma membrane junctions [35,46a,63,84]. Further, specific mutations in the region of residues 81–85 reduce or disrupt CRAC current [46a,94,95]. What is unclear is whether this region contacts STIM1 directly during channel gating. Coimmunoprecipitation and pulldown experiments have demonstrated an interaction between STIM1 and the ORAI1 fragments ORAI1(48–91) and ORAI1(65–87) [32,65], and a fluorescence polarization assay established that STIM1CT can bind the synthetic peptide ORAI1(66–91) [46a]. However, it has neither been proven nor disproven that such an interaction takes place in the context of the intact ORAI1 channel. Recent data indicate that the same mutations at residues 81–85 that compromise STIM1-activated current also block the constitutive current carried by an ORAI1 channel mutated in residues 261–265, the cytoplasmic hinge region following TM4 [46b]. Because the channel mutated at residues 261–265 adopts an open, Ca2+-selective conformation in the absence of STIM1, these findings suggest that the N-terminal ORAI1 peptide interacts with another region of ORAI1, rather than with STIM1, during channel gating.
3.5.2. Gating of the ORAI1 Channel
STIM binding causes movements of ORAI1 transmembrane helices TM1 and TM4 that can be detected directly or inferred from biochemical experiments [46a,87,88]. The TM1 helix movements are part of the gating movement that opens the channel. The presence of a gate in the external region of the pore was first evident from the fact that the G98C sulfhydryl was inaccessible to a cysteine-modifying methanethiosulfonate reagent in resting ORAI1G98C/E106D, but became accessible after store depletion [48]. This result indicated that a barrier to ion flux lies external to G98 and implied that STIM1 can trigger a movement in the vicinity of V102. Along those lines, replacement of either V102 or F99 with a small polar residue results in a constitutive and nonselective ion flux through the channel (Reference 48; Yamashita and Prakriya, personal communication). The simplest view is that these constitutively conducting channels are leaky channels that remain in the resting conformation. In silico simulations support this interpretation, indicating that water molecules in the narrowest part of the pore are less constrained in the V102A channel than in the wild-type channel and that the reduced local constraints on water reduce the energy barrier for Na+ ions to pass through this region [96]. The simulations show that the change in Na+ permeability occurs without appreciable displacement or rotation of the transmembrane helices. Experimentally, a truncated ORAI1 channel that deletes the entire N-terminal cytoplasmic region (residues 1–88) and replaces R91 with glycine is a closed channel, offering direct evidence for a gate within the nonpolar pore-lining segment of TM1, L95-V102 [46a].
The helix bundle consisting of the N-terminal cytoplasmic extensions of TM1 helices in the dOrai structure is a possible second location for the channel gate [43,90,98]. Blockade of the R91C channel by Cd2+ and by diamide cross-linking [48,96] has been taken as evidence for such an internal gate, although the chemical cross-linking pattern of the ORAI1 region from S90C to L96C suggests that the helices have considerable flexibility [42] and hence that the helix bundle observed in the dOrai crystal structure might be just one representative of an ensemble of conformations. Hydrophobic residues substituted at R91, most notably the SCID mutation R91W [21], can block ORAI1 channel current and can even override the constitutive conductance of the V102C channel, presumably due to stabilization of the bundle of helices [48,90,97]. A key question, given the multiple positive charges on the individual wild-type helices in residues 77–91, is whether the helix bundle is stably “closed” in wild-type ORAI1. One conceivable way to stabilize the helix bundle would be by association of intracellular anions with the basic region of the pore, plausible evidence for which is seen in the dOrai structure [43]. Assuming that there is in fact a second, internal gate in the closed ORAI1 channel, the expectation is that both gates must open in a concerted way when STIM activates the channel.
Gating of ORAI1 involves conformational changes in other parts of the channel [86,87], including at residue P245 in the middle of the TM4 helix [88] and at a hinge region in the TM4 cytoplasmic extension [46b,87]. It seems very probable that all the ORAI1 transmembrane helices rearrange during gating. However, judging from the relatively small movements that have been detected in the pore-lining TM1 helix due to gating (References 41 and 42; Yamashita and Prakriya, personal communication), these rearrangements are likely to be subtle.
