

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Personal Habits and Indoor Combustions. Lyon (FR): International Agency for Research on Cancer; 2012. (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 100E.)

Tobacco smoking was considered by previous IARC Working Groups in 1986, 1987 and 2002 (IARC, 1986, 1987, 2004a). Since that time, new data have become available, these have been incorporated into the Monograph, and taken into consideration in the present evaluation.
1. Exposure Data
1.1. Smoked tobacco products
Smoked forms of tobacco include various kinds of cigarettes (manufactured, hand-rolled, filtered, un-filtered and flavoured), cigars and pipes. While cigarette smoking, particularly manufactured cigarettes, is by far the main form of tobacco smoked globally, in some countries other forms of smoked tobacco are dominant (IARC, 2004a). In India, for example, bidis (made of coarse and uncured tobacco) account for about 60% of smoked tobacco products whereas cigarettes account for 20% (Ray & Gupta, 2009; IIPS, 2010). Water pipes, another form of smoked tobacco known by other various names such as gaza, hookah, narghile, shisha, hubble-bubble, are commonly smoked in the Eastern Mediterranean region, in some parts of Asia including India, and in North Africa (Asma et al., 2009).
1.2. Chemical composition of tobacco smoke
1.2.1 Smoke from cigarettes
One cubic cm of fresh, un-aged cigarette mainstream smoke [the smoke emerging from the mouth end of a cigarette during smoking] has about 4 × 109 particles with a mean diameter of about 0.2 µm (Borgerding & Klus, 2005). The size of the particles increases as the smoke ages. Temperatures in the burning cone of the cigarette are about 800 °C during the smoulder period between puffs and increase to 910–920 °C at the periphery of the cone during puffing (Borgerding & Klus, 2005). Hydrogen is generated in the glowing cone, resulting in an oxygen deficient reducing atmosphere (Borgerding & Klus, 2005). The approximate composition of mainstream smoke of a plain cigarette is summarized in Table 1.1 (Borgerding & Klus, 2005). The total particulate matter, after subtraction of the amounts of nicotine and water, is referred to as ‘tar’.
Table 1.1
Approximate chemical composition of mainstream smoke generated by a plain cigarette.
Over 5300 compounds have been identified in tobacco smoke (Rodgman & Perfetti, 2009). Classes of compounds include but are not limited to neutral gases, carbon and nitrogen oxides, amides, imides, lactams, carboxylic acids, lactones, esters, aldehydes, ketones, alcohols, phenols, amines, N-nitrosamines, N-heterocyclics, aliphatic hydrocarbons, monocyclic and polycyclic aromatic hydrocarbons (PAHs), nitriles, anhydrides, carbohydrates, ethers, nitro compounds and metals (Rodgman & Perfetti, 2009).
The addictive properties of tobacco smoke are attributed to nicotine, the principal tobacco alkaloid in smoke (Hukkanen et al., 2005). Minor tobacco alkaloids include nornicotine, anatabine and anabasine (Hukkanen et al., 2005). The tobacco alkaloids are not generally considered carcinogenic, but are accompanied by carcinogens in each puff of smoke.
There are over 70 carcinogens in tobacco smoke that have been evaluated by the IARC Monographs programme as having sufficient evidence for carcinogenicity in either laboratory animals or humans (IARC, 2004a). The different chemical classes of carcinogens and representatives of each are presented in Table 1.2 (IARC, 2004a). Sixteen of these – benzo[a]pyrene (BaP), 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and N′-nitrosonornicotine (NNN), 2-naphthylamine, 4-aminobiphenyl, formaldehyde, 1,3-butadiene, benzene, vinyl chloride, ethylene oxide, arsenic, beryllium, nickel compounds, chromium VI, cadmium, and polonium-210 – are classified as carcinogenic to humans (Group 1). Structures of some representative carcinogens in cigarette smoke are shown in Fig. 1.1. There are other likely carcinogens in cigarette smoke that have not been evaluated by the IARC Monographs programme. These include, for example, PAHs with incompletely characterized occurrence levels and carcinogenic activities; over 500 PAHs have been identified (Rodgman & Perfetti, 2006).
Table 1.2
Tobacco smoke carcinogens evaluated in the IARC Monographs.

Fig. 1.1
Structures of some representative tobacco smoke carcinogens
PAHs, tobacco-specific N-nitrosamines, aromatic amines, aldehydes and certain volatile organics likely contribute significantly to the carcinogenic activity of tobacco smoke (Hecht, 2003).
In the early part of the 20th century, PAHs were identified as carcinogenic constituents of coal tar (Phillips, 1983). They are products of incomplete combustion of all organic matter and occur, always as complex mixtures, in tars, soots, broiled foods, vehicle engine exhaust and tobacco smoke. PAHs are generally locally acting carcinogens, and some, such as the prototypic compound BaP, have strong carcinogenic activity on mouse skin and in rodent lung. Heterocyclic analogues of PAHs also occur in cigarette smoke. Concentrations of individual PAHs in mainstream cigarette smoke are generally in the range of 1–50 ng per cigarette (IARC, 2004a).
Among the carcinogenic N-nitrosamines in tobacco smoke are tobacco-specific N-nitrosamines, which are derived from, and structurally related to, the tobacco alkaloids. Two of the most important of these are NNK and NNN (Hecht & Hoffmann, 1988). Levels of NNK and NNN in cigarette smoke vary depending on tobacco type and other factors, but are frequently in the range of 50–200 ng per cigarette (IARC, 2004a).
Aromatic amines were first identified as human carcinogens from industrial exposures in the dye industry in the early part of the 20th century. They include the well known human bladder carcinogens 2-naphthylamine and 4-aminobiphenyl which, along with other isomers, are found in cigarette smoke, but their levels are generally quite low (1–20 ng per cigarette) (IARC, 2004a).
Aldehydes such as formaldehyde and acetaldehyde occur widely in the human environment and are also found in human blood. Concentrations of acetaldehyde and formaldehyde in cigarette smoke are far higher than those of PAHs, N-nitrosamines or aromatic amines but their carcinogenic activities are weak (Hecht, 2003). Cigarette mainstream smoke typically contains 10–30 µg formaldehyde/cigarette and 800–900 µg acetaldehyde/cigarette (IARC, 2004a).
Volatile hydrocarbons in cigarette smoke include 1,3-butadiene, a powerful multiorgan carcinogen in the mouse, and benzene, a known human leukaemogen. 1,3-Butadiene (20–40 µg/cigarette) and benzene (12–50 µg/cigarette) are two of the most prevalent strong carcinogens in cigarette smoke (IARC, 2004a).
In summary, cigarette smoke is an exceedingly complex mixture which contains over 5300 compounds including multiple toxicants and carcinogens.
1.2.2 Smoke from other tobacco products
Some constituents have been measured in roll-your-own cigarettes, and their levels are comparable to or higher than those in commercial brands. Carcinogen and toxicant levels expressed per unit are higher in cigars than in cigarettes because of their larger size, and in some instances are also higher per litre of smoke. Levels of nicotine and tobacco-specific nitrosamines were comparable in bidis and commercial Indian cigarettes; bidis also contain high levels of eugenol, as do kreteks. Levels of NNK and NNN in chuttas were considerably higher than in standard cigarettes (IARC, 2004a).
1.3. Prevalence of tobacco smoking
1.3.1 Data collection and methods
Data on smoking tobacco are available from WHO's Global Infobase (www.who.int/infobase) and the WHO Global Health Observatory (www.who.int/gho/en) – repositories of information on tobacco use and other risk factors in young people (13–15 years old) and adults (aged 15 years and over). The data span several years and are acquired from government reports, journals and unpublished sources. WHO has in the recent past used and modelled these data to produce estimates of tobacco smoking prevalence, published in the WHO Reports on the Global Tobacco Epidemic. For a complete explanation of methods used, the reader is referred to the Technical Note on Prevalence in the 3rd WHO Report on the Global Tobacco Epidemic (WHO, 2011). The six WHO regions are: EMRO, Eastern Mediterranean Region; EURO, European Region; AFRO, African Region; WPRO, Western Pacific Region; SEARO, South East Asian Region; AMRO, Region of the Americas. A listing of the countries in each region can be viewed at http://www.who.int/about/structure/en/index.html.
1.3.2 Distribution of smokers by WHO region and country
WHO estimates that in 2009, there was about 1.1 billion adult smokers worldwide, representing nearly a quarter (22%) of the global adult population (WHO, 2011). A disaggregation by the six WHO regions (Fig. 1.2) shows that over a third of smokers worldwide live in WPRO (highly influenced by the People’s Republic of China), followed by SEARO, which has around a fifth of the world’s smokers (influenced by India and Indonesia).
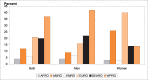
Fig. 1.2
Proportion of adult smokers by WHO region in 2009
The number of smokers in any country is a function of both the prevalence of smoking and the size of the population. A further disaggregation of the regions by country shows that a few countries account for a large proportion of tobacco smokers. Ranked in descending order of the number of smokers, the five countries of China, India, United States of America (USA), Russian Federation and Indonesia account for about 52% of adult smokers in the world, with China and India alone accounting for 40% (Fig. 1.3). Furthermore, nearly two-thirds of the world’s smokers live in only ten countries of the world.
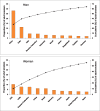
Fig. 1.3
Proportion and cumulative percentage of smokers in high-burden countries, in men (A) and women (B) in 2009
1.3.3 Distribution of smokers by sex
With a global average smoking prevalence of 36%, men account for just over 80% of all smokers. The male adult prevalence is 4–5 times that for women, at 8%. This difference varies across WHO regions. Smoking among men, concentrated in the five countries of China, India, Indonesia, Russian Federation and USA (Fig. 1.3), accounts for about 56% of global smoking among men. Women smokers are mostly concentrated in EURO and AMRO. These two regions account for 40% and 26% of all women smokers globally, respectively. The prevalences for women in these two regions are about half of those in men, whereas the difference is substantially greater in the other regions. Just as men smoke more than women everywhere, so too among young people, boys generally smoke more than girls. There is an increasing concern, however, that the gap may diminish, not because of a reduction in boys prevalence but because of an increase in the proportion of girls who are taking up smoking (Warren et al., 2006).
1.3.4 The four stage smoking model
(a) The four stages of tobacco use
Lopez et al. (1994) used trend data on smoking prevalence and tobacco attributable mortality to show the evolution of tobacco use in a country. Four stages of smoking and attributable mortality have been identified to represent the growth and eventual decline of smoking among men and women (Fig. 1.4).
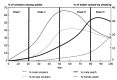
Fig. 1.4
The four stages of the tobacco epidemic
Stage 1 is characterized by low smoking prevalence in men (less than 15%) and very low in women (less than 10%). Death and disease from smoking are not apparent in this phase, as nearly all health effects from smoking are related to past smoking habits and their cumulative effects rather than current smoking. In Stage 2, smoking prevalence in men rapidly increases while it increases more slowly in women. Towards the end of this stage, smoking prevalence in men typically peaks to lie at 50–60%, with 10% of deaths in men attributable to smoking; deaths in women are comparatively fewer. After a protracted period of high smoking prevalence, Stage 3 shows a decline in smoking prevalence in men to around 40%. Smoking prevalence in women peaks and then begins to decline; towards the end of this stage the gap between men's and women's prevalence starts to narrow. However, smoking attributable deaths in men increase from around 10% to 25–30% within a span of three decades; in women the deaths are increasing but are still quite low. In the final Stage 4, smoking prevalences in both men and women continue to decline albeit relatively slowly in comparison with Stage 3, with the gap substantially narrowing to lie at around five percentage points, and as little as one percentage point in some countries. In Stage 4, smoking mortality in men peaks to between 30–35% and then declines to below 30% at the end of this period. In women, the health effects from past smoking persist, with increasing mortality, but remain lower than in men, and recently have begun to decline in some countries.
(b) Smoking prevalence worldwide
Using prevalence data for men and women collected in 2006 for 140 countries, WHO determined at which stage of the tobacco epidemic countries are in the model of Lopez et al. (1994). In Fig. 1.5, countries have been ranked by smoking prevalence in men in ascending order for Stages 1 and 2, and then in descending order for Stages 3 and 4. (Smoking prevalence in men is almost always higher than in women, with a few exceptions observed in the fourth stage.) While most countries fit the classification, there are a few exceptions, most of which in the last stage. Prevalence between Stage 3 and Stage 4 is discontinuous in both sexes. This is due to the classification followed, which puts countries with a relatively narrow difference in prevalence between men and women in Stage 4 even though their prevalence is largely comparable with those in Stage 3.
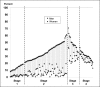
Fig. 1.5
Prevalence of smoking in 140 countries in 2009, staged according to the model by Lopez
Most African countries fall in the first stage of the smoking model, characterized by low smoking prevalence in men and very low prevalence in women. Three of the five high burden countries fall in stage 2 (India, Indonesia and China), with the rest comprising a combination of countries from Africa, South East Asia, eastern Europe and the Middle East. At this stage smoking prevalences in women continue to remain very low, most countries having a prevalence in adult women of less than 10%.
Stage 3 includes the fourth high burden country (Russian Federation), along with countries in eastern Europe, South America and western Europe, which fall at the end of Stage 3. Stage 4 is populated entirely by the developed countries of western Europe, North America and Oceania. The USA, the fifth high burden country, fall in the last stage as a result of the relatively small difference in the smoking prevalence between men and women compared to the other intermediate stages. As mentioned before, Stage 4 includes countries where the smoking prevalence is higher in women than in men, with a small (< 8%) difference.
(c) Age-specific prevalence
Age-specific prevalence for men and women aged 15 years or older is presented for six representative countries for current smoking (Fig. 1.6). There are wide variations in age-specific prevalence between these countries. In men, prevalence varies from less than 10% to 75% in the 15–19 years age range to lie between 10% and 55% in the oldest age range. Prevalence among women varies from less than 1% to as high as 45% in young adults (15–19 years). Unlike men, prevalence in women tends to converge after age 50, lying within 15 percentage points. Prevalence in women is almost always lower than in men in all age groups.
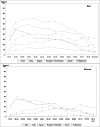
Fig. 1.6
Age-specific rates of smoking prevalence, in men and women in 2009
Initiation of smoking is shifting, and is taking place at earlier ages in both developed and developing countries. In developed countries, quitting smoking is also shifting to occur at a younger age, whereas in developing countries there is no such evidence.
(d) Smoking in youth
Information on smoking habits in youth are collected from a variety of youth surveys that include the Global Youth Tobacco Survey (GYTS), Global school-based Student Health Survey (GSHS) and Health Behaviour in School Aged Children (HBSC). Some countries have their own youth surveys, or have them as part of a general health or household survey, such as the Student Survey in Argentina, the Youth Smoking Survey in Canada, and New Zealand's Tobacco Survey.
The GYTS is a school-based survey designed to monitor tobacco use among youths aged 13 to 15 years. The GYTS uses a standard set of questions and sampling methods in over 160 countries. The survey has core questions that span seven thematic areas pertinent to tobacco. In addition to these, countries can include country-specific questions that allow assessment of tobacco use unique to the country. To assess prevalence of smoking, students are asked to report their smoking habits for both cigarettes and other tobacco products that they may have consumed over the past 30 days. Since its inception in 1999, the GYTS has covered over 2 million students. Although most GYTS are national surveys, in some countries they are limited to subnational locations. Further, countries conduct the GYTS in different years, rendering comparison for the same year difficult.
Prevalence of current tobacco use [including smokeless tobacco] in youth in 2004–09 for fourteen high burden low and middle income countries is shown in Fig. 1.7. The Russian Federation has the highest prevalence of current tobacco use among the high burden countries for which national data are available. Further, in the Americas and Europe the difference in prevalence between boys and girls is smaller than in other regions. In contrast, in Egypt, India and Thailand, prevalences in boys are significantly higher than in girls.
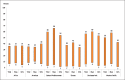
Fig. 1.7
Prevalence of current tobacco use in youth for selected countries, 2005–2009
Fig. 1.8 shows the range of current tobacco use by WHO region for boys and for girls and for both sexes combined. There are wide variations in current tobacco use within each region. The largest variations are observed in EMRO and SEARO irrespective of sex, reflecting potentially disparate initiation rates in countries within the region. In AFRO, the range of current tobacco use between boys and girls is virtually the same. In some countries (e.g. Argentina, Peru, Sierra Leone, Bulgaria, Croatia, Cook Islands, New Zealand), tobacco use in girls exceeds that in boys; but overall boys and girls show remarkably similar propensity to take up tobacco use.
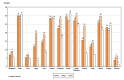
Fig. 1.8
Range of prevalence of current tobacco use in youth, 2005–2009, by WHO region
Warren et al. (2006) present global estimates and regional averages for current tobacco smoking in youth using GYTS data spanning 1999–2005. Their estimates show that one in five boys and one in seven girls currently smoke tobacco. Prevalence of current smoking for both boys and girls combined was highest in AMRO (22.2%) and lowest in WPRO (11.4%). AMRO have the highest average for current tobacco smoking for boys (24%) and for girls (20.4%) whereas the lowest prevalence was in WPRO among boys (15%) and in SEARO among girls (7.1%).
1.4. Regulations and policies: the WHO Framework Convention on Tobacco Control
The WHO Framework Convention on Tobacco Control (WHO FCTC) – the first multilateral evidence-based treaty on tobacco control – articulates tobacco control measures available to countries to counter the growing tobacco epidemic. This treaty, which entered into force in 2005, represents one of the most universal treaties in the United Nations history. In 2008, the WHO launched MPOWER, a technical assistance package comprised of six strategies that reflects one or more of the WHO FCTC measures and helps countries meet their commitments to the WHO FCTC.
2. Cancer in Humans
2.1. Introduction
The available knowledge on the relationship between tobacco smoking and a variety of human cancers is based primarily on epidemiological evidence. An immense amount of such evidence has been obtained, and only a small proportion can be referred to here. The cancers considered to be causally related to tobacco smoking in the previous IARC Monograph on tobacco smoking (IARC, 2004a) included lung, oral cavity, nasal cavity and paranasal sinuses, nasopharynx, oropharynx, hypopharynx, larynx, oesophagus (adenocarcinoma and squamous cell carcinoma), upper aerodigestive tract combined, stomach, pancreas, liver, kidney (body and pelvis), ureter, urinary bladder, cervix and myeloid leukaemia. In addition, it was concluded that there was evidence suggesting lack of carcinogenicity for cancers of the breast and of the endometrium.
Since 2002, there have been additional cohort and case–control studies on the relationship of tobacco smoking in different forms to these and other cancers in many countries. A large body of evidence has been obtained from cohort studies with respect to different cancer sites and types of tobacco product. These cohort studies are described briefly in Table 2.1 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.1.pdf), listed by country. Case–control studies are described in the sections pertaining to cancer sites. More studies are now available from countries and populations that are still at an early stage of the tobacco epidemic. These studies are prone to underestimate the true strengths of the association between tobacco smoking and any specific cancer as the full effect of duration of smoking cannot be evaluated.
2.2. Cancer of the lung
2.2.1 Overview of studies
The main cause of lung cancer in humans is tobacco smoking and most information establishing this fact comes from epidemiological studies in which the assessment of exposure was based on self-reported information on personal smoking habits via self-administered questionnaire or in-person interviews. Since the previous IARC Monograph (IARC, 2004a), numerous studies have been published on the issues of tobacco smoking and sex and racial/ethnic susceptibility, ‘tar’ yields as measured by machine smoking, the relationship between histological changes and the design of cigarettes, dose–response association, genetic susceptibilities and interactions.
2.2.2 Factors affecting risk
Recent epidemiological studies incorporating measures of smoking metabolites in serum or urine are helping to refine our understanding of exposure-response relationships with tobacco smoke. Dose–response evidence has been obtained from three cohort studies (Flanders et al., 2003; Boffetta et al., 2006; Yuan et al., 2009; Table 2.2 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.2.pdf) and four pooled analyses (Lubin & Caporaso, 2006; Lubin et al., 2007a, b, 2008; Table 2.3 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.3.pdf) since the previous IARC Monograph (IARC, 2004a).
The US American Cancer Society Cancer Prevention Study-II (CPS-II) is the largest cohort study on smoking and lung cancer risk using questionnaire assessment of exposure (Flanders et al., 2003). In this study cigarette smoking duration is a much stronger predictor of lung cancer mortality than is cigarette smoking intensity, regardless of age in both men and women. These results are qualitatively similar to those reported by Doll & Peto (1978) and are consistent with IARC (2004a).
In a questionnaire-based assessment of the association of tobacco smoking with lung cancer risk, smokers at higher smoking intensities seem to experience a “reduced potency” per pack such that for equal total exposure, the excess odds ratio per pack–year decreases with intensity (Lubin et al. 2008). Below 15–20 cigarettes/day, the excess odds ratio/pack–year increases with intensity (Lubin & Caporaso, 2006; Lubin et al., 2007a) while above 20 cigarettes/day, there is an ‘inverse-exposure-rate’ effect (Lubin et al., 2007a) suggesting a greater risk for total exposure delivered at lower intensity (or a longer duration) than the equivalent exposure delivered at a higher intensity. The intensity effects were also statistically homogeneous across diverse cancer types, indicating that after accounting for risk from total pack–years, intensity patterns were comparable for cancer of the lung, bladder, oral cavity, pancreas and oesophagus. These analyses suggest that the risk of lung cancer increases with increasing tobacco exposure at all dose levels, but there is some levelling-off effect at the highest intensity of tobacco smoking.
However, when serum cotinine was used as a measure of exposure to tobacco smoking, rather than questionnaire-based data, the odds ratio of lung cancer increased linearly over the full range of exposure from ≤ 5 ng/mL through ≥ 378 ng/mL, with an odds ratio of 55.1 (95% confidence interval (CI): 35.7–85.0) in the highest exposure group. These results suggest that the decreased rate of lung cancer risk at high intensity of tobacco smoke previously described is a statistical artefact. Such an effect may be due to an inaccurate assessment of total tobacco smoke exposure from questionnaire-based studies at high exposure levels (Boffetta et al., 2006). Somewhat similar results were obtained when both 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) and total cotinine in urine were measured in subjects of two large cohort studies from Shanghai men and Singapore men and women (Yuan et al., 2009). Among smokers with comparable smoking histories (as noted in questionnaire data) there is a 9-fold variation in subsequent risk of lung cancer between those with high and those with low levels of total urinary NNAL and cotinine. Thus measurements of urinary cotinine and total NNAL at a single point in time in a smoker could substantially improve the predictive power of a lung cancer assessment model based solely on self-reported smoking history (number of cigarettes/day, number of years of regular smoking). A positive NNAL-lung cancer association of comparable magnitude was observed in both Shanghai and Singapore subjects despite differences in the NNK content of tobacco smoked. The independent association between total urinary cotinine and lung cancer risk, after adjustment for total urinary NNAL and smoking history, suggests that tobacco smoke compounds other than NNK play a role in lung cancer development in smokers. Further, a single measurement of urinary NNAL may closely predict the average level of NNAL measured over a much longer period of time.
2.2.3 Types of tobacco or of cigarette
(a) Tar levels
In a previous IARC Monograph (IARC, 1986), it was concluded on the basis of the case–control, cohort studies and ecological evaluations available at the time that prolonged use of ‘high-tar’ and unfiltered cigarettes is associated with greater risks than prolonged use of filter-tipped and ‘low-tar’ cigarettes. More recently (IARC, 2004a), it has been recognized that the actual quantitative impact of reduced ‘tar’ and filter-tipped cigarettes is difficult to assess because of, respectively, the concomitant increase in tobacco-specific nitrosamines that accompanies the greater use of blend tobacco and the compensatory changes in smoking behaviour by smokers attempting to maintain their accustomed level of nicotine intake. Nevertheless, it was concluded that changes in cigarette types since the 1950s have probably tended to reduce the risk for lung cancer associated with tobacco smoking.
Additional refinement in assessing the health effects associated with smoking cigarettes of various tar content has been possible since the publication of the earlier reports. Compared with smokers of medium tar (15–21 mg) filtered cigarettes risk was higher among men and women who smoked high tar (≥ 22 mg) non-filtered brands but there was no difference in risk among men and women who smoked ‘very low tar’ or ‘low tar’ brands compared with those who smoked ‘medium tar’ brands (Harris et al., 2004). Regardless of tar content of their cigarettes, all current smokers had a far greater risk for lung cancer than people who had stopped smoking or had never smoked (Harris et al., 2004).
(b) Mentholated cigarettes
In the previous IARC Monograph (IARC, 2004a) the conclusion was drawn that there is no additional risk associated with smoking mentholated cigarettes when total consumption (pack–years) was controlled versus non-mentholated ones. Recent evidence supports that conclusion.
Mentholated cigarettes first appeared in the 1920s, but were not widely used until the mid-1950s (Bogen, 1929; Federal Trade Commission, 2001). Since the early 1970s, menthol varieties have accounted for 25–60% of all cigarettes sold in the USA (Federal Trade Commission, 2001). There are strong ethnic differences in the use of menthol cigarettes; more than 60% of Black smokers of both sexes use menthol brands compared to fewer than 25% of White smokers (Royce et al., 1993; Hymowitz et al., 1995). Studies have generally not demonstrated an increased risk of lung cancer for mentholated cigarettes versus non-mentholated cigarettes (Kabat & Hebert, 1994; Carpenter et al., 1999; Brooks et al., 2003; Stellman et al., 2003). Recent evidence also suggests that users of mentholated cigarettes smoke fewer pack–years than those of non-mentholated cigarettes.
The higher incidence of lung cancer among Blacks is an important public health concern but the causes remain unclear. Mentholated cigarette use does not appear to explain the racial disparity observed in lung cancer risk among those having the same total tobacco consumption.
2.2.4 Histology
Compiled databases from IARC and other sources indicated that squamous cell carcinoma rates [per 100000 person-years] among men declined by 30% or more in North America and some European countries between 1980–82 and 1995–97, while changing less dramatically in other areas; small cell carcinoma rates decreased less rapidly. In contrast, the proportion of adenocarcinoma cases rose among men and women in virtually all areas, with the increases among men exceeding 50% in many areas of Europe (Devesa et al., 2005).
Based on a comparison of two large cohort studies initiated by the American Cancer Society (ACS) (CPS-I and CPS-II) in 1960 and 1980, respectively, a stronger association between smoking and adenocarcinoma was observed in recent compared to earlier follow-up periods (Thun & Heath, 1997). Additionally, an association between cigarette smoking and bronchioloalveolar carcinoma was also found in several studies (Falk et al., 1992; Morabia & Wynder, 1992).
A meta-analysis of 8 cohort and 14 case–control studies conducted in Japan among active smokers indicated significant excess lung cancer risks among men for both squamous cell carcinoma (relative risk (RR), 11.7) and adenocarcinoma (RR, 2.30). Among women the risks were 11.3 for squamous cell carcinoma and 1.37 for adenocarcinoma (Wakai et al., 2006).
2.2.5 Population characteristics
(a) Sex
Meta-analyses on sex-specific susceptibility to lung cancer associated with tobacco smoking are presented in Table 2.4 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.4.pdf) and cohort studies in Table 2.5 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.5.pdf).
In the 1990s, two case–control studies indicated that relative risks for lung cancer associated with specific amounts and duration of cigarette smoking may be higher among women than among men (Risch et al., 1993; Zang & Wynder, 1996).
In the large NIH-AARP [National Institutes of Health-American Association of Retired People] cohort (Freedman et al., 2008), smoking was associated with lung cancer risk in both men and women. Age-standardized incidence rates for lung cancer tended to be higher in men than in women with comparable smoking histories (for current smokers and for quitters of less than 10 years), and in cases with squamous cell tumours. However, lung cancer risk was generally similar between men and women.
In a joint analysis, results from the Nurses’ Health Study of women and the Health Professionals Follow-up Study in men (Bain et al., 2004) suggest little difference in lung cancer susceptibility between men and women given equal smoking exposure. The hazard ratio in women ever smokers compared with men was 1.11 (95%CI: 0.95–1.31).
Serum cotinine levels were analysed in lung cancer cases and controls (Boffetta et al., 2006). The lung cancer odds ratios (ORs) estimated for men and women were very similar for those with comparable serum cotinine levels. Other studies that have carefully quantified tobacco exposure via self-administered questionnaire or interview provide additional evidence of a comparable increase in lung cancer risk in the two sexes (Kreuzer et al., 2000; Flanders et al., 2003; Bain et al., 2004).
In a meta-analysis of observational studies on cigarette smoking and cancer from 1961–2003 (conducted on 177 case–control studies, 75 cohort studies and two nested case–control studies), dose–response estimates were available in 44 studies: 19 with estimates for men only, 11 with estimates for women only and 14 with separate estimates for men and women (Gandini et al., 2008). Overall, the risk of lung cancer for men and women increased by 7% for each additional cigarette smoked per day (RR, 1.07; 95%CI: 1.06–1.08). The increased risk appears to be slightly higher in women (RR, 1.08; 95%CI: 1.07–1.10) than in men (RR, 1.07; 95%CI: 1.05–1.08) (P < 0.001; adjusting for study type).
(b) Ethnicity
It has been postulated that susceptibility to lung cancer from tobacco smoking may differ by race and ethnicity (Schwartz & Swanson, 1997; Peto et al., 1999; Stellman et al., 2001; Kiyohara et al., 2004, 2005, 2006; Pinsky, 2006; Wakai et al., 2006; Vineis et al., 2007; Takahashi et al., 2008; Table 2.6 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.6.pdf). Lung cancer incidence rates vary considerable across racial/ethnic groups in the USA and elsewhere. Black men have higher rates than white men, while Hispanics, Asians and American Indians of both sexes have lower rates than whites (Stellman et al., 2003; SEER, 2004). Nutritional habits, smoking patterns, type of tobacco smoked and genetic factors may play a role in such differences between racial and ethnic groups.
The association of tobacco smoking and lung cancer does not appear to be as strong among Japanese as among populations of North America or Europe (Wakai et al., 2006). In a meta-analysis of 8 cohort studies and 14 case–control studies conducted in Japan, the excess lung cancer risks observed for both men (RR, 4.39; 95%CI: 3.92–4.92) and women (RR, 2.79; 95%CI: 2.44–3.20) in both case–control and cohort studies were lower than would have been expected from studies in North America and Europe. The lower lifetime consumption of cigarettes in Japanese, due in part to a later initiation of smoking and a lower consumption per day has been suggested to explain this. Other differences that may have etiological significance include tobacco ingredients, different filters on cigarettes, lifestyle factors including diet, and possibly differences in genetic susceptibility. [The Working Group noted that North American or European populations were not directly included in any of these studies.]
Data from the Asian Pacific Cohort Studies Collaboration, 31 studies involving 480125 persons, evaluated the risk of death from lung cancer associated with smoking habits in Australia, New Zealand and Asia (Huxley et al., 2007b). Among Asian men the hazard ratio was 2.48 versus 9.87 in men in Australia and New Zealand. Among women, the corresponding estimates were 2.35 and 19.33, respectively. [In these studies, Asian populations smoked fewer cigarettes for a shorter period of time compared to those in Australia and New Zealand.]
Based on data from the National Cancer Institute’s Surveillance, Epidemiology, and End Results program (SEER), Chinese women residing in the USA have a fourfold increased risk of lung cancer, and Filipino women a twofold increased risk, compared to that expected based on rates in non-Hispanic whites in the USA with a similar amount of cigarettes smoked (Epplein et al., 2005). Among Chinese women, the increased risk was largely restricted to adenocarcinoma and large cell undifferentiated carcinoma. Chinese females residents of the western US mainland have a much higher risk of lung cancer than would be expected from their tobacco use patterns, just as they do in Asia (Peto et al., 1999; Epplein et al., 2005), the reason for these difference have not been identified. [Controlling for potential confounding factors was limited using aggregate data from SEER.]
Age, sex and race-specific risks of lung cancer mortality among lifetime non-smokers were compared in the two large ACS Cancer Prevention Study cohorts (CPS-I; CPS-II). The mortality rate was higher among African American women than among white women in CPS-II (hazard ratio (HR), 1.43; 95%CI: 1.11–1.36) (Thun et al., 2006). This suggests an inherent susceptibility difference between white and black women but it could also be explained by access to care, diet, or exposure to environmental carcinogens.
The risk for lung cancer associated with cigarette smoking in 183813 African-American, Japanese-American, Latino, native Hawaiian and white men and women was examined in the Multiethnic Cohort Study in the USA (Haiman et al., 2006). Information on demographic factors, smoking status, cigarettes/day smoked, years of smoking, years since quitting, diet, occupations, educational level and racial and ethnic group were collected for all subjects through a self-administered questionnaire at enrolment. Information about age of smoking initiation and cessation rates were collected on a subgroup of 5090 study subjects. Incident lung cancer cases were identified by linkage to the SEER cancer registries covering California and Hawaii. Among those who smoked no more than 10 cigarettes/day and those who smoked 11–20 cigarettes/day, relative risks ranged from 0.21 to 0.39 (P < 0.001) among Japanese Americans and Latinos and from 0.45 to 0.57 (P < 0.001) among whites as compared with African Americans. However, at levels exceeding 30 cigarettes/day, differences between racial/ethnic groups were no longer significant. The differences in lung cancer risk by racial group associated with smoking were observed for both men and women and for all histological types of lung cancer. These findings could not be explained by differences between populations in other known or suspected risk factors, including diet, occupation, and education level or by age at starting smoking or cessation of smoking.
Polymorphisms in glutathione-S-transferase (GST), GSTM1, GSTT1 and GSTP1 genes in humans are associated with reduction of enzymatic activity towards several substrates, including those found in tobacco smoke. In a population based case–control study involving early-onset lung cancer, African Americans carrying at least one G allele at the GSTP1 locus were more likely to have lung cancer compared with African Americans without a G allele after adjustment for age, sex, pack–years of smoking and a history of lung cancer in a first degree relative (OR, 2.9; 95%CI: 1.29–6.20). African Americans with either one or two risk genotypes at the GSTM1 (i.e. null genotype) and GSTP1 loci were at increased risk of having lung cancer compared with those having fully functional GSTM1 and GSTP1 genes (one risk genotype: OR, 2.8; 95%CI: 1.1–7.2 and two risk genotypes: OR, 4.0; 95%CI: 1.3–12.2). No significant single gene associations between GSTM1, GSTT1 and GSTP1 and early-onset lung cancer were observed in Caucasians, after adjusting for age, sex, pack–years and a family history of lung cancer (Cote et al., 2005).
The cytochrome P450 (CYP) superfamily of enzymes catalyses one of the first steps in the metabolism of carcinogens such as polycylic aromatic hydrocarbons, nitroaromatics and arylamines. A population-based case–control study of lung cancer in the metropolitan Detroit area found that neither CYP1A1 MspI nor CYP1A1 Ile462Val was associated with lung cancer susceptibility among Caucasians or African Americans. Among Caucasians, however, CYP1B1 Leu432 Val was significantly associated with lung cancer susceptibility (OR for at least one Val allele, 2.87; 95%CI: 1.63–5.07). Individuals with both this polymorphism and exposure to second-hand tobacco smoke were at particularly high risk for lung cancer. Combinations of particular CYP1B1 polymorphisms appeared to increase risk, although no combination differed significantly from the risk associated with CYP1B1 Leu432 Val alone (Cote et al., 2005; Wenzlaff et al., 2005).
The hypothesis that polymorphisms in TP53 may modulate the risk for lung cancer associated with tobacco smoke was evaluated in a case–control study of lung cancer in Baltimore, Maryland. African-Americans with Pro-T-A-G-G haplotype (combining the polymorphisms TP53_01 (rs1042522), TP53_65 (rs9895829), TP53_66 (re2909430), TP53_16 (rs1625895), and TP_11 (rs12951053)) had both an increased risk for lung cancer (HR, 2.32; 95%CI: 1.38–4.10) and a worsened lung cancer prognosis (HR, 2.38; 95%CI: 0.38–4.10) compared with those having the Arg-T-A-G-T haplotype. No association of TP53 polymorphisms with lung cancer was observed in Caucasians (Mechanic et al., 2007). Common genetic variation in TP53 could modulate lung cancer pathways in African Americans. Differences in lung cancer susceptibility may exist based on race, tobacco exposure and selected genetic polymorphisms (Mechanic et al., 2007).
2.2.6 Interactions
(a) Diet and exercise
Antioxidant vitamins, carotenoids, isothiocyanates, total dietary vegetables and fruit, and physical exercise have been associated with a decreased risk for cancer in some studies but the overall protective effect of diet and exercise account for only a small fraction of the total risk associated with tobacco smoking.
The association of fruit and vegetable with lung cancer incidence among both smokers and non-smokers was evaluated in the European Prospective Investigation into Cancer and Nutrition (EPIC). In current smokers lung cancer risk was significantly decreased with higher vegetable consumption, the hazard ratio being 0.78 (95%CI: 0.62–0.98) per 100 g increase in daily vegetable consumption, and 0.90 (95%CI: 0.81–0.99) per 100 g fruit (Linseisen et al., 2007). While overall consumption of fruits and vegetables was not found to be protective of lung cancer in the NIH-AARP Diet and Health Study, higher consumption of several botanical subgroups (i.e. rosaceae, convolvulaceae, and umbelliferae) was significantly inversely associated with risk, but only in men (Wright et al., 2008).
Cruciferous vegetables (i.e. broccoli, cabbage, cauliflower, Brussels sprouts, kale) are rich in isothiocyanates and have been hypothesized to have anticancer properties that may contribute to reduced risk for lung cancer. Isothiocyanates may inhibit the bioactivation of procarcinogens found in tobacco smoke such as polycyclic aromatic hydrocarbons and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (Hecht, 2000). Isothiocyanates may also enhance excretion of carcinogenic metabolites before they can damage DNA (Gasper et al., 2005). Furthermore, sulforaphane, a major isothiocyanate found in broccoli, can induce cell cycle arrest and apoptosis (Seow et al., 2005). GSTM1 and GSTT1 encode isoenzymes that play an important role in xenobiotic metabolism (Hecht, 2000). Individuals with homozygous deletion of GSTM1 and GSTT1, or both may metabolize isothiocyanates less efficiently and may be more intensely exposed to isothiocyanates after consumption of cruciferous vegetables. Epidemiological evidence from 30 studies on the association between lung cancer and either total cruciferous vegetable consumption (6 cohort and 12 case–control studies) or specific cruciferous vegetables (1 cohort and 11 case–control studies) was recently evaluated (Lam et al., 2009). The risk for lung cancer among those in the highest category of total cruciferous vegetable intake was 22% lower in case–control studies (pooled OR, 0.78; 95%CI: 0.70–0.88) and 17% lower in cohort studies (pooled RR, 0.83; 95%CI: 0.62–1.08). The strongest inverse association of total cruciferous vegetable intake with lung cancer was seen among individuals with GSTM1 and GSTT1 double null genotypes (OR, 0.41; 95%CI: 0.26–0.65; p for interaction = 0.01). The inverse association was observed in both smokers and non-smokers.
The potential role of vitamin A in the development of lung cancer attracted early research interest (Bjelke, 1975). Carotenoids were thought to have anti-cancer properties and early evidence from case–control studies tended to support an inverse association of lung cancer incidence with β-carotene intake and with serum concentrations of β-carotene. However, the case–control design is not ideal for assessing the effect of serum carotenoids as a risk factor for lung cancer risk since the disease is likely to effect serum levels. In a meta-analysis of six randomized clinical trials and 25 prospective observational studies, Gallicchio et al. (2008) computed a pooled relative risk for studies comparing β-carotene supplements with placebo of 1.10 (95%CI: 0.89–1.36). Among observational studies, the pooled relative risk for total carotenoid dietary intake from six studies was 0.86 (95%CI: 0.75–0.99) among current smokers. For dietary intake of β-cryptoxanthin, data from six studies gave a pooled relative risk among smokers of 0.75 (95%CI: 0.58–0.96). No other carotenoids significantly reduced the risk in current, former or never smokers.
Based on a review of the literature, antioxidant vitamins show no clear protective effect on lung cancer risk in smokers or non-smokers, although there was some, albeit inconsistent, evidence pointing to a protective role for vitamin C and E. No clear protective role was observed for vitamin A (Ruano-Ravina et al., 2006).
Increased physical activity has been associated with a reduction in the incidence and mortality from all-site cancer and some site-specific cancers in studies of non-smokers, but less is known about whether physical activity is associated with similar risk reduction in smokers. Several early studies suggested that physical activity is associated with decreased risk of lung cancer in men and women after adjusting for smoking, with risk reductions estimated from 18% (Peterson et al., 2001) to 62% (Kubík et al., 2001). The effect of physical activity on lung cancer risk was assessed in a sample drawn from participants in the Beta-Carotene and Retinol Efficacy Trial. The results suggested that physical activity may play a small role in reducing cancer risk and mortality among those with significant tobacco exposure. The incidence of lung cancer and of all cancer sites combined seemed to be more attenuated by exercise in men than in women, while the attenuation in lung cancer mortality was greater in women than in men. These effects may be more pronounced for younger people and may differ inconsistently by pack–years of smoking (Alfano et al., 2004).
(b) Radon
In a pooled analysis of data from 13 case–control studies of residential radon and lung cancer from nine European countries (7148 cases of lung cancer and 14208 controls), the dose–response relation seemed to be linear with no threshold and remained significant in analyses limited to individuals from homes with measured radon < 200 Bq/m3. The absolute risks of lung cancer by age 75 years at radon concentrations of 0, 100, and 400 Bq/m3 would be about 0.4%, 0.5% and 0.7%, respectively, for lifelong non-smokers, and about 25 times greater (10%, 12% and 16%) for cigarette smokers. These studies show appreciable hazards from residential radon, particularly for smokers and recent ex-smokers (Darby et al., 2005). Similar risks were identified in a pooling project of North American case–control studies (Krewski et al., 2005).
(c) Asbestos
Exposure to asbestos and tobacco smoking are both known causes of lung cancer in humans (Doll & Peto, 1978; de Klerk et al., 1996). Some studies suggest a multiplicative effect [where the effect of asbestos exposure is a multiple of the effect of smoking] (Hammond et al., 1979; Doll & Peto, 1985), and meta-analyses have suggested that the additive model [where asbestos exposure and smoking are independent of each other] is unsound (Lee, 2001; Liddell, 2001). In a recent study of 2935 asbestos miners, persons exposed to asbestos and tobacco who subsequently quit smoking remained at a 90% increased risk of lung cancer up to 20 years after smoking cessation, compared to never-smoker asbestos workers (Reid et al., 2006a).
(d) Genetic polymorphisms
Lung cancer is plausibly caused by the interplay between environmental factors and several low-risk alleles. Attempts in identifying specific single nucleotide polymorphisms (SNPs) responsible for modulating lung cancer risk have yielded few conclusive results. Recent studies have focused on mechanistically plausible polymorphisms in genes coding for enzymes involved in the activation, detoxification and repair of chemical damage caused by tobacco smoke. Genetic association studies indicate that several inherited genetic polymorphisms may be associated with lung cancer risk, but the data from individual studies with low statistical power are conflicting. Evidence from pooled or meta-analyses, along with some individual studies, is briefly summarized below.
(i) Metabolic genes
Most of the 70 carcinogens in tobacco smoke are procarcinogens that must be activated by phase I enzymes and may then be deactivated by phase II enzymes. Polymorphisms that alter the function of the genes involved in the activation or detoxification of tobacco smoke carcinogens can potentially influence an individual’s risk of developing a tobacco-related cancer.
Meta and pooled analyses of 34 case–control, genotype-based studies were conducted to assess the effect of GSTT1 genotypes and smoking on lung cancer risk. No significant interaction was observed (Raimondi et al., 2006). A pooled analysis of 21 case–control studies from the International Collaborative study of Genetic Susceptibility to Environmental Carcinogens showed no evidence of increased risk for lung cancer among carriers of the GSTM1 null genotype and there was no evidence of interaction between GSTM1 genotype and either smoking status or cumulative tobacco consumption (Benhamou et al., 2002). Similarly, in another pooled analysis the summary OR indicated the slow acetylator genotype of N-acetyltransferase 2 (NAT2) detoxification enzyme was not associated with lung cancer risk among Caucasians (Borlak & Reamon-Buettner, 2006). In a pooled analysis to test the hypothesis of interaction among genetic variants in increasing the individual risk for cancer, the cumulative effect of variants in three metabolic genes, CYP1A1, GSTM1 and GSTT1 was assessed. The risk for lung cancer was increased with the combination of CYP1A1*2B or CYP1A1*4 alleles and the double deletion of both GSTM1 and GSTT1 up to an OR of 8.25 (95%CI: 2.29–29.77). The combination including CY1A1*4 among never smokers was associated with an OR of 16.19 (95%CI: 1.90–137). These estimates did not change after adjustment by the number of cigarettes smoked and duration of smoking. The results were consistent across ethnicities and were approximately the same for adenocarcinoma and squamous cell carcinoma (Vineis et al., 2007).
Microsomal epoxide hydrolase 1 (EPHX1) plays an important role in both the activation and detoxification of tobacco-derived carcinogens. Polymorphisms at exons 3 and 4 of the EPHX1 gene have been reported to be associated with variations in EPHX1 activity. In a meta-analysis of 13 case–control studies the low-activity (variant) genotype of EPHX1 polymorphism at exon 3 was associated with decreased risk for lung cancer (OR, 0.65; 95%CI: 0.44–0.96) among whites. In white-populations, the high activity (variant) genotype of EPHX1 polymorphism at exon 4 was associated with a modest increased risk of lung cancer (OR, 1.22; 95%CI: 0.79–1.90) and the predicted low activity was associated with a modest decrease in risk (OR, 0.72; 95%CI: 0.43–1.22) (Kiyohara et al., 2006).
(ii) DNA repair and cell cycle pathways
Data from 14 studies of lung cancer were used in a pooled analysis focusing on 18 sequence variants in 12 DNA repair genes, including APEX1, OGG1, XRCC1, XRCC2, XRCC3, ERCC1, XPD, XPF, XPG, XPA, MGMT and TP53 (Hung et al., 2008a). None of the variants appeared to have a large effect on lung cancer risk. In a recent meta-analysis the X-ray repair cross-complementing protein group 3 (XRCC3) and the xeroderma pigmentosum group D (XPD)/excision repair cross-complementing group 2 (ERCC2) genes were evaluated (Manuguerra et al., 2006). The authors found no association between these genes and the cancer sites investigated (skin, breast and lung). A significant association was identified for XPD/ERCC2 single nucleotide polymorphisms (codons 312 and 751) and lung cancer.
(iii) Nicotine acetylcholine receptor genes
A series of large genome-wide association studies for lung cancer have identified susceptibility loci for lung cancer in chromosome arms 5p, 6p and 15q (Landi et al., 2009). In particular, the susceptibility locus at chromosome region 15q25 includes several genes, including three that encode nicotinic acetylcholine receptor subunits (CHRNA5, CHRNA3 and CHRNB4). Nicotinic acetylcholine receptor subunit genes code for proteins that form receptors present in neuronal and other tissue, in particular alveolar epithelial cells, pulmonary neuroendocrine cells, and lung cancer cell lines (Wang et al., 2001; Minna, 2003) and bind to nicotine and nicotine derivatives including NNN. An association of CHRNA3 and CHRNA5 variants with nicotine dependence has been reported (Saccone et al., 2007; Berrettini et al., 2008). These genes may act, at least partially, upon cigarette smoking. Current smokers with one or two copies of the susceptibility variant are likely to smoke between one and two cigarettes more a day (Spitz et al., 2008). Evidence for an effect of the 15q25 locus among never smokers is conflicting, with an association found in one study in Europe (Hung et al., 2008b) and one in Asia (Wu et al., 2009a), but not in others. Whether genes in the 15q25 locus have an effect on lung cancer beyond their propensity to increase numbers of cigarettes smoked is unclear.
Three genome-wide association studies identified genetic factors that modified disease risk. The first was a genome-wide association analysis to identify genetic polymorphisms associated with lung cancer risk in 1154 lung cancer patients of European ancestry who were current or former smokers and 1137 control subjects who were frequency matched to the lung cancer patients by age, sex, race and smoking status. Two SNPs, rs105173 and rs803419, which mapped to a region of strong linkage disequilibrium within 15q25.1, were strongly associated with risk of lung cancer, with an odds ratio for rs105173 of 1.31 (P = 9.84x10−6). This finding was replicated with an additional 711 case subjects and 632 control subjects from Texas (P = 0.00042) and in 2013 case subjects and 3062 control subjects in the United Kingdom (P = 2.33x10−10). The region of interest encompasses the nicotinic acetylcholine receptor subunit genes CHRNA3 and CHRNA5 (as well as CHRNB4) (Spitz et al., 2008). A second genome-wide association study conducted among 1989 lung cancer cases and 2625 controls from six central European countries confirm these results (Hung et al., 2008a). In a third genome-wide association study of 665 Icelandic, 269 Spanish and 90 Dutch lung cancer cases and 32244 controls a common variant in the nicotinic acetylcholine receptor gene cluster [chromosome region 15q24] was significantly associated with lung cancer risk (OR, 1.31; 95%CI: 0.1.19–1.44). The variant was observed to have a significant effect on the number of cigarettes smoked per day (Thorgeirsson et al., 2008). These studies have all shown a link between this variant and lung cancer risk either through a mechanism involving nicotine dependence or a direct role in downstream signalling pathways that promote carcinogens. Together these results provide compelling evidence of a locus at 15q25 and 15q24 predisposing to lung cancer.
(iv) Alpha(1)-antitrypsin
Alpha(1)-antitrypsin deficiency (α(1)ATD) is one of the most common genetic disorders, especially among European descendents. Recent results suggest that α(1)ATD carriers are at a 70–100% increased risk of lung cancer, accounting for 11% to 12% of patients with lung cancer (Yang et al., 2008). [The specific effect by smoking status was not evaluated.]
(v) Other genes
Mutations in the checkpoint CHEK2 gene have been associated with increased risk of breast, prostate and colon cancer and a decreased risk of lung cancer among those with the I157T missense variant of the CHEK2 gene. In a large Polish case–control study CHEK2 mutations were protective against lung cancer (OR, 0.3; 95%CI: 0.2–0.5) (Cybulski et al., 2008).
The Swedish Family-Cancer Database was used to compare the rate of lung cancers among persons without family history of lung cancer to those with a family history (Li & Hemminki, 2004). A high risk by family history in adenocarcinoma (standardized incidence ratio (SIR), 2.03) and large cell carcinoma (SIR, 2.14) was found, a slightly lower risk among patients with squamous cell carcinoma (SIR, 1.63) and small cell carcinoma (SIR, 1.55). Among siblings, an increased risk was shown for concordant adenocarcinoma and small cell carcinoma at all ages and for all histological types when cancer was diagnosed before age 50. At young age, risks between siblings were higher than those between offspring and parents. These data suggest that a large proportion of lung cancers before age 50 are heritable and due to a high-penetrant recessive gene or genes that predispose to tobacco carcinogen susceptibility.
(e) Viral infection
Data are limited regarding lung cancer risk in human immunodeficiency virus (HIV)-infected persons with modest immune suppression, before the onset of acquired immunodeficiency syndrome (AIDS). Among 57350 HIV-infected persons registered in the USA during 1991–2002 (median CD4 counts 491 cells/mm3), 871 cancers occurred. Risk was elevated for several non-AIDS defining malignancies, including cancer of the lung (SIR, 2.6 [n = 109]) (Engels et al., 2008). [Specific evaluation with smoking status was not performed.]
2.3. Cancers of the upper aerodigestive tract
Evidence relating to cancers of the upper aerodigestive tract obtained from relevant cohort and case–control studies on specific sites is described in Sections 2.3.1 to 2.3.6; studies that looked at several subsites combined are described in Section 2.3.7. The major potential confounders for the relationship between smoking and cancers of the upper aerodigestive tract are alcohol consumption and use of any form of smokeless tobacco, and for some sites infection with human papillomavirus (HPV) (especially HPV16). In general, the studies examined by the Working Group had adjusted for these two confounders when appropriate. Some studies also adjusted for dietary intake, especially of fruits and vegetables, although few reported stratified relative risks.
2.3.1 Cancer of the oral cavity
Tobacco smoking was found to be causally related to oral cancer (IARC, 1986, 2004a). New studies on the relationship between oral cancer and cigarette smoking published since the most recent IARC Monograph (IARC, 2004a) include four cohort studies (Table 2.7 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.7.pdf), and eight case–control studies (Tables 2.8–2.11 online; see below).
(a) Intensity and duration of smoking
Intensity of smoking was measured in almost all cohort (Table 2.7 online) and case–control studies (IARC 2004a; Table 2.8 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.8.pdf and Table 2.9 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.9.pdf). In addition to the number of cigarettes or amount of tobacco smoked daily, cumulative exposure to cigarette smoke was also measured in terms of pack–years, tobacco-years or lifetime tobacco consumption. The link between duration of cigarette consumption and oral cancer was examined in 15 case–control studies. Seven case–control studies also considered age at starting smoking.
One cohort study (McLaughlin et al., 1995) and 14 case–control studies reported a dose-dependent increase in risk with increasing number of cigarettes smoked daily or increasing daily tobacco consumption (Franceschi et al., 1990, 1992, 1999; Nandakumar et al., 1990; Zheng et al., 1990; Choi & Kahyo, 1991; Oreggia et al., 1991; Bundgaard et al., 1995; Zheng et al., 1997; De Stefani et al., 1998; Hayes et al., 1999; De Stefani et al., 2007; Subapriya et al., 2007; Muwonge et al. 2008). Whenever analysed, the trend was always statistically significant (Franceschi et al., 1990, 1992; Oreggia et al., 1991; Bundgaard et al., 1995; McLaughlin et al. 1995; Hayes et al., 1999; Subapriya et al., 2007), except in the study of Muwonge et al. (2008) which also included bidi smokers.
Bundgaard et al. (1995) used lifetime tobacco consumption divided into four categories and reported a positive, significant trend after adjustment for life-time consumption of alcohol and other risk factors. A positive trend was also found in all studies that have analysed consumption in pack–years or tobacco-years (Zheng et al., 1990; Maier et al., 1992a; Macfarlane et al., 1995; Hung et al., 1997; Zheng et al., 1997; De Stefani et al., 1998, 2007; Applebaum et al., 2007; Muwonge et al., 2008), except Muwonge et al. (2008).
Ten studies (Franceschi et al., 1990, 1992; Nandakumar et al., 1990; Zheng et al., 1990; Choi & Kahyo, 1991; Oreggia et al., 1991; Zheng et al., 1997; De Stefani et al., 1998, 2007; Znaor et al., 2003; Subapriya et al., 2007; Muwonge et al., 2008) classified the duration of smoking in up to four categories, and all but one (Nandakumar et al., 1990) reported increased relative risks and a positive trend.
Of six studies that considered age at start of smoking (Franceschi et al., 1990, 1992; Choi & Kahyo, 1991; Oreggia et al., 1991; Zheng et al., 1997; Balaram et al., 2002) two reported a statistically significant trend of increasing risk with decreasing age at starting (Franceschi et al., 1990, 1992).
(b) Cessation of smoking
Three cohort studies (McLaughlin et al., 1995; Freedman et al., 2007a; Friborg et al. 2007) and nine case–control studies (Zheng et al., 1990; Choi & Kahyo, 1991; Oreggia et al., 1991; Franceschi et al., 1992; Ko et al., 1995; Zheng et al., 1997; De Stefani et al., 1998, 2007; Schildt et al., 1998; Balaram et al., 2002; Pacella-Norman et al., 2002; Muwonge et al. 2008) estimated risks for former smokers which were always lower than those for current smokers and in five studies almost reached unity (Zheng et al., 1990; Choi & Kahyo, 1991; Zheng et al., 1997; Schildt et al., 1998; Muwonge et al., 2008). Twelve case–control studies examined the risk by years since quitting and all reported a negative trend, with relative risks compared with those in non-smokers decreasing to near unity after 10 or more years (Franceschi et al., 1990, 1992; De Stefani et al., 1998, 2007; Schlecht et al., 1999a; Table 2.7 online; Table 2.10 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.10.pdf).
(c) Type of cigarette
The effect of the type of cigarette smoked was examined in several case–control studies (Table 2.11 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.11.pdf). The characteristics of the cigarettes included the presence of a filter, the type of tobacco, the tar content and whether the product was manufactured or hand-rolled. Two studies reported a statistically significantly higher risk for black than for blond tobacco (Oreggia et al., 1991; De Stefani et al., 1998, 2007). Similarly, a much higher risk was found for hand-rolled cigarettes than for manufactured cigarettes, and plain cigarettes had a much higher risk than filter-tipped cigarettes (De Stefani et al., 1998, 2007). In one study the differences between black and blond tobacco and between hand-rolled and manufactured cigarettes persisted after stratification by duration of smoking (De Stefani et al., 1998). Smoking cigarettes with a high-tar content led to higher risks than smoking cigarettes with a low-tar content (Franceschi et al., 1992) and the same trend was observed for cigarettes without filter compared to cigarettes with filter (De Stefani et al., 2007).
(d) Sex
Sex-specific effects were examined in two case–control studies (Zheng et al., 1990; Hayes et al., 1999). In both studies, the relative risks for all categories of intensity, duration of smoking and pack–years were higher for women than for men. [The Working Group noted that the background risk of oral cancer is considerably lower in women than men. Thus, the higher relative risk estimates in women than men indicate a higher proportionate contribution from smoking in women than men, rather than higher absolute risk.]
2.3.2 Cancer of the pharynx
Tobacco smoking was considered to be an important cause of oropharyngeal and hypopharyngeal cancers in the previous IARC Monographs on tobacco smoking (IARC, 1986, 2004a). Since then, results available from three cohort (Table 2.12 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.12.pdf) and seven case–control studies (Table 2.13 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.13.pdf and Table 2.14 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.14.pdf) provide further support for the association. Many studies, however, combine cancers of the oral cavity and pharynx (see Section 2.3.7). This section summarizes the evidence from all eight cohort and 21 case–control studies that reported results specifically on oropharyngeal and hypopharyngeal cancer, or on pharyngeal cancer in general; the latter may include data on nasopharyngeal cancer.
The risk for pharyngeal cancer was significantly increased in smokers in four cohort studies (Doll et al., 2005; McLaughlin et al.,1995; Freedman et al., 2007a; Friborg et al., 2007) and all but one of the case–control studies (Rao et al., 1999). The trend of increasing risk associated with increasing daily or cumulative consumption of cigarettes was evident from all these studies, particularly those from Europe (Brugere et al., 1986; Tuyns et al., 1988; Franceschi et al., 1990, 1999; Maier et al., 1994; Escribano Uzcudun et al., 2002; Vlajinac et al., 2006), India (Znaor et al., 2003; Sapkota et al., 2007), Uruguay (De Stefani et al., 1998, 2007) and the USA (McLaughlin et al., 1995; Applebaum et al., 2007), and less strongly so in studies from Canada (Elwood et al., 1984) and the Republic of Korea (Choi & Kahyo, 1991). The multicentre study in Europe, North and South America of Hashibe et al. (2007c) showed increased risks according to frequency (cigarettes/day) and duration (years) in never drinkers. Applebaum et al. (2007) found a relationship between increasing risk of pharyngeal cancer and increased pack–years of smoking in subjects with negative HPV16 serology but not in those with positive HPV16 serology (p value for interaction = 0.007).
In two case–control studies the risk increased with decreasing age at starting smoking (Franceschi et al., 1990; Choi & Kahyo, 1991,), but adjustment was not made for duration and intensity of smoking. In a case–control study from Spain (Escribano Uzcudun et al., 2002) the risk increased with the age of starting smoking.
Former smokers had consistently lower relative risks than did current smokers in both cohort (McLaughlin et al., 1995; Freedman et al., 2007a) and case–control studies (Choi & Kahyo, 1991; De Stefani et al., 1998; Vlajinac et al., 2006). In comparison with non-smokers, the relative risks for former smokers who had quit smoking for more than 10 years were between 2 and 4 (Franceschi et al., 1990; De Stefani et al., 1998; La Vecchia et al., 1999), whereas the relative risks for current smokers in these studies were 10–14. In one study in Brazil (Schlecht et al., 1999a), relative risks for former smokers who had stopped smoking for more than 10 years approached 1, whereas that for current smokers was just below 6. Consumption of black tobacco, hand-rolled cigarettes or plain cigarettes resulted in a higher risk for pharyngeal cancer than consumption of blond tobacco, manufactured cigarettes or filter-tipped cigarettes (De Stefani et al., 1998; 2007).
2.3.3 Cancer of the nasal cavity and accessory sinuses
In the Life Span Study in Japan (Akiba, 1994) the association of tobacco use with sinonasal cancer was examined. A total of 26 cases of sinonasal cancer were identified among 61505 adults during follow-up. Relative risk estimates, adjusted for sex, location, population group, atomic bomb exposure, year of birth and attained age, were 2.9 (95%CI: 0.5–) and 4.0 (95%CI: 1.2–) for former and current smokers, respectively, when compared with non-smokers [upper confidence limits were not reported]. The cohort of 34439 British doctors followed up to 50 years (Doll et al., 2005) showed increased risk for current smokers and smokers of more than 25 cigarettes per day, but only six deaths from nasal cavity and sinuses cancers were observed (Table 2.15 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.15.pdf).
A total of nine case–control studies of nasal cavity and sinus cancers have been conducted. When histological types were combined, all studies found an increased risk associated with cigarette smoking, but only one was statistically significant (Caplan et al., 2000). In seven studies, dose–response in terms of intensity of smoking (cigarettes/day), duration of smoking or pack–years was considered. A positive significant trend was found in five studies (Brinton et al., 1984; Hayes et al., 1987; Fukuda & Shibata, 1990; Zheng et al., 1993; Caplan et al., 2000) and suggested in the other two (Strader et al., 1988; Zheng et al., 1992c).
One study (Zheng et al., 1993a) found a significant decrease in risk for sinonasal cancer associated with increasing number of years since cessation of smoking. In a previous study, the same authors had found a negative, non-significant association (Zheng et al., 1992c).
Five studies analysed squamous-cell carcinomas and adenocarcinomas separately (Brinton et al., 1984; Hayes et al., 1987; Strader et al., 1988; Zheng et al., 1992c; ’t Mannetje et al., 1999). In all studies, there was a significantly increased risk for squamous-cell carcinomas, whereas the risk was generally not increased for adenocarcinomas.
2.3.4 Cancer of the nasopharynx
(a) Cohort studies
The risk for nasopharyngeal carcinoma has been examined in relation to tobacco use in six cohort studies, three of them reported since the last evaluation (IARC 2004a; Table 2.16 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.16.pdf). In one study, conducted in a low-risk area (Chow et al., 1993a), a significant increase in risk among smokers and suggestive positive dose–response relationships by duration of smoking and age at starting smoking were found. In another study, conducted in Province of Taiwan, China, an area in which nasopharyngeal cancer area is endemic, a similarly increased risk was found, but it was not statistically significant (Liaw & Chen, 1998). Doll et al. (2005) identified a risk only for smokers of more than 25 cigarettes per day, however, this result was based on only four deaths. Friborg et al. (2007) in Singapore found statistically significant increased risk of nasopharyngeal cancer only for those smoking for 40 years or more. Hsu et al. (2009) in Taiwan, China observed increased statistically significant risks only for those smoking for 30 years or more and those with cumulative exposure of 30 pack–years or more.
(b) Case–control studies
The study designs and the results of the case–control studies on the association of nasopharyngeal carcinoma with cigarette smoking reported since the previous IARC Monograph (IARC, 2004a) are given in Table 2.17 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.17.pdf) and Table 2.18 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.18.pdf), one being a nested case–control analysis within a cohort study (Marsh et al., 2007).
In total, 14 informative case–control studies were available. In almost all of these, the risk for nasopharyngeal carcinoma was higher in smokers than in non-smokers. In Taiwan, China (Cheng et al., 1999) high risks were statistically significant only for duration of smoking of 20 years or more. In the five studies conducted in the USA (Mabuchi et al., 1985; Nam et al., 1992; Zhu et al., 1995; Vaughan et al., 1996; Marsh et al., 2007), where the incidence of nasopharyngeal carcinoma is low, the relative risks for current smokers ranged between 2 and 4, but were not statistically significant in the two studies (Mabuchi et al., 1985; Marsh et al., 2007). In a study conducted in Shanghai, an area of China in which nasopharyngeal carcinoma is not endemic (Yuan et al., 2000), the relative risk was just below 2. In one study from the Philippines there was a sevenfold increase in risk after more than 30 years of smoking (West et al., 1993). The four studies (Lin et al., 1973; Yu et al., 1990; Ye et al., 1995; Cao et al., 2000) conducted in areas of China in which nasopharyngeal carcinoma is endemic (Taiwan, China, Guangzhou, and Sihui) found relative risks for ever smoking ranging between 2 and 5. In the study from the North of Africa (Feng et al., 2009) the only statistically significant increased risk was found for differentiated nasopharyngeal cancer in those that had smoked more than 22 cigarettes/day. [The result, based only on three cases, is very unstable (RR, 313; 95%CI: 1.94–50336).]
A statistically significant dose–response relationship was detected in seven studies that evaluated the effects of daily or cumulative exposure to tobacco smoke (Yu et al., 1990; Nam et al., 1992; Zhu et al., 1995; Vaughan et al., 1996; Cao et al., 2000; Yuan et al., 2000; Feng et al., 2009) and was suggestive in two others (Lin et al., 1973; West et al., 1993). In two studies the risk of nasopharyngeal carcinoma decreased with increasing time since quitting smoking (Nam et al., 1992; Vaughan et al., 1996).
In the remaining studies, six from areas in which nasopharyngeal carcinoma is endemic (Ng, 1986; Yu et al., 1986; Sriamporn et al., 1992; Zheng et al., 1994; Cheng et al., 1999; Feng et al., 2009; Guo et al., 2009) and seven from areas in which it was not endemic (Henderson et al., 1976; Lanier et al., 1980; Mabuchi et al., 1985; Ning et al., 1990; Armstrong et al., 2000, Marsh et al., 2007), the relative risks for nasopharyngeal carcinoma for ever smoking were not significantly increased (Lanier et al., 1980; Mabuchi et al., 1985; Cheng et al., 1999) or were close to 1.0 (Henderson et al., 1976; Ng, 1986; Yu et al., 1986; Ning et al., 1990; Sriamporn et al., 1992; Zheng et al., 1994; Guo et al., 2009).
In the two studies that distinguished between different histological types, relative risks were higher for keratinized (squamous-cell) carcinoma than for unkeratinized carcinoma (Zhu et al., 1995; Vaughan et al., 1996).
In the three studies in which men and women were analysed separately (Lin et al., 1973; Nam et al., 1992; Yuan et al., 2000), the relative risks were found to increase similarly in both sexes in two studies (Nam et al., 1992; Yuan et al., 2000) and were higher among women in the study of Lin et al. (1973).
2.3.5 Cancer of the oesophagus
In the previous IARC Monograph (IARC, 2004a), both histological subtypes of oesophageal cancer (squamous-cell carcinoma and adenocarcinoma) were considered to be causally related to cigarette smoking. Many more epidemiological studies have since been conducted, and results of these studies further support this conclusion.
(a) Squamous cell carcinoma and unspecified cancer of the oesophagus
Since the previous IARC Monograph (IARC, 2004a), there have been reports on 9 cohort studies (Table 2.19 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.19.pdf) and 22 case–control studies (Tables 2.20–2.23; see below), making 30 cohort and 55 case–control studies in all. All showed that the risk of oesophageal squamous cell carcinoma was associated with cigarette smoking. In one study (Li et al., 1989), the elevated risk was observed only in an area with a relatively low incidence of oesophageal cancer. However, two later studies in the same area, Lin County, China, found a twofold increase in risk for oesophageal cancer among smokers (Gao et al., 1994; Lu et al., 2000).
In most cohort studies and in most case–control studies with relatively large sample sizes (IARC, 2004a; Table 2.19 online; Table 2.20 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.20.pdf; Table 2.21 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.21.pdf), the risk for oesophageal cancer was shown to increase with increasing duration of smoking (11 cohort and 32 case–control studies) or number of cigarettes smoked daily (18 cohort and 31 case–control studies), and to decrease with increasing age at starting smoking (12 case–control studies). In comparison with pharyngeal and laryngeal cancers, relative risks for oesophageal cancer estimated by duration and by intensity of smoking were somewhat lower (see Sections 2.3.2 and 2.3.6, respectively).
Ten cohort and 20 case–control studies (IARC, 2004a; Table 2.19 online; Table 2.22 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.22.pdf) investigated the effect of smoking cessation on risk of oesophageal cancer. Although not all studies analysed the trend, all found a decreasing relative risk with increasing number of years since quitting. In some studies, the risk first started to decrease after 10 years of cessation (Brown et al., 1988; Rolón et al., 1995; Gammon et al., 1997; Castellsagué et al., 1999; Freedman et al., 2007b; Bosetti et al., 2008) or after 30 years of cessation (Pandeya et al., 2008).
When comparing the types of tobacco smoked (Table 2.23 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.23.pdf), consumption of black tobacco resulted in a higher risk for oesophageal cancer than did consumption of blond tobacco (De Stefani et al., 1990; Rolón et al., 1995; Castellsagué et al., 1999; Launoy et al., 2000; Vioque et al., 2008). Similarly, smoking untipped cigarettes generally resulted in a higher risk than smoking filter-tipped cigarettes (Vaughan et al., 1995; Gammon et al., 1997; Castellsagué et al., 1999).
Two studies from the USA reported risks separately for blacks and whites. After adjustment for alcohol consumption, age and income, risks were very similar for former and current smokers and for the number of cigarettes smoked per day and duration of smoking (Brown et al., 1994a; Brown et al., 2001).
(b) Adenocarcinoma of the oesophagus
Two decades ago it was noted that incidence rates for adenocarcinoma of the oesophagus and gastric cardia had increased steadily in the USA, whereas the incidence rate for squamous-cell carcinoma of the oesophagus had remained relatively stable (Blot et al., 1991). An increase in the incidence of adenocarcinoma of the distal oesophagus and cardia was also noted in the United Kingdom (Powell & McConkey, 1990), and in several other countries. Since 1990, several studies have focused on the risk factors for adenocarcinoma of the oesophagus. Since the last evaluation (IARC, 2004a) one cohort study (Freedman et al., 2007b) and three case–control studies (Table 2.24 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.24.pdf; Table 2.25 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.25.pdf) have been reported, totaling 13 case–control studies on the association of cigarette smoking and adenocarcinoma of the oesophagus.
(i) Intensity and duration of smoking
Ten studies, three that included only cases of adenocarcinoma of the oesophagus (Menke-Pluymers et al., 1993; Gammon et al., 1997; Wu et al., 2001), three that included cases of adenocarcinoma of the oesophagus, gastro-oesophageal junction and gastric cardia combined (Kabat et al., 1993; Brown et al., 1994b; Vaughan et al., 1995), and four that stratified by histology (Lindblad et al., 2005; Freedman et al., 2007b; Hashibe et al., 2007a; Pandeya et al., 2008), showed a significant positive association of adenocarcinoma of the oesophagus with cigarette smoking. The relative risks were somewhat lower than those for squamous cell carcinoma of the oesophagus. Three studies, one in China (Gao et al., 1994), one in Sweden (Lagergren et al., 2000), and one in the USA (Zhang et al., 1996), reported similarly elevated relative risks, but some of these risks were not statistically significant, probably because of relatively small numbers of cases.
Of those studies that reported risks adjusted for alcohol consumption, a positive, significant dose–response relationship was found with intensity of smoking (Kabat et al., 1993; Brown et al., 1994b; Gammon et al., 1997; Hashibe et al., 2007a), duration of smoking (Gammon et al., 1997; Pandeya et al., 2008) and/or pack–years (Vaughan et al., 1995; Zhang et al., 1996; Gammon et al., 1997; Pandeya et al., 2008).
(ii) Cessation of smoking
Ten studies provided point estimates for former smokers. In eight, relative risks were lower in former smokers than in current smokers, although they remained elevated (Kabat et al., 1993; Gao et al., 1994; Vaughan et al., 1995; Gammon et al., 1997; Wu et al., 2001; Lindblad et al., 2005; Freedman et al., 2007b; Pandeya et al., 2008), and were increased in the other studies (Lagergren et al., 2000; Hashibe et al., 2007a). The decrease in relative risk associated with years since cessation was weak, but a significant trend was found in two out of six studies (Gammon et al., 1997; Wu et al., 2001).
(iii) Confounding
With the exception of two studies (Levi et al., 1990; Wu et al., 2001), all studies adjusted for alcohol intake as a potential confounder. Three more recent studies also adjusted for fruit and vegetables intake (Freedman et al., 2007b; Hashibe et al., 2007a; Pandeya et al., 2008). Ten of these studies were conducted in the USA (Kabat et al., 1993; Brown et al., 1994b; Vaughan et al., 1995; Zhang et al., 1996; Gammon et al., 1997; Freedman et al., 2007b) the Netherlands (Menke-Pluymers et al., 1993), the United Kingdom (Lindblad et al., 2005), central and eastern Europe (Hashibe et al., 2007a) and Australia (Pandeya et al., 2008), where chewing of betel quid with tobacco or use of other forms of smokeless tobacco are not likely confounders. One study conducted in Sweden was adjusted for snuff use (Lagergren et al., 2000).
(iv) Sex
Kabat et al. (1993) examined risks for men and women separately and observed similar patterns in both sexes, although risks among current smokers and heavy smokers were somewhat higher for women than for men. Lindblad et al. (2005) also found higher risks in women than in men, but they were not statistically significant.
2.3.6 Cancer of the larynx
Laryngeal cancer is one of the cancers most strongly associated with cigarette smoking (IARC, 1986, 2004a). Since the previous IARC Monograph, more epidemiological evidence has become available to strengthen this conclusion.
(a) Potential confounders
Other causes of laryngeal cancer include alcohol consumption, some occupational exposures (e.g. sulphuric acid; IARC, 2012a) and possibly some dietary habits. In investigating associations between smoking and laryngeal cancer, potential confounding by alcohol consumption has been considered in most of the studies.
(b) Intensity and duration of smoking
Cohort and case–control studies have been carried out in Asia, Europe, North and South America, and South Africa. In all, the risk for laryngeal cancer was consistently higher in smokers, and a positive significant trend was observed with increasing duration and intensity of smoking (IARC, 2004a; Table 2.26 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.26.pdf; Table 2.27 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.27.pdf; Table 2.28 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.28.pdf).
