9.1 HISTORICAL
The full-blown expression of hypothyroidism is known as myxedema. Adult myxedema escaped serious attention until Gull described it in 1874 1 . That it was a state resembling the familiar endemic cretinism, but coming on in adult life, was what chiefly impressed Gull. Ord 2 invented the term myxedema in 1873. The disorder arising from surgical removal of the thyroid gland (cachexia strumipriva) was described in 1882 by Reverdin 3 of Geneva and in 1883 by Kocher of Berne 4 . After Gull's description, myxedma aroused enormous interest, and in 1883 the Clinical Society of London appointed a committee to study the disease and report its findings 5 . The committee's report, published in 1888, contains a significant portion of what is known today about the clinical and pathologic aspects of myxedema. It is referred to in the following discussion as the Report on Myxedema. The final conclusions of the 200-page volume are penetrating. They are as follows:
1. That myxedema is a well-defined disease.
2. That the disease affects women much more frequently than men, and that the subjects are for the most part of middle age.
3. That clinical and pathological observations, respectively, indicate in a decisive way that the one condition common to all cases is destructive change of the thyroid gland.
4. That the most common form of destructive change of the thyroid gland consists in the substitution of a delicate fibrous tissue for the proper glandular structure.
5. That the interstitial development of fibrous tissue is also observed very frequently in the skin, and, with much less frequency, in the viscera, the appearances presented by this tissue being suggestive of an irritative or inflammatory process.
6. That pathological observation, while showing cause for the changes in the skin observed during life, for the falling off the hair, and the loss of the teeth, for the increased bulk of body, as due to the excess of subcutaneous fat, affords no explanation of the affections of speech, movement, sensation, consciousness, and intellect, which form a large part of the symptoms of the disease.
7. That chemical examination of the comparatively few available cases fails to show the general existence of an excess of mucin in the tissues adequately corresponding to the amount recorded in the first observation, but that this discrepancy may be, in part, attributed to the fact that tumefaction of the integuments, although generally characteristic of myxedema, varies considerably throughout the course of the disease, and often disappears shortly before death.
8. That in experiments made upon animals, particularly on monkeys, symptoms resembling in a very close and remarkable way those of myxedema have followed complete removal of the thyroid gland, performed under antiseptic precautions, and with, as far as could be ascertained, no injury to the adjacent nerves or to the trachea.
9. That in such experimental cases a large excess of mucin has been found to be present in the skin, fibrous tissues, blood, and salivary glands; in particular the parotid gland, normally containing no mucin, has presented that substance in quantities corresponding to what would be ordinarily found in the submaxillary gland.
10. That following removal of the thyroid gland in man in an important proportion of the cases, symptoms exactly corresponding with those of myxedema subsequently develop.
11. That in a considerable number of cases the operation has not been known to have been followed by such symptoms, the apparent immunity being in many cases probably due to the presence and subsequent development of accessory thyroid glands, or to accidentally incomplete removal, or to insufficiently long observation of the patients after operation.
12. That, whereas injury to the trachea, atrophy of the trachea, injury of the recurrent laryngeal nerves, injury of the cervical sympathetic, and endemic influences, have been by various observers supposed to be the true cases of experimental or of operative myxedema (cachexia strumipriva), there is, in the first place, no evidence to show that, of the numerous and various surgical operations performed on the neck and throat, involving various organs and tissues, any, save those in which the thyroid gland has been removed, have been followed by the symptoms under consideration; that in many of the operations on man, and in most, if not all, of the experimental operations made by Professor Horsley on monkeys and other animals, this procedure avoided all injury of surrounding parts, and was perfectly antiseptic; that myxedema has followed removal of the thyroid gland in persons neither living in nor having lived in localities the seat of endemic cretinism; that, therefore, the positive evidence on this point vastly outweighs the negative; and that it appears strongly proved that myxedema is frequently produced by the removal, as well as by the pathological destruction, of the thyroid gland.
13. That whereas, according to Clause 2, in myxedema women are much more numerously affected than men, in the operative form of myxedema no important numerical difference is observed.
14. That a general review of symptoms and pathology leads to the belief that the disease described under the name of myxedema, as observed in adults, is practically the same disease as that named sporadic cretinism when affecting children; that myxedema is probably identical with cachexia strumipriva; and that a very close affinity exists between myxedema and endemic cretinism.
15. That while these several conditions appear, in the main, to depend on, or to be associated with, destruction or loss of the function of the thyroid gland, the ultimate cause of such destruction or loss is at present not evident.
9.2 DEFINITION AND EPIDEMIOLOGY OF HYPOTHYROIDISM
Hypothyroidism is traditionally defined as deficient thyroidal production of thyroid hormone. The term primary hypothyroidism indicates decreased thyroidal secretion of thyroid hormone by factors affecting the thyroid gland itself; the fall in serum concentrations of thyroid hormone causes an increased secretion of TSH resulting in elevated serum TSH concentrations. Decreased thyroidal secretion of thyroid hormone can also be caused by insufficient stimulation of the thyroid gland by TSH, due to factors directly interfering with pituitary TSH release (secondary hypothyroidism) or indirectly by diminishing hypothalamic TRH release (tertiary hypothyroidism); in clinical practice it is not always possible to discriminate between secondary and tertiary hypothyroidism, which are consequently often referred to as central hypothyroidism. In rare cases, symptoms and signs of thyroid hormone deficiency are caused by the inability of tissues to respond to thyroid hormone by mutations in the nuclear thyroid hormone receptor TRß; this condition, known as thyroid hormone resistance (see Ch. 16 ), is associated with an increased thyroidal secretion of thyroid hormones and increased thyroid hormone concentrations in serum in an attempt of the body to overcome the resistance to thyroid hormone. Mutations in other genes involved with extrathyroidal metabolism and action of thyroid hormones in target tissues may also cause a hypothyroid state. Such cases could be labelled as peripheral (extrathyroidal) hypothyroidism. It thus seems more appropriate to define hypothyroidism as thyroid hormone deficiency in target tissues, irrespective of its cause.
9.2.1. GRADES OF HYPOTHYROIDISM
Hypothyroidism is a graded phenomenon, ranging from very mild cases in which biochemical abnormalities are present but the individual hardly notices symptoms and signs of thyroid hormone deficiency, to very severe cases in which the danger exists to slide down into a life-threatening myxedema coma. In the development of primary hypothyroidism, the transition from the euthyroid to the hypothyroid state is first detected by a slightly elevated serum TSH, caused by a minor decrease in thyroidal secretion of T4 which doesn't give rise to subnormal serum T4 concentrations. The reason for maintaining T4 values within the reference range is the exquisite sensitivity of the pituitary thyrotroph for even very small decreases of serum T4, as exemplified by the log-linear relationship between serum TSH and serum FT4 1 . A further decline in T4 secretion results in serum T4 values below the lower normal limit and even higher TSH values, but serum T3 concentrations remain within the reference range. It is only in the last stage that subnormal serum T3 concentrations are found, when serum T4 has fallen to really very low values associated with markedly elevated serum TSH concentrations (Figure 9-1). Hypothyroidism is thus a graded phenomenon, in which the first stage of subclinical hypothyroidism may progress via mild hypothyroidism towards overt hypothyroidism (Table 9-1) 3 .

Figure 9-1. I
ndividual and median values of thyroid function tests in patients with various grades of hypothyroidism. Discontinuous horizontal lines represent upper limit (TSH) and lower limit (FT4,T3) of the normal reference ranges. (Reproduced with permission) (2)
Table 9-1
Grades of hypothyroidism.
Maintenance of a normal serum T3 concentration until a relatively late stage in the development of hypothyroidism obviously serves as an appropriate mechanism of the body to counteract the impact of diminishing production of T4. It is accomplished by a preferential thyroidal secretion of T3: the increased secretion of TSH enhances the synthesis of T3 more than that of T4 and stimulates thyroidal 5'-monodeiodination of T4 into T3 4,5 . It explains why sometimes a slightly elevated serum T3 is found in the early stage of development of hypothyroidism. About 80% of the daily production rate of T3 is generated in extrathyroidal tissues via the conversion of T4 into T3. The peripheral tissues also have a defense mechanism against developing hypothyroidism by increasing the overall fractional conversion rate of T4 into T3 6 .
9.2.2. EPIDEMIOLOGY OF HYPOTHYROIDISM
Thyroid hormone resistance syndromes are seldom the cause of hypothyroidism; the number of registered patients approximates one thousand (see Ch. 16 ). Central hypothyroidism is also rare; its precise prevalence is unknown, but has been estimated as 0.005% in the general population 7 . Primary hypothyroidism, in contrast, is a very prevalent disease worldwide. It can be endemic in iodine-deficient regions (see Ch. 20 ), but it is also a common disease in iodine-replete areas as evident from prevalence and incidence figures reported in a number of population-based studies 8-14 . The most extensive data has been obtained from the Whickham Survey, a study of 2779 adults randomly selected of the general population in Great Britain who were evaluated between 1972 and 1974 and again twenty years later 8,9 . Most striking are the high prevalence of thyroid microsomal (peroxidase) antibodies and of (subclinical) hypothyroidism, and the marked female preponderance (Table 9-2).
The mean incidence of spontaneous hypothyroidism in women was 3.5/1000 survivors/year, that of hypothyroidism after destructive treatment for thyrotoxicosis 0.6/1000 survivors/year; similar figures were obtained in those who had deceased during follow-up. The hazard rate (the probability to develop hypothyroidism) increased with age; the mean age at diagnosis of hypothyroidism in women was 60 years. Studies from other countries like the USA 10,11 , Japan 12 and Sweden 13 report essentially similar data.
Of particular interest are risk factors for development of hypothyroidism. In women survivors of the Whickham Survey, the risk of developing overt hypothyroidism was 4.3% per year if both raised serum TSH and thyroid antibodies were present initially, 2.6% per year if raised serum TSH was present alone, and 2.1% per year if thyroid antibodies were present alone 9 . At the time of follow-up twenty years later, hypothyroidism had developed in these three groups in 55%, 33% and 27% respectively, but only in 4% if initial serum TSH was normal and thyroid antibodies were absent. The probability of developing hypothyroidism already increases at a rise in serum TSH above 2 mU/L as shown in Figure 9-2 , in thyroid antibody positive as well as in thyroid antibody negative women; it also increases with higher titres of thyroid microsomal antibodies 9 , 15 . These data are confirmed by two other more recent large population-based longitudinal surveys with a mean follow-up of 11-13 years. A figure almost identical to figure 9.2 was obtained in an Austalian study, in which the odds of hypothyroidism increased at TSH >2.5 mU/L, being always higher in the presence of TPO antibodies 16 . Increasing TSH values within the reference range of 0.2-4.5 mU/L gradually increased the risk of future hypothyroidism in the Norwegian HUNT study: odds ratio’s were significantly higher at baseline TSH >1.5 mU/L in women and at TSH > 2.0 in men 17 .
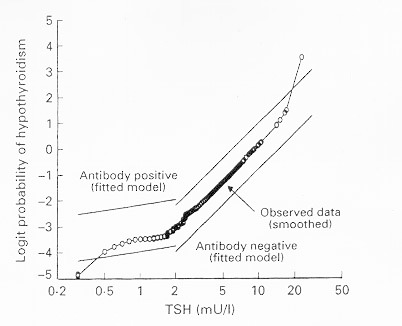
Figure 9-2
Logit probability (log odds) for the development of hypothyroidism as a function of TSH values at first survey during a 20-year follow-up of 912 women in the Whickham Survey. (Reproduced with permission)(9).
9.3 CAUSES OF HYPOTHYROIDISM
A variety of functional or structural disorders may lead to hypothyroidism, the severity of which depends on the degree and duration of thyroid hormone deprivation. A classification according to etiology appears in Table 9-3 . The two principal categories are primary (or thyroprivic) hypothyroidism caused by an inherent inability of the thyroid gland to supply a sufficient amount of the hormone, and central (or trophoprivic) hypothyroidism due to inadequate stimulation of an intrinsically normal thyroid gland resulting from a defect at the level of the pituitary (secondary hypothyroidism) or the hypothalamus (tertiary hypothyroidism). In a third (uncommon) form of hypothyroidism, regulation and function of thyroid gland are intact. Instead, manifestations of hormone deprivation arise from a disorder in the target tissues that reduces their responsiveness to the hormone (peripheral tissue resistance to thyroid hormone) or that inactivates the hormone (in massive infantile hemangiomas).