Detecting gating movements experimentally is important for further analyses of ORAI1 channel function, but the extremely small single-channel current has impeded the use of conventional electrophysiological approaches. Two new approaches have circumvented this roadblock. A Tb3+ luminescence assay that utilizes the intrinsic ability of Tb3+ to bind to the E106 Ca2+ site in ORAI1 channels clearly reports the gating movement at the mouth of the channel triggered by recombinant STIM1 [46a]. In cells, ORAI1 channels with genetically fused calcium indicators have been successful in measuring Ca2+ entry at the internal mouth of single ORAI1 channels [99]. These complementary tools can be used to study ORAI1 channel gating by STIM1 in parallel measurements made in cells and made with purified recombinant STIM1 and ORAI1 in vitro.
3.6. Conclusions
In summary, ER store depletion is a primary event in Ca2+ signaling that results in CRAC current. We have presented a glimpse of our current understanding of the structural changes that lead to activation of the ER Ca2+ sensor STIM1, its interaction with the plasma membrane Ca2+ channel ORAI1, and the gating of ORAI1. We have also pointed out where current knowledge of each of these processes is incomplete. Challenges such as resolving the structure of the entire STIM1 molecule in its resting state and its activated state, precisely defining the STIM-ORAI binding sites that control recruitment and gating of the channel, and solving the structure of the open ORAI1 channel physiologically gated by STIM1 will be met by further biophysical and biochemical studies of STIM1 and ORAI.
References
- 1.
- McDonough, P.M., Button, D.C. (1989) Measurement of cytoplasmic calcium concentration in cell suspensions: Correction for extracellular Fura-2 through use of Mn2+ and probenecid. Cell Calcium. 10:171-180. [PubMed: 2720761]
- 2.
- Clapham, D.E. (2007) Calcium signaling. Cell. 131:1047-1058. [PubMed: 18083096]
- 3.
- Koch, G.L. (1990) The endoplasmic reticulum and calcium storage. Bioessays. 12:527-531. [PubMed: 2085319]
- 4.
- Verkhratsky, A. (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 85:201-279. [PubMed: 15618481]
- 5.
- Putney, J.W.Jr. (1986) A model for receptor-regulated calcium entry. Cell Calcium. 7:1-12. [PubMed: 2420465]
- 6.
- Takemura, H., Hughes, A.R., Thastrup, O., Putney, J.W. (1989) Activation of calcium entry by the tumour promoter thapsigargin in parotid acinar cells. J Biol Chem. 264:12266-12271. [PubMed: 2663854]
- 7.
- Berridge, M.J. (2002) The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium. 32:235-249. [PubMed: 12543086]
- 8.
- Bird, G.S., Hwang, S.Y., Smyth, J.T., Fukushima, M., Boyles, R.R., Putney, J.W.Jr. (2009) STIM1 is a calcium sensor specialized for digital signaling. Curr Biol. 19:1724-1729. [PMC free article: PMC3552312] [PubMed: 19765994]
- 9.
- Lewis, R.S., Cahalan, M.D. (1989) Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1:99-112. [PMC free article: PMC361429] [PubMed: 2519622]
- 10.
- Hoth, M., Penner, R. (1993) Calcium release-activated calcium current in rat mast cells. J Physiol. 465:359-386. [PMC free article: PMC1175434] [PubMed: 8229840]
- 11.
- Hoth, M., Penner, R. (1992) Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 355:353-356. [PubMed: 1309940]
- 12.
- Zweifach, A., Lewis, R.S. (1993) Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci USA. 90:6295-6299. [PMC free article: PMC46915] [PubMed: 8392195]
- 13.
- Hofer, A.M., Fasolato, C., Pozzan, T. (1998) Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: A study using simultaneous measurements of ICRAC and intraluminal [Ca2+]. J Cell Biol. 140:325-334. [PMC free article: PMC2132570] [PubMed: 9442108]
- 14.
- Putney, J.W., Bird, G.S. (1993) The inositol phosphate-calcium signaling system in nonexcitable cells. Endocr Rev. 14:610-631. [PubMed: 8262009]
- 15.