In most case–control studies, the relative risks for laryngeal cancer were near to or greater than 10 for smokers who had smoked for longer than 40 years (Falk et al., 1989; Zheng et al., 1992b) or had smoked more than 20 cigarettes per day (Tuyns et al., 1988; Falk et al., 1989; Choi & Kahyo, 1991; Zatonski et al., 1991; Muscat & Wynder, 1992; Zheng et al., 1992b; Hedberg et al., 1994; Sokić et al., 1994; Talamini et al., 2002). Cancer of the larynx in non-smokers is so rare that several studies used as the reference category light smokers (Herity et al., 1982; Olsen et al., 1985a; De Stefani et al., 1987; Zatonski et al., 1991; López-Abente et al., 1992; Maier & Tisch, 1997), or former smokers (Hashibe et al., 2007b). Consequently, relative risks were lower in these studies, although the increases were still statistically significant.
Three case–control studies reported odds ratios for cancer of the larynx that increased with decreasing age of starting smoking (Franceschi et al., 1990; Zatonski et al., 1991; Talamini et al., 2002).
(c) Cessation of smoking
The risk for cancer of the larynx declines rather rapidly after cessation of smoking (IARC, 2004a; Table 2.29 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.29.pdf). No detectable higher risk compared with never-smokers was seen among subjects who had quit smoking for at least 10 years (Franceschi et al., 1990; Ahrens et al., 1991; Schlecht et al., 1999a, b; Bosetti et al., 2006; Hashibe et al., 2007b).
(d) Types of tobacco or of cigarette
Some investigators considered the role of type of tobacco (IARC, 2004a; Table 2.30 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.30.pdf). An average 2.5-fold higher risk was observed in smokers of black tobacco compared to smokers of blond tobacco (De Stefani et al., 1987; Tuyns et al., 1988; López-Abente et al., 1992). Smoking untipped cigarettes also led to a higher risk than smoking filter-tipped cigarettes (Wynder & Stellman, 1979; Tuyns et al., 1988; Falk et al., 1989). Those that smoke cigarettes only had higher risks of larynx cancer than those that smoke cigars only (Hashibe et al., 2007b).
(e) Subsites
Six studies investigated the risk for glottic and supraglottic cancer separately (Olsen et al., 1985a; Tuyns et al., 1988; López-Abente et al., 1992; Maier et al., 1992b; Muscat & Wynder, 1992; Sapkota et al., 2007). The cancer risk increased with increasing amount smoked per day and with cumulative exposure for both subsites (IARC, 2004a; Table 2.28 online). In addition, the observed relative risks were higher for supraglottic cancer than for glottic cancer (Maier et al., 1992b; Sapkota et al., 2007).
(f) Sex
Few studies investigated sex-specific effects. In one cohort study (Raitiola & Pukander, 1997) similar risks were found for men and women, whereas in two case–control studies (Zheng et al., 1992b; Tavani et al., 1994), the relative risks for women were up to 10-fold higher than for the corresponding categories in men, though a small number of cases were involved. However, Freedman et al. (2007a) observed higher relative risks in men than women (Table 2.26 online). One study looked at women only and found higher risks of laryngeal cancer in former and current smokers relative to non-smokers, and also according to the number of cigarettes per day with a clear dose–response effect (P < 0.001) (Gallus et al., 2003b).
2.3.7 Cancer of the upper aerodigestive tract combined
In epidemiological studies, especially in cohort studies in which there are few cases at some sites, investigators often combine cancers of the oral cavity, pharynx, larynx and oesophagus and term these ‘cancer of the upper aerodigestive tract’. This section summarizes the data from 19 cohort studies (IARC, 2004a; Table 2.31 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.31.pdf), and 40 case–control studies (IARC, 2004a; Tables 2.32–2.35; see below).
(a) Intensity and duration of smoking
In all but two cohort studies from Japan (Kono et al., 1987; Akiba, 1994), the risk for cancer of the upper aerodigestive tract was strongly associated with cigarette smoking. Relative risks increased with increasing daily cigarette consumption (Hammond & Horn, 1958; Doll et al., 1980, 1994; Akiba & Hirayama, 1990; Kuller et al., 1991; Chyou et al., 1995; Engeland et al., 1996; Murata et al., 1996; Yuan et al., 1996; Kjaerheim et al., 1998; Liaw & Chen, 1998; Yun et al., 2005; Freedman et al., 2007a), duration of smoking (Chyou et al., 1995; Yun et al. 2005; Friborg et al., 2007) or pack–years (Liaw & Chen, 1998; Freedman et al., 2007a).
The main characteristics and results of the case–control studies are presented in IARC (2004a), and in Table 2.32 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.32.pdf) and Table 2.33 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.33.pdf), respectively. Intensity of smoking was measured in most of these studies. The link between duration of smoking and cancer of the upper aerodigestive tract was examined in 20 case–control studies (Blot et al., 1988; Merletti et al., 1989; Barra et al., 1991; De Stefani et al., 1992, 2007; Franceschi et al., 1992; Day et al., 1993; Mashberg et al., 1993; Kabat et al., 1994; Lewin et al., 1998; Bosetti et al., 2000a; Garrote et al., 2001; Gallus et al., 2003a; Lissowska et al., 2003; Znaor et al., 2003; Castellsagué et al., 2004; Menvielle et al., 2004a, b; Rodriguez et al., 2004; Hashibe et al., 2007c; Sapkota et al., 2007). Nine also considered age at starting smoking (Blot et al., 1988; Merletti et al., 1989; Barra et al., 1991; Franceschi et al., 1992; Day et al., 1993; Lewin et al., 1998; Garrote et al. 2001; Lissowska et al. 2003; Menvielle et al. 2004a).
In all but one study (Rao et al., 1999) there was an increased risk for cancer of the upper aerodigestive tract associated with cigarette smoking. A clear dose–response relationship was seen with increasing daily tobacco consumption and duration of smoking as well as with decreasing age at starting smoking in most of the studies examined.
(b) Cessation of smoking
Twelve cohort studies (Doll et al., 1980, 1994; Tomita et al., 1991; Akiba, 1994; Chyou et al., 1995; Engeland et al., 1996; Nordlund et al., 1997; Kjaerheim et al., 1998; Yun et al., 2005; Freedman et al., 2007a; Friborg et al., 2007; Ide et al., 2008) provided point estimates for former smokers (IARC 2004a; Table 2.31 online). The relative risks for former smokers were always lower than those for current smokers.
In 16 case–control studies the relative risk by years since quitting was examined and generally a statistically significant negative trend was found (Table 2.34 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.34.pdf).
(c) Types of cigarette
The characteristics studied in several case–control studies included the use of a filter, the type of tobacco, the tar content and whether the product was manufactured or hand-rolled (IARC, 2004a; Table 2.35 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.35.pdf). Consumption of black tobacco, cigars, untipped cigarettes, hand-rolled cigarettes, or cigarettes with a high-tar yield generally resulted in a higher risk than consumption of blond tobacco (Merletti et al., 1989; Castellsagué et al., 2004; De Stefani et al., 2007), filter-tipped cigarettes (Merletti et al., 1989; Mashberg et al., 1993; Kabat et al., 1994; Lissowska et al., 2003; De Stefani et al., 2007), manufactured cigarettes (De Stefani et al., 1992, 2007) or low-tar cigarettes (Franceschi et al., 1992). Two studies from India (Znaor et al., 2003; Sapkota et al. 2007) revealed higher risks of bidi smoking related to cigarettes smoking.
(d) Sex
Sex-specific effects were analysed in four cohort studies (IARC 2004a; Table 2.31 online). In three cohort studies (Hammond & Seidman, 1980; Akiba & Hirayama, 1990; Freedman et al., 2007a) a higher relative risk was found for male smokers than for female smokers; however, Ide et al. (2008) detected a higher risk among women in a study with a small number of cases.
In three case–control studies (Blot et al., 1988; Kabat et al., 1994; Muscat et al., 1996) the relative risks were higher for women than for men in all categories of intensity of smoking (number of cigarettes per day), cumulative exposure (cumulative tar consumption, pack–years, duration of smoking) and age at starting smoking, as well as for former smokers. However, the trends in men were always in the same direction and of the same order of magnitude. An exception to the pattern was that in one study (Merletti et al., 1989) the relative risk for smoking filter-tipped cigarettes was higher than that for smoking untipped cigarettes for women.
Overall, the strength of association by sex was generally similar, especially when taking into account the fact that women generally underreport levels of smoking and that most studies included many fewer women than men.
(e) Ethnicity
Relative risks were reported separately for blacks and whites in a large case–control study from the USA (Day et al., 1993). Relative risks adjusted for alcohol consumption, sex and other relevant variables were very similar for the number of cigarettes smoked per day, years of cigarette smoking, age at starting smoking and number of years since stopping smoking.
2.4. Cancer of the stomach
2.4.1 Overview of studies
In the previous IARC Monograph (IARC, 2004a) it was concluded that there was sufficient evidence that tobacco smoking causes cancer of the stomach. Three meta-analyses have since examined the evidence for gastric cancer in 42 independent cohort studies published between 1958 and July 2007 (Ladeiras-Lopes et al., 2008), in 46 case–control studies published between 1997 and June 2006 (La Torre et al., 2009), and in 10 cohort and 16 case–control studies conducted in Japanese populations published between 1966 and March 2005 (Nishino et al., 2006; Table 2.36 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.36.pdf). For current smokers compared to never smokers, the risk for stomach cancer was found to be statistically significantly increased by 53% (Ladeiras-Lopes et al., 2008), 56% (Nishino et al., 2006), and 57% when considering high quality case–control studies (La Torre et al., 2009), with moderate to high heterogeneity.
Since the previous IARC Monograph (IARC, 2004a), the association between cigarette smoking and stomach cancer risk (15 studies) and mortality (4 studies) has been examined in 19 cohort studies (Table 2.37 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.37.pdf). Eleven of these were conducted in Asia (Sasazuki et al., 2002; Jee et al., 2004; Koizumi et al., 2004; Wen et al., 2004; Fujino et al., 2005; Sauvaget et al., 2005; Tran et al., 2005; Kurosawa et al., 2006; Kim et al., 2007; Sung et al., 2007; Shikata et al., 2008), seven in Europe (Simán et al., 2001; González et al., 2003; Doll et al., 2005; Lindblad et al., 2005; Sjödahl et al., 2007; Batty et al., 2008; Zendehdel et al., 2008) and one in the USA (Freedman et al., 2007a). Only the updated British Doctors’ study (Doll et al., 2005) and the most recent studies (Shikata et al., 2008; Zendehdel et al., 2008) were not included in the meta-analysis of cohort studies (Ladeiras-Lopes et al., 2008). Elevated risks in current smokers were found in all studies. The reported association of current smoking with mortality in the four cohort studies conducted in Taiwan, China (Wen et al., 2004), Japan (Kurosawa et al., 2006) and the United Kingdom (Doll et al., 2005; Batty et al., 2008) was comparable to that with incidence.
In addition, the association between smoking and stomach cancer risk has been reported in 37 case–control studies since the previous IARC Monograph, of which 22 are hospital-based and 15 population-based. With the exception of three studies (Campos et al., 2006; García-González et al., 2007; Suwanrungruang et al., 2008; Table 2.38 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.38.pdf), all these studies were included in the meta-analysis conducted by (La Torre et al., 2009).
2.4.2 Factors affecting risk
(a) Intensity and duration
Clear evidence has been provided by the meta-analyses as well as by the additional cohort studies that the risk for stomach cancer increases significantly with increasing daily cigarette consumption, duration or pack–years of smoking, although individual studies did not always find statistically significant dose–response relationships. In one meta-analysis based on 21 cohort studies, the risk for stomach cancer increased statistically significantly by 53% with consumption of approximately 20 cigarettes per day (Ladeiras-Lopes et al., 2008). Using trend estimation analysis as proposed by Greenland & Longnecker (1992), the authors found an increase in relative risk from 1.3 for the lowest consumption to 1.7 for smoking 30 cigarettes per day.
(b) Cessation of smoking
Risk for stomach cancer has been generally found to be lower in former smokers than in current smokers. In six of the cohort studies decreasing risk with increasing years since stopping smoking was found although none found statistically significant dose–response relationships (González et al., 2003; Koizumi et al., 2004; Sauvaget et al., 2005; Freedman et al., 2007a; Kim et al., 2007; Zendehdel et al., 2008). Risk in former smokers was comparable to never smokers after quitting for 5 years (Kim et al., 2007), 10 years (González et al., 2003; Sauvaget et al., 2005; Freedman et al., 2007a) or 15 years (Koizumi et al., 2004).
2.4.3 Subsites
The effect of current smoking on the risk for stomach cancer by subsite was assessed in ten cohort studies. Elevated risks were found for both cardia and non-cardia cancers. In six studies higher risks were found for cancer of the gastric cardia than for cancer of the distal stomach (Simán et al. 2001; González et al., 2003; Freedman et al., 2007a; Sung et al., 2007; Shikata et al., 2008; Zendehdel et al., 2008), three studies found no difference (Sasazuki et al., 2002; Lindblad et al., 2005; Tran et al., 2005), and in one study higher risk for cancer in the antrum rather than the body or the cardia was found (Koizumi et al., 2004). A meta-analysis yielded statistically significant summary relative risks of 1.87 for cardia cancers and 1.60 for non-cardia cancers based on nine cohort studies (Ladeiras-Lopes et al., 2008). However, there was substantial heterogeneity across studies for cardia cancers. For case–controls studies, the corresponding odds ratios were 2.05 (95%CI: 1.50–2.81) and 2.04 (95%CI: 1.66–2.50), respectively, with greater heterogeneity for non-cardia cancers. Criteria for the classification by subsite were not always described (Simán et al., 2001; Koizumi et al., 2004; Lindblad et al., 2005; Tran et al., 2005) and some studies included tumours located in the upper third of the stomach in the group of cardia cancer (Sasazuki et al., 2002; Sung et al., 2007; Shikata et al. 2008).
In three studies risk estimates for smoking associated stomach cancer were estimated by histological type (Sasazuki et al., 2002; Koizumi et al., 2004; Shikata et al., 2008). The relative risks were 2.1 (95%CI: 1.2–3.6), 1.6 (95%CI: 1.1–2.3) and 2.3 (95%CI: 1.3–4.1) for the differentiated type, respectively, and 0.6 (95%CI: 0.3–1.1), 2.1 (95%CI: 1.1–4.1), and 1.3 (95%CI: 0.5–3.5) for the non-differentiated type, respectively.
2.4.4 Population characteristics
In four of the additional cohort studies risk was reported separately for men and women (González et al., 2003; Jee et al., 2004; Fujino et al., 2005; Kim et al., 2007), in three studies only for men (Koizumi et al., 2004; Tran et al., 2005; Sung et al., 2007) and in one mortality study for men as well as for women (Wen et al., 2004). Generally, the relative risks were smaller in women than in men. For all stomach cancers, risk in current smokers compared to never smokers was found to be significantly increased by 62% in men (based on 18 studies) and by 20% in women (based on nine studies) in the meta-analysis of cohort studies (Ladeiras-Lopes et al., 2008). The men–women differences were independent of exposure level but could be explained by the sex difference in the distribution by histological type and other factors associated with socioeconomic status.
Ethnicity does not appear to modify the effect of smoking on stomach cancer risk. In the meta-analysis of case–control studies risk in current smokers was increased by 78% in Caucasians and by 48% in Asians (La Torre et al., 2009). The summary risk based on the cohort studies increased by 46% and 47% in Caucasian and Asian studies, respectively. In a meta-regression analysis including the variables sex, population, and fruit and vegetable consumption, sex but not origin of the population showed significant differences in risk estimates (Ladeiras-Lopes et al., 2008).
2.4.5 Bias and confounding
Generally, most cohort studies have relied on baseline information and did not update the exposure information, possibly leading to misclassification of smoking status. Most of the recent cohort studies have accounted for confounding by alcohol consumption (Fujino et al., 2005; Lindblad et al., 2005; Sjödahl et al., 2007; Sung et al., 2007) as well as fruit and vegetable consumption (González et al., 2003; Koizumi et al., 2004; Freedman et al. 2007a) and still observed significantly increased risk of stomach cancer in current smokers.
2.4.6 Helicobacter pylori infection
The association between tobacco smoking and stomach cancer could be confounded or modified by the effect of H. pylori infection, an established risk factor for stomach cancer. In three case–control studies (Zaridze et al., 2000; Brenner et al., 2002; Wu et al. 2003), and two cohort studies (Simán et al., 2001; Shikata et al., 2008) the joint effects and possible interaction between H. pylori status and smoking in relation to risk for stomach cancer was investigated. Among subjects who had H. pylori infection, the risk for stomach cancer was higher in current smokers than in non-smokers by 1.6 to 2.7 fold, providing evidence for a causal effect of tobacco smoking independently of H. pylori infection. Smoking was associated with risk elevations of the same order of magnitude among subjects without H. pylori infection. Smoking and H. pylori therefore may act synergistically, leading to very high risks in current smokers with H. pylori infection compared to non-smokers without H. pylori infection. In one study that examined risk by subsite an effect of smoking independent of H. pylori infection for gastric cardia as well as distal gastric cancer was found (Wu et al., 2003). In none of the studies was there statistically significant evidence for interaction.
2.5. Cancer of the pancreas
2.5.1 Overview of studies
Previous IARC Monographs (IARC, 1986, 2004a) concluded that exposure to tobacco smoke caused cancer of the pancreas. Additional evidence has come from a pooled analysis of eight cohort studies with almost 1500 incident cases of pancreatic cancer and an equal number of controls (Lynch et al., 2009) as well as a meta-analysis of 82 independent studies (42 case–control studies, 40 cohort studies) published between 1950 and 2007 (Iodice et al., 2008; Table 2.39 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.39.pdf). In the meta-analysis 74% and 20% significant increased risks for current and former smokers, respectively, were found with significant heterogeneity of effect regarding current smoking across studies. Adjustment for confounders explained some of the heterogeneity (Iodice et al., 2008). A similar significant risk elevation of 77% for current smokers was found in the pooled analysis, without study heterogeneity (Lynch et al., 2009). For former smokers, risk was increased non-significantly by 9%.
Since the previous IARC Monograph (IARC, 2004a), a total of 15 cohort studies have reported on the association between cigarette smoking and pancreatic cancer incidence (8 studies) and mortality (5 studies) or both (one study) (Table 2.40 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.40.pdf), two of which were included in the pooled analysis (Coughlin et al., 2000; Vrieling et al., 2009). Excluding case–control studies that did not report odds ratios for current smokers, there were three additional case–control studies (Duell et al., 2002; Inoue et al., 2003; Alguacil & Silverman, 2004; Table 2.41 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.41.pdf). The effect of cigar and pipe smoking on pancreatic cancer was also examined in the ACS Cancer Prevention Study II regarding mortality (Shapiro et al., 2000; Henley et al., 2004) and in the Kaiser Permanente Medical Care Program regarding incidence (Iribarren et al., 1999). All the additional studies showed an increased risk for pancreatic cancer associated with tobacco smoking, generally higher in current than in former smokers. The reported risk estimates were not always statistically significant, predominantly due to the small size of some studies and therefore lack of statistical precision.
2.5.2 Factors affecting risks
(a) Intensity and duration
Clear evidence has been provided by the meta-analysis, the pooled analysis as well as the additional studies that the risk for cancer of the pancreas increases significantly with increasing daily cigarette consumption, duration and pack–years of smoking (Coughlin et al., 2000; Gapstur et al., 2000; Nilsen & Vatten, 2000; Nilsson et al., 2001; Isaksson et al., 2002; Doll et al., 2005; Yun et al., 2005; Ansary-Moghaddam et al., 2006; Gallicchio et al., 2006; Vrieling et al., 2009). In the meta-analysis risk of pancreatic cancer increased significantly by 62% with an increase of 20 cigarettes per day (based on 45 studies) and by 16% with a 10-year increase in smoking duration (based on 16 studies), but with significant study heterogeneity. In the pooled analysis, the excess odds ratio per pack–years generally declined with increasing smoking intensity (Lynch et al., 2009).
(b) Cessation of smoking
A reduction in risk in former smokers who had stopped smoking for at least 10 years was found in the meta-analysis (Iodice et al., 2008) and the pooled study (Lynch et al., 2009). In some cohort studies risk was already comparable to never smokers five years after quitting (Boyle et al., 1996; Fuchs et al., 1996; Nilsen & Vatten, 2000; Vrieling et al., 2009).
(c) Types of tobacco
In non–cigarette smokers, mortality from pancreatic cancer was increased although not statistically significantly so in cigar smokers in the CPS-II cohort study (Shapiro et al., 2000) as well as a large case–control study (Alguacil & Silverman, 2004) but was less clearly elevated in the smaller Kaiser Permanente cohort study (Iribarren et al., 1999). There was a significantly increased mortality for current cigar smokers who reported inhaling cigar smoke (Shapiro et al., 2000). Pipe smoking was also found to be associated with an increased risk of cancer of the pancreas, which was stronger in those who reported that they inhaled the smoke (Henley et al., 2004). A limitation of the cohort studies is that smoking habits were reported only at baseline, misclassification of smoking exposure is likely to underestimate the associated risks. In the meta-analysis there was a significant increase in risk of 47% associated with current cigar and/or pipe smoking (18 studies) and a non-significant risk elevation of 29% with former cigar and/or pipe smoking (5 studies) (Iodice et al., 2008).
2.5.3 Population characteristics
The effect of sex on pancreatic cancer risk was investigated in two cohort studies (Nilsen & Vatten, 2000; Larsson et al., 2005) and on pancreatic cancer mortality in four cohort studies (Coughlin et al., 2000; Gapstur et al., 2000; Nilsson et al., 2001; Lin et al., 2002a). The relative risks were comparable between men and women and no consistent evidence for an effect modification by sex was observed.
Ethnicity does not appear to modify the association of smoking with pancreatic cancer risk. The roughly twofold elevated risk in current smokers compared to never smokers was observed both in studies of Caucasians (Lynch et al., 2009) and of Asians (Lin et al., 2002a; Jee et al., 2004; Yun et al., 2005; Li et al., 2006). In populations of the Asia-Pacific Region, there was also no difference in the strength of association between Asia and Australia/New Zealand (Ansary-Moghaddam et al., 2006).
2.5.4 Confounding factors
In two large cohort studies the risk estimates for pancreatic cancer associated with cigarette smoking were not substantially influenced by adjustment for further potential confounding factors, including diabetes, body mass index (BMI), alcohol and dietary intake (Coughlin et al., 2000; Vrieling et al., 2009).
2.6. Cancer of the colorectum
2.6.1 Overview of studies
In the previous IARC Monograph (IARC, 2004a) it was not possible to conclude that the association between tobacco smoking and colorectal cancer is casual, principally because of concern about confounding by other risk factors. That evaluation was based on a total of 60 epidemiologic studies, although only few were specifically designed to study the effects of smoking. Studies have however shown consistently that cigarette smoking is a risk factor for colorectal adenomatous polyps, which are recognized precursor lesions of colorectal cancer (Hill, 1978). To explain this discrepancy, Giovannucci et al. (1994) hypothesized that a long induction period is required for tobacco to play a role in colorectal carcinogenesis, which would not be captured by studies with shorter follow-up time.
Four recent meta-analyses consistently showed a strong association between cigarette smoking and colorectal cancer (Botteri et al., 2008a; Liang et al., 2009; Huxley et al., 2009; Tsoi et al., 2009).
2.6.2 Cohort studies
Since the previous IARC Monograph (IARC, 2004a), 22 additional cohort studies have investigated the association between tobacco smoke and colorectal cancer (Table 2.42 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.42.pdf). [Studies that did not provide point estimates of risk (Andersen et al., 2009; Hansen et al., 2009; Murphy et al., 2009) and included prevalent colorectal cancer in patients with other diagnosis (Chan et al., 2007) are excluded from this review]. Seven of the studies were conducted in Europe, nine in Asia and five in the USA. In eleven studies, risk estimates were reported solely for colorectal cancer (Tiemersma et al., 2002a; Limburg et al., 2003; Otani et al., 2003; Colangelo et al., 2004; Sanjoaquin et al., 2004; Lüchtenborg et al., 2005a; Kim et al., 2006; Akhter et al., 2007; Huxley, 2007a; Kenfield et al., 2008; Hannan et al., 2009), five studies separately for colon cancer and rectal cancer (Shimizu et al., 2003; Wakai et al., 2003; Jee et al., 2004; Yun et al., 2005; Batty et al., 2008) and five studies both for colorectal cancer as well as for colon and rectal cancers (Terry et al., 2002a; van der Hel et al., 2003a; Doll et al., 2005; Paskett et al., 2007; Tsong et al., 2007; Gram et al., 2009). Six studies were restricted to women (Terry et al., 2002a; Limburg et al., 2003; van der Hel et al., 2003a; Paskett et al., 2007; Kenfield et al., 2008; Gram et al., 2009), and two studies to men (Doll et al., 2005; Yun et al., 2005; Akhter et al., 2007). One study reported both colorectal incidence and mortality (Limburg et al., 2003) and three studies only reported colorectal cancer mortality (Doll et al., 2005; Huxley, 2007a; Batty et al., 2008; Kenfield et al., 2008).
(a) Smoking status
Virtually all studies reported elevated risk associated with smoking, although results were not always statistically significant. The largest meta-analysis based on 36 prospective studies with data from a total of 3007002 subjects found that compared to never smokers, current smokers had a 15% significantly higher risk of developing colorectal cancer and 27% significantly higher risk of colorectal cancer mortality (Liang et al., 2009; Table 2.43 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.43.pdf). In former smokers, colorectal cancer risk was also significantly elevated by 20% whereas colorectal cancer mortality was non-significantly increased by 20%. The risk estimates were not significantly different between colon and rectal cancer for current smokers (RR, 1.10 versus 1.19) and for former smokers (RR, 1.10 versus 1.20). There was no heterogeneity among colorectal cancer studies and no evidence for publication bias. Comparable risk elevations in current and former smokers were found in the other meta-analyses. For current smokers, the risk for colorectal cancer increased significantly by 16% when using data from 22 cohort studies (Huxley et al., 2009), by 20% based on 28 cohort studies (Tsoi et al., 2009), and by 7% based on data from 45 cohort and case–control studies (Botteri et al., 2008a). In the latter meta-analysis a 17% significantly higher risk of colorectal cancer in former smokers was found.
(b) Intensity of smoking
All but three of the recent 21 cohort studies (van der Hel et al., 2003a; Jee et al., 2004; Sanjoaquin et al., 2004) investigated dose–response relationships, using at least one of number of cigarettes smoked, duration of smoking, pack–years of smoking, age at smoking initiation, time since smoking cessation. In two further studies (Tiemersma et al., 2002a; Batty et al., 2008) these parameters were examined separately in current and former smokers, as by Chao et al. (2000). Statistically significant dose–response trends with amount smoked daily were reported for colorectal cancer (Lüchtenborg et al., 2005a; Akhter et al., 2007; Paskett et al., 2007; Kenfield et al., 2008), for colon cancer (Paskett et al., 2007), and for rectal cancer (Paskett et al., 2007; Tsong et al., 2007). The dose–response of daily cigarette consumption and colorectal cancer was assessed in two meta-analyses (Liang et al., 2009; Tsoi et al., 2009) and both found statistically significant relationships. Based on eleven studies, Liang et al. (2009) found that risk for colorectal cancer increased significantly by 17% with an increase of 20 cigarettes/day and by 38% with an increase of 40 cigarettes/day, while colorectal cancer mortality increased by 41% and 98%, respectively (Table 2.43 online). The risk elevation associated with an increase of 20 cigarettes/day was greater for rectal than for colon cancer (13% versus 3%) but this difference was not statistically significant.
(c) Duration of smoking
In addition to two previously reported studies (Hsing et al., 1998; Chao et al., 2000), thirteen studies have examined duration of smoking and colorectal cancer risk. A statistically significant trend of increasing risk with increasing duration was found for colorectal (Limburg et al., 2003; Kim et al., 2006; Paskett et al., 2007; Gram et al., 2009), for colon cancer (Paskett et al., 2007) and for rectal cancer (Terry et al., 2002a; Paskett et al., 2007; Tsong et al., 2007). In one study, increasing duration of smoking was significantly associated with risk for colorectal cancer solely in former smokers (Tiemersma et al., 2002a). Based on eight studies (Terry et al., 2002a; Tiemersma et al., 2002a; Limburg et al., 2003; Lüchtenborg et al., 2005a; Kim et al., 2006; Akhter et al., 2007; Paskett et al., 2007; Tsong et al., 2007), a meta-analysis for duration of smoking and colorectal cancer incidence yielded highly significant results (Liang et al., 2009). Risk was increased by 9.4% with a 20-year increase in smoking duration and 19.7% with a 40-year increase. Smoking duration was also significantly associated with risk for rectal cancer but not for colon cancer. In another meta-analysis where dose–response relationship was modelled, a nonlinear increase in risk with increasing duration was observed (Botteri et al., 2008a). The risk started to increase after approximately 10 years of smoking and reached statistical significance after 30 years.
(d) Pack–years
Since the previous IARC Monograph, the association of colorectal cancer with pack–years of cigarette smoking has been evaluated in six studies (Limburg et al., 2003; Otani et al., 2003; Shimizu et al., 2003; Wakai et al., 2003; Kim et al., 2006; Gram et al., 2009). In addition to the previously reported significant results (Giovannucci et al., 1994; Heineman et al., 1994; Chao et al., 2000; Stürmer et al., 2000), a statistically significant trend of increasing risk with increasing pack–years was found for colorectal cancer in two studies (Limburg et al., 2003; Gram et al., 2009), and for colon cancer in one study (Gram et al., 2009). In their dose–response analysis of pack–years and colorectal incidence, Liang et al. (2009) included five studies (Giovannucci et al., 1994; Stürmer et al., 2000; Limburg et al., 2003; Otani et al., 2003; Kim et al., 2006) and found a statistically significant trend of increasing risk with increasing pack–years of smoking for colorectal cancer but not specifically for colon or rectal cancer. Risk for colorectal cancer increased by 27% for an increase of 35 pack–years and by 50% for an increase of 60 pack–years.
(e) Age at initiation
In nine of the cohort studies the age at smoking initiation in relation to colorectal cancer (eight studies) or colon and rectal cancer (four studies) was investigated. In four studies a statistically significant trend of increasing risk with decreasing age at initiation of smoking for colorectal cancer was found (Limburg et al., 2003; Kim et al., 2006; Akhter et al., 2007; Gram et al., 2009) and for colon cancer (Gram et al., 2009) and rectal cancer (Tsong et al., 2007). In one meta-analysis (Liang et al., 2009), a highly significant association was found for age at smoking initiation and colorectal cancer incidence based on six studies (Limburg et al., 2003; Kim et al., 2006; Akhter et al., 2007; Paskett et al., 2007; Tsong et al., 2007; Gram et al., 2009). Risk for colorectal cancer was reduced by 2.2% for a 5-year delay in smoking initiation and by 4.4% for a 10-year delay.
(f) Smoking cessation
The effect of smoking cessation by years since stopping was assessed in seven studies, six for colorectal cancer (Tiemersma et al., 2002a; Lüchtenborg et al., 2005a, 2007; Paskett et al., 2007; Kenfield et al., 2008; Gram et al., 2009; Hannan et al., 2009) and three for colon and/or rectal cancer (Wakai et al., 2003; Paskett et al., 2007; Gram et al., 2009). In one study a statistically significant trend in risk reduction with years since quitting was found both overall as well as separately for men and for women (Hannan et al., 2009).
(g) Population characteristics
It has been suggested that the association between smoking and colorectal cancer may be stronger in men than in women. In the three recent cohort studies reporting sex–specific results (Shimizu et al., 2003; Wakai et al., 2003; Colangelo et al., 2004), this was only observed in studies in Japan (Shimizu et al., 2003; Wakai et al., 2003), but could be attributed to the very low prevalence of smoking in women. The studies restricted to women have generally shown associations with cigarette smoking that were of comparable magnitude to those observed in men (Terry et al., 2002a; Limburg et al., 2003; van der Hel et al., 2003a; Paskett et al., 2007; Kenfield et al., 2008; Gram et al., 2009).
Recent studies have been carried out either in Europe and in USA, with predominantly Caucasian study subjects, or in Asia, mostly in Japan and in the Republic of Korea. The results from these studies suggest no differences in the association between tobacco smoking and colorectal cancer between different ethnic groups.
(h) Subsites
Smoking and risks for colon cancer and for rectal cancer were investigated in eleven of the 21 additional studies. Risk patterns are generally consistent between colon and rectal cancer (Otani et al., 2003; van der Hel et al., 2003a; Wakai et al., 2003; Jee et al., 2004; Yun et al., 2005; Batty et al., 2008). In some studies, dose–response relationships were stronger for rectal cancer than for colon cancer (Terry et al., 2002a; Paskett et al., 2007) or were statistically significant only for rectal cancer (Shimizu et al., 2003; Doll et al., 2005; Tsong et al., 2007). In a meta-analysis (Liang et al., 2009) the association was stronger for rectal cancer than for colon cancer in the subset of cohort studies that differentiated cancer by site. Most dose–response variables were not associated with colon cancer incidence whereas associations were stronger for rectal cancer incidence and statistically significant with longer duration of smoking, albeit based only on a small number of studies. In one cohort study the increased risk associated with smoking was more apparent for proximal than for distal colon cancer (Lüchtenborg et al., 2005a), which was not found in an earlier study (Heineman et al., 1994).
(i) Confounding and effect modification
Smokers have been shown to be more likely than non-smokers to be physically inactive, to use alcohol, to have lower consumption of fruits and vegetables and higher consumption of fat and meat, and they are less likely to be screened for colorectal cancer (Le Marchand et al., 1997; Ghadirian et al., 1998; Nkondjock & Ghadirian, 2004; Reid et al., 2006b; Mutch et al., 2009).
Few potential confounders were considered in the cohort studies evaluated in the previous IARC Monograph (IARC, 2004a). Of the cohort studies published since, all except three (van der Hel et al., 2003a; Jee et al., 2004; Doll et al., 2005) considered two or more potential confounders. In eleven of the recent studies adjustments were made for physical activity, alcohol consumption, overweight/obesity (Terry et al., 2002a; Limburg et al., 2003; Otani et al., 2003; Wakai et al., 2003; Yun et al., 2005; Akhter et al., 2007; Ashktorab et al., 2007; Paskett et al., 2007; Tsong et al., 2007; Kenfield et al., 2008; Hannan et al., 2009), and seven also adjusted for dietary habits (e.g. intake of fruits and vegetables, dietary fibres, fat, red meat). Among the studies with the latter adjustments, eight (Giovannucci et al., 1994; Chao et al., 2000; Stürmer et al., 2000; Limburg et al., 2003; Yun et al., 2005; Akhter et al., 2007; Paskett et al., 2007; Hannan et al., 2009) found significant dose–response relationships with at least one of the smoking variables. In two studies a significant association of smoking with colorectal cancer risk was observed after accounting for history of colonoscopy (Paskett et al., 2007; Hannan et al., 2009). Risk factors in multivariable analyses in several studies were level of education, use of menopausal hormone therapy, family history and regular aspirin use. The association between smoking and colorectal cancer was not modified by these other characteristics, or by alcohol consumption in two studies (Otani et al., 2003; Tsong et al., 2007). Therefore, confounding factors do not seem to affect the observed significant increase in risk for colorectal cancer associated with tobacco smoking and the dose–response relationships with smoking variables.
When considering other types of smoking, it is generally found that cigar and pipe smoking are less associated with socioeconomic class and other life-style habits than cigarette smoking. Therefore, it is logical to assume that, for these types of smoking, risk associations derived from epidemiologic studies may be less prone to potential confounding. In all the cohort studies reviewed in the previous IARC Monograph (IARC, 2004a) an elevated, though not always statistically significant, risk was consistently reported for cancers of the colon and the rectum associated with exclusive pipe and/or cigar smoking.
Infection with JC virus has been proposed as a potential risk factor for colon cancer (Rollison et al., 2009) but results still need further validation.
Three cohort studies assessed possible modifying effects by genetic susceptibility. Rapid acetylator phenotype (as determined by polymorphisms of the NAT2 gene involved in metabolism of heterocyclic aromatic amines) was found to increase the risk for colorectal cancer in smokers, in one (van der Hel et al., 2003a) but not in another study (Tiemersma et al., 2002a). For genes involved in the metabolism of polycyclic aromatic hydrocarbons such as GSTM1 or GSTT1, no statistical contribution to the risk of colorectal cancer associated with smoking was observed (Tiemersma et al., 2002a; Lüchtenborg et al., 2005a).
2.6.3 Case–control studies
Thirty-one case–control studies were included in the previous IARC Monograph (IARC, 2004a). Although results were inconsistent with respect to risk association in ever versus former and current smokers, a dose–response relationship with smoking variables was found in some studies. Since then, seventeen case–control studies investigating the association between tobacco smoke and colorectal cancer risk have been published, seven carried out in Asia, four in Europe, five in North America and one in Hawaii (Table 2.44 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.44.pdf). Six studies reported solely for colorectal cancer (Ateş et al., 2005; Chia et al., 2006; Verla-Tebit et al., 2006; Lüchtenborg et al., 2007; Steinmetz et al., 2007; Wu et al., 2009b), four separately for colon and rectal cancer (Ji et al., 2002; Sharpe et al., 2002; Minami & Tateno, 2003; Goy et al., 2008), two for colorectal cancer as well as for colon and rectal cancer (Ho et al., 2004; Gao et al., 2007; Wei et al., 2009), three for colon cancer only (Diergaarde et al., 2003; Kim et al., 2003; Hu et al., 2007) and one for rectal cancer only (Slattery et al., 2003). Nine of the studies reported risk estimates separately for men and for women.
(a) Smoking status
Most case–control studies considered the effects of current and former smoking separately. A positive association between smoking and colorectal cancer was found in virtually all the studies, although the results were generally not statistically significant. Statistically significant increased risk was reported in current smokers for colorectal cancer (Chia et al., 2006; Wu et al., 2009b), for rectal cancer (Slattery et al., 2003; Ho et al., 2004), and in former smokers for colorectal cancer both in men and women combined (Chia et al., 2006) and in women only (Lüchtenborg et al., 2007). Five studies, which did not focus on the main effects of smoking, only evaluated risks for ever smoking (Diergaarde et al., 2003; Kim et al., 2003; Ateş et al., 2005; Gao et al., 2007; Hu et al., 2007); none of these reported significant risk estimates.
(b) Intensity of smoking
Nine case–control studies investigated dose–response relationships considering at least one smoking variable. Number of cigarettes smoked daily was evaluated in seven studies, three for colorectal cancer (Verla-Tebit et al., 2006; Lüchtenborg et al., 2007; Wu et al., 2009b), two for colon and rectal cancer (Ji et al., 2002; Minami & Tateno, 2003), one for rectal cancer (Slattery et al., 2003) and one for colorectal cancer and both subsites (Ho et al., 2004). Statistically significant positive trends of increasing risk with increasing number of cigarettes smoked daily were found for colorectal cancer in only one study (Wu et al., 2009b).
(c) Duration of smoking, pack–years, age at initiation, smoking cessation
Duration of smoking was examined in several studies in relation to colorectal cancer (Ho et al., 2004; Chia et al., 2006; Verla-Tebit et al., 2006; Lüchtenborg et al., 2007; Wu et al., 2009b) and/or to colorectal cancer by subsite (Ji et al., 2002; Minami & Tateno, 2003; Ho et al., 2004). A statistically significant trend with increasing number of years smoked was found in two of the five studies of colorectal cancer (Chia et al., 2006; Wu et al., 2009b). In one study, increasing duration of smoking was significantly associated with risk for rectal cancer in ever smokers but not in current smokers (Ho et al., 2004). In only one earlier case–control study was a significant association in ever smokers with increasing number of years of smoking for colon as well as rectal cancer found (Newcomb et al., 1995).
Duration of smoking exposure was assessed by pack–years of smoking in seven studies (Ji et al., 2002; Slattery et al., 2003; Chia et al., 2006; Verla-Tebit et al., 2006; Lüchtenborg et al., 2007; Goy et al., 2008; Wu et al., 2009b) and by age at smoking initiation in three studies (Ji et al., 2002; Slattery et al., 2003; Wu et al., 2009b). All four studies that evaluated pack–years of smoking with respect to colorectal cancer risk found statistically significant associations. Two studies found a significant association with increasing pack–years in men and women combined; when investigated separately, the increasing trend was statistically significant only in women (Verla-Tebit et al., 2006) or only in men (Wu et al., 2009b). In one study a statistically significant trend with pack–years of smoking in both men and women was found only with non-filtered cigarettes (Lüchtenborg et al., 2007); the relative risk was significant for colon as well as rectal cancer and was greater for rectal cancer.
In two studies a non-significant trend of decreasing risk with increasing time since stopped smoking was found (Verla-Tebit et al., 2006; Lüchtenborg et al., 2007).
(d) Subsites and molecular subtypes
A stronger association between tobacco smoking and rectal cancer compared with colon cancer has generally been observed in the studies that reported risk estimates by cancer site. In a recent meta-analysis including both cohort and case–control studies, higher smoking-related risk estimates for rectal cancer were found than for proximal and distal colon cancer (Botteri et al., 2008a). Stronger relative risk in ever smokers, but not in current smokers, was found for proximal compared to distal tumours in one recent study (Hu et al., 2007).
Colorectal cancer is a multipathway disease. A molecular approach to its classification utilizes: (1) the type of genetic instability, specifically microsatellite instability, and (2) the presence of DNA methylation or the CpG island methylator phenotype (CIMP) (Jass, 2007). Smoking has been associated with microsatellite instability in sporadic colon cancer. Higher risk for microsatellite-unstable than for microsatellite-stable tumours was found in four studies (Slattery et al., 2000; Yang et al., 2000; Chia et al., 2006; Campbell et al., 2009). The observed twofold risk elevation for colorectal cancer showing microsatellite instability is similar in order of magnitude to that found for colorectal polyps. In only one small study similar risk estimates for stable and unstable tumours were found (Diergaarde et al., 2003). Microsatellite instability is characteristic of hereditary nonpolyposis colorectal cancer syndrome and smoking has been associated with colorectal cancer in patients with this syndrome (Watson et al., 2004; Diergaarde et al., 2007). Among sporadic colorectal tumours with microsatellite instability, about 11–28% carry somatic genetic mutations. In addition, the association of colon cancer with smoking was increased two to threefold when widespread CIMP and/or BRAF mutation, irrespective of microsatellite instability status, was present (Samowitz et al., 2006). These data indicate that the association with MSI-high tumours may be attributed to the association of smoking with CIMP and BRAF mutation.
(e) Effect modification
Effect modification by genetic polymorphisms in enzymes metabolizing tobacco smoke constituents could provide further evidence for a causal association between smoking and colorectal cancer. Most studies that have investigated modification of colorectal cancer risk associated with smoking by genetic polymorphisms of xenobiotic enzymes were too small to be informative (Inoue et al., 2000; Smits et al., 2003; Jin et al., 2005; Tranah et al., 2005; van den Donk et al., 2005; Tijhuis et al., 2008). Studies on the possible differential effect by acetylation status have reported stronger association of tobacco smoking (in terms of pack–years) with colorectal cancer risk in slow acetylators phenotypes (Lilla et al., 2006), and with rectal cancer in rapid acetylators phenotypes (Curtin et al., 2009). Furthermore, CYP1A1 and GSTM1 variant alleles were found to greatly affect colon cancer or rectal cancer risk in smokers (Slattery et al., 2004).
2.6.4 Colorectal polyps
Colorectal adenomas and possibly some hyperplastic polyps are considered precursors of colorectal cancer. The epidemiologic evidence on the relationship between cigarette smoking and colorectal polyps has been generally consistent. Since the previous IARC Monograph (IARC, 2004a), twelve further independent studies have investigated this association (Table 2.45 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.45.pdf). All studies found a significantly increased risk for polyps in association with one or more smoking variables. A recent meta-analysis including 42 studies reported a statistically significant positive association between smoking and colorectal adenomas (Botteri et al., 2008b). The meta-analysis, which included several studies that did not explicitly report relative risks for tobacco smoking (Cardoso et al., 2002; Voskuil et al., 2002; Sparks et al., 2004; Gong et al., 2005; Jiang et al., 2005; Kim et al., 2005; Mitrou et al., 2006; Otani et al., 2006; Skjelbred et al., 2006), found a twofold risk elevation for colorectal adenomas in current smokers and a 50% increase in former smokers. The association had been previously found to be equally strong in men and women. In one of two recent studies, there was no difference in the results for men and women separately (Tranah et al., 2004) but significantly greater effects in women were found in the other (Hermann et al., 2009).
Significant positive trends with number of cigarettes per day were found in four (Ji et al., 2006; Larsen et al., 2006; Stern et al., 2006; Shrubsole et al., 2008) of five studies (Tiemersma et al., 2004). Dose–response with duration of smoking was assessed in four studies (Ji et al., 2002; Tiemersma et al., 2004; Stern et al., 2006; Shrubsole et al., 2008) and with pack–years of smoking in five studies (Hoshiyama et al., 2000; Ulrich et al., 2001; Tranah et al., 2004; Ji et al., 2006; Shrubsole et al., 2008; Omata et al., 2009). All nine studies found statistically significant trends, which were consistent with those for adenomas and hyperplastic polyps when reported separately (Ulrich et al., 2001; Ji et al., 2006; Shrubsole et al., 2008). Ever smokers were estimated to have a 13% (95%CI: 9–18%) increasing risk of presenting with adenomatous polyps for every additional 10 pack–years smoked in comparison to never smokers, based on data from 19 studies (Botteri et al., 2008b).
Decreasing risks with years since quitting smoking were found in four studies (Ulrich et al., 2001; Tiemersma et al., 2004; Ji et al., 2006; Shrubsole et al., 2008), statistically significant so in the latter three studies. In comparison to never smokers, former smokers retained moderately elevated risk for colorectal polyps even 20 years after quitting smoking. One study examined both dose metrics (cigarettes per day, duration, and pack–years) and recency of tobacco use: in subjects who had quit smoking for at least 20 years, only the heaviest users of tobacco still had modest excess risks (Ji et al., 2006).
It has been proposed that the association between cigarette smoking and polyps may be stronger with non-progressing adenomas, such as those that are smaller and less villous but the hypothesis is not supported in most studies (Anderson et al., 2003; Toyomura et al., 2004; Ji et al., 2006; Skjelbred et al., 2006). In one study a clearly higher risk for large and multiple adenomas in every anatomic site of the colon was found in a dose–response manner (Toyomura et al., 2004). A meta-analysis found that the combined risk estimate for high-risk adenomas associated with smoking was greater than that for low-risk adenomas and that the difference was statistically significant for current smokers but not former smokers (Botteri et al., 2008b). In addition, a stronger association of smoking with hyperplastic polyps than with adenomas was found in some studies (Ulrich et al., 2001; Ji et al., 2006; Shrubsole et al., 2008) but not in another (Erhardt et al., 2002). The risk associated with smoking may be even higher in subjects presenting with concurrent benign hyperplastic and adenomatous polyps (Ji et al., 2006; Shrubsole et al., 2008).
Relative risk estimates for tobacco smoking and polyps generally range between 2 and 3 whereas those for colorectal cancer range between 1.2 and 1.4. One possible explanation is the effect dilution due to the inclusion of a high proportion of individuals with precursor lesions in the unscreened control groups in most colorectal cancer studies (Terry & Neugut, 1998). Some indirect evidence for this hypothesis is provided by the meta-analysis of colorectal adenomas, which showed that the smoking-associated risk for adenomas was significantly higher in studies including subjects who had undergone complete colonoscopy in comparison to those in which some or all controls had undergone incomplete examination (i.e. only sigmoidoscopy) (Abrams et al., 2008; Botteri et al., 2008b).
It is also possible that smoking is associated with a subset of colorectal cancers so that relative risk estimates for colorectal cancer as a whole are diluted. The pattern of risk observed for colorectal cancer by microsatellite instability status and for type of colorectal polyps suggests that the traditional (non-serrated) adenoma-carcinoma sequence may proceed through a hyperplastic polyps-mixed polyps-serrated adenoma progression and that smoking may be more strongly related to the development of these subtypes (Jass et al., 2000; Hawkins & Ward, 2001). More recently, a BRAF mutation was shown to be a specific marker for the serrated polyp neoplasia pathway originating from a hyperplastic polyp, in which the CIMP-high develops early and the microsatellite instability carcinoma develops late (O’Brien et al., 2006). The findings of strong associations between smoking and colon cancer with CIMP and/or BRAF mutation, irrespective of microsatellite status, are compatible with this observation (Samowitz et al., 2006).
2.7. Hepatocellular carcinoma
2.7.1. Overview of studies
In the previous IARC Monograph (IARC, 2004a), a causal relationship between liver cancer (hepatocellular carcinoma) and smoking was established. Two case–control and one cohort studies have been published since (Table 2.46 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.46.pdf). Overall, most cohort studies and the largest case–control studies, most notably those that included community controls, showed a moderate association between tobacco smoking and risk for hepatocellular carcinoma.
Confounding from alcohol has been addressed in the best studies. The association between alcohol drinking and hepatocellular carcinoma is strong, and alcohol intake is frequently misclassified, leading to potential residual confounding. However an association with smoking has been demonstrated also among non-drinkers.
A meta-analysis was based on 38 cohort studies and 58 case–control studies (Lee et al., 2009). Compared to never smokers, the meta-relative risks adjusted for appropriate confounders were 1.51 (95%CI: 1.37–1.67) for current smokers and 1.12 (0.78–1.60) for former smokers. The increased liver cancer risk among current smokers appeared to be consistent in strata of different regions, study designs, study sample sizes, and publication periods. The association with smoking was observed in non-alcohol-drinkers (RR, 1.34; 95%CI: 0.92–1.94 in men and 1.31; 95%CI: 0.70–2.44 in women). Further supportive evidence is provided by the association between smoking and liver cancer observed among Chinese women and Japanese women, in whom alcohol drinking is extremely rare (Li et al., 2011). One difficulty is that sometimes studies do not specify the histology of liver cancer (hepatocellular versus intra-hepatic biliary tract).
In the update of the Whitehall study (Batty et al., 2008) (a cohort of 17363 government employees in London, followed-up for 38 years), the hazard ratio for death from liver cancer was 1.03 (0.49–2.16) in former smokers and 1.43 (0.69–2.95) in current smokers (based on 57 deaths). In the 50-year follow-up of the British doctors cohort (Doll et al., 2005), there were 74 deaths from liver cancer. Death rates per 100000 per year were 4.4 in never smokers, 10.7 in smokers of 1–14 cigarettes/day, 2.6 in smokers of 15–24 cigarettes/day, and 31.3 in smokers of ≥ 25 cigarettes/day.
2.7.2. Factors affecting risks
(a) Dose–response relationship
Most studies, including the recent ones (Table 2.46 online), show a dose–response relationship with the number of cigarettes smoked and with smoking duration, with exceptions such as Franceschi et al. (2006) and some older studies from Asia. Relative risk estimates increased to 2.0 after 20 years of smoking.
(b) Cessation
Though former smokers tend to have lower relative risks than current smokers, there were no consistent patterns of risks after cessation, including in the recent studies (Table 2.46 online).
2.7.3 Interaction with hepatitis B or C
Infection with hepatitis B virus (HBV) is one of the major causes of liver cancer worldwide, whereas hepatis C virus (HCV) infection causes a large fraction of liver cancer in Japan, Northern Africa and southern Europe. While many studies, most notably from Asia, have found no attenuation of the association between smoking and liver cancer after adjustment/stratification for markers of HBV or HCV infection, an apparent interaction between smoking and HBV or HCV infection has been reported. The increase in risk for liver cancer associated with cigarette smoking appears to be greater among HBV carriers than among uninfected persons in some studies (Tu et al., 1985), but not in others (Kuper et al., 2000a). Two recent reports (Franceschi et al., 2006; Hassan et al., 2008a) studied possible interactions between smoking and hepatitis virus infection and both reported an apparent interaction between smoking and hepatitis C infection. Interactions between smoking and hepatitis B infection were not found among men in one study (Hassan et al., 2008a) and the rarity of HBsAg prevented the evaluation of HBV and smoking in the other (Franceschi et al., 2006; Table 2.46 online). In the meta-analysis by Lee et al. (2009) adjustment for HBV reduced the relative risks in both men and women, while adjustment for HCV did not change the risk in women and increased it in men.
2.8. Renal cell carcinoma
2.8.1. Overview of studies
The previous IARC Monograph (IARC, 2004a) concluded that renal-cell carcinoma is associated with tobacco smoking in both men and women. Four case–control studies and no cohort studies have become available since then (Table 2.47 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.47.pdf). Overall these confirm the previous evidence, though with some conflicting results. In particular, both the study by Hu et al. (2005) in Canada and the multicentre European study by Brennan et al. (2008) do not show a clear effect of smoking. In contrast, the study by Theis et al. (2008) shows an increased risk with smoking duration (irregular, levelling-off after 40 years) and a statistically significant dose–response relationship with pack–years.
In the update of the Whitehall study (Batty et al., 2008) (a cohort of 17363 government employees in London, followed for 38 years), the hazard ratio for deaths from kidney cancer was 0.64 (0.32–1.26) for former smokers, and 1.29 (0.69–2.41) for current smokers (based on 68 deaths). In the 50-year follow-up of the British doctor cohort (Doll et al., 2005) there were 140 deaths from kidney cancer. Mortality rates per 100000 per year were 9.3 in never smokers, 16.4 in smokers of 1–14 cigarettes/day, 16.6 in smokers of 15–24 cigarettes/day, and 15.5 in smokers of ≥ 25 cigarettes/day (age-adjusted).
Hunt et al. (2005) performed a meta-analysis based on 19 case–control studies and 5 cohort studies (total 8032 cases in case–control and 1326 in cohort studies). The relative risk for smoking men was 1.54 (1.42–1.68), and for smoking women was 1.22 (1.09–1.36). A dose–response relationship was found in both men and women. The association observed was more convincing in population-based compared to hospital-based studies.
2.8.2. Confounding
Hypertension is a well established risk factor for kidney cancer but the association with smoking is only indirect. Potential confounding from hypertension was considered only by Brennan et al. (2008).
Other potential confounders such as BMI have been appropriately addressed in most studies.
2.8.3. Cessation
Most studies reviewed in the previous Monograph showed a lower risk for former smokers compared to current smokers, with a significant negative trend with increasing number of years since quitting (IARC, 2004a). In case–control study on smoking cessation and renal-cell carcinoma, the decrease in risk became significant only after 30 years of quitting (Parker et al., 2003). In the meta-analysis (Hunt et al., 2005), former smokers were at reduced risk after 10 years or more of quitting. A clear decline in risk after cessation was also reported by Theis et al. (2008). [The Working Group noted the poor quality of the study, considering the low response rate among controls.]
2.9. Cancer of the lower urinary tract (including cancer of the bladder, ureter, and renal pelvis)
2.9.1 Overview of the studies
The previous IARC Monograph (IARC, 2004a) clearly identified a causal relationship of smoking with transitional-cell carcinomas and squamous-cell carcinomas of the bladder, ureter and renal pelvis both in men and women. Two new case–control studies (Cao et al., 2005; Samanic et al., 2006; Table 2.48 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.48.pdf) and two cohort studies (Bjerregaard et al., 2006; Alberg et al., 2007; Table 2.49 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.49.pdf) have been reported since then in addition to updates of cohort studies with longer follow-up.
In the update of the Whitehall study (Batty et al., 2008) (a cohort of 17363 government employees in London, followed-up for 38 years), the hazard ratio for death from bladder cancer was 0.98 (0.62–1.54) in former smokers and 1.66 (1.06–2.59) in current smokers (based on 164 deaths). In the 50-year follow-up of the British doctors cohort (Doll et al., 2005), there were 220 deaths from bladder cancer. Death rates per 100000 per year were 13.7 in never smokers, 37.7 in smokers of 1–14 cigarettes/day, 31.8 in smokers of 15–24 cigarettes/day, and 51.4 in smokers of ≥ 25 cigarettes/day. All the new studies confirm the existence of a dose–response relationship with the number of cigarettes smoked and with duration, and a decline in relative risk with time since quitting smoking, compared to non-quitters.
2.9.2 Types of tobacco
The risk of lower urinary tract cancer was more strongly associated with smoking air-cured (black) tobacco than smoking flue-cured (blond) tobacco in several studies (IARC, 2004a). The stronger association with air-cured (black) than blond tobacco among current smokers has not been clearly confirmed in a re-analysis of the Spanish multicentre case–control study (Samanic et al., 2006). Relative risks in current smokers were 7.3 (4.9–10.9) in black tobacco smokers and 5.8 (3.4–10.0) in blond tobacco smokers; in former smokers, 4.2 (2.9–6.0) for black tobacco and 1.8 (1.0–3.2) for blond tobacco (Table 2.48 online). The effect of cessation was more pronounced in blond tobacco smokers than in black tobacco smokers, suggesting potentially different mechanisms of action of the two types of tobacco. Air-cured (black) tobacco is richer in arylamines.
2.9.3 Gene–environment interactions
A large number of studies have considered gene–environment interactions between tobacco smoking and genetic polymorphisms, including DNA repair genes (Vineis et al., 2009) and genes involved in carcinogen metabolism (Malats, 2008; Dong et al., 2008). Overall, there is evidence that the slow acetylator variant of the NAT2 gene is involved in bladder carcinogenesis and may interact with smoking. The meta-relative risk for NAT2 slow acetylator and bladder cancer was 1.46 (95%CI: 1.26–1.68; P = 2.5 × 10−7), based on 36 studies and 5747 cases (Dong et al., 2008). Similar but weaker evidence has been provided for GSTM1 (Malats, 2008).
The extent of interaction between NAT2 variants and smoking is still unclear. In one study the NAT2 acetylation status was found to modulate the association of bladder cancer and cigarette smoking through smoking intensity and not smoking duration (Lubin et al., 2007). Studies are not consistent concerning the three-way association between smoking intensity, NAT2 and bladder cancer. Some studies found greater effects at a lower level of exposure and others the opposite (Malats, 2008). Genome-wide association studies have indicated 8q24 as a region that may confer high risk for bladder cancer (Kiemeney et al., 2008).
2.10. Myeloid leukaemia (acute and chronic)
Myeloid leukaemia in adults was observed to be causally related to cigarette smoking in the previous IARC Monograph (IARC, 2004a). Risk increased with amount of tobacco smoked in a substantial number of adequate studies, with evidence of a dose–response relationship. Biological plausibility for a causal relationship of smoking with myeloid leukaemia is provided by the finding of known leukaemogens in tobacco smoke, one of which (benzene) is present in relatively large amounts. No evidence was found for an association with acute lymphocytic leukaemia.
One recently published cohort study included information on acute and chronic myeloid leukaemias (Fernberg et al.., 2007), based on 372 incident cases. A weak association was found between acute myeloid leukaemia and intensity of smoking, and a statistically significant association with current smoking (RR, 1.5; 95%CI: 1.06–2.11). No association was found with chronic myeloid leukaemia.
In the update of the Whitehall study (Batty et al.., 2008) (a cohort of 17363 government employees in London, followed-up for 38 years), the hazard ratio for mortality from myeloid leukaemias (acute plus chronic) was 5.08 (95%CI: 1.78–14.5) for current smokers, and 3.84 (95%CI: 1.35–11.0) for former smokers (based on 66 deaths). In the 50-year follow-up of the British doctors cohort (Doll et al.., 2005), there were 100 deaths from myeloid leukaemias. The mortality rates per 100000 per year were 6.3 in never smokers, 2.8 in smokers of 1–14 cigarettes/day, 14.0 in smokers of 15–24, and 18.3 in smokers of ≥ 25 cigarettes/day (age-adjusted).
2.11. Other leukaemias and lymphomas
2.11.1 Non-Hodgkin lymphoma
Six cohort studies have been published on the association between non-Hodgkin lymphoma and smoking, all reviewed in the previous IARC Monograph (IARC, 2004a). In five of these, no increased risk among smokers was evident (Doll et al., 1994; McLaughlin et al., 1995; Adami et al., 1998; Herrinton & Friedman, 1998; Parker et al., 2000). However, in one study, men who had ever smoked cigarettes had a twofold increase in risk for non-Hodgkin lymphoma, and the risk was still higher among the heaviest smokers (Linet et al., 1992). Data from case–control studies generally also fail to support an effect of smoking on the incidence of non-Hodgkin lymphoma (Peach & Barnett, 2001; Stagnaro et al., 2001; Schöllkopf et al., 2005; Bracci & Holly, 2005; Table 2.50 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.50.pdf). Reanalysis of data of an Italian study (Stagnaro et al., 2004) found a statistically significant association (OR, 1.4; 95%CI: 1.1–1.7) for blond tobacco exposure and non-Hodgkin lymphoma risk.
Three studies and a pooled analysis have examined histological subtypes of non-Hodgkin lymphoma. In one cohort study in women, smoking was associated with increased risk for follicular non-Hodgkin lymphoma (Parker et al., 2000). Similarly, two other studies reported a weak positive association between smoking and risk for follicular lymphoma, but no effect for other histological types (Herrinton & Friedman, 1998; Stagnaro et al., 2001). A large pooled analysis based on nine North-American and European case–control studies found an overall odds ratio of 1.07 (95%CI: 1.0–1.15) for smokers; the association was particularly strong for follicular lymphoma (OR, 1.31; 95%CI: 1.12–1.52) (Morton et al., 2005).
2.11.2 Hodgkin lymphoma
In the previous IARC Monograph (IARC, 2004a) seven studies on the association between Hodgkin lymphoma and smoking were examined and null or weakly positive associations were noted. Among studies published since, a positive association was observed in two case–control (Willett et al., 2007; Kanda et al., 2009) and three cohort studies (Nieters et al., 2006; Lim et al., 2007; Nieters et al., 2008), while one study found no clear association (Monnereau et al., 2008). Several other recent studies also reported a positive association, but with some internal inconsistencies. In a European multicentre case–control study, no association was observed between tobacco and Hodgkin lymphoma for subjects below age 35 years, whereas for older subjects, ever-smokers experienced a doubled risk of Hodgkin lymphoma as compared to never smokers (Besson et al., 2006). In contrast, a positive association was observed in young adults participating in the International Twin Study (Cozen et al., 2009). A positive association was observed in a Scandinavian case–control study, but without a clear dose–response (Hjalgrim et al., 2007). In a case–control study addressing infectious precursors, particularly Epstein-Barr virus (EBV), an increased risk for EBV-positive Hodgkin lymphoma was found among current smokers (Glaser et al., 2004; Table 2.50 online).
Several of the above studies found positive associations for Hodgkin lymphoma while also demonstrating null or inverse associations with non-Hodgkin lymphoma (Nieters et al., 2006; Lim et al., 2007; Nieters et al., 2008; Kanda et al., 2009).
2.11.3 Multiple myeloma
In the previous IARC Monograph (IARC, 2004a), the large majority of studies on tobacco smoking and risk for multiple myeloma evaluated showed no clear association. More recently, two case–control studies found a positive association (Vlajinac et al., 2003; Nieters et al., 2006), whereas no clear association was observed in another case–control study (Monnereau et al., 2008) or in a cohort study in Sweden (Fernberg et al., 2007; Table 2.51 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.51.pdf).
2.12. Cancer of the breast
Approximately 150 epidemiological studies have been published on the relationship between breast cancer and active and passive smoking. The results from these studies have been comprehensively examined in peer-reviewed literature (Palmer & Rosenberg, 1993; Terry et al., 2002a; Johnson et al., 2002; Johnson, 2005; Terry & Goodman, 2006; Miller et al., 2007). The previous IARC Monograph (IARC, 2004a) considered studies conducted through June 2002 and concluded that there is evidence suggesting lack of carcinogenicity of tobacco smoking in humans for cancers of the female breast.
Other consensus reviews have drawn different conclusions, based partly on the availability of new data, and partly on differences in interpretation:
- The 2001 US Surgeon General Report on Women and Smoking (Department of Health & Human Services, 2001) concluded that tobacco smoking does not appear to appreciably affect breast cancer risk overall. However, several issues were not entirely resolved, including whether starting to smoke at an early age increases risk, whether certain subgroups defined by genetic polymorphisms are differentially affected by smoking, and whether exposure to second-hand smoke affects risk.
- The 2004 US Surgeon General report on “The Health Consequences of Smoking” (Department of Health & Human Services, 2004) concluded the evidence is suggestive of no causal relationship between tobacco smoking and breast cancer.
- The 2009 Canadian Expert Panel on Tobacco Smoke and Breast Cancer Risk (Collishaw et al., 2009) concludes that based on the weight of evidence from epidemiological and toxicological studies and understanding of biological mechanisms, the associations between tobacco smoking and both pre- and post-menopausal breast cancer are consistent with causality.
The lack of agreement in the conclusions from these groups is not surprising, given that the observed associations are weaker and less consistent for breast cancer than for other tobacco-related cancers. Furthermore, several methodological considerations could either obscure a small increase in risk caused by tobacco smoking, or alternatively introduce a spurious association where no causal relationship exists.
2.12.1 Methodological and related issues
The principal concerns about studies of tobacco smoking and breast cancer are the following: timing of exposure, the relevant disease endpoint, the potential for confounding by factors associated with both smoking and the occurrence/detection of breast cancer, the hypothesis that tobacco smoking may have opposing effects on breast cancer risk (protective and detrimental), and the hypothesis that some women may be genetically more susceptible to develop breast cancer from smoking, and that increased risk in these subgroups may be obscured in analyses of average risk in the population.
(a) Misclassification of exposure
Self-reported information on tobacco smoking is generally considered more reliable than questionnaire information on exposure to second-hand tobacco smoke. However, studies of tobacco smoking have not uniformly considered the duration of smoking (years), the average amount smoked (cigarettes/day), or the timing of initiation in relation to first full-term pregnancy. Only one (Al-Delaimy et al., 2004) of the seven available cohort studies updated the information on smoking behaviour during follow-up. Whereas some exposure variables, such as age at initiation and age at first full-term pregnancy remain constant over time, others, such as smoking status, duration and age at cessation do not. Furthermore, the average age at initiation and duration of smoking are highly correlated with birth cohort and attained age. While the number of years of smoking before first full term pregnancy has been proposed as a potentially relevant measure of exposure, the range of this variable is constrained except among women whose first pregnancy occurs at an older age, which is itself an independent risk factor for breast cancer.
(b) Specificity of disease endpoints
Breast cancer is not a single disease. Accordingly, some researchers have postulated that exposure to tobacco smoke (from tobacco smoking or second-hand tobacco smoke) could differentially affect certain clinical subtypes of breast such as pre- or post-menopausal cancers or tumours with or without hormonal receptors. It is also possible that smoking might affect the survival of women with breast cancer, whether or not it affects incidence rates. Most published studies have measured incidence rates as the endpoint, although some have measured mortality rates or effects on survival.
(c) Confounding
Alcohol consumption is positively correlated with tobacco smoking (Marshall et al., 1999) and is an established cause of breast cancer (IARC, 2010a; Monograph on Consumption of Alcoholic Beverages in this Volume). Most epidemiologic studies attempt to control for alcohol consumption using questionnaire information on usual drinking patterns. This approach is vulnerable to residual confounding, because self-reported data on lifetime alcohol consumption leave room for misclassification. Potential confounding by alcohol consumption is of greater concern for current than for former smokers, since, on average, current smokers drink more than former smokers (Reynolds et al., 2004a, b). One study by the Collaborative Group on Hormonal Factors and Breast Cancer (Hamajima et al., 2002) controlled rigorously for alcohol consumption by restricting the analysis of smoking and breast cancer to women who reported drinking no alcohol.
Conversely, mammography screening can be a negative confounder in studies of tobacco smoking and breast cancer incidence. Few studies of tobacco smoking in relation to breast cancer have controlled for mammography screening. Current smokers report a lower frequency of mammographic screening than never-smokers, whereas health conscious former smokers report higher screening rates (Gross et al., 2006). Mammography screening affects the detection rather than the occurrence of breast cancer; it detects some tumours that might otherwise never have been recognized and allows earlier diagnosis of others, thereby increasing breast cancer incidence in the short-term. The consequence of uncontrolled confounding by mammography screening would be to underestimate an association between current smoking and breast cancer incidence, and to overestimate the association in former smokers. Confounding by screening would be expected to have the opposite effect in studies of breast cancer mortality.
Other correlates of tobacco smoking might also confound a potential association between tobacco smoking and breast cancer, although their net effect is likely to be smaller and harder to predict than confounding by alcohol and mammography screening. Women who smoke undergo menopause about two to three years earlier than never-smokers (Baron et al., 1990). The effect of this may be partly or wholly offset by the greater likelihood of girls who experience early menarche to initiate smoking in early adolescence (Jean et al., 2011). There is no documentation that smokers and never-smokers differ with respect to average years of ovulation. Tobacco smoking also has a complex relationship to body mass index. Post-menopausal women who smoke are less likely to be overweight or obese than former or never smokers, but overweight adolescent girls are more likely to begin smoking for weight control (Fine et al., 2004). Similarly complex relationships exist between smoking and physical activity. Current smokers report less physical activity than either former or never smokers (Kaczynski et al., 2008; Trost et al., 2002), but only a small proportion of the population engages in the vigorous physical activity that is needed to protect against breast cancer. The socioeconomic correlates of smoking have changed over time. Women who attended college during the 1960s and 1970s were more likely to initiate smoking than less educated women, but subsequently college-educated women have been more likely to quit. Thus, the potential for confounding by reproductive patterns and use of post-menopausal hormone treatment varies by birth cohort and differs for current and former smokers.
Most epidemiological studies have attempted to control for factors that might confound the relationship between breast cancer and tobacco smoking using questionnaire information collected on these factors. None of the published studies have been able to control for all of the potential confounders, however. Most studies lack data on screening behaviour and have limited information on alcohol consumption, use of post-menopausal hormones, and physical activity.
(d) Potential anti-estrogenic effects of tobacco smoking
Indirect evidence suggests that tobacco smoking may have anti-estrogenic effects that might offset the adverse effects of tobacco smoke carcinogens on breast cancer risk. Baron et al. (1990) pointed to observations suggesting lower estrogen activity levels in women who smoke compared to those who do not. Smokers have lower risk of endometrial cancer (Department of Health & Human Services, 2004), higher risk of osteoporosis (Jensen et al., 1985; Jensen & Christiansen, 1988), earlier age at natural menopause (Baron et al., 1990) and lower mammography density (Roubidoux et al., 2003) than women who do not smoke. Smoking also attenuates the effects of hormone replacement therapy (HRT) on lipid profiles (Jensen & Christiansen, 1988) and serum estrone (McDivit et al., 2008). No difference in serum concentrations of estradiol and estrone between post-menopausal smokers and non-smokers have been reported in several studies (Cassidenti et al., 1992; Khaw et al., 1988; Berta et al., 1991; Longcope et al., 1986; Berta et al., 1992; Cauley et al., 1989; Friedman et al., 1987; Key et al., 1991). However, smokers have been observed to have higher levels of androgens (Cassidenti et al., 1992) (specifically androstenedione) (Khaw et al., 1988; Cauley et al., 1989; Friedman et al., 1987; Key et al., 1991), prolactin (Berta et al., 1991), and unbound serum estradiol (Cassidenti et al., 1992).
(e) Genetically susceptible subgroups
Certain subgroups of women may have greater risk of breast cancer when exposed to tobacco smoke because of genetic or other factors affecting cancer susceptibility. Potential interactions between inherited polymorphisms and tobacco smoking have been studied for selected candidate genes that affect carcinogen metabolism, modulation of oxidative damage, immune responses, and DNA repair (see Sections 2.12.4b and 4.2).
2.12.2 Analytical studies
Over 130 epidemiological studies on tobacco smoking and breast cancer were reviewed.
(a) Incidence in current and former smokers
Since the previous IARC Monograph (IARC, 2004a), seven reports on cohort studies (Al-Delaimy et al., 2004; Reynolds et al., 2004a; Gram et al., 2005; Hanaoka et al., 2005; Olson et al., 2005; Cui et al., 2006; Ha et al., 2007) have been published on breast cancer incidence in relation to tobacco smoking (Table 2.52 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.52.pdf). Breast cancer incidence was significantly associated with current tobacco smoking in three studies (Reynolds et al., 2004a; Olson et al., 2005; Cui et al., 2006), with relative risk estimates among the larger studies ranging from 1.12 (95%CI: 0.92–1.37) (Al-Delaimy et al., 2004) to 1.32 (95%CI:1.10–1.57) (Reynolds et al., 2004a). Former smoking was significantly associated with risk in only one cohort (Al-Delaimy et al., 2004), with relative risk estimates across all of the cohorts ranging from 1.00 (95%CI: 0.93–1.08) (Cui et al., 2006) to 1.18 (95%CI: 1.02–1.36) (Al-Delaimy et al., 2004). The association with breast cancer is stronger in current than in former smokers in four of the seven cohort studies (Reynolds et al., 2004a; Hanaoka et al., 2005; Olson et al., 2005; Cui et al., 2006), although the confidence intervals overlap widely in all but one (Cui et al., 2006). [The Working group noted that three cohort studies (Gram et al., 2005; Hanaoka et al., 2005; Olson et al., 2005) provided data on both the age-adjusted and the multivariate-adjusted risk estimates for current and former smoking. None of these showed attenuation of the estimate associated with current smoking, and two (Hanaoka et al., 2005; Olson et al., 2005) reported somewhat stronger estimates when adjusted for established risk factors besides age. None of the studies adjusted for the frequency of mammography screening. Residual confounding by screening and incomplete control for other risk factors would be expected to cause underestimation of the association with current smoking, and overestimation of the association with former smoking.]
Since the previous IARC Monograph (IARC, 2004a), a total of 12 case–control studies on tobacco smoking and breast cancer incidence have been published (Table 2.53 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.53.pdf). Results from the case–control studies are less consistent than those from the cohort studies. Six studies (Li et al., 2004; Mechanic et al., 2006; Magnusson et al., 2007; Prescott et al., 2007; Roddam et al., 2007; Slattery et al., 2008) differentiated between current and former smokers, while the six other reports (Band et al., 2002; Lash & Aschengrau, 2002; Gammon et al., 2004; Rollison et al., 2008; Ahern et al., 2009; Young et al., 2009) specify only ever or never smokers. Only one study (Li et al., 2004) reported a borderline significant increase in risk associated with current smoking, and two studies (Band et al., 2002; Rollison et al., 2008) with ever smoking.
None of the six case–control studies that presented data on breast cancer incidence separately for current and former smokers found a significant difference in risk between the two smoking categories; the relative risk estimates were higher for former than for current smokers in four of the studies (Mechanic et al., 2006; Prescott et al., 2007; Roddam et al., 2007; Slattery et al., 2008) and identical in the fifth (Magnusson et al., 2007).