The most common cause of hypothyroidism is destruction of the thyroid gland by disease or as a consequence of vigorous ablative therapies to control thyrotoxicosis. Primary hypothyroidism may also result from inefficient hormone synthesis caused by inherited biosynthetic defects (see Ch. 16 ), a deficient supply of iodine (see Ch. 20 ), or inhibition of hormonogenesis by various drugs and chemicals (see Ch. 5 ). In such instances, hypothyroidism is typically associated with thyroid gland enlargement (goitrous hypothyroidism).
Table 9-3
Causes of hypothyroidism.
9.3.1. CENTRAL HYPOTHYROIDISM
Hypothalamic disorders cause reduced TSH secretion by impairing the production or transport of TRH to the pituitary gland. Hypothyroidism may occur because the pituitary secretes TSH in insufficient quantities, or secretes TSH with an abnormal glycosylation pattern which reduces the biologic activity of TSH 1,2,3 . Treatment with oral TRH restores the biologic activity of TSH, suggesting that deficient hypothalamic TRH release induces both quantitative and qualitative abnormalities of TSH secretion. TSH molecules with reduced biologic activity may retain their immunologic reactivity in TSH immunoassays, explaining the sometimes observed slightly increased values of serum TSH (up to 10 mU/l) in central hypothyroidism 18, 23 .
The term central hypothyroidism is preferred because it is not always possible to distinguish between hypothalamic and pituitary causes. Central hypothyroidism is also associated with a decreased nocturnal TSH surge (due to loss of the nocturnal increase in TSH pulse amplitude under preservation of the nighttime increase in TSH pulse frequency), which further hampers maintenance of a normal thyroid function 4,5 .
Central hypothyroidism is a relatively rare condition occurring about equally in both sexes. Congenital cases of central hypothyroidism are due to structural lesions like pituitary hypoplasia, midline defects and Rathke's pouch cysts, or to functional defects in TSH biosynthesis and release like loss-of-function' mutations in genes encoding for the TRH receptor 6 , the TSH-beta subunit 7,8 , pituitary-specific transcription factors ( POU1F1 , PROP1, LHX3, LHX4 or HESX1), and LEPR or IGSF1 9 . Familial hypothyroidism due to TSHβ gene mutations follows an autosomal mode of inheritance. The β-subunit (118 aa) heterodimerizes noncovalently with the α-subunit through a segment called the seat-belt (aa 88-105). The described mutations of the TSHβ gene hamper dimerization with the α-subunit and thereby the correct secretion of the mature TSH heterodimer: Q42X and Q29X introduce a premature stop codon resulting in a truncated TSHβ subunit, G29R is a nonsense mutation preventing dimer formation, and C105V, 114X is a frameshift mutation causing disruption of one of the two disulfide bridges stabilizing the seat belt region 7,8,19,20 . Plasma TSH levels are variable, the TSH response to TRH is impaired but PRL secretion is normal, and plasma glycoprotein hormone α-subunits are high 19 . Mutations in pituitary transcription factors like POU1F1 and PROP1 are associated with deficiencies of TSH, GH and PRL 9 . Loss-of-function mutations in the membrane glycoprotein IGSF1 cause an X-linked syndrome characterized by central hypothyroidism, hypoprolactinemia, delayed puberty, macroorchidism and increased body weight; it is hypothesized that central hypothyroidism in these cases is secondary to an associated impairment in pituitary TRH signalling 32,33 . Cases of central hypothyroidism in childhood are mostly caused by craniopharyngioma (TSH deficiency in 53%) or cranial irradiation for brain tumors like dysgerminoma (TSH deficiency in 6%) or hematological malignancies 24 . Prophylactic cranial irradiation of the central nervous system in children with acute lymphoblastic leukaemia did not have an adverse effect on thyroid function within a median follow-up time of 8 years 21 .
Central hypothyroidism in adults is most frequently due to pituitary macroadenomas and pituitary surgery or irradiation 22 . The occurrence of TSH deficiency occurs usually after loss of GH and gonadotropin secretion. Return to euthyroidism is sometimes observed after selective adenomectomy 10 . Radiotherapy of brain tumors or pituitary adenomas is followed by hypothyroidism in up to 65%; the onset of hypothyroidism may be seen many years after radiotherapy 11,12 . Less common causes of adult central hypothyroidism are head injury 13, 25 , ischemic necrosis due to postpartum hemorrhage (Sheehan's syndrome), pituitary apoplexy, infiltrative diseases, and lymphocytic hypophysitis 14 . Lymphocytic hypophysitis seems to be an autoimmune disease; it occurs predominantly in women, especially during and after pregnancy, and the clinical picture is characterized by a pituitary mass and hypopituitarism 26 . A systematic review of articles published between 2000 and 2007 reported frequencies of anterior hypopituitarism in adults in the chronic phase after traumatic brain injury or subarachnoid hemorrhage (27): TSH deficiency was observed in 5.9% (95% CI 1.3-10.5) after subarachnoid hemmorrhage, and in 4.1% (95% CI 2.9-5.7) after traumatic brain injury. In prospective studies after traumatic brain injury TSH deficiency was observed in 3.9%, 6.8%, 2.1% and 4.2% at the acute phase and after 3, 6 and 12 months respectively (27).
Dopamine infusion inhibits the release of TSH, which may decrease T4 production rate by 56% 15 . Supraphysiological amounts of endogenous or exogenous glucocorticoids also dampen the release of TSH, but give seldom rise to decreased serum T4 values. The same is true for treatment with long-acting somatostatin analogs. A transient decrease of TSH secretion can be observed after withdrawal of TSH-suppressive doses of L-thyroxine, which may last up to 6 weeks 16 .
A new and novel cause of iatrogenic central hypothyroidism is from the administration of the RXR-selective ligand, bexarotene (Targretin). This medication is highly effective in cutaneous T cell lymphoma, but as reported by Sherman et al, up to 70% of patients treated with daily doses > 300 mg/m 2 had symptoms and signs of hypothyroidism. This was associated with reduction of serum TSH to an average of 0.05 mU/l, and reduction of free T4 from 12.9 pmol/l to 5.8 pmol/l 17 . A single dose of bexarotene rapidly and significantly suppresses serum TSH in healthy subjects, without an effect on serum prolactin or cortisol, suggesting a specific effect on thyrotropes (28). In vitro studies have shown that activity of the TSHβ subunit gene promoter is suppressed by 9-cis-retinoic acid and bexarotene 17 , but other studies have not confirmed this 29 . Rexinoids may further increase thyroid hormone metabolism through deiodination, sulfation and possibly glucuronidation (30,31).The condition can be appropriately treated by administration of thyroid hormone. (17)
9.3.2 CHRONIC AUTOIMMUNE THYROIDITIS
Chronic autoimmune thyroiditis may eventually cause hypothyroidism, mainly via destruction of thyrocytes (see also Ch. 7 ). In goitrous autoimmune hypothyroidism (the classical variant originally described by Hashimoto) the histology of the thyroid gland is characterized by massive lymphocytic infiltration with formation of germinal centers and oxyphilic changes of thyrocytes. In atrophic myxedema fibrosis is predominant, next to lymphocytic infiltration. The diffuse Hashimoto goiter has a peculiar firm consistency like rubber; the goiter may regress with time but can persist in many cases 1 . In some instances the patient presents with an initial transient hyperthyroid stage, called Hashitoxicosis'. The term Hashimoto's disease is generally used to indicate auto-immune destruction of thyrocytes which may eventually result in hypothyroidism although many cases remain euthyroid (see also Ch. 8 ). The serological hallmark of Hashimoto's disease is the presence of high titers of thyroid peroxidase (TPO) autoantibodies, formerly known as thyroid microsomal antibodies. The opposite of Hashimoto's disease is Graves' disease characterized by the presence of TSH receptor stimulating antibodies resulting in hyperthyroidism. The two disease entities frequently overlap, and can be viewed as the opposite ends of a continuous spectrum of autoimmune thyroid disease. Indeed, many patients with Graves' disease have TPO antibodies, and some case reports mention classical features of Graves' disease like exophthalmos and pretibial myxedema in the presence of hypothyroidism without any previous thyrotoxicosis 2 . TSH receptor blocking antibodies do occur in Hashimoto's disease, contributing to thyroid atrophy and hypothyroidism; they are more prevalent in Japanese than in Caucasian patients 3,4 . TSH receptor antibodies in Hashimoto's disease are negatively correlated to serum FT4 and thyroid size 5 .
The clinical manifestation of Hashimoto's disease with respect to thyroid function and thyroid size depends on the net effect of the various immunologic effector mechanisms involved in chronic autoimmune thyroiditis. Genetic and environmental factors may modulate the expression of the disease (6). Autoimmune hypothyroidism is associated with a number of single nucleotide polymorphisms in susceptibility genes (HLA-DR3, CTLA-4, PTPN22, Tg) (7). The prevalence of Hashimoto’s thyroiditis is higher in regions with a high ambient iodine intake than in iodine-deficient areas 8,9,10 . Smoking to a certain extent protects against the development of TPO antibodies and overt autoimmune hypothyroidism (11,12,13).
9.3.3 REVERSIBLE AUTOIMMUNE HYPOTHYROIDISM
Chronic autoimmune thyroiditis. Conventional wisdom has it that ‘once hypothyroid means always hypothyroid'. Indeed, the vast majority of patients with hypothyroidism due to chronic autoimmune thyroiditis require life-long thyroxine replacement therapy, but spontaneous recovery does occur in about 5% 1 . Return to the euthyroid state is apparently more frequent in countries like Japan, where - at a high ambient iodine intake - restriction of dietary iodine alone may induce a remission 2 .Conditions that increase the likelihood of spontaneous recovery are the presence of a goiter, a relatively high thyroidal radioiodine uptake, and a preserved increase of T3 after the administration of TRH during thyroxine treatment 3,4,5 .The spontaneous evolution from hypothyroidism back to euthyroidism has been related to the disappearance of TSH receptor blocking antibodies 6 . Changes in the titers of co-existing TSH receptor blocking and stimulating antibodies explain the sometimes observed alternating course of hypothyroidism and hyperthyroidism in the same subject 7 .
Silent thyroiditis and postpartum thyroiditis. Silent or painless thyroiditis and postpartum thyroiditis are variant forms of chronic autoimmune thyroiditis. The autoimmune reaction causes a mainly T-cell mediated destructive thyroiditis, which however is self-limiting. The characteristic course of the disease is thus first a thyrotoxic stage due to the release of stored hormone from the disrupted follicles, followed by a hypothyroid stage during the recovery towards a normal thyroid architecture; usually euthyroidism is restored within a few months (see also Ch. 8 ). In many cases the disease remains unnoticed, as clinical symptoms and signs are mostly limited. In the postpartum period it is also quite natural to attribute emerging complaints - especially if they are nonspecific in nature - to the aftermath of pregnancy and the work load of having a baby. Postpartum thyroiditis is, however, a rather common event, with an incidence of 4-6% as evident from several population-based studies 8,9 . The incidence in type I diabetes mellitus is four times higher, up to 25% 10 . Postpartum thyroiditis can be predicted to a certain extent from the presence of TPO antibodies in the serum of pregnant women in the first trimester: a titer of >100 kU/l at 2 weeks has a positive predictive value of 0.50 and a negative predictive value of 0.98 in this respect 9 . The titer of TPO antibodies decreases in the second and third trimester, and increases again in the postpartum period . Women who have experienced postpartum thyroiditis, have a 40% risk to develop again postpartum thyroiditis after a following pregnancy. About 20-30% of women with postpartum thyroiditis will develop permanent hypothyroidism within 5 years; the risk is higher in women with high titers of TPO-antibodies 11 . A subset of women with postpartum thyroiditis experience only a thyrotoxic phase; they are less at risk for later development of hypothyroidism 12 . Maternal TPO antibodies are associated with depression in the postpartum period 13 and with impaired child development 14 . A low maternal FT4 concentration during early pregnancy is also associated with impaired psychomotor development in infancy 15,16 .
