Clinical Description
Hyperphosphatemic familial tumoral calcinosis (HFTC) is characterized most commonly by tumoral calcinosis or ectopic calcifications (typically in periarticular soft tissues exposed to repetitive trauma or prolonged pressure) and hyperostosis (typically manifesting as painful swelling overlying the diaphyses of long bones). Onset of lesions typically occurs in the first two decades of life. The dental phenotype unique to HFTC includes enamel hypoplasia, short and bulbous roots, pulp chamber and canal obliterations, and pulp stones. Less frequently reported findings include large and small vessel calcifications, testicular microlithiasis, and angioid streaks of the retina.
HFTC results from a relative deficiency of or resistance to the phosphate-regulating hormone FGF23, leading to hyperphosphatemia due to increased renal phosphate reabsorption and elevated or inappropriately normal 1,25D production, which promotes gastrointestinal absorption of phosphorus and calcium (for more details see Pathophysiology).
In a review of the medical literature, Rafaelsen et al [2014] found 56 individuals with HFTC from 35 different families with a molecularly confirmed diagnosis of HFTC (i.e., biallelic germline loss-of-function pathogenic variants in FGF23, GALNT3, or KL).
Since the Rafaelsen et al [2014] review, 14 additional individuals with GALNT3-associated HFTC [Krstevska et al 2012, Favia et al 2014, Finer et al 2014, Demellawy et al 2015, Masi et al 2015, Vieira et al 2015, Jost et al 2016, Ramnitz et al 2016] and five individuals with FGF23-associated HFTC [Shah et al 2014, Keskar et al 2015, Jost et al 2016] have been reported.
For unknown reasons, the manifestations of HFTC vary among family members with the same pathogenic variants and genetic background and similar biochemical profiles. One member may have profound, extensive tumoral calcinosis while others are symptom free [Ramnitz et al 2016]. Of note, since HFTC has been reported in many pedigrees with multigenerational consanguinity, some phenotypic features found in association with HFTC may represent the expression of other autosomal recessive disorders.
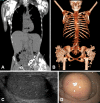
Tumoral calcinosis represents ectopic calcification in the skin and/or subcutaneous tissue in periarticular locations consisting primarily of hydroxyapatite [Boskey et al 1983, Slavin et al 1993] and – in some reports – calcium carbonate [Annamunthodo 1960]. The lesions often occur at sites of repeat trauma and/or pressure, with the lateral hips being the most frequently reported site. However, the clinical spectrum of tumoral calcinosis is widely variable, ranging from no lesions [Ramnitz et al 2016], to isolated eyelid calcifications [Ichikawa et al 2006], to periarticular calcifications of significant size ().
These lesions can be extremely painful and debilitating; some may progress in size, perforate the skin, and drain liquid hydroxyapatite (also known as "milk of calcium"), which is often confused with purulent drainage. Such lesions often heal poorly.
Depending on the size and location, tumoral calcinosis can significantly impair range of motion or lead to frozen joints.
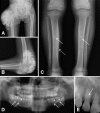
Hyperostosis typically presents as painful swelling in the areas overlying the diaphyses of the long bones (especially the tibiae), and can be the first observed manifestation of HFTC [Clarke et al 1984, Frishberg et al 2005, Ichikawa et al 2007a, Olauson et al 2008, Ichikawa et al 2010, Joseph et al 2010, Favia et al 2014, Jost et al 2016].
Some patients experience repeat episodes of diaphysitis (inflammation of the shaft of a long bone) involving a number of sites including the ulna, tibia, metacarpal bones, and radius [Clarke et al 1984, Campagnoli et al 2006b, Gok et al 2009].
Biopsies of hyperostosis lesions demonstrate reactive new bone surrounded by fibroblastic stroma infiltrated with plasma cells, lymphocytes, and polymorphonuclear cells; cultures are negative [Clarke et al 1984, Ichikawa et al 2010].
Vascular calcifications, identified in affected individuals ranging in age from three to 58 years, are rarely the presenting feature of HFTC. While the true frequency is unknown, the risk of vascular calcifications in HFTC is likely increased over that of the general population.
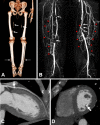
The calcifications are often incidental findings on radiologic studies, and patients are often asymptomatic. Some patients have significant symptoms of peripheral vascular insufficiency such as pain, cold extremities, and decreased peripheral pulses. Small to large vessel calcifications have been identified in variable anatomic locations including the cerebral vasculature, lower-extremity vasculature (), coronary arteries (), mesenteric arteries, renal arteries, carotid arteries, iliac vessels, and aorta [Martinez et al 1990, Li Voon Chong et al 1999, Ichikawa et al 2007c, Lammoglia & Mericq 2009, Rafaelsen et al 2014, Masi et al 2015, Ramnitz et al 2016]. The most severe reported consequences of vascular calcifications were below- and above-the-knee amputations in one man and digit amputations in the man's two affected sisters [Shah et al 2014].
Dental phenotype characteristic of HFTC includes enamel hypoplasia, short and bulbous roots, pulp chamber and canal obliterations, and pulp stones () [Lyles et al 1985, Burkes et al 1991, Benet-Pagès et al 2005, Laleye et al 2008, Krstevska et al 2012, Favia et al 2014].
Dental findings, reported to be the presenting sign in some individuals [Dumitrescu et al 2009, Vieira et al 2015], may be the most common imaging findings of HFTC as they can occur in the absence of calcinosis.
Renal calcifications have been described in two individuals, one of whom had mild impairment in renal function [Chefetz et al 2005, Bergwitz et al 2009].
Inflammation. The cause and role of inflammation in the pathogenesis of the calcific tumors is unclear. Microscopy of the lesions reveals macrophage engulfment of hydroxyapatite crystals, which probably release inflammatory cytokines, the likely etiology of the observed systemic inflammation [Ramnitz et al 2016].
Five individuals have had elevated markers of inflammation; i.e., C-reactive protein (CRP) and/or erythrocyte sedimentation rate (ESR) [Garringer et al 2006, Masi et al 2009, Masi et al 2015, Ramnitz et al 2016]. In one individual clinical symptoms of inflammation included recurrent acute inflammatory polyarthritis of the shoulders, hands, and feet [Garringer et al 2006]. Two others experienced increased fatigue, inflammation-mediated anemia of chronic disease, and thrombocytosis; one also had intermittent fevers and a cutaneous inflammatory calcific reaction [Ramnitz et al 2016].
Testicular microlithiasis was reported in a boy age 14 years. The ultrasound examination showed increased echogenicity with scattered hyperechogenic lesions. A biopsy of one testicle revealed diffuse intratubular calcifications; oligo-azoospermia was noted on spermiogram [Campagnoli et al 2006b]. Similar findings were reported in a man age 45 years [Garringer et al 2007].
Angioid streaks of the retina were reported in four individuals with HFTC [McPhaul & Engel 1961, Yancovitch et al 2011]. One had symptomatic sudden onset of blurred vision attributed to choroidal neovascularization secondary to angioid streaks 14 years after the initial diagnosis of HFTC [McGrath et al 2010].