(b) Years of cessation
When the relative risk for breast cancer incidence in former smokers is examined by years since cessation in cohort studies (Table 2.54 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.54.pdf), the point estimates do not consistently decrease with longer time since cessation. In none of the four cohort studies (London et al., 1989b; Egan et al., 2002; Reynolds et al., 2004a; Cui et al., 2006) and in only one (Li et al., 2005) of the five case–control studies (Chu et al., 1990; Gammon et al., 1998; Johnson et al., 2000; Kropp & Chang-Claude, 2002; Li et al., 2005) that formally tested for trend (Table 2.55 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.55.pdf) was there a statistically significant decrease in relative risk observed with longer time since cessation. Only one study has reported data on breast cancer mortality in relation to years since quitting or age at cessation (Calle et al., 1994). A statistically significant inverse trend in the relative risk estimates was reported with both years since quitting (p trend = 0.04) and younger age at cessation (p trend = 0.02). [The Working Group noted that the inverse trends in the relative risk of dying from breast cancer observed in this study are weaker than those observed with most other cancers designated as causally associated with smoking.]
(c) Duration of smoking and age at initiation
Tables 2.56–2.61 (see below for links) list the published epidemiologic studies that relate breast cancer incidence to duration of tobacco smoking, age at initiation and/or timing relative to first full term pregnancy.
Longer duration of smoking is associated with higher breast cancer incidence in five of seven cohort studies (Table 2.56 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.56.pdf). A similar trend is seen inconsistently among the 33 case–control studies that report relative risk estimates by duration of smoking (Table 2.57 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.57.pdf). Among the 18 studies that reported a formal test of trend, eight studies (Gammon et al., 1998; Johnson et al., 2000; Reynolds et al., 2004a; Gram et al., 2005; Li et al., 2005; van der Hel et al., 2005; Cui et al., 2006; Mechanic et al., 2006) reported a statistically significant or borderline increase in the relative risk of incident breast cancer with the duration of smoking; seven studies (Ewertz, 1990; Palmer et al., 1991; Egan et al., 2002; Al-Delaimy et al., 2004; Lissowska et al., 2006; Magnusson et al., 2007; Prescott et al., 2007) reported no trend, and one study (Brinton et al., 1986) reported an inverse relationship.
Thirty studies, including cohort (Tables 2.58 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.58.pdf) and case–control studies (Table 2.59 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.59.pdf) related breast cancer incidence to age at smoking initiation. Fifteen of these (Chu et al., 1990; Ewertz, 1990; Palmer et al., 1991; Nordlund et al., 1997; Gammon et al., 1998; Johnson et al., 2000; Egan et al., 2002; Kropp & Chang-Claude, 2002; Gram et al., 2005; Cui et al., 2006; Lissowska et al., 2006; Ha et al., 2007; Lissowska et al., 2007; Magnusson et al., 2007; Prescott et al., 2007; Slattery et al., 2008) reported a formal test of trend. Among these, only two (Gram et al., 2005; Ha et al., 2007) found a statistically significant or borderline significantly higher risk in women who began smoking at a younger ages; twelve studies (Chu et al., 1990; Ewertz, 1990; Palmer et al., 1991; Nordlund et al., 1997; Gammon et al., 1998; Johnson et al., 2000; Egan et al., 2002; Cui et al., 2006; Lissowska et al., 2006; Magnusson et al., 2007; Prescott et al., 2007; Slattery et al., 2008) found no relationship with age at initiation, and one (Kropp & Chang-Claude, 2002) reported higher risk among women who began smoking later. [The Working Group noted that at least two studies (Cui et al., 2006; Slattery et al., 2008) appear to have included never-smokers in the tests of trend and that the categories that define age at initiation differ across studies.]
The relative risk of incident breast cancer according to the timing of smoking initiation relative to first full-term pregnancy was reported in 21 studies, of cohort (Table 2.60 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.60.pdf) and case–control (Table 2.61 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.61.pdf) design. For nine studies (Hunter et al., 1997; Egan et al., 2002; Al-Delaimy et al., 2004; Reynolds et al., 2004a; Li et al., 2005; Cui et al., 2006; Prescott et al., 2007; Rollison et al., 2008; Young et al., 2009) categorical data on years of smoking before first pregnancy are presented, whereas for 12 (Lash & Aschengrau, 1999; Innes & Byers, 2001; Band et al., 2002; Kropp & Chang-Claude, 2002; Lash & Aschengrau, 2002; Fink & Lash, 2003; Lawlor et al., 2004; Gram et al., 2005; Olson et al., 2005; Lissowska et al., 2006; Magnusson et al., 2007; Slattery et al., 2008) whether smoking was initiated before or after the initial pregnancy was considered. Breast cancer incidence is consistently higher when smoking began before or during first pregnancy in most (Hunter et al., 1997; Lash & Aschengrau, 1999; Innes & Byers, 2001; Band et al., 2002; Egan et al., 2002; Al-Delaimy et al., 2004; Reynolds et al., 2004a; Gram et al., 2005; Li et al., 2005; Olson et al., 2005; Cui et al., 2006; Slattery et al., 2008; Young et al., 2009) but not all (Kropp & Chang-Claude, 2002; Lash & Aschengrau, 2002; Fink & Lash, 2003; Prescott et al., 2007) studies that tested this. [The Working Group noted that the number of years of smoking before first pregnancy is highly correlated with age at first full-term pregnancy, which is itself an independent risk factor for breast cancer.]
It has been argued that some studies, and especially cohort studies, may underestimate the true association between tobacco smoking and breast cancer risk by ignoring or underestimating lifetime exposure to second-hand tobacco smoke of those in the referent group (California Environmental Protection Agency, 2005; Johnson, 2005; Collishaw et al., 2009). This criticism is based on the hypothesis that exposure to second-hand smoke may confer almost the same degree of breast cancer risk as tobacco smoking. Under this hypothesis, the inclusion of women exposed to second-hand smoke in the referent group dilutes the contrast between exposed and unexposed women in studies of tobacco smoking, and causes underestimation of the association between tobacco smoking and breast cancer. In several case–control studies the association between breast cancer and tobacco smoking strengthened when the referent group was defined as women with “never active, never-passive” exposure to tobacco smoke (Morabia et al., 1996; Lash & Aschengrau, 1999; Johnson et al., 2000; Kropp & Chang-Claude, 2002). In contrast, a stronger association between tobacco smoking and breast cancer risk, when women exposed only to second-hand smoke are excluded from the referent group, has not been observed in cohort studies (Egan et al., 2002; Reynolds et al., 2004a). Debate continues over whether the case–control studies should be considered “of highest quality” because they provide “lifetime exposure assessment” (Collishaw et al., 2009) or whether the cohort studies are more credible, because prospectively-collected exposure data are not susceptible to the recall bias that can affect retrospective studies.
(d) Survival and mortality from breast cancer
The relationship between smoking and the natural history of breast cancer has been examined in several studies (Daniell, 1988; Ewertz et al., 1991; Daniell et al., 1993; Scanlon et al., 1995; Yu et al., 1997; Manjer et al., 2000; Murin & Inciardi, 2001; Holmes et al., 2007). In cross-sectional analyses, Daniell et al. (1993) found that smokers with breast cancer had more and larger lymph node metastases than non-smokers, after controlling for primary tumour size and other variables. Further, a case–control study (Murin & Inciardi, 2001) and a retrospective cohort study (Scanlon et al., 1995) found smoking to be associated with an increased risk of developing pulmonary metastases from breast cancer. However, these studies could not definitively distinguish lung metastases from primary lung cancers.
Five cohort studies have focused specifically upon the association of tobacco smoking with either breast cancer survival (Ewertz et al., 1991; Yu et al., 1997; Manjer et al., 2000; Holmes et al., 2007) or breast cancer death rates (Calle et al., 1994). A study of 1774 Danish women showed no association between smoking and breast cancer survival (Ewertz et al., 1991), as did a study of 5056 women with breast cancer in the Nurse’s Health Study (Holmes et al., 2007). In contrast, follow-up of 792 women with in situ or invasive breast cancer detected in a screening study in Malmø, Sweden found a crude relative risk for smokers and ex-smokers, compared to never smokers, of 1.44 (95%CI: 1.01–2.06) and of 1.13 (95%CI: 0.66–1.94), respectively (Manjer et al., 2000). The relative risk associated with smoking remained significant after adjustment for age and stage at diagnosis (RR, 2.14; 95%CI: 1.47–3.10). A study based on the ACS Cancer Prevention Study II reported an association between current smoking and increased breast cancer death rates after six years of follow-up (Table 2.56 online; Calle et al., 1994). Risk of death attributed to breast cancer was positively and significantly related to the duration of current smoking reported at the time of enrolment. However, the authors acknowledge that mortality studies cannot exclude biases arising from the effect of smoking on overall death rates, which could increase the potential for prevalent breast cancer to be coded as the underlying cause of death on the death certificate (Calle et al., 1994).
2.12.3 Subtypes
(a) Pre- versus post-menopausal
Since the previous IARC Monograph (IARC, 2004a), 19 case–control studies have published data on tobacco smoking in relation to pre- and post-menopausal breast cancer (Table 2.62 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.62.pdf). The results are inconsistent. Of the 12 studies that provide information separately for current smokers (Schechter et al., 1985; Brinton et al., 1986; Rohan & Baron, 1989; Ewertz, 1990; Baron et al., 1996; Gammon et al., 1998; Millikan et al., 1998; Johnson et al., 2000; Zheng et al., 2002; Magnusson et al., 2007; Slattery et al., 2008), only five (Schechter et al., 1985; Johnson et al., 2000; Magnusson et al., 2007; Slattery et al., 2008) found a stronger association with pre- than with post-menopausal breast cancer. The other analyses show either similar associations (Brinton et al., 1986; Ewertz, 1990; Baron et al., 1996; Gammon et al., 1998; Millikan et al., 1998; Zheng et al., 2002) or a stronger association with post-menopausal breast cancer (Rohan & Baron, 1989; Millikan et al., 1998; Johnson et al., 2000; Zheng et al., 2002).
(b) Hormone receptor status
Two cohort studies (London et al., 1989a; Manjer et al., 2001), one case–control study (Morabia et al., 1998) and a case series (Yoo et al., 1997) have examined the association between quantitative measures of cigarette smoking and breast cancer risk according to estrogen receptor (ER) status. In one of the cohort studies (Manjer et al., 2001), a statistically significant increased risk (RR, 1.6) of ER negative tumours associated with current smoking was found but no clear association between smoking and ER positive tumours, and no difference in the association with progestogen receptor (PR)-positive and PR-negative tumours. In the other three studies there was no clear difference in the association related to ER or PR receptor status.
2.12.4 Susceptible populations
More than 30 studies and meta-analyses (Alberg et al., 2004; Terry & Goodman, 2006; Ambrosone et al., 2008; Collishaw et al., 2009) have evaluated whether a family history of breast cancer and/or inherited polymorphisms in various genes may confer greater susceptibility to develop breast cancer from exposure to tobacco smoke. These are described below in relation to the measure indicating potential susceptibility.
(a) Family history
In two studies, whether a family history of breast cancer modifies susceptibility to develop breast cancer from tobacco smoking has been examined. Couch et al. (2001) measured breast cancer incidence among female family members in a cohort of breast cancer cases diagnosed between 1944 and 1952 at the University of Minnesota. Sisters and daughters in families with at least three breast and/or ovarian cancers were at 2.4 fold higher risk for breast cancer (95%CI: 1.2–5.1) if they smoked compared to never-smokers. No dose–response was observed in relation to pack–years of smoking.
Suzuki et al. (2007) reported a statistically significant interaction between family history of breast cancer and smoking history in a hospital-based case–control study of 3861 breast cancer cases treated at a large cancer centre in Japan between 1988 and 2000. A family history of breast cancer in the absence of smoking was associated with a relative risk of 1.44 (95%CI: 1.21–1.71); the relative risk estimate was 1.95 (95%CI: 1.36–2.81) in women who reported < 30 pack–years of tobacco smoking, and 4.33 (95%CI: 1.65–11.40) in women who reported > 30 pack–years of smoking.
[The Working group noted that Japanese women who smoked during this time period may have differed from never-smokers in other characteristics related to breast cancer. Besides its strong correlation with female smoking, “Westernization” might be associated with delayed childbearing, smaller families, higher body mass index, and greater use of post-menopausal hormones.]
(b) Genetic polymorphisms
Studies of breast cancer, smoking and low penetrance genetic polymorphisms are summarized in Table 2.63 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.63.pdf). The candidate genes in these studies are involved in carcinogen metabolism [N-acetyltransferases (NAT1, NAT2), cytochrome P450s (CYP1A1, CYP1B1, CYP2E2), GSTs], host responses to oxidative stress (superoxide dismutase) or to infectious organisms (myeloperoxidase and immunoglobulin binding protein) and DNA repair (O6-methylguanine DNA methyltransferase, nucleotide excision repair).
The most consistent associations with breast cancer risk have been observed among long-term smokers with the NAT2 slow acetylation genotype (Terry & Goodman, 2006). NAT2 slow acetylation genotype is thought to confer less capability to detoxify tobacco smoke carcinogens and is associated with an increase in breast cancer risk (Ambrosone et al., 1996, 2008). Approximately 50–60% of Caucasian women are reported to be slow acetylators.
Table 2.63 (online) lists 15 studies of polymorphisms in NAT2, of which 9 were included in a pooled analysis and 13 in a meta-analysis (Ambrosone et al., 2008). [The study by Delfino et al. (2000) was excluded from these analyses because cases included women with benign breast disease; the study by Lilla et al. (2005) was not considered because it is based on the same population as that by Chang-Claude et al. (2002).] The meta-analysis found a statistically significant association between ever tobacco smoking and breast cancer risk among women with the NAT2 slow acetylator genotype (meta-RR, 1.27; 95%CI: 1.16–1.40) but not in those with rapid acetylator genotype (meta-RR, 1.05; 95%CI: 0.95–1.17). Pack–years of tobacco smoking was significantly associated with increasing breast cancer risk among women with NAT2 slow acetylator genotype (meta-RR for ever smokers, 1.44; 95%CI: 1.23–1.68, for > 20 pack–years versus never smokers), but not among rapid acetylators (Ambrosone et al., 2008). No main effect was seen between NAT2 status and breast cancer risk (meta-RR, 1.0; 95%CI: 0.93–1.07). In contrast to an earlier meta-analysis (Alberg et al., 2004), this study observed no difference in risk for pre- or post-menopausal breast cancer. The pooled analysis of nine studies (Ambrosone et al., 2008) reported pooled risk estimates for pre- and post-menopausal women of 1.49 (95%CI: 1.08–2.04) and 1.42 (95%CI: 1.16–1.74), respectively, among women with slow NAT2 genotype and at least 20 pack–years of smoking compared to never-smokers. The corresponding values for women with rapid acetylator genotype were 1.29 (95%CI: 0.89–1.86) and 0.88 (95%CI: 0.69–1.13). A statistically significant interaction was observed between pack–years of smoking as a continuous variable and NAT2 genotype (p interaction = 0.03).
A population-based case–control study published after the meta-analysis by Ambrosone et al. compared the prevalence of the NAT2 genotypes and their joint effect with smoking on breast cancer risk in Hispanic and non-Hispanic white women (Baumgartner et al., 2009). Non-Hispanic white women were more likely (P < 0.001) than Hispanics to have a slow (41.7% versus 33.5%) or very slow (19.0% versus 11.1%) NAT2 acetylator status. Breast cancer risk was significantly increased in non-Hispanic smoking white women with a very slow acetylator genotype (RR, 2.46; 95%CI: 1.07–5.65 for current versus never).
[The Working Group noted that publication bias remains a concern in the studies of NAT2 published to date. All of the studies included in the meta-analysis by Ambrosone et al. were published between 1996 and 2006; some among them (Morabia et al., 2000; Sillanpää et al., 2007) reported very strong associations that seem inconsistent with the rest of the data. Because genetic studies often examine multiple genes, it is plausible that studies that find no main effect with NAT2 have not examined this association or that null results for smoking have not been published.]
Fewer studies with less consistent findings have been published on polymorphisms in other genes such as NAT1, CYP1A1, GST, NOS3, MPO, MnSOD2 and various DNA repair genes (Table 2.63 online).
2.12.5 High penetrance genes & prognosis
At least seven studies have examined the hypothesis that tobacco smoking may modify breast cancer risk among women who carry BRCA1 and BRCA2 mutations (Brunet et al., 1998; Ghadirian et al., 2004; Colilla et al., 2006; Gronwald et al., 2006; Nkondjock et al., 2006; Breast Cancer Family Registry, 2008; Ginsburg et al., 2009). The results have been inconsistent. A recent case–control study of women under age 50 years who were carriers of mutations in BRCA1 or BRCA2 reported increased risk for breast cancer associated with as little as five pack–years of smoking. Compared to non-smokers, the risk associated with five or more pack–years of smoking was 2.3 (95%CI: 1.6–3.5) for BRCA1 mutation carriers and 2.6 (95%CI: 1.8–3.9) for BRCA2 mutation carriers (Breast Cancer Family Registry, 2008). In contrast, six other studies reported no increased risk among BRCA1 or BRCA2 carriers who smoke. The Canadian Panel review (Collishaw et al., 2009) postulated that the five previous studies (Brunet et al., 1998; Ghadirian et al., 2004; Colilla et al., 2006; Gronwald et al., 2006; Nkondjock et al., 2006) may have failed to observe a relationship because they included prevalent cases. However, a sixth study published since the Canadian panel review is also negative (Ginsburg et al., 2009).
2.13. Cancer of the cervix
The association between smoking and cervical cancer has been examined in many epidemiological studies over the past few decades.
Since the previous IARC Monograph (IARC, 2004a), additional epidemiological studies have been published. Study design and results of the case–control studies restricted to HPV positive women or that adjusted for HPV status are presented in Table 2.64 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.64.pdf) and Table 2.65 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.65.pdf). Cohort studies and pooled analyses are presented in Table 2.66 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.66.pdf) and Table 2.67 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.67.pdf), respectively. Table 2.68 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.68.pdf) and Table 2.69 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.69.pdf) present additional cohort studies and pooled analyses on tobacco smoking and cervical, cervical intraepithelia neoplasia and carcinoma in situ, with our without controlling for HPV status, respectively.
2.13.1. Dose–response relationship
A positive association between smoking and incidence of cervical squamous-cell carcinoma, which account for approximately 90% of all cervical cancers, has been shown consistently over several decades in many epidemiological studies of various designs conducted across different geographic regions. Dose–response associations with smoking intensity and duration were noted in many of the studies where such associations were examined (Berrington de González et al., 2004; Appleby et al., 2006). Conversely, no clear association was found among former smokers. For adenocarcinoma of the cervix, which usually account for less than 10% of the total of all types of cervical cancer, there appears to be no clear association with smoking (Berrington de González et al., 2004).
2.13.2. Interaction with HPV positivity
Epidemiological studies of smoking and cervical cancer increasingly have considered the effects of HPV infection, which is recognized as the main etiological factor for invasive and pre-invasive cervical neoplasia worldwide (IARC, 1995, 2012b). HPV infection has been considered not only with respect to possible effect modification (Hellberg & Stendahl, 2005; Gunnell et al., 2006), but also to confounding, as both HPV infection and smoking habits are directly associated with number of sexual partners and other indications of high-risk sexual behaviours (Sikström et al., 1995; Wang et al. 2004; Hellberg & Stendahl, 2005; McIntyre-Seltman et al., 2005; Syrjänen et al., 2007). Although there have been exceptions (Syrjänen et al., 2007), recent studies have generally continued to show that statistical adjustment for the potential confounding effects of HPV infection, or restricting studies to women with high risk HPV infection (Plummer et al., 2003), does not appreciably alter the finding of a positive association or its magnitude (McIntyre-Seltman et al., 2005; Appleby et al., 2006; Tolstrup et al., 2006; Tsai et al., 2007; Nishino et al., 2008; Kapeu et al., 2009).
Statistical adjustment for the potentially confounding effect of HPV infection was usually based on the measured presence of HPV DNA in cervical cells or anti-HPV serum antibodies in multivariate analytical models; as noted above, studies have also restricted their analyses to HPV-positive cases and controls. As there is currently no reliable marker of persistent HPV infection, case–control studies based on a cross-sectional measurement of HPV cannot distinguish between transient and persistent infections (Franco et al., 1999). Tobacco smoking is suspected to facilitate acquisition or persistence of an HPV infection through a reduced number of Langerhans cells and CD4 lymphocytes, which are markers of local immune response in the cervix (Vaccarella et al., 2008). In addition, smoking may affect innate immunity (Ferson et al., 1979). Current smokers have been shown to have a slightly higher HPV prevalence than non-smokers in a broad range of world populations after adjustment for life-time number of sexual partners (OR, 1.18; 95%CI: 1.01–1.39) (Vaccarella et al., 2008). Studies have evaluated the effect of smoking on HPV persistence. One study shows lower probability of HPV clearance among ever smokers (Giuliano et al., 2002) but a few others found no relationship (Molano et al., 2003; Richardson et al., 2005).
2.14. Cancer of the endometrium
2.14.1 Overview of studies
To date, at least 42 epidemiological studies have examined the association between smoking and endometrial cancer, 25 reviewed in the previous IARC Monograph (IARC, 2004a) and 17 published since then (Petridou et al., 2002; Folsom et al., 2003; Furberg & Thune, 2003; Newcomb & Trentham-Dietz, 2003; Beral et al., 2005; Matthews et al., 2005; Viswanathan et al., 2005; Okamura et al., 2006; Strom et al., 2006; Trentham-Dietz et al., 2006; Weiss et al., 2006a; Al-Zoughool et al., 2007; Bjørge et al., 2007; Lacey et al., 2007; Loerbroks et al., 2007; Setiawan et al., 2007; Lindemann et al., 2008). Study design and results of the additional studies are presented separately for the case–control studies (Table 2.70 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.70.pdf and Table 2.71 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.71.pdf, respectively) and for the cohort studies (Table 2.72 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.72.pdf and Table 2.73 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.73.pdf, respectively).
(a) Cohort studies
The majority of the 13 cohort studies (Engeland et al., 1996; Terry et al., 1999, 2002b; Folsom et al., 2003; Furberg & Thune 2003; Beral et al., 2005; Viswanathan et al., 2005; Al-Zoughool et al., 2007; Bjørge et al., 2007; Lacey et al., 2007; Loerbroks et al., 2007; Setiawan et al., 2007; Lindemann et al., 2008) suggest a decreased risk among current smokers, including the largest study with over 9000 cases (Bjørge et al., 2007). In five of these studies quantitative smoking measures have been examined in relation to endometrial cancer risk (Terry et al., 1999, 2002b; Viswanathan et al., 2005; Al-Zoughool et al., 2007; Loerbroks et al., 2007). Of these, one (Terry et al., 1999) found a 50% reduced risk among current smokers in the highest level of intensity (11 cigarettes per day or more) compared with non-smokers, but the number of cases was low and the confidence intervals correspondingly wide. A more recent and larger cohort study (Terry et al., 2002b) found a statistically significant 40% reduced risk among current smokers of more than 20 cigarettes per day, but showed somewhat weaker and statistically non-significant reductions in risk with smoking of long duration or high cumulative consumption (i.e. pack–years). In contrast, the risk among former smokers was similar to that among never smokers. The largest of these studies generally showed decreasing risk of endometrial cancer with increasing smoking intensity, duration, and pack–years of consumption (Viswanathan et al., 2005). Three studies examined the association between time since smoking cessation and endometrial cancer risk. Two of these studies suggested a positive association with time since quitting (compared with non-smokers) (Viswanathan et al., 2005; Loerbroks et al., 2007), whereas one found no association (Terry et al., 2002b).
(b) Case–control studies
The results of 17 population-based case–control studies (Smith et al., 1984; Tyler et al., 1985; Franks et al., 1987; Elliott et al., 1990; Rubin et al., 1990; Brinton et al., 1993; Goodman et al., 1997; Shields et al., 1999; Jain et al., 2000; McCann et al., 2000; Newcomer et al., 2001; Weiderpass & Baron, 2001; Newcomb & Trentham-Dietz, 2003; Matthews et al., 2005; Strom et al., 2006; Trentham-Dietz et al., 2006; Weiss et al., 2006a), that have included between 46 and 1304 endometrial cancer cases, generally have shown reductions in risk among current smokers compared with never smokers (although the magnitude of the reduction in risk has varied somewhat); results among former smokers compared with never smokers were equally variable, albeit somewhat weaker overall. The results of eight hospital-based case–control studies (Kelsey et al., 1982; Lesko et al., 1985; Levi et al., 1987; Stockwell & Lyman, 1987; Koumantaki et al., 1989; Austin et al., 1993; Petridou et al., 2002; Okamura et al., 2006), which included between 83 and 1374 endometrial cancer cases, are somewhat consistent with those of population-based studies. They showed moderate (e.g. 30–40%) reduction in risks among current compared with never smokers, and unaltered risks (or perhaps a small 10–20% reduction in risk) in former compared with never smokers. The largest of the hospital-based studies (Stockwell & Lyman, 1987), with 1374 cases and 3921 controls, found both former and current smokers to be at moderately (approximately 30%) reduced risk of endometrial cancer. To date, six population-based case–control studies (Tyler et al., 1985; Lawrence et al., 1987, 1989; Brinton et al., 1993; Newcomer et al., 2001; Weiderpass & Baron, 2001) have examined quantitative measures of smoking in relation to endometrial cancer risk, generally showing inverse associations to be strongest among current smokers of high intensity or long duration.
2.14.2 Confounders
Whereas the majority of these studies adjusted their relative risk estimates for potentially confounding variables, such as BMI, HRT, parity, diabetes, and age at menopause, studies that did not adjust for these variables tended to show similar inverse associations. Within individual studies, statistical adjustment for the effects of BMI and other covariates often made little difference, although some attenuation of relative risk estimates has been noted (Weiderpass & Baron, 2001; Terry et al., 2002c).
2.14.3 Effect modification
The association between smoking and endometrial cancer risk according to factors that are known determinants of endogenous hormone concentrations, and which may counteract or augment possible tobacco-related hormonal changes, have been examined in several studies. These factors include menopausal status, HRT and BMI. Effect modification can reflect true underlying differences in the association across strata (for example, if cigarette smoking acts to reduce or modify estrogen concentrations differently in one group compared with another), but can also reflect methodological factors, such as differences that occur by chance or through the varying prevalence of confounding variables.
(a) Menopausal status
Although endometrial cancer is rare among pre-menopausal women, several studies have examined the association between cigarette smoking and endometrial cancer risk according to menopausal status, because the effect of smoking (if any) might vary according to the underlying hormonal milieu. The studies have included two cohort studies (Terry et al., 2002b; Al-Zoughool et al., 2007), five population-based case–control studies (Smith et al., 1984; Franks et al., 1987; Lawrence et al., 1987; Brinton et al., 1993; Weiderpass & Baron, 2001), and four hospital-based case–control studies (Lesko et al., 1985; Levi et al., 1987; Stockwell & Lyman, 1987; Koumantaki et al., 1989). In all but one of these studies, a study of early stage endometrial cancer (Lawrence et al., 1987), the inverse association was (to varying degrees) stronger among post-menopausal than pre-menopausal women. Among pre-menopausal women, the relative risk estimates for cigarette smoking have been inconsistent, sometimes showing increased risks with certain measures of cigarette smoking (Smith et al., 1984; Stockwell & Lyman, 1987; Koumantaki et al., 1989; Brinton et al., 1993; Al-Zoughool et al., 2007), sometimes showing decreased risks (Lawrence et al., 1987; Levi et al., 1987; Brinton et al., 1993; Terry et al., 2002b), and sometimes showing practically no association (Lesko et al., 1985; Weiderpass & Baron, 2001; Al-Zoughool et al., 2007). In analyses limited to post-menopausal women, on the other hand, all showed between 10% and 80% reduced risks of endometrial cancer with the various smoking measures.
(b) Hormone replacement therapy
Given the possibility that cigarette smoking affects hormone concentrations mostly among women who are taking HRT (Jensen et al., 1985; Jensen & Christiansen, 1988; Cassidenti et al., 1990), the inverse association between tobacco smoking and endometrial cancer risk might be stronger among HRT users than among non-users. However, the results of studies that have examined the association between smoking and endometrial cancer risk according to HRT use have been equivocal (Weiss et al., 1980; Franks et al., 1987; Lawrence et al., 1987; Levi et al., 1987; Terry et al., 2002b; Beral et al., 2005). Whereas in two studies (Franks et al., 1987; Levi et al., 1987) a larger reduction in risk among smokers taking HRT than among smokers not taking HRT was observed, in two other studies (Lawrence et al., 1987; Terry et al., 2002b) there was no difference in the association according to HRT status. A cohort study that examined associations only among women using HRT showed no clear association among users of continuous combined HRT and cyclic combined HRT, but some suggestion of increased risk among smokers who used tibolone (perhaps more clearly among former smokers) (Beral et al., 2005). Thus, although effect modification by HRT status is biologically plausible, the available epidemiological evidence is equivocal.
(c) Relative body weight
Obesity is an established risk factor for endometrial cancer (IARC, 2002). Smokers tend to have a lower BMI than non-smokers, although former smokers tend to have a higher BMI than current or never smokers (Baron et al., 1990). Two case–control studies have examined the association between cigarette smoking and endometrial cancer risk according to BMI, one population-based (Elliott et al., 1990) and one hospital-based (Levi et al., 1987). Neither of these studies found clear differences in the association between smoking and endometrial cancer risk according to BMI. In a population-based case–control study of early stage endometrial cancer (Lawrence et al., 1987), the inverse association with cigarette smoking tended to become stronger with increasing absolute rather than relative body weight.
2.14.4 Gene polymorphisms
Cigarette smoking and estrogen are both thought to influence cancer risk through pathways that are under the control of specific genes, such as those involved in the formation of bulky DNA adducts by estrogen metabolites (Cavalieri et al., 2000) and both bulky and non-bulky adducts formed by carcinogens in tobacco smoke (Terry & Rohan, 2002). Therefore, studies have been conducted to examine the association between smoking and endometrial cancer risk according to genes that repair these types of DNA damage. In a moderately-sized population-based case–control study no clear effect modification according to certain polymorphisms in the XPA and XPC genes, both of which are involved in the nucleotide excision repair of bulky DNA adducts and may influence endometrial cancer risk, were found (Weiss et al., 2005, 2006b). A nested case–control study also showed no clear effect modification according to three polymorphisms in CYP1A1 (McGrath et al., 2007), a gene that encodes microsomal CYP1A1, which contributes to aryl hydrocarbon hydroxylase activity, catalysing the metabolism of PAHs and other carcinogens found in tobacco smoke (Masson et al., 2005). In another nested case–control study some evidence was found that the association between smoking and endometrial cancer may vary according to a polymorphism (Ile143Val) in O6-methylguanine DNA methyltransferase (MGMT). Overall, studies that address the association between smoking and endometrial cancer risk according to genotype are scarce.
2.15. Cancer of the prostate
Many epidemiological studies have examined the association between cigarette smoking and prostate cancer risk, and most have shown no consistent association (Hickey et al., 2001; Levi & La Vecchia, 2001; Batty et al., 2008; Butler et al., 2009; Huncharek et al., 2010; Table 2.74 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.74.pdf; Table 2.75 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.75.pdf). However, questions remain regarding whether smoking may alter risk in various population subgroups, for example, those defined by certain genotypes, and whether any association with smoking may be stronger for, or limited to, advanced tumours or prostate cancer mortality. Regarding this latter issue, the majority of epidemiological studies, including several large, long-term cohort studies, have reported a positive association between smoking and prostate cancer mortality (Rohrmann et al., 2007; Zu & Giovannucci, 2009). Several studies that examined smoking in relation to both prostate cancer incidence and mortality tend to show positive results only for the latter (Rohrmann et al., 2007; Zu & Giovannucci, 2009). Given the largely null results with respect to prostate cancer incidence, the latter findings suggest that smoking is less likely to be a causal agent in prostate cancer initiation than an agent that acts on existing tumours to promote their progression (Zu & Giovannucci, 2009).
A recent review of smoking and prostate cancer that focused specifically on aggressive and fatal tumours, considered the findings from 14 cohort studies (Zu & Giovannucci, 2009). Nine of these studies showed statistically significant increased risk with at least one smoking measure, and five showed increased risks that were not statistically significant for any measure. Only one study showed no association with any measure of tobacco consumption. Seven studies of various designs examined smoking with respect to indicators of cancer aggressive behaviour at the time of diagnosis. In these studies smoking was associated positively with tumour grade, risk of regional, distant, extraprostatic or metastatic disease, Gleason score, and biochemical outcome (failure) after prostate brachytherapy and in several dose–response associations with the respective endpoint were demonstrated. In one study smoking cessation was associated with a decline in risk compared with that among current smokers.
The association between smoking and prostate cancer risk according to genotype and other potentially effect-modifying factors have been examined in several studies. For example, in a population-based case–control study tobacco use was a risk factor for prostate cancer primarily among men with high BMI (Sharpe & Siemiatycki, 2001). The results of a cohort study in Switzerland suggest that risk of prostate cancer mortality is increased in smokers, particularly those with low plasma vitamin E levels (Eichholzer et al., 1999). These latter associations, as well as those regarding several genotypes that may modify the association (Mao et al., 2004; Nock et al., 2006; Quiñones et al., 2006; Yang et al., 2006; Iguchi et al., 2009; Kesarwani et al., 2009), have yet to be fully clarified.
[The Working Group noted that several of the studies of smoking and prostate cancer mortality did not demonstrate clear dose–response associations with risk, and noted the possibility of bias due to confounding by screening behaviour. However, in the Health Professionals Follow-up Study, screening behaviour was not found to differ appreciably between smokers and non-smokers. In an analysis limited to men with a negative digital rectal examination in the prior two years, stronger associations were found between smoking and metastatic prostate cancer risk among high intensity smokers (RR, 4.2; 95%CI: 1.6–10.9) (Zu & Giovannucci, 2009). This finding was evidence against bias from screening behaviour.]
2.16. Cancer of the ovary
2.16.1 Overview of studies
A total of over 30 epidemiological studies have investigated the association between tobacco smoking and ovarian cancer risk. Of these, 24 were case–control studies (IARC, 2004a; Table 2.76 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.76.pdf; Table 2.77 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.77.pdf) and six were cohort studies (IARC, 2004a; Table 2.78 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.78.pdf; Table 2.79 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.79.pdf). Most studies showed no statistically significant association between a measure of smoking and risk for ovarian cancer overall (Newhouse et al., 1977; Smith et al., 1984; Tzonou et al., 1984; Baron et al., 1986; Stockwell & Lyman, 1987; Whittemore et al., 1988; Hartge et al., 1989; Polychronopoulou et al., 1993; Engeland et al., 1996; Goodman et al., 2001; Goodman & Tung, 2003; Pan et al., 2004; Zhang et al., 2004; Kurian et al., 2005; Niwa et al., 2005; Baker et al., 2006; Huusom et al., 2006; Fujita et al., 2008; Lurie et al., 2008; Nagle et al., 2008; Tworoger et al., 2008); some showed positive associations (Doll et al., 1980; Tverdal et al., 1993; Kuper et al., 2000b; Marchbanks et al., 2000; Green et al., 2001; Modugno et al., 2002; Gram et al., 2008; Rossing et al., 2008) and one (Riman et al., 2004) showed an inverse association.
2.16.2 Histological subtypes
Differences in ovarian cancer risk factor profiles have been observed according to histological type, on the basis of which it has been suggested that mucinous and non-mucinous tumours are etiologically distinct diseases (Risch et al., 1996). Epidemiological studies that have considered histological type tend to support a positive association primarily between cigarette smoking and mucinous ovarian tumours (Kuper et al., 2000b; Marchbanks et al., 2000; Green et al., 2001; Modugno et al., 2002; Pan et al., 2004; Zhang et al., 2004; Kurian et al., 2005; Tworoger et al., 2008). In contrast, two studies showed no clear association between smoking and risk of mucinous or non-mucinous ovarian tumours (Riman et al., 2004; Baker et al., 2006). In addition, one early case–control study (Newhouse et al., 1977), with 300 ovarian cancer cases and with both population and hospital controls, found no clear association with “ever” compared with “never” smoking, and reported no differences according to histological type.
A pooled analysis of 10 case–control studies (Kurian et al., 2005) with 254 cases of mucinous and 1580 non-mucinous tumours found an increased risk of mucinous tumours among current smokers (RR, 2.4; 95%CI: 1.5–3.8), a positive association that was not observed with other histological types. Former smokers in that analysis did not have an increased risk of any histological type of ovarian cancer. This type of dose–response, whereby current smokers have a higher risk than former smokers, was observed in most, but not all, studies of mucinous ovarian cancer (Tables 2.77 and 2.79 online). Overall, the positive association between cigarette smoking and risk of mucinous ovarian tumours is generally consistent across both case–control and cohort studies conducted among various populations. In contrast, associations with smoking have been mostly null with respect to non-mucinous ovarian tumours, suggesting that recall bias is unlikely to explain the association with mucinous tumours.
[The Working Group considered the possibility that women who smoke may come to medical attention more frequently. This raises the possibility of detection bias, because mucinous tumours, benign or malignant, tend to be quite large and could be more easily detected on routine physical exam or testing. However, the Working Group felt that detection bias would not account for the association entirely.
2.17. Cancer of the thyroid
The previous IARC Monograph (IARC, 2004a) noted inconsistent associations between smoking and thyroid cancer risk. In 2003, a pooled analysis of 14 case–control studies showed that smoking was inversely associated with thyroid cancer risk (Mack et al., 2003; Table 2.80 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.80.pdf; Table 2.81 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.81.pdf). The sample consisted of 2725 thyroid cancer cases (2247 women, 478 men) and 4776 controls (3699 women, 1077 men). The inverse association was stronger among current smokers (RR, 0.6; 9% CI: 0.6–0.7) than former smokers (RR, 0.9; 9% CI: 0.8–1.1) and were similar in both men and women, for both papillary and follicular thyroid cancers, as well as by age and region. An inverse association between smoking and thyroid cancer risk was also found in a subsequent case–control study (Nagano et al., 2007). In contrast, two case–control studies (Zivaljevic et al., 2004; Bufalo et al., 2006) reported no clear association between smoking and thyroid cancer risk (no risk ratio estimates were reported; hence, data are not shown in the tables) and a cohort study with 169 incident cases of thyroid cancer, also found no clear association with any qualitative or quantitative smoking measure (Navarro Silvera et al., 2005; Table 2.82 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.82.pdf; Table 2.83 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.83.pdf).
2.18. Other cancers
The cancers reviewed in this section generally have low incidence and mortality rates and are not considered to be strongly associated with cigarette smoking. This raises the possibility of preferential reporting of positive associations in epidemiological studies.
2.18.1 Cancer of the salivary gland
Studies of smoking and cancers of the salivary gland reviewed in the previous IARC Monograph (IARC, 2004a) were sparse and their results were inconsistent (Spitz et al., 1990; Swanson & Burns, 1997; Hayes et al., 1999). A few additional studies also show inconsistent results (Kotwall, 1992; Pinkston & Cole, 1996; Horn-Ross et al., 1997; Vories & Ramirez, 1997; Muscat & Wynder, 1998). Studies that focused specifically on Warthin’s tumour [papillary cystadenoma lymphomatosum or adenolymphoma, a benign tumour of the parotid gland] tend to show strong positive associations with smoking (Kotwall, 1992; Pinkston & Cole, 1996; Vories & Ramirez, 1997). One study (Pinkston & Cole, 1996) compared the risk for Warthin’s tumour with that for other salivary gland tumours and found that smoking increased risk significantly only for Warthin’s tumour.
2.18.2 Cancer of the small intestine
Epidemiological studies (all of case–control design) reviewd in the previous IARC Monograph (IARC, 2004a) have been inconsistent in showing a positive association between smoking and cancers of the small intestine (Chow et al., 1993b; Chen et al., 1994; Wu et al., 1997; Negri et al., 1999; Kaerlev et al., 2002). A more recent study showed no clear association (Hassan et al., 2008b).
2.18.3 Cancers of the gallbladder and extra-hepatic bile ducts
Epidemiological studies of smoking and risk of cancers of the gallbladder and extra-hepatic bile ducts reviewed in the previous IARC Monograph (IARC, 2004a) tended to show null, weak, or moderately strong positive associations. More recent studies also tend to show either no clear association with biliary tract carcinoma/extra-hepatic cholangiocarcinoma (Shaib et al., 2007; Welzel et al., 2007) or suggest positive associations with gallbladder/biliary cancers (Pandey & Shukla, 2003; Yagyu et al., 2008; Grainge et al., 2009). Attention should be paid to potential confounders, especially BMI, when considering the results of epidemiological studies of risk of cancers of the gallbladder and extra-hepatic bile ducts. Recent studies that statistically adjusted for BMI, on gallbladder disease risk (Grainge et al., 2009) or on extrahepatic biliary tract carcinoma risk (Ahrens et al., 2007), showed a positive and null association with smoking, respectively. To date, there are too few studies with adequate control for potentially confounding factors to determine any clear pattern.
2.18.4 Soft-tissue sarcoma
As reported in the previous IARC Monograph (IARC, 2004a), one cohort study found an association between cigarette smoking and mortality from soft-tissue sarcoma after 26 years of follow-up but no dose–response relationship with the number of cigarettes/day, duration of smoking or pack–years (Zahm et al., 1992). No effect of cigarette smoking was detected in an Italian hospital-based case–control study (Franceschi & Serraino, 1992).
2.18.5 Cancer of the skin
(a) Melanoma
Several case–control studies found no difference in the prevalence of tobacco smoking between patients with malignant melanoma and controls, and one study found an inverse association (IARC, 2004a). An inverse association with smoking was also found in the US Radiologic Technologists cohort Study (Freedman et al., 2003a). In that study, smoking for at least 30 years compared with never smoking was inversely related to melanoma risk (RR, 0.6; 95%CI: 0.3–1.3), though risk was not associated with number of cigarettes/day. An inverse association was also observed in a cohort of Swedish construction workers (Odenbro et al., 2007). In this study, the risk for malignant melanoma was reduced in a dose-dependant manner for both cigarette and pipe smokers. The possibility that smoking may reduce the risk for melanoma should, therefore, be considered.
(b) Non-melanoma skin cancer
Four studies showed a positive association between smoking and non-melanoma skin cancer risk (De Stefani et al., 1995; Wojno, 1999; Smith & Randle, 2001; Boyd et al., 2002), and two found no clear association (van Dam et al., 1999; Corona et al., 2001). When distinguishing between histological subtypes, tobacco smoking was linked to the incidence of squamous-cell carcinoma of the skin in most studies, whereas the results for basal cell carcinoma remain inconsistent (Zak-Prelich et al., 2004). No clear association between smoking and risk for basal cell carcinoma was found in a cohort study (Freedman et al., 2003b).
2.18.6 Cancer of the penis
Case–control studies of smoking and penile cancer (Hellberg et al., 1987; Daling et al., 1992, 2005; Maden et al., 1993; Harish & Ravi, 1995; Table 2.84 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.84.pdf; Table 2.85 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.85.pdf) and reviews of studies of smoking and penile cancer and population surveys (Dillner et al., 2000; Favorito et al., 2008; Bleeker et al., 2009; Table 2.86 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.86.pdf; Table 2.87 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.87.pdf) consistently showed a positive association. In most studies there was a dose–response relationship, with higher risks among those with increased smoking intensity and/or duration. A study in Brazil showed a positive correlation with penile tumour grade (Favorito et al., 2008). Based on the two reviews (Dillner et al., 2000; Bleeker et al., 2009), relative risks were generally increased twofold to fivefold among smokers.
Most studies did not adjust for HPV infection. In one case–control study (Daling et al., 2005), current smoking was associated with a 160% increased risk of HPV-positive penile cancer (n = 75), and a 180% increased risk of HPV-negative penile cancer (n = 19), suggesting no important effect modification.
2.18.7 Cancer of the testis
Studies reviewed in the previous IARC Monograph (IARC, 2004a) showed no association between cigarette smoking and risk for testicular cancer. More recently, two case–control studies showed positive associations with smoking, one in Canada (Srivastava & Kreiger, 2004) and one in the Czech Republic (Dusek et al., 2008).
2.18.8 Cancer of the central nervous system
A recent meta-analysis was conducted on smoking in relation to glioma risk (Mandelzweig et al., 2009), which included 17 epidemiological studies (6 cohort and 11 case–control). It was concluded that smoking is not associated with risk of glioma, despite a small significant increased risk seen in cohort studies. A recent cohort study found no association between smoking and carcinoma of the brain (Batty et al., 2008). There have been no consistent associations of smoking with other CNS tumours (IARC, 2004a). In a population-based case–control study in the USA, smoking was associated with increased risk of intracranial meningioma in men (OR, 2.1; 95%CI: 1.1–4.2) but not in women (Phillips et al., 2005).
2.18.9 Cancer of the adrenal gland
Data on risk factors for adrenal carcinoma are sparse. In the US Veterans’ Study there was a fivefold increase in risk among current cigarette smokers during 26 years of follow-up, with risk being particularly high among those who smoked most intensely (Chow et al., 1996). Other forms of tobacco use were associated with a statistically non-significant increase in risk. A case–control study in the USA found a twofold increase in risk for adrenal cancer among heavy smokers in men, but not in women (Hsing et al., 1996).
2.19. Bidi smoking
2.19.1 Cancer of the oral cavity
(a) Overview of studies
The association between cancers of oral cavity and bidi smoking has been examined in 10 case–control studies conducted in India (Sankaranarayanan et al., 1989a, b, 1990a; Rao et al., 1994; Rao & Desai, 1998; Dikshit & Kanhere, 2000; Balaram et al., 2002; Znaor et al., 2003; Subapriya et al., 2007; Muwonge et al., 2008; Table 2.88 available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.88.pdf). In these studies both cases and controls were interviewed and analyses were restricted to men, except for the studies by Balaram et al. (2002) and Subapriya et al. (2007), because very few women smoked among study subjects.
Three hospital-based case–control studies considered cancers of subsites of the oral cavity (gingiva, tongue and floor of the mouth, buccal and labial mucosa) (Sankaranarayanan et al., 1989a, b, 1990a). All three studies showed a higher oral cancer risk for bidi smoking. In one early study an unadjusted relative risk of 1.6 (95%CI: 1.3–2.0) for oral cancer in bidi smokers was reported (Rao et al., 1994). [The Working Group noted that the study had several deficiencies, particularly in the selection of controls that resulted in cigarette smoking apparently being protective for oral cancer.] In another early study (Rao & Desai, 1998) relative risks were estimated after stratification by age and place of residence. Bidi smoking was a significant risk factor for cancer of the base of the tongue (RR, 5.9; 95%CI: 4.2–8.2) but not significant for cancer of the anterior tongue. Relative risk for bidi smoking adjusted for alcohol drinking, illiteracy, non-vegetarian diet and tobacco chewing showed significant risk for cancer of the base of the tongue (RR, 4.7; 95%CI: 3.5–6.3) but not for cancer of the anterior tongue. In a population-based case–control study a relative risk of 1.5 (95%CI: 0.9–2.4), adjusted for age and tobacco quid chewing for smokers (bidis and/or cigarettes), was found (Dikshit & Kanhere, 2000).
Two hospital-based multi centre case–control studies on cancer of the oral cavity were conducted in southern India. One included 309 cases and 292 controls (Balaram et al., 2002). The risk for oral cavity cancer among those who smoked < 20 bidis per day was 2.0 (95%CI: 1.1–3.8) and 2.5 (95%CI: 1.4–4.4) for ≥ 20 per day. The second study included 1563 cases and 3638 controls and found a risk for bidi smoking only of 2.2 (95%CI: 1.75–2.63) compared to never smokers, adjusted for age, centre, level of education, alcohol consumption and chewing (Znaor et al., 2003).
In a hospital-based case–control study with 388 oral squamous cell carcinoma cases (202 men and 186 women) and an equal number of age and sex-matched controls the effect of lifestyle factors (tobacco chewing, smoking and alcohol drinking, diet and dental care) on the risk of oral cancer was evaluated (Subapriya et al., 2007). Both cases and controls were interviewed using a structured questionnaire. The risk estimate for bidi smoking based on 22 cases (84 cases included in the model) and 22 controls was 4.6 (95%CI not given).
Data from a randomized control trial conducted between 1996 and 2004 in Trivandrum, southern India were used in a nested case–control analysis with 282 (163 men and 119 women) incident oral cancer cases and 1410 matched population controls aged 35 years and over (Muwonge et al., 2008). Oral cancer risk among men, adjusted for education and religion, was 1.9 (95%CI: 1.1–3.2) for bidi smokers only compared to never smokers. No association was found between mixed smoking of bidi and cigarette and risk of oral cancer.
Rahman et al. (2003) performed a meta-analysis to investigate the relationship between bidi smoking and oral cancer. They identified 12 case–control studies published in English during 1996–2002 with quantitative information on bidi smoking and oral cancer. Of these, ten studies were conducted in India, one in Sri Lanka and one in Pakistan. All cases were confirmed histologically and exposure data were collected by direct interview. In these studies ORs were not adjusted for tobacco chewing or alcohol drinking. The OR for bidi smokers compared to never smokers based on random effects model was 3.1 (95%CI: 2.0 –5.0). The ORs ranged from 2.0 to 3.6 in different regions of India: studies conducted in Mumbai had an OR of 3.6 (95%CI: 1.6 –7.9), in central India 2.7 (95%CI: 1.6–4.6), in Kerala 2.0 (95%CI: 1.5–2.9) and in Bangalore 2.0 (95%CI: 1.1–3.7).
(b) Dose–response evidence
The trends in relative risks by intensity and duration of bidi smoking were both statistically significant in two studies (Rao et al., 1994; Rao & Desai, 1998). A meta-analysis based on three studies on duration of bidi smoking and on five studies on number of bidi sticks per day, showed a dose–response relationship for duration of bidi smoking but not for number of sticks used per day (Rahman et al., 2003).
In a nested case–control analysis (Muwonge et al., 2008) a dose–response relationship was observed for duration of bidi smoking (P = 0.045). [It is not clear if the analysis was restricted to bidi smokers only (n = 40 men) and if smokers with combined smoking habits (bidi and cigarette) were excluded. Moreover, ORs for the dose–response analysis were not reported.]
2.19.2 Cancer of the pharynx
Five case–control studies, two hospital-based (Wasnik et al., 1998; Rao et al., 1999), one population-based (Dikshit & Kanhere, 2000) and two multicentric studies (Znaor et al., 2003; Sapkota et al., 2007) were conducted on cancers of oropharynx and hypopharynx in India (Table 2.88 online). In all these studies, analyses were restricted to men because very few women smoked among study subjects.
Wasnik et al. (1998) conducted a case–control study on oropharyngeal cancers with cases and controls were matched on age and sex. Odds ratios for tobacco smoking, predominantly in the form of bidi and/or chillum, were 2.3 (95%CI: 1.2–3.7) after adjustment for tobacco chewing and outdoor occupation. [The Working Group noted some problems with the data analysis.]
Rao et al. (1999) reported a relative risk for bidi smoking adjusted for alcohol, illiteracy, diet and tobacco chewing of 4.7 (3.6–6.3) for oropharyngeal cancer and of 2.8 (2.1–3.7) for cancer of the hypopharynx. Dikshit & Kanhere (2000) found an odds ratio for oropharyngeal cancer among bidi smokers only of 7.9 (95%CI: 5.1–12.4).
Znaor et al. (2003) reported a risk for bidi smoking only for pharyngeal cancer of 4.7 (95%CI: 3.5–6.3) and for combined bidi and cigarette smoking of 3.6 (95%CI: 2.55–4.98). Sapkota et al. (2007) reported an odds ratio for hypopharyngeal cancer of 6.8 (95%CI: 4.6–10.0) for bidi smokers compared to never smokers.
A dose–response relationship was observed for intensity and duration of bidi smoking for both cancers of oropharynx and hypopharynx (Rao et al., 1999; Dikshit & Kanhere, 2000; Sapkota et al., 2007).
2.19.3 Cancer of the lung
One cohort study (Jayalekshmy et al., 2008), one population-based case–control study (Dikshit & Kanhere, 2000) and two hospital-based case–control studies (Gupta et al., 2001; Gajalakshmi et al., 2003) in India (Table 2.88 online) have investigated the relationship between bidi smoking and lung cancer. In all these studies both cases and controls were interviewed and analyses were restricted to men because very few women smoked among study subjects. One hospital-based case–control study in Chiang Mai, Thailand, looked at the association between lung cancer and khii yoo, hand-rolled cigars. The risk for lung cancer for khii yoo smoking was 1.2 in men and 1.5 in women, P > 0.05 (Simarak et al., 1977).
In the population based case–control study by Dikshit & Kanhere (2000) the age-adjusted relative risk for lung cancer among bidi smokers only was 11.6 (95%CI: 6.4–21.3).
Gupta et al. (2001) reported an odds ratio for bidi smoking of 5.8 (95%CI: 3.4–9.7) from a hospital-based case–control study of lung cancer conducted in Chandigarh. Gajalakshmi et al. (2003) conducted a case–control study in two centres in which all subjects were interviewed by trained social investigators with standard questionnaires. Odds ratios were adjusted for age, educational level, centre, chewing and alcohol habit. The odds ratios of lung cancer for former and current bidi smokers were 3.4 (95%CI: 2.1 –5.4) and 5.3 (95%CI: 3.8–7.3), respectively. Odds ratios for former and current smokers of cigarette and bidi combined were 4.0 (95%CI: 2.5–6.6) and 9.1 (95%CI: 6.2–13.2), respectively.
Baseline data of a cohort of 359 619 residents in Kerala, India was collected by direct interview using standardized questionnaires during 1990–97 (Jayalekshmy et al., 2008). After excluding rare earth workers, those who died, were diagnosed with cancer before 1997 or died within three years of interview, there were 65 829 bidi-smoking men aged 30–84 years old. Two hundred and twelve lung cancer cases were identified by the Karunagappally Cancer Registry between 1997 and 2004. The relative risk for lung cancer for current compared to never bidi smokers calculated by Poisson regression analysis and adjusted for age, religion and education was 3.9 (95%CI: 2.6–6.0; P < 0.001). The risk was lower among former than among current smokers.
(a) Dose–response evidence
Lung cancer risks increased with increasing bidi smoking intensities. The highest odds ratio was found for 9 pack–years (3.9; 95%CI: 2.1–7.1) (Gupta et al., 2001). In a cohort study Jayalekshmy et al. (2008) found increased lung cancer incidence with increasing number of bidi sticks smoked per day (P < 0.001) and with increasing duration of bidi smoking (P < 0.001). [The number of lung cancer cases was small in each category, resulting in wide confidence intervals.] Gajalakshmi et al. (2003) also reported increased risk with duration and intensity of bidi smoking.
(b) Cessation of smoking
In two case–control studies (Gupta et al., 2001; Gajalakshmi et al., 2003) there was a clear decreasing trend in risk for years since quitting. Gajalakshmi et al. (2003) reported that lung cancer risk of former bidi smokers fell to 0.4 (0.1–1.2) after quitting for more than 15 years. The cohort study conducted in Kerala did not have the power to assess the risk associated with stopping bidi smoking (Jayalekshmy et al., 2008).
2.19.4 Cancer of the larynx
Two hospital based case–control studies (Sankaranarayanan et al., 1990b; Rao et al., 1999) showed a higher risk for bidi smokers (Table 2.88 online). The relative risk was adjusted for age and religion in Sankaranarayanan et al. (1990b) study and for alcohol use, illiteracy, vegetarian/non-vegetarian diet and tobacco chewing in Rao et al. (1999) study. A multicentre case–control study on laryngeal cancer was conducted in four Indian centres using standardized questionnaires adjusting risks for centre, age, socioeconomic status, alcohol consumption, tobacco snuffing and tobacco chewing (Sapkota et al., 2007). Compared to never smokers bidi smokers had a higher risk for cancers of the supraglottis (OR, 7.5; 95%CI: 3.8–14.7), glottis (OR, 5.3; 95%CI: 3.2–8.9) and rest of larynx (OR, 9.6; 95%CI: 5.6–16.4).
All levels of intensity and duration of bidi smoking were associated with significant relative risk estimates and dose–response for laryngeal cancer (Sankaranarayanan et al., 1990b; Rao et al., 1999). A strong dose–response relationship was observed for duration and frequency of bidi smoking for cancers of supraglottis, glottis and rest of larynx (Sapkota et al., 2007).
2.19.5 Cancer of the oesophagus
Three hospital-based case–control studies and one multicentre study (Sankaranarayanan et al., 1991; Nandakumar et al., 1996; Nayar et al., 2000; Znaor et al. 2003) showed increased risk for oesophageal cancer among bidi smokers in India (Table 2.88 online). A significantly elevated risk for all three segments of the oesophagus was reported (Nandakumar et al., 1996). One study (Nayar et al., 2000) adjusted for chewing of betel leaf with tobacco and low consumption of vegetables other than leafy vegetables. The multicentre case–control study conducted in two centres in South India found an increased risk for oesophageal cancer for bidi smoking only (OR, 3.3; 95%CI: 2.45–4.39) (Znaor et al., 2003). Odds ratios were adjusted for age, centre, level of education, alcohol consumption and chewing. Only men were analysed in all the above studies.
Significant effects were noted in men for all levels of intensity and for duration of more than 20 years of bidi smoking (Sankaranarayanan et al., 1991).
2.19.6 Cancer of the stomach
In a hospital-based case–control study the association between stomach cancer and bidi smoking was analysed as part of a multicentre study (Gajalakshmi & Shanta, 1996). Cases and controls were matched on age, sex, religion and mother tongue. The odds ratio for stomach cancer for current bidi smokers only was 3.2 (95%CI: 1.8–5.7) and for current smokers of any type of tobacco was 2.7 (95%CI: 1.8–4.1).
Table 2.88 (online) summarizes the studies published since the last IARC Monograph (IARC, 2004a). A hospital-based case–control study of stomach cancer included 170 stomach cancer cases (121 men and 49 women) and 2184 controls (1309 men and 875 women) aged 30–75 years (Rao et al., 2002). The association between bidi smoking and stomach cancer was not significant (RR, 0.8; 95%CI: 0.5–1.2) in a univariate analysis. The risk increased with increase in lifetime exposure to bidi smoking and was highly significant (P < 0.001).
One study investigated stomach cancer risk in association with smoking of meiziol, a local cigarette in Mizoram, India (Phukan et al., 2005). Statistically significant higher risks were seen for smokers of combined users of tobacco (cigarette and meiziol), with an odds ratio of 3.1 (95%CI: 2.0–11.1). Among users of a single type of tobacco, higher risks were seen for meiziol smokers (OR, 2.2; 95%CI: 1.3–9.3) in the multivariate model in comparison to cigarette smokers. Overall, the excess risk was limited to smokers of > 10 meiziols per day.
2.20. Synergistic effects of tobacco smoking and alcohol drinking
This section addresses the combined effects of smoking and alcohol consumption on cancers of oral cavity, pharynx, larynx and oesophagus, which have been examined extensively. For the purposes of this report interdependence of effects is termed effect modification, and synergism and antagonism are used to describe the consequences of the interdependence of disease risk when both risk factors are present (Rothman & Greenland, 1998). The studies varied in their methods and in the approaches used to assess effect modification, which ranged from descriptive to formal estimation of interaction terms in multivariate models. Study designs of the case–control and cohort studies are presented in Table 2.89 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.89.pdf) and Table 2.90 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.90.pdf), respectively; and the results for both study designs are presented in Table 2.91 (available at http://monographs.iarc.fr/ENG/Monographs/vol100E/100E-01-Table2.91.pdf).
2.20.1 Cancers of the upper aerodigestive tract
It was noted in the previous IARC Monograph (IARC, 2004a) with relatively large numbers of cases and controls that the pattern of increasing cancer risk with increasing alcohol consumption is strong (Mashberg et al., 1993; Kabat et al., 1994). For cancers of the oral cavity, recent evidence comes from seven case–control studies and one cohort study. The pattern of odds ratios for smoking, across categories of alcohol consumption, is consistent with synergism. In four case–control studies with relatively large numbers of cases and controls (more than 200 cases and equivalent number of controls), the pattern of increasing cancer risks with increasing alcohol consumption was strong (Schlecht et al., 1999b; Znaor et al., 2003; Castellsagué et al., 2004; Hashibe et al., 2009). In the cohort study from Taiwan, China (Yen et al., 2008) similar strong risks were also observed. In all four case–control studies in which the estimate of formal statistical interaction was examined, the tests were statistically significant (Schlecht et al., 1999b; Znaor et al., 2003; Castellsagué et al., 2004; Hashibe et al., 2009). In two case–control studies from India (Znaor et al., 2003; Muwonge et al., 2008) and in the cohort study from Taiwan, China (Yen et al., 2008) the interaction of tobacco smoking, alcohol and betel quid chewing was examined. In general, the results suggested increasing risks when betel quid chewing was included in the model.
Five case–control studies and one cohort study examined the effect of interaction between tobacco and alcohol in pharyngeal cancer. The results from case–control studies were similar to those observed for oral cancer (Olsen et al., 1985b; Choi & Kahyo, 1991; Schlecht et al., 1999b; Znaor et al., 2003; Hashibe et al., 2009). In a Singapore cohort study (Friborg et al., 2007) the pattern of odds ratios for smoking across categories of alcohol consumption was consistent with synergism for oropharyngeal but not for nasopharyngeal cancer.
Two cohort and fourteen case–control studies reported on joint effects of tobacco smoking and alcohol drinking on the risk for oesophageal cancer. Since multiple logistic regression models were used for analysing most of these studies, some of them tested likelihood ratio test for departure from multiplicativity of the individual effects of tobacco and alcohol. Generally, the positive results were stronger for squamous cell carcinoma. However, these tests for interaction are inadequate to assess synergy. Four studies from India and Taiwan, China, included betel quid chewing to the joint effect analysis of tobacco smoking and alcohol consumption and the results suggested increasing risks of oesophageal cancer.
Most of the twenty case–control studies of laryngeal cancer provided strong evidence for synergism of tobacco smoking and alcohol consumption. Only Zheng et al. (1992) did not find consistent evidence with synergism. In several studies, tests for interaction were carried out and reported to be ‘non significant.’ These were tests for departure from the multiplicative models, typically multiple logistic regression models, used to analyse the case–control data, and not tests for departure from additive model.
Several studies (14 case–control, 3 cohort) reported on cancer of the ‘mixed upper aerodigestive tract’, comprising studies on squamous cell carcinomas, regardless of specific sites. These studies also provided strong evidence for synergism.
The Working Group considers that there is strong evidence of tobacco smoking and alcohol consumption interaction on the incidence of upper aerodigestive tract cancers, as well as with regard to cancer of specific subsites of this anatomical region.
2.21. Synthesis
2.21.1. Lung
Tobacco smoking is the major cause of lung cancer, primarily from cigarettes. Duration of smoking is the strongest determinant of lung cancer in smokers. Risk also increases in proportion to the number of cigarettes smoked. The strong dose– and duration–response relationships between lung cancer and tobacco smoking have been confirmed more recently in both questionnaire-based and biomarker-based studies. Tobacco smoking increases the risk of all histological types of lung cancer.
Differences in the intensity and/or duration of tobacco smoking may explain, in part, the lower lung cancer risks in Asian populations relative to whites. However, several studies of genetic polymorphisms among African-American and Caucasian populations provide some preliminary evidence supporting the hypothesis of a racial/ethnic disparity in susceptibility.
The results from observational studies do not provide strong support that a higher intake or a greater circulating concentration of carotenoids reduce lung cancer risk, particular in light of the elevated risk of lung cancer observed in the randomized trials of β-carotene supplementation. Residual confounding from smoking and the possibility that carotenoid measurements are serving as markers for a diet rich in total fruit and vegetables mitigate the likelihood of any protective role for total carotenoids or β-cryptoxanthins.
The specific genes that are responsible for enhanced lung cancer risk remain poorly understood, in spite of hundreds of candidate gene studies. Single-gene studies conducted to date have several limitations which contribute to inconclusive results, including small sample size and associated low power to detect moderate risks when allele frequencies are low.
2.21.2 Upper areodigestive tract
(a) Oral cavity
Tobacco smoking is causally associated with cancer of the oral cavity in both men and women. Since the previous Monograph, additional evidence has accumulated that further confirms the association. Risk increases with duration and intensity of smoking, and decreases after quitting.
(b) Pharynx
Tobacco smoking is an important cause of oropharyngeal and hypopharyngeal cancers. The risk increases with increasing duration and intensity of smoking and decreases with increasing time since quitting.
(c) Nasal cavity and accessory sinuses
The evidence of an association between tobacco smoking and sinonasal cancer is based on the results from case–control studies, each of which may be subject to different sources of bias. However, presence of a dose–response relationship in most studies, the decrease in risk associated with time since quitting, the consistently higher risks for squamous-cell carcinoma than for adenocarcinoma and the lack of potential confounders support the existence of a causal association.
(d) Nasopharynx
Although the interpretation of the results is complicated by small sample sizes in several studies, by different criteria used for the selection of controls and by the control groups in some studies including smoking-related diseases, the combined evidence shows an association between tobacco smoking and nasopharyngeal carcinoma in both endemic and non-endemic areas. Most studies that adjusted for known and suspected causes of nasopharyngeal carcinoma such as intake of Chinese-style salted fish, other dietary factors, alcohol drinking and family history of nasopharyngeal carcinoma, suggested only a limited confounding effect of these factors. Adjustment for infection with Epstein–Barr virus (human herpes virus 4), a major cause of nasopharyngeal carcinoma worldwide, was possible in just one of the available studies. However, it is unlikely that confounding by infection with Epstein–Barr virus would explain the observed association between tobacco smoking and risk for nasopharyngeal carcinoma.
(e) Oesophagus
Several well conducted case–control studies found a statistically significant higher risk for adenocarcinoma of the oesophagus in smokers than in nonsmokers. Positive dose–response relationships obtained using various indicators of amount smoked support a causal association, which is further corroborated by the findings of decreasing risks after smoking cessation. Several of these studies reported relative risks adjusted for alcohol consumption and other potential confounders. Further risk factors, such as chewing betel quid with tobacco or use of other forms of smokeless tobacco, have not been considered in these populations, but are not likely to be strong confounders. Studies from Australia, China and Europe also found increased risks for smokers.
(f) Larynx
Laryngeal cancer is one of the cancers most strongly associated with cigarette smoking. Recent epidemiological evidence strengthens this conclusion.
2.21.3 Stomach
The additional epidemiologic data showing a consistent association of stomach cancer with tobacco smoking in both men and women greatly strengthens the previous conclusion of a causal association. There was insufficient evidence for differential risks between cardia and non-cardia stomach cancer. Confounding and effect modification by H. pylori has not been found.
2.21.4 Pancreas
The additional data supports the previous evaluation that cancer of the pancreas is causally associated with tobacco smoking. The risk increases with increasing daily consumption levels and duration of smoking and decreases with increasing time since cessation of smoking. The risk remains elevated after accounting for potential confounding factors.
2.21.5 Colorectum
At the previous evaluation, there was already some evidence from prospective cohort and case–control studies that the risk of colorectal cancer is increased among tobacco smokers. However, inadequate adjustment for various potential confounders was considered to possibly account for some of the small increase in risk that appears to be associated with smoking. Since then, an appreciable amount of data has accumulated to support a causal association with smoking. In virtually all the cohort studies published since elevated risk associated with smoking was found, although not always statistically significant. More than half of the cohort studies that assessed dose–response relationships found statistically significant increasing risks with increasing daily cigarette consumption, duration of smoking and/or pack–years of smoking. Risk of colorectal cancer decreased with increasing delay in smoking initiation and years since cessation of smoking. A meta-analysis based on 36 cohort studies with data from a total of 3 million subjects found a significantly 15% increased risk of colorectal cancer and 27% higher risk of colorectal cancer mortality in current smokers compared to never smokers. A stronger association with smoking for rectal cancer than for colon cancer was found in the meta-analysis of the subset of cohort studies that differentiated colorectal cancer by site. Risk for colorectal cancer increased significantly by 17% and by 38% with 20 cigarettes and 40 cigarettes/day, respectively, and was elevated by 9.4% and by 19.7% with a 20-year and a 40-year duration of smoking, respectively. While these results are persuasive, this meta-analysis could not correct for the potential confounders in the individual studies. Convincing evidence has been provided by three large cohort studies that adjusted for at least four important potential confounders (i.e. physical activity, alcohol consumption, body mass index and dietary intake of fruits and vegetables and/or meat); two studies also adjusted for history of colonoscopy. Significant dose–response relationships were found with one or more of the smoking variables, for risk of colorectal cancer and/or colon cancer and/or rectal cancer. Earlier cohort studies may not have been able to establish the association because of insufficient follow-up time and a limited number of cases. Updated results of several large cohort studies, which now show clearly significant increased risk of colorectal cancer associated with smoking, provide support for the lag-time hypothesis for smoking and colorectal risk.
Recent evidence suggests that smoking may be associated with the subtype of colorectal cancer characterized by microsatellite instability, and by CIMP status and BRAF mutation. For this subtype, the magnitude of risk associated with smoking reaches the twofold risk elevation consistently observed for colorectal adenomas and supported by a recent meta-analysis. Smoking has been associated with a stronger risk for hyperplastic polyps than for adenomas. Also, CIMP positivity and BRAF mutations have been associated with hyperplastic polyps, particularly serrated polyps. These data suggest that smoking may be associated primarily with a subtype of colorectal cancer that develops through a hyperplastic (serrated) polyp progression. The association with smoking may therefore be diluted when considering colon cancers overall.
2.21.6 Liver
Recent studies on smoking and hepatocellular carcinoma supports the established causal relationship. Supporting evidence comes from the consistency of the findings across regions (with the best evidence coming from Asian studies), and the observations of an association among non-drinkers and after controlling for hepatitis B or C virus infection.
2.21.7 Kidney
Recent evidence supports a causal association between kidney cancer and smoking. After adjustment for body mass index and hypertension, current and former smokers still had a greater risk for renal-cell cancer. A dose–response relationship with the number of cigarettes smoked has been noted in most studies, and a few also noted a reduction in risk after cessation.
2.21.8 Urinary bladder
Tobacco smoking is causally associated to bladder cancer, based on a large number of case–control and cohort studies that showed statistically significant associations not explained by confounding or bias. Risk increased with the duration of smoking and the number of cigarettes smoked. Also, stopping smoking at any age avoids the further increase in risk incurred by continued smoking. The evidence supporting a modulating role by NAT2 polymorphisms is convincing.
2.21.9 Myeloid leukaemia
There is evidence for a causal association of tobacco smoking with myeloid leukaemia.
2.21.10 Breast
New evidence from cohort and case–control studies and from meta-analyses of genetic polymorphisms has become available since the previous IARC Monograph (IARC, 2004a). Results from seven new cohort studies consistently show a small overall association between current smoking and breast cancer incidence, with relative risk estimates ranging from 1.1–1.3 in studies with at least 100 exposed cases. The overall association is weaker than that observed with other cancers that have been designated as causally related to smoking, and the dose–response relationships (with years of smoking, cigarettes smoked per day, age at initiation) are correspondingly small.
Emerging evidence from case–control studies suggests that inherited polymorphisms in the NAT2 gene, which encode the slow acetylator phenotype, may modify (increase) the association between smoking and breast cancer. The p-value for interaction with pack–years of smoking as a continuous variable is statistically significant (P = 0.03) and another small study published since this meta-analysis supports the conclusion. The potential for publication bias remains of concern.
It is biologically plausible that tobacco smoke could be causally related to breast cancer risk. There are multiple chemicals in tobacco smoke that are known to cause mammary cancer in rodents. These substances reach the breast in humans; some are stored in adipose tissue, and some can be detected in nipple aspirate and DNA adducts.
Hypotheses have been proposed to explain why numerous well conducted epidemiological studies have generally not observed strong or consistent associations between tobacco smoking and breast cancer. Underlying all of these is the theory that tobacco smoking may have both protective and detrimental effects on breast cancer risk, which cancel each other out and which could explain the atypical dose–response relationship that has been reported between tobacco smoke and breast cancer from some studies.
2.21.11 Cervix
The largely positive findings observed in studies of cohort design, the relatively high consistency of positive associations found for squamous-cell carcinoma of the cervix (but not adenocarcinomas) across all epidemiological studies, including those with adjustment for a wide range of potentially confounding variables, and the positive associations observed in studies restricted to HPV-positive individuals, all argue against the observed positive association being due to recall or selection bias or confounding.
2.21.12 Endometrium
The results of epidemiological studies to date, including recent studies, largely show inverse associations of smoking with risk of postmenopausal endometrial cancer. However, the Working Group noted the few studies of premenopausal cancer that were less consistent, as well as indications of an increased risk among smokers in a recent multicentre European study.
2.21.13 Prostate
Many epidemiological studies have examined the association between cigarette smoking and prostate cancer risk, and most have shown no consistent association. The question remains whether smoking may alter risk in various population subgroups.
2.21.14 Ovary
A causal association between cigarette smoking and risk for mucinous ovarian tumours is indicated by 1) the consistency of the positive association across the large majority of ten pooled case–control studies and ten additional independent epidemiological studies of both case–control and cohort design, 2) the relatively strong magnitude of the association (typically greater than a doubling of risk among current smokers), 3) the tendency to show dose–response associations with risk, such that current smokers generally have higher risk than former smokers and the dose–response observed with measures of smoking intensity in some (but not all) studies, and 4) the specificity of the positive association with the mucinous histological type, which argues against recall bias as an explanation of the findings.
2.21.15 Thyroid
A pooled analysis of 14 case–control studies showed that smoking was inversely associated with thyroid cancer risk. Similar inverse associations were also observed in two subsequent case–control studies.
2.21.16 Other sites
There is inconsistent or sparse evidence for an association between tobacco smoking and other cancer sites that were considered by the Working Group.
2.21.17 Bidi smoking
Overall, bidi smoking increases the risk for cancers of the oral cavity, oropharynx, hypopharynx, larynx, lung, oesophagus and stomach.