Cytokine-induced thyroiditis. Cytokines are heavily involved in immune reactions (see Ch. 7 ), and it is thus not surprising that treatment with pharmacological doses of cytokines may induce autoimmune diseases in susceptible subjects. Treatment with interleukin-2 or interferon-α (IFNα) of patients with malignant tumors or hepatitis B or C is causally related to the occurrence of TPO-antibodies and the development of abnormal thyroid function 17,18,19,20 . The incidence is about 5-20%. Two types of IFNα-induced hypothyroidism have been recognized: autoimmune and non-autoimmune (21). Interferon-α induced autoimmune hypothyroidism is characterized by the presence of TPO antibodies. Elevated TPO antibodies before start of IFNα therapy increases the risk (positive predictive value 68% for the development of overt autoimmune hypothyroidism) (22), but TPO antibodies may develop de novo during IFNα treatment in 10-40% (23) Interferon-α induced non-autoimmune hypothyroidism is a destructive thyroiditis: a self-limited inflammatory disorder of the thyroid gland, characterized by an early thyrotoxic phase caused by the release of preformed thyroid hormones, and a late hypothyroid phase with complete resolution in most cases. IFNα has many immune effects including activation of immune cells, switching the immune response to Th1 pathways, downregulation of Treg cells, induction of cytokine release, and induction of MHC I expression on thyroid cells, all likely involved in the pathogenesis of IFNα induced autoimmune hypothyroidism (although it remains less well understood why IFNα can also induce Graves’ hyperthyroidism) (23). IFNα also exerts a direct toxic effect on thyrocytes, possibly involved in IFNα induced non-autoimmune hypothyroidism (26). IFNα-induced thyroiditis is most common in patients with chronic hepatitis C. It has therefore been hypothesized that hepatitis C virus could trigger thyroiditis by infecting thyroid cells. Supporting this view is the finding that the hepatitis C virus E2 protein can bind to CD81 molecules on thyroid cells and provoke IL-8 secretion (24).The prevalence of autoimmune and non-autoimmune types is about similar. Treatment is with levothyroxine, with no need to stop IFNα therapy. L-T4 replacement requirements may increase if patients are treated with a second course of interferon, or may decrease or end after completion of the IFNα course (19,25). It is recommended to screen all patients before starting IFNα therapy (TSH, TPO-Ab). If TSH is normal and TPO antibodies negative, TSH monitoring every three months is recommended until completion of IFNα treatment. If TPO antibodies are present, TSH monitoring every two months might be useful (23).
9.3.4 POSTOPERATIVE AND POSTRADIATION HYPOTHYROIDISM
Surgery. An important cause of hypothyroidism is surgical removal of the gland. Up to 40 percent of patients who undergo thyroidectomy for Graves' disease develop hypothyroidism (1). Most patients become hypothyroid in the first year after surgery; immediate postoperative hypothyroidism may resolve spontaneously by 6 months. After the first year the cumulative incidence of hypothyroidism rises by 1-2% per year. The frequency of hypothyroidism depends on the zeal of the surgeon and on other factors, such as the function of the thyroid remnant or the presence of active thyroiditis. Its occurrence correlates with the presence of antibodies to thyroid antigens. Thus, progressive destruction of residual tissue by thyroiditis may be the pathogenic mechanism. Hypothyroidism after surgical removal of multinodular goiter is less common (about 15%). Myxedema occurs almost invariably after subtotal thyroidectomy for Hashimoto's thyroiditis and after removal of lingual thyroids.
Radioiodine. A leading cause of hypothyroidism is radioactive iodine (RAI) treatment of Graves' disease. The frequency with which hypothyroidism supervenes RAI therapy is dependent on multiple factors, the principal one being the dose of RAI administered. The incidence of hypothyroidism 10 years after treatment is reported as high as 70 percent 1 . Hypothyroidism frequently develops already in the first year after treatment (with spontaneous return to euthyroidism in some patients), but it may not be manifest until years later in others. Its cumulative occurrence after the first year continues to rise with 0.5-2% annually, and it has been suggested that virtually all patients treated in this way will eventually become hypothyroid. Various treatment schedules have been devised with the hope of diminishing the incidence of RAI-induced hypothyroidism 2,3 , but in general, a lower incidence of hypothyroidism is invariably associated with a higher prevalence of persistent thyrotoxicosis that requires retreatment 3,4 . Inadvertent administration of RAI during gestation may cause neonatal hypothyroidism when given to the mother during the last two trimesters and also occasionally in the first trimester of pregnancy 5 . Hypothyroidism occurs less often (6-13 %) after 131I treatment of toxic nodular goiter 6,7 .
External irradiation. Hypothyroidism may supervene after therapeutic irradiation of the neck for any of a number of malignant diseases. It is particularly common (25-50%) after irradiation for Hodgkins' and non-Hodgkins' lymphoma, especially when the thyroid has not been shielded during mantle field irradiation and when iodine-containing X-ray contrast agents have been used prior to radiotherapy 8 . External radiotherapy for head and neck cancer (e.g. laryngeal carcinoma) carries an actuarial risk of 15% for developing overt hypothyroidism three years after treatment 10 . Elevated TSH values are even more common, with a 5-year incidence rate of 48% in another study with a median follow-up of 4,4 years 11 . Total body irradiation with subsequent bone marrow transplantation for acute leukemia or aplastic anemia may cause (subclinical) hypothyroidism in about 25%, usually occurring after one year and transient in half of the patients 9 . Probably because of radiation damage, subclinical or overt hypothyroidism is common among surviving bone marrow transplant recipients: there is a greater risk among younger patients, and need for life-long surveillance (12).
9.3.5 INFILTRATIVE AND INFECTIOUS DISEASES
The production of hypothyroidism by infiltrative disease is mentioned for completeness, despite the rarity of these conditions. Among these rare causes of primary hypothyroidism are sarcoidosis, cystinosis 1 (up to 86% in adults), progressive systemic sclerosis and amyloidosis 2 . Hypothyroidism is a frequent sequela of invasive fibrous thyroiditis of Riedel, occurring in 30-40% of the patients.
Hypothyroidism due to infectious disease is equally rare (3). Infection of the thyroid gland is somewhat more frequent in immunocompromised patients and in subjects with pre-existent thyroid abnormalities. Hypothyroidism in the recovery phase of subacute thyroiditis of De Quervain - a condition most likely related to a previous viral infection- is in contrast a very common event (see Ch. 19 ).
9.3.6 CONGENITAL HYPOTHYROIDISM
Congenital hypothyroidism can be permanent or transient in nature. Transient cases might be caused by transplacental passage of TSH receptor blocking antibodies, or iodine excess. Permanent cases are caused by either loss of functional tissue (mostly thyroid dysgenesis), by functional defects in thyroid hormone biosynthesis (‘loss of function' mutations in genes encoding for the TSH-R, NIS, Tg, TPO, DUOX2 and its maturation factor DUOXA2, or DEHAL), or by thyroid hormone resistance (TRα and TRβ1 mutations). For full discussion: see Ch. 15 and 16 .
9.3.7 IODINE DEFICIENCY AND IODINE EXCESS
Hypothyroidism caused by iodine deficiency is discussed in Ch. 20 . It is remarkable that hypothyroidism can also be caused by iodine excess, a condition described in the literature as ‘iodide-induced myxedema'. It can be explained by autoregulatory mechanisms operative in the thyroid gland. Inorganic iodide in excess of daily doses of 500-1000 µg inhibits organification of iodide; this phenomenon is known as the Wolff-Chaikoff effect. Usually an escape from the Wolff-Chaikoff effect occurs after several weeks. An unidentified iodinated product of the organification process (presumably an iodinated lipid) seems to be involved, which inhibits thyroidal iodide transport: consequently, the intrathyroidal iodine concentration falls below the level required for inhibition of organification 1 . Failure to escape from the Wolff-Chaikoff effect may produce hypothyroidism and this occurs preferentially in subjects with pre-existent subtle organification defects. Indeed patients with chronic autoimmune thyroiditis, previous subacute or postpartum thyroiditis, or previous radioiodine or surgical therapy are prone to iodide-induced hypothyroidism 2 ,3 .
Sources of iodine excess are an iodine-rich diet (e.g. seaweed ) and iodine-containing drugs like potassium iodide, some vitamin preparations, kelp tablets, topical antiseptics, radiographic contrast agents, and amiodarone. Amiodarone contains 39% of iodine by weight; large quantities of iodine are released during the biotransformation of the drug, giving rise to a 45-60 times higher iodine exposure than the optimal daily iodine intake of 150-300 µg recommended by the WHO.Amiodarone-induced hypothyroidism occurs predominantly in the first 18 months of treatment, especially in females with pre-existent thyroid antibodies 4 . Its incidence is higher in regions with a high ambient iodine intake than in areas with a lower iodine intake (22% and 5% respectively) 5 .
Mild iodide fortification of salt in Denmark increased average urinary iodide from the 45-61ug/l range up to 86-93ug/l. This cautious iodization of salt was accompanied by a moderate increase in the baseline incidence rate of overt hypothyroidism (38/100,000/yr) by 20-30%. This occurred primarily in young and middle-aged subjects with previous moderate iodine deficiency (6).
9.3.8 DRUG-INDUCED HYPOTHYROIDISM
A variety of therapeutic drugs can lead to moderate or even severe hypothyroidism (see also Ch. 9.8.3 ). The common antithyroid drugs (carbimazole, methimazole, and propylthiouracil) if given in sufficient quantity will cause hypothyroidism. This is also theoretically possible with agents that can block the uptake of iodide by the thyroid, such as perchlorate or thiocyanate, although these are rarely given. In susceptible individuals, primarily those with a history of autoimmune thyroid disease such as Hashimoto's or Graves' disease or in patients who have had either radiation or surgical trauma to the thyroid gland, large doses of iodide can cause goitrous hypothyroidism 1,2 (see also Ch. 9.3.7 ). While this is now less common, since iodides are no longer given for chronic pulmonary disease and lipid-soluble contrast agents are no longer used in diagnostic procedures, the problem may arise with patients taking iodine supplements or natural foods with high iodine content. Lithium has similar effects to those of iodide; it inhibits thyroid hormone release as well as hormone synthesis 3 . While lithium-induced hypothyroidism is more common in patients with underlying autoimmune disease, it has been reported in individuals with apparently normal thyroid glands. Long-term treatment with lithium results in goiter in about 50%, in subclinical hypothyroidism in about 20%, and in overt hypothyroidism also in 20% 4 . There are a large number of organic compounds that may impair thyroid function. These include phenol derivatives such as resorcinol, benzoic acid compounds such as para-aminosalicylic acid, the oral sulfonylurea compounds, phenylbutazone, aminoglutethimide, and a number of other agents 5 . Industrial pollution with polychlorinated biphenyls can also cause goitrous hypothyroidism 6 . In workers exposed to perchlorate, serum TSH and thyroid volume were not affected (7). Also in healthy volunteers, a 6-month exposure to perchlorate at doses up to 3 mg/day had no effect on thyroid function (8). Environmental low-level perchlorate exposure was ubiquitous in pregnant women but did not affect thyroid function (9).