- Zweifach, A., Lewis, R.S. (1995) Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and -independent mechanisms. J Biol Chem. 270:14445-14451. [PubMed: 7540169]
- 16.
- Fierro, L., Parekh, A.B. (1999) Fast calcium-dependent inactivation of calcium release-activated calcium current (CRAC) in RBL-1 cells. J Membr Biol. 168:9-17. [PubMed: 10051685]
- 17.
- Prakriya, M., Lewis, R.S. (2003) CRAC channels: Activation, permeation, and the search for a molecular identity. Cell Calcium. 33:311-321. [PubMed: 12765678]
- 18.
- Roos, J., DiGregorio, P.J., Yeromin, A.V., Ohlsen, K., Lioudyno, M., Zhang, S., Safrina, O., et al. (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 169:435-445. [PMC free article: PMC2171946] [PubMed: 15866891]
- 19.
- Liou, J., Kim, M.L., Heo, W.D., Jones, J.T., Myers, J.W., Ferrell, J.E.Jr., Meyer, T. (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 15:1235-1241. [PMC free article: PMC3186072] [PubMed: 16005298]
- 20.
- Yeromin, A.V., Roos, J., Stauderman, K.A., Cahalan, M.D. (2004) A store-operated calcium channel in Drosophila S2 cells. J Gen Physiol. 123:167-182. [PMC free article: PMC2217434] [PubMed: 14744989]
- 21.
- Feske, S., Gwack, Y., Prakriya, M., Srikanth, S., Puppel, S.H., Tanasa, B., Hogan, P.G., Lewis, R.S., Daly, M., Rao, A. (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 441:179-185. [PubMed: 16582901]
- 22.
- Vig, M., Peinelt, C., Beck, A., Koomoa, D.L., Rabah, D., Koblan-Huberson, M., Kraft, S., et al. (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 312:1220-1223. [PMC free article: PMC5685805] [PubMed: 16645049]
- 23.
- Zhang, S.L., Yeromin, A.V., Zhang, X.H., Yu, Y., Safrina, O., Penna, A., Roos, J., Stauderman, K.A., Cahalan, M.D. (2006) Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci USA. 103:9357-9362. [PMC free article: PMC1482614] [PubMed: 16751269]
- 24.
- Peinelt, C., Vig, M., Koomoa, D.L., Beck, A., Nadler, M.J., Koblan-Huberson, M., Lis, A., Fleig, A., Penner, R., Kinet, J.P. (2006) Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat Cell Biol. 8:771-773. [PMC free article: PMC5685802] [PubMed: 16733527]
- 25.
- Soboloff, J., Spassova, M.A., Tang, X.D., Hewavitharana, T., Xu, W., Gill, D.L. (2006) Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 281:20661-20665. [PubMed: 16766533]
- 26.
- Mercer, J.C., Dehaven, W.I., Smyth, J.T., Wedel, B., Boyles, R.R., Bird, G.S., Putney, J.W.Jr. (2006) Large store-operated calcium selective currents due to coexpression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 281:24979-24990. [PMC free article: PMC1633822] [PubMed: 16807233]
- 27.
- Wu, M.M., Buchanan, J., Luik, R.M., Lewis, R.S. (2006) Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 174(6):803-813. [PMC free article: PMC2064335] [PubMed: 16966422]
- 28.
- Zhang, S.L., Yu, Y., Roos, J., Kozak, J.A., Deerinck, T.J., Ellisman, M.H., Stauderman, K.A., Cahalan, M.D. (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 437:902-905. [PMC free article: PMC1618826] [PubMed: 16208375]
- 29.
- Prakriya, M., Feske, S., Gwack, Y., Srikanth, S., Rao, A., Hogan, P.G. (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature. 443:230-233. [PubMed: 16921383]
- 30.
- Luik, R.M., Wu, M.M., Buchanan, J., Lewis, R.S. (2006) The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 174:815-825. [PMC free article: PMC2064336] [PubMed: 16966423]
- 31.
- Xu, P., Lu, J., Li, Z., Yu, X., Chen, L., Xu, T. (2006) Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem Biophys Res Commun. 350:969-976. [PubMed: 17045966]
- 32.