3. Cancer in Experimental Animals
3.1. Mainstream tobacco smoke
3.1.1 Mouse
There have been multiple studies of the carcinogenic potential of tobacco smoke in mice (Table 3.1). Lifetime exposure of several mouse strains to cigarette smoke failed to result in the production of lung tumours (Harris & Negroni, 1967; Otto & Elmenhorst, 1967; Henry & Kouri, 1986). However, studies involving lifetime exposure of C57BL mice to a mixture of flue-cured or air-cured cigarette smoke or to the gas phase of flue-cured cigarette smoke led to significant increases in the number of lung tumours (adenomas) (Harris et al., 1974). Similarly, lifetime exposure of Snell’s mice to the gas phase of cigarette smoke led to an increased incidence of lung adenocarcinomas (Leuchtenberger & Leuchtenberger, 1970). Exposure of B6C3F1 female mice to smoke for lifetime led to increased incidence of lung adenomas, bronchiolar papillomas and lung adenocarcinomas in smoke-exposed mice. In addition, the occurrence of squamous cell carcinomas of the nasal cavity in smoke-exposed mice was increased (Hutt et al., 2005). In a recent study, Swiss mice were exposed whole-body to cigarette smoke for 120 days, starting within 12 hours of the birth. Smoke-exposed mice developed microscopic lung tumours beginning only 75 days after birth and reaching an overall incidence of 78.3% after 181–230 days. The mean lung tumour multiplicity was 6.1 and 13.6 tumours per mouse in males and females, respectively. In addition, malignant tumours, some of which may have had a metastatic origin, were detected in the urinary tract of smoke-exposed mice (Balansky et al., 2007).
Table 3.1
Carcinogenicity in response to mainstream tobacco smoke in animals.
3.1.2 Rat
Several studies have evaluated the carcinogenic potential of mainstream tobacco smoke in rats (Table 3.1). Exposure of Wistar rats to cigarette smoke for lifetime did not increase the lung tumour incidence (Davis et al., 1975). In contrast, exposure of Fischer 344 rats to a mixture of non-filter cigarette smoke for 128 weeks resulted in an increased incidence of nasal and lung tumours. There was also an increase in subcutaneous sarcomas at forelimb ulceration sites (Dalbey et al., 1980). CDF rats were exposed to low-dose cigarette smoke (LCS) or high-dose cigarette smoke (HCS) for 126 weeks. The incidence of lung tumours was significantly higher only in female rats that received HCS (Finch et al., 1995). In a recent study, Fischer 344 rats received whole body exposure to smoke containing either 100 mg (LCS) or 250 mg (HCS) total particulate matter/m3 for 30 months. This led to significant increases in the incidence of lung and nasal cavity tumours in male rats treated with HCS but not with LCS. In female rats, there were significant increases in the incidence of lung adenomas in animals treated with HCS and of all lung tumours in animals treated with both LCS and HCS. There was also a significant increase in the occurrence of nasal cavity tumours in female rats treated with HCS (Mauderly et al., 2004).
3.1.3 Hamster
Four studies have evaluated the ability of mainstream tobacco smoke to induce tumours in hamsters (Table 3.1). Syrian golden hamsters were exposed to either a mixture of German reference cigarette smoke or of dark air-cured cigarette smoke for lifetime. There were increases in the incidence of laryngeal carcinomas in hamsters exposed to both smoke preparations (Dontenwill et al., 1973). In a subsequent study, hamsters were exposed to a mixture of German reference cigarette smoke containing 1.5 mg nicotine, 0.173 mg phenol and 12.7 mL carbon monoxide/cigarette for lifetime. The incidence of laryngeal tumours in smoke-exposed hamsters was higher than in controls (Dontenwill et al., 1977). BIO male hamsters exposed to a mixture of US reference smoke for 100 weeks developed laryngeal and nasopharyngeal tumours (Bernfeld et al., 1974). In a subsequent study, male BIO hamsters exposed to smoke from commercial British filter cigarettes developed higher incidence of laryngeal tumours than controls (Bernfeld et al., 1979).
3.2. Co-administration of tobacco smoke with known carcinogens and other agents
Study design and results of the studies on co-administration of tobacco smoke with known carcinogens and other agents are summarized in Table 3.2.
Table 3.2
Carcinogenicity in response to exposure to mainstream tobacco smoke in conjunction with exposure to known carcinogens or other agents in animals.
3.2.1 Rat
(a) Benzo[a]pyrene
Wistar rats received a single intratracheal instillation of 2 mg benzo[a]pyrene followed by lifetime exposure to cigarette smoke. This treatment led to a low incidence of lung tumours that was not significantly higher than in controls (Davis et al., 1975). In another study Wistar rats were given intratracheal instillations of benzo[a]pyrene mixed with ferric oxide and exposed to cigarette smoke either during initiation and post-initiation or only after treatment with benzo[a]pyrene/ferric oxide (post-initiation). Inhalation of cigarette smoke during the initiation and post-initiation phases of carcinogenesis resulted in a higher lung tumour (squamous-cell carcinoma) multiplicity than that seen in rats exposed during the post-initiation phase only (Gupta et al., 1990).
(b) Radon progeny
Sprague-Dawley rats were exposed to radon progeny at cumulative doses of 4000, 500 or 100 work-level-months (WLM), with or without concurrent exposure to cigarette smoke by inhalation for one year. Rats exposed to 4000 WLM radon progeny, without exposure to smoke, developed lung carcinomas (17/50). Thirty four carcinomas were seen in 50 rats exposed to radon and cigarette smoke. The 500 WLM radon progeny group exposed to radon only had 2/28 lung carcinomas as compared with 8/30 rats exposed to radon and cigarette smoke. No tumours were observed in rats treated with 100 WLM radon and one carcinoma was seen among 30 rats exposed to 100 WLM radon and cigarette smoke. Seventy five percent of the lung tumours were squamous-cell carcinomas, 20% were adenocarcinomas, and the remainder were undifferentiated carcinomas (Chameaud et al., 1982).
(c) Plutonium oxide
CDF®/CrlBR rats were exposed to either filtered air or mainstream cigarette smoke at concentrations of either 100 or 250 mg total particulate matter/m3 (LCS and HCS groups, respectively). At 12 weeks, rats were removed from smoke chambers and exposed nose-only to plutonium oxide (239PuO2) then returned to the smoke chambers one week later for 30 months of continuous exposure to either filtered air or cigarette smoke. The incidence and multiplicity of lung tumours (adenocarcinomas, squamous-cell carcinomas, adenosquamous carcinomas) in animals exposed to both concentrations of cigarette smoke and 239PuO2 were higher than those in animals exposed to 239PuO2, LCS or HCS alone (Finch et al., 1995).
3.2.2. Hamster
(a) 7,12-Dimethylbenz[a]anthracene
Groups of 160 Syrian golden hamsters received 7,12-dimethylbenz[a]anthracene (DMBA) intratracheally, followed by cigarette smoke for life, or treated with cigarette smoke or DMBA only. A total of 32 squamous-cell carcinomas of the larynx were observed in animals treated with both DMBA and cigarette smoke, in comparison with 17 in hamsters exposed to cigarette smoke only and none in hamsters treated with DMBA alone (Dontenwill et al., 1973). Similar results were reported from other experiments in which Syrian golden hamsters were exposed to DMBA and cigarette smoke (Hoffmann et al., 1979).
(b) N-Nitrosodiethylamine
Groups of hamsters received a single subcutaneous injection of N-nitrosodiethylamine (NDEA) and then were exposed to smoke from unfiltered cigarettes, filtered cigarettes and sham smoke. Controls were exposed to either unfiltered cigarette smoke, filtered cigarette smoke or sham smoke. In the NDEA-smoke-treated groups, epithelial hyperplasias and/or papillomas of the larynx were induced at higher frequency than in controls (Takahashi et al., 1992). Hamsters exposed to cigarette smoke in air also received 12 weekly subcutaneous injections of NDEA (total dose, 10 mg/hamster). Treatment with NDEA only resulted in both benign and malignant tumours of the respiratory tract, and co-exposure to cigarette smoke potentiated the development of tumours in the nasal cavity (Harada et al., 1985).
3.3. Smoke condensates
Study design and results of the studies on administration of tobacco smoke condensates are summarized in Table 3.3.
Table 3.3
Carcinogenicity in response to exposure to cigarette-smoke condensate in animals.
3.3.1 Skin application
(a) Mouse
Cigarette-smoke condensate produces both benign and malignant tumours on mouse skin. The carcinogenic potency of the cigarette-smoke condensate depends upon tobacco variety, composition of cigarette paper and the presence of additives (Wynder et al., 1957; Gargus et al., 1976; Gori, 1976).
(b) Rabbit
Cigarette-smoke condensate induced skin papillomas and carcinomas when applied to the ears of rabbits for lifetime (Graham et al., 1957).
3.3.2 Intrapulmonary administration
Injection of 24 mg cigarette-smoke condensate into the lungs of female Osborne Mendel rats led to the development of squamous cell carcinomas (Stanton et al., 1972). These observations were confirmed by Dagle et al. (1978) who observed a dose-dependent incidence of lung carcinomas when cigarette-smoke condensate prepared from two types of cigarettes were given.
3.3.3 Initiation-promotion skin painting studies
Cigarette-smoke condensate and its fractions can act as skin co-carcinogens in Swiss and SENCAR mice when tested in conjunction with croton oil (Hoffmann & Wynder, 1971) or DMBA (Wynder & Hoffmann, 1961; Meckley et al., 2004a, b; Hayes et al., 2007).
3.3.4 Bidi smoke
Swiss albino mice administered 1 mg bidi smoke condensate in dimethyl sulfoxide (DMSO) by oral gavage developed haemangiomas (4/15), stomach carcinoma (1/15), and esophageal carcinoma (1/15), whereas no tumours were observed in controls (Pakhale et al., 1988).
3.4. Synthesis
Mainstream tobacco smoke induced lung tumours in mice, lung and nasal cavity tumours in rats and laryngeal carcinomas in hamsters.
Co-administration of tobacco smoke with benzo[a]pyrene, radon progeny and plutonium resulted in higher lung tumour responses in rats than administration of either agent alone. Hamsters exposed to cigarette smoke and either DMBA or NDEA had higher lung tumour responses compared to cigarette smoke, DMBA or NDEA alone.
Topical application of cigarette-smoke condensate led to the development of skin tumours in mice and rabbits; intrapulmonary administration of cigarette-smoke condensate induced squamous cell carcinomas in rat lung.
4. Other Relevant Data
4.1. Overview of the mechanistic evidence for the carcinogenicity of tobacco
4.1.1 Conceptual model of the carcinogenesis of tobacco and tobacco smoke
A conceptual model for understanding mechanisms by which tobacco smoke causes cancer is shown in Fig. 4.1 (Hecht, 1999, 2003). This model also applies to smokeless tobacco and other forms of smoked tobacco and, in theory, to second-hand tobacco smoke since it contains all of the same carcinogens and toxicants as mainstream cigarette smoke, although at lower doses.
Fig. 4.1
Conceptual model for understanding mechanisms of tobacco carcinogenesis
The major accepted mechanistic pathway is summarized in the central track of Fig. 4.1. Smokers inhale carcinogens which, either directly or after metabolism, covalently bind to DNA, forming DNA adducts. DNA adducts are central to chemical carcinogenesis because they can cause miscoding and permanent mutations. If these mutations occur in critical regions of oncogenes and tumour suppressor genes, which are essential in growth control, the result can be loss of normal cellular proliferation mechanisms, genomic instability, and cancer. A study that sequenced 623 cancer-related genes in 188 human lung adenocarcinomas validated this premise by finding multiple somatic mutations in critical growth control genes, consistent with the chronic bombardment of cellular DNA by tobacco smoke carcinogens and their metabolically activated forms (Ding et al., 2008).
Each step of this conceptual model is considered in detail below.
Most people begin smoking cigarettes when they are teenagers, and become addicted to nicotine. Nicotine is not generally considered to be a carcinogen (Schuller, 2009), but it is accompanied in each puff of each cigarette by a complex mixture of carcinogens and toxicants. There are over 60 carcinogens in cigarette smoke that have been evaluated in the previous IARC Monograph as having sufficient evidence for carcinogenicity in laboratory animals (IARC, 2004a), sixteen of which are considered to be carcinogenic to humans (Group 1). There are also many other carcinogens and potential carcinogens in cigarette smoke that have not been evaluated (Rodgman & Perfetti, 2006; see Section 1.1). Structures of tobacco smoke constituents and biomarkers discussed here are presented in Fig. 4.2.

Fig. 4.2
Structures of compounds discussed in the text
Numerous studies demonstrate the uptake of tobacco smoke carcinogens and toxicants by smokers, and showed higher levels of their metabolites in urine and blood of smokers than non-smokers (Sections 4.1.1 and 4.1.2). There are substantial differences in carcinogen exposure among people because of the number and types of cigarettes they smoke and the ways in which they smoke them. These differences can be monitored in part by biomarkers of exposure such as urinary metabolites or haemoglobin adducts (Section 4.1.2). Haemoglobin adducts of multiple aromatic amines and volatile carcinogens have been demonstrably related to tobacco (Hatsukami et al., 2006a). There may also be differences in carcinogen exposure due to genetic variations (Section 4.2).
The body’s response to cigarette smoke constituents is similar to its response to pharmaceutical agents and other foreign compounds. Drug metabolizing enzymes, most frequently CYPs, convert these compounds to more water soluble forms, facilitating excretion. During this natural protective attempt, some reactive intermediates are formed. These intermediates are frequently electrophilic (electron seeking, or bearing a partial or full positive charge). Electrophilic intermediates may react with water, generally resulting in detoxification, or may covalently bind to nucleophilic (electron rich) sites in DNA, forming DNA adducts (Guengerich, 2001; Jalas et al., 2005), which are critical in the carcinogenic process (see Section 4.1.3c). CYP1A1 and CYP1B1, repeatedly shown to be inducible by cigarette smoke via interactions of smoke compounds with the aryl hydrocarbon receptor (AhR), are particularly important in the metabolic activation of PAHs, while CYP2A13 is critical for the metabolism of NNK (Nebert et al., 2004; Jalas et al., 2005). The inducibility of certain CYPs may be a critical aspect of cancer susceptibility in smokers (Nebert et al., 2004). CYP1A2, CYP2A6, CYP2E1 and CYP3A4 are also important in the metabolism of cigarette smoke carcinogens to DNA binding intermediates (Jalas et al., 2005), and aldo-keto reductase enzymes, also induced by tobacco smoke (Quinn et al., 2008), are involved in the metabolism of NNK, BaP and other tobacco smoke carcinogens. Competing with this process of “metabolic activation” resulting in DNA binding is the intended metabolic detoxification, which leads to harmless excretion of carcinogen metabolites, and is also catalysed by CYPs and a variety of other enzymes including GSTs, uridine diphosphate-glucuronosyl transferases (UGTs), and arylsulfatases. The relative amounts of carcinogen metabolic activation and detoxification differ among individuals. It is widely hypothesized that this balance will affect cancer risk with those having higher activation and lower detoxification capacity being the most susceptible. This premise is supported in part by molecular epidemiologic studies of polymorphisms, or variants in more than 1% of the population, in certain genes coding for these enzymes (Vineis et al., 2003; Carlsten et al., 2008).
DNA adducts are thought to be a critical lesion in carcinogenesis. Many investigations demonstrate the presence of DNA adducts in human tissues, and some of these are summarized in Section 4.1.2c. There is massive evidence, particularly from studies which use relatively non-specific DNA adduct measurement methods, that DNA adduct levels in the lung and other tissues of smokers are higher than in non-smokers, and some epidemiologic data link these higher adduct levels to increased cancer risk (IARC, 2004b; Veglia et al., 2008). However, there is much more limited evidence from studies using specific carcinogen-derived DNA adducts as biomarkers (Pfeifer et al., 2002). Oxidative DNA damage has also been observed, and this may result partially from exposure to metals in cigarette smoke (Stavrides, 2006).
Cellular DNA repair systems can excise DNA adducts and restore normal DNA structure (Christmann et al., 2003). These complex multiple systems include direct base repair by alkyltransferases, removal of DNA damage by base and nucleotide excision repair, mismatch repair, and double strand repair. If these DNA repair systems are unsuccessful in fixing the damage, then the DNA adducts can persist, increasing the probability of a permanent mutation. There are polymorphisms in genes coding for some DNA repair enzymes. If these variants lead to deficient DNA repair, the probability of cancer development can increase (Vineis et al., 2009).
DNA adducts can cause miscoding during replication when DNA polymerase enzymes misread the DNA adduct and consequently insert the wrong base opposite to it. There is some specificity in the relationship between specific DNA adducts formed from cigarette smoke carcinogens and the types of mutations which they cause. G to T and G to A mutations have often been observed (Section 4.1.3) (Hecht, 1999). Extensive studies have characterized the mutations which occur because of specific carcinogen-DNA adducts (Delaney & Essigmann, 2008). Mutations have been reported in the KRAS oncogene in lung cancer and in the TP53 tumour suppressor gene in a variety of cigarette smoke-induced cancers (Ahrendt et al., 2001; Pfeifer et al., 2002; Ding et al., 2008). The cancer causing role of these genes has been firmly established in animal studies (Lubet et al., 2000; Johnson et al., 2001). A selection and promotion process may also play a role in the final mutation spectrum seen in genes in smoking-associated tumours (Rodin & Rodin, 2005; Sudo et al., 2008).
Urinary mutagenicity, sister chromatid exchanges, micronuclei in buccal cells, and other genetic effects have been consistently observed in smokers at higher levels than in non-smokers (IARC, 2004a; Proia et al., 2006). In addition to mutations, numerous cytogenetic changes are observed in lung cancer, and chromosome damage throughout the field of the aerodigestive tract is strongly associated with cigarette smoke exposure. Mutations resulting from DNA adducts can cause loss of normal cellular growth control functions, via a complex process of signal transduction pathways, ultimately resulting in genomic instability, cellular proliferation and cancer (Ding et al., 2008). Apoptosis, or programmed cell death, is a protective process, and can remove cells which have DNA damage, thus serving as a counterbalance to these mutational events. The balance between apoptotic mechanisms and those suppressing apoptosis will have a major impact on tumour growth.
While the central track of Fig. 4.1 is the major pathway by which tobacco smoke carcinogens cause cancer, other mechanisms also contribute, as indicated in the top and bottom tracks (Hecht, 2003). Nicotine, NNK, and NNN bind to nicotinic and other cellular receptors, resulting in activation of serine/threonine kinase Akt (also known as protein kinase B), protein kinase A, and other changes. Nicotine and NNK increase expression of survivin, an inhibitor of apoptosis in normal human bronchial epithelial cells, and survivin mRNA is detected in bronchial brush samples from heavy smokers (Jin et al., 2008). This can cause decreased apoptosis, increased angiogenesis, and increased transformation (Heeschen et al., 2001; West et al., 2003). Thus, although nicotine is not carcinogenic, it may enhance carcinogenicity in various ways (Schuller, 2009). Cigarette smoke also contains well established oxidants, co-carcinogens, tumour promoting fractions, and inflammatory agents, as well as cilia-toxic compounds such as acrolein, which impede clearance. Many studies demonstrate the co-carcinogenic and cytotoxic effects of catechol, an important constituent of cigarette smoke. An epigenetic pathway frequently observed in tobacco-induced cancers is enzymatic methylation of promoter regions of genes such as p16 and FHIT [fragile histidine triad gene, a gene coding for a dinucleoside 5′, 5′′′- P1, P3-triphosphate hydrolase, a putative tumour suppressor protein] resulting in gene silencing, which are also strongly implicated in tobacco-induced lung cancer (D’Agostini et al., 2006; Bhutani et al., 2008). When this occurs in tumour suppressor genes, the result can be unregulated proliferation (Belinsky, 2005). Inflammation due to smoking is associated with tumour promotion and activation of factors such as NFκB. Inflammation also plays a role in chronic obstructive pulmonary disease (COPD), which in turn is an independent risk factor for lung cancer (Smith et al., 2006; Turner et al., 2007; Lee et al., 2008a).
This conceptual model can be applied to smokeless tobacco products. Smokeless tobacco products have much lower levels of carcinogens and toxicants that result from combustion, so the effects of these agents are not seen to a significant extent. The most prevalent strong carcinogens in smokeless tobacco are the tobacco-specific nitrosamines; other nitrosamines, PAHs, aldehydes and metals are also present, and there are large amounts of some inorganic salts that may contribute to inflammation (IARC, 2007a; Stepanov et al., 2008). An additional factor in carcinogenesis by betel quid with tobacco is the basic pH resulting from addition of slaked lime to the quid, leading to oxidative damage and inflammation (IARC, 2004b).
Multiple studies demonstrate that tobacco-specific nitrosamines are absorbed and metabolised in smokeless tobacco users (IARC, 2007a).
There is evidence for DNA adduct formation in oral tissues of smokeless tobacco users, and sister chromatid exchanges, chromosomal aberrations, and micronuclei – consequences of DNA adduct formation – have been reported (Proia et al., 2006; Warnakulasuriya & Ralhan, 2007). Many studies have demonstrated RAS and TP53 mutations in smokeless tobacco users (Warnakulasuriya & Ralhan, 2007) consistent with the conceptual framework.
Oxidative stress and reactive oxygen species could play a significant role in cancer induction in smokeless tobacco users, particularly at high pH (Boffetta et al., 2008). Chronic local inflammation and irritation induced by smokeless tobacco and its constituents could have a tumour promoting or co-carcinogenic effect (Boffetta et al., 2008). Upregulation of cyclooxygenase-2, involved in prostaglandin synthesis and inflammation, has been observed in animal studies upon exposure to smokeless tobacco (Boffetta et al., 2008). Smokeless tobacco products have relatively high levels of sodium chloride (NaCl), which could contribute to inflammation, tumour promotion, and co-carcinogenesis. Cancer of the oral cavity is strongly associated with tobacco smoking (IARC, 2004a) or chewing (IARC, 2007a) and alcoholic beverage drinking (IARC, 2010a) However only a fraction of exposed subjects develop tumours, which suggests that other exposures such as HPV may be independently involved or act as cofactors. HPV is known to infect the oral cavity of healthy individuals and several HPV-related lesions have been characterized (IARC, 2007b). Herpes simplex virus has also been shown to enhance the carcinogenicity of smokeless tobacco products in animal studies (Park et al., 1986). These factors may contribute significantly to the local carcinogenic effects characteristic of smokeless tobacco use.
4.1.2 Absorption, distribution, metabolism and excretion
There are examples of toxicant and carcinogen metabolism and excretion for representatives of virtually every major class of compounds; some of these are summarized in Table 4.1. Nicotine and five of its urinary metabolites – cotinine, 3′-hydroxycotinine and their glucuronides, and nicotine glucuronide – comprise about 73–96% of the nicotine dose (Hukkanen et al., 2005), and are found in blood, sweat, hair and toenails (Al Delaimy, 2002; Hukkanen et al., 2005; Stepanov et al., 2007; Al Delaimy & Willett, 2008). Metabolites of various polycyclic aromatic hydrocarbons including pyrene, phenanthrene, fluorene, and benzo[a]pyrene have been quantified in human urine and are higher in smokers than in non-smokers (Hecht, 2002; Hecht et al., 2005a; Jacob et al., 2007; Hansen et al., 2008). Metabolites of tobacco-specific nitrosamines – NNAL and its glucuronides (total NNAL) from NNK; and NNN and its glucuronides (total NNN) from NNN – are present in human urine (Hecht, 2002; Stepanov & Hecht, 2005; Hecht et al., 2008a; Stepanov et al., 2008). Total NNAL has also been quantified in blood and toenails (Hecht et al., 2002; Stepanov et al., 2007). Aromatic amine-haemoglobin adducts have been frequently measured in human blood, and their levels increase with smoking (Hecht, 2002; Hatsukami et al., 2006a). Mercapturic acids of several tobacco smoke compounds such as benzene, 1,3-butadiene, acrolein, and ethylene oxide are present in human urine and are related to smoking (Carmella et al., 2009). Haemoglobin adducts of acrylonitrile and related compounds are elevated in smokers’ blood, and levels of metals such as Cd are increased in smokers’ urine (Carmella et al., 2002; IARC, 2004b).
Table 4.1
Examples of toxicant or carcinogen metabolites in tobacco users.
All of the metabolites listed in Table 4.1 are elevated in cigarette smokers; in studies of second-hand smoke exposure, only nicotine metabolites and urinary total NNAL are consistently increased in exposed versus non-exposed subjects, although one very large study also observed an increase in PAH metabolites (Pirkle et al., 2006; Hecht, 2008; Suwan-ampai et al., 2009). Smokeless tobacco users have significantly raised levels of nicotine metabolites and tobacco-specific nitrosamine metabolites compared to non-tobacco users (Hecht et al., 2007).
4.1.3 Biomarkers
Tobacco carcinogen biomarkers are quantifiable entities that can be specifically related to tobacco carcinogens. Specificity to a given carcinogen is critical because tobacco carcinogens vary widely in their potency and target organs.
Considering the mechanistic framework outlined in Fig. 4.1, one could visualize various types of biomarkers. Currently, biomarkers of carcinogen/toxicant dose, reflecting the second box of the central track of Fig. 4.1, are by far the most extensively used and validated. The second most common are measurements of DNA adducts (or protein adducts as their surrogates), but fewer of these have both practical utility and validation with respect to tobacco carcinogen specificity.
The use of tobacco carcinogen biomarkers bypasses many uncertainties in estimation of dose. The most commonly used estimation of dose is self-reported number of cigarettes/day, but this is not a very good marker. It may not be reported accurately and it provides no information on the way in which the cigarettes were smoked, which is critical when one considers the common phenomenon of smoker’s compensation. Brand information together with machine smoking measurements of specific components is another way of obtaining a measure of dose. However, machine smoking measurements are known to have limitations and the application of a given machine smoking protocol to a given smoker requires smoking topography measurements for that smoker. A disadvantage of tobacco carcinogen biomarkers is that they are affected to some extent by individual differences in metabolism, which may complicate interpretation of dose.
(a) Urinary biomarkers
Probably the most practical and, to date, the most extensively applied tobacco carcinogen biomarkers are urinary metabolites of tobacco carcinogens, and these have been comprehensively reviewed (Hecht, 2002; IARC, 2004a). Advantages include the ready availability of samples, and concentrations in urine that are easily quantifiable using modern analytical chemistry methods, most frequently liquid chromatography-tandem mass spectrometry (LC-MS/MS). The urinary metabolites listed in Table 4.1 have all been used as biomarkers and all are validated with respect to exposure in cigarette smokers (Carmella et al., 2009). Total nicotine equivalents (the sum of nicotine and the five metabolites in Table 4.1) is a particularly effective way of estimating nicotine dose from tobacco products.
Total NNAL, the sum of NNAL and its glucuronides, is a highly useful biomarker of NNK exposure (Hecht, 2002, 2003; Hatsukami et al., 2006a). The tobacco-specificity of NNK, and therefore total NNAL, is a key feature of this biomarker because studies in which it is applied are not confounded by other environmental or dietary exposures. It also has a considerably longer half-life than cotinine and several other urinary biomarkers. Total NNAL has been used in numerous studies that estimated uptake of NNK in smokers under varying circumstances. In one example, smokers reduced their number of cigarettes smoked per day, but there was not a corresponding decrease in NNK uptake due to compensation (Hecht et al., 2004). In another study, NNK and PAH uptake, estimated by total NNAL and 1-hydroxypyrene, respectively, were compared in smokers of regular, light, and ultra-light cigarettes, and found to be similar, consistent with epidemiologic studies that demonstrate no protection against lung cancer in smokers of light compared to regular cigarettes (Hecht et al., 2005b). Other studies evaluated NNK uptake in smokers who switched from their current cigarette brand to products advertised as being less hazardous, but the results generally did not support these claims (Hatsukami et al., 2004). One of the most useful applications of total NNAL has been in studies of non-smokers exposed to second-hand tobacco smoke (Hecht, 2003). The sensitivity and specificity of this biomarker are ideal for such studies, and it is the most commonly elevated tobacco carcinogen biomarker in non-smokers exposed to second-hand smoke. Total NNAL has also found utility in establishing NNK uptake in smokeless tobacco users (Hecht et al., 2002, 2007, 2008a, b; Hecht, 2008)
The relationship of urinary total NNAL to lung cancer was demonstrated in a study of stored urine samples collected years before diagnosis of lung cancer from smokers in Shanghai, China and Singapore (Yuan et al., 2009). There was a significant relationship between total NNAL and lung cancer incidence, after correction for numbers of cigarettes smoked per day and duration of smoking. An 8.5 fold increased risk for lung cancer was observed for those smokers in the highest tertile of total NNAL and cotinine, relative to smokers with the same smoking history but in the lowest tertiles of total NNAL and cotinine. Urinary biomarkers were also used to demonstrate higher uptake of nicotine and NNK per cigarette in smokers with polymorphisms in the nicotinic acetylcholine genes associated with lung cancer in genome-wide association studies (see Section 4.2; Le Marchand et al., 2008). Collectively, these results indicate that urinary total NNAL is not only a biomarker of exposure, but also a biomarker of risk for lung cancer.
(b) Serum and saliva metabolites
Serum and saliva metabolites have been used as biomarkers much less often than urine metabolites. The most frequently measured tobacco smoke toxicant in serum and saliva is cotinine, documented as a useful biomarker of cigarette smoking in many studies (Lee, 1999; Hukkanen et al., 2005). Total NNAL can be readily quantified in serum and its levels remain relatively constant in a given smoker sampled at bimonthly intervals over a one year period. Consistent with the results described above, one study showed a significant relationship between total NNAL in prospectively collected serum samples from smokers and lung cancer risk (Church et al., 2009). Other biomarkers that have been measured in serum include cadmium, benzene, styrene and r-1,t-2,3,c-4-tetrahydroxy-1,2,3,4-tetrahydrophenanthrene (PheT) (IARC, 2004a; Church et al., 2009).
(c) DNA adducts
Fig. 4.3 presents an overview of metabolism and DNA adduct formation from eight tobacco smoke compounds (clockwise from top left): BaP, NNK, N-nitrosodimethylamine (NDMA), NNN, acrolein, ethylene oxide, acetaldehyde and 4-aminobiphenyl. Evidence exists for DNA adduct formation from each of these carcinogens in smokers, based on studies carried out with tissues or blood cells. DNA adduct biomarkers have been applied mainly in studies of smokers, and there is far less evidence from studies of second-hand tobacco smoke or smokeless tobacco use.
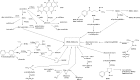
Fig. 4.3
Overview of metabolism and DNA adduct formation from eight tobacco smoke constituents
The structures of DNA adducts of tobacco smoke carcinogens have been characterized in detail, but a complete description of these structures is beyond the scope of this section. Selected DNA adduct structures are shown in Fig. 4.4. A major DNA adduct of BaP results from trans- addition of the benzo[a]pyrene diol epoxide (BPDE) to the N2-position of dG (Szeliga & Dipple, 1998). Pyridyloxobutyl (POB)-DNA adducts of NNK and NNN are formed at the 7- and O6-positions of deoxyguanosine dG, the O2-position of thymidine, and the O2-position of deoxycytidine (Hecht, 2008). They can be measured in part as 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB) released upon hydrolysis. Metabolic activation of NNK also leads to 7-methyl-dG and O6-methyl-dG, identical to the DNA adducts formed from NDMA and other DNA methylating agents (Hecht, 2008). Ethylating agents and ethylene oxide in cigarette smoke also alkylate dG (Zhao et al., 1999; Singh et al., 2005). Acrolein and crotonaldehyde react with DNA to produce exocyclic 1,N2-dG adducts, while acetaldehyde forms a Schiff base adduct with the exocyclic N2 amino group of dG. There is evidence for the presence of all these DNA adducts in tissues or blood cells of smokers, but there are also many studies in which these specific adducts have been sought but not found (Boysen & Hecht, 2003).
Fig. 4.4
Structures of some DNA adducts of tobacco smoke constituents
Measurement of these DNA adducts as biomarkers potentially can provide the most direct link between cellular exposure and cancer, because DNA adducts are so critical in carcinogenesis. However, it is challenging because their levels are extremely low, frequently ranging from 1 per 106 to 1 per 108 normal bases, and the tissue or blood samples containing them are usually available in only small quantities. Fortunately, the routine detection of amol levels [attomole, equivalent to 10 moles] of DNA adducts by conventional LC-MS/MS techniques is now feasible (Singh & Farmer, 2006). There are still relatively few examples of quantitation of specific DNA adducts of tobacco carcinogens in tissues of smokers using mass spectrometry, high pressure liquid chromatography (HPLC)-fluorescence, HPLC with electrochemical detection, or postlabelling techniques (Pfeifer et al., 2002). A much larger body of work has used the highly sensitive, but relatively non-specific 32P-postlabelling and immunoassay methods of DNA adduct detection. Although the adducts detected using 32P-postlabelling are often referred to as “aromatic DNA adducts,” there is strong evidence that they are not related to PAHs (Arif et al., 2006). Adduct levels are generally higher in lung tissues of smokers than non-smokers while studies using blood DNA have produced varied results. Adducts have also been detected in the larynx, oral and nasal mucosa, bladder, cervix, breast, pancreas, stomach, placenta, foetal tissue, cardiovascular tissues, sputum, and sperm of smokers (IARC, 2004a). A meta-analysis of the relationship of DNA adduct levels in smokers to cancer, as determined by 32P-postlabelling in the majority of studies or enzyme linked immunosorbent assay (ELISA), demonstrated a positive relationship in current smokers (Veglia et al., 2003; 2008).
(d) Protein adducts
Carcinogen-haemoglobin (Hb) and serum albumin adducts are regarded as surrogates for DNA adduct measurements. Although these proteins are not targets for carcinogenesis, virtually all carcinogens that react with DNA will also react with protein. Advantages of haemoglobin adducts include the ready availability of haemoglobin from blood and the relatively long lifetime of the erythrocyte in humans – 120 days –,which provides an opportunity for adducts to accumulate. Studies on protein adducts in smokers have been comprehensively reviewed (IARC, 2004a).
Haemoglobin adducts of aromatic amines are a highly informative type of carcinogen biomarker, with levels that are consistently higher in smokers than non-smokers, particularly for 3-aminobiphenyl and 4-aminobiphenyl-Hb adducts. Haemoglobin binds aromatic amines efficiently because heme accelerates the rate of nitrosoarene formation from the hydroxylamine, which is produced metabolically from the aromatic amine by CYP1A2 (Fig. 4.3; Skipper & Tannenbaum, 1990). Binding of the nitrosoarene occurs at the β-93 cysteine residue of human haemoglobin; the adduct is hydrolysed releasing the free amine, which is quantified by GC-MS (Skipper & Tannenbaum, 1990). Adduct levels are clearly related to cigarette smoking (Skipper & Tannenbaum, 1990). Adducts that form at the terminal valine of haemoglobin are also useful biomarkers: examples include those derived from ethylene oxide, acrylonitrile and acrylamide (Bergmark, 1997; Fennell et al., 2000). Ethylated N-terminal valine of haemoglobin is also higher in smokers than in non-smokers (Carmella et al., 2002).
HPB-releasing Hb adducts of NNK and NNN have been quantified in studies of smokers and smokeless tobacco users (IARC, 2004a, 2007a). These adducts are thought to be tobacco-specific, but some studies report their presence in non-smokers (Falter et al., 1994; Schlöbe et al., 2008).
4.1.4 Genetic and related effects
(a) Mutagenicity and cytogenetic effects
Tobacco smoke and its condensates are mutagenic in a wide variety of test systems from bacteria to mammalian cells in culture to rodents and humans (DeMarini, 2004; IARC, 2004a; Husgafvel-Pursiainen, 2004). In bacterial systems, the heterocyclic amines and aromatic amines in condensates account for much of the frameshift mutagenicity, whereas the PAHs and nitrosamines may account for some of the base-substitution mutagenicity (DeMarini et al., 1995). G to T is the predominant class of base-substitution mutation induced by condensates in experimental systems and found in oncogenes and tumour-suppressor genes in smoking-associated lung tumours (IARC, 2004a). The genotoxic potencies of a variety of condensates in several genotoxicity assays likely have only qualitative value with regard to health risk assessment (DeMarini et al., 2008). This is consistent with findings that smokers of low- or high-tar cigarettes have similar urinary levels of lung carcinogens (Hecht et al., 2005b; Hatsukami et al., 2006b) and similar risks for lung cancer (Harris et al., 2004).
In rodents, cigarette smoke induces sister chromatid exchange and micronuclei in bone marrow and lung cells. Human newborns of smoking mothers have increased frequencies of HPRT mutations, chromosomal translocations, and DNA strand breaks. Sperm of smokers has increased frequencies of aneuploidy, DNA adducts, strand breaks, and oxidative damage. Cigarette smoke also causes germ-cell mutations in mice (Yauk et al., 2007). Collectively, these data suggest that smoking is likely a germ-cell mutagen in humans. Smoking produces mutagenic urine and somatic-cell mutations in humans, including HPRT mutations, sister chromatid exchange, microsatellite instability and DNA damage in a variety of tissues. Genotoxic effects have been found in eight organ sites at which tobacco smoke causes cancer in humans (DeMarini, 2004; IARC, 2004a).
(b) Mutations in TP53, KRAS and related genes
Gene mutation data from a variety of databases, including the IARC Cancer TP53 Mutation Database (http://www-p53.iarc.fr/), have been collated in the Genetic Alterations in Cancer (GAC) database (http://dir-apps.niehs.nih.gov/gac/) so that mutations in a variety of genes in various cancerous tissues can be compared. An assessment of the Gene Alterations in Cancer database showed that at least three genes were mutated more frequently in lung tumours from smokers than non-smokers (Lea et al., 2007): TP53 (39 versus 26%), K-RAS (20 versus 3%), and loss of heterozygosity at FHIT (57 versus 27%). Thus, genes in the cell cycle (TP53), cell signalling (KRAS) and apoptotic (FHIT) pathways are mutated more frequently in smoking- rather than in nonsmoking-associated lung tumours. Genomic sequencing of lung tumours has identified other mutated genes that are associated with smoking; ten times more genes are mutated in lung tumours from smokers compared to non-smokers (Ding et al., 2008).
GC to TA transversions were the predominant class of base-substitution mutation found in TP53 and KRAS genes in lung tumours from smokers, with the frequency of this mutation in TP53 being 30% in smokers versus 22% in non-smokers. In smoking-associated oral cancers, the percentage of GC to TA mutations in TP53 was 15% versus 2%, respectively. This mutation spectrum is consistent with that produced by a variety of known carcinogens present in tobacco smoke (IARC, 2004a). At the codon level, the most frequently mutated codons in TP53 in lung tumours of smokers were 157, 175, 245, 248, and 273, all of which occur in the DNA-binding domain of the protein; among these codons, only 273 was mutated in lung tumours from non-smokers. Only three of these codons (157, 245 and 273) were mutated in smoking-associated larynx tumours, and only codon 157 was mutated in smoking-associated oral tumours. Thus, the mutational specificity at TP53 is different among smoking- and nonsmoking-associated tumours and among smoking-associated tumours at various organs (Lea et al., 2007). Thus, different pathways are involved in the development of different types of tumours (Le Calvez et al., 2005; Mounawar et al., 2007; Subramanian & Govindan, 2008).
4.1.5 Effects on gene expression profile
As indicated in a review by Sen et al. (2007) involving microarray analysis of 18 studies in human smokers, 7 in smoke-exposed rodents, and 3 in condensate-exposed mammalian cells, smoking generally upregulated a wide variety of genes, especially those involved in the stress response, phase I metabolism, and immune response. Genes that were consistently expressed differentially in smokers (as assessed in alveolar macrophages, lung cells or peripheral lymphocytes) included metallothioneins, heat-shock proteins, superoxide dismutase, glutathione transferase, heme oxygenase, CYP genes (1A2, 1A1 and 1B1), interleukins and chemokines.
Spira et al. (2004) analysed global gene expression in bronchial epithelial cells and found that the expression levels of metabolizing and antioxidant genes had reverted to control levels after two years of smoking cessation. However, expression of potential oncogenes and tumour-suppressor genes never reverted to never-smoker levels even after years of smoking cessation. Consistently, expression of microRNAs is generally downregulated by cigarette smoke (Izzotti et al., 2009). As discussed below, smoking also altered methylation patterns and gene expression in smoking-associated tumours.
4.1.6 Other effects associated with carcinogenesis
(a) Proliferation, differentiation, apoptosis, and inflammation
As noted above, the signal-transduction pathways in lung tumours from smokers are distinctly different from those of non-smokers (Mountzios et al., 2008). Fig 4.5 shows details of signalling pathways that are deregulated by tobacco smoke. The involvement of high frequencies of mutated K-RAS and TP53 genes in smoking-associated lung tumours results in altered regulation of cell proliferation, differentiation, cytoskeletal organization and protein trafficking. Cigarette smoking activates NF-κB, which induces pro-inflammatory cytokine expression and induces growth factors and proliferative signals (Mountzios et al., 2008). This gene also influences the expression of the anti-apoptotic gene BCL2 and pro-apoptotic gene BAX. Smoking produces chronic inflammation, which promotes cancer (Walser et al., 2008). Smoking results in high levels of reactive oxygen species, which damage epithelial and endothelial cells and impair their function. In smoking-associated lung cancer, elevated levels of cyclooxygenase-2 (COX-2) and prostaglandin (PGE2) indicate apoptosis resistance, proliferation, immunosuppression, angiogenesis, invasion, and epithelial-mesenchymal transition (Walser et al., 2008).
Fig. 4.5
General scheme of some cell-signalling pathways that are deregulated by tobacco smoke in lung carcinogenesis
(b) Endogenous nitrosation
Intragastric formation of N-nitroso compounds, measured using urinary nitrosamino acids excreted in urine, was increased in smokers compared to non-smokers (Hoffmann & Brunnemann, 1983). Two recent studies demonstrated that NNN forms endogenously in some users of nicotine replacement therapy products (Stepanov et al., 2009a, b).
(c) Hormonal changes
These are described in Section 4.3.2a.
4.2. Polymorphisms in carcinogen-metabolizing genes
4.2.1 Introduction
It has been long proposed that the known variation among individuals in their capacity to activate and detoxify carcinogens may be associated with increased susceptibility to cancer, and that polymorphisms of carcinogen-metabolising genes may play a significant role. The most intensively studied genes involved in the metabolism of carcinogens include the various CYP genes, the GST genes and the NAT genes. Other relevant xenobiotic-metabolising genes, such as EPHX, sulfotransferase (SULT), UGT, myeloperoxidase (MPO), and NAD(P)H quinone oxidoreductase-1 (NQO1) genes, have also been studied. Recently, extensive pooled studies and reviews have been published on polymorphisms of carcinogen-metabolising genes and their role in cancer susceptibility, especially in tobacco-related lung cancer and cancers at other sites. Similarly, various biomarkers of exposure and genotoxicity that are presumed to provide a mechanistic basis for such associations have been comprehensively investigated in relation to these polymorphisms. A brief overview based largely on reviews and the meta- and pooled analyses is presented here.
4.2.2 Genetic polymorphisms of carcinogen metabolism: some central genes
(a) CYP genes
CYPs comprise the principal enzyme system catalysing various phase I oxidation reactions, including metabolic activation and detoxification of many carcinogenic substances in tobacco smoke such as PAHs. Of the various CYP enzymes expressed in humans, many of those belonging to CYP1 to CYP3 families play a role in carcinogen metabolism, producing highly reactive DNA-damaging metabolites as well as detoxified metabolites (Guengerich & Shimada, 1998; Lang & Pelkonen, 1999; Ingelman-Sundberg, 2004). CYPs have evolved into a wide superfamily with close to 60 different active genes currently identified; most of these genes exhibit polymorphism (www.cypalleles.ki.se).
(i) CYP1A1
Several allelic variants of the human CYP1A1 gene are currently known (www.cypalleles.ki.se). The major variant forms of the CYP1A1 gene (wildtype allele CYP1A1*1) mostly frequently studied for association to cancer susceptibility include the following two alleles: (i) CYP1A1*2A allele (m1 allele; Msp I) and (ii) CYP1A1*2B (Cascorbi et al., 1996) or CYP1A1*2C (www.cypalleles.ki.se) allele (m2 allele; Ile462Val). Importantly, the CYP1A1 m1 allele and m2 allele are in complete linkage disequilibrium in Caucasians (Kawajiri, 1999; Bartsch et al., 2000). In addition, CYP1A1*4 allele (m4; Thr461Asn) (Cascorbi et al., 1996), and CYP1A1*3 (m3) allele found in African-Americans but not in Caucasians or Asians (Crofts et al., 1993) are included in some studies (Bartsch et al., 2000).
In smoking-related lung cancer, the various CYP1A1 polymorphisms as well as the differences in the frequencies of the rare variant alleles between ethnicities contribute to the differences in findings. There are collective analyses of data predominantly indicating an overall mild to moderate effect of CYP1A1 polymorphisms on lung cancer risk (Kawajiri, 1999; Bartsch et al., 2000; Houlston, 2000; Le Marchand et al., 2003; Vineis et al., 2003; Vineis et al., 2004; Lee et al., 2008a; Shi et al., 2008). In many reviews and meta- or pooled analyses the increased risk associated with CYP1A1 polymorphism has most clearly been seen in Asian populations (Kawajiri, 1999; Le Marchand et al., 2003; Vineis et al., 2003; Lee et al., 2008a; Shi et al., 2008).
Multiple studies have also analysed the gene-gene interactions between CYP1A1, GSTM1 and GSTT1 polymorphisms and lung cancer (d’Errico et al., 1999; Houlston, 1999; Benhamou et al., 2002; Bolt & Thier, 2006; Raimondi et al., 2006; Ye et al., 2006; Carlsten et al., 2008). Some of the analyses have indicated that the elevated risk for lung cancer may be more pronounced for some CYP1A1/GSTM1 null genotype combinations (Le Marchand et al., 1998; Bartsch et al., 2000; Vineis et al., 2004, 2007; Lee et al., 2008a; Shi et al., 2008).
(ii) CYP1A2
CYP1A2 is highly inducible and metabolises, including deacetylation reactions, many tobacco smoke carcinogens such as aromatic and heterocyclic amines and nitro-aromatic compounds, and tobacco-specific nitrosamines such as NNK (Nebert et al., 2004; Jalas et al., 2005; IARC, 2007a). A few major variant alleles have been described (www.cypalleles.ki.se), some of which may have been reported to influence inducibility (Nakajima et al., 1999; Ingelman-Sundberg et al., 2007). Overall, the phenotype-genotype relations have not been well established for CYP1A2, although current evidence points towards contribution of genetic variation (Murayama et al., 2004; Ingelman-Sundberg et al., 2007); data on possible associations with tobacco related cancer are sparse (Agundez, 2004; Nebert & Dalton, 2006).
(iii) CYP2A6
Several aspects of smoking behaviour are likely to be influenced by CYP2A6 genetic variation, which influences nicotine metabolism (Malaiyandi et al., 2005; Mwenifumbo & Tyndale, 2007). The most important functionally altered allele is CYP2A6*4 (gene deletion), which confers a poor-metabolizer phenotype in homozygous individuals (Malaiyandi et al., 2005; Ingelman-Sundberg et al., 2007; Mwenifumbo & Tyndale, 2007). In some studies, polymorphic variants of CYP2A6 gene have been implicated in susceptibility to smoking-related cancers (Gambier et al., 2005; Malaiyandi et al., 2005; Nakajima, 2007). In line with this, the accumulated data have suggested that CYP2A6 polymorphism may affect cancer risk in smokers but not in non-smokers (Tan et al., 2001; Kamataki et al., 2005; Malaiyandi et al., 2005; Canova et al., 2009).
(iv) CYP2A13
From human CYPs, CYP2A13 is the primary form involved in the metabolic activation of the tobacco-specific nitrosamines NNK and NNN (Jalas et al., 2005; IARC, 2007a). The CYP2A13 gene exhibits polymorphism in humans (Zhang et al., 2002; Jalas et al., 2005), and experimental studies suggest that some of the polymorphisms may affect the hydroxylation of NNN and NNK (Jalas et al., 2005; Schlicht et al., 2007). However, the data on possible effects of these polymoprhisms on the risk of tobacco-related cancers in humans are still limited (Wang et al., 2003; Song et al., 2009; Timofeeva et al., 2009).
(v) CYP2D6
The CYP2D6 gene shows high variability in expression. The enzyme is not inducible, and therefore genetic variation largely contributes to the interindividual variation in enzyme activity. Currently, more than 100 different functional CYP2D6 gene variants have been described, and these are divided into alleles causing abolished, decreased, normal, and ultrarapid enzyme activity (Ingelman-Sundberg, 2005; Ingelman-Sundberg et al., 2007). The most important null alleles leading to poor-metabolizer phenotype are CYP2D6*4 (splice defect) and CYP2D6*5 (gene deletion) (Ingelman-Sundberg, 2005; Ingelman-Sundberg et al., 2007).
A large series of studies have been carried out over the past 20 years on the association between CYP2D6 polymorphism and susceptibility to lung cancer and to some other tobacco-related cancers (Wolf & Smith, 1999). Despite some indication of an association between CYP2D6 poor-metabolizer and decreased risk for lung cancer, no major role for CYP2D6 in carcinogen metabolism or a molecular basis for such an association have been discovered (Wolf & Smith, 1999; Ingelman-Sundberg, 2005).
(vi) Other CYP genes
CYP1B1 allelic variants that affect the catalytic activity have been described but they have been studied to a lesser extent for the association with susceptibility to smoking-related cancers (Thier et al., 2003). Some positive findings have been reported on head and neck cancer (Ko et al., 2001), and lung cancer (Zienolddiny et al., 2008).
Several polymorphisms have been characterized in the CYP2E1 gene and several positive associations with the risk of different cancers have been reported, in particular for cancers of the upper aerodigestive tract, lung and gastro-intestinal tract (Section 2.19). CYP2E1 may also play an important role in the interaction of the carcinogenic effects of alcohol and tobacco (Section 4.4).
From the human CYP3A locus (CYP3A4, CYP3A5 and CYP3A7), the CYP3A4*1B allele has been associated with lung cancer and prostate cancer in some studies but not in all (Dally et al., 2003; Rodriguez-Antona & Ingelman-Sundberg, 2006). However, the role of these variants in relation to tobacco smoking is unknown.
(b) GSTM1 and other GST genes
Polymorphic GST genes have long been proposed to modify susceptibility to lung cancer (Seidegård et al., 1986; Ketterer et al., 1992). The polymorphic genes encoding the various classes of cytosolic GST enzymes include the GSTM1 and GSTM3 genes (mu class), the GSTP1 gene (pi class), and the GSTT1 gene (theta class). The gene deletion (null) allele of the GSTM1 gene (GSTM1*0) and of the GSTT1 gene (GSTT1*0) have been the most intensively studied polymorphisms in relation to increased susceptibility to cancer (Strange et al., 2001; Bolt & Thier, 2006; McIlwain et al., 2006). For the GSTP1 gene, the form most abundantly present in lung tissue, genetic variation in exon 5 (GSTP1*2; Ile105Val), in exon 6 (Ala114Val), as well as a combination of these, are the variations most frequently studied for cancer susceptibility (Watson et al., 1998; Cote et al., 2009).
Numerous reviews, meta- and pooled analyses have been published over the past 15 years or so for the GST genes with systematic assessments covering altogether tens of thousands of cases and controls. For the GSTM1 null genotype, such analyses have largely provided negative, suggestive or at most moderately positive results for an association with an increased risk for lung cancer (d’Errico et al., 1999; Houlston, 1999; Benhamou et al., 2002; Ye et al., 2006; Carlsten et al., 2008). The larger the studies, the less significant the estimates for the role of GSTM1 emerge in systematic analysis (Ye et al., 2006; Carlsten et al., 2008). Also the varying allele frequencies related to ethnic background affect the findings for GSTM1 as well as for many other genes (Garte et al., 2001; Ye et al., 2006; Carlsten et al., 2008; Lee et al., 2008a).
In a meta-analysis of the association between the GSTT1 gene polymorphism and lung cancer no association between GSTT1 null genotype and risk for lung cancer in Caucasians was observed, but a positive association was found for Asians (Raimondi et al., 2006). A significant association for either Caucasians or Asians was also not found in a pooled analysis (Raimondi et al., 2006). A meta-analysis found no significant association between lung cancer risk and the GSTP1 Ile105Val polymorphism; but the pooled analysis suggested an overall statistically significant mild association between lung cancer and homozygosity or heterozygosity for the Val105 allele (Cote et al., 2009).
A recent body of epidemiologic data suggests an inverse association between cruciferous vegetables/isothiocyanates intake and cancers of the colorectum, lung and breast; the studies also provide evidence that this protective effect is greater among individuals who possess the GSTM1 or T1 null genotype, who would be expected to accumulate higher levels of isothiocyanates at the target tissue level, a pre-requisite for their enzyme-inducing effects (Seow et al., 2005). The association between isothiocyanates and cancer, and its modification by GSTM1 and GSTT1 status, is most consistent for lung cancer and appears to be strongest among current smokers who possess the combined GSTM1 and GSTT1 null genotypes (London et al., 2000a; Spitz et al., 2000; Zhao et al., 2001; Brennan et al., 2005; Seow et al., 2005).
(c) NAT1 and NAT2 genes
The pooled and meta-analyses carried out on NAT1 and NAT2 polymorphisms and bladder cancer risk have consistently reported significantly increased risk for NAT2 slow acetylators (Dong et al., 2008; Malats, 2008; see also Section 2.9). Data on NAT1 fast acetylators are inconsistent, as are the studies suggesting an increased risk for NAT2 rapid acetylator status. Additionally, genotypes for other genes, specially GSTM1, have also been implicated (Vineis et al. 2001; García-Closas et al., 2005; Hein, 2006; Sanderson et al., 2007; Dong et al., 2008; Malats, 2008).
In a recent large study on tobacco-related lung cancer and upper aerodigestive cancers, the NAT genes, in particular NAT*10 haplotype, emerged from a set of 16 genes as involved in the risk (McKay et al., 2008). When more than one hundred single nucleotide polymorphisms for 31 genes involved in phase I or phase II metabolism or in antioxidant defence were investigated, only four of the previously reported polymorphisms of the GSTP1, EPHX1 and superoxide dismutase SOD2 genes and the NAT1 fast acetylator phenotype remained significantly associated with risk of non-small cell lung cancer after correction for multiple testing (Zienolddiny et al., 2008).
In breast cancer, several recent meta-analyses of epidemiological studies have suggested increased risk among smokers with the NAT2 slow acetylator genotype; such an association has been observed especially among long-term smokers and post-menopausal women (Terry & Goodman, 2006; Ambrosone et al., 2008; Ochs-Balcom et al., 2007; Baumgartner et al., 2009).
In all, the role of the NAT gene polymorphisms in tobacco-related cancers, with the exceptions of increased risk of bladder cancer and possibly breast cancer in NAT2 slow acetylators, remains largely open due to the incomplete understanding of phenotype-genotype relationships, and the interplay between these two genes and their polymorphisms (Hein, 2002, 2006).
(d) Others
Genes coding for EPHX, UGT and SULT enzymes, mainly but not exclusively involved in detoxification reactions, exhibit polymophisms with numerous gene variants discovered (Mackenzie et al., 1997; London et al., 2000b; Glatt et al., 2001; Burchell, 2003). Additional polymorphic genes studied for their significance in cancer susceptibility are the NQO1 and MPO genes, with NQO1 playing a dual role in the detoxification and activation of procarcinogens, and MPO converting lipophilic carcinogens into hydrophilic forms (Nebert et al., 2002). All these genes have been studied for their possible association with tobacco-related cancer risk to a varying extent and with variable outcomes (London et al., 2000b; Bamber et al., 2001; Garte, 2001; To-Figueras et al., 2001; Tiemersma et al., 2002b; Guillemette, 2003; Wells et al., 2004; Kiyohara et al., 2005; Moreno et al., 2005; Nagar & Remmel, 2006; Gallagher et al., 2007).
4.2.3 Biomarkers of tobacco carcinogenesis and polymorphic genes of carcinogen metabolism
A myriad of studies have investigated association between various biomarkers of tobacco-related carcinogenesis and genetic variation of genes involved in carcinogen metabolism. For involvement in increased cancer susceptibility, a large variety of intermediate biomarker have been studied, including PAH metabolites in urine, urinary mutagenicity, DNA and protein adducts, cytogenetic alterations, HPRT mutant lymphocytes, as well as somatic mutations of the tumour suppressor gene TP53 and KRAS oncogene occurring in cancer tissue.
(a) PAH metabolites and mutagenicity in urine
(i) PAH metabolites in urine
Increased excretion of 1-hydroxypyrene in urine in association with the GSTM1 null genotype has been reported in many studies on individuals with occupational or environmental exposure to PAHs (Yang et al., 1999; Alexandrie et al., 2000; Lee et al., 2001; Kuljukka-Rabb et al., 2002; Kato et al., 2004). The associations seen between GSTT1 polymorphism and the PAH metabolites are somewhat more variable. Similarly, the joint effect of GSTM1 and GSTT1 null genotypes, as well as the effects of some other genes of xenobiotic metabolism, such as EPHX, CYP1A1, CYP1A2 and the aryl hydrocarbon receptor (AhR) gene have been either positive or negative (Yang et al., 1999; Alexandrie et al., 2000; Lee et al., 2001; Zhang et al., 2001; Kuljukka-Rabb et al., 2002; Yang et al., 2003; Chen et al., 2007; Cocco et al., 2007; Bin et al., 2008).
Another PAH metabolite studied in this context is phenanthrene, the simplest PAHs with a bay region, a feature closely associated with carcinogenicity. A study quantified ratios of urinary products of metabolic activation (such as PheT) and detoxification (such as phenanthrols, HOPhe) of phenanthrene in 346 smokers, who were also genotyped for 11 polymorphisms in genes involved in PAHs metabolism, including the CYP1A1 and GSTM1 genes. A significant association between the presence of the CYP1A1 Ile462Val polymorphism and high PheT/3-HOPhe ratios was found, particularly in combination with the GSTM1 null polymorphism (Hecht et al., 2006).
Overall, the data on the influence of genetic variation in PAHs metabolism on the levels of the urinary metabolite biomarkers are variable, and currently inconclusive.
(ii) Urinary mutagenicity
One relatively early line of research investigated the relationship between urinary mutagenicity and genetic variation in activation or detoxification genes. These studies, however, have seldom been focused on smokers only but rather on other sources of exposure (Pavanello & Clonfero, 2000).
In some studies, NAT2 slow acetylator genotype either alone or in combination with GSTM1 null genotype has been associated with increased urinary mutagenicity in the Salmonella test in individuals with occupational, environmental or medicinal PAH-related exposure, or in smokers (Vineis & Malats, 1999; Pavanello & Clonfero, 2000). In another study, CYP1A2 activity, but not NAT2, GSTM1 or GSTT1 genotypes influenced urinary mutagen excretion in smokers (Pavanello et al., 2002). A further study also suggested contribution of the CYP1A2 gene variation to increased urinary mutagenicity in heavy smokers (Pavanello et al., 2005). Associations with variants of other xenobiotic-metabolising genes (such as EPHX1) have also been reported, with somewhat complex results (Kuljukka-Rabb et al., 2002).
(b) DNA adducts
The relationship between the variants of polymorphic genes of carcinogen metabolism and tobacco smoke-related DNA adduct formation has been addressed in an abundant number of studies among smokers, occupationally exposed groups, and patients with smoking-related cancer. In addition, multiple in vitro studies on this relationship have been carried out (Bartsch et al., 2000; Pavanello & Clonfero, 2000; Alexandrov et al., 2002; Wiencke, 2002).
The intensive efforts to study the relationship between CYP1A1 and GSTM1 gene polymorphism and the level of aromatic-hydrophobic/bulky PAH-DNA adducts in human lungs have so far provided little evidence for a role of a single metabolic genotype or their combinations on DNA adduct formation, with largely weak, non-significant or contradictory results. However, a trend of increasing adduct levels in subjects with the CYP1A1*2-GSTM1*0 genotype combination has been observed, which was reinforced when BPDE-DNA adducts were specifically assessed. These results suggest a gene-gene interaction, supported by biological data from other studies (Bartsch et al., 2000; Alexandrov et al., 2002; Wiencke, 2002). Such gene-gene interaction lends support to the increased risk for lung cancer found in carriers of these genotypes in Japanese, among whom the frequency of the variant CYP1A1 allele is much higher (Bartsch et al., 2000; Alexandrov et al., 2002).
A wide selection of genes and genotypes included in the various studies have made it difficult to assess the overall role of the polymorphisms of GSTM1 and other genes alone or in combination. Differences between the studies in the types of adducts determined, the various tissues, cell types and cancers studied, detection methods, variation in sources and types of exposure, sample size, gender differences, and sometimes poor knowledge regarding the alleles, genotypes and haplotypes under study also contribute to the large variability seen in these studies (d’Errico et al., 1999; Hemminki et al., 2001; Alexandrov et al., 2002; Wiencke, 2002).
(c) Cytogenetic biomarkers of genotoxicity
(i) Chromosome aberrations and sister chromatid exchanges
Early studies investigating whether homozygosity for the GSTM1 null allele affects prevalence of cytogenetic changes in lymphocytes of smokers reported positive results (Seidegård et al., 1990; van Poppel et al., 1992; Cheng et al., 1995). Since then, studies have investigated the association between genetic polymorphisms of xenobiotic-metabolising genes and cytogenetic biomarkers in smokers and in some occupational groups (Rebbeck, 1997; Autrup, 2000; Pavanello & Clonfero, 2000; Norppa, 2003, 2004).
Collectively, the reported findings are in support of increased susceptibility of smokers to chromosomal effects in association with GSTM1 and GSTT1 null variants deficient in detoxification of tobacco smoke carcinogens. Exposure to genotoxicants generated from other environmental sources (e.g. polluted air, diet, endogenous sources such as reactive oxygen species) may contribute to the observed associations, and it is likely that other polymorphic metabolic genes such as NAT2 may be involved (Pavanello & Clonfero, 2000; Norppa, 2001, 2003).
(ii) Micronucleus induction
The relationship between formation of micronuclei and genetic polymorphisms of carcinogen metabolism has been addressed in a wide range of human population studies (Norppa, 2003, 2004). Induction of micronuclei in smokers may be little, if at all, affected by GSTM1, GSTT1 or NAT2 genotypes. In contrast, the NAT1 rapid genotype appears to show an association with increased susceptibility to smoking-related micronuclei (Norppa, 2004).
A recent review evaluated more than seventy human studies on genetic polymorphisms and micronucleus frequency detected either in peripheral blood lymphocytes or exfoliated cells in populations exposed to various genotoxic agents. There were no significant genotype effects involved in micronucleus induction in smokers (Iarmarcovai et al., 2008). The relationship between genetic polymorphisms and micronucleus formation is complex, and is influenced to a variable extent by several genes of xenobiotic metabolism and DNA repair, as well as the variety of chromosomal alterations known to contribute to micronucleus formation (Iarmarcovai et al., 2008).
(iii) Chromosomal damage induced in vitro
The effects of genotypes or genotype combinations in vitro on the induction of various cytogenetic endpoints by tobacco-smoke carcinogens and their metabolites have been studied, initially focused on the GSTM1 and GSTT1 null genotypes (Norppa, 2001, 2004). In a study investigating NNK in vitro, lymphocytes from GSTM1 null donors were more sensitive to induction of chromosomal aberrations and sister chromatid exchanges by NNK than lymphocytes from GSTM1 positive donors (Salama et al., 1999).
(d) Gene mutations
(i) HPRT mutant lymphocytes
Associations between the frequencies of HPRT mutant T-lymphocytes in populations exposed to genotoxic agents, such as smokers, and the polymorphism of xenobiotic-metabolising genes have been studied. In the early studies, positive, weak, or negative associations were reported for GSTM1 null genotype, and negative findings were published for NAT2 slow acetylator genotype in occupationally exposed or non-exposed subjects (Rebbeck, 1997; Vineis & Malats, 1999). When healthy, non-smoking and occupationally non-exposed young adults were studied for HPRT mutant frequency and polymorphisms in CYP1A1, GSTM1 and NAT2 genes, none of these polymorphisms, analysed individually, were found to influence the HPRT mutant frequency (Davies et al., 1999). A significant interaction between the GSTM1 null genotype and NAT2 slow acetylator was associated with higher mutant frequency, but no other genotype combinations (Davies et al., 1999). Some later studies have reported variable associations between HPRT mutant frequency and polymorphisms for either individual genes (GSTM1, GSTT1 or EPHX1) or some of the genotypes in combination among exposed (Viezzer et al., 1999; Abdel-Rahman et al., 2001, 2003).
(ii) Mutations of the TP53 gene and other cancer-related genes
Whether the frequency of somatic mutations detected in tumour tissue in cancer-related genes, primarily the TP53 tumour suppressor gene and KRAS oncogene, may be modified by polymorphisms in carcinogen metabolizing genes was first investigated assessing the effects of the GSTM1 genotype, alone or in combination with other genetic polymorphisms. Several, but not all, such studies showed significant association between GSTM1 null genotype and either the frequency or type of TP53 mutations in smoking-induced lung cancer or other cancer type (Rebbeck, 1997; Vineis & Malats, 1999; Autrup, 2000). Fewer studies examined the association between TP53 mutations and GSTT1 polymorphism, and some results suggested the involvement of both null genotypes (Vineis & Malats, 1999; Autrup, 2000).
In smokers with non-small cell lung cancer, the risk of mutation was found to be the highest among the homozygous carriers of the CYP1A1 rare allele CYP1A1 MspI (Ile462Val) who also exhibited the GSTM1 null genotype (Kawajiri et al., 1996). Similarly, positive associations between K-RAS mutations and homozygosity for the CYP1A1 rare allele were observed; the risk of mutation was enhanced when the CYP1A1 susceptible genotype was combined with GSTM1 null genotype (Kawajiri et al., 1996). In another study, also carried out in a Japanese study population, K-RAS mutations occurred with greater frequency in lung adenocarcinoma smoking patients and of the GSTM1 null genotype as compared with the GSTM1 positive genotype (Noda et al., 2004).
Many of the studies that assessed NAT2 acetylator genotypes have found non-significant associations with the frequency or type of TP53 mutation in bladder, lung, or other cancers (Vineis & Malats, 1999; Autrup, 2000). A study on bladder cancer did not find an overall association between TP53 mutation frequency and GSTM1, GSTT1, GSTP1 or NAT2 genotypes. However, among patients with TP53 mutations, transversion mutations were more frequent in those with GSTM1 null genotype as compared to those with GSTM1 positive genotype; no significant associations were found for the NAT2 gene (Ryk et al., 2005).
In rectal cancer, overall negative results for an association between TP53 or KRAS mutations and GSTM1 and NAT2 polymorphisms among smokers and non-smokers exposed to tobacco smoke were found (Curtin et al., 2009). An interaction of second-hand tobacco smoke and NAT2 was found in TP53 mutation positive tumours but not in smokers (Curtin et al., 2009). Earlier, an increased risk of TP53 transversion mutations among GSTM1 positive individuals who smoked cigarettes was found in colon cancer (Slattery et al., 2002).
A statistically significant association was observed between the GSTT1 null genotype and TP53 mutation status of breast tumour in one study (Gudmundsdottir et al., 2001), while in another larger study none of the genotypes for CYP1B1, GSTM1, GSTT1 and GSTP1 genes alone were associated with somatic TP53 mutations (Van Emburgh et al., 2008).
In summary, data from various cancer types on the association between genetic polymorphisms of carcinogen-metabolizing genes and somatic mutations of the TP53 and K-RAS genes vary widely and do not permit to conclude (Rebbeck, 1997; Vineis & Malats, 1999; Autrup, 2000).
4.3. Site-specific mechanisms of carcinogenicity of tobacco smoke
4.3.1 Sites with sufficient evidence of carcinogenicity of tobacco smoking
(a) Lung
The conceptual model presented in Section 4.1 (Fig. 4.1) depicts the main mechanistic steps by which cigarette smoke causes cancer. Smokers inhale into their lungs carcinogens which, either directly or after metabolism, covalently bind to DNA, forming DNA adducts (see Section 4.1, Fig. 4.3). Tobacco smoke contains multiple strong lung carcinogens such as NNN, NNK, PAHs, 1,3-butadiene and cadmium. Levels of tobacco smoke-related DNA adducts, mainly 32P-postlabelled aromatic-hydrophobic/PAH-related bulky DNA adducts, in the lung are higher in smokers than in non-smokers (Phillips, 2002; IARC, 2004a; Hecht, 2008). Higher levels of DNA adducts have further been linked to increased risk for cancer in pooled and meta-analyses (IARC, 2004a; Veglia et al., 2008).
Mutations in TP53 and K-RAS genes, two central genes of human carcinogenesis, are more frequently mutated in smokers' lung cancer as compared to lung cancer from non-smokers (DeMarini, 2004; IARC, 2004a; Lea et al., 2007; Ding et al., 2008; see Section 4.1.3). In particular, TP53 but also to some extent K-RAS mutations found in smoking-associated lung tumours exhibit mutational specificity that is consistent with the pattern produced by PAH diol epoxides in experimental studies and different from that observed in non-smokers' lung cancer (Pfeifer et al., 2002; DeMarini, 2004; IARC, 2004a; Le Calvez et al., 2005; Section 4.1.3). Keeping with such exposure-specific mutation profile, lung cancer in non-smokers exposed to second-hand tobacco smoke shows mutational similarity to smokers' lung cancer, although less data are available (Husgafvel-Pursiainen, 2004; IARC, 2004a; Le Calvez et al., 2005; Subramanian & Govindan, 2008). The different pathways of lung carcinogenesis for smokers and non-smokers are likely to involve somatic mutations and other genetic alterations in a larger set of genes that are critical in controlling normal cellular growth via signal transduction (Bode & Dong, 2005; Lea et al., 2007; Ding et al., 2008).
Smoking-related lung carcinogenesis also involves a multitude of other alterations influencing the complex pathogenic pathways involved in lung cancer development, such as increased inflammation, aberrant apoptosis, increased angiogenesis, tumour progression and tumour metastasis (Wolff et al., 1998; Heeschen et al., 2001; Schuller, 2002; West et al., 2003; Smith et al., 2006; Lee et al., 2008b; Section 4.1.5). Continued exposure to toxicants, genotoxicants, carcinogens, co-carcinogens and tumour promoters present in tobacco smoke has major effects on biological processes at all steps of multistep tumourigenesis of human lung (Hecht, 2003, 2008; Section 4.1). For example, nicotine in tobacco smoke is currently not described as a full carcinogen, but it exerts its biological effects via binding to nicotinic and other cellular receptors and likely enhances cell transformation and carcinogenicity through mechanisms not yet defined (Heeschen et al., 2001; West et al., 2003).
Numerous studies have provided evidence that the human genome may contain one or several loci that confer susceptibility to lung cancer. There are low-penetrance genes involved in the metabolism of tobacco smoke carcinogens, DNA repair and cell cycle control that may influence individual susceptibility to lung cancer (Spitz et al., 2006). The role of the polymorphisms of these various classes of genes in lung carcinogenesis requires a systematic evaluation of the genetic evidence with stringent criteria (Ioannidis, 2008; Risch & Plass, 2008; Vineis et al., 2009; Sections 4.1 and 4.2). Recently, genome-wide association studies have identified a susceptibility locus at chromosome 15q25.1 (Amos et al., 2008; Hung et al., 2008; Thorgeirsson et al., 2008). The identity or function of the gene is not yet known, nor is the mechanism through which it may predispose to lung cancer. It is however likely that lung cancer susceptibility is related to the nicotine receptor gene residing at 15q25.1, and there is some evidence suggesting that it may be related to increased uptake of nicotine and NNK per cigarette (Le Marchand et al., 2008).
In addition to genetic alterations, a growing body of evidence shows that epigenetic mechanisms, such as aberrant DNA methylation, histone modifications and RNA-mediated gene silencing are involved in cancer development (Jones & Baylin, 2007; Cortez & Jones, 2008). In lung carcinogenesis, gene promoter-associated (CpG island-specific) hypermethylation is an early and frequent event causing transcriptional inactivation of genes involved in regulation of cellular growth and differentiation (Belinsky, 2004). For example, several studies have indicated that the tumour suppressor gene p16 (p16INK4a/CDKN2A), a cell cycle regulator, is among the genes most frequently inactivated by aberrant methylation in lung cancer from smokers (Belinsky, 2004), with differences seen between smokers and never-smokers (Toyooka et al., 2006). Significant associations have been established between smoking and promoter hypermethylation of tumour suppressor genes in lung tumours from smokers, and in plasma, serum or sputum DNA from cancer-free smokers (Belinsky, 2004; Belinsky et al., 2005, 2006; Toyooka et al., 2006).
(b) Oral cavity
PAHs can be carcinogenic at the site of application, which could include the human oral cavity. DMBA, a highly carcinogenic PAH not present in tobacco or tobacco smoke, is a standard model compound for induction of oral tumours in the hamster cheek pouch; less is known about the effects on the oral cavity of PAHs that do occur in tobacco products (Shklar, 1972; Rao, 1984; Vairaktaris et al., 2008). A mixture of NNN and NNK induced oral tumours in rats when applied locally (Hecht et al., 1986), and DNA adduct formation from NNN, NNK and NNAL has been observed in the rat oral cavity (Zhang et al., 2009a, b). HPB-releasing DNA adducts from NNK and/or NNN have been reported in exfoliated oral cells from smokers and smokeless tobacco users (Heling et al., 2008) and HPB-releasing heamoglobin adducts are elevated in smokeless tobacco users (IARC, 2007a). Unidentified DNA adduct levels are consistently elevated in oral cells and tissues from smokers compared to non-smokers (IARC, 2004a). Mutations in the TP53 gene have been observed in oral tumours from smokers and smokeless tobacco users (IARC, 2006b, 2007a; Warnakulasuriya & Ralhan, 2007). Tobacco-associated genetic mutations including micronuclei, gene mutations, DNA polymorphisms, and chromosomal abnormalities have been reported in studies of buccal cells from smokers and smokeless tobacco users (Proia et al., 2006). The use of lime by betel quid chewers is associated with enhanced oxidative damage that could play a role in inflammation or tumour promotion (IARC, 2004b).
(c) Larynx and nasopharynx
Hamsters exposed to cigarette smoke by inhalation consistently developed benign and malignant tumours of the larynx; tumours were produced by inhalation of the particulate phase, but not the gas phase of cigarette smoke (IARC, 1986). In related studies in which hamsters were treated with DMBA by intratracheal instillation followed by exposure to cigarette smoke, a significantly higher incidence of laryngeal tumours was observed than in hamsters exposed only to cigarette smoke or to DMBA (IARC, 1986). Collectively, these results indicate an initiation-promotion mechanism for the production of laryngeal tumours, and are consistent with the results of experiments in which tobacco smoke condensate is applied to mouse skin (IARC, 1986). The combined data implicate PAHs and tumour promoters in tobacco smoke as potential etiologic agents for cancer of the larynx in hamsters. Levels of DNA adducts measured by non-specific methods were higher in larynx tissue from smokers than from non-smokers (IARC, 2004a). Analyses of mutations in the TP53 gene from tumours of the larynx in smokers show a pattern similar to that observed in lung tumours, and both are consistent with the pattern produced by PAH diol epoxides (IARC, 2006b). The available data are consistent with the conceptual framework illustrated in Fig. 4.1 (Szyfter et al., 1999).