Tyrosine kinase inhibitors freqently affect thyroid gland function and thyroid hormone metabolism. Imatinib and motesanib therapy has no adverse thyroid effects when a normal thyroid gland is in situ, but may require an increase in the replacement dose of L-T4 in hypothyroid patients (see section 9.8.3). In contrast, sunitinib and sorafenib therapy (applied in gastrointestinal stromal tumors and renal cell carcinoma) gives rise to primary hypothyroidism in a high proportion of patients (10). In the first study on 42 euthyroid patients treated by sunitinib, 36% had persistent hypothyroidism requiring L-T4 treatment, 17% had TSH between 5 and 7 mU/L, and 10% had suppressed TSH levels (11). In a prospective study among 59 patients, 61% were found to have a transient or permanently elevated TSH, and 27% required L-T4 replacement (12). The probability of hypothyroidism increases with each time and each cycle of treatment. Serum TSH increases at the end of the ON phase and is near the mormal range at the end of the OFF phase, leading to intermittent hypothyroidism. After several treatment cycles a permanent hypothyroidism occurs. It is uncertain if thyroid function tests return to normal after definitive withdrawal of sunitinib therapy.How sunitinib reduce thyroid gland function, is incompletely understood. Inhibition of thyroid radioiodine uptake has been observed (13) but in vitro experiments showed no effect on sodium-iodide transporters in thyroid cells (14). Impairment of thyroid peroxidase activity is shown in vitro (15), but still to be confirmed in vivo. Destructive thyroiditis has also been proposed (11,16). Recent case studies report marked reduction of thyroid volume and blood flow during sunitinib (17,18). A unifying hypothesis is that sunitinib (inhibiting vascular endothelial growth factor receptors as a major mechanism of action on tumors) causes regresion of the thyroid vascular bed resulting in impaired thyroid function(19).Vasoconstriction of thyroid vessels could reduce glandular uptake of radioiodine. Reduced thyroid perfusion could cause apoptosis of thyroid cells, resulting in thyroiditis in some patients. Sorafenib therapy in patients with metastatic renal cell carcinoma was associated with TSH elevations in 18% after 2-4 months, and one quarter of them developed thyroglobulin antibodies (20). TSH may be suppressed before the development of elevated TSH levels, suggesting destructive thyroiditis (21). Hypothyroidism would persist after sorafenib withdrawal. Sorafenib also has anti-angiogenic effects. It has been postulated that thyroid toxicity is restricted to tyrosine kinase inhibitors targeting key kinase receptors in angiogenic pathways, but not other kinase receptors (22,23).
9.3.9. CONSUMPTIVE HYPOTHYROIDISM (MASSIVE INFANTILE HEMANGIOMA)
Severe hypothyroidism has been described in a few infants with massive hemangiomas, due to high levels of activity of type 3 iodothyronine deiodinase in the hemangioma tissue 1 . Type 3 deiodinase inactivates T4 by conversion into reverse T3 (explaining the paradoxically high serum rT3 concentrations in these hypothyroid patients), and T3 by conversion into 3,3’-diiodothyronine. The high level of expression of type 3 deiodinase is likely induced by growth factors. The infants have no evidence of thyroid gland disease, and their hypothyroidism is apparently caused by an increased rate of thyroid hormone degradation in extra-thyroidal tissues outstripping the rate of thyroid hormone production: a nice example of “consumptive” hypothyroidism. This type of “peripheral” hypothyroidism has also been observed in a young adult 3 , in an athyreotic adult on levothyroxine 5 , and in a 54-yr patient with a large malignant solitary fibrous tumor expressing functional type 3 deiodinase activity 4 Surgical removal of the tumor restores euthyroidism.
9.4 PATHOLOGY OF HYPOTHYROIDISM
The characteristic pathologic finding in hypothyroidism is a peculiar mucinous nonpitting edema (myxedema), which is most obvious in the dermis but can be present in many organs. The myxedema is due to accumulation of hyaluronic acid and other glycosaminoglycans in interstitial tissue; these hydrophilic molecules attract much water 1 . The deposits of glycosaminoglycans have been related to loss of the inhibitory effects of thyroid hormone on the synthesis of hyaluronate, fibronectin and collagen by fibroblasts 2,3 .
The skin is distinctly abnormal. There is hyperkeratotic plugging of sweat glands and hair follicles. The dermis is edematous, and the collagen fibers are separated, swollen, and frayed. Extracellular material that appears eosinophilic or basophilic in hematoxylin and eosin stains, or that appears pink (metachromatic) with toluidine blue, or takes the periodic acid-Schiff (PAS) stain for mucopolysaccharides is much increased in the dermis. A sparse mononuclear cell infiltrate may be found about the blood vessels.
Skeletal muscle cells are swollen and appear grossly to be pale and edematous. Frequently microscopic examination reveals no significant abnormality. Alternatively, the normal striations are lost, and degenerative foci are seen in the cells. The fibers are separated in these degenerative foci by accumulations of a basophilic, PAS-positive homogenous infiltrate. This infiltrate may appear as a semilunar deposit under the sarcolemma.
The heart may be dilated and hypertrophied. Interstitial edema and an increase in fibrous tissue are present. The individual muscle cells may show the same changes seen in skeletal muscle. The serous cavities may all contain abnormal amounts of fluid with a normal or high protein content. The liver may appear normal or may show evidence of edema. Central congestive fibrosis in the absence of congestive heart failure has been described. The mitochondria tend to be spherical and their limiting membranes smooth, whereas those of the liver in thyrotoxicosis vary in shape and have wrinkled outer membranes 4 . The skeleton may be unusually dense on radiographic examination. In children, bone maturation is usually retarded, and typical epiphyseal dysgenesis of hypothyroidism is present 5 . The brain may show atrophy of cells, gliosis, and foci of degeneration. Deposition of mucinous material and round bodies containing glycogen (neural myxedematous bodies) has been found in the cerebellum of patients with long-standing myxedema and ataxia 6 . In uncorrected congenital hypothyroidism , the brain retains infantile characteristics. There is neuronal hypoplasia, retarded myelination, and decreased vascularity (see Ch. 15 ). The blood vessels often show prominent atherosclerosis. Whether this condition is more severe than might be anticipated on the basis of the patient's age and sex remains an unsettled question. In the intestinal tract there is an accumulation of mast cells and interstitial mucoid material, especially near the basement membrane. The smooth muscle cells may show lesions similar to those seen in skeletal muscle. The mucosa of the stomach, small bowel, and large bowel may be atrophic. The rest of the gastrointestinal tract, especially the colon, may be very dilated (myxedema megacolon). The uterus typically has a proliferative or atrophic endometrium in premenopausal women.
The kidney is grossly normal. Light and electron microscopic studies of renal biopsy samples have demonstrated thickening of the glomerular and tubular basement membranes, proliferation of the endothelial and mesangial cells, intracellular inclusions, and extracellular deposition of amorphous material with characteristics of acid mucopolysaccharides 8,9 .
In the pituitary in primary myxedema there is an increase in a class of cells that can be identified by the iron-periodic, acid Schiff, or aldehyde fuchsin staining techniques 10 . These are referred to variously as gamma cells, sparsely granulated basophils, or amphophils. Presumably they are derived from basophilic cells or chromophobes and are active in secreting TSH. Acidophilic cells are decreased. Patients who are congenitally hypothyroid and those who are hypothyroid during childhood may develop pituitary fossa enlargement. Occasionally prolonged hypothyroidism leads to sella enlargement in the adolescent and adult, and pituitary tumors have been described 11 . In these glands acidophils are virtually absent. In pituitary hypothyroidism the pituitary may be replaced by fibrous and cystic structures, granulomas, or neoplasia. Occasionally hypothyroidism due to deficient TSH secretion occurs in patients having the empty sella syndrome or because of isolated TSH or TRH deficiency. The adrenals may be normal or their cortex may be atrophied. The combination of adrenal cortical atrophy and hypothyroidism is known as Schmidt's syndrome and is thought to be of autoimmune etiology. Bloodworth found clinical evidence for hypothyroidism in 9 of 35 patients with Addison's disease; in 8 there was fibrosis of thethyroid, with atrophy in 4. The adrenal medulla appeared normal 12 . The ovaries and parathyroids have shown no definite abnormalities. The testes may show Leydig cells with involutionary nucleus and cytoplasm, hyalinization, or involution of the tubular cells, and proliferation of intertubular connective tissue in hypothyroidism with onset before puberty. Onset after maturity, in one case, led to similar changes that were restricted to the tubules.
The pancreatic islets are usually normal, although hyperplasia was present in one of our autopsied cases.
9.5 SYSTEMIC MANIFESTATIONS OF HYPOTHYROIDISM
The clinical expression of thyroid hormone deficiency varies considerably between individuals, depending on the cause, duration and severity of the hypothyroid state. Characteristically, there is a slowing of physical and mental activity, and of many organ functions.
9.5.1 ENERGY AND NUTRIENT METABOLISM
Energy metabolism. Thyroid hormone deficiency slows metabolism, resulting in a decrease of resting energy expenditure, oxygen consumption, and utilization of substrates. Reduced thermogenesis is related to the characteristic cold intolerance of hypothyroid patients. Measurement of the resting energy expenditure is rarely performed nowadays. In patients with complete athyreosis it falls between 35 and 45 percent below normal. In Addison's disease, the BMR may fall to -25 or -30 percent and, in hypopituitarism to below - 50 percent. The failure to find a metabolic rate as low as - 35 percent, when the clear-cut picture of myxedema is present, is very unusual. The effect of thyroid hormone deficiency on appetite and energy intake is not precisely known but energy expenditure certainly decreases, leading to a slight net gain in energy stores. Body weight increases on average by 10% due to an increase if body fat and retention of water and salt. An increase of adipose tissue mass results in an increase of serum leptin, which mediates a decrease in energy intake while energy disposal increases, eventually leading to a reduction in adipose tissue mass. Interactions between leptin and thyroid hormone have thus attracted much interest , especially because prolonged fasting in rodents decreases leptin and inhibits the hypothalamic-pituitary-thyroid axis resulting in a fall of serum TSH and serum T4. In hypothyroid patients, an increase, no change, or a decrease in plasma leptin concentrations has been reported (1-4,48). Whether thyroid hormone regulates leptin secretion independent of body mass index and body fat, remains controversial. In one study, leptin concentrations expressed as standard deviation scores (Z-scores) from the mean value of female controls matched for body mass index and age, were lower in hypothyroid and higher in thyrotoxic women, whereas Z-scores did not deviate from the expected values after restoration of the euthyroid state 1 . Thyroid hormone apparently modulates serum leptin only to a small extent. Ghrelin, a gastric peptide that plays a role in appetite stimulation and energy balance, is elevated in hypothyroid patients in most studies with a return to normal after L-T4 treatment (4-6). . It appears that leptin is mainly involved in thyroid hormone effects on energy homeostasis, whereas ghrelin may serve a compensatory physiological role (9).Serum adiponectin and resistin concentrations are not changed in hypothyroidism relative to controls (3,7,8,49). Serum obestatin and visfatin are increased in hypothyroidism; visfatin levels had a direct relationship with insulin resistance and body mass index (50,51).
Protein metabolism. The effect of hypothyroidism on protein metabolism is complex, and its effect on the concentration of a given protein difficult to predict. In general, both the synthesis and the degradation of proteins are reduced, but hypothyroid patients are in positive nitrogen balance. Despite both a decrease in the rate of albumin synthesis and degradation, the total exchangeable albumin pool increases in myxedema 10 . The albumin is distributed in a much larger volume, suggesting enhanced permeability of capillary walls. A synthesis of thyroid hormone-responsive proteins is clearly reduced in the hypothyroid state, whereas that of proteins such as TSH or glycosaminoglycans may be increased under the same circumstances 11,12 . Comparative studies of protein translation by hepatic ribosomes from T3-treated hypothyroid rats show that the mRNA's from some proteins are increased and others are decreased. Most of these proteins have not been identified. Treatment of myxedema is accompanied by mobilization of extracellular protein and a marked but temporary negative nitrogen balance, reflecting the mobilization of extracellular protein 13 . In a later phase there is an increase in urinary potassium and phosphorus together with nitrogen in amounts suggesting that cellular protein is also being metabolized 14 .