- Park, C.Y., Hoover, P.J., Mullins, F.M., Bachhawat, P., Covington, E.D., Raunser, S., Walz, T., Garcia, K.C., Dolmetsch, R.E., Lewis, R.S. (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 136:876-890. [PMC free article: PMC2670439] [PubMed: 19249086]
- 33.
- Liou, J., Fivaz, M., Inoue, T., Meyer, T. (2007) Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 104:9301-9306. [PMC free article: PMC1890489] [PubMed: 17517596]
- 34.
- Stathopulos, P.B., Li, G.Y., Plevin, M.J., Ames, J.B., Ikura, M. (2006) Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J Biol Chem. 281:35855-35862. [PubMed: 17020874]
- 35.
- Li, Z., Lu, J., Xu, P., Xie, X., Chen, L., Xu, T. (2007) Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem. 282:29448-29456. [PubMed: 17702753]
- 36.
- Hogan, P.G., Lewis, R.S., Rao, A. (2010) Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 28:491-533. [PMC free article: PMC2861828] [PubMed: 20307213]
- 37.
- Gudlur, A., Zhou, Y., Hogan, P.G. (2013) STIM-ORAI interactions that control the CRAC channel. Curr Top Membr. 71:33-58. [PubMed: 23890110]
- 38.
- Yeromin, A.V., Zhang, S.L., Jiang, W., Yu, Y., Safrina, O., Cahalan, M.D. (2006) Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 443:226-229. [PMC free article: PMC2756048] [PubMed: 16921385]
- 39.
- Gwack, Y., Srikanth, S., Feske, S., Cruz-Guilloty, F., Oh-hora, M., Neems, D.S., Hogan, P.G., Rao, A. (2007) Biochemical and functional characterization of Orai proteins. J Biol Chem. 282:16232-16243. [PubMed: 17293345]
- 40.
- Mignen, O., Thompson, J.L., Shuttleworth, T.J. (2008) Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol. 586:419-425. [PMC free article: PMC2375595] [PubMed: 18006576]
- 41.
- McNally, B.A., Yamashita, M., Engh, A., Prakriya, M. (2009) Structural determinants of ion permeation in CRAC channels. Proc Natl Acad Sci. 106:22516-22521. [PMC free article: PMC2792162] [PubMed: 20018736]
- 42.
- Zhou, Y., Ramachandran, S., Oh-Hora, M., Rao, A., Hogan, P.G. (2010) Pore architecture of the ORAI1 store-operated calcium channel. Proc Natl Acad Sci USA. 107:4896-4901. [PMC free article: PMC2841875] [PubMed: 20194792]
- 43.
- Hou, X., Pedi, L., Diver, M.M., Long, S.B. (2012) Crystal structure of the calcium release-activated calcium channel Orai. Science. 338:1308-1313. [PMC free article: PMC3695727] [PubMed: 23180775]
- 44.
- Amcheslavsky, A., Wood, M.L., Yeromin, A.V., Parker, I., Freites, J.A., Tobias, D.J., Cahalan, M.D. (2015) Molecular biophysics of Orai store-operated Ca2+ channels. Biophys J. 108:237-246. [PMC free article: PMC4302196] [PubMed: 25606672]
- 45.
- Yamashita, M., Navarro-Borelly, L., McNally, B.A., Prakriya, M. (2007) Orai1 mutations alter ion permeation and Ca2+-dependent fast inactivation of CRAC channels: Evidence for coupling of permeation and gating. J Gen Physiol. 130:525-540. [PMC free article: PMC2151669] [PubMed: 17968026]
- 46.
- a. Gudlur, A., Quintana, A., Zhou, Y., Hirve, N., Mahapatra, S., Hogan, P.G. (2014) STIM1 triggers a gating rearrangement at the extracellular mouth of the ORAI1 channel. Nat Commun. 5:5164; [PMC free article: PMC4376667] [PubMed: 25296861]
b. Zhou, Y., Cai, X., Loktionova, N.A., Wang, X., Nwokonko, R.M., Wang, X., Wang, Y., Rothberg, B.S., Trebak, M., Gill, D.L. (2016) The STIM1 binding site nexus remotely controls Orai1 channel gating. Nat Commun. 7:13725. [PMC free article: PMC5155162] [PubMed: 27929067] - 47.