Formaldehyde, a constituent of cigarette smoke, causes nasopharyngeal cancer in humans (IARC, 2006a). A recent study demonstrates a 10-fold higher level of the formaldehyde-DNA adduct N6-hydroxymethyldeoxyadenosine in leukocytes of smokers compared to non-smokers, suggesting its possible involvement in nasopharyngeal cancer in smokers (Wang et al., 2009). Acetaldehyde, another carcinogenic constituent of tobacco smoke, which also forms genotoxic adducts (Section 4.1), may also contribute to the development of these forms of head and neck cancer.
(d) Oesophagus
Nitrosamines are probably the most effective oesophageal carcinogens known, with particularly strong activity in the rat (Lijinsky, 1992). NNN and NDEA are both present in cigarette smoke, and levels of NNN greatly exceed those of NDEA (IARC, 2004a). NNN is also present in considerable quantities in smokeless tobacco and betel quid containing tobacco (IARC, 2004a, 2007a). Thus, NNN is a likely candidate as a causative agent for esophageal cancer in smokers, smokeless tobacco users, and chewers of betel-quid with tobacco. While considerable mechanistic data are available from studies of NNN in laboratory animals (Hecht, 1998; Wong et al., 2005; Lao et al., 2007; Zhang et al., 2009a), there are little comparable data in humans.
Increased acetaldehyde production derived both from tobacco smoke and from microbial alcohol oxidation may play a role in the synergistic carcinogenic action of alcohol and smoking on oesophagus, as well as on other upper aerodigestive locations (Homann et al., 2000; Salaspuro & Salaspuro, 2004; Lee et al., 2007a).
(e) Stomach
Hypermethylation of the E-cadherin 1 gene (CDH1) was observed preferentially in gastric tumours from smokers rather than non-smokers (Poplawski et al., 2008). CDH1 can act as a tumour-suppressor gene, preventing cells from growing and dividing in an uncontrolled way to form a cancerous tumour. Because the protein encoded by this gene helps cells stick together, altered regulation may lead to metastasis.
Boccia et al. (2007) found an increased risk for stomach cancer among smokers who had the SULT1A1 His genotype, and Lee et al. (2006) found an increased risk for those who had the m2 allelic variant of CYP1A1. A nested case–control study found that smokers had an increased risk of gastric cancer if they carried at least one variant allele A in Ex7+129 C > A (Thr461Asn, m4) of CYP1A1 (Agudo et al., 2006). Stomach cancer tissue from smokers had higher levels of stable DNA adducts than did those from non-smokers; however, the number of non-smokers was quite small (Dyke et al., 1992).
(f) Pancreas
NNK and its metabolite NNAL are the only pancreatic carcinogens known to be present in tobacco and tobacco smoke. NNK was detected in the pancreatic juice of 15 of 18 samples from smokers, at levels significantly higher than in non-smokers; NNAL and NNN were also detected in some samples (Prokopczyk et al., 2002). DNA adducts of NNK and NNAL were present in pancreatic tissue of rats treated with these nitrosamines (Zhang et al., 2009b), but were not detected in most human pancreatic tissue samples (Prokopczyk et al., 2005).
(g) Colorectum
Tobacco smoke contains heterocyclic amines, such as 2-amino-1-methyl-6-phenylimidazo[4,5,6]pyridine (PhIP), which are intestinal carcinogens in rats and mutate the adenomatous polyposis coli (Apc) gene in mice (Møllersen et al., 2004). The APC gene is frequently mutated and has altered expression in human colon cancer (Samowitz et al., 2007; Samowitz, 2008). A recent model of colon cancer by Sweeney et al. (2009) suggests that this disease can develop via at least three independent mechanistic pathways. One pathway is initiated by methylation of MINT (methylation in tumour) markers that proceeds down a pathway predisposing to microsatellite instability, followed by methylation of the mismatch repair gene mutL homologue 1 (MHL1) and the tumour-suppressor gene TP16, followed by mutation in BRAF (a homologue of a viral raf oncogen). A second independent pathway is initiated with a mutation in the APC gene, followed by a mutation in the TP53 gene. A third independent pathway involves only KRAS2 mutations. One study found BPDE-DNA adducts at a higher frequency in colon DNA from smokers than from non-smokers (Alexandrov et al., 1996). Mutations or epigenetic changes in some or all of these genes have been found in smoking-associated colon or colorectal tumours.
Microsatellite instability, which is the expansion or contraction of short nucleotide repeats, occurs in approximately 10–15% of sporadic colorectal cancer, and is usually associated with smoking and hypermethylation of the promoter of the mismatch repair gene MLH1 (Samowitz, 2008). Smoking-associated colorectal tumours also have high frequencies of methylation at CpG islands (Samowitz, 2008).
In a case–control study of colorectal cancer, Kasahara et al. (2008) found that the genetic polymorphism APEX1/APE1 (apurinic/apyrimidinic endonuclease-1) Asp148Glu, which is a gene involved in DNA repair, was associated with risk for colorectal cancer among smokers but not non-smokers. Other studies have also found associations between polymorphisms in the DNA repair genes XRCC1 and smoking and risk for colorectal cancer (Stern et al., 2007; Campbell et al., 2009).
(h) Liver
Tobacco smoke contains liver carcinogens such as furan and certain nitrosamines. Liver tumours exhibit increased expression of C-MYC, P16INK4A, epidermal growth factor receptor (EGFR), telomerase, transforming growth factor-α (TGF-α), insulin-like growth factor-2 (IGF-2) and RAF oncogene (Abou-Alfa, 2006). Smokers show altered expression of some of these genes or of genes in the same or similar pathways (Sen et al., 2007). A genome-wide association study found that SNP rs1447295 in the 8q24 chromosome was positively associated with liver cancer among ever-smokers (Park et al., 2008). Thus, tobacco smoke appears to have epigenetic effects on the liver that may contribute to hepatocellular carcinoma.
(i) Urinary bladder
Tobacco smoke contains aromatic amines such as 4-aminobiphenyl and 2-naphthylamine, which are human bladder carcinogens (see IARC, 2012a). In bladder tumours, smoking was associated with a more than twofold increase risk of methylation of the promoter region of the P16INK4A gene and of the soluble Frizzled receptor protein (SFRP) gene (Marsit et al., 2006). In addition, Tang et al. (2009) suggested that epigenetic silencing of Wnt antagonists through hypermethylation may play a role in smoking-related invasive bladder cancer (Tang et al., 2009). SNP rs6983267 of the 8q24 chromosome was inversely associated with bladder cancer among ever-smokers (Park et al., 2008). Smokers generally have mutagenic urine and smoking is associated with specific cytogenetic changes and DNA breaks in bladder tumours (DeMarini, 2004). Smoking-associated stable DNA adducts have been found in bladder tissue or exfoliated urothelial cells, supporting a role for DNA damage in smoking-associated bladder cancer (Phillips, 2002).
(j) Cervix
The cervical mucus of smokers is more mutagenic than that of non-smokers, and cervical epithelia of smokers have higher frequencies of micronuclei than those of non-smokers (DeMarini, 2004). Several studies have found increased levels of DNA adducts in cervical tissue from smokers relative to non-smokers, suggesting a role for smoking-associated DNA damage in cervical cancer (Phillips, 2002).
(k) Ovary
It has been observed that the inverse associations reported for serous and endometrioid tumours with respect to parity and oral contraceptives did not hold for the mucinous tumours. Based on these observations, Risch et al. (1996) suggested that mucinous ovarian tumours may be etiologically unrelated to the other types of epithelial tumours. Whereas mucinous elements such as gastric or intestinal type glands may be seen in mature teratomas, a form of germ cell neoplasia, overall mucinous tumours are classified as surface epithelial tumours because transitions among the subtypes may be observed. The major difference between mucinous and serous tumours is their biologic behaviour. Mucinous carcinomas of the ovary are slow growing tumours that appear to develop from their benign counterparts. The fact that the transitions between the benign, borderline, and malignant form of the disease can be seen in the same tumour suggests that over time, there is a progression from benign to malignant (Riopel et al., 1999). K-ras mutational analysis, for example, demonstrates a heterogeneous distribution of the mutation within different parts of the same neoplasm, suggesting that acquisition of the K-ras mutation occurs in malignant transformation (Mandai et al., 1998). Serous carcinomas seem to develop de novo rather than from a benign pre-existing lesion; alternatively, the rate of progression is rapid and the precursor lesion is obliterated before the detection of the tumour. In some data, current smoking is associated with a shorter interval to detection of mucinous than non-mucinous tumours. Because the mucinous tumour is slow growing, smoking could contribute to the malignant progression of the adenoma-carcinoma sequence, as the benign form of the tumour may have been present for some time.
(l) Leukaemia
Tobacco smoke contains known leukaemogens such as benzene, 1,3-butadiene and formaldehyde (IARC, 2012a). The mechanisms of leukaemogenesis are currently not well understood. Data indicate that leukaemogenic agents, such as benzene, cause toxicity to the haemotopoietic system, as well as genotoxicity at low levels, and that genetic polymorphisms may be involved in these processes (Aksoy, 1989; Lan et al., 2004; Garte et al., 2008; Hosgood et al., 2009; Lau et al., 2009; Rappaport et al., 2009). Recent studies suggest the importance in carcinogen-related leukaemogenesis of damage to haematopoietic stem/progenitor cells circulating in the peripheral blood, or, alternatively, damage to primitive pluripotent progenitor cells present in other tissues (Zhang et al., 2009c). In these two models, damaged stem/progenitor cells would then travel to the bone marrow and become initiated leukaemic stem cells. Mechanisms considered central in these models are: disruption of bone marrow DNA, through e.g. formation of DNA adducts, DNA–protein crosslinks, the action of free radicals or active states of oxygen; intercalation of metals within the DNA structure; or inhibition of enzymes involved in cell division (Zhang et al., 2007, 2009c).
4.3.2 Sites with limited evidence of carcinogenicity or evidence suggesting lack of carcinogenicity
(a) Breast
(i) Carcinogenic pathway
Carcinogens found in tobacco smoke pass through the alveolar membrane and into the blood stream, by means of which they can be transported to the breast via plasma lipoproteins (Yamasaki & Ames, 1977; Shu & Bymun, 1983; Plant et al., 1985). Tobacco smoke contains known rodent mammary carcinogens, including PAHs and aromatic amines (IARC, 1986, 2004a; el-Bayoumy, 1992; Ambrosone & Shields, 1999; Ambrosone, 2001; Hoffmann et al., 2001) which, due to their lipophilicity, can be stored in breast adipose tissue (Obana et al., 1981; Morris & Seifter, 1992) and then metabolized and activated by human mammary epithelial cells (MacNicoll et al., 1980). Tobacco smoke constituents reach the breast as demonstrated by the detection of cotinine in breast fluid (Petrakis et al., 1978). There is evidence suggesting the presence of mutagenic arylamines (Thompson et al., 2002) and PAHs (Zanieri et al., 2007) in human breast milk. Cigarette smoke condensate has been shown to transform normal human breast epithelial cells in vitro (Narayan et al., 2004), perhaps by blocking long-patch base excision repair (Kundu et al., 2007). Transformation and cytogenetic effects have been observed in human mammary epithelial cells after exposure to chemical carcinogens such as PAHs or arylamine (Mane et al., 1990; Eldridge et al., 1992; Calaf & Russo, 1993).
The formation of specific adducts from PAHs and aromatic amines has been observed in human breast epithelial cells in vitro, and unspecified-DNA adducts have been found in exfoliated ductal epithelial cells in human breast milk (Gorlewska-Roberts et al., 2002; Thompson et al., 2002).
Mutations in the TP53 tumour suppressor gene have been found in 15–30% of breast cancers (Goldman & Shields, 1998; Olivier & Hainaut, 2001). An increased prevalence and altered spectrum of TP53 mutations in breast tumours have been observed among current smokers compared with never smokers (Conway et al., 2002). The breast tumours with the most pronounced smoking-related mutational pattern (for example, a greater number of G:C→T:A transversions) were from women who had smoked for more than 20 years, although total TP53 mutations were not associated with smoking duration (Conway et al., 2002). This increased frequency of G to T transversions in smokers versus non-smokers is also observed in the IARC TP53 database (IARC, 2006b; Van Emburgh et al., 2008).
Recent meta-analyses of epidemiological studies tend to show positive associations of breast cancer with long-term smoking among NAT2 slow acetylators, especially among post-menopausal women (who are more likely than pre-menopausal women to be very long-term smokers). Firozi et al. (2002) showed that breast tissue from NAT2 slow acetylators had significantly higher levels of the diagonal radioactive zone (smoking-related) DNA adduct pattern than that from fast acetylators.
High rates of breast cancer in women exposed to ionizing radiation during adolescence (aged 10–19 years at exposure) (Tokunaga et al., 1987) suggested that the adolescent breast may also be sensitive to the DNA-damaging effects of other exposures. This might also be true for the genotoxic compounds contained in tobacco smoke. Although some studies have supported such association, the results have been sparse and mixed. In addition, it is difficult to separate the effects of early life exposure to tobacco and smoking duration (Terry & Rohan, 2002).
Early age at first full-term pregnancy has been associated with reduced breast cancer risk (Kelsey et al., 1993), hypothetically due to terminal differentiation of the breast epithelium that occurs late in the first trimester. It has been suggested that in the early stages of pregnancy, when growth-promoting hormone levels are high, but before terminal differentiation (Montelongo et al., 1992), the breast may be particularly susceptible to the cancer-promoting chemicals in tobacco smoke. Several epidemiological studies compared measures of smoking before and after a first full-term pregnancy. Although suggestive, the data did not consistently show an increased risk for breast cancer among women who smoked before a first full-term pregnancy (Adami et al., 1988; Hunter et al., 1997; Band et al., 2002; Egan et al., 2003; Gram et al., 2005; Li et al., 2005; Olson et al., 2005; Cui et al., 2006). Smoking was associated with a 50% increased risk among women with slow NAT2 acetylation genotype (Egan et al., 2003). Overall, studies of risk in association with the timing of smoking relative to a first pregnancy are inconclusive; nevertheless, the breast tissue appears to have a greater susceptibility to the carcinogenic chemicals in tobacco smoke before compared to after terminal differentiation of breast epithelium.
(ii) Estrogenic pathway
The “anti-estrogenic” mechanism through which tobacco smoking may inhibit breast cancer progression is unclear. Estrogen is a known risk factor for breast cancer and several hypotheses have been proposed: earlier age at menopause among smokers, a reduction in the gastrointestinal absorption or distribution of estrogen, enhanced metabolism of estradiol to inactive catechol estrogens, increased binding of estrogens by serum sex hormone-binding globulin, lowered levels of estrogen derived from adipose tissue (Baron, 1984; Baron et al., 1990; Terry & Rohan, 2002). Several studies of cigarette smoking and mammographically-defined breast density showed lower measures of breast density in current smokers than in non-smokers (Sala et al., 2000; Vachon et al., 2000; Warwick et al., 2003; Jeffreys et al., 2004; Modugno et al., 2006; Bremnes et al., 2007; Butler et al., 2008). Since exposure to estrogen has been associated positively with breast density, a strong risk factor for breast cancer (McCormack & dos Santos Silva, 2006), the results of these studies are consistent with an anti-estrogenic effect of cigarette smoking. Although smokers and non-smokers may have the same concentrations of estrogens overall, it may be the type rather than the absolute levels of circulating estrogens that is important. Smokers might have a lower concentration of more biologically active estrogens, primarily 16-α-hydroxyestrone (16α-OHE1) (Michnovicz et al., 1986, 1988; Berta et al., 1992; Berstein et al., 2000; Terry et al., 2002b). Estrogen can be metabolized along three pathways, to 16α-OHE1 or to 2-OHE1 or to 4-OHE1. 16α-OHE1 and 4-OHE1 have been observed to increase mammary epithelial cell proliferation rates in experimental studies (Schütze et al., 1993, 1994; IARC, 2007c). In contrast, 2-OHE1 might decrease epithelial cell proliferation rates (Bradlow et al., 1996; Muti et al., 2000). If cigarette smoking increases estradiol 2-hydroxylation, as has been suggested (Michnovicz et al., 1986), thereby increasing the ratio of 2-OHE1:16-α-OHE1, an inverse association between smoking and breast cancer risk might be observed. However, only one study has directly examined 2-hydroxylation in relation to cigarette smoking (Michnovicz et al., 1986). Using injected radiolabelled estradiol, a 50% increased estradiol 2-hydroxylation was found in premenopausal women who smoked at least 15 cigarettes/day compared with non-smokers. Two studies of urinary estrogens found increased excretion of 2-OHE1 and decreased excretion of estriol among smokers (Michnovicz et al., 1988; Berstein et al., 2000), which may also support the hypothesis that smoking decreases the formation of active estrogen metabolites along the 16α-hydroxylation pathway. However, the ratio of urinary 2-OHE1:16α-OHE1 was not related to breast cancer risk in the one case–control study that examined the association (Ursin et al., 1999). The 4-hydroxylation of estrogens is catalysed by CYP1B1, which is induced by tobacco smoke (Nebert et al., 2004). This has been postulated as an additional pathway that could lead to formation of DNA adducts via catechol estrogen-quinones (Gaikwad et al., 2008) and oxidative/DNA damage via redox-cycling (Zhu & Conney, 1998). The ratio of 2-OHE1:4-OHE1 has been studied in relation to breast cancer risk and smoking in one study (Berstein et al., 2000). Smokers carrying the CYP1B1 Val allele [associated with high hydroxylation activity] had a significantly higher risk for breast cancer compared to never smokers with the Leu/Leu [wildtype] genotype (Saintot et al., 2003).
(b) Endometrium
Exogenous estrogens unopposed by progesterone have been shown to increase the risk for endometrial cancer through increased mitotic activity of endometrial cells, increased number of DNA replication errors, and somatic mutations resulting in the malignant phenotype (IARC, 2007c, 2012c). Hence, factors associated with estrogen absorption or metabolism may alter the risk of this malignancy. Several investigators have hypothesized that cigarette smoking might be have anti-estrogenic effects, and through this mechanism reduce the risk of endometrial cancer (Baron, 1984; Baron et al., 1990; Terry et al., 2002b, 2004a).
Whether mediated through changes in the amount of adipose tissue, altered age at menopause, or anti-estrogenic effects, blood hormone concentrations might be an important link between smoking and the reduced risk of endometrial cancer observed in most epidemiological studies. The estrogens that have typically been studied in relation to cigarette smoking include estrone, sex hormone binding globulin (SHBG)-bound estradiol, and estriol. Blood concentrations of androgens, typically androstenedione and dehydroepiandrosterone sulfate (DHEAS), have also been studied, because these are biological precursors of estrone. Studies that have examined blood concentrations of SHBG are less common, and studies of unbound (free) estradiol are scarce.
Studies of cigarette smoking and blood hormone concentrations have been conducted mostly among post-menopausal women who were not taking HRT. Of these studies, nine examined serum (Friedman et al., 1987; Cauley et al., 1989; Slemenda et al., 1989; Schlemmer et al., 1990; Cassidenti et al., 1992; Austin et al., 1993; Law et al., 1997) or plasma (Khaw et al., 1988; Longcope & Johnston, 1988) estrone, ten examined serum (Friedman et al., 1987; Cauley et al., 1989; Slemenda et al., 1989; Schlemmer et al., 1990; Key et al., 1991; Cassidenti et al., 1992; Austin et al., 1993; Law et al., 1997) or plasma (Khaw et al., 1988; Longcope & Johnston, 1988) estradiol, and two examined serum (Cassidenti et al., 1992) or plasma (Longcope & Johnston, 1988) free estradiol. These studies consistently showed little or no association between smoking and blood estrogen concentrations among post-menopausal women who were not taking hormone replacement therapy. Among pre-menopausal women, three studies (Longcope & Johnston, 1988; Key et al., 1991; Berta et al., 1992) found no clear association between cigarette smoking and estrogen concentrations. Studies that adjusted hormone measurements for the effects of BMI (and other covariates) showed similar results to those that did not, suggesting that BMI is not a strong confounder of this association.
In two studies the association between cigarette smoking and blood estrogen concentrations after randomization of women to groups receiving either estradiol or placebo were examined (Jensen & Christiansen, 1988; Cassidenti et al., 1990). In a small study of 25 post-menopausal women, unbound estradiol was significantly lower among smokers than non-smokers both at baseline and shortly after taking micronized estradiol orally (Cassidenti et al., 1990). No important differences were observed between smokers and non-smokers in serum concentrations of either estrone or bound estradiol. In contrast, in a study in which 110 post-menopausal women were randomized to take hormones (either orally or percutaneously) or a placebo (Jensen & Christiansen, 1988), smokers had lower concentrations of both estrone and bound estradiol than non-smokers after oral (but not percutaneous) hormone treatment for at least one year (concentrations of free estrogens were not examined). These results indicate that smoking might affect the absorption or metabolism of hormones used in replacement therapy.
Of the five studies that have examined the association between cigarette smoking and serum (Lapidus et al., 1986; Cassidenti et al., 1992; Law et al., 1997) or plasma (Khaw et al., 1988; Longcope & Johnston, 1988) SHBG, none found any clear association. However, one of these studies (Khaw et al., 1988) found an inverse association between smoking and the ratio of bound estradiol to SHBG, a measure of estrogen activity. In this context, Cassidenti et al. (1990) found unbound (but not SHBG-bound) estradiol was significantly lower among smokers than non-smokers both at baseline and after taking oral estradiol, suggesting an increased SHBG-binding capacity in the women who smoked.
In post-menopausal women, androgens are the major source of estrone, converted through aromatization in fat deposits. Thus, adiposity is positively correlated with estrogen concentrations in post-menopausal women. Of the nine studies in which blood concentrations of androstenedione were examined in smokers (Friedman et al., 1987; Khaw et al., 1988; Longcope & Johnston, 1988; Cauley et al., 1989; Slemenda et al., 1989; Schlemmer et al., 1990; Cassidenti et al., 1992; Austin et al., 1993; Law et al., 1997), higher circulating concentrations were found among current than among never or former smokers in all studies. However, there was no clear variation in blood estrone concentrations by smoking status, suggesting a reduced conversion of androstenedione to estrone among smokers. Of the five studies where cigarette smoking and DHEAS concentrations were examined, three (Khaw et al., 1988; Cassidenti et al., 1992; Law et al., 1997) found increased blood concentrations among current smokers, one (Friedman et al., 1987) found also an increase that was not statistically significant, whereas another (Key et al., 1991) found no clear differences according to smoking status.
Cigarette smoking and urinary estrogen concentrations have been examined in seven studies (MacMahon et al., 1982; Michnovicz et al., 1986; Trichopoulos et al., 1987; Michnovicz et al., 1988; Berta et al., 1992; Key et al., 1996; Berstein et al., 2000). Of these, three found no major differences according to smoking status (Trichopoulos et al., 1987; Michnovicz et al., 1988; Berta et al., 1992). The remaining four studies all showed lower urinary estriol concentrations among smokers than among non-smokers, but mixed results for urinary estrone and estradiol. Two of these studies (Michnovicz et al., 1988; Berstein et al., 2000) showed higher concentrations of 2-hydroxyestrone among smokers, than non-smokers but only after estrogen treatment in Berstein et al. (2000).
Age at natural menopause varies substantially under the influence of genetic and environmental factors (McKinlay, 1996). A relatively early age at menopause has been associated with reduced risk of endometrial cancer (Kelsey et al., 1982; Baron, 1984; Baron et al., 1990; Akhmedkhanov et al., 2001). A one year decrease in age at menopause has been associated approximately with a 7% decrease in risk (Kelsey et al., 1982). It has been proposed that cigarette smoking decreases the age at natural menopause (Baron et al., 1990), more clearly with qualitative than quantitative smoking measures (Parente et al., 2008), and thus might reduce endometrial cancer risk through reduced exposure to endogenous estrogens. On average, smokers have menopause approximately 1 to 1.5 years earlier than non-smokers (Terry et al., 2002b, 2004a). Adjustment for obesity and other covariates did not alter the results (Terry et al., 2002b).
4.4. Mechanistic considerations of the interaction of ethanol and tobacco carcinogens
The combined effects of alcoholic beverages and tobacco on the risk for cancer incidence and mortality have been widely studied in human populations. When tested for multiplicative and additive interactions, synergistic effects of alcoholic beverages and tobacco have been found, especially for oropharyngeal and oesophageal cancers (Homann et al., 2000; Castellsagué et al., 2004; Salaspuro & Salaspuro, 2004; Lee et al., 2005a; Lee et al., 2007b).
Data support at least four possible mechanisms for the modifying effects of alcoholic beverages on cancer risk due to tobacco.
- Alcohol may have a local permeabilizing effect on penetration of the oral mucosa by tobacco carcinogens (Du et al., 2000), particularly important in the case of oropharyngeal and oesophageal cancer.
- CYP2E1 and other CYPs may both activate and detoxify carcinogens present in tobacco smoke, including NDMA, NDEA, NNK, benzene and other tobacco-derived carcinogens in two ways: CYP induction increases metabolic activation of tobacco carcinogens leading to enhanced formation of proximate reactive chemical species at target sites; and alteration of phase II conjugation/detoxification enzymes by ethanol may also occur, changing the effective dose at the target site.
- Competitive inhibition of CYP metabolism leads to reduced central hepatic and gastrointestinal clearance thus increasing dose delivery of carcinogens to peripheral target tissues (reviewed in Meskar et al., 2001).
- Effects of acetaldehyde derived by microbial alcohol oxidation and from the tobacco smoke (Homann et al., 2000; Salaspuro & Salaspuro, 2004).
Supportive evidence for ii) and iii) is briefly presented below.
4.4.1 Effects of induction of CYPs by ethanol
(a) CYP2E1
Ethanol induces CYP2E1 in the human liver and in all species tested. Over 70 substrates of CYP2E1 have been compiled (Raucy & Carpenter, 1993; Guengerich et al., 1994; Djordjević et al., 1998; Klotz & Ammon, 1998; Cederbaum, 2006). Among those are tobacco carcinogens such as benzene, vinyl chloride, NDMA, NDEA and N-nitrosopyrrolidine, as well as many low-molecular-weight compounds. Induction of CYP2E1 by ethanol generated increased levels of toxic metabolites from the metabolism of many of these chemicals (Novak & Woodcroft, 2000). Pyridine, a constituent of tobacco smoke and substrate of CYP2E1, generates DNA damaging products by redox-cycling (Kim & Novak, 1990).
In humans, in addition to the prominent CYP2E1 expression in the centrilobular regions of the liver, the enzyme is also detectable in the kidney cortex and, at lower levels, in organs such as the oropharynx, nasal mucosa, ovary, testis, small intestine, colon and pancreas (Ingelman-Sundberg et al., 1994; Lieber, 1999, 2004).
In rats, ethanol induced CYP2E1 in epithelia of the cheek, tongue and oesophagus (Shimizu et al., 1990). As a result of CYP2E1 induction by ethanol in the upper respiratory tract and possibly of inhibition of carcinogen clearance, hamsters had a significant increase of nasal cavity and tracheal tumours after intraperitoneal injection of N-nitrosopyrrolidine (McCoy et al., 1981). Thus, induction of CYP2E1 by ethanol may participate in the genesis of cancers at several sites via metabolic activation of tobacco carcinogens into reactive species in target tissues.
(b) Other xenobiotic-activating CYPs
In addition to CYP2E1, several CYPs, including CYP3A4 and probably CYP1A2 in humans, and CYP1A1, 2B1 and 3A in rat liver, may be induced by ethanol. Of particular interest are members of the CYP3A family, which have wide substrate specificity and have been implicated in the activation of several known or suspected human carcinogens, including those derived from tobacco (Wojnowski & Kamdem, 2006). Both CYP3A4 and CYP1A2 metabolize NNK (Jalas et al., 2005). Based on the Michaelis constant (Km) data (IARC, 2007a), the relative efficiencies in NNK metabolism by human CYP are (from greatest catalyst to least): 2A13 > 2B6 > 2A6 > 1A2 ~1A1 > 2D6 ~2E1 ~3A4. As the amount of CYP enzymes with overlapping substrate specificity that participate in nitrosamine metabolism varies according to organ and species, it is difficult to determine their individual contribution at target sites.
4.4.2 Effects of inhibition of CYPs by ethanol
Ethanol is a competitive inhibitor of CYP2E1 (reviewed in Anderson, 1992). It also inhibits the activities of CYP1A1, 2B6 and 2C19 but not those of CYP1A2.
Direct inhibition of CYPs by ethanol in target tissues may reduce metabolic activation of xenobiotics and hence local toxic and tumorigenic effects. Thus CYP inhibition in the liver could increase extrahepatic exposure to genotoxic metabolites from tobacco carcinogens that are substrates for these CYP enzymes. This mechanism is supported by several studies.
Ethanol caused a fivefold increase in oesophageal DNA adducts in rats induced by NDEA (Swann, 1984). In monkeys, O6-methylguanine-DNA adducts after an oral dose of NDMA with or without ethanol were increased by co-exposure to ethanol in all tissues except the liver (Anderson et al., 1996). Effects were seen in the oesophagus (17-fold increase), colonic mucosa (12-fold), pancreas (sixfold), urinary bladder (11-fold), ovary (ninefold), uterus (eightfold), brain (ninefold), spleen (13-fold) and nasal mucosa (fivefold). In these studies, ethanol treatment was acute, so that enzyme induction was improbable, and the oesophagus was not directly exposed to either ethanol or carcinogen. This indicates that a systemic interaction, most likely inhibition of hepatic carcinogen clearance, was responsible for the observed effects in the oesophagus and other extrahepatic tissues. The 17-fold increase in DNA adducts in the monkey oesophagus is similar to the 18-fold increased risk for human oesophageal cancers in tobacco smokers combined with heavy alcohol drinking (Tuyns et al., 1977).
The relevance of increased genotoxic effects in extrahepatic target sites by ethanol is confirmed by many rodent experiments. Oral dosing of mice with NDMA in ethanol resulted in nasal cavity tumours (olfactory neuroblastoma) that were not seen with NDMA or ethanol alone (Griciute et al., 1981). Ethanol in the drinking-water led to a ninefold increase in oesophageal tumours in rats induced by NDEA (Aze et al., 1993). Ethanol given by gavage to nursing dams together with NDMA or NNK (Chhabra et al., 2000) increased O6-methylguanine-DNA adducts in maternal mammary glands, by 10-fold with NDMA and to a lesser extent with NNK. In the suckling infants, DNA adducts were detected in the lungs and kidneys after maternal exposure to NDMA and increased about fourfold after maternal co-treatment with ethanol. In mice, ethanol given with NDMA in the drinking-water resulted in a fourfold increase in lung tumours, but had no significant effect when NDMA was given intragastrically, intraperitoneally, subcutaneously or intravenously (Anderson, 1992). These negative findings support that direct inhibition of hepatic carcinogen clearance by ethanol is the main operative mechanism.
There is indirect evidence that ethanol can inhibit the in vivo clearance of the carcinogen NDMA in humans: individuals with chronic renal failure showed detectable blood and urine levels of NDMA, which were increased by consumption of ethanol (Dunn et al., 1990). Other studies that involved sources of NDMA from tobacco smoke, diet or pharmaceuticals are consistent with ethanol reducing its clearance rate in humans (Anderson, 1992).
Other possible modifying effects of ethanol in tobacco-related tumorigenesis are presented in Section 4 of the Monograph on Consumption of Alcoholic Beverages in this Volume.
4.5. Synthesis
4.5.1. Mechanisms of tobacco-related carcinogenesis
The pathways by which tobacco products cause cancer essentially recapitulate established mechanisms of carcinogenesis by individual compounds, which were elaborated by landmark studies during the second half of the 20th century. These studies demonstrate that most carcinogens, either directly or after metabolism catalyzed by multiple cytochrome P450 enzymes, react with nucleophilic sites in DNA to form covalent binding products called adducts (a contraction for “addition products”). These DNA adducts, if left unrepaired by cellular DNA repair enzymes, persist and cause mistakes during DNA replication leading to incorporation of the wrong base in a DNA strand and consequent permanent mutations. If these permanent mutations occur in important regions of critical growth control genes such as the oncogene KRAS or the tumor suppressor gene p53, cellular growth processes can become severely unregulated and cancer can result. Multiple studies of mutations in KRAS, p53, and other growth control genes in lung tumours from smokers, some of which report thousands of mutations, are fully consistent with this overall concept.
It is the complexity of tobacco carcinogenesis which challenges investigators to identify specific mechanisms that fully explain the ways in which tobacco products cause each type of cancer. There are over 70 established carcinogens in cigarette smoke, and analyses of smokers’ urine and blood clearly demonstrate higher uptake of these compounds in smokers than in non-smokers. The urine of smokers is consistently mutagenic. Similar considerations apply to smokeless tobacco users, although there are fewer identified carcinogens. Multiple DNA adducts are present in the lungs and other tissues of smokers, and sister chromatid exchanges as well as other genetic effects are consistently observed. But much less is known about the specifics of the process. Only relatively few DNA adducts in smokers’ lungs have been structurally characterized and the relationship between specific adducts and the consequent mutations in critical genes is still somewhat unsettled.
There are other processes which contribute to cancer induction by tobacco products, based on multiple studies in both laboratory animals and humans. These include inflammation, tumor promotion, oxidative damage, co-carcinogenesis, and direct activation of cellular growth pathways by constituents of smoke. Many studies demonstrate the involvement of these processes in tobacco carcinogenesis but the details by which they interact with the DNA damage pathways and their roles in specific cancers caused by tobacco products are still not fully understood.
4.5.2 Genetic polymorphisms
Multiple studies have been carried out on the role of genetic polymorphisms of xenobiotic metabolism in smoking-related carcinogenesis in humans. These studies have covered various cancer types, with lung cancer representing one of the most intensively studied. The polymorphic genes, their variant forms, and the genotype combinations investigated in these studies have similarly been numerous. In addition to the associations with increased risk of cancer, much data have accumulated on relationships between the polymorphisms and the various biomarkers of tobacco carcinogenesis in non-cancer control populations, whether smokers or non-smokers, in subjects with work-related exposure or in patients with other cancers.
Despite the massive body of research, many observations remain ambiguous. Some associations between genetic polymorphism and increased risk for cancer, such as for the GSTM1 null genotype, alone or in combination with CYP1A1 polymorphism, in lung cancer, or the NAT2 slow acetylator genotype in bladder cancer and breast cancer appear stronger and more consistent, but not without controversies. Similarly, the data on the various biomarkers of tobacco-related carcinogenesis exhibit inconsistencies.
The variability in the data is at least partially likely due to differences between the studies in the genes and gene variants included (many of which are still of unknown functional or regulatory consequence), in the types of cancer studied, in levels and sources of exposure, in ethnic backgrounds, in sex, in histological types and in the features of the genome such as haplotype blocks and copy number variation resulting in linkage disequilibrium. In addition, gene-gene interactions and gene-environment interactions are likely to contribute to the discrepancies in current data. Mechanisms of tobacco-related carcinogenesis also involve genes from numerous other classes, such as those encoding for DNA repair proteins and many other regulators of cell cycle and growth. In addition; there are well described mechanistic pathways of carcinogenesis mediated via epigenetic alterations and genetic instability, to mention a few.
4.5.3. Site-specific mechanisms
The Working Group reviewed the mechanistic evidence relative to specific target sites for which there is sufficient evidence of carcinogenicity in humans, i.e. lung, oral cavity, oesophagus, larynx and nasopharynx, pancreas, stomach, liver, urinary bladder, leukaemia, cervix and ovary. Genotoxic effects have been found in eight organ sites at which tobacco smoke causes cancer in humans (DeMarini, 2004).
Sites with limited evidence of carcinogenicity or evidence suggesting lack of carcinogenicity in humans include the breast and the endothelium and relevant mechanisms are presented below.
Breast — There are several plausible mechanisms by which smoking may increase breast cancer risk, and some data support such an effect, including the increased risk among long-term smokers with NAT2 slow genotype. Despite the overall lack of clear association in epidemiological studies, and the potential anti-estrogenic effects of smoking, the possibility that smoking increases breast cancer risk is biologically plausible.
Endothelium — The mechanisms by which cigarette smoking reduces the risk for endometrial cancer among current smokers, mainly among postmenopausal, remain unclear.
4.5.4 Interaction of ethanol and tobacco carcinogens
Data in rodents and non-human primates on the relationships between a) inhibition of hepatic clearance of nitrosamines by ethanol, b) the formation of promutagenic DNA adducts and c) tumours in extra-hepatic targets, likely also pertain in humans.
5. Evaluation
There is sufficient evidence in humans for the carcinogenicity of tobacco smoking.
Tobacco smoking causes cancers of the lung, oral cavity, naso-, oro- and hypopharynx, nasal cavity and accesory sinuses, larynx, oesophagus, stomach, pancreas, colorectum, liver, kidney (body and pelvis), ureter, urinary bladder, uterine cervix and ovary (mucinous), and myeloid leukaemia. Also, a positive association has been observed between tobacco smoking and cancer of the female breast. For cancers of the endometrium (post-menopausal) and of the thyroid, there is evidence suggesting lack of carcinogenicity.
There is sufficient evidence in humans for the carcinogenicity of parental smoking. Parental smoking causes hepatoblastoma in children. Also, a positive association has been observed between parental smoking and childhood leukaemia (particularly acute lymphocytic leukaemia).
There is sufficient evidence in experimental animals for the carcinogenicity of tobacco smoke and of tobacco smoke condensates.
Tobacco smoking is carcinogenic to humans (Group 1).
References
- Abdel-Rahman SZ, Ammenheuser MM, Ward JB Jr. Human sensitivity to 1,3-butadiene: role of microsomal epoxide hydrolase polymorphisms. Carcinogenesis. 2001;22:415–423. [PubMed: 11238181] [CrossRef]
- Abdel-Rahman SZ, El-Zein RA, Ammenheuser MM, et al. Variability in human sensitivity to 1,3-butadiene: Influence of the allelic variants of the microsomal epoxide hydrolase gene. Environ Mol Mutagen. 2003;41:140–146. [PubMed: 12605384] [CrossRef]
- Abou-Alfa GK. Hepatocellular carcinoma: molecular biology and therapy. Semin Oncol. 2006;33 Suppl 11:S79–S83. [PMC free article: PMC1861815] [PubMed: 17178294] [CrossRef]
- Abrams JA, Terry MB, Neugut AI. Cigarette smoking and the colorectal adenoma-carcinoma sequence. Gastroenterology. 2008;134:617–619. [PubMed: 18242224] [CrossRef]
- Adami HO, Lund E, Bergström R, Meirik O. Cigarette smoking, alcohol consumption and risk of breast cancer in young women. Br J Cancer. 1988;58:832–837. [PMC free article: PMC2246867] [PubMed: 3224085]
- Adami J, Nyrén O, Bergström R, et al. Smoking and the risk of leukemia, lymphoma, and multiple myeloma (Sweden). Cancer Causes Control. 1998;9:49–56. [PubMed: 9486463] [CrossRef]
- Agudo A, Sala N, Pera G, et al. Polymorphisms in metabolic genes related to tobacco smoke and the risk of gastric cancer in the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev. 2006;15:2427–2434. [PubMed: 17164366] [CrossRef]
- Agundez JA. Cytochrome P450 gene polymorphism and cancer. Curr Drug Metab. 2004;5:211–224. [PubMed: 15180491] [CrossRef]
- Ahern TP, Lash TL, Egan KM, Baron JA. Lifetime tobacco smoke exposure and breast cancer incidence. Cancer Causes Control. 2009;20:1837–1844. [PubMed: 19533391] [CrossRef]
- Ahrendt SA, Decker PA, Alawi EA, et al. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92:1525–1530. [PubMed: 11745231] [CrossRef]
- Ahrens W, Jöckel KH, Patzak W, Elsner G. Alcohol, smoking, and occupational factors in cancer of the larynx: a case-control study. Am J Ind Med. 1991;20:477–493. [PubMed: 1785612] [CrossRef]
- Ahrens W, Timmer A, Vyberg M, et al. Risk factors for extrahepatic biliary tract carcinoma in men: medical conditions and lifestyle: results from a European multicentre case-control study. Eur J Gastroenterol Hepatol. 2007;19:623–630. [PubMed: 17625430] [CrossRef]
- Akhmedkhanov A, Zeleniuch-Jacquotte A, Toniolo P. Role of exogenous and endogenous hormones in endometrial cancer: review of the evidence and research perspectives. Ann N Y Acad Sci. 2001;943:296–315. [PubMed: 11594550]
- Akhter M, Nishino Y, Nakaya N, et al. Cigarette smoking and the risk of colorectal cancer among men: a prospective study in Japan. Eur J Cancer Prev. 2007;16:102–107. [PubMed: 17297385] [CrossRef]
- Akiba S. Analysis of cancer risk related to longitudinal information on smoking habits. Environ Health Perspect. 1994;102 Suppl 8:15–19. [PMC free article: PMC1566541] [PubMed: 7851325]
- Akiba S, Hirayama T. Cigarette smoking and cancer mortality risk in Japanese men and women–results from reanalysis of the six-prefecture cohort study data. Environ Health Perspect. 1990;87:19–26. [PMC free article: PMC1567850] [PubMed: 2269225] [CrossRef]
- Aksoy M. Hematotoxicity and carcinogenicity of benzene. Environ Health Perspect. 1989;82:193–197. [PMC free article: PMC1568112] [PubMed: 2676498] [CrossRef]
- Al-Delaimy WK. Hair as a biomarker for exposure to tobacco smoke. Tob Control. 2002;11:176–182. [PMC free article: PMC1759008] [PubMed: 12198265] [CrossRef]
- Al-Delaimy WK, Cho E, Chen WY, et al. A prospective study of smoking and risk of breast cancer in young adult women. Cancer Epidemiol Biomarkers Prev. 2004;13:398–404. [PubMed: 15006915]
- Al-Delaimy WK, Willett WC. Measurement of tobacco smoke exposure: comparison of toenail nicotine biomarkers and self-reports. Cancer Epidemiol Biomarkers Prev. 2008;17:1255–1261. [PubMed: 18483348] [CrossRef]
- Al-Zoughool M, Dossus L, Kaaks R, et al. Risk of endometrial cancer in relationship to cigarette smoking: results from the EPIC study. Int J Cancer. 2007;121:2741–2747. [PubMed: 17657712] [CrossRef]
- Alberg AJ, Daudt A, Huang HY, et al. N-acetyltransferase 2 (NAT2) genotypes, cigarette smoking, and the risk of breast cancer. Cancer Detect Prev. 2004;28:187–193. [PubMed: 15225898] [CrossRef]
- Alberg AJ, Kouzis A, Genkinger JM, et al. A prospective cohort study of bladder cancer risk in relation to active cigarette smoking and household exposure to secondhand cigarette smoke. Am J Epidemiol. 2007;165:660–666. [PubMed: 17204516] [CrossRef]
- Alexandrie AK, Warholm M, Carstensen U, et al. CYP1A1 and GSTM1 polymorphisms affect urinary 1-hydroxypyrene levels after PAH exposure. Carcinogenesis. 2000;21:669–676. [PubMed: 10753202] [CrossRef]
- Alexandrov K, Cascorbi I, Rojas M, et al. CYP1A1 and GSTM1 genotypes affect benzo[a]pyrene DNA adducts in smokers’ lung: comparison with aromatic/hydrophobic adduct formation. Carcinogenesis. 2002;23:1969–1977. [PubMed: 12507920] [CrossRef]
- Alexandrov K, Rojas M, Kadlubar FF, et al. Evidence of anti-benzo[a]pyrene diolepoxide-DNA adduct formation in human colon mucosa. Carcinogenesis. 1996;17:2081–2083. [PubMed: 8824539] [CrossRef]
- Alfano CM, Klesges RC, Murray DM, et al. Physical activity in relation to all-site and lung cancer incidence and mortality in current and former smokers. Cancer Epidemiology, Biomarkers & Prevention. 2004;13:2233–2241. [PubMed: 15598785]
- Alguacil J, Silverman DT. Smokeless and other noncigarette tobacco use and pancreatic cancer: a case-control study based on direct interviews. Cancer Epidemiol Biomarkers Prev. 2004;13:55–58. [PubMed: 14744733] [CrossRef]
- Ambrosone CB. Impact of genetics on the relationship between smoking and breast cancer risk. Journal of Women's Cancer. 2001;3:17–22.
- Ambrosone CB, Freudenheim JL, Graham S, et al. Cigarette smoking, N-acetyltransferase 2 genetic polymorphisms, and breast cancer risk. JAMA. 1996;276:1494–1501. [PubMed: 8903261] [CrossRef]
- Ambrosone CB, Kropp S, Yang J, et al. Cigarette smoking, N-acetyltransferase 2 genotypes, and breast cancer risk: pooled analysis and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2008;17:15–26. [PubMed: 18187392] [CrossRef]
- Ambrosone CB, Shields PG (1999). Smoking as a risk factor for breast cancer. In: Breast cancer: molecular genetics, pathogenesis, and therapeutics. Bowcock AM, editor. Tetowa, NJ: Humana Press, pp. 519–536,.
- Amos CI, Wu X, Broderick P, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. [PMC free article: PMC2713680] [PubMed: 18385676] [CrossRef]
- Andersen V, Agerstjerne L, Jensen D, et al. The multidrug resistance 1 (MDR1) gene polymorphism G-rs3789243-A is not associated with disease susceptibility in Norwegian patients with colorectal adenoma and colorectal cancer; a case control study. BMC Med Genet. 2009;10:18. [PMC free article: PMC2662819] [PubMed: 19250544] [CrossRef]
- Anderson JC, Attam R, Alpern Z, et al. Prevalence of colorectal neoplasia in smokers. Am J Gastroenterol. 2003;98:2777–2783. [PubMed: 14687832] [CrossRef]
- Anderson LM (1992). Modulation of nitrosamine metabolism by ethanol: implications for cancer risk. In: Alcohol and Cancer. Watson RR, editor. Boca Raton, FL: CRC Press, pp. 17–54.
- Anderson LM, Souliotis VL, Chhabra SK, et al. N-nitrosodimethylamine-derived O6-methylguanine in DNA of monkey gastrointestinal and urogenital organs and enhancement by ethanol. Int J Cancer. 1996;66:130–134. [PubMed: 8608956] [CrossRef]
- Ansary-Moghaddam A, Huxley R, Barzi F, et al. Asia Pacific Cohort Studies Collaboration. The effect of modifiable risk factors on pancreatic cancer mortality in populations of the Asia-Pacific region. Cancer Epidemiol Biomarkers Prev. 2006;15:2435–2440. [PubMed: 17164367] [CrossRef]
- Applebaum KM, Furniss CS, Zeka A, et al. Lack of association of alcohol and tobacco with HPV16-associated head and neck cancer. J Natl Cancer Inst. 2007;99:1801–1810. [PubMed: 18042931] [CrossRef]
- Appleby P, Beral V, Berrington de González A, et al. International Collaboration of Epidemiological Studies of Cervical Cancer. Carcinoma of the cervix and tobacco smoking: collaborative reanalysis of individual data on 13,541 women with carcinoma of the cervix and 23,017 women without carcinoma of the cervix from 23 epidemiological studies. Int J Cancer. 2006;118:1481–1495. [PubMed: 16206285] [CrossRef]
- Arif JM, Dresler C, Clapper ML, et al. Lung DNA adducts detected in human smokers are unrelated to typical polyaromatic carcinogens. Chem Res Toxicol. 2006;19:295–299. [PubMed: 16485906] [CrossRef]
- Armstrong RW, Imrey PB, Lye MS, et al. Nasopharyngeal carcinoma in Malaysian Chinese: occupational exposures to particles, formaldehyde and heat. Int J Epidemiol. 2000;29:991–998. [PubMed: 11101539] [CrossRef]
- Ashktorab H, Begum R, Akhgar A, et al. Folate status and risk of colorectal polyps in African Americans. Dig Dis Sci. 2007;52:1462–1470. [PubMed: 17372834] [CrossRef]
- Asma S, Bettcher DW, Samet J et al. (2009). Tobacco. In: Oxford Textbook Of Public Health. Detels R, editor. Oxford: Oxford University Press.
- Ateş NA, Tamer L, Ateş C, et al. Glutathione S-transferase M1, T1, P1 genotypes and risk for development of colorectal cancer. Biochem Genet. 2005;43:149–163. [PubMed: 15932063] [CrossRef]
- Austin H, Drews C, Partridge EE. A case-control study of endometrial cancer in relation to cigarette smoking, serum estrogen levels, and alcohol use. Am J Obstet Gynecol. 1993;169:1086–1091. [PubMed: 8238164]
- Autrup H. Genetic polymorphisms in human xenobiotica metabolizing enzymes as susceptibility factors in toxic response. Mutat Res. 2000;464:65–76. [PubMed: 10633178]
- Aze Y, Toyoda K, Furukawa F, et al. Enhancing effect of ethanol on esophageal tumor development in rats by initiation of diethylnitrosamine. Carcinogenesis. 1993;14:37–40. [PubMed: 8425269] [CrossRef]
- Bain C, Feskanich D, Speizer FE, et al. Lung cancer rates in men and women with comparable histories of smoking. J Natl Cancer Inst. 2004;96:826–834. [PubMed: 15173266]
- Baker JA, Odunuga OO, Rodabaugh KJ, et al. Active and passive smoking and risk of ovarian cancer. Int J Gynecol Cancer. 2006;16 Suppl 1:211–218. [PubMed: 16515593] [CrossRef]
- Balansky R, Ganchev G, Iltcheva M, et al. Potent carcinogenicity of cigarette smoke in mice exposed early in life. Carcinogenesis. 2007;28:2236–2243. [PubMed: 17522065] [CrossRef]
- Balaram P, Sridhar H, Rajkumar T, et al. Oral cancer in southern India: the influence of smoking, drinking, paan-chewing and oral hygiene. Int J Cancer. 2002;98:440–445. [PubMed: 11920597] [CrossRef]
- Bamber DE, Fryer AA, Strange RC, et al. Phenol sulphotransferase SULT1A1*1 genotype is associated with reduced risk of colorectal cancer. Pharmacogenetics. 2001;11:679–685. [PubMed: 11692076] [CrossRef]
- Band PR, Le ND, Fang R, Deschamps M. Carcinogenic and endocrine disrupting effects of cigarette smoke and risk of breast cancer. Lancet. 2002;360:1044–1049. [PubMed: 12383984] [CrossRef]
- Baron JA. Smoking and estrogen-related disease. Am J Epidemiol. 1984;119:9–22. [PubMed: 6362403]
- Baron JA, Byers T, Greenberg ER, et al. Cigarette smoking in women with cancers of the breast and reproductive organs. J Natl Cancer Inst. 1986;77:677–680. [PubMed: 3462409]
- Baron JA, La Vecchia C, Levi F. The antiestrogenic effect of cigarette smoking in women. Am J Obstet Gynecol. 1990;162:502–514. [PubMed: 2178432]
- Baron JA, Newcomb PA, Longnecker MP, et al. Cigarette smoking and breast cancer. Cancer Epidemiol Biomarkers Prev. 1996;5:399–403. [PubMed: 9162307]
- Barra S, Barón AE, Franceschi S, et al. Cancer and non-cancer controls in studies on the effect of tobacco and alcohol consumption. Int J Epidemiol. 1991;20:845–851. [PubMed: 1800421] [CrossRef]
- Bartsch H, Nair U, Risch A, et al. Genetic polymorphism of CYP genes, alone or in combination, as a risk modifier of tobacco-related cancers. Cancer Epidemiol Biomarkers Prev. 2000;9:3–28. [PubMed: 10667460]
- Batty GD, Kivimaki M, Gray L, et al. Cigarette smoking and site-specific cancer mortality: testing uncertain associations using extended follow-up of the original Whitehall study. Ann Oncol. 2008;19:996–1002. [PubMed: 18212091] [CrossRef]
- Baumgartner KB, Schlierf TJ, Yang D, et al. N-acetyltransferase 2 genotype modification of active cigarette smoking on breast cancer risk among hispanic and non-hispanic white women. Toxicol Sci. 2009;112:211–220. [PMC free article: PMC2782353] [PubMed: 19692670]
- Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4:707–717. [PubMed: 15343277]
- Belinsky SA. Silencing of genes by promoter hypermethylation: key event in rodent and human lung cancer. Carcinogenesis. 2005;26:1481–1487. [PubMed: 15661809] [CrossRef]
- Belinsky SA, Klinge DM, Dekker JD, et al. Gene promoter methylation in plasma and sputum increases with lung cancer risk. Clin Cancer Res. 2005;11:6505–6511. [PubMed: 16166426]
- Belinsky SA, Liechty KC, Gentry FD, et al. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006;66:3338–3344. [PubMed: 16540689]
- Benhamou S, Lee WJ, Alexandrie AK, et al. Meta- and pooled analyses of the effects of glutathione S-transferase M1 polymorphisms and smoking on lung cancer risk. Carcinogenesis. 2002;23:1343–1350. [PubMed: 12151353] [CrossRef]
- Beral V, Bull D, Reeves G., Million Women Study Collaborators. Endometrial cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2005;365:1543–1551. [PubMed: 15866308] [CrossRef]
- Bergmark E. Hemoglobin adducts of acrylamide and acrylonitrile in laboratory workers, smokers and nonsmokers. Chem Res Toxicol. 1997;10:78–84. [PubMed: 9074806] [CrossRef]
- Bernfeld P, Homburger F, Russfield AB. Strain differences in the response of inbred Syrian hamsters to cigarette smoke inhalation. J Natl Cancer Inst. 1974;53:1141–1157. [PubMed: 4279301]
- Bernfeld P, Homburger F, Soto E, Pai KJ. Cigarette smoke inhalation studies in inbred Syrian golden hamsters. J Natl Cancer Inst. 1979;63:675–689. [PubMed: 288930]
- Berrettini W, Yuan X, Tozzi F, et al. Alpha-5/alpha-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol Psychiatry. 2008;13:368–373. [PMC free article: PMC2507863] [PubMed: 18227835] [CrossRef]
- Berrington de González A, Sweetland S, Green J. Comparison of risk factors for squamous cell and adenocarcinomas of the cervix: a meta-analysis. Br J Cancer. 2004;90:1787–1791. [PMC free article: PMC2409738] [PubMed: 15150591]
- Berstein LM, Tsyrlina EV, Kolesnik OS, et al. Catecholestrogens excretion in smoking and non-smoking postmenopausal women receiving estrogen replacement therapy. J Steroid Biochem Mol Biol. 2000;72:143–147. [PubMed: 10775805] [CrossRef]
- Berta L, Fortunati N, Gennari P, et al. Influence of cigarette smoking on pituitary and sex hormone balance in healthy premenopausal women. Fertil Steril. 1991;56:788–789. [PubMed: 1915961]
- Berta L, Frairia R, Fortunati N, et al. Smoking effects on the hormonal balance of fertile women. Horm Res. 1992;37:45–48. [PubMed: 1398476] [CrossRef]
- Besson H, Brennan P, Becker N, et al. Tobacco smoking, alcohol drinking and Hodgkin's lymphoma: A European multicenter case-control study (Epilymph). Br J Cancer. 2006;95:378–384. [PMC free article: PMC2360649] [PubMed: 16819547]
- Bhutani M, Pathak AK, Fan YH, et al. Oral epithelium as a surrogate tissue for assessing smoking-induced molecular alterations in the lungs. Cancer Prev Res (Phila). 2008;1:39–44. [PMC free article: PMC4183362] [PubMed: 19138934]
- Bin P, Leng S, Cheng J, et al. Association of aryl hydrocarbon receptor gene polymorphisms and urinary 1-hydroxypyrene in polycyclic aromatic hydrocarbon-exposed workers. Cancer Epidemiol Biomarkers Prev. 2008;17:1702–1708. [PubMed: 18628420] [CrossRef]
- Bjelke E. Dietary vitamin A and human lung cancer. Int J Cancer. 1975;15:561–565. [PubMed: 1140863] [CrossRef]
- Bjerregaard BK, Raaschou-Nielsen O, Sørensen M, et al. Tobacco smoke and bladder cancer–in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer. 2006;119:2412–2416. [PubMed: 16894557] [CrossRef]
- Bjørge T, Engeland A, Tretli S, Weiderpass E. Body size in relation to cancer of the uterine corpus in 1 million Norwegian women. Int J Cancer. 2007;120:378–383. [PubMed: 17066451] [CrossRef]
- Bleeker MC, Heideman DA, Snijders PJ, et al. Penile cancer: epidemiology, pathogenesis and prevention. World J Urol. 2009;27:141–150. [PubMed: 18607597] [CrossRef]
- Blot WJ, Devesa SS, Kneller RW, Fraumeni JF Jr. Rising incidence of adenocarcinoma of the esophagus and gastric cardia. JAMA. 1991;265:1287–1289. [PubMed: 1995976] [CrossRef]
- Blot WJ, McLaughlin JK, Winn DM, et al. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988;48:3282–3287. [PubMed: 3365707]
- Boccia S, Sayed-Tabatabaei FA, Persiani R, et al. Polymorphisms in metabolic genes, their combination and interaction with tobacco smoke and alcohol consumption and risk of gastric cancer: a case-control study in an Italian population. BMC Cancer. 2007;7:206. [PMC free article: PMC2194718] [PubMed: 17996038]
- Bode AM, Dong Z. Signal transduction pathways in cancer development and as targets for cancer prevention. Prog Nucleic Acid Res Mol Biol. 2005;79:237–297. [PubMed: 16096030]
- Boffetta P, Clark S, Shen M, et al. Serum cotinine level as predictor of lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2006;15:1184–1188. [PubMed: 16775179] [CrossRef]
- Boffetta P, Hecht SS, Gray N, et al. Smokeless tobacco and cancer. Lancet Oncol. 2008;9:667–675. [PubMed: 18598931] [CrossRef]
- Bogen E. The composition of cigarets and cigaret smoke. Journal of the American Medical Association. 1929;93:1110–1114.
- Bolt HM, Thier R. Relevance of the deletion polymorphisms of the glutathione S-transferases GSTT1 and GSTM1 in pharmacology and toxicology. Curr Drug Metab. 2006;7:613–628. [PubMed: 16918316] [CrossRef]
- Bono R, Vincenti M, Saglia U, et al. Tobacco smoke and formation of N-(2-hydroxyethyl)valine in human hemoglobin. Arch Environ Health. 2002;57:416–421. [PubMed: 12641182] [CrossRef]
- Borgerding M, Klus H. Analysis of complex mixtures–cigarette smoke. Exp Toxicol Pathol. 2005;57 Suppl 1:43–73. [PubMed: 16092717] [CrossRef]
- Borlak J, Reamon-Buettner SM. N-acetyltransferase 2 (NAT2) gene polymorphisms in colon and lung cancer patients. BMC Med Genet. 2006;7:58. [PMC free article: PMC1533812] [PubMed: 16827944] [CrossRef]
- Bosetti C, Gallus S, Peto R, et al. Tobacco smoking, smoking cessation, and cumulative risk of upper aerodigestive tract cancers. Am J Epidemiol. 2008;167:468–473. [PubMed: 18056925] [CrossRef]
- Bosetti C, Garavello W, Gallus S, La Vecchia C. Effects of smoking cessation on the risk of laryngeal cancer: an overview of published studies. Oral Oncol. 2006;42:866–872. [PubMed: 16931120] [CrossRef]
- Bosetti C, Negri E, Franceschi S, et al. Risk factors for oral and pharyngeal cancer in women: a study from Italy and Switzerland. Br J Cancer. 2000;82:204–207. a. [PMC free article: PMC2363177] [PubMed: 10638990]
- Botteri E, Iodice S, Bagnardi V, et al. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300:2765–2778. a. [PubMed: 19088354] [CrossRef]
- Botteri E, Iodice S, Raimondi S, et al. Cigarette smoking and adenomatous polyps: a meta-analysis. Gastroenterology. 2008;134:388–395. b. [PubMed: 18242207] [CrossRef]
- Boyd AS, Shyr Y, King LE Jr. Basal cell carcinoma in young women: an evaluation of the association of tanning bed use and smoking. J Am Acad Dermatol. 2002;46:706–709. [PubMed: 12004311] [CrossRef]
- Boyle P, Maisonneuve P, Bueno de Mesquita B, et al. Cigarette smoking and pancreas cancer: a case control study of the search programme of the IARC. Int J Cancer. 1996;67:63–71. [PubMed: 8690527] [CrossRef]
- Boysen G, Hecht SS. Analysis of DNA and protein adducts of benzo[a]pyrene in human tissues using structure-specific methods. Mutat Res. 2003;543:17–30. [PubMed: 12510015] [CrossRef]
- Bracci PM, Holly EA. Tobacco use and non-Hodgkin lymphoma: results from a population-based case-control study in the San Francisco Bay Area, California. Cancer Causes Control. 2005;16:333–346. [PubMed: 15953976]
- Bradlow HL, Telang NT, Sepkovic DW, Osborne MP. 2-hydroxyestrone: the ‘good’ estrogen. J Endocrinol. 1996;150 Suppl:S259–S265. [PubMed: 8943806]
- Breast Cancer Family Registry. Kathleen Cuningham Consortium for Research into Familial Breast Cancer (Australasia) Ontario Cancer Genetics Network (Canada). Smoking and risk of breast cancer in carriers of mutations in BRCA1 or BRCA2 aged less than 50 years. Breast Cancer Res Treat. 2008;109:67–75. [PMC free article: PMC4093798] [PubMed: 17972172] [CrossRef]
- Bremnes Y, Ursin G, Bjurstam N, Gram IT. Different measures of smoking exposure and mammographic density in postmenopausal Norwegian women: a cross-sectional study. Breast Cancer Res. 2007;9:R73. [PMC free article: PMC2242671] [PubMed: 17963507] [CrossRef]
- Brennan P, Hsu CC, Moullan N, et al. Effect of cruciferous vegetables on lung cancer in patients stratified by genetic status: a mendelian randomisation approach. Lancet. 2005;366:1558–1560. [PubMed: 16257343]
- Brennan P, van der Hel O, Moore LE, et al. Tobacco smoking, body mass index, hypertension, and kidney cancer risk in central and eastern Europe. Br J Cancer. 2008;99:1912–1915. [PMC free article: PMC2600689] [PubMed: 19034282] [CrossRef]
- Brenner H, Arndt V, Bode G, et al. Risk of gastric cancer among smokers infected with Helicobacter pylori. Int J Cancer. 2002;98:446–449. [PubMed: 11920598] [CrossRef]
- Brinton LA, Barrett RJ, Berman ML, et al. Cigarette smoking and the risk of endometrial cancer. Am J Epidemiol. 1993;137:281–291. [PubMed: 8452136]
- Brinton LA, Blot WJ, Becker JA, et al. A case-control study of cancers of the nasal cavity and paranasal sinuses. Am J Epidemiol. 1984;119:896–906. [PubMed: 6731431]
- Brinton LA, Schairer C, Stanford JL, Hoover RN. Cigarette smoking and breast cancer. Am J Epidemiol. 1986;123:614–622. [PubMed: 3953540]
- Brooks DR, Palmer JR, Strom BL, Rosenberg L. Menthol cigarettes and risk of lung cancer. Am J Epidemiol. 2003;158:609–616, discussion 617–620. [PubMed: 14507595] [CrossRef]
- Brown LM, Blot WJ, Schuman SH, et al. Environmental factors and high risk of esophageal cancer among men in coastal South Carolina. J Natl Cancer Inst. 1988;80:1620–1625. [PubMed: 3193480] [CrossRef]
- Brown LM, Hoover R, Silverman D, et al. Excess incidence of squamous cell esophageal cancer among US Black men: role of social class and other risk factors. Am J Epidemiol. 2001;153:114–122. [PubMed: 11159155] [CrossRef]
- Brown LM, Hoover RN, Greenberg RS, et al. Are racial differences in squamous cell esophageal cancer explained by alcohol and tobacco use? J Natl Cancer Inst. 1994;86:1340–1345. a. [PubMed: 8064893] [CrossRef]
- Brown LM, Silverman DT, Pottern LM, et al. Adenocarcinoma of the esophagus and esophagogastric junction in white men in the United States: alcohol, tobacco, and socioeconomic factors. Cancer Causes Control. 1994;5:333–340. b. [PubMed: 8080945] [CrossRef]
- Brugere J, Guenel P, Leclerc A, Rodriguez J. Differential effects of tobacco and alcohol in cancer of the larynx, pharynx, and mouth. Cancer. 1986;57:391–395. [PubMed: 3942973] [CrossRef]
- Brunet JS, Ghadirian P, Rebbeck TR, et al. Effect of smoking on breast cancer in carriers of mutant BRCA1 or BRCA2 genes. J Natl Cancer Inst. 1998;90:761–766. [PubMed: 9605646] [CrossRef]
- Bufalo NE, Leite JL, Guilhen ACT, et al. Smoking and susceptibility to thyroid cancer: an inverse association with CYP1A1 allelic variants. Endocr Relat Cancer. 2006;13:1185–1193. [PubMed: 17158763] [CrossRef]
- Bundgaard T, Wildt J, Frydenberg M, et al. Case-control study of squamous cell cancer of the oral cavity in Denmark. Cancer Causes Control. 1995;6:57–67. [PubMed: 7718736] [CrossRef]
- Burchell B. Genetic variation of human UDP-glucuronosyltransferase: implications in disease and drug glucuronidation. Am J Pharmacogenomics. 2003;3:37–52. [PubMed: 12562215]
- Butler LM, Gold EB, Greendale GA, et al. Menstrual and reproductive factors in relation to mammographic density: the Study of Women’s Health Across the Nation (SWAN). Breast Cancer Res Treat. 2008;112:165–174. [PMC free article: PMC2664291] [PubMed: 18066689] [CrossRef]
- Butler LM, Wang R, Wong AS, et al. Cigarette smoking and risk of prostate cancer among Singapore Chinese. Cancer Causes Control. 2009;20:1967–1974. [PubMed: 19579052]
- Calaf G, Russo J. Transformation of human breast epithelial cells by chemical carcinogens. Carcinogenesis. 1993;14:483–492. [PubMed: 8453725] [CrossRef]
- California Environmental Protection Agency (2005). Health effects assessment for ETS: final. Sacramento, CA: California Environmental Protection Agency, Office of Environmental Health Hazard Assesment.
- Calle EE, Miracle-McMahill HL, Thun MJ, Heath CW Jr. Cigarette smoking and risk of fatal breast cancer. Am J Epidemiol. 1994;139:1001–1007. [PubMed: 8178779]
- Campbell PT, Curtin K, Ulrich CM, et al. Mismatch repair polymorphisms and risk of colon cancer, tumour microsatellite instability and interactions with lifestyle factors. Gut. 2009;58:661–667. [PMC free article: PMC2903215] [PubMed: 18523027] [CrossRef]
- Campos F, Carrasquilla G, Koriyama C, et al. Risk factors of gastric cancer specific for tumor location and histology in Cali, Colombia. World J Gastroenterol. 2006;12:5772–5779. [PMC free article: PMC4100656] [PubMed: 17007041]
- Canova C, Hashibe M, Simonato L, et al. Genetic associations of 115 polymorphisms with cancers of the upper aerodigestive tract across 10 European countries: the ARCAGE project. Cancer Res. 2009;69:2956–2965. [PubMed: 19339270] [CrossRef]
- Cao SM, Liu Q, Huang TB. Analysis for risk factors of nasopharyngeal carcinoma in Sihui city. Cancer. 2000;19:987–989. [chinese]
- Cao W, Cai L, Rao JY, et al. Tobacco smoking, GSTP1 polymorphism, and bladder carcinoma. Cancer. 2005;104:2400–2408. [PubMed: 16240451] [CrossRef]
- Caplan LS, Hall HI, Levine RS, Zhu K. Preventable risk factors for nasal cancer. Ann Epidemiol. 2000;10:186–191. [PubMed: 10813512] [CrossRef]
- Cardoso MA, Maruta LM, Kida AA, et al. Micronutrients and the risk of colorectal adenomas: a case-control study in São Paulo, Brazil. IARC Sci Publ. 2002;156:361–363. [PubMed: 12484206]
- Carlsten C, Sagoo GS, Frodsham AJ, et al. Glutathione S-transferase M1 (GSTM1) polymorphisms and lung cancer: a literature-based systematic HuGE review and meta-analysis. Am J Epidemiol. 2008;167:759–774. [PubMed: 18270371] [CrossRef]
- Carmella SG, Chen M, Han S, et al. Effects of smoking cessation on eight urinary tobacco carcinogen and toxicant biomarkers. Chem Res Toxicol. 2009;22:734–741. [PMC free article: PMC2704054] [PubMed: 19317515] [CrossRef]
- Carmella SG, Chen M, Villalta PW, et al. Ethylation and methylation of hemoglobin in smokers and non-smokers. Carcinogenesis. 2002;23:1903–1910. [PubMed: 12419839] [CrossRef]
- Carpenter CL, Jarvik ME, Morgenstern H, et al. Mentholated cigarette smoking and lung-cancer risk. Ann Epidemiol. 1999;9:114–120. [PubMed: 10037555] [CrossRef]
- Cascorbi I, Brockmöller J, Roots I. A C4887A polymorphism in exon 7 of human CYP1A1: population frequency, mutation linkages, and impact on lung cancer susceptibility. Cancer Res. 1996;56:4965–4969. [PubMed: 8895751]
- Cassidenti DL, Pike MC, Vijod AG, et al. A reevaluation of estrogen status in postmenopausal women who smoke. Am J Obstet Gynecol. 1992;166:1444–1448. [PubMed: 1534446]
- Cassidenti DL, Vijod AG, Vijod MA, et al. Short-term effects of smoking on the pharmacokinetic profiles of micronized estradiol in postmenopausal women. Am J Obstet Gynecol. 1990;163:1953–1960. [PubMed: 2256508]
- Castellsagué X, Muñoz N, De Stefani E, et al. Independent and joint effects of tobacco smoking and alcohol drinking on the risk of esophageal cancer in men and women. Int J Cancer. 1999;82:657–664. [PubMed: 10417762] [CrossRef]
- Castellsagué X, Quintana MJ, Martínez MC, et al. The role of type of tobacco and type of alcoholic beverage in oral carcinogenesis. Int J Cancer. 2004;108:741–749. [PubMed: 14696101] [CrossRef]
- Cauley JA, Gutai JP, Kuller LH, et al. The epidemiology of serum sex hormones in postmenopausal women. Am J Epidemiol. 1989;129:1120–1131. [PubMed: 2729251]
- Cavalieri E, Frenkel K, Liehr JG, et al. Estrogens as endogenous genotoxic agents–DNA adducts and mutations. J Natl Cancer Inst Monogr. 2000;(27):75–93. [PubMed: 10963621]
- Cederbaum AI. CYP2E1–biochemical and toxicological aspects and role in alcohol-induced liver injury. Mt Sinai J Med. 2006;73:657–672. [PubMed: 16878272]
- Chameaud J, Perraud R, Chrétien J, et al. Lung carcinogenesis during in vivo cigarette smoking and radon daughter exposure in rats. Recent Results Cancer Res. 1982;82:11–20. [PubMed: 7111833]
- Chan AO, Jim MH, Lam KF, et al. Prevalence of colorectal neoplasm among patients with newly diagnosed coronary artery disease. JAMA. 2007;298:1412–1419. [PubMed: 17895457]
- Chang-Claude J, Kropp S, Jäger B, et al. Differential effect of NAT2 on the association between active and passive smoke exposure and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2002;11:698–704. [PubMed: 12163321]
- Chao A, Thun MJ, Jacobs EJ, et al. Cigarette smoking and colorectal cancer mortality in the cancer prevention study II. J Natl Cancer Inst. 2000;92:1888–1896. [PubMed: 11106680] [CrossRef]
- Chen B, Hu Y, Jin T, et al. The influence of metabolic gene polymorphisms on urinary 1-hydroxypyrene concentrations in Chinese coke oven workers. Sci Total Environ. 2007;381:38–46. [PubMed: 17498780] [CrossRef]
- Chen CC, Neugut AI, Rotterdam H. Risk factors for adenocarcinomas and malignant carcinoids of the small intestine: preliminary findings. Cancer Epidemiol Biomarkers Prev. 1994;3:205–207. [PubMed: 8019367]
- Cheng TJ, Christiani DC, Xu X, et al. Glutathione S-transferase mu genotype, diet, and smoking as determinants of sister chromatid exchange frequency in lymphocytes. Cancer Epidemiol Biomarkers Prev. 1995;4:535–542. [PubMed: 7549811]
- Cheng YJ, Hildesheim A, Hsu MM, et al. Cigarette smoking, alcohol consumption and risk of nasopharyngeal carcinoma in Taiwan. Cancer Causes Control. 1999;10:201–207. [PubMed: 10454065] [CrossRef]
- Chhabra SK, Anderson LM, Perella C, et al. Coexposure to ethanol with N-nitrosodimethylamine or 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone during lactation of rats: marked increase in O6-methylguanine-DNA adducts in maternal mammary gland and in suckling lung and kidney. Toxicol Appl Pharmacol. 2000;169:191–200. [PubMed: 11097872] [CrossRef]
- Chia VM, Newcomb PA, Bigler J, et al. Risk of microsatellite-unstable colorectal cancer is associated jointly with smoking and nonsteroidal anti-inflammatory drug use. Cancer Res. 2006;66:6877–6883. [PubMed: 16818666] [CrossRef]
- Choi SY, Kahyo H. Effect of cigarette smoking and alcohol consumption in the aetiology of cancer of the oral cavity, pharynx and larynx. Int J Epidemiol. 1991;20:878–885. [PubMed: 1800426] [CrossRef]
- Chow WH, Hsing AW, McLaughlin JK, Fraumeni JF Jr. Smoking and adrenal cancer mortality among United States veterans. Cancer Epidemiol Biomarkers Prev. 1996;5:79–80. [PubMed: 8850265]
- Chow WH, Linet MS, McLaughlin JK, et al. Risk factors for small intestine cancer. Cancer Causes Control. 1993;4:163–169. b. [PubMed: 8481495] [CrossRef]
- Chow WH, McLaughlin JK, Hrubec Z, et al. Tobacco use and nasopharyngeal carcinoma in a cohort of US veterans. Int J Cancer. 1993;55:538–540. a. [PubMed: 8406978] [CrossRef]
- Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. [PubMed: 14599765] [CrossRef]
- Chu SY, Stroup NE, Wingo PA, et al. Cigarette smoking and the risk of breast cancer. Am J Epidemiol. 1990;131:244–253. [PubMed: 2404408]
- Church TR, Anderson KE, Caporaso NE, et al. A prospectively measured serum biomarker for a tobacco-specific carcinogen and lung cancer in smokers. Cancer Epidemiol Biomarkers Prev. 2009;18:260–266. [PMC free article: PMC3513324] [PubMed: 19124507] [CrossRef]
- Chyou PH, Nomura AM, Stemmermann GN. Diet, alcohol, smoking and cancer of the upper aerodigestive tract: a prospective study among Hawaii Japanese men. Int J Cancer. 1995;60:616–621. [PubMed: 7860134] [CrossRef]
- Cocco P, Moore PS, Ennas MG, et al. Effect of urban traffic, individual habits, and genetic polymorphisms on background urinary 1-hydroxypyrene excretion. Ann Epidemiol. 2007;17:1–8. [PubMed: 16406813] [CrossRef]
- Colangelo LA, Gapstur SM, Gann PH, Dyer AR. Cigarette smoking and colorectal carcinoma mortality in a cohort with long-term follow-up. Cancer. 2004;100:288–293. [PubMed: 14716762] [CrossRef]
- Colilla S, Kantoff PW, Neuhausen SL, et al. The joint effect of smoking and AIB1 on breast cancer risk in BRCA1 mutation carriers. Carcinogenesis. 2006;27:599–605. [PubMed: 16244359] [CrossRef]
- Collishaw NE, Boyd NF, Cantor KP et al. (2009). Canadian expert panel on tobacco smoke and breast cancer risk. Toronto: Ontario Tobacco Research Unit. [PubMed: 21148114]
- Conway K, Edmiston SN, Cui L, et al. Prevalence and spectrum of p53 mutations associated with smoking in breast cancer. Cancer Res. 2002;62:1987–1995. [PubMed: 11929815]
- Cooper CS, Grover PL, Sims P (1983). The metabolism and activation of benzo[a]pyrene. In: Progress in Drug Metabolism, Vol. 7. Bridges JW, Chasseaud LF, editors. London: Wiley & Sons, pp. 295–396.