Carbohydrate metabolism. Glucose is absorbed from the intestine at a slower rate than normal. Fasting plasma glucose and fasting insulin levels are mostly similar to control values (8,15) although sometimes slightly lower glucose and higher insulin values than normal are reported 16,17,18 . Glycosylated hemoglobin is normal (21). The occurrence of hypoglycemia in hypothyroid patients should alert the physician to concomitant diseases (e.g. hypopituitarism). The development of hypothyroidism in patients with insulin-dependent diabetes mellitus may require lowering of the insulin dose to counteract the decreased rate of insulin degradation (22).The oral glucose tolerance test usually shows no abnormalities but a peak value that remains elevated at 2 hours can be observed (15,16,17), probably related to slow gastric motility and delayed absorption. Insulin response to an oral glucose load is variable; sometimes it is higher than in controls (15,18). When sugar is given intravenously, the glucose disappearance rate is prolonged although the peak value is normal in magnitude and in time of occurrence; the insulin response is blunted and slightly delayed 19 . Their exists fair evidence that hypothyroidism is associated with some degree of insulin resistance. The HOMA index (Homeostasis Model of Assessment) reflects the insulin resistance in the fasting state (mainly insulin resistance in the liver), while the Matsuda index reflects insulin sensitivity in the postprandrial state (mainly insulin sensitivity in the peripheral tissues). The HOMA index was found to be normal (5,8,15) or increased (18, 20) in hypothyroid patients vs euthyroid controls, whereas the Matsuda index was decreased and correlated positively with serum FT4 (15,18). The data suggest that insulin resistance might be present in some patients in the fasting state, but more frequently in the postprandrial state. Several other studies point into the same direction. In isolated monocytes derived from hypothyroid patients, insulin-stimulated rates of glucose transport are decreased due to impaired translocation of GLUT4 glucose transporters on the plasma membrane 18 . Hypothyroid patients as compared to euthyroid controls, also have lower postprandrial glucose uptake in muscles and adipose tissue 15 . Euglycemic hyperinsulinemic clamp studies in hypothyroid patients show an increase in insulin sensitivity after restoration of the euthyroid state (21).
Lipid metabolism. Biosynthesis of fatty acids and lipolysis are reduced. Changes in serum lipids are listed in Table 9-4 . The lipid changes bear in general a reciprocal relationship to the level of thyroid activity.The increased serum cholesterol in hypothyroidism may represent an alteration in a substrate steady-state level caused by a transient proportionally greater retardation in degradation than in synthesis 23,24,25 . The increase of serum cholesterol is largely accounted for by an increase of LDL-cholesterol, which is cleared less efficiently from the circulation due to a decreased T3-dependent gene expressing of the hepatic LDL-receptor. there is also evidence that the increase of LDL-cholesterol is also mediated via non-LDL receptor pathways by inducing Cyp7a1 expression and stimulating the conversion and excretion of cholesterol as bile acids 26,27,28,29 ,52,53 .
Table 9-4
Changes in serum lipids in hypothyroidism.
Interestingly, the LDL particles of hypothyroid patients are also susceptible to increased oxidizability 30 . The increase of HDL2- but not of HDL3-cholesterol 31,32,33 is due to a diminished activity of cholesteryl ester transfer protein 34,35 and hepatic lipase (which is involved in the conversion of HDL2 to HDL3). The changes in plasma LDL-and HDL-cholesterol are related to changes in free thyroxine, not to polymorphisms in LDL receptor or cholesteryl ester transfer protein genes 36 . Serum levels of apolipoprotein B and AI are increased but apolipoprotein AII levels are not. The sometimes present modest increase of serum triglycerides has been related to a decreased lipoprotein lipase activity in adipose tissue, suggesting hypertriglyceridemia in hypothyroidism is caused by a decreased clearance by adipose tissue (15). Another study however suggests the combination of observed normal lipolysis, low lipid oxidation rates and high triglyceride concentrations is compatible with increased triglyceride synthesis (37).An oral lipid tolerance test indicates postprandial lipemia (defined as an increase of triglycerides by 80% or more) is more frequent in hypothyroid patients than controls(38). Free fatty acids concentrations in serum are mostly normal, but decreased and increased values have also been reported (37,39). Lipoprotein(a) levels are also found to be normal in most studies (31,33,40) Remnant particles in serum (reflecting chylomicron and VLDL remnants) are less effectively cleared in hypothyroidism 41,42 . Taken together, the changes in plasma lipids in hypothyroidism result in an atherogenic lipid profile, although this has been disputed (43). Several studies do indicate, however, increased oxidative stress in hypothyroid patients, as evident from higher levels of serum malondialdehyde and nitric oxide and lower levels of the anti-oxidant paraoxonase in serum relative to controls (44,45).The observed increased oxidative stress is independent of body mass index (46). In subcutaneous fat biopsies of hypothyroid patients the mRNA expression of uncoupling protein-2 (UCP2) is decreased; UCP2 mRNA was related to lipid oxidation rate, basal free fatty acids, and serum T3 (47). UCP2 is a determinant of fat oxidation pathways, and may be involved in changes in metabolic pathways in thyroid disease.
9.5.2 FACIES AND INTEGUMENT
In the Report on Myxedema there is a detailed analysis of the symptoms of 109 patients described as "cretinoid," "expressionless," "heavy," "apathetic," "masklike," "vacant," "stolid," "good-tempered," "blunted," and "large-featured." The face is expression less when at rest, but it is not masklike, as in Parkinson's disease. When spoken to, the person with myxedema usually responds with a smile, which spreads after a latent period very slowly over the face. The patient is good-tempered but not entirely apathetic. The face is not vacant, as that of psychopathic patient may be. The features (except for the tongue) are not large, as in acromegaly. The face is expressionless at rest, puffy, pale, and often with a yellowish or old ivory tint. It is seldom as puffy as the classic facies of chronic renal failure. The skin of the face is parchment-like. In spite of the swelling it may be traced with fine wrinkles, particularly in pituitary myxedema. The swelling sometimes gives it a round or moonlike appearance (Fig. 9-3) .

Figure 9-3
(A) The classic torpid facies of severe myxedema in a man. The face appears puffy, and the eyelids are edematous. The skin is thickened and dry. (B) The facies in pituitary myxedema is often characterized by skin of normal thickness, covered by fine wrinkles. Puffiness is usually less than in primary myxedema. The eyelids are often edematous. The palpebral fissure may be narrwowed because of blepharoptosis, due to diminished tone of the sympathetic nervous fibers to Müller's levator palpebral superious muscle and is the opposite of the lid retraction seen in thyrotoxicosis. The modest measurable exophthalmos seen in some patients with myxedema is presumably related to accumulation of the same mucous edema in the orbit as is seen elsewhere. It is not progressive and carries no threat to vision, as in the ophthalmopathy of Graves' disease. The tongue is usually large, occasionally to the point of clumsiness. Sometimes a patient will complain of this problem. Sometimes it is smooth, as in pernicious anemia (of course, pernicious anemia may coexist). Patients do not usually complain of soreness of the tongue, as they may in pernicious anemia. When anemia is marked, the tongue may be pale, but more often it is red, in contrast to the pallid face.
The voice is husky, low-pitched, and coarse. The speech is deliberate and slow. Often there is difficulty in articulation. Certain words are stumbled over and slurred, much as they are during alcoholic intoxication. The enlargement of the tongue, and possibly some thickness of the lips, may be responsible. The hair, both of the head and elsewhere, is dry, brittle, and sparse, and lacks shine. It varies in texture from coarse to normal. Its growth is retarded and it falls out readily. The eyebrows often are practically gone. Their disappearance begins at the lateral margin, giving rise to Queen Anne's sign. It should be noted, however, that this sign is not uncommon in elderly euthyroid women. In men the beard becomes sparse, and its rate of growth becomes greatly retarded. Haircuts are necessary only at long intervals. A shave a week is sufficient. The scalp is dry and scaly.
The skin is cool as a result of decreased metabolism as well as cutaneous vasoconstriction. It is dry due to reduced secretion by sweat and sebaceous glands.Scaling is common but rarely assumes the appearance characteristic of ichthyosis. The tissues beneath it seem thick, but usually do not pit on pressure. In the lower extremities, pitting edema is not uncommon. Subcutaneous fat may be increased, with the formation of definite fat pads, especially above the clavicles, but is conspicuously absent in the more advanced form of the disease (myxedematous cachexia).Retardation in the rate of healing of surgical wounds and of ulcerations, such as leg ulcers, has been described in myxedema. The nails are thickened and brittle. These changes are probably dependent, as are those of skin and hair, on retardation in growth. Nails require paring only at greatly lengthened intervals.
The hands and feet have a broad appearance, due to thickening of subcutaneous tissue. However, there is no bony overgrowth, so that they bear no resemblance to the extremities in acromegaly. Unusual coldness of the arms and legs is sometimes a subject of complaint. The palms are cool and dry. The characteristic skin changes are due to an increased amount of normal glycosaminoglycans and protein. The glycosaminoglycans are demonstrated by metachromasia after staining with toluidine blue. An increased concentration of glycosaminoglycans, composed principally of hyaluronic acid and chondroitin sulfuric acid, occurs in histologically similar skin lesions found in hyperthyroidism (pretibial myxedema). This excess accumulation of normal intercellular material represents not only an alteration in steady-state equilibrium but an actual increase in the synthesis and accumulation of glycosaminoglycan 1 .The glycosaminoglycans are long-chain polymers of D-glucuronic acid and N-acetyl-D-glucosamine, forming hyaluronic acid, or of L-iduronic acid and N-acetyl-D-galactosamine sulfate, forming chondroitin sulfate B. They exist free and in ionic or covalent linkage to protein. These mucoproteins comprise part of the normal nonfibrillar intercellular matrix, the ground substance holding cells together. As they are characteristically hygroscopic, they presumably hold in bound form the nonpitting water comprising the mucous edema. The total amount of exchangeable sodium is increased in myxedema despite a slight reduction in its plasma concentration 2 .
The sodium is extravascular and probably in the interstitial spaces. The diuresis seen after giving thyroid hormone to a hypothyroid subject occurs coincidentally with a decrease in tissue metachromasia and a temporary negative nitrogen balance 3 , and with this condition the extravascular sodium is mobilized and excreted. Studies with human skin fibroblasts have suggested that thyroid hormone inhibits the synthesis of hyaluronate. The mechanism for this effect has not been identified, but the thyroid hormone levels required to produce it in vitro are in the physiologic range 1 , 4 . Although similar deposits of mucopolysaccharides are found in the orbits of patients with the ophthalmopathy of Graves' disease and in the areas of localized myxedema, this striking observation has unfortunately not provided any basic understanding of the phenomenon, either in this condition or in primary myxedema (5).
In summary, in hypothyroidism the skin is dry, pale, thick, and rough with scales, and it feels cold. Hashimoto’s thyroiditis is associated with vitiligo (RR 25.6 with 95% CI 13.3-44.2 in women, and 15.8 with 95% CI 0.40-85.2 in men) (6). Thyroid autoimmunity may also be associated with chronic urticaria (7,8).
9.5.3 NERVOUS SYSTEM
Studies using 31P nuclear magnetic resonance spectroscopy of the frontal lobe of adult hypothyroid patients report reversible alterations in phosphate metabolism, suggesting impairment of mitochondrial metabolism 1 . Thyroid hormone receptors are present in human brain. These and other findings indicate the adult human brain as a thyroid hormone responsive organ, and provide a biologic basis for the very prevalent neurologic and neurobehavioral symptoms in adult hypothyroid patients 2 (Table 9-5).