- Frischauf, I., Zayats, V., Deix, M., Hochreiter, A., Jardin, I., Muik, M., Lackner, B., et al. (2015) A calcium-accumulating region, CAR, in the channel Orai1 enhances Ca2+ permeation and SOCE-induced gene transcription. Sci Signal. 8:ra131. [PMC free article: PMC5117258] [PubMed: 26696631]
- 48.
- McNally, B.A., Somasundaram, A., Yamashita, M., Prakriya, M. (2012) Gated regulation of CRAC channel ion selectivity by STIM1. Nature. 482:241-245. [PMC free article: PMC3276717] [PubMed: 22278058]
- 49.
- Varnai, P., Toth, B., Toth, D.J., Hunyady, L., Balla, T. (2007) Visualization and manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1-Orai1 complex. J Biol Chem. 282:29678-29690. [PubMed: 17684017]
- 50.
- Stathopulos, P.B., Schindl, R., Fahrner, M., Zheng, L., Gasmi-Seabrook, G.M., Muik, M., Romanin, C., Ikura, M. (2013) STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat Commun. 4:2963. [PMC free article: PMC3927877] [PubMed: 24351972]
- 51.
- Oritani, K., Kincade, P.W. (1996) Identification of stromal cell products that interact with pre-B cells. J Cell Biol. 134:771-782. [PMC free article: PMC2120935] [PubMed: 8707854]
- 52.
- Manji, S.S., Parker, N.J., Williams, R.T., van Stekelenburg, L., Pearson, R.B., Dziadek, M., Smith, P.J. (2000) STIM1: A novel phosphoprotein located at the cell surface. Biochim Biophys Acta. 1481:147-155. [PubMed: 11004585]
- 53.
- Williams, R.T., Senior, P.V., Van Stekelenburg, L., Layton, J.E., Smith, P.J., Dziadek, M.A. (2002) Stromal interaction molecule 1 (STIM1), a transmembrane protein with growth suppressor activity, contains an extracellular SAM domain modified by N-linked glycosylation. Biochim Biophys Acta. 1596:131-137. [PubMed: 11983428]
- 54.
- Spassova, M.A., Soboloff, J., He, L.P., Xu, W., Dziadek, M.A., Gill, D.L. (2006) STIM1 has a plasma membrane role in the activation of store-operated Ca2+ channels. Proc Natl Acad Sci USA. 103:4040-4045. [PMC free article: PMC1449642] [PubMed: 16537481]
- 55.
- Mignen, O., Thompson, J.L., Shuttleworth, T.J. (2007) STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J Physiol. 579:703-715. [PMC free article: PMC2151373] [PubMed: 17158173]
- 56.
- Lefkimmiatis, K., Srikanthan, M., Maiellaro, I., Moyer, M.P., Curci, S., Hofer, A.M. (2009) Store-operated cyclic AMP signaling mediated by STIM1. Nat Cell Biol. 11:433-442. [PubMed: 19287379]
- 57.
- Park, C.Y., Shcheglovitov, A., Dolmetsch, R. (2010) The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science. 330:101-105. [PubMed: 20929812]
- 58.
- Collins, S.R., Meyer, T. (2011) Evolutionary origins of STIM1 and STIM2 within ancient Ca2+ signaling systems. Trends Cell Biol. 21:202-211. [PMC free article: PMC3175768] [PubMed: 21288721]
- 59.
- Cahalan, M.D. (2009) STIMulating store-operated Ca2+ entry. Nat Cell Biol. 11:669-677. [PMC free article: PMC2721799] [PubMed: 19488056]
- 60.
- Soboloff, J., Rothberg, B.S., Madesh, M., Gill, D.L. (2012) STIM proteins: Dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 13:549-565. [PMC free article: PMC3458427] [PubMed: 22914293]
- 61.
- Stathopulos, P.B., Zheng, L., Li, G.Y., Plevin, M.J., Ikura, M. (2008) Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 135:110-122. [PubMed: 18854159]
- 62.