- Corona R, Dogliotti E, D’Errico M, et al. Risk factors for basal cell carcinoma in a Mediterranean population: role of recreational sun exposure early in life. Arch Dermatol. 2001;137:1162–1168. [PubMed: 11559211]
- Cortez CC, Jones PA. Chromatin, cancer and drug therapies. Mutat Res. 2008;647:44–51. [PMC free article: PMC2631123] [PubMed: 18691602]
- Cote ML, Chen W, Smith DW, et al. Meta- and pooled analysis of GSTP1 polymorphism and lung cancer: a HuGE-GSEC review. Am J Epidemiol. 2009;169:802–814. [PMC free article: PMC2727222] [PubMed: 19240225] [CrossRef]
- Cote ML, Kardia SL, Wenzlaff AS, et al. Combinations of glutathione S-transferase genotypes and risk of early-onset lung cancer in Caucasians and African Americans: a population-based study. Carcinogenesis. 2005;26:811–819. [PubMed: 15661806] [CrossRef]
- Couch FJ, Cerhan JR, Vierkant RA, et al. Cigarette smoking increases risk for breast cancer in high-risk breast cancer families. Cancer Epidemiol Biomarkers Prev. 2001;10:327–332. [PubMed: 11319172]
- Coughlin SS, Calle EE, Patel AV, Thun MJ. Predictors of pancreatic cancer mortality among a large cohort of United States adults. Cancer Causes Control. 2000;11:915–923. [PubMed: 11142526] [CrossRef]
- Cozen W, Hamilton AS, Zhao P, et al. A protective role for early oral exposures in the etiology of young adult Hodgkin lymphoma. Blood. 2009;114:4014–4020. [PMC free article: PMC2774542] [PubMed: 19738032]
- Crofts F, Cosma GN, Currie D, et al. A novel CYP1A1 gene polymorphism in African-Americans. Carcinogenesis. 1993;14:1729–1731. [PubMed: 8104732] [CrossRef]
- Cui Y, Miller AB, Rohan TE. Cigarette smoking and breast cancer risk: update of a prospective cohort study. Breast Cancer Res Treat. 2006;100:293–299. [PubMed: 16773435] [CrossRef]
- Curtin K, Samowitz WS, Wolff RK, et al. Somatic alterations, metabolizing genes and smoking in rectal cancer. Int J Cancer. 2009;125:158–164. [PMC free article: PMC2782655] [PubMed: 19358278] [CrossRef]
- Cybulski C, Masojc B, Oszutowska D, et al. Constitutional CHEK2 mutations are associated with a decreased risk of lung and laryngeal cancers. Carcinogenesis. 2008;29:762–765. [PubMed: 18281249] [CrossRef]
- D’Agostini F, Izzotti A, Balansky R, et al. Early loss of Fhit in the respiratory tract of rodents exposed to environmental cigarette smoke. Cancer Res. 2006;66:3936–3941. [PubMed: 16585223] [CrossRef]
- d’Errico A, Malats N, Vineis P, Boffetta P. Review of studies of selected metabolic polymorphisms and cancer. IARC Sci Publ. 1999;148:323–393. [PubMed: 10493265]
- Dagle GE, McDonald KE, Smith LG, Stevens DL Jr. Pulmonary carcinogenesis in rats given implants of cigarette smoke condensate in beeswax pellets. J Natl Cancer Inst. 1978;61:905–910. [PubMed: 278868]
- Dalbey WE, Nettesheim P, Griesemer R, et al. Chronic inhalation of cigarette smoke by F344 rats. J Natl Cancer Inst. 1980;64:383–390. [PubMed: 6928229]
- Daling JR, Madeleine MM, Johnson LG, et al. Penile cancer: importance of circumcision, human papillomavirus and smoking in in situ and invasive disease. Int J Cancer. 2005;116:606–616. [PubMed: 15825185] [CrossRef]
- Daling JR, Sherman KJ, Hislop TG, et al. Cigarette smoking and the risk of anogenital cancer. Am J Epidemiol. 1992;135:180–189. [PubMed: 1311142]
- Dally H, Edler L, Jäger B, et al. The CYP3A4*1B allele increases risk for small cell lung cancer: effect of gender and smoking dose. Pharmacogenetics. 2003;13:607–618. [PubMed: 14515059] [CrossRef]
- Daniell HW. Increased lymph node metastases at mastectomy for breast cancer associated with host obesity, cigarette smoking, age, and large tumor size. Cancer. 1988;62:429–435. [PubMed: 3383142] [CrossRef]
- Daniell HW, Tam E, Filice A. Larger axillary metastases in obese women and smokers with breast cancer–an influence by host factors on early tumor behavior. Breast Cancer Res Treat. 1993;25:193–201. [PubMed: 8369520] [CrossRef]
- Darby S, Hill D, Auvinen A, et al. Radon in homes and risk of lung cancer: collaborative analysis of individual data from 13 European case-control studies. BMJ. 2005;330:223. [PMC free article: PMC546066] [PubMed: 15613366] [CrossRef]
- Davies MJ, Turner JG, Vives-Bauza C, Rumsby PC. Investigation of mutant frequency at the HPRT locus and changes in microsatellite sequences in healthy young adults. Mutat Res. 1999;431:317–323. [PubMed: 10635997]
- Davis BR, Whitehead JK, Gill ME, et al. Response of rat lung to inhaled tobacco smoke with or without prior exposure to 3,4-benzpyrene (BP) given by intratracheal instillation. Br J Cancer. 1975;31:469–484. [PMC free article: PMC2009453] [PubMed: 1156528]
- Day GL, Blot WJ, Austin DF, et al. Racial differences in risk of oral and pharyngeal cancer: alcohol, tobacco, and other determinants. J Natl Cancer Inst. 1993;85:465–473. [PubMed: 8445674] [CrossRef]
- de Klerk NH, Musk AW, Eccles JL, et al. Exposure to crocidolite and the incidence of different histological types of lung cancer. Occup Environ Med. 1996;53:157–159. [PMC free article: PMC1128437] [PubMed: 8704855] [CrossRef]
- De Stefani E, Boffetta P, Deneo-Pellegrini H, et al. The effect of smoking and drinking in oral and pharyngeal cancers: a case-control study in Uruguay. Cancer Lett. 2007;246:282–289. [PubMed: 16624486] [CrossRef]
- De Stefani E, Boffetta P, Oreggia F, et al. Smoking patterns and cancer of the oral cavity and pharynx: a case-control study in Uruguay. Oral Oncol. 1998;34:340–346. [PubMed: 9861338] [CrossRef]
- De Stefani E, Correa P, Oreggia F, et al. Risk factors for laryngeal cancer. Cancer. 1987;60:3087–3091. [PubMed: 3677031] [CrossRef]
- De Stefani E, Espasandin J, Ronco A, et al. 1995Tobacco smoking and the risk of non-melanoma skin cancer. Tob Control 4(2)175–179.PMCID: PMC1759430 10.1136/tc.4.2.175. [CrossRef]
- De Stefani E, Muñoz N, Estève J, et al. Mate drinking, alcohol, tobacco, diet, and esophageal cancer in Uruguay. Cancer Res. 1990;50:426–431. [PubMed: 2295081]
- De Stefani E, Oreggia F, Rivero S, Fierro L. Hand-rolled cigarette smoking and risk of cancer of the mouth, pharynx, and larynx. Cancer. 1992;70:679–682. [PubMed: 1623483] [CrossRef]
- Delaney JC, Essigmann JM. Biological properties of single chemical-DNA adducts: a twenty year perspective. Chem Res Toxicol. 2008;21:232–252. [PMC free article: PMC2821157] [PubMed: 18072751] [CrossRef]
- Delfino RJ, Smith C, West JG, et al. Breast cancer, passive and active cigarette smoking and N-acetyltransferase 2 genotype. Pharmacogenetics. 2000;10:461–469. [PubMed: 10898115] [CrossRef]
- DeMarini DM. Genotoxicity of tobacco smoke and tobacco smoke condensate: a review. Mutat Res. 2004;567:447–474. [PubMed: 15572290] [CrossRef]
- DeMarini DM, Gudi R, Szkudlinska A, et al. Genotoxicity of 10 cigarette smoke condensates in four test systems: comparisons between assays and condensates. Mutat Res. 2008;650:15–29. [PubMed: 18006367]
- DeMarini DM, Shelton ML, Levine JG. Mutation spectrum of cigarette smoke condensate in Salmonella: comparison to mutations in smoking-associated tumors. Carcinogenesis. 1995;16:2535–2542. [PubMed: 7586163] [CrossRef]
- Department of Health and Human Services (2001). Women and Smoking: A Report of the Surgeon General. Washington: Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health.
- Department of Health and Human Services (2004). The Health Consequences of Smoking: A Report of the Surgeon General. Atlanta, Georgia: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health.
- Devesa SS, Bray F, Vizcaino AP, Parkin DM. International lung cancer trends by histologic type: male:female differences diminishing and adenocarcinoma rates rising. Int J Cancer. 2005;117:294–299. [PubMed: 15900604] [CrossRef]
- Diergaarde B, Braam H, Vasen HF, et al. Environmental factors and colorectal tumor risk in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol. 2007;5:736–742. [PubMed: 17544999]
- Diergaarde B, Vrieling A, van Kraats AA, et al. Cigarette smoking and genetic alterations in sporadic colon carcinomas. Carcinogenesis. 2003;24:565–571. [PubMed: 12663519] [CrossRef]
- Dikshit RP, Kanhere S. Tobacco habits and risk of lung, oropharyngeal and oral cavity cancer: a population-based case-control study in Bhopal, India. Int J Epidemiol. 2000;29:609–614. [PubMed: 10922335] [CrossRef]
- Dillner J, von Krogh G, Horenblas S, Meijer CJLM. Etiology of squamous cell carcinoma of the penis. Scand J Urol Nephrol Suppl. 2000;34:189–193. [PubMed: 11144896] [CrossRef]
- Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. [PMC free article: PMC2694412] [PubMed: 18948947] [CrossRef]
- Djordjević D, Nikolić J, Stefanović V. Ethanol interactions with other cytochrome P450 substrates including drugs, xenobiotics, and carcinogens. Pathol Biol (Paris). 1998;46:760–770. [PubMed: 9922992]
- Doll R, Gray R, Hafner B, Peto R. Mortality in relation to smoking: 22 years’ observations on female British doctors. Br Med J. 1980;280:967–971. [PMC free article: PMC1601142] [PubMed: 7417764] [CrossRef]
- Doll R, Peto R. Cigarette smoking and bronchial carcinoma: dose and time relationships among regular smokers and lifelong non-smokers. J Epidemiol Community Health. 1978;32:303–313. [PMC free article: PMC1060963] [PubMed: 744822] [CrossRef]
- Doll R, Peto R (1985). Effects on health of exposure to asbestos. London: Health and Safety Commission.
- Doll R, Peto R, Boreham J, Sutherland I. Mortality from cancer in relation to smoking: 50 years observations on British doctors. Br J Cancer. 2005;92:426–429. [PMC free article: PMC2362086] [PubMed: 15668706]
- Doll R, Peto R, Wheatley K, et al. Mortality in relation to smoking: 40 years’ observations on male British doctors. BMJ. 1994;309:901–911. [PMC free article: PMC2541142] [PubMed: 7755693]
- Dong LM, Potter JD, White E, et al. Genetic susceptibility to cancer: the role of polymorphisms in candidate genes. JAMA. 2008;299:2423–2436. [PMC free article: PMC2772197] [PubMed: 18505952] [CrossRef]
- Dontenwill W, Chevalier HJ, Harke HP, et al. Investigations on the effects of chronic cigarette-smoke inhalation in Syrian golden hamsters. J Natl Cancer Inst. 1973;51:1781–1832. [PubMed: 4765388]
- Dontenwill W, Chevalier HJ, Harke HP, et al. Untersuchungen über den Effekt der chronischen Zigarettenrauchinhalation beim syrischen Goldhamster und über die Bedeutung des Vitamin A auf die bei Berauchung gefundenen Organveründerungen. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol. 1977;89:153–180. [PubMed: 143143] [CrossRef]
- Du X, Squier CA, Kremer MJ, Wertz PW. Penetration of N-nitrosonornicotine (NNN) across oral mucosa in the presence of ethanol and nicotine. J Oral Pathol Med. 2000;29:80–85. [PubMed: 10718403] [CrossRef]
- Duell EJ, Holly EA, Bracci PM, et al. A population-based, case-control study of polymorphisms in carcinogen-metabolizing genes, smoking, and pancreatic adenocarcinoma risk. J Natl Cancer Inst. 2002;94:297–306. [PubMed: 11854392]
- Dunn SR, Simenhoff ML, Lele PS, et al. N-nitrosodimethylamine blood levels in patients with chronic renal failure: modulation of levels by ethanol and ascorbic acid. J Natl Cancer Inst. 1990;82:783–787. [PubMed: 2325149] [CrossRef]
- Dusek L, Abrahamova J, Lakomy R, et al. Multivariate analysis of risk factors for testicular cancer: a hospital-based case-control study in the Czech Republic. Neoplasma. 2008;55:356–368. [PubMed: 18505349]
- Dyke GW, Craven JL, Hall R, Garner RC. Smoking-related DNA adducts in human gastric cancers. Int J Cancer. 1992;52:847–850. [PubMed: 1459723] [CrossRef]
- Egan KM, Newcomb PA, Titus-Ernstoff L, et al. Association of NAT2 and smoking in relation to breast cancer incidence in a population-based case-control study (United States). Cancer Causes Control. 2003;14:43–51. [PubMed: 12708724] [CrossRef]
- Egan KM, Stampfer MJ, Hunter D, et al. Nurses’ Health Study. Active and passive smoking in breast cancer: prospective results from the Nurses’ Health Study. Epidemiology. 2002;13:138–145. [PubMed: 11880753] [CrossRef]
- Eichholzer M, Stähelin HB, Lüdin E, Bernasconi F. Smoking, plasma vitamins C, E, retinol, and carotene, and fatal prostate cancer: seventeen-year follow-up of the prospective basel study. Prostate. 1999;38:189–198. [PubMed: 10068343] [CrossRef]
- el-Bayoumy K. Environmental carcinogens that may be involved in human breast cancer etiology. Chem Res Toxicol. 1992;5:585–590. [PubMed: 1445997] [CrossRef]
- Eldridge SR, Gould MN, Butterworth BE. Genotoxicity of environmental agents in human mammary epithelial cells. Cancer Res. 1992;52:5617–5621. [PubMed: 1394185]
- Elliott EA, Matanoski GM, Rosenshein NB, et al. Body fat patterning in women with endometrial cancer. Gynecol Oncol. 1990;39:253–258. [PubMed: 2258066] [CrossRef]
- Elwood JM, Pearson JC, Skippen DH, Jackson SM. Alcohol, smoking, social and occupational factors in the aetiology of cancer of the oral cavity, pharynx and larynx. Int J Cancer. 1984;34:603–612. [PubMed: 6500740] [CrossRef]
- Engeland A, Andersen A, Haldorsen T, Tretli S. Smoking habits and risk of cancers other than lung cancer: 28 years’ follow-up of 26,000 Norwegian men and women. Cancer Causes Control. 1996;7:497–506. [PubMed: 8877046] [CrossRef]
- Engels EA, Biggar RJ, Hall HI, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. International Journal of Cancer. 2008;123:187–194. [PubMed: 18435450] [CrossRef]
- Epplein M, Schwartz SM, Potter JD, Weiss NS. Smoking-adjusted lung cancer incidence among Asian-Americans (United States). Cancer Causes Control. 2005;16:1085–1090. [PubMed: 16184474] [CrossRef]
- Erhardt JG, Kreichgauer HP, Meisner C, et al. Alcohol, cigarette smoking, dietary factors and the risk of colorectal adenomas and hyperplastic polyps–a case control study. Eur J Nutr. 2002;41:35–43. [PubMed: 11990006] [CrossRef]
- Escribano Uzcudun A, Rabanal Retolaza I, García Grande A, et al. Pharyngeal cancer prevention: evidence from a case–control study involving 232 consecutive patients. J Laryngol Otol. 2002;116:523–531. [PubMed: 12238672]
- Ewertz M. Smoking and breast cancer risk in Denmark. Cancer Causes Control. 1990;1:31–37. [PubMed: 2102274] [CrossRef]
- Ewertz M, Gillanders S, Meyer L, Zedeler K. Survival of breast cancer patients in relation to factors which affect the risk of developing breast cancer. Int J Cancer. 1991;49:526–530. [PubMed: 1917153] [CrossRef]
- Falk RT, Pickle LW, Brown LM, et al. Effect of smoking and alcohol consumption on laryngeal cancer risk in coastal Texas. Cancer Res. 1989;49:4024–4029. [PubMed: 2736543]
- Falk RT, Pickle LW, Fontham ET, et al. Epidemiology of bronchioloalveolar carcinoma. Cancer Epidemiology, Biomarkers & Prevention. 1992;1:339–344. [PubMed: 1339048]
- Falter B, Kutzer C, Richter E. Biomonitoring of hemoglobin adducts: aromatic amines and tobacco-specific nitrosamines. Clin Investig. 1994;72:364–371. [PubMed: 8086771] [CrossRef]
- Favorito LA, Nardi AC, Ronalsa M, et al. Epidemiologic study on penile cancer in Brazil. Int Braz J Urol. 2008;34:587–591, discussion 591–593. [PubMed: 18986562] [CrossRef]
- Federal Trade Commission (2001). Cigarette report for 1999. C. Department. Washington D.C.
- Feng BJ, Khyatti M, Ben-Ayoub W, et al. Cannabis, tobacco and domestic fumes intake are associated with nasopharyngeal carcinoma in North Africa. Br J Cancer. 2009;101:1207–1212. [PMC free article: PMC2768108] [PubMed: 19724280] [CrossRef]
- Fennell TR, MacNeela JP, Morris RW, et al. Hemoglobin adducts from acrylonitrile and ethylene oxide in cigarette smokers: effects of glutathione S-transferase T1-null and M1-null genotypes. Cancer Epidemiol Biomarkers Prev. 2000;9:705–712. [PubMed: 10919741]
- Fernberg P, Odenbro A, Bellocco R, et al. Tobacco use, body mass index, and the risk of leukemia and multiple myeloma: a nationwide cohort study in Sweden. Cancer Res. 2007;67:5983–5986. [PubMed: 17575169] [CrossRef]
- Ferson M, Edwards A, Lind A, et al. Low natural killer-cell activity and immunoglobulin levels associated with smoking in human subjects. Int J Cancer. 1979;23:603–609. [PubMed: 457307] [CrossRef]
- Finch GL, Nikula KJ, Barr EB et al. (1995). Effects of combined exposure of F344 rats to radiation and chronically inhaled cigarette smoke. In: Annual Report of the Inhalation Toxicology Research Institute. ITRI-149. Bice DE, Hahn FF, Hoover R et al.,editors.
- Fine LJ, Philogene GS, Gramling R, et al. Prevalence of multiple chronic disease risk factors. 2001 National Health Interview Survey. Am J Prev Med. 2004;27 Suppl:18–24. [PubMed: 15275670] [CrossRef]
- Fink AK, Lash TL. A null association between smoking during pregnancy and breast cancer using Massachusetts registry data (United States). Cancer Causes Control. 2003;14:497–503. [PubMed: 12946045] [CrossRef]
- Firozi PF, Bondy ML, Sahin AA, et al. Aromatic DNA adducts and polymorphisms of CYP1A1, NAT2, and GSTM1 in breast cancer. Carcinogenesis. 2002;23:301–306. [PubMed: 11872636] [CrossRef]
- Flanders WD, Lally CA, Zhu BP, et al. Lung cancer mortality in relation to age, duration of smoking, and daily cigarette consumption: results from Cancer Prevention Study II. Cancer Res. 2003;63:6556–6562. [PubMed: 14559851]
- Folsom AR, Demissie Z, Harnack L., Iowa Women’s Health Study. Glycemic index, glycemic load, and incidence of endometrial cancer: the Iowa women’s health study. Nutr Cancer. 2003;46:119–124. [PubMed: 14690786] [CrossRef]
- Franceschi S, Barra S, La Vecchia C, et al. Risk factors for cancer of the tongue and the mouth. A case-control study from northern Italy. Cancer. 1992;70:2227–2233. [PubMed: 1394055] [CrossRef]
- Franceschi S, Levi F, La Vecchia C, et al. Comparison of the effect of smoking and alcohol drinking between oral and pharyngeal cancer. Int J Cancer. 1999;83:1–4. [PubMed: 10449598] [CrossRef]
- Franceschi S, Montella M, Polesel J, et al. Hepatitis viruses, alcohol, and tobacco in the etiology of hepatocellular carcinoma in Italy. Cancer Epidemiol Biomarkers Prev. 2006;15:683–689. [PubMed: 16614109] [CrossRef]
- Franceschi S, Serraino D. Risk factors for adult soft tissue sarcoma in northern Italy. Ann Oncol. 1992;3 Suppl 2:S85–S88. [PubMed: 1622876]
- Franceschi S, Talamini R, Barra S, et al. Smoking and drinking in relation to cancers of the oral cavity, pharynx, larynx, and esophagus in northern Italy. Cancer Res. 1990;50:6502–6507. [PubMed: 2208109]
- Franco EL, Villa LL, Sobrinho JP, et al. Epidemiology of acquisition and clearance of cervical human papillomavirus infection in women from a high-risk area for cervical cancer. J Infect Dis. 1999;180:1415–1423. [PubMed: 10515798] [CrossRef]
- Franks AL, Kendrick JS, Tyler CW Jr. Postmenopausal smoking, estrogen replacement therapy, and the risk of endometrial cancer. Am J Obstet Gynecol. 1987;156:20–23. [PubMed: 3799753]
- Freedman DM, Sigurdson A, Doody MM, et al. Risk of melanoma in relation to smoking, alcohol intake, and other factors in a large occupational cohort. Cancer Causes Control. 2003;14:847–857. a. [PubMed: 14682442] [CrossRef]
- Freedman DM, Sigurdson A, Doody MM, et al. Risk of basal cell carcinoma in relation to alcohol intake and smoking. Cancer Epidemiol Biomarkers Prev. 2003;12:1540–1543. b. [PubMed: 14693751]
- Freedman ND, Abnet CC, Leitzmann MF, et al. Prospective investigation of the cigarette smoking-head and neck cancer association by sex. Cancer. 2007;110:1593–1601. a. [PubMed: 17724671] [CrossRef]
- Freedman ND, Abnet CC, Leitzmann MF, et al. A prospective study of tobacco, alcohol, and the risk of esophageal and gastric cancer subtypes. Am J Epidemiol. 2007;165:1424–1433. b. [PubMed: 17420181] [CrossRef]
- Freedman ND, Leitzmann MF, Hollenbeck AR, et al. Cigarette smoking and subsequent risk of lung cancer in men and women: analysis of a prospective cohort study. Lancet Oncol. 2008;9:649–656. [PMC free article: PMC2601691] [PubMed: 18556244] [CrossRef]
- Friborg JT, Yuan JM, Wang R, et al. A prospective study of tobacco and alcohol use as risk factors for pharyngeal carcinomas in Singapore Chinese. Cancer. 2007;109:1183–1191. [PubMed: 17315158]
- Friedman AJ, Ravnikar VA, Barbieri RL. Serum steroid hormone profiles in postmenopausal smokers and nonsmokers. Fertil Steril. 1987;47:398–401. [PubMed: 2951278]
- Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med. 1996;156:2255–2260. [PubMed: 8885826] [CrossRef]
- Fujino Y, Mizoue T, Tokui N, et al. JACC Study Group. Cigarette smoking and mortality due to stomach cancer: findings from the JACC Study. J Epidemiol. 2005;15 Suppl 2:S113–S119. [PMC free article: PMC8639042] [PubMed: 16127222] [CrossRef]
- Fujita M, Tase T, Kakugawa Y, et al. Smoking, earlier menarche and low parity as independent risk factors for gynecologic cancers in Japanese: a case-control study. Tohoku J Exp Med. 2008;216:297–307. [PubMed: 19060444] [CrossRef]
- Fukuda K, Shibata A. Exposure-response relationships between woodworking, smoking or passive smoking, and squamous cell neoplasms of the maxillary sinus. Cancer Causes Control. 1990;1:165–168. [PubMed: 2102286] [CrossRef]
- Furberg AS, Thune I. Metabolic abnormalities (hypertension, hyperglycemia and overweight), lifestyle (high energy intake and physical inactivity) and endometrial cancer risk in a Norwegian cohort. Int J Cancer. 2003;104:669–676. [PubMed: 12640672] [CrossRef]
- Gaikwad NW, Yang L, Muti P, et al. The molecular etiology of breast cancer: evidence from biomarkers of risk. Int J Cancer. 2008;122:1949–1957. [PMC free article: PMC4422386] [PubMed: 18098283]
- Gajalakshmi CK, Shanta V. Lifestyle and risk of stomach cancer: a hospital-based case-control study. Int J Epidemiol. 1996;25:1146–1153. [PubMed: 9027518] [CrossRef]
- Gajalakshmi V, Hung RJ, Mathew A, et al. Tobacco smoking and chewing, alcohol drinking and lung cancer risk among men in southern India. Int J Cancer. 2003;107:441–447. [PubMed: 14506745] [CrossRef]
- Gallagher CJ, Muscat JE, Hicks AN, et al. The UDP-glucuronosyltransferase 2B17 gene deletion polymorphism: sex-specific association with urinary 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol glucuronidation phenotype and risk for lung cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:823–828. [PubMed: 17416778] [CrossRef]
- Gallicchio L, Boyd K, Matanoski G, et al. Carotenoids and the risk of developing lung cancer: a systematic review. Am J Clin Nutr. 2008;88:372–383. [PubMed: 18689373]
- Gallicchio L, Kouzis A, Genkinger JM, et al. Active cigarette smoking, household passive smoke exposure, and the risk of developing pancreatic cancer. Prev Med. 2006;42:200–205. [PubMed: 16458957] [CrossRef]
- Gallus S, Altieri A, Bosetti C, et al. Cigarette tar yield and risk of upper digestive tract cancers: case-control studies from Italy and Switzerland. Ann Oncol. 2003;14:209–213. a. [PubMed: 12562646] [CrossRef]
- Gallus S, Bosetti C, Franceschi S, et al. Laryngeal cancer in women: tobacco, alcohol, nutritional, and hormonal factors. Cancer Epidemiol Biomarkers Prev. 2003;12:514–517. b. [PubMed: 12814996]
- Gambier N, Batt AM, Marie B, et al. Association of CYP2A6*1B genetic variant with the amount of smoking in French adults from the Stanislas cohort. Pharmacogenomics J. 2005;5:271–275. [PubMed: 15940289] [CrossRef]
- Gammon MD, Eng SM, Teitelbaum SL, et al. Environmental tobacco smoke and breast cancer incidence. Environ Res. 2004;96:176–185. [PubMed: 15325878] [CrossRef]
- Gammon MD, Schoenberg JB, Ahsan H, et al. Tobacco, alcohol, and socioeconomic status and adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst. 1997;89:1277–1284. [PubMed: 9293918] [CrossRef]
- Gammon MD, Schoenberg JB, Teitelbaum SL, et al. Cigarette smoking and breast cancer risk among young women (United States). Cancer Causes Control. 1998;9:583–590. [PubMed: 10189043] [CrossRef]
- Gandini S, Botteri E, Iodice S, et al. Tobacco smoking and cancer: a meta-analysis. International Journal of Cancer. 2008;122:155–164. [PubMed: 17893872] [CrossRef]
- Gao CM, Takezaki T, Wu JZ, et al. CYP2E1 Rsa I polymorphism impacts on risk of colorectal cancer association with smoking and alcohol drinking. World J Gastroenterol. 2007;13:5725–5730. [PMC free article: PMC4171258] [PubMed: 17963298]
- Gao YT, McLaughlin JK, Blot WJ, et al. Risk factors for esophageal cancer in Shanghai, China. I. Role of cigarette smoking and alcohol drinking. Int J Cancer. 1994;58:192–196. [PubMed: 8026880] [CrossRef]
- Gapstur SM, Gann PH, Lowe W, et al. Abnormal glucose metabolism and pancreatic cancer mortality. JAMA. 2000;283:2552–2558. [PubMed: 10815119] [CrossRef]
- García-Closas M, Malats N, Silverman D, et al. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. 2005;366:649–659. [PMC free article: PMC1459966] [PubMed: 16112301] [CrossRef]
- García-González MA, Lanas A, Quintero E, et al. Spanish Gastroenterological Association AEG. Gastric cancer susceptibility is not linked to pro-and anti-inflammatory cytokine gene polymorphisms in whites: a Nationwide Multicenter Study in Spain. Am J Gastroenterol. 2007;102:1878–1892. [PubMed: 17640324] [CrossRef]
- Gargus JL, Powers MB, Habermann RT et al. (1976). Mouse dermal bioassays of cigarette smoke condensates. In: Report No. 1. Toward Less Hazardous Cigarettes. The First Set of Experimental Cigarettes (DHEW Publ. No. (NIH)76–905). Washington DC: Public Health Service, National Institutes of Health, National Cancer Institute, pp. 85–94.
- Garrote LF, Herrero R, Reyes RM, et al. Risk factors for cancer of the oral cavity and oro-pharynx in Cuba. Br J Cancer. 2001;85:46–54. [PMC free article: PMC2363910] [PubMed: 11437401] [CrossRef]
- Garte S. Metabolic susceptibility genes as cancer risk factors: time for a reassessment? Cancer Epidemiol Biomarkers Prev. 2001;10:1233–1237. [PubMed: 11751439]
- Garte S, Gaspari L, Alexandrie AK, et al. Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiol Biomarkers Prev. 2001;10:1239–1248. [PubMed: 11751440]
- Garte S, Taioli E, Popov T, et al. Genetic susceptibility to benzene toxicity in humans. J Toxicol Environ Health A. 2008;71:1482–1489. [PubMed: 18836923]
- Gasper AV, Al-Janobi A, Smith JA, et al. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. Am J Clin Nutr. 2005;82:1283–1291. [PubMed: 16332662]
- Ghadirian P, Lubinski J, Lynch H, et al. Smoking and the risk of breast cancer among carriers of BRCA mutations. Int J Cancer. 2004;110:413–416. [PubMed: 15095307] [CrossRef]
- Ghadirian P, Maisonneuve P, Perret C, et al. Epidemiology of sociodemographic characteristics, lifestyle, medical history, and colon cancer: a case-control study among French Canadians in Montreal. Cancer Detect Prev. 1998;22:396–404. [PubMed: 9727620] [CrossRef]
- Ginsburg O, Ghadirian P, Lubinski J, et al. Hereditary Breast Cancer Clinical Study Group. Smoking and the risk of breast cancer in BRCA1 and BRCA2 carriers: an update. Breast Cancer Res Treat. 2009;114:127–135. [PMC free article: PMC3033012] [PubMed: 18483851] [CrossRef]
- Giovannucci E, Rimm EB, Stampfer MJ, et al. A prospective study of cigarette smoking and risk of colorectal adenoma and colorectal cancer in U.S. men. J Natl Cancer Inst. 1994;86:183–191. [PubMed: 8283490]
- Giuliano AR, Sedjo RL, Roe DJ, et al. Clearance of oncogenic human papillomavirus (HPV) infection: effect of smoking (United States). Cancer Causes Control. 2002;13:839–846. [PubMed: 12462549] [CrossRef]
- Glaser SL, Keegan TH, Clarke CA, et al. Smoking and Hodgkin lymphoma risk in women United States. Cancer Causes Control. 2004;15:387–397. [PubMed: 15141139]
- Glatt H, Boeing H, Engelke CE, et al. Human cytosolic sulphotransferases: genetics, characteristics, toxicological aspects. Mutat Res. 2001;482:27–40. [PubMed: 11535246]
- Goldman R, Shields PG. Molecular epidemiology of breast cancer. In Vivo. 1998;12:43–48. [PubMed: 9575425]
- Gong Z, Xie D, Deng Z, et al. The PPARgamma Pro12Ala polymorphism and risk for incident sporadic colorectal adenomas. Carcinogenesis. 2005;26:579–585. [PubMed: 15564289] [CrossRef]
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer. 2003;107:629–634. [PubMed: 14520702] [CrossRef]
- Goodman MT, Hankin JH, Wilkens LR, et al. Diet, body size, physical activity, and the risk of endometrial cancer. Cancer Res. 1997;57:5077–5085. [PubMed: 9371506]
- Goodman MT, McDuffie K, Kolonel LN, et al. Case-control study of ovarian cancer and polymorphisms in genes involved in catecholestrogen formation and metabolism. Cancer Epidemiol Biomarkers Prev. 2001;10:209–216. [PubMed: 11303589]
- Goodman MT, Tung KH. Active and passive tobacco smoking and the risk of borderline and invasive ovarian cancer (United States). Cancer Causes Control. 2003;14:569–577. [PubMed: 12948288] [CrossRef]
- Gori GB (1976). Report on the second set of experimental cigarettes. In: Report No. 2. Toward Less Hazardous Cigarettes. The Second Set of Experimental Cigarettes (DHEW Publ. No. (NIH) 76–1111). Washington DC: Public Health Service, National Institutes of Health, National Cancer Institute.
- Gorlewska-Roberts K, Green B, Fares M, et al. Carcinogen-DNA adducts in human breast epithelial cells. Environ Mol Mutagen. 2002;39:184–192. [PubMed: 11921188] [CrossRef]
- Goy J, Rosenberg MW, King WD. Health risk behaviors: examining social inequalities in bladder and colorectal cancers. Ann Epidemiol. 2008;18:156–162. [PubMed: 18191762]
- Graham EA, Croninger AB, Wynder EL. Experimental production of carcinoma with cigarette tar. IV. Successful experiments with rabbits. Cancer Res. 1957;17:1058–1066. [PubMed: 13489709]
- Grainge MJ, West J, Solaymani-Dodaran M, et al. The antecedents of biliary cancer: a primary care case-control study in the United Kingdom. Br J Cancer. 2009;100:178–180. [PMC free article: PMC2634685] [PubMed: 19018260] [CrossRef]
- Gram IT, Braaten T, Adami HO, et al. Cigarette smoking and risk of borderline and invasive epithelial ovarian cancer. Int J Cancer. 2008;122:647–652. [PubMed: 17918152]
- Gram IT, Braaten T, Lund E, et al. Cigarette smoking and risk of colorectal cancer among Norwegian women. Cancer Causes Control. 2009;20:895–903. [PMC free article: PMC2694321] [PubMed: 19274482] [CrossRef]
- Gram IT, Braaten T, Terry PD, et al. Breast cancer risk among women who start smoking as teenagers. Cancer Epidemiol Biomarkers Prev. 2005;14:61–66. [PubMed: 15668477]
- Green A, Purdie D, Bain C, et al. Cigarette smoking and risk of epithelial ovarian cancer (Australia). Cancer Causes Control. 2001;12:713–719. [PubMed: 11562111] [CrossRef]
- Greenland S, Longnecker MP. Methods for trend estimation from summarized dose-response data, with applications to meta-analysis. Am J Epidemiol. 1992;135:1301–1309. [PubMed: 1626547]
- Griciute L, Castegnaro M, Bereziat JC. Influence of ethyl alcohol on carcinogenesis with N-nitrosodimethylamine. Cancer Lett. 1981;13:345–352. [PubMed: 7306961] [CrossRef]
- Gronwald J, Byrski T, Huzarski T, et al. Influence of selected lifestyle factors on breast and ovarian cancer risk in BRCA1 mutation carriers from Poland. Breast Cancer Res Treat. 2006;95:105–109. [PubMed: 16261399] [CrossRef]
- Gross CP, Filardo G, Singh HS, et al. The relation between projected breast cancer risk, perceived cancer risk, and mammography use. Results from the National Health Interview Survey. J Gen Intern Med. 2006;21:158–164. [PMC free article: PMC1484644] [PubMed: 16390511]
- Gudmundsdottir K, Tryggvadottir L, Eyfjord JE. GSTM1, GSTT1, and GSTP1 genotypes in relation to breast cancer risk and frequency of mutations in the p53 gene. Cancer Epidemiol Biomarkers Prev. 2001;10:1169–1173. [PubMed: 11700265]
- Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol. 2001;14:611–650. [PubMed: 11409933] [CrossRef]
- Guengerich FP, Shimada T. Activation of procarcinogens by human cytochrome P450 enzymes. Mutat Res. 1998;400:201–213. [PubMed: 9685642]
- Guengerich FP, Shimada T, Yun CH, et al. Interactions of ingested food, beverage, and tobacco components involving human cytochrome P4501A2, 2A6, 2E1, and 3A4 enzymes. Environ Health Perspect. 1994;102 Suppl 9:49–53. [PMC free article: PMC1566791] [PubMed: 7698084]
- Guillemette C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J. 2003;3:136–158. [PubMed: 12815363] [CrossRef]
- Gunnell AS, Tran TN, Torrång A, et al. Synergy between cigarette smoking and human papillomavirus type 16 in cervical cancer in situ development. Cancer Epidemiol Biomarkers Prev. 2006;15:2141–2147. [PubMed: 17057029] [CrossRef]
- Guo X, Johnson RC, Deng H, et al. Evaluation of nonviral risk factors for nasopharyngeal carcinoma in a high-risk population of Southern China. Int J Cancer. 2009;124:2942–2947. [PMC free article: PMC4406046] [PubMed: 19296536] [CrossRef]
- Gupta D, Boffetta P, Gaborieau V, Jindal SK. Risk factors of lung cancer in Chandigarh, India. Indian J Med Res. 2001;113:142–150. [PubMed: 11558323]
- Gupta MP, Khanduja KL, Koul IB, Sharma RR. Effect of cigarette smoke inhalation on benzo[a]pyrene-induced lung carcinogenesis in vitamin A deficiency in the rat. Cancer Lett. 1990;55:83–88. [PubMed: 2265418] [CrossRef]
- Ha M, Mabuchi K, Sigurdson AJ, et al. Smoking cigarettes before first childbirth and risk of breast cancer. Am J Epidemiol. 2007;166:55–61. [PubMed: 17426039] [CrossRef]
- Haiman CA, Stram DO, Wilkens LR, et al. Ethnic and racial differences in the smoking-related risk of lung cancer. N Engl J Med. 2006;354:333–342. [PubMed: 16436765]
- Hamajima N, Hirose K, Tajima K, et al. Collaborative Group on Hormonal Factors in Breast Cancer. Alcohol, tobacco and breast cancer–collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br J Cancer. 2002;87:1234–1245. [PMC free article: PMC2562507] [PubMed: 12439712] [CrossRef]
- Hammond EC, Horn D. Smoking and death rates: report on forty-four months of follow-up of 187,783 men. 2. Death rates by cause. J Am Med Assoc. 1958;166:1294–1308. [PubMed: 12308037]
- Hammond EC, Seidman H. Smoking and cancer in the United States. Prev Med. 1980;9:169–174. [PubMed: 7383981] [CrossRef]
- Hammond EC, Selikoff IJ, Seidman H. Asbestos exposure, cigarette smoking and death rates. Ann N Y Acad Sci. 1979;330 1 Health Hazard:473–490. [PubMed: 294198] [CrossRef]
- Hanaoka T, Yamamoto S, Sobue T, et al. Japan Public Health Center-Based Prospective Study on Cancer and Cardiovascular Disease Study Group. Active and passive smoking and breast cancer risk in middle-aged Japanese women. Int J Cancer. 2005;114:317–322. [PubMed: 15540214] [CrossRef]
- Hannan LM, Jacobs EJ, Thun MJ. The association between cigarette smoking and risk of colorectal cancer in a large prospective cohort from the United States. Cancer Epidemiol Biomarkers Prev. 2009;18:3362–3367. [PubMed: 19959683] [CrossRef]
- Hansen AM, Mathiesen L, Pedersen M, Knudsen LE. Urinary 1-hydroxypyrene (1-HP) in environmental and occupational studies–a review. Int J Hyg Environ Health. 2008;211:471–503. [PubMed: 18222724] [CrossRef]
- Hansen RD, Krath BN, Frederiksen K, et al. GPX1 Pro(198)Leu polymorphism, erythrocyte GPX activity, interaction with alcohol consumption and smoking, and risk of colorectal cancer. Mutat Res. 2009;664:13–19. [PubMed: 19428376]
- Harada T, Kitazawa T, Maita K, Shirasu Y. Effects of vitamin C on tumor induction by diethylnitrosamine in the respiratory tract of hamsters exposed to cigarette smoke. Cancer Lett. 1985;26:163–169. [PubMed: 3978606] [CrossRef]
- Harish K, Ravi R. The role of tobacco in penile carcinoma. Br J Urol. 1995;75:375–377. [PubMed: 7735804]
- Harris JE, Thun MJ, Mondul AM, Calle EE. Cigarette tar yields in relation to mortality from lung cancer in the cancer prevention study II prospective cohort, 1982–1998. BMJ (Clinical Research Ed.). 2004;328:72–78. [PMC free article: PMC314045] [PubMed: 14715602] [CrossRef]
- Harris RJ, Negroni G. Production of lung carcinomas in C57BL mice exposed to a cigarette smoke and air mixture. Br Med J. 1967;4:637–641. [PMC free article: PMC1749206] [PubMed: 4293822] [CrossRef]
- Harris RJ, Negroni G, Ludgate S, et al. The incidence of lung tumours in c57bl mice exposed to cigarette smoke: air mixtures for prolonged periods. Int J Cancer. 1974;14:130–136. [PubMed: 4617705] [CrossRef]
- Hartge P, Schiffman MH, Hoover R, et al. A case-control study of epithelial ovarian cancer. Am J Obstet Gynecol. 1989;161:10–16. [PubMed: 2750791]
- Hashibe M, Boffetta P, Janout V, et al. Esophageal cancer in Central and Eastern Europe: tobacco and alcohol. Int J Cancer. 2007;120:1518–1522. a. [PubMed: 17205526] [CrossRef]
- Hashibe M, Boffetta P, Zaridze D, et al. Contribution of tobacco and alcohol to the high rates of squamous cell carcinoma of the supraglottis and glottis in Central Europe. Am J Epidemiol. 2007;165:814–820. b. [PubMed: 17244634] [CrossRef]
- Hashibe M, Brennan P, Benhamou S, et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst. 2007;99:777–789. c. [PubMed: 17505073] [CrossRef]
- Hashibe M, Brennan P, Chuang SC, et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev. 2009;18:541–550. [PMC free article: PMC3051410] [PubMed: 19190158] [CrossRef]
- Hassan MM, Phan A, Li D, et al. Risk factors associated with neuroendocrine tumors: A U.S.-based case-control study. Int J Cancer. 2008;123:867–873. b. [PubMed: 18491401] [CrossRef]
- Hassan MM, Spitz MR, Thomas MB, et al. Effect of different types of smoking and synergism with hepatitis C virus on risk of hepatocellular carcinoma in American men and women: case-control study. Int J Cancer. 2008;123:1883–1891. a. [PMC free article: PMC2673571] [PubMed: 18688864] [CrossRef]
- Hatsukami DK, Benowitz NL, Rennard SI, et al. Biomarkers to assess the utility of potential reduced exposure tobacco products. Nicotine Tob Res. 2006;8:600–622. a. [PMC free article: PMC6615727] [PubMed: 16920658]
- Hatsukami DK, Le CT, Zhang Y, et al. Toxicant exposure in cigarette reducers versus light smokers. Cancer Epidemiol Biomarkers Prev. 2006;15:2355–2358. b. [PMC free article: PMC6512339] [PubMed: 17164356] [CrossRef]
- Hatsukami DK, Lemmonds C, Zhang Y, et al. Evaluation of carcinogen exposure in people who used “reduced exposure” tobacco products. J Natl Cancer Inst. 2004;96:844–852. [PubMed: 15173268]
- Hawkins NJ, Ward RL. Sporadic colorectal cancers with microsatellite instability and their possible origin in hyperplastic polyps and serrated adenomas. J Natl Cancer Inst. 2001;93:1307–1313. [PubMed: 11535705]
- Hayes JR, Meckley DR, Stavanja MS, et al. Effect of a flue-curing process that reduces tobacco specific nitrosamines on the tumor promotion in SENCAR mice by cigarette smoke condensate. Food Chem Toxicol. 2007;45:419–430. [PubMed: 17070977] [CrossRef]
- Hayes RB, Bravo-Otero E, Kleinman DV, et al. Tobacco and alcohol use and oral cancer in Puerto Rico. Cancer Causes Control. 1999;10:27–33. [PubMed: 10334639] [CrossRef]
- Hayes RB, Kardaun JW, de Bruyn A. Tobacco use and sinonasal cancer: a case-control study. Br J Cancer. 1987;56:843–846. [PMC free article: PMC2002409] [PubMed: 3435710]
- Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:559–603. [PubMed: 9625726] [CrossRef]
- Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. [PubMed: 10413421] [CrossRef]
- Hecht SS. Inhibition of carcinogenesis by isothiocyanates. Drug Metab Rev. 2000;32:395–411. [PubMed: 11139137] [CrossRef]
- Hecht SS. Human urinary carcinogen metabolites: biomarkers for investigating tobacco and cancer. Carcinogenesis. 2002;23:907–922. [PubMed: 12082012] [CrossRef]
- Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat Rev Cancer. 2003;3:733–744. [PubMed: 14570033] [CrossRef]
- Hecht SS. Progress and challenges in selected areas of tobacco carcinogenesis. Chem Res Toxicol. 2008;21:160–171. [PMC free article: PMC2556958] [PubMed: 18052103] [CrossRef]
- Hecht SS, Carmella SG, Edmonds A, et al. Exposure to nicotine and a tobacco-specific carcinogen increase with duration of use of smokeless tobacco. Tob Control. 2008;17:128–131. b. [PMC free article: PMC3889131] [PubMed: 18375734] [CrossRef]
- Hecht SS, Carmella SG, Murphy SE, et al. Similar exposure to a tobacco-specific carcinogen in smokeless tobacco users and cigarette smokers. Cancer Epidemiol Biomarkers Prev. 2007;16:1567–1572. [PubMed: 17684130] [CrossRef]
- Hecht SS, Carmella SG, Stepanov I, et al. Metabolism of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone to its biomarker total NNAL in smokeless tobacco users. Cancer Epidemiol Biomarkers Prev. 2008;17:732–735. a. [PubMed: 18349296] [CrossRef]
- Hecht SS, Carmella SG, Ye M, et al. Quantitation of metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone after cessation of smokeless tobacco use. Cancer Res. 2002;62:129–134. [PubMed: 11782369]
- Hecht SS, Carmella SG, Yoder A, et al. Comparison of polymorphisms in genes involved in polycyclic aromatic hydrocarbon metabolism with urinary phenanthrene metabolite ratios in smokers. Cancer Epidemiol Biomarkers Prev. 2006;15:1805–1811. [PubMed: 17035385] [CrossRef]
- Hecht SS, Chen M, Yoder A, et al. Longitudinal study of urinary phenanthrene metabolite ratios: effect of smoking on the diol epoxide pathway. Cancer Epidemiol Biomarkers Prev. 2005;14:2969–2974. a. [PubMed: 16365018] [CrossRef]
- Hecht SS, Hoffmann D. Tobacco-specific nitrosamines, an important group of carcinogens in tobacco and tobacco smoke. Carcinogenesis. 1988;9:875–884. [PubMed: 3286030] [CrossRef]
- Hecht SS, Murphy SE, Carmella SG, et al. Effects of reduced cigarette smoking on the uptake of a tobacco-specific lung carcinogen. J Natl Cancer Inst. 2004;96:107–115. [PubMed: 14734700]
- Hecht SS, Murphy SE, Carmella SG, et al. Similar uptake of lung carcinogens by smokers of regular, light, and ultralight cigarettes. Cancer Epidemiol Biomarkers Prev. 2005;14:693–698. b. [PubMed: 15767351] [CrossRef]
- Hecht SS, Rivenson A, Braley J, et al. Induction of oral cavity tumors in F344 rats by tobacco-specific nitrosamines and snuff. Cancer Res. 1986;46:4162–4166. [PubMed: 3731083]
- Hedberg K, Vaughan TL, White E, et al. Alcoholism and cancer of the larynx: a case-control study in western Washington (United States). Cancer Causes Control. 1994;5:3–8. [PubMed: 8123776] [CrossRef]
- Heeschen C, Jang JJ, Weis M, et al. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med. 2001;7:833–839. [PubMed: 11433349] [CrossRef]
- Hein DW. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res. 2002;506–507:65–77. [PubMed: 12351146]
- Hein DW. N-acetyltransferase 2 genetic polymorphism: effects of carcinogen and haplotype on urinary bladder cancer risk. Oncogene. 2006;25:1649–1658. [PMC free article: PMC1434721] [PubMed: 16550165] [CrossRef]
- Heineman EF, Zahm SH, McLaughlin JK, Vaught JB. Increased risk of colorectal cancer among smokers: results of a 26-year follow-up of US veterans and a review. Int J Cancer. 1994;59:728–738. [PubMed: 7989109] [CrossRef]
- Heling A-K, Schutte-Borkovec K, Heppel C et al. (2008). Pyridyloxobutylated DNA adducts in oral mucosa of nonsmokers, smokers and snuff dippers in relation to other biomarkers of exposure. AACR Meeting Abstracts, April 2008, Abstract 1883.
- Hellberg D, Stendahl U. The biological role of smoking, oral contraceptive use and endogenous sexual steroid hormones in invasive squamous epithelial cervical cancer. Anticancer Res. 2005;25:3041–3046. [PubMed: 16080563]
- Hellberg D, Valentin J, Eklund T, Nilsson S. Penile cancer: is there an epidemiological role for smoking and sexual behaviour? Br Med J (Clin Res Ed). 1987;295:1306–1308. [PMC free article: PMC1248379] [PubMed: 3120988] [CrossRef]
- Hemminki K, Koskinen M, Zhao C. DNA adducts as a marker for cancer risk? Int J Cancer. 2001;92:923–925. [PubMed: 11351318] [CrossRef]
- Henderson BE, Louie E, SooHoo Jing J, et al. Risk factors associated with nasopharyngeal carcinoma. N Engl J Med. 1976;295:1101–1106. [PubMed: 980005]
- Henley SJ, Thun MJ, Chao A, Calle EE. Association between exclusive pipe smoking and mortality from cancer and other diseases. J Natl Cancer Inst. 2004;96:853–861. [PubMed: 15173269] [CrossRef]
- Henry CJ, Kouri RE. Chronic inhalation studies in mice. II. Effects of long-term exposure to 2R1 cigarette smoke on (C57BL/Cum x C3H/AnfCum)F1 mice. J Natl Cancer Inst. 1986;77:203–212. [PubMed: 3459913]
- Herity B, Moriarty M, Daly L, et al. The role of tobacco and alcohol in the aetiology of lung and larynx cancer. Br J Cancer. 1982;46:961–964. [PMC free article: PMC2011218] [PubMed: 7150489]
- Hermann S, Rohrmann S, Linseisen J (2009). Lifestyle factors, obesity and the risk of colorectal adenomas in EPIC-Heidelberg. Cancer Causes Control. [PubMed: 19466571]
- Herrinton LJ, Friedman GD. Cigarette smoking and risk of non-Hodgkin’s lymphoma subtypes. Cancer Epidemiol Biomarkers Prev. 1998;7:25–28. [PubMed: 9456239]
- Hickey K, Do KA, Green A. Smoking and prostate cancer. Epidemiol Rev. 2001;23:115–125. [PubMed: 11588835]
- Hill M. Etiology of the adenoma-carcinoma sequence. Major Probl Pathol. 1978;10:153–162. [PubMed: 616868]
- Hjalgrim H, Ekström-Smedby K, Rostgaard K, et al. Cigarette smoking and risk of Hodgkin lymphoma: a population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2007;16:1561–1566. [PubMed: 17684129]
- Ho JW, Lam TH, Tse CW, et al. Smoking, drinking and colorectal cancer in Hong Kong Chinese: a case-control study. Int J Cancer. 2004;109:587–597. [PubMed: 14991582] [CrossRef]
- Hoffmann D, Brunnemann KD. Endogenous formation of N-nitrosoproline in cigarette smokers. Cancer Res. 1983;43:5570–5574. [PubMed: 6616484]
- Hoffmann D, Hoffmann I, El-Bayoumy K. The less harmful cigarette: a controversial issue. a tribute to Ernst L. Wynder. Chem Res Toxicol. 2001;14:767–790. [PubMed: 11453723] [CrossRef]
- Hoffmann D, Rivenson A, Hecht SS, et al. Model studies in tobacco carcinogenesis with the Syrian golden hamster. Prog Exp Tumor Res. 1979;24:370–390. [PubMed: 395571]
- Hoffmann D, Wynder EL. A study of tobacco carcinogenesis. XI. Tumor initiators, tumor accelerators, and tumor promoting activity of condensate fractions. Cancer. 1971;27:848–864. [PubMed: 5574075] [CrossRef]
- Holmes MD, Murin S, Chen WY, et al. Smoking and survival after breast cancer diagnosis. Int J Cancer. 2007;120:2672–2677. [PubMed: 17278091] [CrossRef]
- Homann N, Tillonen J, Meurman JH, et al. Increased salivary acetaldehyde levels in heavy drinkers and smokers: a microbiological approach to oral cavity cancer. Carcinogenesis. 2000;21:663–668. [PubMed: 10753201] [CrossRef]
- Horn-Ross PL, Ljung BM, Morrow M. Environmental factors and the risk of salivary gland cancer. Epidemiology. 1997;8:414–419. [PubMed: 9209856] [CrossRef]
- Hosgood HD 3rd, Zhang L, Shen M, et al. Association between genetic variants in VEGF, ERCC3 and occupational benzene haematotoxicity. Occup Environ Med. 2009;66:848–853. [PMC free article: PMC2928224] [PubMed: 19773279]
- Hoshiyama Y, Kono S, Sasaba T, et al. Relation of Cigarette Smoking, Alcohol Use, and Dietary Habits to Colon Adenomas: A Case-Control Study in Saitama, Japan. Asian Pac J Cancer Prev. 2000;1:139–146. [PubMed: 12718681]
- Houlston RS. Glutathione S-transferase M1 status and lung cancer risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 1999;8:675–682. [PubMed: 10744127]
- Houlston RS. CYP1A1 polymorphisms and lung cancer risk: a meta-analysis. Pharmacogenetics. 2000;10:105–114. [PubMed: 10761998] [CrossRef]
- Hsing AW, McLaughlin JK, Chow WH, et al. Risk factors for colorectal cancer in a prospective study among U.S. white men. Int J Cancer. 1998;77:549–553. [PubMed: 9679757] [CrossRef]
- Hsing AW, Nam JM, Co Chien HT, et al. Risk factors for adrenal cancer: an exploratory study. Int J Cancer. 1996;65:432–436. [PubMed: 8621222] [CrossRef]
- Hsu WL, Chen JY, Chien YC, et al. Independent effect of EBV and cigarette smoking on nasopharyngeal carcinoma: a 20-year follow-up study on 9,622 males without family history in Taiwan. Cancer Epidemiol Biomarkers Prev. 2009;18:1218–1226. [PubMed: 19336547] [CrossRef]
- Hu J, Morrison H, Mery L, et al. Canadian Cancer Registries Epidemiology Research Group. Diet and vitamin or mineral supplementation and risk of colon cancer by subsite in Canada. Eur J Cancer Prev. 2007;16:275–291. [PubMed: 17554200] [CrossRef]
- Hu J, Ugnat AM., Canadian Cancer Registries Epidemiology Research Group. Active and passive smoking and risk of renal cell carcinoma in Canada. Eur J Cancer. 2005;41:770–778. [PubMed: 15763654] [CrossRef]
- Hukkanen J, Jacob P 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57:79–115. [PubMed: 15734728] [CrossRef]
- Huncharek M, Haddock KS, Reid R, Kupelnick B. Smoking as a risk factor for prostate cancer: a meta-analysis of 24 prospective cohort studies. Am J Public Health. 2010;100:693–701. [PMC free article: PMC2836346] [PubMed: 19608952] [CrossRef]
- Hung HC, Chuang J, Chien YC, et al. Genetic polymorphisms of CYP2E1, GSTM1, and GSTT1; environmental factors and risk of oral cancer. Cancer Epidemiol Biomarkers Prev. 1997;6:901–905. [PubMed: 9367063]
- Hung RJ, Christiani DC, Risch A, et al. International lung cancer consortium: pooled analysis of sequence variants in DNA repair and cell cycle pathways. Cancer Epidemiology, Biomarkers & Prevention. 2008;17:3081–3089. a. [PMC free article: PMC2756735] [PubMed: 18990748] [CrossRef]
- Hung RJ, McKay JD, Gaborieau V, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. [PubMed: 18385738] [CrossRef]
- Hung RJ, McKay JD, Gaborieau V, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. b. [PubMed: 18385738] [CrossRef]
- Hunt JD, van der Hel OL, McMillan GP, et al. Renal cell carcinoma in relation to cigarette smoking: meta-analysis of 24 studies. Int J Cancer. 2005;114:101–108. [PubMed: 15523697] [CrossRef]
- Hunter DJ, Hankinson SE, Hough H, et al. A prospective study of NAT2 acetylation genotype, cigarette smoking, and risk of breast cancer. Carcinogenesis. 1997;18:2127–2132. [PubMed: 9395212] [CrossRef]
- Husgafvel-Pursiainen K. Genotoxicity of environmental tobacco smoke: a review. Mutat Res. 2004;567:427–445. [PubMed: 15572289] [CrossRef]
- Hutt JA, Vuillemenot BR, Barr EB, et al. Life-span inhalation exposure to mainstream cigarette smoke induces lung cancer in B6C3F1 mice through genetic and epigenetic pathways. Carcinogenesis. 2005;26:1999–2009. [PubMed: 15944214] [CrossRef]
- Huusom LD, Frederiksen K, Høgdall EV, et al. Association of reproductive factors, oral contraceptive use and selected lifestyle factors with the risk of ovarian borderline tumors: a Danish case-control study. Cancer Causes Control. 2006;17:821–829. [PubMed: 16783610] [CrossRef]
- Huxley R., Asia Pacific Cohort Studies Collaboration. The role of lifestyle risk factors on mortality from colorectal cancer in populations of the Asia-Pacific region. Asian Pac J Cancer Prev. 2007;8:191–198. a. [PubMed: 17696730]
- Huxley R, Jamrozik K, Lam TH, et al. Asia Pacific Cohort Studies Collaboration. Impact of smoking and smoking cessation on lung cancer mortality in the Asia-Pacific region. Am J Epidemiol. 2007;165:1280–1286. b. [PubMed: 17369610] [CrossRef]
- Huxley RR, Ansary-Moghaddam A, Clifton P, et al. The impact of dietary and lifestyle risk factors on risk of colorectal cancer: a quantitative overview of the epidemiological evidence. Int J Cancer. 2009;125:171–180. [PubMed: 19350627] [CrossRef]
- Hymowitz N, Corle D, Royce J, et al. Smokers’ baseline characteristics in the COMMIT trial. Prev Med. 1995;24:503–508. [PubMed: 8524726] [CrossRef]
- IARC. Tobacco smoking. IARC Monogr Eval Carcinog Risk Chem Hum. 1986;38:1–421.
- IARC. Overall evaluations of carcinogenicity: an updating of IARC Monographs volumes 1 to 42. IARC Monogr Eval Carcinog Risks Hum Suppl. 1987;7:1–440. [PubMed: 3482203]
- IARC. Human papillomaviruses. IARC Monogr Eval Carcinog Risks Hum. 1995;64:1–378. [PMC free article: PMC5366848] [PubMed: 16755705]
- IARC (2002). IARC handbook of cancer prevention, vol. 6. Weight control and physical activity. IARC Press, Lyon: International Agency for Research on Cancer.
- IARC. Tobacco smoke and involuntary smoking. IARC Monogr Eval Carcinog Risks Hum. 2004;83:1–1438. a. [PMC free article: PMC4781536] [PubMed: 15285078]
- IARC. Betel-quid and areca-nut chewing and some areca-nut derived nitrosamines. IARC Monogr Eval Carcinog Risks Hum. 2004;85:1–334. b. [PMC free article: PMC4781453] [PubMed: 15635762]
- IARC. Formaldehyde, 2-butoxyethanol and 1-tert-butoxypropan-2-ol. IARC Monogr Eval Carcinog Risks Hum. 2006;88:1–478. a. [PMC free article: PMC4781641] [PubMed: 17366697]
- IARC (2006b). TP53 mutation database. http://www.p53.iarc.fr/index.htlm.
- IARC. Smokeless tobacco and some tobacco-specific N-nitrosamines. IARC Monogr Eval Carcinog Risks Hum. 2007;89:1–592. a. [PMC free article: PMC4781254] [PubMed: 18335640]
- IARC. Human papillomaviruses. IARC Monogr Eval Carcinog Risks Hum. 2007;90:1–636. b. [PMC free article: PMC4781057] [PubMed: 18354839]
- IARC. Combined estrogen-progestogen contraceptives and combined estrogen-progestogen menopausal therapy. IARC Monogr Eval Carcinog Risks Hum. 2007;91:1–528. c. [PMC free article: PMC4781221] [PubMed: 18756632]
- IARC. 1,3-Butadiene, ethylene oxide and vinyl halides (vinyl fluoride, vinyl chloride and vinyl bromide). IARC Monogr Eval Carcinog Risks Hum. 2008;97:1–510. [PMC free article: PMC4782275] [PubMed: 20232717]
- IARC. Alcohol consumption and ethyl carbamate. IARC Monogr Eval Carcinog Risks Hum. 2010;96:1–1428. a. [PMC free article: PMC4781168] [PubMed: 21735939]
- IARC. Some Aromatic Amines, Organic Dyes, and Related Exposures. IARC Monogr Eval Carcinog Risks Hum. 2010;99:1–692. b. [PMC free article: PMC5046080] [PubMed: 21528837]
- IARC. Pharmaceuticals. IARC Monogr Eval Carcinog Risks Hum. 2012;100A:1–437. c.
- IARC. Biological agents. IARC Monogr Eval Carcinog Risks Hum. 2012;100B:1–475. b.
- IARC. Chemical agents and related occupations. IARC Monogr Eval Carcinog Risks Hum. 2012;100F a. [PMC free article: PMC4781612] [PubMed: 23189753]
- Iarmarcovai G, Bonassi S, Botta A, et al. Genetic polymorphisms and micronucleus formation: a review of the literature. Mutat Res. 2008;658:215–233. [PubMed: 18037339] [CrossRef]
- Ide R, Mizoue T, Fujino Y, et al. JACC Study Group. Cigarette smoking, alcohol drinking, and oral and pharyngeal cancer mortality in Japan. Oral Dis. 2008;14:314–319. [PubMed: 18449960] [CrossRef]
- Iguchi T, Sugita S, Wang CY, et al. MnSOD genotype and prostate cancer risk as a function of NAT genotype and smoking status. In Vivo. 2009;23:7–12. [PMC free article: PMC2670457] [PubMed: 19368118]
- IIPS; International Institute for Population Sciences (2010).* Mumbai and Ministry of Health and Family Welfare, Government of India. Global Adult Tobacco Survey India (GATS India). New Delhi, India: Ministry of Health and Family Welfare, Government of India.
- Ingelman-Sundberg M. Human drug metabolising cytochrome P450 enzymes: properties and polymorphisms. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:89–104. [PubMed: 14574440] [CrossRef]
- Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5:6–13. [PubMed: 15492763] [CrossRef]
- Ingelman-Sundberg M, Ronis MJ, Lindros KO, et al. Ethanol-inducible cytochrome P4502E1: regulation, enzymology and molecular biology. Alcohol Alcohol Suppl. 1994;2:131–139. [PubMed: 8974327]
- Ingelman-Sundberg M, Sim SC, Gomez A, Rodriguez-Antona C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther. 2007;116:496–526. [PubMed: 18001838] [CrossRef]
- Innes KE, Byers TE. Smoking during pregnancy and breast cancer risk in very young women (United States). Cancer Causes Control. 2001;12:179–185. [PubMed: 11246847] [CrossRef]
- Inoue H, Kiyohara C, Marugame T, et al. Cigarette smoking, CYP1A1 MspI and GSTM1 genotypes, and colorectal adenomas. Cancer Res. 2000;60:3749–3752. [PubMed: 10919645]
- Inoue M, Tajima K, Takezaki T, et al. Epidemiology of pancreatic cancer in Japan: a nested case-control study from the Hospital-based Epidemiologic Research Program at Aichi Cancer Center (HERPACC). Int J Epidemiol. 2003;32:257–262. [PubMed: 12714546] [CrossRef]
- Ioannidis JP. Calibration of credibility of agnostic genome-wide associations. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:964–972. [PubMed: 18361430]
- Iodice S, Gandini S, Maisonneuve P, Lowenfels AB. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks Arch Surg. 2008;393:535–545. [PubMed: 18193270] [CrossRef]
- Iribarren C, Tekawa IS, Sidney S, Friedman GD. Effect of cigar smoking on the risk of cardiovascular disease, chronic obstructive pulmonary disease, and cancer in men. N Engl J Med. 1999;340:1773–1780. [PubMed: 10362820] [CrossRef]
- Isaksson B, Jonsson F, Pedersen NL, et al. Lifestyle factors and pancreatic cancer risk: a cohort study from the Swedish Twin Registry. Int J Cancer. 2002;98:480–482. [PubMed: 11920604] [CrossRef]
- Izzotti A, Calin GA, Arrigo P, et al. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009;23:806–812. [PMC free article: PMC2653990] [PubMed: 18952709] [CrossRef]
- Jacob P 3rd, Wilson M, Benowitz NL. Determination of phenolic metabolites of polycyclic aromatic hydrocarbons in human urine as their pentafluorobenzyl ether derivatives using liquid chromatography-tandem mass spectrometry. Anal Chem. 2007;79:587–598. [PubMed: 17222024] [CrossRef]
- Jain MG, Howe GR, Rohan TE. Nutritional factors and endometrial cancer in Ontario, Canada. Cancer Control. 2000;7:288–296. [PubMed: 10832115]
- Jalas JR, Hecht SS, Murphy SE. Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco specific carcinogen. Chem Res Toxicol. 2005;18:95–110. [PubMed: 15720112] [CrossRef]
- Jass JR. Molecular heterogeneity of colorectal cancer: Implications for cancer control. Surg Oncol. 2007;16 Suppl 1:S7–S9. [PubMed: 18023574] [CrossRef]
- Jass JR, Young J, Leggett BA. Hyperplastic polyps and DNA microsatellite unstable cancers of the colorectum. Histopathology. 2000;37:295–301. [PubMed: 11012735]
- Jayalekshmy PA, Akiba S, Nair MK, et al. Bidi smoking and lung cancer incidence among males in Karunagappally cohort in Kerala, India. Int J Cancer. 2008;123:1390–1397. [PubMed: 18623085] [CrossRef]
- Jean RT, Wilkinson AV, Spitz MR, et al. 2011Psychosocial risk and correlates of early menarche in Mexican-American girls. Am J Epidemiol 1731203–1210.Epub 2011 Mar 31. [PMC free article: PMC3121322] [PubMed: 21454827]
- Jee SH, Samet JM, Ohrr H, et al. Smoking and cancer risk in Korean men and women. Cancer Causes Control. 2004;15:341–348. [PubMed: 15141135] [CrossRef]
- Jeffreys M, Warren R, Gunnell D, et al. Life course breast cancer risk factors and adult breast density (United Kingdom). Cancer Causes Control. 2004;15:947–955. [PubMed: 15577297]
- Jensen J, Christiansen C. Effects of smoking on serum lipoproteins and bone mineral content during postmenopausal hormone replacement therapy. Am J Obstet Gynecol. 1988;159:820–825. [PubMed: 2972209]
- Jensen J, Christiansen C, Rødbro P. Cigarette smoking, serum estrogens, and bone loss during hormone-replacement therapy early after menopause. N Engl J Med. 1985;313:973–975. [PubMed: 4047104] [CrossRef]
- Ji BT, Dai Q, Gao YT, et al. Cigarette and alcohol consumption and the risk of colorectal cancer in Shanghai, China. Eur J Cancer Prev. 2002;11:237–244. [PubMed: 12131657] [CrossRef]
- Ji BT, Weissfeld JL, Chow WH, et al. Tobacco smoking and colorectal hyperplastic and adenomatous polyps. Cancer Epidemiol Biomarkers Prev. 2006;15:897–901. [PubMed: 16702367] [CrossRef]
- Jiang J, Suzuki S, Xiang J, et al. Plasma carotenoid, alpha-tocopherol and retinol concentrations and risk of colorectal adenomas: A case-control study in Japan. Cancer Lett. 2005;226:133–141. [PubMed: 15885891] [CrossRef]
- Jin MJ, Chen K, Song L, et al. The association of the DNA repair gene XRCC3 Thr241Met polymorphism with susceptibility to colorectal cancer in a Chinese population. Cancer Genet Cytogenet. 2005;163:38–43. [PubMed: 16271954] [CrossRef]
- Jin Q, Menter DG, Mao L, et al. Survivin expression in normal human bronchial epithelial cells: an early and critical step in tumorigenesis induced by tobacco exposure. Carcinogenesis. 2008;29:1614–1622. [PMC free article: PMC2516487] [PubMed: 18635526] [CrossRef]
- Johnson KC. Accumulating evidence on passive and active smoking and breast cancer risk. Int J Cancer. 2005;117:619–628. [PubMed: 15929073] [CrossRef]
- Johnson KC, Goodman M, Kelsh M, et al. Passive and active smoking as possible confounders in studies of breast cancer risk. Epidemiology. 2002;13:610–611, author reply 610–611. [PubMed: 12192238] [CrossRef]
- Johnson KC, Hu J, Mao Y., Canadian Cancer Registries Epidemiology Research Group. Passive and active smoking and breast cancer risk in Canada, 1994–97. Cancer Causes Control. 2000;11:211–221. [PubMed: 10782655] [CrossRef]
- Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. [PubMed: 11323676] [CrossRef]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. [PMC free article: PMC3894624] [PubMed: 17320506] [CrossRef]
- Kabat GC, Chang CJ, Wynder EL. The role of tobacco, alcohol use, and body mass index in oral and pharyngeal cancer. Int J Epidemiol. 1994;23:1137–1144. [PubMed: 7721514] [CrossRef]
- Kabat GC, Hebert JR. Use of mentholated cigarettes and oropharyngeal cancer. Epidemiology. 1994;5:183–188. [PubMed: 8172993]
- Kabat GC, Ng SKC, Wynder EL. Tobacco, alcohol intake, and diet in relation to adenocarcinoma of the esophagus and gastric cardia. Cancer Causes Control. 1993;4:123–132. [PubMed: 8481491]
- Kaczynski AT, Manske SR, Mannell RC, Grewal K. Smoking and physical activity: a systematic review. Am J Health Behav. 2008;32:93–110. [PubMed: 18021037]
- Kadlubar FF, Beland FA (1985). Chemical properties of ultimate carcinogenic metabolites of arylamines and arylamides. In: Polycyclic Hydrocarbons and Carcinogenesis. Harvey RG, editor. Washington, D.C.: American Chemical Society.