Neurologic symptoms and signs. We are aware of no characteristic motor phenomena other than those due to weakness and to syndromes that seem to represent cerebellar dysfunction. A tendency to poor coordination was noted originally by the Myxoedema Commission. Jellinek and Kelly 3 described a series of myxedematous patients with ataxia, intention tremor, nystagmus, and dysdiadochokinesia. Ataxia has been noted in 8 percent of a large series of hypothyroid patients 4 . The delayed relaxation phase of the deep tendon reflexes is a well-known manifestation. Patients may have intention tremor, nystagmus, and an inability to make rapid alternating movements. In fact, this inability has long been used as a test for myxedema. The cause of this syndrome is not apparent, although deposition of mucinous material in the cerebellar tissue may be of pathogenetic importance. Whatever the cause is, it is important that these symptoms show a prompt and definite decrease after replacement therapy with thyroid hormone 5 . Sensory phenomena are common. Numbness, tingling, and painful paresthesias are frequent 6 and are especially common in hypothyroidism after surgery or 131I therapy. Paresthesias were present in 79 percent of one series of 109 patients. A metachromatic infiltrate has been found in the lateral femoral cutaneous nerve and sural nerve, together with axon cylinder degeneration 7 . Nerve conduction time is usually normal. Murray and Simpson 8 found that in some hypothyroid patients signs of median nerve pressure were present, apparently because of encroachment on the nerve by myxedematous infiltrates in the carpal tunnel 9,10 . A recent study reports carpal tunnel syndrome in 29% and signs of sensorimotor axonal neuropathy in 42% 22 . Deafness is a very characteristic and troublesome symptom of hypothyroidism. Both nerve and conduction deafness and combinations of the two have been reported, and vestibular abnormalities have also been demonstrated 37 . Serous otitis media is not uncommon. Two-thirds of patients complain of dizziness, vertigo, or tinnitus occasionally: these problems again suggest damage to the eighth nerve or labyrinth, or possibly to the cerebellum. Whatever type of deafness is present, there is marked improvement after thyroid therapy. Acute thyroxine depletion caused by total thyroidectomy has no deleterious effects on hearing up to 6 weeks 11 . Acquired hearing loss in association with adult-onset hypothyroidism should be distinguished from the sensorineural deafness of Pendred's syndrome. In the latter, treatment of hypothyroidism does not correct the hearing defect. Night blindness is not uncommon. It is caused by a deficiency in the pigment retinene, which is required for the adaptation to dark. Uncorrected deficiency of thyroid hormone during neonatal life causes not only more profound neurologic abnormalities but also irreversible damage (see Ch. 15 ). Hashimoto’s encephalopathy is a condition in which otherwise unexplained central nervous system dysfunction is observed in patients with Hashimoto’s disease and positive TPO-antibodies. The condition responds to glucocorticoids.A causal relation to thyroid autoimmunity is believed probable, but remains uncertain 26,27 . EEG abnormalities can be present, again depending on the severity and duration of the hypothyroidism. There may be absence of alpha waves and presence of low-amplitude theta and delta waves. Visual and auditory evoked potentials may be delayed as a consequence of abnormal cerebral cortical metabolism. Sleep apnea is not uncommon 15 . It has been difficult to assign a causal role for the myopathy versus the coexistent obesity in some of the reported cases. However, the muscular dysfunction may extend to the diaphragm and intercostal muscles, thus impairing the ventilatory mechanism.
Mental Symptoms. The mental picture in patients with overt hypothyroidism usually is one of extreme complacency. Memory is undoubtedly impaired, and attention and the desire to think are reduced . The emotional level seems definitely low, and irritability is decreased. Except in the terminal stage, reasoning power is preserved. Questions are answered intelligently, but slowly and without enthusiasm, and often with evidence of amusement. In a minority of patients, nervousness and apprehension are present. Cognitive tests of patients with moderate to severe hypothyroidism indicate difficulties in performing calculations, recent memory loss, reduced attention span, and slow reaction time 14,28 . Failing memory correlates inversely with serum T3 and T4 (23). Hypothyroidism may give rise rarely to reversible dementia, associated with reversible cerebral hypoperfusion (24). Recent studies indicate that hypothyroid-related memory deficits are not attributable to attentional deficit but rather to specific retrieval deficits (29). Hypothyroid patients showed prolongation of latencies only in the early ERP (event-related potential) components compared to controls, with speeding of sensory and cognitive processing after treatment (30).The cognitive impairment in hypothyroidism seems to be predominantly mnemonic in nature, possibly reflecting a specific defect in hippocampal memory (31). Imaging studies (functional MRI) have linked poorer memory states to specific brain areas and to reduced hippocampal volume 38,39 .
Psychiatric syndromes. The typical somnolence of severe hypothyroidism may suggest the psychiatric diagnosis of depression or dementia 16 . Patients are generally akinetic, though isolated case reports appear of patients who became hypomanic and agitated or garrulous (myxedema wit) as manifestations of this condition. Psychosis with hallucinations may occur (myxedema madness) 32 . Depression is so often associated with hypothyroidism that thyroid function tests should be performed in the evaluation of any patient presenting with this symptom. Central 5-hydroxytryptamine activity is reduced in hypothyroid patients 12 , and T3 supplementation might increase the efficacy of antidepressant drugs 13 although large randomized clinical trials in patients with major depression have produced conflicting results (32,33). At times, the depression in hypothyroidism is more severe than any of the other clinical manifestations of the disease. Because hypothyroidism is so readily treated, it is an especially important cause to eliminate in any patient with major depression. If the condition is due to hypothyroidism, it will resolve with time and appropriate treatment 17,18 . Patients hospitalized with hypothyroidism have a greater risk of readmission with depression or bipolar disorder than control patients 35 .
Cerebral blood flow, oxygen consumption, and glucose consumption have been reported to be diminished in proportion to the drop in metabolism in the rest of the body 19 , but older studies found unaltered glucose and oxygen use by the brain in either hypo- or hyperthyroid animals or humans 20 . In one study, cerebral cortical perfusion was little changed with treatment, but there was a decided fall in cerebrovascular resistance 21 . More recent studies indicate a generalized decrease in regional cerebral blood flow of 24% and in cerebral glucose metabolism of 12%, indicating that brain activity is globally reduced in severe hypothyroidism without the regional modifications usually observed in primary depression 25 . In 2009 neuropsychiatric symptoms of hypothyroid patients were studied in relation to changes in relative regional cerebral glucose metabolism after L-T4 treatment (36). Reduction of behavioral complaints during L-T4 therapy was associated with restoration of metabolic activity in brain areas that are integral to the regulation of affect and recognition.The findings suggest that thyroid hormone modulates regional glucose metabolism and psychiatric symptoms in the mature brain.. Experimental animal studies have shown that adult hypothyroidism in rats potentiates fear memory and also increases vulnerability to develop emotional memories. The findings further suggested that enhanced corticosterone signalling in the amygdala was involved in the pathophysiological mechanisms of fear memory potentiation 40 . Recent developments in brain imaging techniques thus provide novel insights in the relationship between hypothyroidism and mood disorders 41,42 .
9.5.4 CARDIOVASCULAR SYSTEM
Table 9-6
Cardiovascular manifestations of hypothyroidism.
Understanding of the cellular mechanisms of thyroid hormone action on the cardiovascular system has made it possible to explain to a large extent the decrease of cardiac output and cardiac contractility, the diastolic hypertension, the increased systemic vascular resistance, and the rhythm disturbances that result from hypothyroidism (Table 9-6) (37,38). Hypothyroidism decreases tissue thermogenesis by 5-8%, and increases resistance in peripheral arterioles through the direct effect of T3 on vascular smooth muscle cells. Diastolic blood pressure rises, and the afterload of the heart increases. Cardiac chronotropy and inotropy is reduced, resulting in a decrease of cardiac output to < 4.5 L/min. Thyroid hormone is an important regulator of cardiac gene expression, and many of the cardiac manifestations of hypothyroidism are associated with alterations in T3-mediated gene expression. T3 regulates positively sarcoplasmatic reticulum Ca2+-ATPase and negatively its inhibitor phospholamban, which together function in intracellular calcium cycling and thereby regulate diastolic function. The reduced expression of sarcoplasmatic reticulum Ca2+-ATPase and the increased expression of phospholamban in the hypothyroid heart explains the slowing of the isovolumic relaxation phase of diastolic function, typical for hypothyroidism.T3 regulates positively α-myosin heavy chain (the fast myosin with higher ATPase activity), and negatively β-myosin heavy chain (the slow myosin). In the hypothyroid heart, the expression of α-myosin heavy chain is decreased and of β-myosin heavy chain increased. T3 positively regulates the ion channels sodium potassium ATPase (Na+,K+-ATPase), the voltage-gated potassium channels (Kv1.5, Kv4.2, Kv4.3), whereas T3 negatively regulates the sodium-calcium exchanger (Na+/Ca2+exchanger).Together these channels coordinate the electrochemical responses of the myocardium. T3 positively regulates the β1-adrenergic receptors. The pacemaker-related genes, hyperpolarization-activated cyclic-nucleotide-gated channels 2 and 4, are transcriptionally regulated by thyroid hormone.
Systemic changes. Pulse rate and stroke volume are diminished in hypothyroidism, and cardiac output is accordingly decreased, often to one-half the normal value 1 . Myocardial contractility is reduced, but there is also a steep decline in the circulatory load, so that the circulation rarely fails until very late in the disease 2 . The speed of shortening is slowed, but the total force is not much modified. 3 . Myocardial adenyl cyclase levels are reduced 4 . The decrease in pulse rate occurs more or less in parallel with that of the metabolism. Stroke volume is reduced more than pulse rate at any given level, and is therefore the major determinant of the low cardiac output. Since the reduction in cardiac output is usually proportional to the decreased oxygen consumption by the tissues, the arteriovenous (AV) oxygen difference is normal or may be slightly increased. Slow peripheral circulation, and therefore more complete extraction of oxygen, as well as anemia, may be responsible for the increased AV oxygen difference. Myocardial oxygen consumption is decreased, usually more than blood supply to the myocardium, so that angina is infrequent. In some patients a reduction in cardiac output greater than the decline in oxygen consumption indicates specific cardiac damage from the myxedema 5 .Venous pressure is normal, but peripheral resistance is increased. Restoration of the euthyroid state normalizes peripheral vascular resistance. Changes in peripheral vascular resistance are not related to plasma adrenomedullin, but altered atrial natriuretic peptide secretion and adrenergic tone may contribute 29,39 . Central arterial stiffness is increased in hypothyroidism 30 , and arterial blood pressure is often mildly increased. It varies widely, but diastolic hypertension is usually restored to normal after treatment 6,7,31 .
Cardiomegaly. The heart in hypothyroidism has been a focus of much controversy. The term Myxodemherz was introduced by Zondek in 1918 8 . It embraced dilatation of the left and right sides of the heart, slow, indolent heart action with normal blood pressure, and lowering of the P and T waves of the electrocardiogram. Zondek found that after treatment with thyroid hormone there was a return of the dilated heart to somewhere near normal size, a more rapid pulse without change in blood pressure, and gradual return of the P and T waves to normal. These findings have been confirmed and extended. Indeed, occasional severely hypothyroid patients without underlying heart disease have congestive heart failure or low cardiac output reversed by thyroid hormone administration 7,9,10 . Therefore, congestive heart failure or impaired cardiac output relative to metabolic needs can be caused by hypothyroidism.Patients with untreated hypothyroidism are indeed at increased risk of heart failure 40 . Microscopic examination discloses myxedematous changes of the myocardial fibers.The cause of the cardiac enlargement has been disputed. Clearly, it is not due to hypertrophy alone, since it would not disappear so rapidly with treatment. One factor may be a decrease in contractility of the heart muscle. This decrease would require a lengthening of muscle fibers in order to perform the required work. Disappearance of interstitial fluid alone could account for only part of the observed shrinkage. Since the treatment of myxedema restores the hypothyroid heart to normal, there is apparently little permanent structural damage 9,10 . Cardiac glycosides will not improve the function of the heart in uncomplicated myxedema. Although the drug is efficacious if heart failure has been produced by coincident organic disease, myxedematous patients with coincident heart disease and congestive heart failure may tolerated digoxin poorly, just as they do morphine. This poor tolerance probably represents delayed metabolism, rather than myocardial sensitivity to the drug. The plasma concentration of digoxin is higher than in the normal subject at the same dose level, and smaller doses are required. When the heart in myxedema does not return to a normal size under thyroid hormone administration, hypertrophy due to some other disease is present as a complication. The return in size to normal under treatment is slow and progressive, requiring between 3 weeks and 10 months for completion.
Pericardial effusion. Gordon 11 long ago called attention to the occurrence of pericardial effusion in myxedema and explained the increase in the transverse diameter of the heart shadow on this basis. Effusion must frequently play a role in the increase in the size of the heart shadow, but it has amazingly little effect on cardiodynamics. The presence of fluid may be reflected in the right ventricular pressure contour, but tamponade, although reported, is rare 12,13 ,41 . Effusions of the pericardium, pleura, and peritoneum are common findings in hypothyroidism 14 . The protein of the effusion may be high or in the range of transudates. In 11 patients with tamponade studied, pericardial fluid protein ranged from 2.2 to 7.6 g/dl 12 . Occasionally, the fluid is high in cholesterol, with a "gold paint" appearance 13 . The hypothyroid heart responds normally to exercise 5,7 . Graettinger et al. 1 found that after exercise the low resting cardiac output increased normally with an increase of stroke volume and usually, of pulse rate. Their patients had slightly elevated resting pulmonary artery and right ventricular pressures and a diastolic dip in right ventricular pressure, all compatible with pericardial effusion. They doubt that myxedema alone can ever produce congestive heart failure, and believe that the recorded abnormalities represent not myocardial disease but pericardial effusion.