- Huang, G.N., Zeng, W., Kim, J.Y., Yuan, J.P., Han, L., Muallem, S., Worley, P.F. (2006) STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol. 8:1003-1010. [PubMed: 16906149]
- 63.
- Muik, M., Frischauf, I., Derler, I., Fahrner, M., Bergsmann, J., Eder, P., Schindl, R., et al. (2008) Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 283:8014-8022. [PubMed: 18187424]
- 64.
- Zhang, S.L., Kozak, J.A., Jiang, W., Yeromin, A.V., Chen, J., Yu, Y., Penna, A., Shen, W., Chi, V., Cahalan, M.D. (2008) Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J Biol Chem. 283:17662-17671. [PMC free article: PMC2427323] [PubMed: 18420579]
- 65.
- Zhou, Y., Meraner, P., Kwon, H.T., Machnes, D., Oh-hora, M., Zimmer, J., Huang, Y., Stura, A., Rao, A., Hogan, P.G. (2010) STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat Struct Mol Biol. 17:112-116. [PMC free article: PMC2902271] [PubMed: 20037597]
- 66.
- Yuan, J.P., Zeng, W., Dorwart, M.R., Choi, Y.J., Worley, P.F., Muallem, S. (2009) SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 11:337-343. [PMC free article: PMC2663385] [PubMed: 19182790]
- 67.
- Kawasaki, T., Lange, I., Feske, S. (2009) A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 385:49-54. [PMC free article: PMC2821023] [PubMed: 19433061]
- 68.
- Zhou, Y., Srinivasan, P., Razavi, S., Seymour, S., Meraner, P., Gudlur, A., Stathopulos, P.B., Ikura, M., Rao, A., Hogan, P.G. (2013) Initial activation of STIM1, the regulator of store-operated calcium entry. Nat Struct Mol Biol. 20:973-981. [PMC free article: PMC3784406] [PubMed: 23851458]
- 69.
- Ma, G., Wei, M., He, L., Liu, C., Wu, B., Zhang, S.L., Jing, J., et al. (2015) Inside-out Ca2+ signalling prompted by STIM1 conformational switch. Nat Commun. 6:7826. [PMC free article: PMC4509486] [PubMed: 26184105]
- 70.
- Yang, X., Jin, H., Cai, X., Li, S., Shen, Y. (2012) Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc Natl Acad Sci USA. 109:5657-5662. [PMC free article: PMC3326449] [PubMed: 22451904]
- 71.
- Fahrner, M., Muik, M., Schindl, R., Butorac, C., Stathopulos, P., Zheng, L., Jardin, I., Ikura, M., Romanin, C. (2014) A coiled-coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1). J Biol Chem. 289:33231-33244. [PMC free article: PMC4246082] [PubMed: 25342749]
- 72.
- Luik, R.M., Wang, B., Prakriya, M., Wu, M.M., Lewis, R.S. (2008) Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 454:538-542. [PMC free article: PMC2712442] [PubMed: 18596693]
- 73.
- Hogan, P.G. (2015) The STIM1-ORAI1 microdomain. Cell Calcium. 58:357-367. [PMC free article: PMC4564343] [PubMed: 26215475]
- 74.
- Brandman, O., Liou, J., Park, W.S., Meyer, T. (2007) STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 131:1327-1339. [PMC free article: PMC2680164] [PubMed: 18160041]
- 75.
- Huang, Y., Zhou, Y., Wong, H.C., Chen, Y., Chen, Y., Wang, S., Castiblanco, A., Liu, A., Yang, J.J. (2009) A single EF-hand isolated from STIM1 forms dimer in the absence and presence of Ca2+. FEBS J. 276:5589-5597. [PMC free article: PMC2866080] [PubMed: 19694801]
- 76.
- Furukawa, Y., Teraguchi, S., Ikegami, T., Dagliyan, O., Jin, L., Hall, D., Dokholyan, N.V., et al. (2014) Intrinsic disorder mediates cooperative signal transduction in STIM1. J Mol Biol. 426:2082-2097. [PubMed: 24650897]
- 77.
- Muik, M., Fahrner, M., Schindl, R., Stathopulos, P., Frischauf, I., Derler, I., Plenk, P., et al. (2011) STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 30:1678-1689. [PMC free article: PMC3101990] [PubMed: 21427704]
- 78.