- Kaerlev L, Teglbjaerg PS, Sabroe S, et al. The importance of smoking and medical history for development of small bowel carcinoid tumor: a European population-based case-control study. Cancer Causes Control. 2002;13:27–34. [PubMed: 11899115]
- Kamataki T, Fujieda M, Kiyotani K, et al. Genetic polymorphism of CYP2A6 as one of the potential determinants of tobacco-related cancer risk. Biochem Biophys Res Commun. 2005;338:306–310. [PubMed: 16176798] [CrossRef]
- Kanda J, Matsuo K, Kawase T, et al. 2009Association of alcohol intake and smoking with malignant lymphoma risk in Japanese: a hospital-based case-control study at Aichi Cancer Center. Cancer Epidemiol Biomarkers Prev 182436–2441.Epub 2009 Sep 1. [PubMed: 19723911]
- Kapeu AS, Luostarinen T, Jellum E, et al. Is smoking an independent risk factor for invasive cervical cancer? A nested case-control study within Nordic biobanks. Am J Epidemiol. 2009;169:480–488. [PubMed: 19074773] [CrossRef]
- Kasahara M, Osawa K, Yoshida K, et al. Association of MUTYH Gln324His and APEX1 Asp148Glu with colorectal cancer and smoking in a Japanese population. J Exp Clin Cancer Res. 2008;27:49. [PMC free article: PMC2575194] [PubMed: 18823566] [CrossRef]
- Kato M, Loomis D, Brooks LM, et al. Urinary biomarkers in charcoal workers exposed to wood smoke in Bahia State, Brazil. Cancer Epidemiol Biomarkers Prev. 2004;13:1005–1012. [PubMed: 15184257]
- Kawajiri K. CYP1A1. IARC Sci Publ. 1999;148:159–172. [PubMed: 10493257]
- Kawajiri K, Eguchi H, Nakachi K, et al. Association of CYP1A1 germ line polymorphisms with mutations of the p53 gene in lung cancer. Cancer Res. 1996;56:72–76. [PubMed: 8548778]
- Kelsey JL, Gammon MD, John EM. Reproductive factors and breast cancer. Epidemiol Rev. 1993;15:36–47. [PubMed: 8405211]
- Kelsey JL, LiVolsi VA, Holford TR, et al. A case-control study of cancer of the endometrium. Am J Epidemiol. 1982;116:333–342. [PubMed: 7114042]
- Kenfield SA, Stampfer MJ, Rosner BA, Colditz GA. Smoking and smoking cessation in relation to mortality in women. JAMA. 2008;299:2037–2047. [PMC free article: PMC2879642] [PubMed: 18460664] [CrossRef]
- Kesarwani P, Singh R, Mittal RD. Association of GSTM3 intron 6 variant with cigarette smoking, tobacco chewing and alcohol as modifier factors for prostate cancer risk. Arch Toxicol. 2009;83:351–356. [PubMed: 18668224] [CrossRef]
- Ketterer B, Harris JM, Talaska G, et al. The human glutathione S-transferase supergene family, its polymorphism, and its effects on susceptibility to lung cancer. Environ Health Perspect. 1992;98:87–94. [PMC free article: PMC1519626] [PubMed: 1486868] [CrossRef]
- Key TJ, Pike MC, Baron JA, et al. Cigarette smoking and steroid hormones in women. J Steroid Biochem Mol Biol. 1991;39 4A:529–534. [PubMed: 1832941] [CrossRef]
- Key TJ, Pike MC, Brown JB, et al. Cigarette smoking and urinary oestrogen excretion in premenopausal and post-menopausal women. Br J Cancer. 1996;74:1313–1316. [PMC free article: PMC2075917] [PubMed: 8883424]
- Khaw KT, Tazuke S, Barrett-Connor E. Cigarette smoking and levels of adrenal androgens in postmenopausal women. N Engl J Med. 1988;318:1705–1709. [PubMed: 2967432] [CrossRef]
- Kiemeney LA, Thorlacius S, Sulem P, et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet. 2008;40:1307–1312. [PMC free article: PMC4539560] [PubMed: 18794855] [CrossRef]
- Kim CS, Kim MC, Cheong HK, et al. 2005[The association of obesity and left colonic adenomatous polyps in Korean adult men]J Prev Med Pub Health 38415–419.PMID:16358826. [PubMed: 16358826]
- Kim HJ, Lee SM, Choi NK, et al. Smoking and colorectal cancer risk in the Korean elderly. J Prev Med Pub Health. 2006;39:123–129. [ ][Article in Korean] [PubMed: 16615266]
- Kim JI, Park YJ, Kim KH, et al. hOGG1 Ser326Cys polymorphism modifies the significance of the environmental risk factor for colon cancer. World J Gastroenterol. 2003;9:956–960. [PMC free article: PMC4611404] [PubMed: 12717837]
- Kim SG, Novak RF. Role of P450IIE1 in the metabolism of 3-hydroxypyridine, a constituent of tobacco smoke: redox cycling and DNA strand scission by the metabolite 2,5-dihydroxypyridine. Cancer Res. 1990;50:5333–5339. [PubMed: 2167153]
- Kim Y, Shin A, Gwack J, et al. Cigarette smoking and gastric cancer risk in a community-based cohort study in Korea. J Prev Med Public Health. 2007;40:467–474. [PubMed: 18063902] [CrossRef]
- Kiyohara C, Yoshimasu K, Shirakawa T, Hopkin JM. Genetic polymorphisms and environmental risk of lung cancer: a review. Rev Environ Health. 2004;19:15–38. [PubMed: 15186038]
- Kiyohara C, Yoshimasu K, Takayama K, Nakanishi Y. NQO1, MPO, and the risk of lung cancer: a HuGE review. Genet Med. 2005;7:463–478. [PubMed: 16170238] [CrossRef]
- Kiyohara C, Yoshimasu K, Takayama K, Nakanishi Y. EPHX1 polymorphisms and the risk of lung cancer: a HuGE review. Epidemiology. 2006;17:89–99. [PubMed: 16357600]
- Kjaerheim K, Gaard M, Andersen A. The role of alcohol, tobacco, and dietary factors in upper aerogastric tract cancers: a prospective study of 10,900 Norwegian men. Cancer Causes Control. 1998;9:99–108. [PubMed: 9486469] [CrossRef]
- Klotz U, Ammon E. Clinical and toxicological consequences of the inductive potential of ethanol. Eur J Clin Pharmacol. 1998;54:7–12. [PubMed: 9591923] [CrossRef]
- Ko Y, Abel J, Harth V, et al. Association of CYP1B1 codon 432 mutant allele in head and neck squamous cell cancer is reflected by somatic mutations of p53 in tumor tissue. Cancer Res. 2001;61:4398–4404. [PubMed: 11389067]
- Ko YC, Huang YL, Lee CH, et al. Betel quid chewing, cigarette smoking and alcohol consumption related to oral cancer in Taiwan. J Oral Pathol Med. 1995;24:450–453. [PubMed: 8600280] [CrossRef]
- Koizumi Y, Tsubono Y, Nakaya N, et al. Cigarette smoking and the risk of gastric cancer: a pooled analysis of two prospective studies in Japan. Int J Cancer. 2004;112:1049–1055. [PubMed: 15386347] [CrossRef]
- Kono S, Ikeda M, Tokudome S, et al. Cigarette smoking, alcohol and cancer mortality: a cohort study of male Japanese physicians. Jpn J Cancer Res. 1987;78:1323–1328. [PubMed: 3123436]
- Kotwall CA. Smoking as an etiologic factor in the development of Warthin’s tumor of the parotid gland. Am J Surg. 1992;164:646–647. [PubMed: 1334381] [CrossRef]
- Koumantaki Y, Tzonou A, Koumantakis E, et al. A case-control study of cancer of endometrium in Athens. Int J Cancer. 1989;43:795–799. [PubMed: 2714884] [CrossRef]
- Kreuzer M, Boffetta P, Whitley E, et al. Gender differences in lung cancer risk by smoking: a multicentre case-control study in Germany and Italy. Br J Cancer. 2000;82:227–233. [PMC free article: PMC2363175] [PubMed: 10638994]
- Krewski D, Lubin JH, Zielinski JM, et al. Residential radon and risk of lung cancer: a combined analysis of 7 North American case-control studies. Epidemiology. 2005;16:137–145. [PubMed: 15703527]
- Kropp S, Chang-Claude J. Active and passive smoking and risk of breast cancer by age 50 years among German women. Am J Epidemiol. 2002;156:616–626. [PubMed: 12244030] [CrossRef]
- Kubík A, Zatloukal P, Boyle P, et al. A case-control study of lung cancer among Czech women. Lung Cancer. 2001;31:111–122. [PubMed: 11165390]
- Kuljukka-Rabb T, Nylund L, Vaaranrinta R, et al. The effect of relevant genotypes on PAH exposure-related biomarkers. J Expo Anal Environ Epidemiol. 2002;12:81–91. [PubMed: 11859435] [CrossRef]
- Kuller LH, Ockene JK, Meilahn E, et al. MRFIT Research Group. Cigarette smoking and mortality. Prev Med. 1991;20:638–654. [PubMed: 1758843] [CrossRef]
- Kundu CN, Balusu R, Jaiswal AS, et al. Cigarette smoke condensate-induced level of adenomatous polyposis coli blocks long-patch base excision repair in breast epithelial cells. Oncogene. 2007;26:1428–1438. [PubMed: 16924228] [CrossRef]
- Kuper H, Titus-Ernstoff L, Harlow BL, Cramer DW. Population based study of coffee, alcohol and tobacco use and risk of ovarian cancer. Int J Cancer. 2000;88:313–318. b. [PubMed: 11004686] [CrossRef]
- Kuper H, Tzonou A, Kaklamani E, et al. Tobacco smoking, alcohol consumption and their interaction in the causation of hepatocellular carcinoma. Int J Cancer. 2000;85:498–502. a. [PubMed: 10699921] [CrossRef]
- Kurian AW, Balise RR, McGuire V, Whittemore AS. Histologic types of epithelial ovarian cancer: have they different risk factors? Gynecol Oncol. 2005;96:520–530. [PubMed: 15661246] [CrossRef]
- Kurosawa M, Kikuchi S, Xu J, Inaba Y. Highly salted food and mountain herbs elevate the risk for stomach cancer death in a rural area of Japan. J Gastroenterol Hepatol. 2006;21:1681–1686. [PubMed: 16984589] [CrossRef]
- La Torre G, Chiaradia G, Gianfagna F, et al. Smoking status and gastric cancer risk: an updated meta-analysis of case-control studies published in the past ten years. Tumori. 2009;95:13–22. [PubMed: 19366050]
- La Vecchia C, Franceschi S, Bosetti C, et al. Time since stopping smoking and the risk of oral and pharyngeal cancers. J Natl Cancer Inst. 1999;91:726–728. [PubMed: 10218516] [CrossRef]
- Lacey JV Jr, Leitzmann MF, Chang SC, et al. Endometrial cancer and menopausal hormone therapy in the National Institutes of Health-AARP Diet and Health Study cohort. Cancer. 2007;109:1303–1311. [PubMed: 17315161] [CrossRef]
- Ladeiras-Lopes R, Pereira AK, Nogueira A, et al. Smoking and gastric cancer: systematic review and meta-analysis of cohort studies. Cancer Causes Control. 2008;19:689–701. [PubMed: 18293090] [CrossRef]
- Lagergren J, Bergström R, Lindgren A, Nyrén O. The role of tobacco, snuff and alcohol use in the aetiology of cancer of the oesophagus and gastric cardia. Int J Cancer. 2000;85:340–346. [PubMed: 10652424] [CrossRef]
- Lam TK, Gallicchio L, Lindsley K, et al. Cruciferous vegetable consumption and lung cancer risk: a systematic review. Cancer Epidemiol Biomarkers Prev. 2009;18:184–195. [PMC free article: PMC2735794] [PubMed: 19124497] [CrossRef]
- Lan Q, Zhang L, Li G, et al. Hematotoxicity in workers exposed to low levels of benzene. Science. 2004;306:1774–1776. [PMC free article: PMC1256034] [PubMed: 15576619] [CrossRef]
- Landi MT, Chatterjee N, Yu K, et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am J Hum Genet. 2009;85:679–691. [PMC free article: PMC2775843] [PubMed: 19836008] [CrossRef]
- Lang M, Pelkonen O. Metabolism of xenobiotics and chemical carcinogenesis. IARC Sci Publ. 1999;(148):13–22. [PubMed: 10493245]
- Lanier A, Bender T, Talbot M, et al. Nasopharyngeal carcinoma in Alaskan Eskimos Indians, and Aleuts: a review of cases and study of Epstein-Barr virus, HLA, and environmental risk factors. Cancer. 1980;46:2100–2106. [PubMed: 6253051] [CrossRef]
- Lao Y, Yu N, Kassie F, et al. Analysis of pyridyloxobutyl DNA adducts in F344 rats chronically treated with (R)- and (S)-N’-nitrosonornicotine. Chem Res Toxicol. 2007;20:246–256. [PMC free article: PMC2518847] [PubMed: 17305408] [CrossRef]
- Lapidus L, Lindstedt G, Lundberg PA, et al. Concentrations of sex-hormone binding globulin and corticosteroid binding globulin in serum in relation to cardiovascular risk factors and to 12-year incidence of cardiovascular disease and overall mortality in postmenopausal women. Clin Chem. 1986;32:146–152. [PubMed: 3940696]
- Larsen IK, Grotmol T, Almendingen K, Hoff G. Lifestyle as a predictor for colonic neoplasia in asymptomatic individuals. BMC Gastroenterol. 2006;6:5. [PMC free article: PMC1374667] [PubMed: 16412216] [CrossRef]
- Larsson SC, Permert J, Håkansson N, et al. Overall obesity, abdominal adiposity, diabetes and cigarette smoking in relation to the risk of pancreatic cancer in two Swedish population-based cohorts. Br J Cancer. 2005;93:1310–1315. [PMC free article: PMC2361517] [PubMed: 16288300] [CrossRef]
- Lash TL, Aschengrau A. Active and passive cigarette smoking and the occurrence of breast cancer. Am J Epidemiol. 1999;149:5–12. [PubMed: 9883788]
- Lash TL, Aschengrau A. A null association between active or passive cigarette smoking and breast cancer risk. Breast Cancer Res Treat. 2002;75:181–184. [PubMed: 12243511] [CrossRef]
- Lau A, Belanger CL, Winn LM. In utero and acute exposure to benzene: investigation of DNA double-strand breaks and DNA recombination in mice. Mutat Res. 2009;676:74–82. [PubMed: 19486867]
- Launoy G, Milan C, Faivre J, et al. Tobacco type and risk of squamous cell cancer of the oesophagus in males: a French multicentre case-control study. Int J Epidemiol. 2000;29:36–42. [PubMed: 10750601] [CrossRef]
- Law MR, Cheng R, Hackshaw AK, et al. Cigarette smoking, sex hormones and bone density in women. Eur J Epidemiol. 1997;13:553–558. [PubMed: 9258568] [CrossRef]
- Lawlor DA, Ebrahim S, Smith GD. Smoking before the birth of a first child is not associated with increased risk of breast cancer: findings from the British Women’s Heart and Health Cohort Study and a meta-analysis. Br J Cancer. 2004;91:512–518. [PMC free article: PMC2409831] [PubMed: 15226777] [CrossRef]
- Lawrence C, Tessaro I, Durgerian S, et al. Smoking, body weight, and early-stage endometrial cancer. Cancer. 1987;59:1665–1669. [PubMed: 3828966] [CrossRef]
- Lawrence C, Tessaro I, Durgerian S, et al. Advanced-stage endometrial cancer: contributions of estrogen use, smoking, and other risk factors. Gynecol Oncol. 1989;32:41–45. [PubMed: 2909447] [CrossRef]
- Le Calvez F, Mukeria A, Hunt JD, et al. TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res. 2005;65:5076–5083. [PubMed: 15958551] [CrossRef]
- Le Marchand L, Derby KS, Murphy SE, et al. Smokers with the CHRNA lung cancer-associated variants are exposed to higher levels of nicotine equivalents and a carcinogenic tobacco-specific nitrosamine. Cancer Res. 2008;68:9137–9140. [PMC free article: PMC2587068] [PubMed: 19010884] [CrossRef]
- Le Marchand L, Guo C, Benhamou S, et al. Pooled analysis of the CYP1A1 exon 7 polymorphism and lung cancer (United States). Cancer Causes Control. 2003;14:339–346. [PubMed: 12846365] [CrossRef]
- Le Marchand L, Sivaraman L, Pierce L, et al. Associations of CYP1A1, GSTM1, and CYP2E1 polymorphisms with lung cancer suggest cell type specificities to tobacco carcinogens. Cancer Res. 1998;58:4858–4863. [PubMed: 9809991]
- Le Marchand L, Wilkens LR, Kolonel LN, et al. Associations of sedentary lifestyle, obesity, smoking, alcohol use, and diabetes with the risk of colorectal cancer. Cancer Res. 1997;57:4787–4794. [PubMed: 9354440]
- Lea IA, Jackson MA, Li X, et al. Genetic pathways and mutation profiles of human cancers: site- and exposure-specific patterns. Carcinogenesis. 2007;28:1851–1858. [PMC free article: PMC2131731] [PubMed: 17693665] [CrossRef]
- Lee CH, Wu DC, Lee JM, et al. Carcinogenetic impact of alcohol intake on squamous cell carcinoma risk of the oesophagus in relation to tobacco smoking. Eur J Cancer. 2007;43:1188–1199. a. [PubMed: 17383866] [CrossRef]
- Lee CH, Wu DC, Lee JM, et al. Anatomical subsite discrepancy in relation to the impact of the consumption of alcohol, tobacco and betel quid on esophageal cancer. Int J Cancer. 2007;120:1755–1762. b. [PubMed: 17230518] [CrossRef]
- Lee CY, Lee JY, Kang JW, Kim H. Effects of genetic polymorphisms of CYP1A1, CYP2E1, GSTM1, and GSTT1 on the urinary levels of 1-hydroxypyrene and 2-naphthol in aircraft maintenance workers. Toxicol Lett. 2001;123:115–124. [PubMed: 11641039] [CrossRef]
- Lee JM, Yanagawa J, Peebles KA, et al. Inflammation in lung carcinogenesis: new targets for lung cancer chemoprevention and treatment. Crit Rev Oncol Hematol. 2008;66:208–217. b. [PMC free article: PMC2483429] [PubMed: 18304833] [CrossRef]
- Lee K, Cáceres D, Varela N, et al. Allelic variants of cytochrome P4501A1 (CYP1A1), glutathione S transferase M1 (GSTM1) polymorphisms and their association with smoking and alcohol consumption as gastric cancer susceptibility biomarkers. Rev Med Chil. 2006;134:1107–1115. [PubMed: 17171211]
- Lee KM, Kang D, Clapper ML, et al. CYP1A1, GSTM1, and GSTT1 polymorphisms, smoking, and lung cancer risk in a pooled analysis among Asian populations. Cancer Epidemiol Biomarkers Prev. 2008;17:1120–1126. a. [PubMed: 18463401] [CrossRef]
- Lee KW, Kuo WR, Tsai SM, et al. Different impact from betel quid, alcohol and cigarette: risk factors for pharyngeal and laryngeal cancer. Int J Cancer. 2005;117:831–836. a. [PubMed: 15957167] [CrossRef]
- Lee PN (1999). Uses and abuses of cotinine as a marker of tobacco smoke exposure. In: Analytical Determination of Nicotine and Related Compounds and Their Metabolites. Gorrod JW, Jacob P III, editors. Amsterdam: Elsevier, pp. 669–719 .
- Lee PN. Relation between exposure to asbestos and smoking jointly and the risk of lung cancer. Occup Environ Med. 2001;58:145–153. [PMC free article: PMC1740104] [PubMed: 11171926] [CrossRef]
- Lee YCA, Cohet C, Yang YC, et al. Meta-analysis of epidemiologic studies on cigarette smoking and liver cancer. Int J Epidemiol. 2009;38:1497–1511. [PubMed: 19720726] [CrossRef]
- Lesko SM, Rosenberg L, Kaufman DW, et al. Cigarette smoking and the risk of endometrial cancer. N Engl J Med. 1985;313:593–596. [PubMed: 4022045] [CrossRef]
- Leuchtenberger C, Leuchtenberger R (1970). Effects of chronic inhalation of whole fresh cigarette smoke and of its gas phase on pulmonary tumorigenesis in Snell's mice. In: Morphology of Experimental Respiratory Carcinogenesis. Proceedings of a Biology Division. Nettesheim P, Hanna MGJ, Deathevage JWJ, editors. Oak Ridge National Laboratory Conference, US Atomic Energy Commission, pp. 329–346.
- Levi F, La Vecchia C. Tobacco smoking and prostate cancer: time for an appraisal. Ann Oncol. 2001;12:733–738. [PubMed: 11484946] [CrossRef]
- Levi F, la Vecchia C, Decarli A. Cigarette smoking and the risk of endometrial cancer. Eur J Cancer Clin Oncol. 1987;23:1025–1029. [PubMed: 3665988] [CrossRef]
- Levi F, Ollyo JB, La Vecchia C, et al. The consumption of tobacco, alcohol and the risk of adenocarcinoma in Barrett’s oesophagus. Int J Cancer. 1990;45:852–854. [PubMed: 2335388] [CrossRef]
- Lewin F, Norell SE, Johansson H, et al. Smoking tobacco, oral snuff, and alcohol in the etiology of squamous cell carcinoma of the head and neck: a population-based case-referent study in Sweden. Cancer. 1998;82:1367–1375. [PubMed: 9529030]
- Li CI, Malone KE, Daling JR. The relationship between various measures of cigarette smoking and risk of breast cancer among older women 65–79 years of age (United States). Cancer Causes Control. 2005;16:975–985. [PubMed: 16132806] [CrossRef]
- Li JY, Ershow AG, Chen ZJ, et al. A case-control study of cancer of the esophagus and gastric cardia in Linxian. Int J Cancer. 1989;43:755–761. [PubMed: 2714880] [CrossRef]
- Li W, Ray RM, Gao DL, et al. Occupational risk factors for pancreatic cancer among female textile workers in Shanghai, China. Occup Environ Med. 2006;63:788–793. [PMC free article: PMC2078009] [PubMed: 16847032] [CrossRef]
- Li X, Hemminki K. Inherited predisposition to early onset lung cancer according to histological type. International Journal of Cancer. 2004;112:451–457. [PubMed: 15382071] [CrossRef]
- Li Y, Jiang Y, Zhang M, et al. Drinking behaviour among men and women in China: the 2007 China Chronic Disease and Risk Factor Surveillance. Addiction. 2011;106:1946–1956. [PubMed: 21771141]
- Li Y, Millikan RC, Bell DA, et al. Cigarette smoking, cytochrome P4501A1 polymorphisms, and breast cancer among African-American and white women. Breast Cancer Res. 2004;6:R460–R473. [PMC free article: PMC468667] [PubMed: 15217514] [CrossRef]
- Liang PS, Chen TY, Giovannucci E. Cigarette smoking and colorectal cancer incidence and mortality: systematic review and meta-analysis. Int J Cancer. 2009;124:2406–2415. [PubMed: 19142968] [CrossRef]
- Liaw KM, Chen CJ. Mortality attributable to cigarette smoking in Taiwan: a 12-year follow-up study. Tob Control. 1998;7:141–148. [PMC free article: PMC1759691] [PubMed: 9789932] [CrossRef]
- Liddell FD. The interaction of asbestos and smoking in lung cancer. Ann Occup Hyg. 2001;45:341–356. [PubMed: 11418084]
- Lieber CS. Microsomal ethanol-oxidizing system (MEOS): the first 30 years (1968–1998)–a review. Alcohol Clin Exp Res. 1999;23:991–1007. [PubMed: 10397283] [CrossRef]
- Lieber CS. The discovery of the microsomal ethanol oxidizing system and its physiologic and pathologic role. Drug Metab Rev. 2004;36:511–529. [PubMed: 15554233] [CrossRef]
- Lijinsky W (1992). Chemistry and biology of N-Nitroso compounds. Monographs on Cancer Research. Cambridge University Press.
- Lilla C, Risch A, Kropp S, Chang-Claude J. SULT1A1 genotype, active and passive smoking, and breast cancer risk by age 50 years in a German case-control study. Breast Cancer Res. 2005;7:R229–R237. [PMC free article: PMC1064130] [PubMed: 15743503] [CrossRef]
- Lilla C, Verla-Tebit E, Risch A, et al. Effect of NAT1 and NAT2 genetic polymorphisms on colorectal cancer risk associated with exposure to tobacco smoke and meat consumption. Cancer Epidemiol Biomarkers Prev. 2006;15:99–107. [PubMed: 16434594]
- Lim U, Morton LM, Subar AF, et al. Alcohol, smoking, and body size in relation to incident Hodgkin's and non-Hodgkin's lymphoma risk. Am J Epidemiol. 2007;166:697–708. [PubMed: 17596266]
- Limburg PJ, Vierkant RA, Cerhan JR, et al. Cigarette smoking and colorectal cancer: long-term, subsite-specific risks in a cohort study of postmenopausal women. Clin Gastroenterol Hepatol. 2003;1:202–210. [PubMed: 15017492]
- Lin TM, Chen KP, Lin CC, et al. Retrospective study on nasopharyngeal carcinoma. J Natl Cancer Inst. 1973;51:1403–1408. [PubMed: 4762926]
- Lin Y, Tamakoshi A, Kawamura T, et al. JACC Study Group. Japan Collaborative Cohort. A prospective cohort study of cigarette smoking and pancreatic cancer in Japan. Cancer Causes Control. 2002;13:249–254. a. [PubMed: 12020106] [CrossRef]
- Lindblad M, Rodríguez LA, Lagergren J. Body mass, tobacco and alcohol and risk of esophageal, gastric cardia, and gastric non-cardia adenocarcinoma among men and women in a nested case-control study. Cancer Causes Control. 2005;16:285–294. [PubMed: 15947880] [CrossRef]
- Lindemann K, Vatten LJ, Ellstrøm-Engh M, Eskild A. Body mass, diabetes and smoking, and endometrial cancer risk: a follow-up study. Br J Cancer. 2008;98:1582–1585. [PMC free article: PMC2391095] [PubMed: 18362938] [CrossRef]
- Linet MS, McLaughlin JK, Hsing AW, et al. Is cigarette smoking a risk factor for non-Hodgkin’s lymphoma or multiple myeloma? Results from the Lutheran Brotherhood Cohort Study. Leuk Res. 1992;16:621–624. [PubMed: 1635380] [CrossRef]
- Linseisen J, Rohrmann S, Miller AB, et al. Fruit and vegetable consumption and lung cancer risk: updated information from the European Prospective Investigation into Cancer and Nutrition (EPIC). International Journal of Cancer. 2007;121:1103–1114. [PubMed: 17487840] [CrossRef]
- Lissowska J, Brinton LA, Garcia-Closas M. Re: more data regarding the effects of passive smoking on breast cancer risk among younger women. Int J Cancer. 2007;120:2517–2518. [PubMed: 17290385] [CrossRef]
- Lissowska J, Brinton LA, Zatonski W, et al. Tobacco smoking, NAT2 acetylation genotype and breast cancer risk. Int J Cancer. 2006;119:1961–1969. [PubMed: 16721725] [CrossRef]
- Lissowska J, Pilarska A, Pilarski P, et al. Smoking, alcohol, diet, dentition and sexual practices in the epidemiology of oral cancer in Poland. Eur J Cancer Prev. 2003;12:25–33. [PubMed: 12548107] [CrossRef]
- Loerbroks A, Schouten LJ, Goldbohm RA, van den Brandt PA. Alcohol consumption, cigarette smoking, and endometrial cancer risk: results from the Netherlands Cohort Study. Cancer Causes Control. 2007;18:551–560. [PMC free article: PMC1914283] [PubMed: 17437180] [CrossRef]
- London SJ, Colditz GA, Stampfer MJ, et al. Prospective study of relative weight, height, and risk of breast cancer. JAMA. 1989;262:2853–2858. a. [PubMed: 2810620] [CrossRef]
- London SJ, Colditz GA, Stampfer MJ, et al. Prospective study of smoking and the risk of breast cancer. J Natl Cancer Inst. 1989;81:1625–1631. b. [PubMed: 2795691] [CrossRef]
- London SJ, Smart J, Daly AK. Lung cancer risk in relation to genetic polymorphisms of microsomal epoxide hydrolase among African-Americans and Caucasians in Los Angeles County. Lung Cancer. 2000;28:147–155. b. [PubMed: 10717332] [CrossRef]
- London SJ, Yuan JM, Chung FL, et al. Isothiocyanates, glutathione S-transferase M1 and T1 polymorphisms, and lung-cancer risk: a prospective study of men in Shanghai, China. Lancet. 2000;356:724–729. a. [PubMed: 11085692] [CrossRef]
- Longcope C, Baker R, Johnston CC Jr. Androgen and estrogen metabolism: relationship to obesity. Metabolism. 1986;35:235–237. [PubMed: 3456482] [CrossRef]
- Longcope C, Johnston CC Jr. Androgen and estrogen dynamics in pre- and postmenopausal women: a comparison between smokers and nonsmokers. J Clin Endocrinol Metab. 1988;67:379–383. [PubMed: 3392163] [CrossRef]
- Lopez AD, Collishaw N, Piha T. A descriptive model of the cigarette epidemic in developed countries. Tobacco Control. 1994;3:242–247. [CrossRef]
- López-Abente G, Pollán M, Monge V, Martínez-Vidal A. Tobacco smoking, alcohol consumption, and laryngeal cancer in Madrid. Cancer Detect Prev. 1992;16:265–271. [PubMed: 1473116]
- Lu J, Lian S, Sun X, et al. A case-control study on the risk factors of esophageal cancer in Linzhou. Zhonghua Liu Xing Bing Xue Za Zhi. 2000;21:434–436. [PubMed: 11860829]
- Lubet RA, Zhang Z, Wiseman RW, You M. Use of p53 transgenic mice in the development of cancer models for multiple purposes. Exp Lung Res. 2000;26:581–593. [PubMed: 11195457] [CrossRef]
- Lubin JH, Alavanja MC, Caporaso N, et al. Cigarette smoking and cancer risk: modeling total exposure and intensity. Am J Epidemiol. 2007;166:479–489. a. [PubMed: 17548786] [CrossRef]
- Lubin JH, Caporaso N, Hatsukami DK, et al. The association of a tobacco-specific biomarker and cigarette consumption and its dependence on host characteristics. Cancer Epidemiology, Biomarkers & Prevention. 2007;16:1852–1857. b. [PubMed: 17855705] [CrossRef]
- Lubin JH, Caporaso NE. Cigarette smoking and lung cancer: modeling total exposure and intensity. Cancer Epidemiology, Biomarkers & Prevention. 2006;15:517–523. [PubMed: 16537710] [CrossRef]
- Lubin JH, Kogevinas M, Silverman D, et al. Evidence for an intensity-dependent interaction of NAT2 acetylation genotype and cigarette smoking in the Spanish Bladder Cancer Study. Int J Epidemiol. 2007;36:236–241. [PubMed: 17510079] [CrossRef]
- Lubin JH, Virtamo J, Weinstein SJ, Albanes D. Cigarette smoking and cancer: intensity patterns in the alpha-tocopherol, beta-carotene cancer prevention study in Finnish men. Am J Epidemiol. 2008;167:970–975. [PubMed: 18250081] [CrossRef]
- Lüchtenborg M, Weijenberg MP, Kampman E, et al. Cigarette smoking and colorectal cancer: APC mutations, hMLH1 expression, and GSTM1 and GSTT1 polymorphisms. Am J Epidemiol. 2005;161:806–815. a. [PubMed: 15840612] [CrossRef]
- Lüchtenborg M, White KK, Wilkens L, et al. Smoking and colorectal cancer: different effects by type of cigarettes? Cancer Epidemiol Biomarkers Prev. 2007;16:1341–1347. [PubMed: 17626999] [CrossRef]
- Lurie G, Wilkens LR, Thompson PJ, et al. Genetic polymorphisms in the Paraoxonase 1 gene and risk of ovarian epithelial carcinoma. Cancer Epidemiol Biomarkers Prev. 2008;17:2070–2077. [PMC free article: PMC2729507] [PubMed: 18708400] [CrossRef]
- Lynch SM, Vrieling A, Lubin JH, et al. Cigarette smoking and pancreatic cancer: a pooled analysis from the pancreatic cancer cohort consortium. Am J Epidemiol. 2009;170:403–413. [PMC free article: PMC2733861] [PubMed: 19561064] [CrossRef]
- Mabuchi K, Bross DS, Kessler II. Cigarette smoking and nasopharyngeal carcinoma. Cancer. 1985;55:2874–2876. [PubMed: 3995493] [CrossRef]
- Macfarlane GJ, Zheng T, Marshall JR, et al. Alcohol, tobacco, diet and the risk of oral cancer: a pooled analysis of three case-control studies. Eur J Cancer B Oral Oncol. 1995;31B:181–187. [PubMed: 7549758] [CrossRef]
- Mack WJ, Preston-Martin S, Dal Maso L, et al. A pooled analysis of case-control studies of thyroid cancer: cigarette smoking and consumption of alcohol, coffee, and tea. Cancer Causes Control. 2003;14:773–785. [PubMed: 14674742] [CrossRef]
- Mackenzie PI, Owens IS, Burchell B, et al. The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence. Pharmacogenetics. 1997;7:255–269. [PubMed: 9295054]
- MacMahon B, Trichopoulos D, Cole P, Brown J. Cigarette smoking and urinary estrogens. N Engl J Med. 1982;307:1062–1065. [PubMed: 7121516]
- MacNicoll AD, Easty GC, Neville AM, et al. Metabolism and activation of carcinogenic polycyclic hydrocarbons by human mammary cells. Biochem Biophys Res Commun. 1980;95:1599–1606. [PubMed: 6774726] [CrossRef]
- Maden C, Sherman KJ, Beckmann AM, et al. History of circumcision, medical conditions, and sexual activity and risk of penile cancer. J Natl Cancer Inst. 1993;85:19–24. [PubMed: 8380060] [CrossRef]
- Magnusson C, Wedrén S, Rosenberg LU. Cigarette smoking and breast cancer risk: a population-based study in Sweden. Br J Cancer. 2007;97:1287–1290. [PMC free article: PMC2360473] [PubMed: 17912245] [CrossRef]
- Maier H, Dietz A, Gewelke U, et al. Tobacco and alcohol and the risk of head and neck cancer. Clin Investig. 1992;70:320–327. a. [PubMed: 1521046] [CrossRef]
- Maier H, Gewelke U, Dietz A, et al. Laryngeal cancer and occupation — results of the Heidelberg laryngeal cancer study. HNO. 1992;40:44–51. b[ ] [PubMed: 1568886]
- Maier H, Sennewald E, Heller GF, Weidauer H. Chronic alcohol consumption–the key risk factor for pharyngeal cancer. Otolaryngol Head Neck Surg. 1994;110:168–173. [PubMed: 7906410]
- Maier H, Tisch M. Epidemiology of laryngeal cancer: results of the Heidelberg case-control study. Acta Otolaryngol Suppl. 1997;527:160–164. [PubMed: 9197510] [CrossRef]
- Malaiyandi V, Sellers EM, Tyndale RF. Implications of CYP2A6 genetic variation for smoking behaviors and nicotine dependence. Clin Pharmacol Ther. 2005;77:145–158. [PubMed: 15735609] [CrossRef]
- Malats N. Genetic epidemiology of bladder cancer: scaling up in the identification of low-penetrance genetic markers of bladder cancer risk and progression. Scand J Urol Nephrol Suppl. 2008;42:131–140. [PubMed: 18815927] [CrossRef]
- Mandai M, Konishi I, Kuroda H, et al. Heterogeneous distribution of K-ras-mutated epithelia in mucinous ovarian tumors with special reference to histopathology. Hum Pathol. 1998;29:34–40. [PubMed: 9445131] [CrossRef]
- Mandelzweig L, Novikov I, Sadetzki S (2009). Smoking and risk of glioma: a meta-analysis. Cancer Causes Control. [PubMed: 19568697]
- Mane SS, Purnell DM, Hsu IC. Genotoxic effects of five polycyclic aromatic hydrocarbons in human and rat mammary epithelial cells. Environ Mol Mutagen. 1990;15:78–82. [PubMed: 2307152] [CrossRef]
- Manjer J, Andersson I, Berglund G, et al. Survival of women with breast cancer in relation to smoking. Eur J Surg. 2000;166:852–858. [PubMed: 11097150] [CrossRef]
- Manjer J, Malina J, Berglund G, et al. Smoking associated with hormone receptor negative breast cancer. Int J Cancer. 2001;91:580–584. [PubMed: 11251985] [CrossRef]
- Manuguerra M, Saletta F, Karagas MR, et al. XRCC3 and XPD/ERCC2 single nucleotide polymorphisms and the risk of cancer: a HuGE review. Am J Epidemiol. 2006;164:297–302. [PubMed: 16707649] [CrossRef]
- Mao GE, Morris G, Lu QY, et al. Glutathione S-transferase P1 Ile105Val polymorphism, cigarette smoking and prostate cancer. Cancer Detect Prev. 2004;28:368–374. [PubMed: 15542263] [CrossRef]
- Marchbanks PA, Wilson H, Bastos E, et al. Cigarette smoking and epithelial ovarian cancer by histologic type. Obstet Gynecol. 2000;95:255–260. [PubMed: 10674590] [CrossRef]
- Marsh GM, Youk AO, Buchanich JM, et al. Work in the metal industry and nasopharyngeal cancer mortality among formaldehyde-exposed workers. Regul Toxicol Pharmacol. 2007;48:308–319. [PubMed: 17544557] [CrossRef]
- Marshall JR, Hastrup JL, Ross JS. Mismeasurement and the resonance of strong confounders: correlated errors. Am J Epidemiol. 1999;150:88–96. [PubMed: 10400558]
- Marsit CJ, Karagas MR, Schned A, Kelsey KT. Carcinogen exposure and epigenetic silencing in bladder cancer. Ann N Y Acad Sci. 2006;1076:810–821. [PubMed: 17119258] [CrossRef]
- Mashberg A, Boffetta P, Winkelman R, Garfinkel L. Tobacco smoking, alcohol drinking, and cancer of the oral cavity and oropharynx among U.S. veterans. Cancer. 1993;72:1369–1375. [PubMed: 8339227] [CrossRef]
- Masson LF, Sharp L, Cotton SC, Little J. Cytochrome P-450 1A1 gene polymorphisms and risk of breast cancer: a HuGE review. Am J Epidemiol. 2005;161:901–915. [PubMed: 15870154] [CrossRef]
- Matthews CE, Xu WH, Zheng W, et al. Physical activity and risk of endometrial cancer: a report from the Shanghai endometrial cancer study. Cancer Epidemiol Biomarkers Prev. 2005;14:779–785. [PubMed: 15824143] [CrossRef]
- Mauderly JL, Gigliotti AP, Barr EB, et al. Chronic inhalation exposure to mainstream cigarette smoke increases lung and nasal tumor incidence in rats. Toxicol Sci. 2004;81:280–292. [PubMed: 15213336] [CrossRef]
- McCann SE, Freudenheim JL, Marshall JR, et al. Diet in the epidemiology of endometrial cancer in western New York (United States). Cancer Causes Control. 2000;11:965–974. [PubMed: 11142531] [CrossRef]
- McCormack VA, dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:1159–1169. [PubMed: 16775176] [CrossRef]
- McCoy GD, Hecht SS, Katayama S, Wynder EL. Differential effect of chronic ethanol consumption on the carcinogenicity of N-nitrosopyrrolidine and N’-nitrosonornicotine in male Syrian golden hamsters. Cancer Res. 1981;41:2849–2854. [PubMed: 7248945]
- McDivit AM, Greendale GA, Stanczyk FZ, Huang M-H. Effects of alcohol and cigarette smoking on change in serum estrone levels in postmenopausal women randomly assigned to fixed doses of conjugated equine estrogens with or without a progestin. Menopause. 2008;15:382–385. [PubMed: 18000469] [CrossRef]
- McGrath M, Hankinson SE, De Vivo I. Cytochrome P450 1A1, cigarette smoking, and risk of endometrial cancer (United States). Cancer Causes Control. 2007;18:1123–1130. [PubMed: 17717632] [CrossRef]
- McIlwain CC, Townsend DM, Tew KD. Glutathione S-transferase polymorphisms: cancer incidence and therapy. Oncogene. 2006;25:1639–1648. [PMC free article: PMC6361140] [PubMed: 16550164] [CrossRef]
- McIntyre-Seltman K, Castle PE, Guido R, et al. ALTS Group. Smoking is a risk factor for cervical intraepithelial neoplasia grade 3 among oncogenic human papillomavirus DNA-positive women with equivocal or mildly abnormal cytology. Cancer Epidemiol Biomarkers Prev. 2005;14:1165–1170. [PubMed: 15894667] [CrossRef]
- McKay JD, Hashibe M, Hung RJ, et al. Sequence variants of NAT1 and NAT2 and other xenometabolic genes and risk of lung and aerodigestive tract cancers in Central Europe. Cancer Epidemiol Biomarkers Prev. 2008;17:141–147. [PubMed: 18199719] [CrossRef]
- McKinlay SM. The normal menopause transition: an overview. Maturitas. 1996;23:137–145. [PubMed: 8735352] [CrossRef]
- McLaughlin JK, Hrubec Z, Blot WJ, Fraumeni JF Jr. Smoking and cancer mortality among U.S. veterans: a 26-year follow-up. Int J Cancer. 1995;60:190–193. [PubMed: 7829214] [CrossRef]
- Mechanic LE, Bowman ED, Welsh JA, et al. Common genetic variation in TP53 is associated with lung cancer risk and prognosis in African Americans and somatic mutations in lung tumors. Cancer Epidemiology, Biomarkers & Prevention. 2007;16:214–222. [PubMed: 17301252] [CrossRef]
- Mechanic LE, Millikan RC, Player J, et al. Polymorphisms in nucleotide excision repair genes, smoking and breast cancer in African Americans and whites: a population-based case-control study. Carcinogenesis. 2006;27:1377–1385. [PubMed: 16399771] [CrossRef]
- Meckley D, Hayes JR, Van Kampen KR, et al. A responsive, sensitive, and reproducible dermal tumor promotion assay for the comparative evaluation of cigarette smoke condensates. Regul Toxicol Pharmacol. 2004;39:135–149. a. [PubMed: 15041145] [CrossRef]
- Meckley DR, Hayes JR, Van Kampen KR, et al. Comparative study of smoke condensates from 1R4F cigarettes that burn tobacco versus ECLIPSE cigarettes that primarily heat tobacco in the SENCAR mouse dermal tumor promotion assay. Food Chem Toxicol. 2004;42:851–863. b. [PubMed: 15046832] [CrossRef]
- Menke-Pluymers MB, Hop WC, Dees J, et al. The Rotterdam Esophageal Tumor Study Group. Risk factors for the development of an adenocarcinoma in columnar-lined (Barrett) esophagus. Cancer. 1993;72:1155–1158. [PubMed: 8339208] [CrossRef]
- Menvielle G, Luce D, Goldberg P, et al. Smoking, alcohol drinking and cancer risk for various sites of the larynx and hypopharynx. A case-control study in France. Eur J Cancer Prev. 2004;13:165–172. a. [PubMed: 15167214] [CrossRef]
- Menvielle G, Luce D, Goldberg P, Leclerc A. Smoking, alcohol drinking, occupational exposures and social inequalities in hypopharyngeal and laryngeal cancer. Int J Epidemiol. 2004;33:799–806. b. [PubMed: 15155704] [CrossRef]
- Merletti F, Boffetta P, Ciccone G, et al. Role of tobacco and alcoholic beverages in the etiology of cancer of the oral cavity/oropharynx in Torino, Italy. Cancer Res. 1989;49:4919–4924. [PubMed: 2758421]
- Meskar A, Plee-Gautier E, Amet Y, et al. Alcohol-xenobiotic interactions. Role of cytochrome P450 2E1. Pathol Biol (Paris). 2001;49:696–702. [PubMed: 11762131] [CrossRef]
- Michnovicz JJ, Hershcopf RJ, Naganuma H, et al. Increased 2-hydroxylation of estradiol as a possible mechanism for the anti-estrogenic effect of cigarette smoking. N Engl J Med. 1986;315:1305–1309. [PubMed: 3773953]
- Michnovicz JJ, Naganuma H, Hershcopf RJ, et al. Increased urinary catechol estrogen excretion in female smokers. Steroids. 1988;52:69–83. [PubMed: 2854668] [CrossRef]
- Miller MD, Marty MA, Broadwin R, et al. California Environmental Protection Agency. The association between exposure to environmental tobacco smoke and breast cancer: a review by the California Environmental Protection Agency. Prev Med. 2007;44:93–106. [PubMed: 17027075] [CrossRef]
- Millikan RC, Pittman GS, Newman B, et al. Cigarette smoking, N-acetyltransferases 1 and 2, and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 1998;7:371–378. [PubMed: 9610785]
- Minami Y, Tateno H. Associations between cigarette smoking and the risk of four leading cancers in Miyagi Prefecture, Japan: a multi-site case-control study. Cancer Sci. 2003;94:540–547. [PubMed: 14529588] [CrossRef]
- Minna JD. Nicotine exposure and bronchial epithelial cell nicotinic acetylcholine receptor expression in the pathogenesis of lung cancer. J Clin Invest. 2003;111:31–33. [PMC free article: PMC151841] [PubMed: 12511585]
- Mitrou PN, Watson MA, Loktionov AS, et al. MTHFR (C677T and A1298C) polymorphisms and risk of sporadic distal colorectal adenoma in the UK Flexible Sigmoidoscopy Screening Trial (United Kingdom). Cancer Causes Control. 2006;17:793–801. [PubMed: 16783607] [CrossRef]
- Modugno F, Ness RB, Cottreau CM. Cigarette smoking and the risk of mucinous and nonmucinous epithelial ovarian cancer. Epidemiology. 2002;13:467–471. [PubMed: 12094103] [CrossRef]
- Modugno F, Ngo DL, Allen GO, et al. Breast cancer risk factors and mammographic breast density in women over age 70. Breast Cancer Res Treat. 2006;97:157–166. [PubMed: 16362132] [CrossRef]
- Molano M, Van den Brule A, Plummer M, et al. Determinants of clearance of human papillomavirus infections in Colombian women with normal cytology: a population-based, 5-year follow-up study. Am J Epidemiol. 2003;158:486–494. [PubMed: 12936904]
- Møllersen L, Vikse R, Andreassen A, et al. Adenomatous polyposis coli truncation mutations in 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced intestinal tumours of multiple intestinal neoplasia mice. Mutat Res. 2004;557:29–40. [PubMed: 14706516]
- Monnereau A, Orsi L, Troussard X, et al. Cigarette smoking, alcohol drinking, and risk of lymphoid neoplasms: results of a French case-control study. Cancer Causes Control. 2008;19:1147–1160. [PubMed: 18781390]
- Montelongo A, Lasunción MA, Pallardo LF, Herrera E. Longitudinal study of plasma lipoproteins and hormones during pregnancy in normal and diabetic women. Diabetes. 1992;41:1651–1659. [PubMed: 1446807] [CrossRef]
- Morabia A, Bernstein M, Héritier S, Khatchatrian N. Relation of breast cancer with passive and active exposure to tobacco smoke. Am J Epidemiol. 1996;143:918–928. [PubMed: 8610705]
- Morabia A, Bernstein M, Ruiz J, et al. Relation of smoking to breast cancer by estrogen receptor status. Int J Cancer. 1998;75:339–342. [PubMed: 9455790] [CrossRef]
- Morabia A, Bernstein MS, Bouchardy I, et al. Breast cancer and active and passive smoking: the role of the N-acetyltransferase 2 genotype. Am J Epidemiol. 2000;152:226–232. [PubMed: 10933269] [CrossRef]
- Morabia A, Wynder EL. Relation of bronchioloalveolar carcinoma to tobacco. BMJ. 1992;304:541–543. [PMC free article: PMC1881389] [PubMed: 1313719] [CrossRef]
- Moreno V, Glatt H, Guino E, et al. Bellvitge Colorectal Cancer Study Group. Polymorphisms in sulfotransferases SULT1A1 and SULT1A2 are not related to colorectal cancer. Int J Cancer. 2005;113:683–686. [PubMed: 15455379] [CrossRef]
- Morris JJ, Seifter E. The role of aromatic hydrocarbons in the genesis of breast cancer. Med Hypotheses. 1992;38:177–184. [PubMed: 1513270] [CrossRef]
- Morton LM, Hartge P, Holford TR, et al. Cigarette smoking and risk of non-Hodgkin lymphoma: a pooled analysis from the International Lymphoma Epidemiology Consortium (interlymph). Cancer Epidemiol Biomarkers Prev. 2005;14:925–933. [PubMed: 15824165]
- Mounawar M, Mukeria A, Le Calvez F, et al. Patterns of EGFR, HER2, TP53, and KRAS mutations of p14arf expression in non-small cell lung cancers in relation to smoking history. Cancer Res. 2007;67:5667–5672. [PubMed: 17575133] [CrossRef]
- Mountzios G, Fouret P, Soria J-C. Mechanisms of Disease: signal transduction in lung carcinogenesis – a comparison of smokers and never-smokers. Nat Clin Pract Oncol. 2008;5:610–618. [PubMed: 18628738] [CrossRef]
- Murata M, Takayama K, Choi BCK, Pak AWP. A nested case-control study on alcohol drinking, tobacco smoking, and cancer. Cancer Detect Prev. 1996;20:557–565. [PubMed: 8939341]
- Murayama N, Soyama A, Saito Y, et al. Six novel nonsynonymous CYP1A2 gene polymorphisms: catalytic activities of the naturally occurring variant enzymes. J Pharmacol Exp Ther. 2004;308:300–306. [PubMed: 14563787] [CrossRef]
- Murin S, Inciardi J. Cigarette smoking and the risk of pulmonary metastasis from breast cancer. Chest. 2001;119:1635–1640. [PubMed: 11399684] [CrossRef]
- Murphy G, Shu XO, Gao YT et al. (2009). Family cancer history affecting risk of colorectal cancer in a prospective cohort of Chinese women. Cancer Causes Control. [PMC free article: PMC2843785] [PubMed: 19418234]
- Muscat JE, Richie JP Jr, Thompson S, Wynder EL. Gender differences in smoking and risk for oral cancer. Cancer Res. 1996;56:5192–5197. [PubMed: 8912856]
- Muscat JE, Wynder EL. Tobacco, alcohol, asbestos, and occupational risk factors for laryngeal cancer. Cancer. 1992;69:2244–2251. [PubMed: 1562970] [CrossRef]
- Muscat JE, Wynder EL. A case/control study of risk factors for major salivary gland cancer. Otolaryngol Head Neck Surg. 1998;118:195–198. [PubMed: 9482552] [CrossRef]
- Mutch MG, Schoen RE, Fleshman JW, et al. A multicenter study of prevalence and risk factors for aberrant crypt foci. Clin Gastroenterol Hepatol. 2009;7:568–574. [PubMed: 19418605] [CrossRef]
- Muti P, Bradlow HL, Micheli A, et al. Estrogen metabolism and risk of breast cancer: a prospective study of the 2:16alpha-hydroxyestrone ratio in premenopausal and postmenopausal women. Epidemiology. 2000;11:635–640. [PubMed: 11055622] [CrossRef]
- Muwonge R, Ramadas K, Sankila R, et al. Role of tobacco smoking, chewing and alcohol drinking in the risk of oral cancer in Trivandrum, India: a nested case-control design using incident cancer cases. Oral Oncol. 2008;44:446–454. [PubMed: 17933578] [CrossRef]
- Mwenifumbo JC, Tyndale RF. Genetic variability in CYP2A6 and the pharmacokinetics of nicotine. Pharmacogenomics. 2007;8:1385–1402. [PubMed: 17979512] [CrossRef]
- Nagano J, Mabuchi K, Yoshimoto Y, et al. A case-control study in Hiroshima and Nagasaki examining non-radiation risk factors for thyroid cancer. J Epidemiol. 2007;17:76–85. [PMC free article: PMC7058453] [PubMed: 17545694] [CrossRef]
- Nagar S, Remmel RP. Uridine diphosphoglucuronosyltransferase pharmacogenetics and cancer. Oncogene. 2006;25:1659–1672. [PubMed: 16550166] [CrossRef]
- Nagle CM, Olsen CM, Webb PM, et al. Australian Cancer Study Group. Australian Ovarian Cancer Study Group. Endometrioid and clear cell ovarian cancers: a comparative analysis of risk factors. Eur J Cancer. 2008;44:2477–2484. [PubMed: 18707869] [CrossRef]
- Nakajima M. Smoking behavior and related cancers: the role of CYP2A6 polymorphisms. Curr Opin Mol Ther. 2007;9:538–544. [PubMed: 18041664]
- Nakajima M, Yokoi T, Mizutani M, et al. Genetic polymorphism in the 5′-flanking region of human CYP1A2 gene: effect on the CYP1A2 inducibility in humans. J Biochem. 1999;125:803–808. [PubMed: 10101295]
- Nam JM, McLaughlin JK, Blot WJ. Cigarette smoking, alcohol, and nasopharyngeal carcinoma: a case-control study among U.S. whites. J Natl Cancer Inst. 1992;84:619–622. [PubMed: 1556772] [CrossRef]
- Nandakumar A, Anantha N, Pattabhiraman V, et al. Importance of anatomical subsite in correlating risk factors in cancer of the oesophagus–report of a case–control study. Br J Cancer. 1996;73:1306–1311. [PMC free article: PMC2074521] [PubMed: 8630297]
- Nandakumar A, Thimmasetty KT, Sreeramareddy NM, et al. A population-based case-control investigation on cancers of the oral cavity in Bangalore, India. Br J Cancer. 1990;62:847–851. [PMC free article: PMC1971507] [PubMed: 2245179]
- Narayan S, Jaiswal AS, Kang D, et al. Cigarette smoke condensate-induced transformation of normal human breast epithelial cells in vitro. Oncogene. 2004;23:5880–5889. [PubMed: 15208684] [CrossRef]
- Navarro Silvera SA, Miller AB, Rohan TE. Risk factors for thyroid cancer: a prospective cohort study. Int J Cancer. 2005;116:433–438. [PubMed: 15818623] [CrossRef]
- Nayar D, Kapil U, Joshi YK, et al. Nutritional risk factors in esophageal cancer. J Assoc Physicians India. 2000;48:781–787. [PubMed: 11273469]
- Nebert DW, Dalton TP. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat Rev Cancer. 2006;6:947–960. [PubMed: 17128211] [CrossRef]
- Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. [PubMed: 15028720] [CrossRef]
- Nebert DW, Roe AL, Vandale SE, et al. NAD(P)H:quinone oxidoreductase (NQO1) polymorphism, exposure to benzene, and predisposition to disease: a HuGE review. Genet Med. 2002;4:62–70. [PubMed: 11882782] [CrossRef]
- Negri E, Bosetti C, La Vecchia C, et al. Risk factors for adenocarcinoma of the small intestine. Int J Cancer. 1999;82:171–174. [PubMed: 10389747] [CrossRef]
- Newcomb PA, Storer BE, Marcus PM. Cigarette smoking in relation to risk of large bowel cancer in women. Cancer Res. 1995;55:4906–4909. [PubMed: 7585528]
- Newcomb PA, Trentham-Dietz A. Patterns of postmenopausal progestin use with estrogen in relation to endometrial cancer (United States). Cancer Causes Control. 2003;14:195–201. [PubMed: 12749724] [CrossRef]
- Newcomer LM, Newcomb PA, Trentham-Dietz A, Storer BE. Hormonal risk factors for endometrial cancer: modification by cigarette smoking (United States). Cancer Causes Control. 2001;12:829–835. [PubMed: 11714111] [CrossRef]
- Newhouse ML, Pearson RM, Fullerton JM, et al. A case control study of carcinoma of the ovary. Br J Prev Soc Med. 1977;31:148–153. [PMC free article: PMC479015] [PubMed: 588853]
- Ng TP. A case-referent study of cancer of the nasal cavity and sinuses in Hong Kong. Int J Epidemiol. 1986;15:171–175. [PubMed: 3721678] [CrossRef]
- Nieters A, Deeg E, Becker N. Tobacco and alcohol consumption and risk of lymphoma: results of a population-based case-control study in Germany. Int J Cancer. 2006;118:422–430. [PubMed: 16080191]
- Nieters A, Rohrmann S, Becker N, et al. Smoking and lymphoma risk in the European prospective investigation into cancer and nutrition. Am J Epidemiol. 2008;167:1081–1089. [PubMed: 18321867]
- Nilsen TI, Vatten LJ. A prospective study of lifestyle factors and the risk of pancreatic cancer in Nord-Trøndelag, Norway. Cancer Causes Control. 2000;11:645–652. [PubMed: 10977109] [CrossRef]
- Nilsson S, Carstensen JM, Pershagen G. Mortality among male and female smokers in Sweden: a 33 year follow up. J Epidemiol Community Health. 2001;55:825–830. [PMC free article: PMC1763319] [PubMed: 11604439] [CrossRef]
- Ning JP, Yu MC, Wang QS, Henderson BE. Consumption of salted fish and other risk factors for nasopharyngeal carcinoma (NPC) in Tianjin, a low-risk region for NPC in the People’s Republic of China. J Natl Cancer Inst. 1990;82:291–296. [PubMed: 2299678] [CrossRef]
- Nishino K, Sekine M, Kodama S, et al. Cigarette smoking and glutathione S-transferase M1 polymorphism associated with risk for uterine cervical cancer. J Obstet Gynaecol Res. 2008;34:994–1001. [PubMed: 19012698]
- Nishino Y, Inoue M, Tsuji I, et al. Research Group for the Development and Evaluation of Cancer Prevention Strategies in Japan. Tobacco smoking and gastric cancer risk: an evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jpn J Clin Oncol. 2006;36:800–807. [PubMed: 17210611] [CrossRef]
- Niwa Y, Wakai K, Suzuki S, et al. JACC Study Group. Cigarette smoking and the risk of ovarian cancer in the Japanese population: findings from the Japanese Collaborate Cohort study. J Obstet Gynaecol Res. 2005;31:144–151. [PubMed: 15771641] [CrossRef]
- Nkondjock A, Ghadirian P. Dietary carotenoids and risk of colon cancer: case-control study. Int J Cancer. 2004;110:110–116. [PubMed: 15054875] [CrossRef]
- Nkondjock A, Robidoux A, Paredes Y, et al. Diet, lifestyle and BRCA-related breast cancer risk among French-Canadians. Breast Cancer Res Treat. 2006;98:285–294. [PubMed: 16541324] [CrossRef]
- Nock NL, Liu X, Cicek MS, et al. Polymorphisms in polycyclic aromatic hydrocarbon metabolism and conjugation genes, interactions with smoking and prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2006;15:756–761. [PubMed: 16614120] [CrossRef]
- Noda N, Matsuzoe D, Konno T, et al. Risk for K-ras gene mutations in smoking-induced lung cancer is associated with cytochrome P4501A1 and glutathione S-transferase micro1 polymorphisms. Oncol Rep. 2004;12:773–779. [PubMed: 15375499]
- Nordlund LA, Carstensen JM, Pershagen G. Cancer incidence in female smokers: a 26-year follow-up. Int J Cancer. 1997;73:625–628. [PubMed: 9398036] [CrossRef]
- Norppa H. Genetic polymorphisms and chromosome damage. Int J Hyg Environ Health. 2001;204:31–38. [PubMed: 11725342] [CrossRef]
- Norppa H. Genetic susceptibility, biomarker respones, and cancer. Mutat Res. 2003;544:339–348. [PubMed: 14644336] [CrossRef]
- Norppa H. Cytogenetic biomarkers and genetic polymorphisms. Toxicol Lett. 2004;149:309–334. [PubMed: 15093278] [CrossRef]
- Novak RF, Woodcroft KJ. The alcohol-inducible form of cytochrome P450 (CYP 2E1): role in toxicology and regulation of expression. Arch Pharm Res. 2000;23:267–282. [PubMed: 10976571] [CrossRef]
- O’Brien MJ, Yang S, Mack C, et al. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol. 2006;30:1491–1501. [PubMed: 17122504]
- Obana H, Hori S, Kashimoto T, Kunita N. Polycyclic aromatic hydrocarbons in human fat and liver. Bull Environ Contam Toxicol. 1981;27:23–27. [PubMed: 7296033] [CrossRef]
- Ochs-Balcom HM, Wiesner G, Elston RC. A meta-analysis of the association of N-acetyltransferase 2 gene (NAT2) variants with breast cancer. Am J Epidemiol. 2007;166:246–254. [PubMed: 17535831]
- Odenbro A, Gillgren P, Bellocco R, et al. The risk for cutaneous malignant melanoma, melanoma in situ and intraocular malignant melanoma in relation to tobacco use and body mass index. Br J Dermatol. 2007;156:99–105. [PubMed: 17199574] [CrossRef]
- Okamura C, Tsubono Y, Ito K, et al. Lactation and risk of endometrial cancer in Japan: a case-control study. Tohoku J Exp Med. 2006;208:109–115. [PubMed: 16434833] [CrossRef]
- Olivier M, Hainaut P. TP53 mutation patterns in breast cancers: searching for clues of environmental carcinogenesis. Semin Cancer Biol. 2001;11:353–360. [PubMed: 11562177] [CrossRef]
- Olsen J, Sabreo S, Fasting U. Interaction of alcohol and tobacco as risk factors in cancer of the laryngeal region. J Epidemiol Community Health. 1985;39:165–168. a. [PMC free article: PMC1052426] [PubMed: 4009100] [CrossRef]
- Olsen J, Sabroe S, Ipsen J. Effect of combined alcohol and tobacco exposure on risk of cancer of the hypopharynx. J Epidemiol Community Health. 1985;39:304–307. b. [PMC free article: PMC1052462] [PubMed: 4086960] [CrossRef]
- Olson JE, Vachon CM, Vierkant RA, et al. Prepregnancy exposure to cigarette smoking and subsequent risk of postmenopausal breast cancer. Mayo Clin Proc. 2005;80:1423–1428. [PubMed: 16295021] [CrossRef]
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med. 2009;48:123–128. [PubMed: 19182421] [CrossRef]
- Oreggia F, De Stefani E, Correa P, Fierro L. Risk factors for cancer of the tongue in Uruguay. Cancer. 1991;67:180–183. [PubMed: 1985715] [CrossRef]
- Otani T, Iwasaki M, Ikeda S, et al. Serum triglycerides and colorectal adenoma in a case-control study among cancer screening examinees (Japan). Cancer Causes Control. 2006;17:1245–1252. [PubMed: 17111255] [CrossRef]
- Otani T, Iwasaki M, Yamamoto S, et al. Japan Public Health Center-based Prospective Study Group. Alcohol consumption, smoking, and subsequent risk of colorectal cancer in middle-aged and elderly Japanese men and women: Japan Public Health Center-based prospective study. Cancer Epidemiol Biomarkers Prev. 2003;12:1492–1500. [PubMed: 14693743]
- Otto H, Elmenhorst H. Experimental studies on tumour induction with the gas phase of cigarette smoke.] Z Krebsforsch. 1967;70:45–47. [CrossRef]
- Pacella-Norman R, Urban MI, Sitas F, et al. Risk factors for oesophageal, lung, oral and laryngeal cancers in black South Africans. Br J Cancer. 2002;86:1751–1756. [PMC free article: PMC2375408] [PubMed: 12087462] [CrossRef]
- Pakhale SS, Sarkar S, Jayant K, Bhide SV. Carcinogenicity of Indian bidi and cigarette smoke condensate in Swiss albino mice. J Cancer Res Clin Oncol. 1988;114:647–649. [PubMed: 3204112] [CrossRef]
- Palmer JR, Rosenberg L. Cigarette smoking and the risk of breast cancer. Epidemiol Rev. 1993;15:145–156. [PubMed: 8405197]
- Palmer JR, Rosenberg L, Clarke EA, et al. Breast cancer and cigarette smoking: a hypothesis. Am J Epidemiol. 1991;134:1–13. [PubMed: 1853854]
- Pan SY, Ugnat AM, Mao Y, et al. Canadian Cancer Registries Epidemiology Research Group. Association of cigarette smoking with the risk of ovarian cancer. Int J Cancer. 2004;111:124–130. [PubMed: 15185353] [CrossRef]
- Pandey M, Shukla VK. Lifestyle, parity, menstrual and reproductive factors and risk of gallbladder cancer. Eur J Cancer Prev. 2003;12:269–272. [PubMed: 12883378] [CrossRef]
- Pandeya N, Williams GM, Sadhegi S, et al. Associations of duration, intensity, and quantity of smoking with adenocarcinoma and squamous cell carcinoma of the esophagus. Am J Epidemiol. 2008;168:105–114. [PubMed: 18483122] [CrossRef]
- Parente RC, Faerstein E, Celeste RK, Werneck GL. The relationship between smoking and age at the menopause: A systematic review. Maturitas. 2008;61:287–298. [PubMed: 19019585] [CrossRef]
- Park NH, Sapp JP, Herbosa EG. Oral cancer induced in hamsters with herpes simplex infection and simulated snuff dipping. Oral Surg Oral Med Oral Pathol. 1986;62:164–168. [PubMed: 3462612] [CrossRef]
- Park SL, Chang S-C, Cai L, et al. Associations between variants of the 8q24 chromosome and nine smoking-related cancer sites. Cancer Epidemiol Biomarkers Prev. 2008;17:3193–3202. [PMC free article: PMC2664075] [PubMed: 18990762] [CrossRef]
- Parker AS, Cerhan JR, Dick F, et al. Smoking and risk of non-Hodgkin lymphoma subtypes in a cohort of older women. Leuk Lymphoma. 2000;37:341–349. [PubMed: 10752985]
- Parker AS, Cerhan JR, Janney CA, et al. Smoking cessation and renal cell carcinoma. Ann Epidemiol. 2003;13:245–251. [PubMed: 12684190] [CrossRef]
- Paskett ED, Reeves KW, Rohan TE, et al. Association between cigarette smoking and colorectal cancer in the Women’s Health Initiative. J Natl Cancer Inst. 2007;99:1729–1735. [PubMed: 18000222] [CrossRef]
- Pavanello S, Clonfero E. Biological indicators of genotoxic risk and metabolic polymorphisms. Mutat Res. 2000;463:285–308. [PubMed: 11018745] [CrossRef]
- Pavanello S, Pulliero A, Lupi S, et al. Influence of the genetic polymorphism in the 5′-noncoding region of the CYP1A2 gene on CYP1A2 phenotype and urinary mutagenicity in smokers. Mutat Res. 2005;587:59–66. [PubMed: 16188490]
- Pavanello S, Simioli P, Lupi S, et al. Exposure levels and cytochrome P450 1A2 activity, but not N-acetyltransferase, glutathione S-transferase (GST) M1 and T1, influence urinary mutagen excretion in smokers. Cancer Epidemiol Biomarkers Prev. 2002;11:998–1003. [PubMed: 12376499]
- Peach HG, Barnett NE. Critical review of epidemiological studies of the association between smoking and non-Hodgkin’s lymphoma. Hematol Oncol. 2001;19:67–80. [PubMed: 11438976] [CrossRef]
- Penning TM, Drury JE. Human aldo-keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Arch Biochem Biophys. 2007;464:241–250. [PMC free article: PMC2025677] [PubMed: 17537398] [CrossRef]
- Peterson GM, Schmitz K et al. (2001). Physical activity among smokers is inversely associated with lung cancer incidence in Iowa Women's Health Study. 25th Annual Meeting of the American Society of Preventive Oncology.
- Peto R, Chen ZM, Boreham J. Tobacco–the growing epidemic. Nat Med. 1999;5:15–17. [PubMed: 9883828] [CrossRef]
- Petrakis NL, Gruenke LD, Beelen TC, et al. Nicotine in breast fluid of nonlactating women. Science. 1978;199:303–305. [PubMed: 619458] [CrossRef]
- Petridou E, Koukoulomatis P, Dessypris N, et al. Why is endometrial cancer less common in Greece than in other European Union countries? Eur J Cancer Prev. 2002;11:427–432. [PubMed: 12394239] [CrossRef]
- Pfeifer GP, Denissenko MF, Olivier M, et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435–7451. [PubMed: 12379884] [CrossRef]
- Phillips DH. Smoking-related DNA and protein adducts in human tissues. Carcinogenesis. 2002;23:1979–2004. [PubMed: 12507921] [CrossRef]
- Phillips LE, Longstreth WT Jr, Koepsell T, et al. 2005Active and passive cigarette smoking and risk of intracranial meningioma. Neuroepidemiology 24117–122.Epub 2004 Dec 30. [PubMed: 15637448]
- Phukan RK, Zomawia E, Narain K, et al. Tobacco use and stomach cancer in Mizoram, India. Cancer Epidemiol Biomarkers Prev. 2005;14:1892–1896. [PubMed: 16103433]
- Pinkston JA, Cole P. Cigarette smoking and Warthin’s tumor. Am J Epidemiol. 1996;144:183–187. [PubMed: 8678050]
- Pinsky PF. Racial and ethnic differences in lung cancer incidence: how much is explained by differences in smoking patterns? (United States). Cancer Causes Control. 2006;17:1017–1024. [PubMed: 16933052] [CrossRef]
- Pirkle JL, Bernert JT, Caudill SP, et al. Trends in the exposure of nonsmokers in the U.S. population to secondhand smoke: 1988–2002. Environ Health Perspect. 2006;114:853–858. [PMC free article: PMC1480505] [PubMed: 16759984]
- Plant AL, Benson DM, Smith LC. Cellular uptake and intracellular localization of benzo(a)pyrene by digital fluorescence imaging microscopy. J Cell Biol. 1985;100:1295–1308. [PMC free article: PMC2113754] [PubMed: 3980583] [CrossRef]
- Plummer M, Herrero R, Franceschi S, et al. IARC Multi-centre Cervical Cancer Study Group. Smoking and cervical cancer: pooled analysis of the IARC multi-centric case–control study. Cancer Causes Control. 2003;14:805–814. [PubMed: 14682438] [CrossRef]
- Polychronopoulou A, Tzonou A, Hsieh CC, et al. Reproductive variables, tobacco, ethanol, coffee and somatometry as risk factors for ovarian cancer. Int J Cancer. 1993;55:402–407. [PubMed: 8375923] [CrossRef]
- Poplawski T, Tomaszewska K, Galicki M, et al. Promoter methylation of cancer-related genes in gastric carcinoma. Exp Oncol. 2008;30:112–116. [PubMed: 18566573]
- Powell J, McConkey CC. Increasing incidence of adenocarcinoma of the gastric cardia and adjacent sites. Br J Cancer. 1990;62:440–443. [PMC free article: PMC1971432] [PubMed: 2206952]
- Prescott J, Ma H, Bernstein L, Ursin G. Cigarette smoking is not associated with breast cancer risk in young women. Cancer Epidemiol Biomarkers Prev. 2007;16:620–622. [PubMed: 17372262] [CrossRef]
- Preussmann R, Stewart BW (1984). N-Nitroso Carcinogens. In: Chemical Carcinogens, Second Edition, ACS Monograph 182. Searle CE, editor. Washington, DC: American Chemical Society.
- Proia NK, Paszkiewicz GM, Nasca MA, et al. Smoking and smokeless tobacco-associated human buccal cell mutations and their association with oral cancer–a review. Cancer Epidemiol Biomarkers Prev. 2006;15:1061–1077. [PubMed: 16775162] [CrossRef]
- Prokopczyk B, Hoffmann D, Bologna M, et al. Identification of tobacco-derived compounds in human pancreatic juice. Chem Res Toxicol. 2002;15:677–685. [PubMed: 12018989] [CrossRef]
- Prokopczyk B, Leder G, Trushin N, et al. 4-Hydroxy-1-(3-pyridyl)-1-butanone, an indicator for 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced DNA damage, is not detected in human pancreatic tissue. Cancer Epidemiol Biomarkers Prev. 2005;14:540–541. [PubMed: 15734986] [CrossRef]
- Quinn AM, Harvey RG, Penning TM. Oxidation of PAH trans-dihydrodiols by human aldo-keto reductase AKR1B10. Chem Res Toxicol. 2008;21:2207–2215. [PMC free article: PMC2645959] [PubMed: 18788756] [CrossRef]
- Quiñones LA, Irarrázabal CE, Rojas CR, et al. Joint effect among p53, CYP1A1, GSTM1 polymorphism combinations and smoking on prostate cancer risk: an exploratory genotype-environment interaction study. Asian J Androl. 2006;8:349–355. [PubMed: 16625286] [CrossRef]
- Rahman M, Sakamoto J, Fukui T. Bidi smoking and oral cancer: a meta-analysis. Int J Cancer. 2003;106:600–604. [PubMed: 12845659] [CrossRef]
- Raimondi S, Paracchini V, Autrup H, et al. Meta- and pooled analysis of GSTT1 and lung cancer: a HuGE-GSEC review. Am J Epidemiol. 2006;164:1027–1042. [PubMed: 17000715] [CrossRef]
- Raitiola HS, Pukander JS. Etiological factors of laryngeal cancer. Acta Otolaryngol Suppl. 1997;529:215–217. [PubMed: 9288314] [CrossRef]
- Rao AR. Modifying influences of betel quid ingredients on B(a)P-induced carcinogenesis in the buccal pouch of hamster. Int J Cancer. 1984;33:581–586. [PubMed: 6327538] [CrossRef]
- Rao DN, Desai PB. Risk assessment of tobacco, alcohol and diet in cancers of base tongue and oral tongue–a case control study. Indian J Cancer. 1998;35:65–72. [PubMed: 9849026]
- Rao DN, Desai PB, Ganesh B. Alcohol as an additional risk factor in laryngopharyngeal cancer in Mumbai–a case-control study. Cancer Detect Prev. 1999;23:37–44. [PubMed: 9892989] [CrossRef]
- Rao DN, Ganesh B, Dinshaw KA, Mohandas KM. A case-control study of stomach cancer in Mumbai, India. Int J Cancer. 2002;99:727–731. [PubMed: 12115507] [CrossRef]
- Rao DN, Ganesh B, Rao RS, Desai PB. Risk assessment of tobacco, alcohol and diet in oral cancer–a case-control study. Int J Cancer. 1994;58:469–473. [PubMed: 8056441] [CrossRef]
- Rappaport SM, Kim S, Lan Q, et al. Evidence that humans metabolize benzene via two pathways. Environ Health Perspect. 2009;117:946–952. [PMC free article: PMC2702411] [PubMed: 19590688]
- Raucy JL, Carpenter SJ. The expression of xenobiotic-metabolizing cytochromes P450 in fetal tissues. J Pharmacol Toxicol Methods. 1993;29:121–128. [PubMed: 8364226] [CrossRef]
- Ray CS, Gupta P. Bidis and Smokeless Tobacco. Current Science. 2009;96:1324–1334.
