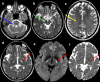
Clinical Description
The phenotypic spectrum of CLCN2-related leukoencephalopathy ranges from childhood onset with mild ataxia, learning disabilities, and headaches to adult onset with mild ataxia and decreased vision [Depienne et al 2013]. Infertility or involuntary childlessness in adulthood has been observed in four adult males [Di Bella et al 2014; Authors, personal observation]. Several individuals with psychiatric concerns (depression or psychosis) and/or auditory abnormalities including mild hearing loss, tinnitus, and vertigo have been observed. Other medical concerns occurring in a few individuals may be rare parts of the phenotype or unrelated findings. The disease course has been reported as stable to slowly progressive; considering that adults were previously normal, extremely slow progression is most likely. All individuals reported to date have remained ambulatory. The oldest known individual with CLCN2-related leukoencephalopathy is age 70 years. No disease-related deaths have been reported to date.
To date, 31 individuals have been identified with biallelic pathogenic variants in CLCN2 [Depienne et al 2013; Di Bella et al 2014; Hanagasi et al 2015; Giorgio et al 2017; Zeydan et al 2017; Guo et al 2019; Hoshi et al 2019; Ngo et al 2020; Ozaki et al 2020; Parayil Sankaran et al 2020; Authors, personal observation]. The following description of the phenotypic features associated with this condition is based on these reports and personal observations.
Motor skills. Initial motor development is normal. At presentation, most affected individuals display signs of mild cerebellar ataxia with action tremor and gait instability; some also show mild signs of spasticity. Affected individuals remain ambulatory and do not require support for walking.
Cognitive skills. Some affected individuals have mild learning problems from early on, but most initially have normal intellect. Mild cognitive decline has been observed in some affected individuals [Guo et al 2019; Authors, personal observation]. One affected individual had severe cognitive impairment.
Psychiatric symptoms. Depression and psychosis or schizophrenia-like symptoms have been observed in several affected individuals [Depienne et al 2013; Authors, personal observation].
Headache. Some affected individuals complain of intermittent severe diffuse headaches.
Vision. Some affected individuals have a retinopathy or optic atrophy leading to mild visual impairment; blindness has been observed in one adult. Some affected individuals have visual field defects at formal testing, indicating subclinical retinopathy. Double vision has also been observed in some.
Auditory abnormality. Individuals with progressive hearing loss [Depienne et al 2013], unilateral mild hearing loss [Zeydan et al 2017], tinnitus [Guo et al 2019] and vertigo/dizziness [Parayil Sankaran et al 2020; Authors, personal observation] have been observed.
Male infertility. One male with azoospermia (but no neurologic dysfunction) was found to have CLCN2-related leukoencephalopathy during a workup for infertility [Di Bella et al 2014]. Three adult males diagnosed with CLCN2-related leukoencephalopathy have had a history of involuntary childlessness [Authors, personal observation]. To date, no affected males with offspring have been observed.
Other. A single individual with paroxysmal kinesigenic dyskinesia [Hanagasi et al 2015] has been reported.
An infant who presented with frequent generalized tonic-clonic seizures at age three months has been reported. Seizures were effectively controlled by anti-seizure medication (phenobarbital and valproate). A boy age 13 years with the exact same homozygous pathogenic variant as the infant did not have epilepsy [Ozaki et al 2020].
Two additional individuals with biallelic pathogenic variants in CLCN2 and epilepsy were identified [Authors, personal observation]. Since epilepsy has a fairly high incidence in the general population and any brain disease in itself enhances the risk of epilepsy, it is not possible to say whether this is related to CLCN2-related leukoencephalopathy. Animal studies are also inconclusive; one study reports interictal epileptic brain activity and a lowered seizure threshold in Clcn2-null mice [Cortez et al 2010], while two other studies report no alteration of seizure threshold in Clcn2-null mice [Bösl et al 2001, Blanz et al 2007]. Therefore, the link between partial or complete loss of chloride channel-2 function and epilepsy remains unresolved.