Electrocardiogram. The electrocardiogram reveals characteristic changes 5 , 7 , 10 , 15-19 . The rate is slow and the voltage is low. The T waves are flattened or inverted. Axis deviation, an increased P-R interval, and widened QRS complexes and prolonged QT interval are seen, but these signs are not diagnostic of myxedema. The pattern reverts toward normal with treatment, but the final pattern depends on the presence or absence of intrinsic myocardial disease. The rare occurrence of complete heart block complicated by Adams-Stokes attacks, with reversion to sinus rhythm after treatment with thyroid hormone, has been reported as has ventricular tachycardia 18,19 .Changes resembling those of ischemic heart disease may be found during exercise: they may indicate an intrinsic anoxia rather than organic narrowing of the coronary vessels 5 , 7 , 10 , 17 .The ECG changes have usually been attributed to the histologic changes in the myocardium. However, removal of pericardial fluid may immediately reverse the pattern toward normal suggesting that the effusion may in part be responsible for the abnormalities.The systolic time intervals are prolonged in hypothyroidism 5 , 7 , 10 , 20 . They can be measured by several techniques and have been expressed as the ratio of the pre-jection period and the left ventricular ejection time or the interval between the onset of the QRS complex of the ECG and the onset of the Korotkoff sound 21,22 . The most obvious effect of thyroid hormone deficiency on the heart is a lengthening of both systolic and early diastolic time characteristics. As evaluated by equilibrium radionuclide angiography, the time to peak emptying rate and the time to peak filling rate are longer in hypothyroid patients than in controls 23 ; the time intervals are negatively related to serum FT4 in the hypothyroid patients. The subtle decrease in early active relaxation and prolongation of contraction without major changes in global systolic function of hypothyroid patients is reversible upon thyroid hormone replacement therapy 24 .
Atherosclerosis. It is frequently suggested that accelerated atherosclerosis occurs in hypothyroidism 32 . Hypothyroidism accelerates atheromatous changes when these are induced experimentally in animals, but data in humans are not complete enough to justify this assertion. Most autopsied myxedematous subjects have severe atherosclerosis, but they are also usually 60 years or more of age. Arterial disease did not appear to be accelerated in patients rendered hypothyroid for therapy of angina pectoris or congestive heart failure 22 , but they have been observed over a relatively short period. Increased coronary arteriosclerosis is found in myxedematous patients with hypertension, but not if they are normotensive 25 (see further § 9.8.4). Nevertheless, the atherogenic profile of serum lipids and increased levels of homocysteine in hypothyroidism might well contribute to a higher prevalence of atherosclerosis in hypothyroid patients. Indeed , a 20-year follow-up study in the UK did observe a higher incidence of ischemic heart disease in subjects with subclinical hypothyroidism 35 . Another population-based study from The Netherlands found subclinical hypothyroidism to be an independent risk factor for aortic atherosclerosis and myocardial infarction; the attributable risk was comparable to that of other known risk factors for coronary artery disease 36 . A Danish nationwide register study found an excess risk of being diagnosed with cardiovascular disease, both before the diagnosis and following the diagnosis of hypothyroidism 42 .The topic is more fully discussed in § 9.10.4.
Angina pectoris. Occasionally angina pectoris is encountered in myxedema under two sets of circumstances. The less common is that in which angina or angina-like pain is present before treatment 26,27,28 . This generally indicates the presence of significant coronary artery disease since there is inadequate myocardial oxygenation despite reduced cardiac output and O2 utilization. Although improvement sometimes occurs with therapy 27 , this should not be undertaken until angiographic evaluation of the coronary arteries has been performed. Angina may also appear for the first time after therapy has been initiated, indicating that coronary flow is inadequate for resumption of normal cardiac function 26,27,28 . Again, this may indicate the presence of a structural lesion in coronary arteries.
9.5.5 RESPIRATORY SYSTEM
Dyspnea is a frequent complaint of myxedematous patients, but it is also a common symptom among well persons. Congestive heart failure of separate origin, pleural effusion, anemia, obesity, or pulmonary disease may be responsible. Some information on pulmonary function in hypothyroidism is available 1-7 . Wilson and Bedell 1 found a normal vital capacity and arterial PCO2 and PO2 in 16 patients. They also found a decreased maximal breathing capacity, decreased diffusion capacity, and decreased ventilatory response to carbon dioxide. Decreased ventilatory drive is present in about one-third of hypothyroid patients, and the response to hypoxia returns rapidly within a week after beginning therapy 6 .The severity of hypothyroidism parallels the incidence of impaired ventilatory drive. Weakness of the respiratory muscles has also been implicated as a cause of alveolar hypoventilation. Patients with myxedema may develop carbon dioxide retention, and carbon dioxide narcosis may be a cause of myxedema coma 3,4 .
Radiologic pulmonary abnormalities suggestive of fibrotic disease are associated with severe hypothyroidism, and may resolve with levothyroxine therapy (8).Myxedematous patients are more subject to respiratory infections.
Obstructive sleep apnea has been documented in hypothyroidism in about 7% and is reversible with therapy 5 , 7 . The prevalence of hypothyroidism in patients seen for snoring or obstructive sleep apnea syndrome is, however, no greater than that seen in the general population 9 . The same authors report little or no improvement in apnea symptoms upon thyroid hormone replacement therapy in the hypothyroid patients. Indeed, when patients are obese, cure rate of obstructive sleep apnea is not impressive. But replacement therapy is reported to be successful in most patients with obstructive sleep apnea, when they were grossly hypothyroid, generally nonobese and had e.g.reduction of macroglossia and goiter size (10). Obstructive sleep apnea in hypothyroidism seems to be caused by pharynx narrowing due to soft tissue infiltration by mucopolysaccharides and protein (10). Altered regulatory control of pharyngeal dilator mucles due to neuropathy may also be involved.
9.5.6 MUSCULOSKELETAL SYSTEM
Muscles. Muscle symptoms like myalgia, muscle weakness, stiffness, cramps and easy fatiguability are very prevalent in hypothyroid patients 23,24 . Weakness in one or more muscles groups is present in 38% as evident from manual muscle strength testing 22 .The symptoms are aggravated by exposure to cold. They are also prominent during the rapid onset of hypothyroidism after surgery or 131I therapy. Impairment of mitochondrial oxidative metabolism provides a biochemical substrate for these complaints, as evident from a rise in the ratio of inorganic phosphate to ATP in resting muscle and an important decrease in phosphocreatine in working hypothyroid muscle with a greater fall in intracellular pH than in controls 1,2 . Transition from fast type II to slow type I muscle fibers is involved in the change of muscle bioenergetics 3 , which is probably multifactorial. One patient with disabling muscle cramps was found to have reduced a-glucosidase activity in a muscle biopsy; after therapy with T4, the symptoms disappeared and the muscle enzyme activity returned to normal 4 . The electromyogram in myxedema may be normal or may demonstrate abnormalities distinct from those seen in myotonia or other muscle disease 5 . Polyphasic action potentials, hyperirritability, repetitive discharge, and low-voltage, short-duration motor unit potentials have been described. In the hypothyroid rat the rate of isometric relaxation is slow, and tension is less than in euthyroid or hypothyroid rat muscle at the same frequency of stimulation. Generalized muscular hypertrophy, accompanied by easy fatigue and slowness of movements, occurs in some cretins and myxedematous children or adults. It has been referred to as the Kocher-Debré-Sémélaigne syndrome in children 6 and as Hoffmann's syndrome in adults 7 . These patients do not have the classic electromyographic findings of myotonia. The myopathy of hypothyroidism is in some patients associated with weakness even though the muscles are hypertrophied.Chronic hypothyroid myopathy with increased muscular volume rarely cause entrapment syndromes 8 . Reflex contraction and relaxation time is prolonged mainly because of the intrinsic alterations in muscle contractility. Nerve conduction time may also be prolonged. Delayed reflex relaxation is characteristic and has been developed into a diagnostic test of thyroid function 9 . As with many other peripheral tissue function tests, there is considerable overlap between normal and mildly hypothyroid ranges. The rate-limiting step in muscle relaxation is the reuptake of calcium by the sarcoplasmic reticulum. In skeletal muscle, this process is dependent on the content of calcium ATPase. Recent studies have indicated that calcium ATPase activity of the fast twitch variety (SERCA-1) is markedly reduced in hypothyroidism 10 , and there is an accompanying impairment of calcium reuptake as a consequence. This occurs at a transcriptional level, since thyroid hormone response elements have been identified in the 5' flanking region of the SERCA-1 calcium ATPase gene 11 . The reduction in calcium ATPase would appear to explain one of the most obvious clinical manifestations of hypothyroidism, namely, delayed relaxation of the deep tendon reflexes. Skeletal muscle type 2 deiodinase activity is low and not influenced by hypothyroidism (25).
Table 9-7
Manifestations of hypothyroidism in the musculoskeletal system.
Joints. At the clinical level, patients with hypothyroidism may present with many manifestations, suggesting rheumatic disease such as arthralgias, joint stiffness, effusions, and carpal tunnel syndrome 12,13 . On the other hand, the symptoms may also suggest polymyalgia rheumatica, or primary myositis. The similarity of the symptoms of hypothyroidism to those of rheumatoid arthritis or osteoarthritis, especially when these are combined with the paresthesias of more severe hypothyroidism, should automatically lead to a consideration of hypothyroidism in any patient presenting with these symptoms. For example, in 5 to 10 percent of patients with carpal tunnel syndrome, primary hypothyroidism may be the cause, due to the accumulation of the hygroscopic glycosaminoglycan in the interstitial space with compression of the median nerve.
Bones. While calcium, phosphate, and bone density are generally normal in hypothyroidism, there is evidence of reduced bone turnover and resistance to the action of parathyroid hormone (PTH) 14-21 . Thus, serum (PTH) levels are elevated 16 . This is presumably the cause of the elevation in 1,25(OH)2-vitamin D3 19 . 25-OH-vitamin D3 levels are normal. The increase in PTH and vitamin D in turn increases calcium absorption. The reduction in glomerular filtration rate (GFR) and reduced bone turnover reduce urinary calcium and hydroxyproline levels and cause subnormal alkaline phosphatase, osteocalcin, and IGF-1 levels 15 . The alkaline phosphatase reduction is particularly important in children, in whom this enzyme is normally elevated due to bone growth. In children delayed linear growth or short stature are well-recognized signs suggesting the possibility of hypothyroidism. In addition, it is well recognized that epiphyseal dysgenesis and the delayed appearance of calcification centers are characteristic of hypothyroidism in infants and children. This subject is discussed in greater detail in Chapter 15 . Hypothyroidism is associated with enhanced susceptibility to fractures 26,27 .
9.5.7 GASTROINTESTINAL SYSTEM
The symptoms from the digestive system are essentially the expression of the slow rate at which the living machinery is turning over. Anorexia, which is common, can reasonably be interpreted as the reflection of a lowered food requirement, and constipation, which is frequently present, is the result of a lowered food intake and decreased peristaltic activity. Although two-third of patients have reported weight gain, it is of modest degree and due largely to the accumulation of fluid rather than fat. Contrary to popular belief, obesity is decidedly not a feature of hypothyroidism.
Complete achlorhydria occurs in more than half of myxedematous patients 1 . As many as 25 percent of patients with myxedema, like those with Hashimoto's thyroiditis, have circulating antibodies directed against the gastric parietal cells. This finding explains, at least in part, the frequency of achlorhydria and impaired absorption of vitamin B12. It is reported that up to 14 percent of patients with idiopathic myxedema have coincident pernicious anemia 2 .