- Rebbeck TR. Molecular epidemiology of the human glutathione S-transferase genotypes GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiol Biomarkers Prev. 1997;6:733–743. [PubMed: 9298582]
- Reid A, de Klerk NH, Ambrosini GL, et al. The risk of lung cancer with increasing time since ceasing exposure to asbestos and quitting smoking. Occup Environ Med. 2006;63:509–512. a. [PMC free article: PMC2078130] [PubMed: 16849527] [CrossRef]
- Reid ME, Duffield-Lillico AJ, Sunga A, et al. Selenium supplementation and colorectal adenomas: an analysis of the nutritional prevention of cancer trial. Int J Cancer. 2006;118:1777–1781. b. [PubMed: 16217756] [CrossRef]
- Reynolds P, Hurley S, Goldberg DE, et al. Active smoking, household passive smoking, and breast cancer: evidence from the California Teachers Study. J Natl Cancer Inst. 2004;96:29–37. a. [PubMed: 14709736] [CrossRef]
- Reynolds P, Hurley SE, Hoggatt K, et al. Correlates of active and passive smoking in the California Teachers Study cohort. J Womens Health (Larchmt). 2004;13:778–790. b. [PubMed: 15385072] [CrossRef]
- Richardson H, Abrahamowicz M, Tellier PP, et al. Modifiable risk factors associated with clearance of type-specific cervical human papillomavirus infections in a cohort of university students. Cancer Epidemiol Biomarkers Prev. 2005;14:1149–1156. [PubMed: 15894665] [CrossRef]
- Riman T, Dickman PW, Nilsson S, et al. Some life-style factors and the risk of invasive epithelial ovarian cancer in Swedish women. Eur J Epidemiol. 2004;19:1011–1019. [PubMed: 15648594] [CrossRef]
- Riopel MA, Ronnett BM, Kurman RJ. Evaluation of diagnostic criteria and behavior of ovarian intestinal-type mucinous tumors: atypical proliferative (borderline) tumors and intraepithelial, microinvasive, invasive, and metastatic carcinomas. Am J Surg Pathol. 1999;23:617–635. [PubMed: 10366144] [CrossRef]
- Risch A, Plass C. Lung cancer epigenetics and genetics. Int J Cancer. 2008;123:1–7. [PubMed: 18425819]
- Risch HA, Howe GR, Jain M, et al. Are female smokers at higher risk for lung cancer than male smokers? A case-control analysis by histologic type. Am J Epidemiol. 1993;138:281–293. [PubMed: 8395141]
- Risch HA, Marrett LD, Jain M, Howe GR. Differences in risk factors for epithelial ovarian cancer by histologic type. Results of a case-control study. Am J Epidemiol. 1996;144:363–372. [PubMed: 8712193]
- Roddam AW, Pirie K, Pike MC, et al. Active and passive smoking and the risk of breast cancer in women aged 36–45 years: a population based case-control study in the UK. Br J Cancer. 2007;97:434–439. [PMC free article: PMC2360334] [PubMed: 17579618] [CrossRef]
- Rodgman A, Perfetti TA. The composition of cigarette smoke: a catalogue of the polycyclic aromatic hydrocarbons. Beitr. Tabakforschung Inti. 2006;22:13–69.
- Rodgman A, Perfetti TA (2009). Alphabetical Component Index. In: The Chemical Components of Tobacco and Tobacco Smoke. Rodgman A, Perfetti TA, editors. Boca Raton, FL: CRC Press, pp. 1483–1784.
- Rodin SN, Rodin AS. Origins and selection of p53 mutations in lung carcinogenesis. Semin Cancer Biol. 2005;15:103–112. [PubMed: 15652455] [CrossRef]
- Rodriguez T, Altieri A, Chatenoud L, et al. Risk factors for oral and pharyngeal cancer in young adults. Oral Oncol. 2004;40:207–213. [PubMed: 14693246] [CrossRef]
- Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer. Oncogene. 2006;25:1679–1691. [PubMed: 16550168] [CrossRef]
- Rohan TE, Baron JA. Cigarette smoking and breast cancer. Am J Epidemiol. 1989;129:36–42. [PubMed: 2910070]
- Rohrmann S, Genkinger JM, Burke A, et al. Smoking and risk of fatal prostate cancer in a prospective U.S. study. Urology. 2007;69:721–725. [PMC free article: PMC2853921] [PubMed: 17445658] [CrossRef]
- Rollison DE, Brownson RC, Hathcock HL, Newschaffer CJ. Case-control study of tobacco smoke exposure and breast cancer risk in Delaware. BMC Cancer. 2008;8:157. [PMC free article: PMC2424067] [PubMed: 18518960] [CrossRef]
- Rollison DE, Helzlsouer KJ, Lee JH, et al. Prospective study of JC virus seroreactivity and the development of colorectal cancers and adenomas. Cancer Epidemiol Biomarkers Prev. 2009;18:1515–1523. [PMC free article: PMC2743003] [PubMed: 19383887] [CrossRef]
- Rolón PA, Castellsagué X, Benz M, Muñoz N. Hot and cold mate drinking and esophageal cancer in Paraguay. Cancer Epidemiol Biomarkers Prev. 1995;4:595–605. [PubMed: 8547825]
- Rossing MA, Cushing-Haugen KL, Wicklund KG, Weiss NS. Cigarette smoking and risk of epithelial ovarian cancer. Cancer Causes Control. 2008;19:413–420. [PubMed: 18080774] [CrossRef]
- Rothman KJ, Greenland S (1998). Modern Epidemiology, 2nd ed. Philadelphia, USA: Lippincott-Raven Publishers.
- Roubidoux MA, Kaur JS, Griffith KA, et al. Relationship of mammographic parenchymal patterns to breast cancer risk factors and smoking in Alaska Native women. Cancer Epidemiol Biomarkers Prev. 2003;12:1081–1086. [PubMed: 14578146]
- Royce JM, Hymowitz N, Corbett K, et al. COMMIT Research Group. Smoking cessation factors among African Americans and whites. Am J Public Health. 1993;83:220–226. [PMC free article: PMC1694582] [PubMed: 8427327] [CrossRef]
- Ruano-Ravina A, Figueiras A, Freire-Garabal M, Barros-Dios JM. Antioxidant vitamins and risk of lung cancer. Curr Pharm Des. 2006;12:599–613. [PubMed: 16472151] [CrossRef]
- Rubin GL, Peterson HB, Lee NC, et al. Estrogen replacement therapy and the risk of endometrial cancer: remaining controversies. Am J Obstet Gynecol. 1990;162:148–154. [PubMed: 2301483]
- Ryk C, Berggren P, Kumar R, et al. Influence of GSTM1, GSTT1, GSTP1 and NAT2 genotypes on the p53 mutational spectrum in bladder tumours. Int J Cancer. 2005;113:761–768. [PubMed: 15499621] [CrossRef]
- Saccone SF, Hinrichs AL, Saccone NL, et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet. 2007;16:36–49. [PMC free article: PMC2270437] [PubMed: 17135278] [CrossRef]
- Saintot M, Malaveille C, Hautefeuille A, Gerber M. Interactions between genetic polymorphism of cytochrome P450–1B1, sulfotransferase 1A1, catechol-o-methyltransferase and tobacco exposure in breast cancer risk. Int J Cancer. 2003;107:652–657. [PubMed: 14520706] [CrossRef]
- Sala E, Warren R, McCann J, et al. Smoking and high-risk mammographic parenchymal patterns: a case-control study. Breast Cancer Res. 2000;2:59–63. [PMC free article: PMC13911] [PubMed: 11056684] [CrossRef]
- Salama SA, Abdel-Rahman SZ, Sierra-Torres CH, et al. Role of polymorphic GSTM1 and GSTT1 genotypes on NNK-induced genotoxicity. Pharmacogenetics. 1999;9:735–743. [PubMed: 10634136] [CrossRef]
- Salaspuro V, Salaspuro M. Synergistic effect of alcohol drinking and smoking on in vivo acetaldehyde concentration in saliva. Int J Cancer. 2004;111:480–483. [PubMed: 15239123] [CrossRef]
- Samanic C, Kogevinas M, Dosemeci M, et al. Smoking and bladder cancer in Spain: effects of tobacco type, timing, environmental tobacco smoke, and gender. Cancer Epidemiol Biomarkers Prev. 2006;15:1348–1354. [PubMed: 16835335] [CrossRef]
- Samowitz WS. Genetic and epigenetic changes in colon cancer. Exp Mol Pathol. 2008;85:64–67. [PubMed: 18482722] [CrossRef]
- Samowitz WS, Albertsen H, Sweeney C, et al. Association of smoking, CpG island methylator phenotype, and V600E BRAF mutations in colon cancer. J Natl Cancer Inst. 2006;98:1731–1738. [PubMed: 17148775] [CrossRef]
- Samowitz WS, Slattery ML, Sweeney C, et al. APC mutations and other genetic and epigenetic changes in colon cancer. Mol Cancer Res. 2007;5:165–170. [PubMed: 17293392] [CrossRef]
- Sanderson S, Salanti G, Higgins J. Joint effects of the N-acetyltransferase 1 and 2 (NAT1 and NAT2) genes and smoking on bladder carcinogenesis: a literature-based systematic HuGE review and evidence synthesis. Am J Epidemiol. 2007;166:741–751. [PubMed: 17675654] [CrossRef]
- Sanjoaquin MA, Appleby PN, Thorogood M, et al. Nutrition, lifestyle and colorectal cancer incidence: a prospective investigation of 10998 vegetarians and non-vegetarians in the United Kingdom. Br J Cancer. 2004;90:118–121. [PMC free article: PMC2395312] [PubMed: 14710217] [CrossRef]
- Sankaranarayanan R, Duffy SW, Day NE, et al. A case-control investigation of cancer of the oral tongue and the floor of the mouth in southern India. Int J Cancer. 1989;44:617–621. b. [PubMed: 2793234] [CrossRef]
- Sankaranarayanan R, Duffy SW, Nair MK, et al. Tobacco and alcohol as risk factors in cancer of the larynx in Kerala, India. Int J Cancer. 1990;45:879–882. b. [PubMed: 2335391] [CrossRef]
- Sankaranarayanan R, Duffy SW, Padmakumary G, et al. Tobacco chewing, alcohol and nasal snuff in cancer of the gingiva in Kerala, India. Br J Cancer. 1989;60:638–643. a. [PMC free article: PMC2247097] [PubMed: 2803939]
- Sankaranarayanan R, Duffy SW, Padmakumary G, et al. Risk factors for cancer of the buccal and labial mucosa in Kerala, southern India. J Epidemiol Community Health. 1990;44:286–292. a. [PMC free article: PMC1060671] [PubMed: 2277249] [CrossRef]
- Sankaranarayanan R, Duffy SW, Padmakumary G, et al. Risk factors for cancer of the oesophagus in Kerala, India. Int J Cancer. 1991;49:485–489. [PubMed: 1917146] [CrossRef]
- Sapkota A, Gajalakshmi V, Jetly DH, et al. Smokeless tobacco and increased risk of hypopharyngeal and laryngeal cancers: a multicentric case-control study from India. Int J Cancer. 2007;121:1793–1798. [PubMed: 17583577] [CrossRef]
- Sasazuki S, Sasaki S, Tsugane S., Japan Public Health Center Study Group. Cigarette smoking, alcohol consumption and subsequent gastric cancer risk by subsite and histologic type. Int J Cancer. 2002;101:560–566. [PubMed: 12237898] [CrossRef]
- Sauvaget C, Lagarde F, Nagano J, et al. Lifestyle factors, radiation and gastric cancer in atomic-bomb survivors (Japan). Cancer Causes Control. 2005;16:773–780. [PubMed: 16132787] [CrossRef]
- Scanlon EF, Suh O, Murthy SM, et al. Influence of smoking on the development of lung metastases from breast cancer. Cancer. 1995;75:2693–2699. [PubMed: 7743472] [CrossRef]
- Schechter MT, Miller AB, Howe GR. Cigarette smoking and breast cancer: a case-control study of screening program participants. Am J Epidemiol. 1985;121:479–487. [PubMed: 4014139]
- Schildt EB, Eriksson M, Hardell L, Magnuson A. Oral snuff, smoking habits and alcohol consumption in relation to oral cancer in a Swedish case-control study. Int J Cancer. 1998;77:341–346. [PubMed: 9663593] [CrossRef]
- Schlecht NF, Franco EL, Pintos J, et al. Interaction between tobacco and alcohol consumption and the risk of cancers of the upper aero-digestive tract in Brazil. Am J Epidemiol. 1999;150:1129–1137. b. [PubMed: 10588073]
- Schlecht NF, Franco EL, Pintos J, Kowalski LP. Effect of smoking cessation and tobacco type on the risk of cancers of the upper aero-digestive tract in Brazil. Epidemiology. 1999;10:412–418. a. [PubMed: 10401876] [CrossRef]
- Schlemmer A, Jensen J, Riis BJ, Christiansen C. Smoking induces increased androgen levels in early post-menopausal women. Maturitas. 1990;12:99–104. [PubMed: 2123959] [CrossRef]
- Schlicht KE, Michno N, Smith BD, et al. Functional characterization of CYP2A13 polymorphisms. Xenobiotica. 2007;37:1439–1449. [PubMed: 17922361]
- Schlöbe D, Hölzle D, Hatz D, et al. 4-Hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in lung, lower esophagus and cardia of sudden death victims. Toxicology. 2008;245:154–161. [PubMed: 18243467] [CrossRef]
- Schöllkopf C, Smedby KE, Hjalgrim H, et al. Cigarette smoking and risk of non-Hodgkin's lymphoma — a population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2005;14:1791–1796. [PubMed: 16030118]
- Schuller HM. Mechanisms of smoking-related lung and pancreatic adenocarcinoma development. Nat Rev Cancer. 2002;2:455–463. [PubMed: 12189387]
- Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat Rev Cancer. 2009;9:195–205. [PubMed: 19194381] [CrossRef]
- Schütze N, Vollmer G, Knuppen R. Catecholestrogens are agonists of estrogen receptor dependent gene expression in MCF-7 cells. J Steroid Biochem Mol Biol. 1994;48:453–461. [PubMed: 8180106] [CrossRef]
- Schütze N, Vollmer G, Tiemann I, et al. Catecholestrogens are MCF-7 cell estrogen receptor agonists. J Steroid Biochem Mol Biol. 1993;46:781–789. [PubMed: 8274412] [CrossRef]
- Schwartz AG, Swanson GM. Lung carcinoma in African Americans and whites. A population-based study in metropolitan Detroit, Michigan. Cancer. 1997;79:45–52. [PubMed: 8988725] [CrossRef]
- SEER (2004). Surveillance research Program, Cancer Statistics Branch. http://www
.seer.cancer .gov/publications/csr.html. - Seidegård J, Pero RW, Markowitz MM, et al. Isoenzyme(s) of glutathione transferase (class Mu) as a marker for the susceptibility to lung cancer: a follow up study. Carcinogenesis. 1990;11:33–36. [PubMed: 2295125] [CrossRef]
- Seidegård J, Pero RW, Miller DG, Beattie EJ. A glutathione transferase in human leukocytes as a marker for the susceptibility to lung cancer. Carcinogenesis. 1986;7:751–753. [PubMed: 3698203] [CrossRef]
- Sen B, Mahadevan B, DeMarini DM. Transcriptional responses to complex mixtures: a review. Mutat Res. 2007;636:144–177. [PubMed: 17888717] [CrossRef]
- Seow A, Vainio H, Yu MC. Effect of glutathione-S-transferase polymorphisms on the cancer preventive potential of isothiocyanates: an epidemiological perspective. Mutat Res. 2005;592:58–67. [PubMed: 16019037] [CrossRef]
- Setiawan VW, Pike MC, Kolonel LN, et al. Racial/ethnic differences in endometrial cancer risk: the multiethnic cohort study. Am J Epidemiol. 2007;165:262–270. [PubMed: 17090617] [CrossRef]
- Shaib YH, El-Serag HB, Nooka AK, et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: a hospital-based case-control study. Am J Gastroenterol. 2007;102:1016–1021. [PubMed: 17324130] [CrossRef]
- Shapiro JA, Jacobs EJ, Thun MJ. Cigar smoking in men and risk of death from tobacco-related cancers. J Natl Cancer Inst. 2000;92:333–337. [PubMed: 10675383] [CrossRef]
- Sharpe CR, Siemiatycki J. Joint effects of smoking and body mass index on prostate cancer risk. Epidemiology. 2001;12:546–551. [PubMed: 11505174] [CrossRef]
- Sharpe CR, Siemiatycki JA, Rachet BP. The effects of smoking on the risk of colorectal cancer. Dis Colon Rectum. 2002;45:1041–1050. [PubMed: 12195188]
- Shi X, Zhou S, Wang Z, et al. CYP1A1 and GSTM1 polymorphisms and lung cancer risk in Chinese populations: a meta-analysis. Lung Cancer. 2008;59:155–163. [PubMed: 17900751] [CrossRef]
- Shields TS, Weiss NS, Voigt LF, Beresford SA. The additional risk of endometrial cancer associated with unopposed estrogen use in women with other risk factors. Epidemiology. 1999;10:733–738. [PubMed: 10535788] [CrossRef]
- Shikata K, Doi Y, Yonemoto K, et al. Population-based prospective study of the combined influence of cigarette smoking and Helicobacter pylori infection on gastric cancer incidence: the Hisayama Study. Am J Epidemiol. 2008;168:1409–1415. [PubMed: 18945691] [CrossRef]
- Shimizu M, Lasker JM, Tsutsumi M, Lieber CS. Immunohistochemical localization of ethanol-inducible P450IIE1 in the rat alimentary tract. Gastroenterology. 1990;99:1044–1053. [PubMed: 2203661]
- Shimizu N, Nagata C, Shimizu H, et al. Height, weight, and alcohol consumption in relation to the risk of colorectal cancer in Japan: a prospective study. Br J Cancer. 2003;88:1038–1043. [PMC free article: PMC2376385] [PubMed: 12671701] [CrossRef]
- Shklar G. Experimental oral pathology in the Syrian hamster. Prog Exp Tumor Res. 1972;16:518–538. [PubMed: 4557243]
- Shrubsole MJ, Wu H, Ness RM, et al. Alcohol drinking, cigarette smoking, and risk of colorectal adenomatous and hyperplastic polyps. Am J Epidemiol. 2008;167:1050–1058. [PubMed: 18304959] [CrossRef]
- Shu HP, Bymun EN. Systemic excretion of benzo(a)pyrene in the control and microsomally induced rat: the influence of plasma lipoproteins and albumin as carrier molecules. Cancer Res. 1983;43:485–490. [PubMed: 6293698]
- Sikström B, Hellberg D, Nilsson S, Mårdh PA. Smoking, alcohol, sexual behaviour and drug use in women with cervical human papillomavirus infection. Arch Gynecol Obstet. 1995;256:131–137. [PubMed: 7574905] [CrossRef]
- Sillanpää P, Heikinheimo L, Kataja V, et al. CYP1A1 and CYP1B1 genetic polymorphisms, smoking and breast cancer risk in a Finnish Caucasian population. Breast Cancer Res Treat. 2007;104:287–297. [PubMed: 17063266] [CrossRef]
- Simán JH, Forsgren A, Berglund G, Florén C-H. Tobacco smoking increases the risk for gastric adenocarcinoma among Helicobacter pylori-infected individuals. Scand J Gastroenterol. 2001;36:208–213. [PubMed: 11252415] [CrossRef]
- Simarak S, de Jong UW, Breslow N, et al. Cancer of the oral cavity, pharynx/larynx and lung in North Thailand: case-control study and analysis of cigar smoke. Br J Cancer. 1977;36:130–140. [PMC free article: PMC2025440] [PubMed: 19028]
- Singh R, Farmer PB. Liquid chromatography-electrospray ionization-mass spectrometry: the future of DNA adduct detection. Carcinogenesis. 2006;27:178–196. [PubMed: 16272169] [CrossRef]
- Singh R, Kaur B, Farmer PB. Detection of DNA damage derived from a direct acting ethylating agent present in cigarette smoke by use of liquid chromatography-tandem mass spectrometry. Chem Res Toxicol. 2005;18:249–256. [PubMed: 15720129] [CrossRef]
- Sjödahl K, Lu Y, Nilsen TI, et al. Smoking and alcohol drinking in relation to risk of gastric cancer: a population-based, prospective cohort study. Int J Cancer. 2007;120:128–132. [PubMed: 17036324] [CrossRef]
- Skipper PL, Tannenbaum SR. Protein adducts in the molecular dosimetry of chemical carcinogens. Carcinogenesis. 1990;11:507–518. [PubMed: 2182215] [CrossRef]
- Skjelbred CF, Saebø M, Wallin H, et al. Polymorphisms of the XRCC1, XRCC3 and XPD genes and risk of colorectal adenoma and carcinoma, in a Norwegian cohort: a case control study. BMC Cancer. 2006;6:67. [PMC free article: PMC1458350] [PubMed: 16542436] [CrossRef]
- Slattery ML, Curtin K, Anderson K, et al. Associations between cigarette smoking, lifestyle factors, and microsatellite instability in colon tumors. J Natl Cancer Inst. 2000;92:1831–1836. [PubMed: 11078760] [CrossRef]
- Slattery ML, Curtin K, Giuliano AR, et al. Active and passive smoking, IL6, ESR1, and breast cancer risk. Breast Cancer Res Treat. 2008;109:101–111. [PMC free article: PMC2532584] [PubMed: 17594514] [CrossRef]
- Slattery ML, Curtin K, Ma K, et al. GSTM-1 and NAT2 and genetic alterations in colon tumors. Cancer Causes Control. 2002;13:527–534. [PubMed: 12195642] [CrossRef]
- Slattery ML, Edwards S, Curtin K, et al. Associations between smoking, passive smoking, GSTM-1, NAT2, and rectal cancer. Cancer Epidemiol Biomarkers Prev. 2003;12:882–889. [PubMed: 14504199]
- Slattery ML, Samowtiz W, Ma K, et al. CYP1A1, cigarette smoking, and colon and rectal cancer. Am J Epidemiol. 2004;160:842–852. [PubMed: 15496536] [CrossRef]
- Slemenda CW, Hui SL, Longcope C, Johnston CC Jr. Cigarette smoking, obesity, and bone mass. J Bone Miner Res. 1989;4:737–741. [PubMed: 2816518]
- Smith CJ, Perfetti TA, King JA. Perspectives on pulmonary inflammation and lung cancer risk in cigarette smokers. Inhal Toxicol. 2006;18:667–677. [PubMed: 16864557] [CrossRef]
- Smith EM, Sowers MF, Burns TL. Effects of smoking on the development of female reproductive cancers. J Natl Cancer Inst. 1984;73:371–376. [PubMed: 6589429]
- Smith JB, Randle HW. Giant basal cell carcinoma and cigarette smoking. Cutis. 2001;67:73–76. [PubMed: 11204609]
- Smits KM, Gaspari L, Weijenberg MP, et al. Interaction between smoking, GSTM1 deletion and colorectal cancer: results from the GSEC study. Biomarkers. 2003;8:299–310. [PubMed: 12944179] [CrossRef]
- Sokić SI, Adanja BJ, Marinković JP, Vlajinac HD. Case-control study of risk factors in laryngeal cancer. Neoplasma. 1994;41:43–47. [PubMed: 8202195]
- Song DK, Xing DL, Zhang LR, et al. Association of NAT2, GSTM1, GSTT1, CYP2A6, and CYP2A13 gene polymorphisms with susceptibility and clinicopathologic characteristics of bladder cancer in Central China. Cancer Detect Prev. 2009;32:416–423. [PubMed: 19303722]
- Sparks R, Bigler J, Sibert JG, et al. TGFbeta1 polymorphism (L10P) and risk of colorectal adenomatous and hyperplastic polyps. Int J Epidemiol. 2004;33:955–961. [PubMed: 15020570] [CrossRef]
- Spira A, Beane J, Shah V, et al. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci U S A. 2004;101:10143–10148. [PMC free article: PMC454179] [PubMed: 15210990] [CrossRef]
- Spitz MR, Amos CI, Dong Q, et al. The CHRNA5–A3 region on chromosome 15q24–25.1 is a risk factor both for nicotine dependence and for lung cancer. J Natl Cancer Inst. 2008;100:1552–1556. [PMC free article: PMC2720751] [PubMed: 18957677] [CrossRef]
- Spitz MR, Duphorne CM, Detry MA, et al. Dietary intake of isothiocyanates: evidence of a joint effect with glutathione S-transferase polymorphisms in lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2000;9:1017–1020. [PubMed: 11045782]
- Spitz MR, Fueger JJ, Goepfert H, Newell GR. Salivary gland cancer. A case-control investigation of risk factors. Arch Otolaryngol Head Neck Surg. 1990;116:1163–1166. [PubMed: 2206501]
- Spitz MR, Wu X, Wilkinson A, Wei D (2006). Cancer of the Lung. In: Cancer Epidemiology and Prevention. Schottenfeld D, Fraumeni JF, editors. New York: Oxford University Press, Chap 33, pp. 638–658.
- Sriamporn S, Vatanasapt V, Pisani P, et al. Environmental risk factors for nasopharyngeal carcinoma: a case-control study in northeastern Thailand. Cancer Epidemiol Biomarkers Prev. 1992;1:345–348. [PubMed: 1305465]
- Srivastava A, Kreiger N. Cigarette smoking and testicular cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:49–54. [PubMed: 14744732] [CrossRef]
- Stagnaro E, Ramazzotti V, Crosignani P, et al. Smoking and hematolymphopoietic malignancies. Cancer Causes Control. 2001;12:325–334. [PubMed: 11456228] [CrossRef]
- Stagnaro E, Tumino R, Parodi S, et al. Non-Hodgkin's lymphoma and type of tobacco smoke. Cancer Epidemiol Biomarkers Prev. 2004;13:431–437. [PubMed: 15006920]
- Stanton MF, Miller E, Wrench C, Blackwell R. Experimental induction of epidermoid carcinoma in the lungs of rats by cigarette smoke condensate. J Natl Cancer Inst. 1972;49:867–877. [PubMed: 4647499]
- Stavrides JC. Lung carcinogenesis: pivotal role of metals in tobacco smoke. Free Radic Biol Med. 2006;41:1017–1030. [PubMed: 16962926] [CrossRef]
- Steinmetz J, Spyckerelle Y, Guéguen R, Dupré C. Alcohol, tobacco and colorectal adenomas and cancer. Case-control study in a population with positive fecal occult blood tests. Presse Med. 2007;36:1174–1182. [PubMed: 17350789]
- Stellman SD, Chen Y, Muscat JE, et al. Lung cancer risk in white and black Americans. Ann Epidemiol. 2003;13:294–302. [PubMed: 12684197] [CrossRef]
- Stellman SD, Takezaki T, Wang L, et al. Smoking and lung cancer risk in American and Japanese men: an international case-control study. Cancer Epidemiol Biomarkers Prev. 2001;10:1193–1199. [PubMed: 11700268]
- Stepanov I, Carmella SG, Briggs A, et al. Presence of the carcinogen N’-nitrosonornicotine in the urine of some users of oral nicotine replacement therapy products. Cancer Res. 2009;69:8236–8240. a. [PMC free article: PMC2783463] [PubMed: 19843845]
- Stepanov I, Carmella SG, Han S, et al. Evidence for endogenous formation of N’-nitrosonornicotine in some long-term nicotine patch users. Nicotine Tob Res. 2009;11:99–105. b. [PMC free article: PMC2734288] [PubMed: 19246447]
- Stepanov I, Hecht SS. Tobacco-specific nitrosamines and their pyridine-N-glucuronides in the urine of smokers and smokeless tobacco users. Cancer Epidemiol Biomarkers Prev. 2005;14:885–891. [PubMed: 15824160] [CrossRef]
- Stepanov I, Hecht SS, Lindgren B, et al. Relationship of human toenail nicotine, cotinine, and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol to levels of these biomarkers in plasma and urine. Cancer Epidemiol Biomarkers Prev. 2007;16:1382–1386. [PubMed: 17627002] [CrossRef]
- Stepanov I, Upadhyaya P, Carmella SG, et al. Extensive metabolic activation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers. Cancer Epidemiol Biomarkers Prev. 2008;17:1764–1773. [PMC free article: PMC2542896] [PubMed: 18628430] [CrossRef]
- Stern MC, Conti DV, Siegmund KD, et al. DNA repair single-nucleotide polymorphisms in colorectal cancer and their role as modifiers of the effect of cigarette smoking and alcohol in the Singapore Chinese Health Study. Cancer Epidemiol Biomarkers Prev. 2007;16:2363–2372. [PubMed: 18006925] [CrossRef]
- Stern MC, Siegmund KD, Conti DV, et al. XRCC1, XRCC3, and XPD polymorphisms as modifiers of the effect of smoking and alcohol on colorectal adenoma risk. Cancer Epidemiol Biomarkers Prev. 2006;15:2384–2390. [PubMed: 17164360] [CrossRef]
- Stockwell HG, Lyman GH. Cigarette smoking and the risk of female reproductive cancer. Am J Obstet Gynecol. 1987;157:35–40. [PubMed: 3605266]
- Strader CH, Vaughan TL, Stergachis A. Use of nasal preparations and the incidence of sinonasal cancer. J Epidemiol Community Health. 1988;42:243–248. [PMC free article: PMC1052733] [PubMed: 2471766] [CrossRef]
- Strange RC, Spiteri MA, Ramachandran S, Fryer AA. Glutathione-S-transferase family of enzymes. Mutat Res. 2001;482:21–26. [PubMed: 11535245]
- Strom BL, Schinnar R, Weber AL, et al. Case-control study of postmenopausal hormone replacement therapy and endometrial cancer. Am J Epidemiol. 2006;164:775–786. [PubMed: 16997897] [CrossRef]
- Stürmer T, Glynn RJ, Lee IM, et al. Lifetime cigarette smoking and colorectal cancer incidence in the Physicians’ Health Study I. J Natl Cancer Inst. 2000;92:1178–1181. [PubMed: 10904092] [CrossRef]
- Subapriya R, Thangavelu A, Mathavan B, et al. Assessment of risk factors for oral squamous cell carcinoma in Chidambaram, Southern India: a case-control study. Eur J Cancer Prev. 2007;16:251–256. [PubMed: 17415096] [CrossRef]
- Subramanian J, Govindan R. Molecular genetics of lung cancer in people who have never smoked. Lancet Oncol. 2008;9:676–682. [PubMed: 18598932] [CrossRef]
- Sudo H, Li-Sucholeiki X-C, Marcelino LA, et al. Fetal-juvenile origins of point mutations in the adult human tracheal-bronchial epithelium: absence of detectable effects of age, gender or smoking status. Mutat Res. 2008;646:25–40. [PubMed: 18824180]
- Sung NY, Choi KS, Park EC, et al. Smoking, alcohol and gastric cancer risk in Korean men: the National Health Insurance Corporation Study. Br J Cancer. 2007;97:700–704. [PMC free article: PMC2360367] [PubMed: 17637680] [CrossRef]
- Suwan-ampai P, Navas-Acien A, Strickland PT, Agnew J. Involuntary tobacco smoke exposure and urinary levels of polycyclic aromatic hydrocarbons in the United States, 1999 to 2002. Cancer Epidemiol Biomarkers Prev. 2009;18:884–893. [PubMed: 19258471] [CrossRef]
- Suwanrungruang K, Sriamporn S, Wiangnon S, et al. Lifestyle-related risk factors for stomach cancer in northeast Thailand. Asian Pac J Cancer Prev. 2008;9:71–75. [PubMed: 18439078]
- Suzuki T, Matsuo K, Wakai K, et al. Effect of familial history and smoking on common cancer risks in Japan. Cancer. 2007;109:2116–2123. [PubMed: 17410537] [CrossRef]
- Swann PF. 1984Effect of ethanol on nitrosamine metabolism and distribution. Implications for the role of nitrosamines in human cancer and for the influence of alcohol consumption on cancer incidence. IARC Sci Publ57501–512. [PubMed: 6533040]
- Swanson GM, Burns PB. Cancers of the salivary gland: workplace risks among women and men. Ann Epidemiol. 1997;7:369–374. [PubMed: 9279445] [CrossRef]
- Sweeney C, Boucher KM, Samowitz WS, et al. Oncogenetic tree model of somatic mutations and DNA methylation in colon tumors. Genes Chromosomes Cancer. 2009;48:1–9. [PMC free article: PMC3742107] [PubMed: 18767147] [CrossRef]
- Syrjänen K, Shabalova I, Petrovichev N, et al. Smoking is an independent risk factor for oncogenic human papillomavirus (HPV) infections but not for high-grade CIN. Eur J Epidemiol. 2007;22:723–735. [PubMed: 17828436] [CrossRef]
- Szeliga J, Dipple A. DNA adduct formation by polycyclic aromatic hydrocarbon dihydrodiol epoxides. Chem Res Toxicol. 1998;11:1–11. [PubMed: 9477220] [CrossRef]
- Szyfter K, Szmeja Z, Szyfter W, et al. Molecular and cellular alterations in tobacco smoke-associated larynx cancer. Mutat Res. 1999;445:259–274. [PubMed: 10575435]
- ’t Mannetje A, Kogevinas M, Luce D, et al. Sinonasal cancer, occupation, and tobacco smoking in European women and men. Am J Ind Med. 1999;36:101–107. [PubMed: 10361593] [CrossRef]
- Takahashi I, Matsuzaka M, Umeda T, et al. Differences in the influence of tobacco smoking on lung cancer between Japan and the USA: possible explanations for the ‘smoking paradox’ in Japan. Public Health. 2008;122:891–896. [PubMed: 18420238] [CrossRef]
- Takahashi M, Imaida K, Mitsumori K, et al. Promoting effects of cigarette smoke on the respiratory tract carcinogenesis of Syrian golden hamsters treated with diethylnitrosamine. Carcinogenesis. 1992;13:569–572. [PubMed: 1576708] [CrossRef]
- Talamini R, Bosetti C, La Vecchia C, et al. Combined effect of tobacco and alcohol on laryngeal cancer risk: a case-control study. Cancer Causes Control. 2002;13:957–964. [PubMed: 12588092] [CrossRef]
- Tan W, Chen GF, Xing DY, et al. Frequency of CYP2A6 gene deletion and its relation to risk of lung and esophageal cancer in the Chinese population. Int J Cancer. 2001;95:96–101. [PubMed: 11241319] [CrossRef]
- Tang Y, Simoneau AR, Liao WX, et al. WIF1, a Wnt pathway inhibitor, regulates SKP2 and c-myc expression leading to G1 arrest and growth inhibition of human invasive urinary bladder cancer cells. Mol Cancer Ther. 2009;8:458–468. [PMC free article: PMC2768341] [PubMed: 19174556] [CrossRef]
- Tavani A, Negri E, Franceschi S, et al. Attributable risk for laryngeal cancer in northern Italy. Cancer Epidemiol Biomarkers Prev. 1994;3:121–125. [PubMed: 8049633]
- Terry MB, Neugut AI. Cigarette smoking and the colorectal adenoma-carcinoma sequence: a hypothesis to explain the paradox. Am J Epidemiol. 1998;147:903–910. [PubMed: 9596467]
- Terry P, Baron JA, Weiderpass E, et al. Lifestyle and endometrial cancer risk: a cohort study from the Swedish Twin Registry. Int J Cancer. 1999;82:38–42. [PubMed: 10360818] [CrossRef]
- Terry P, Rohan TE, Franceschi S et al. (2004a). Endometrial cancer. In: Tobacco: Science, Policy and Public Health. Boyle P, Gray N, Henningfield J et al., editors. Oxford University Press.
- Terry PD, Goodman M. Is the association between cigarette smoking and breast cancer modified by genotype? A review of epidemiologic studies and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:602–611. [PubMed: 16614098] [CrossRef]
- Terry PD, Miller AB, Rohan TE. Prospective cohort study of cigarette smoking and colorectal cancer risk in women. Int J Cancer. 2002;99:480–483. a. [PubMed: 11992421] [CrossRef]
- Terry PD, Miller AB, Rohan TE. A prospective cohort study of cigarette smoking and the risk of endometrial cancer. Br J Cancer. 2002;86:1430–1435. b. [PMC free article: PMC2375370] [PubMed: 11986776]
- Terry PD, Rohan TE. Cigarette smoking and the risk of breast cancer in women: a review of the literature. Cancer Epidemiol Biomarkers Prev. 2002;11:953–971. [PubMed: 12376493]
- Terry PD, Rohan TE, Franceschi S, Weiderpass E. Cigarette smoking and the risk of endometrial cancer. Lancet Oncol. 2002;3:470–480. c. [PubMed: 12147433] [CrossRef]
- Theis RP, Dolwick Grieb SM, Burr D, et al. Smoking, environmental tobacco smoke, and risk of renal cell cancer: a population-based case-control study. BMC Cancer. 2008;8:387. [PMC free article: PMC2633310] [PubMed: 19108730] [CrossRef]
- Thier R, Brüning T, Roos PH, et al. Markers of genetic susceptibility in human environmental hygiene and toxicology: the role of selected CYP, NAT and GST genes. Int J Hyg Environ Health. 2003;206:149–171. [PubMed: 12872524] [CrossRef]
- Thompson PA, DeMarini DM, Kadlubar FF, et al. Evidence for the presence of mutagenic arylamines in human breast milk and DNA adducts in exfoliated breast ductal epithelial cells. Environ Mol Mutagen. 2002;39:134–142. [PubMed: 11921181] [CrossRef]
- Thorgeirsson TE, Geller F, Sulem P, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. [PMC free article: PMC4539558] [PubMed: 18385739] [CrossRef]
- Thun MJ, Heath CW Jr. Changes in mortality from smoking in two American Cancer Society prospective studies since 1959. Prev Med. 1997;26:422–426. [PubMed: 9245660] [CrossRef]
- Thun MJ, Henley SJ, Burns D, et al. Lung cancer death rates in lifelong nonsmokers. J Natl Cancer Inst. 2006;98:691–699. [PubMed: 16705123]
- Tiemersma EW, Bunschoten A, Kok FJ, et al. Effect of SULT1A1 and NAT2 genetic polymorphism on the association between cigarette smoking and colorectal adenomas. Int J Cancer. 2004;108:97–103. [PubMed: 14618622] [CrossRef]
- Tiemersma EW, Kampman E, Bueno de Mesquita HB, et al. Meat consumption, cigarette smoking, and genetic susceptibility in the etiology of colorectal cancer: results from a Dutch prospective study. Cancer Causes Control. 2002;13:383–393. a. [PubMed: 12074508] [CrossRef]
- Tiemersma EW, Kloosterman J, Bunschoten A, et al. Role of EPHX genotype in the associations of smoking and diet with colorectal adenomas. IARC Sci Publ. 2002;156:491–493. b. [PubMed: 12484240]
- Tijhuis MJ, Visker MH, Aarts JM, et al. NQO1 and NFE2L2 polymorphisms, fruit and vegetable intake and smoking and the risk of colorectal adenomas in an endoscopy-based population. Int J Cancer. 2008;122:1842–1848. [PubMed: 18074351] [CrossRef]
- Timofeeva MN, Kropp S, Sauter W, et al. LUCY-Consortium. CYP450 polymorphisms as risk factors for early-onset lung cancer: gender-specific differences. Carcinogenesis. 2009;30:1161–1169. [PubMed: 19414505] [CrossRef]
- To-Figueras J, Gené M, Gómez-Catalán J, et al. Lung cancer susceptibility in relation to combined polymorphisms of microsomal epoxide hydrolase and glutathione S-transferase P1. Cancer Lett. 2001;173:155–162. [PubMed: 11597790] [CrossRef]
- Tokunaga M, Land CE, Yamamoto T, et al. Incidence of female breast cancer among atomic bomb survivors, Hiroshima and Nagasaki, 1950–1980. Radiat Res. 1987;112:243–272. [PubMed: 3685255] [CrossRef]
- Tolstrup J, Munk C, Thomsen BL, et al. The role of smoking and alcohol intake in the development of high-grade squamous intraepithelial lesions among high-risk HPV-positive women. Acta Obstet Gynecol Scand. 2006;85:1114–1119. [PubMed: 16929418] [CrossRef]
- Tomita M, Odaka M, Matsumoto M, et al. Cigarette smoking and mortality among Japanese males in a prospective cohort study. Nippon Koshu Eisei Zasshi. 1991;38:492–497. [PubMed: 1747539]
- Toyomura K, Yamaguchi K, Kawamoto H, et al. Relation of cigarette smoking and alcohol use to colorectal adenomas by subsite: the self-defense forces health study. Cancer Sci. 2004;95:72–76. [PubMed: 14720330] [CrossRef]
- Toyooka S, Tokumo M, Shigematsu H, et al. Mutational and epigenetic evidence for independent pathways for lung adenocarcinomas arising in smokers and never smokers. Cancer Res. 2006;66:1371–1375. [PubMed: 16452191]
- Tran GD, Sun XD, Abnet CC, et al. Prospective study of risk factors for esophageal and gastric cancers in the Linxian general population trial cohort in China. Int J Cancer. 2005;113:456–463. [PubMed: 15455378] [CrossRef]
- Tranah GJ, Giovannucci E, Ma J, et al. Epoxide hydrolase polymorphisms, cigarette smoking and risk of colorectal adenoma in the Nurses’ Health Study and the Health Professionals Follow-up Study. Carcinogenesis. 2004;25:1211–1218. [PubMed: 14988221] [CrossRef]
- Tranah GJ, Giovannucci E, Ma J, et al. APC Asp1822Val and Gly2502Ser polymorphisms and risk of colorectal cancer and adenoma. Cancer Epidemiol Biomarkers Prev. 2005;14:863–870. [PubMed: 15824157] [CrossRef]
- Trentham-Dietz A, Nichols HB, Hampton JM, Newcomb PA. Weight change and risk of endometrial cancer. Int J Epidemiol. 2006;35:151–158. [PubMed: 16278243] [CrossRef]
- Trichopoulos D, Brown J, MacMahon B. Urine estrogens and breast cancer risk factors among post-menopausal women. Int J Cancer. 1987;40:721–725. [PubMed: 3692620] [CrossRef]
- Trost SG, Owen N, Bauman AE, et al. Correlates of adults’ participation in physical activity: review and update. Med Sci Sports Exerc. 2002;34:1996–2001. [PubMed: 12471307] [CrossRef]
- Tsai HT, Tsai YM, Yang SF, et al. 2007Lifetime cigarette smoke and second-hand smoke and cervical intraepithelial neoplasm — a community-based case-control study. Gynecol Oncol 105181–188.Epub 2007 Jan 3. [PubMed: 17204311]
- Tsoi KK, Pau CY, Wu WK, et al. Cigarette smoking and the risk of colorectal cancer: a meta-analysis of prospective cohort studies. Clin Gastroenterol Hepatol. 2009;7:682–688, e1–e5. [PubMed: 19245853]
- Tsong WH, Koh WP, Yuan JM, et al. Cigarettes and alcohol in relation to colorectal cancer: the Singapore Chinese Health Study. Br J Cancer. 2007;96:821–827. [PMC free article: PMC2360085] [PubMed: 17311023]
- Tu JT, Gao RN, Zhang DH, Gu BC. Hepatitis B virus and primary liver cancer on Chongming Island, People’s Republic of China. Natl Cancer Inst Monogr. 1985;69:213–215. [PubMed: 3834335]
- Turner MC, Chen Y, Krewski D, et al. Chronic obstructive pulmonary disease is associated with lung cancer mortality in a prospective study of never smokers. Am J Respir Crit Care Med. 2007;176:285–290. [PubMed: 17478615] [CrossRef]
- Tuyns AJ, Estève J, Raymond L, et al. Cancer of the larynx/hypopharynx, tobacco and alcohol: IARC international case-control study in Turin and Varese (Italy), Zaragoza and Navarra (Spain), Geneva (Switzerland) and Calvados (France). Int J Cancer. 1988;41:483–491. [PubMed: 3356483] [CrossRef]
- Tuyns AJ, Péquignot G, Jensen OM. [Esophageal cancer in Ille-et-Vilaine in relation to levels of alcohol and tobacco consumption. Risks are multiplying] Bull Cancer. 1977;64:45–60. [PubMed: 861389]
- Tverdal A, Thelle D, Stensvold I, et al. Mortality in relation to smoking history: 13 years’ follow-up of 68,000 Norwegian men and women 35–49 years. J Clin Epidemiol. 1993;46:475–487. [PubMed: 8501474] [CrossRef]
- Tworoger SS, Gertig DM, Gates MA, et al. Caffeine, alcohol, smoking, and the risk of incident epithelial ovarian cancer. Cancer. 2008;112:1169–1177. [PubMed: 18213613] [CrossRef]
- Tyler CW Jr, Webster LA, Ory HW, Rubin GL. Endometrial cancer: how does cigarette smoking influence the risk of women under age 55 years having this tumor? Am J Obstet Gynecol. 1985;151:899–905. [PubMed: 3985056]
- Tzonou A, Day NE, Trichopoulos D, et al. The epidemiology of ovarian cancer in Greece: a case-control study. Eur J Cancer Clin Oncol. 1984;20:1045–1052. [PubMed: 6540687] [CrossRef]
- Ulrich CM, Bigler J, Whitton JA, et al. Epoxide hydrolase Tyr113His polymorphism is associated with elevated risk of colorectal polyps in the presence of smoking and high meat intake. Cancer Epidemiol Biomarkers Prev. 2001;10:875–882. [PubMed: 11489754]
- Ursin G, London S, Stanczyk FZ, et al. Urinary 2-hydroxyestrone/16alpha-hydroxyestrone ratio and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 1999;91:1067–1072. [PubMed: 10379970] [CrossRef]
- Vaccarella S, Herrero R, Snijders PJ, et al. IARC HPV Prevalence Surveys (IHPS) Study Group. Smoking and human papillomavirus infection: pooled analysis of the International Agency for Research on Cancer HPV Prevalence Surveys. Int J Epidemiol. 2008;37:536–546. [PubMed: 18316350] [CrossRef]
- Vachon CM, Kuni CC, Anderson K, et al. Association of mammographically defined percent breast density with epidemiologic risk factors for breast cancer (United States). Cancer Causes Control. 2000;11:653–662. [PubMed: 10977110] [CrossRef]
- Vairaktaris E, Spyridonidou S, Papakosta V, et al. The hamster model of sequential oral oncogenesis. Oral Oncol. 2008;44:315–324. [PubMed: 18061531] [CrossRef]
- van Dam RM, Huang Z, Rimm EB, et al. Risk factors for basal cell carcinoma of the skin in men: results from the health professionals follow-up study. Am J Epidemiol. 1999;150:459–468. [PubMed: 10472945]
- van den Donk M, Buijsse B, van den Berg SW, et al. Dietary intake of folate and riboflavin, MTHFR C677T genotype, and colorectal adenoma risk: a Dutch case-control study. Cancer Epidemiol Biomarkers Prev. 2005;14:1562–1566. [PubMed: 15941973] [CrossRef]
- van der Hel OL, Bueno de Mesquita HB, Sandkuijl L, et al. Rapid N-acetyltransferase 2 imputed phenotype and smoking may increase risk of colorectal cancer in women (Netherlands). Cancer Causes Control. 2003;14:293–298. a. [PubMed: 12814209] [CrossRef]
- van der Hel OL, Bueno-de-Mesquita HB, van Gils CH, et al. Cumulative genetic defects in carcinogen metabolism may increase breast cancer risk (The Netherlands). Cancer Causes Control. 2005;16:675–681. [PubMed: 16049806] [CrossRef]
- Van Emburgh BO, Hu JJ, Levine EA, et al. Polymorphisms in drug metabolism genes, smoking, and p53 mutations in breast cancer. Mol Carcinog. 2008;47:88–99. [PMC free article: PMC3722359] [PubMed: 17683074] [CrossRef]
- van Poppel G, de Vogel N, van Balderen PJ, Kok FJ. Increased cytogenetic damage in smokers deficient in glutathione S-transferase isozyme mu. Carcinogenesis. 1992;13:303–305. [PubMed: 1740022] [CrossRef]
- Vaughan TL, Davis S, Kristal A, Thomas DB. Obesity, alcohol, and tobacco as risk factors for cancers of the esophagus and gastric cardia: adenocarcinoma versus squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 1995;4:85–92. [PubMed: 7742727]
- Vaughan TL, Shapiro JA, Burt RD, et al. Nasopharyngeal cancer in a low-risk population: defining risk factors by histological type. Cancer Epidemiol Biomarkers Prev. 1996;5:587–593. [PubMed: 8824359]
- Veglia F, Loft S, Matullo G, et al. Genair-EPIC Investigators. DNA adducts and cancer risk in prospective studies: a pooled analysis and a meta-analysis. Carcinogenesis. 2008;29:932–936. [PubMed: 18343884] [CrossRef]
- Veglia F, Matullo G, Vineis P. Bulky DNA adducts and risk of cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2003;12:157–160. [PubMed: 12582026]
- Verla-Tebit E, Lilla C, Hoffmeister M, et al. Cigarette smoking and colorectal cancer risk in Germany: a population-based case-control study. Int J Cancer. 2006;119:630–635. [PubMed: 16496385] [CrossRef]
- Viezzer C, Norppa H, Clonfero E, et al. Influence of GSTM1, GSTT1, GSTP1, and EPHX gene polymorphisms on DNA adduct level and HPRT mutant frequency in coke-oven workers. Mutat Res. 1999;431:259–269. [PubMed: 10635992]
- Vineis P, Anttila S, Benhamou S, et al. Evidence of gene gene interactions in lung carcinogenesis in a large pooled analysis. Carcinogenesis. 2007;28:1902–1905. [PubMed: 17307802] [CrossRef]
- Vineis P, Malats N. Strategic issues in the design and interpretation of studies on metabolic polymorphisms and cancer. IARC Sci Publ. 1999;148:51–61. [PubMed: 10493248]
- Vineis P, Manuguerra M, Kavvoura FK, et al. A field synopsis on low-penetrance variants in DNA repair genes and cancer susceptibility. J Natl Cancer Inst. 2009;101:24–36. [PubMed: 19116388]
- Vineis P, Marinelli D, Autrup H, et al. Current smoking, occupation, N-acetyltransferase-2 and bladder cancer: a pooled analysis of genotype-based studies. Cancer Epidemiol Biomarkers Prev. 2001;10:1249–1252. [PubMed: 11751441]
- Vineis P, Veglia F, Anttila S, et al. CYP1A1, GSTM1 and GSTT1 polymorphisms and lung cancer: a pooled analysis of gene-gene interactions. Biomarkers. 2004;9:298–305. [PubMed: 15764294] [CrossRef]
- Vineis P, Veglia F, Benhamou S, et al. CYP1A1 T3801C polymorphism and lung cancer: a pooled analysis of 2451 cases and 3358 controls. Int J Cancer. 2003;104:650–657. [PubMed: 12594823] [CrossRef]
- Vioque J, Barber X, Bolumar F, et al. PANESOES Study Group. Esophageal cancer risk by type of alcohol drinking and smoking: a case-control study in Spain. BMC Cancer. 2008;8:221. [PMC free article: PMC2529333] [PubMed: 18673563] [CrossRef]
- Viswanathan AN, Feskanich D, De Vivo I, et al. Smoking and the risk of endometrial cancer: results from the Nurses’ Health Study. Int J Cancer. 2005;114:996–1001. [PubMed: 15645490] [CrossRef]
- Vlajinac HD, Marinkovic JM, Sipetic SB, et al. Case-control study of oropharyngeal cancer. Cancer Detect Prev. 2006;30:152–157. [PubMed: 16647226] [CrossRef]
- Vlajinac HD, Pekmezović TD, Adanja BJ, et al. Case-control study of multiple myeloma with special reference to diet as risk factor. Neoplasma. 2003;50:79–83. [PubMed: 12687283]
- Vories AA, Ramirez SG. Warthin’s tumor and cigarette smoking. South Med J. 1997;90:416–418. [PubMed: 9114834]
- Voskuil DW, Kampman E, Grubben MJ, et al. Meat consumption and meat preparation in relation to colorectal adenomas among sporadic and HNPCC family patients in The Netherlands. Eur J Cancer. 2002;38:2300–2308. [PubMed: 12441267] [CrossRef]
- Vrieling A, Bueno-De-Mesquita B, Boshuizen HC, et al. Fruit and vegetable consumption and pancreatic cancer risk in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer. 2009;124:1926–1934. [PubMed: 19107929]
- Wakai K, Hayakawa N, Kojima M, et al. JACC Study Group. Smoking and colorectal cancer in a non-Western population: a prospective cohort study in Japan. J Epidemiol. 2003;13:323–332. [PMC free article: PMC9727326] [PubMed: 14674660]
- Wakai K, Inoue M, Mizoue T, et al. Research Group for the Development and Evaluation of Cancer Prevention Strategies in Japan. Tobacco smoking and lung cancer risk: an evaluation based on a systematic review of epidemiological evidence among the Japanese population. Jpn J Clin Oncol. 2006;36:309–324. [PubMed: 16735374] [CrossRef]
- Walser T, Cui X, Yanagawa J, et al. Smoking and lung cancer: the role of inflammation. Proc Am Thorac Soc. 2008;5:811–815. [PMC free article: PMC4080902] [PubMed: 19017734] [CrossRef]
- Wang H, Tan W, Hao B, et al. Substantial reduction in risk of lung adenocarcinoma associated with genetic polymorphism in CYP2A13, the most active cytochrome P450 for the metabolic activation of tobacco-specific carcinogen NNK. Cancer Res. 2003;63:8057–8061. [PubMed: 14633739]
- Wang M, Cheng G, Balbo S, et al. 2009Clear differences in levels of a formaldehyde-DNA adduct in leukocytes of smokers and non-smokers. Cancer Research69:7170–7174. [PMC free article: PMC2745488] [PubMed: 19738046]
- Wang SS, Schiffman M, Herrero R, et al. Determinants of human papillomavirus 16 serological conversion and persistence in a population-based cohort of 10 000 women in Costa Rica. Br J Cancer. 2004;91:1269–1274. [PMC free article: PMC2409899] [PubMed: 15292929] [CrossRef]
- Wang Y, Pereira EF, Maus AD, et al. Human bronchial epithelial and endothelial cells express alpha7 nicotinic acetylcholine receptors. Mol Pharmacol. 2001;60:1201–1209. [PubMed: 11723227]
- Warnakulasuriya KA, Ralhan R. Clinical, pathological, cellular and molecular lesions caused by oral smokeless tobacco–a review. J Oral Pathol Med. 2007;36:63–77. [PubMed: 17238967]
- Warren CW, Jones NR, Eriksen MP, Asma S., Global Tobacco Surveillance System (GTSS) collaborative group. Patterns of global tobacco use in young people and implications for future chronic disease burden in adults. Lancet. 2006;367:749–753. [PubMed: 16517275] [CrossRef]
- Warwick J, Pinney E, Warren RM, et al. Breast density and breast cancer risk factors in a high-risk population. Breast. 2003;12:10–16. [PubMed: 14659350] [CrossRef]
- Wasnik KS, Ughade SN, Zodpey SP, Ingole DL. Tobacco consumption practices and risk of oro-pharyngeal cancer: a case-control study in Central India. Southeast Asian J Trop Med Public Health. 1998;29:827–834. [PubMed: 10772572]
- Watson MA, Stewart RK, Smith GB, et al. Human glutathione S-transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19:275–280. [PubMed: 9498276] [CrossRef]
- Watson P, Ashwathnarayan R, Lynch HT, Roy HK. Tobacco use and increased colorectal cancer risk in patients with hereditary nonpolyposis colorectal cancer (Lynch syndrome). Arch Intern Med. 2004;164:2429–2431. [PubMed: 15596632]
- Wei YS, Lu JC, Wang L, et al. Risk factors for sporadic colorectal cancer in southern Chinese. World J Gastroenterol. 2009;15:2526–2530. [PMC free article: PMC2686912] [PubMed: 19469004]
- Weiderpass E, Baron JA. Cigarette smoking, alcohol consumption, and endometrial cancer risk: a population-based study in Sweden. Cancer Causes Control. 2001;12:239–247. [PubMed: 11405329] [CrossRef]
- Weiss JM, Saltzman BS, Doherty JA, et al. Risk factors for the incidence of endometrial cancer according to the aggressiveness of disease. Am J Epidemiol. 2006;164:56–62. a. [PubMed: 16675538] [CrossRef]
- Weiss JM, Weiss NS, Ulrich CM, et al. Interindividual variation in nucleotide excision repair genes and risk of endometrial cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:2524–2530. [PubMed: 16284373] [CrossRef]
- Weiss JM, Weiss NS, Ulrich CM, et al. Nucleotide excision repair genotype and the incidence of endometrial cancer: effect of other risk factors on the association. Gynecol Oncol. 2006;103:891–896. b. [PubMed: 16806437] [CrossRef]
- Weiss NS, Farewall VT, Szekely DR, et al. Oestrogens and endometrial cancer: effect of other risk factors on the association. Maturitas. 1980;2:185–190. [PubMed: 7442552] [CrossRef]
- Wells PG, Mackenzie PI, Chowdhury JR, et al. Glucuronidation and the UDP-glucuronosyltransferases in health and disease. Drug Metab Dispos. 2004;32:281–290. [PubMed: 14977861] [CrossRef]
- Welzel TM, Graubard BI, El-Serag HB, et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: a population-based case-control study. Clin Gastroenterol Hepatol. 2007;5:1221–1228. [PMC free article: PMC2083573] [PubMed: 17689296] [CrossRef]
- Wen CP, Tsai SP, Chen CJ, Cheng TY. The mortality risks of smokers in Taiwan: Part I: cause-specific mortality. Prev Med. 2004;39:528–535. [PubMed: 15313092] [CrossRef]
- Wenzlaff AS, Cote ML, Bock CH, et al. CYP1A1 and CYP1B1 polymorphisms and risk of lung cancer among never smokers: a population-based study. Carcinogenesis. 2005;26:2207–2212. [PubMed: 16051642] [CrossRef]
- West KA, Brognard J, Clark AS, et al. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest. 2003;111:81–90. [PMC free article: PMC151834] [PubMed: 12511591]
- West S, Hildesheim A, Dosemeci M. Non-viral risk factors for nasopharyngeal carcinoma in the Philippines: results from a case-control study. Int J Cancer. 1993;55:722–727. [PubMed: 7503957] [CrossRef]
- Whittemore AS, Wu ML, Paffenbarger RS Jr, et al. Personal and environmental characteristics related to epithelial ovarian cancer. II. Exposures to talcum powder, tobacco, alcohol, and coffee. Am J Epidemiol. 1988;128:1228–1240. [PubMed: 3195564]
- WHO (2011).* Third WHO Report on the Global Tobacco Epidemic. World Health Organization. http://www
.who.int/tobacco /global_report/2011/en/index.html. - Wiencke JK. DNA adduct burden and tobacco carcinogenesis. Oncogene. 2002;21:7376–7391. [PubMed: 12379880] [CrossRef]
- Willett EV, O'Connor S, Smith AG, Roman E. Does smoking and alcohol modify the risk of Epstein-Barr virus-positive or -negative Hodgkin lymphoma. Epidemiology. 2007;18:130–136. [PubMed: 17099321]
- Wojno TH. The association between cigarette smoking and basal cell carcinoma of the eyelids in women. Ophthal Plast Reconstr Surg. 1999;15:390–392. [PubMed: 10588245] [CrossRef]
- Wojnowski L, Kamdem LK. Clinical implications of CYP3A polymorphisms. Expert Opin Drug Metab Toxicol. 2006;2:171–182. [PubMed: 16866606] [CrossRef]
- Wolf CR, Smith G. Cytochrome P450 CYP2D6. IARC Sci Publ. 1999;148:209–229. [PubMed: 10493260]
- Wolff H, Saukkonen K, Anttila S, et al. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res. 1998;58:4997–5001. [PubMed: 9823297]
- Wong HL, Murphy SE, Hecht SS. Cytochrome P450 2A-catalyzed metabolic activation of structurally similar carcinogenic nitrosamines: N’-nitrosonornicotine enantiomers, N-nitrosopiperidine, and N-nitrosopyrrolidine. Chem Res Toxicol. 2005;18:61–69. [PubMed: 15651850] [CrossRef]
- Wright ME, Park Y, Subar AF, et al. Intakes of fruit, vegetables, and specific botanical groups in relation to lung cancer risk in the NIH-AARP Diet and Health Study. Am J Epidemiol. 2008;168:1024–1034. [PMC free article: PMC2631557] [PubMed: 18791192] [CrossRef]
- Wu AH, Crabtree JE, Bernstein L, et al. Role of Helicobacter pylori CagA+ strains and risk of adenocarcinoma of the stomach and esophagus. Int J Cancer. 2003;103:815–821. [PubMed: 12516104] [CrossRef]
- Wu AH, Wan P, Bernstein L. A multiethnic population-based study of smoking, alcohol and body size and risk of adenocarcinomas of the stomach and esophagus (United States). Cancer Causes Control. 2001;12:721–732. [PubMed: 11562112] [CrossRef]
- Wu AH, Yu MC, Mack TM. Smoking, alcohol use, dietary factors and risk of small intestinal adenocarcinoma. Int J Cancer. 1997;70:512–517. [PubMed: 9052748] [CrossRef]
- Wu C, Hu Z, Yu D, et al. Genetic variants on chromosome 15q25 associated with lung cancer risk in Chinese populations. Cancer Res. 2009;69:5065–5072. a. [PubMed: 19491260] [CrossRef]
- Wu IC, Lee CH, Kuo CH, et al. Consumption of cigarettes but not betel quid or alcohol increases colorectal cancer risk. J Formos Med Assoc. 2009;108:155–163. b. [PubMed: 19251551] [CrossRef]
- Wynder EL, Gottlieb S, Wright G. A study of tobacco carcinogenesis. IV. Different tobacco types. Cancer. 1957;10:1206–1209. [PubMed: 13489672] [CrossRef]
- Wynder EL, Graham EA, Croninger AB. Experimental production of carcinoma with cigarette tar. Cancer Res. 1953;13:855–864. [PubMed: 13116124]
- Wynder EL, Hoffmann D. A study of tobacco carcinogenesis. VIII. The role of the acidic fractions as promoters. Cancer. 1961;14:1306–1315. [PubMed: 14008628] [CrossRef]
- Wynder EL, Stellman SD. Impact of long-term filter cigarette usage on lung and larynx cancer risk: a case-control study. J Natl Cancer Inst. 1979;62:471–477. [PubMed: 283277]
- Yagyu K, Kikuchi S, Obata Y, et al. JACC Study Group. Cigarette smoking, alcohol drinking and the risk of gallbladder cancer death: a prospective cohort study in Japan. Int J Cancer. 2008;122:924–929. [PubMed: 17955487] [CrossRef]
- Yamasaki E, Ames BN. Concentration of mutagens from urine by absorption with the nonpolar resin XAD-2: cigarette smokers have mutagenic urine. Proc Natl Acad Sci U S A. 1977;74:3555–3559. [PMC free article: PMC431630] [PubMed: 333441] [CrossRef]
- Yang J, Qian LX, Wu HF, et al. Genetic polymorphisms in the cytochrome P450 1A1 and 2E1 genes, smoking, drinking and prostate cancer susceptibility: a case-control study in a Han nationality population in Southern China. Int J Urol. 2006;13:773–780. [PubMed: 16834659] [CrossRef]
- Yang M, Kim S, Lee E, et al. Sources of polycyclic aromatic hydrocarbon exposure in non-occupationally exposed Koreans. Environ Mol Mutagen. 2003;42:250–257. [PubMed: 14673870] [CrossRef]
- Yang M, Koga M, Katoh T, Kawamoto T. A study for the proper application of urinary naphthols, new biomarkers for airborne polycyclic aromatic hydrocarbons. Arch Environ Contam Toxicol. 1999;36:99–108. [PubMed: 9828267] [CrossRef]
- Yang P, Cunningham JM, Halling KC, et al. Higher risk of mismatch repair-deficient colorectal cancer in alpha(1)-antitrypsin deficiency carriers and cigarette smokers. Mol Genet Metab. 2000;71:639–645. [PubMed: 11136557] [CrossRef]
- Yang P, Sun Z, Krowka MJ, et al. Alpha1-antitrypsin deficiency carriers, tobacco smoke, chronic obstructive pulmonary disease, and lung cancer risk. Arch Intern Med. 2008;168:1097–1103. [PMC free article: PMC2562773] [PubMed: 18504338] [CrossRef]
- Yauk CL, Berndt ML, Williams A, et al. Mainstream tobacco smoke causes paternal germ-line DNA mutation. Cancer Res. 2007;67:5103–5106. [PubMed: 17545587] [CrossRef]
- Ye WM, Ye YN, Lin RD, et al. Case control study on nasopharyngeal cancer in southern region of Fujian province. Chin J Prev Control Chronic Dis. 1995;3:158–161.
- Ye Z, Song H, Higgins JP, et al. Five glutathione s-transferase gene variants in 23,452 cases of lung cancer and 30,397 controls: meta-analysis of 130 studies. PLoS Med. 2006;3:524–534. [PMC free article: PMC1391981] [PubMed: 16509765] [CrossRef]
- Yen TT, Lin WD, Wang CP, et al. The association of smoking, alcoholic consumption, betel quid chewing and oral cavity cancer: a cohort study. Eur Arch Otorhinolaryngol. 2008;265:1403–1407. [PubMed: 18389268] [CrossRef]
- Yoo KY, Tajima K, Miura S, et al. Breast cancer risk factors according to combined estrogen and progesterone receptor status: a case-control analysis. Am J Epidemiol. 1997;146:307–314. [PubMed: 9270409]
- Young E, Leatherdale S, Sloan M, et al. Age of smoking initiation and risk of breast cancer in a sample of Ontario women. Tob Induc Dis. 2009;5:4. [PMC free article: PMC2656478] [PubMed: 19222858] [CrossRef]
- Yu GP, Ostroff JS, Zhang ZF, et al. Smoking history and cancer patient survival: a hospital cancer registry study. Cancer Detect Prev. 1997;21:497–509. [PubMed: 9398990]
- Yu MC, Garabrant DH, Huang TB, Henderson BE. Occupational and other non-dietary risk factors for nasopharyngeal carcinoma in Guangzhou, China. Int J Cancer. 1990;45:1033–1039. [PubMed: 2351484] [CrossRef]
- Yu MC, Ho JH, Lai SH, Henderson BE. Cantonese-style salted fish as a cause of nasopharyngeal carcinoma: report of a case-control study in Hong Kong. Cancer Res. 1986;46:956–961. [PubMed: 3940655]
- Yuan JM, Koh WP, Murphy SE, et al. Urinary levels of tobacco-specific nitrosamine metabolites in relation to lung cancer development in two prospective cohorts of cigarette smokers. Cancer Res. 2009;69:2990–2995. [PMC free article: PMC2664854] [PubMed: 19318550] [CrossRef]
- Yuan JM, Ross RK, Wang XL, et al. Morbidity and mortality in relation to cigarette smoking in Shanghai, China. A prospective male cohort study. JAMA. 1996;275:1646–1650. [PubMed: 8637137] [CrossRef]
- Yuan JM, Wang XL, Xiang YB, et al. Non-dietary risk factors for nasopharyngeal carcinoma in Shanghai, China. Int J Cancer. 2000;85:364–369. [PubMed: 10652428] [CrossRef]
- Yun YH, Jung KW, Bae JM, et al. Cigarette smoking and cancer incidence risk in adult men: National Health Insurance Corporation Study. Cancer Detect Prev. 2005;29:15–24. [PubMed: 15734213] [CrossRef]
- Zahm SH, Heineman EF, Vaught JB. Soft tissue sarcoma and tobacco use: data from a prospective cohort study of United States veterans. Cancer Causes Control. 1992;3:371–376. [PubMed: 1617125] [CrossRef]
- Zak-Prelich M, Narbutt J, Sysa-Jedrzejowska A. Environmental risk factors predisposing to the development of basal cell carcinoma. Dermatol Surg. 2004;30 s2:248–252. [PubMed: 14871217] [CrossRef]
- Zang EA, Wynder EL. Differences in lung cancer risk between men and women: examination of the evidence. J Natl Cancer Inst. 1996;88:183–192. [PubMed: 8632492] [CrossRef]
- Zanieri L, Galvan P, Checchini L, et al. Polycyclic aromatic hydrocarbons (PAHs) in human milk from Italian women: influence of cigarette smoking and residential area. Chemosphere. 2007;67:1265–1274. [PubMed: 17258279] [CrossRef]
- Zaridze D, Borisova E, Maximovitch D, Chkhikvadze V. Alcohol consumption, smoking and risk of gastric cancer: case-control study from Moscow, Russia. Cancer Causes Control. 2000;11:363–371. [PubMed: 10843447] [CrossRef]
- Zatonski W, Becher H, Lissowska J, Wahrendorf J. Tobacco, alcohol, and diet in the etiology of laryngeal cancer: a population-based case-control study. Cancer Causes Control. 1991;2:3–10. [PubMed: 1873431] [CrossRef]
- Zendehdel K, Nyrén O, Luo J, et al. Risk of gastroesophageal cancer among smokers and users of Scandinavian moist snuff. Int J Cancer. 2008;122:1095–1099. [PubMed: 17973262] [CrossRef]
- Zhang J, Ichiba M, Hara K, et al. Urinary 1-hydroxypyrene in coke oven workers relative to exposure, alcohol consumption, and metabolic enzymes. Occup Environ Med. 2001;58:716–721. [PMC free article: PMC1740063] [PubMed: 11600727] [CrossRef]
- Zhang L, Rothman N, Li G, et al. Aberrations in chromosomes associated with lymphoma and therapy-related leukemia in benzene-exposed workers. Environ Mol Mutagen. 2007;48:467–474. [PubMed: 17584886] [CrossRef]
- Zhang L, Steinmaus C, Eastmond DA, et al. Formaldehyde exposure and leukemia: a new meta-analysis and potential mechanisms. Mutat Res. 2009;681:150–168. c. [PubMed: 18674636] [CrossRef]
- Zhang S, Wang M, Villalta PW, et al. Quantitation of pyridyloxobutyl DNA adducts in nasal and oral mucosa of rats treated chronically with enantiomers of N’-nitrosonornicotine. Chem Res Toxicol. 2009;22:949–956. a. [PMC free article: PMC2743010] [PubMed: 19405515] [CrossRef]
- Zhang S, Wang M, Villalta PW, et al. Analysis of pyridyloxobutyl and pyridylhydroxybutyl DNA adducts in extrahepatic tissues of F344 rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem Res Toxicol. 2009;22:926–936. b. [PMC free article: PMC2701567] [PubMed: 19358518] [CrossRef]
- Zhang X, Su T, Zhang QY, et al. Genetic polymorphisms of the human CYP2A13 gene: identification of single-nucleotide polymorphisms and functional characterization of an Arg257Cys variant. J Pharmacol Exp Ther. 2002;302:416–423. [PubMed: 12130698]
- Zhang Y, Coogan PF, Palmer JR, et al. Cigarette smoking and increased risk of mucinous epithelial ovarian cancer. Am J Epidemiol. 2004;159:133–139. [PubMed: 14718214] [CrossRef]
- Zhang ZF, Kurtz RC, Sun M, et al. Adenocarcinomas of the esophagus and gastric cardia: medical conditions, tobacco, alcohol, and socioeconomic factors. Cancer Epidemiol Biomarkers Prev. 1996;5:761–768. [PubMed: 8896886]
- Zhao B, Seow A, Lee EJ, et al. Dietary isothiocyanates, glutathione S-transferase -M1, -T1 polymorphisms and lung cancer risk among Chinese women in Singapore. Cancer Epidemiol Biomarkers Prev. 2001;10:1063–1067. [PubMed: 11588132]
- Zhao C, Tyndyk M, Eide I, Hemminki K. Endogenous and background DNA adducts by methylating and 2-hydroxyethylating agents. Mutat Res. 1999;424:117–125. [PubMed: 10064855]
- Zheng T, Holford T, Chen Y, et al. Risk of tongue cancer associated with tobacco smoking and alcohol consumption: a case-control study. Oral Oncol. 1997;33:82–85. [PubMed: 9231164] [CrossRef]
- Zheng T, Holford TR, Zahm SH, et al. Cigarette smoking, glutathione-s-transferase M1 and t1 genetic polymorphisms, and breast cancer risk (United States). Cancer Causes Control. 2002;13:637–645. [PubMed: 12296511] [CrossRef]
- Zheng TZ, Boyle P, Hu HF, et al. Tobacco smoking, alcohol consumption, and risk of oral cancer: a case-control study in Beijing, People’s Republic of China. Cancer Causes Control. 1990;1:173–179. [PubMed: 2102288] [CrossRef]
- Zheng W, Blot WJ, Shu XO, et al. Diet and other risk factors for laryngeal cancer in Shanghai, China. Am J Epidemiol. 1992;136:178–191. [PubMed: 1415140]
- Zheng W, Blot WJ, Shu XO, et al. Diet and other risk factors for laryngeal cancer in Shanghai, China. Am J Epidemiol. 1992;136:178–191. b. [PubMed: 1415140]
- Zheng W, Blot WJ, Shu XO, et al. A population-based case-control study of cancers of the nasal cavity and paranasal sinuses in Shanghai. Int J Cancer. 1992;52:557–561. c. [PubMed: 1399136] [CrossRef]
- Zheng W, McLaughlin JK, Chow WH, et al. Risk factors for cancers of the nasal cavity and paranasal sinuses among white men in the United States. Am J Epidemiol. 1993;138:965–972. a. [PubMed: 8256781]
- Zheng W, McLaughlin JK, Gridley G, et al. A cohort study of smoking, alcohol consumption, and dietary factors for pancreatic cancer (United States). Cancer Causes Control. 1993;4:477–482. [PubMed: 8218880] [CrossRef]
- Zheng YM, Tuppin P, Hubert A, et al. Environmental and dietary risk factors for nasopharyngeal carcinoma: a case-control study in Zangwu County, Guangxi, China. Br J Cancer. 1994;69:508–514. [PMC free article: PMC1968852] [PubMed: 8123482]
- Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis. 1998;19:1–27. [PubMed: 9472688] [CrossRef]
- Zhu K, Levine RS, Brann EA, et al. A population-based case-control study of the relationship between cigarette smoking and nasopharyngeal cancer (United States). Cancer Causes Control. 1995;6:507–512. [PubMed: 8580298] [CrossRef]
- Zienolddiny S, Campa D, Lind H, et al. A comprehensive analysis of phase I and phase II metabolism gene polymorphisms and risk of non-small cell lung cancer in smokers. Carcinogenesis. 2008;29:1164–1169. [PubMed: 18258609] [CrossRef]
- Zivaljevic V, Vlajinac H, Marinkovic J, et al. Cigarette smoking as a risk factor for cancer of the thyroid in women. Tumori. 2004;90:273–275. [PubMed: 15315303]
- Znaor A, Brennan P, Gajalakshmi V, et al. Independent and combined effects of tobacco smoking, chewing and alcohol drinking on the risk of oral, pharyngeal and esophageal cancers in Indian men. Int J Cancer. 2003;105:681–686. [PubMed: 12740918] [CrossRef]
- Zu K, Giovannucci E. 2009Smoking and aggressive prostate cancer: a review of the epidemiologic evidence. Cancer Causes ControlEpub ahead of print. [PubMed: 19562492]
- TOBACCO SMOKING - Personal Habits and Indoor CombustionsTOBACCO SMOKING - Personal Habits and Indoor Combustions
Your browsing activity is empty.
Activity recording is turned off.
See more...