- Fernandez-Busnadiego, R., Saheki, Y., De Camilli, P. (2015) Three-dimensional architecture of extended synaptotagmin-mediated endoplasmic reticulum-plasma membrane contact sites. Proc Natl Acad Sci USA. 112:E2004-E2013. [PMC free article: PMC4413308] [PubMed: 25787254]
- 79.
- Perni, S., Dynes, J.L., Yeromin, A.V., Cahalan, M.D., Franzini-Armstrong, C. (2015) Nanoscale patterning of STIM1 and Orai1 during store-operated Ca2+ entry. Proc Natl Acad Sci USA. 112:E5533-E5554. [PMC free article: PMC4603497] [PubMed: 26351694]
- 80.
- Korzeniowski, M.K., Manjarres, I.M., Varnai, P., Balla, T. (2010) Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci Signal. 3:ra82. [PMC free article: PMC3408607] [PubMed: 21081754]
- 81.
- Yu, J., Zhang, H., Zhang, M., Deng, Y., Wang, H., Lu, J., Xu, T., Xu, P. (2013) An aromatic amino acid in the coiled-coil 1 domain plays a crucial role in the auto-inhibitory mechanism of STIM1. Biochem J. 454:401-409. [PubMed: 23795811]
- 82.
- Muik, M., Fahrner, M., Derler, I., Schindl, R., Bergsmann, J., Frischauf, I., Groschner, K., Romanin, C. (2009) A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J Biol Chem. 284:8421-8426. [PMC free article: PMC2659200] [PubMed: 19189966]
- 83.
- Covington, E.D., Wu, M.M., Lewis, R.S. (2010) Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol Biol Cell. 21:1897-1907. [PMC free article: PMC2877647] [PubMed: 20375143]
- 84.
- Navarro-Borelly, L., Somasundaram, A., Yamashita, M., Ren, D., Miller, R.J., Prakriya, M. (2008) STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 586:5383-5401. [PMC free article: PMC2655373] [PubMed: 18832420]
- 85.
- Barr, V.A., Bernot, K.M., Srikanth, S., Gwack, Y., Balagopalan, L., Regan, C.K., Helman, D.J., et al. (2008) Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: Puncta and distal caps. Mol Biol Cell. 19:2802-2817. [PMC free article: PMC2441672] [PubMed: 18448669]
- 86.
- McNally, B.A., Somasundaram, A., Jairaman, A., Yamashita, M., Prakriya, M. (2013) The C- and N-terminal STIM1 binding sites on Orai1 are required for both trapping and gating CRAC channels. J Physiol. 591:2833-2850. [PMC free article: PMC3690689] [PubMed: 23613525]
- 87.
- Tirado-Lee, L., Yamashita, M., Prakriya, M. (2015) Conformational changes in the Orai1 C-terminus evoked by STIM1 Binding. PLoS One. 10:e0128622. [PMC free article: PMC4452722] [PubMed: 26035642]
- 88.
- Palty, R., Stanley, C., Isacoff, E.Y. (2015) Critical role for Orai1 C-terminal domain and TM4 in CRAC channel gating. Cell Res. 25:963-980. [PMC free article: PMC4528054] [PubMed: 26138675]
- 89.
- Frischauf, I., Muik, M., Derler, I., Bergsmann, J., Fahrner, M., Schindl, R., Groschner, K., Romanin, C. (2009) Molecular determinants of the coupling between STIM1 and Orai channels: Differential activation of Orai1-3 channels by a STIM1 coiled-coil mutant. J Biol Chem. 284:21696-21706. [PMC free article: PMC2755892] [PubMed: 19506081]
- 90.
- Derler, I., Fahrner, M., Carugo, O., Muik, M., Bergsmann, J., Schindl, R., Frischauf, I., Eshaghi, S., Romanin, C. (2009) Increased hydrophobicity at the N terminus/membrane interface impairs gating of the severe combined immunodeficiency-related ORAI1 mutant. J Biol Chem. 284:15903-15915. [PMC free article: PMC2708886] [PubMed: 19366689]
- 91.
- Lee, K.P., Yuan, J.P., Zeng, W., So, I., Worley, P.F., Muallem, S. (2009) Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc Natl Acad Sci USA. 106:14687-14692. [PMC free article: PMC2732841] [PubMed: 19706554]
- 92.
- Zhou, Y., Wang, X., Wang, X., Loktionova, N.A., Cai, X., Nwokonko, R.M., Vrana, E., Wang, Y., Rothberg, B.S., Gill, D.L. (2015) STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat Commun. 6:8395. [PMC free article: PMC4598629] [PubMed: 26399906]
- 93.
- Yuan, J.P., Zeng, W., Huang, G.N., Worley, P.F., Muallem, S. (2007) STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol. 9:636-645. [PMC free article: PMC2699187] [PubMed: 17486119]
- 94.
- Lis, A., Zierler, S., Peinelt, C., Fleig, A., Penner, R. (2010) A single lysine in the N-terminal region of store-operated channels is critical for STIM1-mediated gating. J Gen Physiol. 136:673-686. [PMC free article: PMC2995155] [PubMed: 21115697]
- 95.
- Bergsmann, J., Derler, I., Muik, M., Frischauf, I., Fahrner, M., Pollheimer, P., Schwarzinger, C., Gruber, H.J., Groschner, K., Romanin, C. (2011) Molecular determinants within N terminus of Orai3 protein that control channel activation and gating. J Biol Chem. 286:31565-31575. [PMC free article: PMC3173129] [PubMed: 21724845]
- 96.
- Dong, H., Fiorin, G., Carnevale, V., Treptow, W., Klein, M.L. (2013) Pore waters regulate ion permeation in a calcium release-activated calcium channel. Proc Natl Acad Sci USA. 110:17332-17337. [PMC free article: PMC3808583] [PubMed: 24101457]
- 97.
- Zhang, S.L., Yeromin, A.V., Hu, J., Amcheslavsky, A., Zheng, H., Cahalan, M.D. (2011) Mutations in Orai1 transmembrane segment 1 cause STIM1-independent activation of Orai1 channels at glycine 98 and channel closure at arginine 91. Proc Natl Acad Sci USA. 108:17838-17843. [PMC free article: PMC3203763] [PubMed: 21987804]
- 98.
- Derler, I., Plenk, P., Fahrner, M., Muik, M., Jardin, I., Schindl, R., Gruber, H.J., Groschner, K., Romanin, C. (2013) The extended transmembrane Orai1 N-terminal (ETON) region combines binding interface and gate for Orai1 activation by STIM1. J Biol Chem. 288:29025-29034. [PMC free article: PMC3790000] [PubMed: 23943619]
- 99.
- Dynes, J.L., Amcheslavsky, A., Cahalan, M.D. (2016) Genetically targeted single-channel optical recording reveals multiple Orai1 gating states and oscillations in calcium influx. Proc Natl Acad Sci USA. 113:440-445. [PMC free article: PMC4720334] [PubMed: 26712003]
Aparna Gudlur
Division of Signaling and Gene Expression
La Jolla Institute for Allergy and Immunology
La Jolla, California
Patrick Hogan
Division of Signaling and Gene Expression
La Jolla Institute for Allergy and Immunology
La Jolla, California
Publication Details
Author Information and Affiliations
Authors
Aparna Gudlur and Patrick Hogan.Copyright
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
Publisher
CRC Press/Taylor & Francis, Boca Raton (FL)
NLM Citation
Gudlur A, Hogan P. Signaling ER Store Depletion to Plasma Membrane Orai Channels. In: Kozak JA, Putney JW Jr., editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. Chapter 3. doi: 10.1201/9781315152592-3


)%2C%20showing%20the%20Ca2%2B%20binding%20site%2C%20a%20proposed%20vestibule%20comprised%20of%20aspartate%20residues%20in%20TM1-TM2%20loop%2C%20nonpolar%20region%20with%20the%20gate%20at%20V102%2C%20and%20a%20part%20of%20the%20basic%20region%20with%20R91.&p=BOOKS&id=531432_fig3_2b.jpg "Click on image to zoom")