Dysphagia or heartburn may be due to disordered esophageal motility 3 . Dyspepsia, nausea or vomiting may be due to delayed gastric emptying. Abdominal discomfort, flatulence and bloating occur in patients with small intestinal bacterial overgrowth: its prevalence (as demonstrated by a positive hydrogen glucose breath test) in hypothroid patients is rather high (54% vs 5% in a control group) (4). Bacterial overgrowth decontamination (by treatment with 1200 mg rifaximin each day for a week) is associated with improved gastrointestinal manifestations. Intestinal transit time is prolonged (5). Constipation may result from diminished motility, with the rare occurrence of fecal impaction. The syndrome of ileus may be seen occasionally 6 , and a megacolon may be evident on radiography 7,8 ; rarely pseudoobstruction develops. Intestinal absorption is slowed. Galactose and glucose tolerance curves show a delayed rise to a lower peak than normal and a delayed return to baseline. Xylose absorption is impaired 9 . Myxedematous ascites is rare 10 . Gallbladder motility is decreased, and the gallbladder may appear distended on x-ray examination 11,12 .
Symptoms or signs of disturbed liver or exocrine pancreatic function are usually not encountered, but chemical examination may suggest disease. Serum glutamine-oxaloacetic transaminase (GOT), lactate dehydrogenase (LDH), and CPK levels are elevated in patients with hypothyroidism 13,15 . The enzymes return to normal over 2 to 4 weeks during treatment. Urinary amylase levels may be increased. CEA levels are also increased and drop with therapy 14 . Serum liver enzyme activities ( alanine aminotransferase ALT and gamma-glutamyltransferase γGT) increase steadily across increasing TSH categories (also with TSH values within the reference range), ranging from mean values of 29 to 41 U/l for ALT and of 36 to 62 U/l for γGT 16 . At TSH levels >10 mU/l, ALT >40 U/l is observed in 24% and γGT >40 U/l in 30%.
Table 9-8
Gastrointestinal manifestations of hypothyroidism.
9.5.8 RENAL FUNCTION, WATER AND ELECTROLYTES
Hypothyroid patients tend to drink small amounts of water and to have diminished urinary output. Clinical evidence of renal failure is not often found, but laboratory examination may disclose certain departures from normal renal function; serum creatinine is raised by 10-20% , normalizing after L-T4 treatment 1,19,23 . Serum cystatin C is strongly influenced by thyroid function, and it may give erroneous results for assessing renal function in hypothyroid patients 19,23,24 .Because of decreased cardiac output and blood volume, renal blood flow is decreased, but it remains the same percentage of cardiac output. The glomerular filtration rate and effective renal plasma flow are decreased, but the filtration fraction is normal or variably altered 2,3,4 ,25 .
Hyponatremia sometimes occurs 26,27 .The response to water loading is variable. Moses et al. 5 reported that deficient diuresis after water loading is a sign of pituitary myxedema, but others, notably Crispell and co-workers 6 have found that severe primary myxedema may be associated with a delayed excretion that is not corrected by cortisone but rather by replacement with thyroid hormone. Perhaps the difference in opinion arised from interpretation of the normal response to water loading. This possibility is suggested by the data of Bleifer et al. 7 , who found a decrease in maximal diuresis in some patients with primary myxedema to below the normal lower limit of urine flow (3 ml/min), but not down to the 1 to 3 ml/min seen in panhypopituitarisn. The role of the antidiuretic hormone vasopressin (AVP) and of solute excretion in producing the decreased response to water loading was unknown. The defect was usually attributed to a decreased glomerular filtration rate, but in some patients inappropriately high levels of serum vasopressin have been demonstrated 8-12 . Since urinary hydroxycorticoid excretion is decreased, the adrenals might be incriminated as responsible for delayed water excretion. Other evidence, however, suggests (see below) that the tissue supply of adrenal cortical hormones is usually normal in myxedema. The diminished free water clearance in hypothyroidism occurs irrespective of the presence of hyponatremia. The inappropriate antiduresis in hypothyroidism was thus not fully understood, and a pure renal mechanism was hypothesized independent of vasopressin 18 . Better understanding has been obtained since the discovery of water channels, the aquaporins. Activation of the vasopressin V2 receptor on the basolateral membrane of the principal cells of the collecting ducts in the kidney by vasopressin (AVP) leads to upregulation of aquaporin-2 (AQP2). It involves shuttling the AQP2 containing vesicles from the cytoplasm to the apical membrane, leading to fast water transport across the bilipid apical membrane (20). Patients with advanced primary hypothyroidism may be hyponatriemic and fail to suppress plasma AVP with an acute water load. Advanced hypothyroidism is associated with a decrease in blood pressure, which is expected to activate baroreceptor-mediated non-osmotic AVP release, and indeed this is the case. In a rat model of hypothyroidism the increase in plasma AVP was associated with upregulation of AQP2 (21). A V2 receptor antagonist reverses the increased AQP2 and the impaired response to an acute water load. In contrast to diuretics, which enhance water and electrolyte excretion, the V2 receptor antagonists (vaptans) increase electrolyte-free water excretion.So far, the use of vaptans in severely hypothyroid patients with hyponatremia has not been reported 28 .
Occasionally, minimal proteinuria is seen. This condition could be due to congestive heart failure or to the increased capillary transudation of protein typical of hypothyroidism.
The total body sodium content is increased. The excessive sodium is presumably bound to extracellular mucopolysaccharides. In spite of reduced renal blood flow and blood volume, the sodium retention is probably not a reflection of altered renal function. In fact, salt loads are usually excreted readily and serum sodium concentrations tend to be low 13 , in contrast to other clinical situations associated with sodium retention, such as congestive heart failure 8 . The dilutional syndrome may be a result of inappropriate secretion of AVP 9-12 , but not in all patients. Thus, the dilutional syndrome in severe myxedema may be due to a resetting of the osmolar receptor, which causes water to be retained at a lower level of plasma osmotic pressure. The various changes in renal function may not return to normal at the same rate after treatment. Weight loss after therapy of hypothyroidism is mainly caused by excretion of excess body water associated with myxoedema 29 .
The serum uric acid level is elevated in hypothyroid men and postmenopausal women, apparently as a consequence of a decrease in renal blood flow characteristic of the disease 14 . No consistent changes in plasma potassium levels have been reported. Total magnesium levels may be elevated and the bound fraction and urinary excretion are reduced 15 . A modest hypocalcemia has been observed in some patients. The significance of low ANF concentrations in hypothyroidism is presently unclear 16 Plasma homocysteine concentrations are increased in hypothyroidism, related to lower folate levels and a lower creatinine clearance in thyroid hormone deficiency; restoration of the euthyroid state decreases plasma homocysteine levels into the normal range 1,17 .
In summary, the effects of hypothyroidism on the kidneys are: decreased glomerular filtration rate, decreased renal plasma flow, decreased sodium reabsorption, decreased renal ability to dilute urine, leading to increased serum creatinine and hyponatremia (22).
9.5.9 REPRODUCTIVE FUNCTION
Male gonads and reproduction 36 Alterations in androgens associated with hypothyroidism are rather complex and are due to the consequences of thyroid hormone deprivation on the production, serum transport and metabolic pathways of these steroids. Primary hypothyroidism results in a decrease in sex hormone binding globulin (SHBG) and thereby in total testosterone concentrations in serum; free testosterone is either normal or reduced (in approximately 60% of hypothyroid males) 1 . Serum estradiol is normal, but dehydroepiandrosterone (DHEA), DHEA sulfate, androstenediol and pregnenolone sulfate are decreased in serum of hypothyroid men compared with controls 2 . The metabolic clearance rate of androgens is decreased, and the conversion ratio of testosterone to androstenedione is reduced 3,4,5 . Serum LH and FSH are normal, but the LH and FSH response to GnRH is impaired. The testicles are histologically immature if hypothyroidism preceded puberty and show tubular involution if onset was after puberty 6 . In children, precocious testicular enlargement with early seminiferous tubular maturation has been reported 7 . This abnormality promptly subsides with the correction of the hypothyroid state, and is explained by spillover of the action of TRH on gonadotropes and of TSH on FSH receptors 8,9 . Libido in men may be decreased, and some men may be impotent. In one study among 14 adult hypothyroid males, a high prevalence was observed of hypoactive sexual desire, delayed ejaculation, and erectile dysfunction; there was significant improvement on L-T4 therapy 10 . In another study 84% of 44 hypothyroid patients had a score of 21 or less on the Sexual Health Inventory for Males (SHIM), compared with only 34% of 71 controls; L-T4 treatment increased SHIM scores and restored normal erectile function 11 . Little is known about the effects of hypothyroidism on human spermatogenesis and fertility. In one study reinduction of hypothyroidism did not lead to seminal changes, when compared with the same patients in the euthyroid state (12). In another small study it was reported that L-T4 treatment was associated with some improvement in sperm count and motility (13). More recently, male spermatogenesis was prospectively investigated in 25 hypothyroid men before and after L-T4 treatment by semen analysis, fructose and acid phosphatase measurements, teratozoospermia index, and acridine orange test (14). It was concluded that hypothyroidism had an adverse effect on human spermatogenesis, with sperm morphology the only parameter that was significantly affected.
Female gonads and reproduction 36 In hypothyroid women SHBG is decreased and thereby also total serum estradiol, estrone, and testosterone, but free estradiol and free testosterone are normal 1,15 . Metabolic clearance rate of estrogens is reduced, and peripheral aromatization and conversion of testosterone to Δ4-androstenedione are increased. LH and FSH are normal, but their responses to GnRH can be blunted or delayed. In children, hypothyroidism sometimes induces precocious puberty with menstruation and breast development 16 . These abnormalities disappear with correction of the hypothyroid state, and are explained by spillover of the action of TRH on gonadotropes and of TSH on FSH receptors 8,9 . The endometrium in premenopausal women is typically proliferative or, less common, atrophic. The proliferative endometrium and low urinary pregnanetriol levels suggest failure of LH production and of ovulation (17). Indeed the pulsatile gonadotropin release in the follicular phase is normal, but the ovulatory surge may not happen 18 . In adult premenopausal hypothyroid women, 77% have regular cycles and 23% irregular periods; corresponding figures in controls are 92% and 8% respectively 19 . Oligomenorrhea and hypermenorrhea/menorrhagia are the most common menstrual disturbances. Menorrhagia is probably due to estrogen breakthrough bleeding secondary to anovulation. Defects in hemostasis factors (see section 9.5.9.11) that occur in hypothyroidism may also contribute. Menstrual disturbances tend to be more severe in women with more severe hypothyroidism. Hypomenorrhea and amenorrhea are less common, at variance with the common belief that amenorrhea is the most frequent symptom. The lower frequency of menstrual abnormalities reported in more recent studies, compared with the older ones, may be attributed to delayed diagnosis and thereby more severe hypothyroidism in the earlier studies 20,21,22,23 .The amenorrhea-galactorrhea syndrome is occasionally found in adult hypothyroid women due to hyperprolactinemia; it is reversible with L-T4 treatment 24 . Although infertility may be a problem in either sex, the literature contains many reports of pregnancy in untreated myxoedematoous women 25,26 with frequent successful outcomes 27 . Studies examining the incidence of infertility in hypothyroid patients are scarce. No prospective controlled studies are available, whereas studies on the prevalence of hypothyroidism in patients presenting at specialized infertility clinics are subject to large selection bias. One study detected primary and secondary infertility in one (6.2%) of 16 overtly hypothyroid women, a frequency comparable to that found in control women 23 . Among 704 infertile women without previous thyroid disorders, 2.3% had increased serum TSH levels, comparable to that found in the general female population of reproductive age 28 . Nevertheless, a recent study shows that serum TSH levels are a significant predictor of fertilization failure in women undergoing in vitro fertilization 29 . Thus, although there is a known association between hypothyroidism and decreased fertility, hypothyroidism does not preclude the possibility to conceive. A particular study reports that 34% of hypothyroid women became pregnant without treatment: 11% of them had overt and 89% subclinical hypothyroidism 30 .
Pregnancy. When treatment has been started during pregnancy, more often than not a normal child is produced. Nevertheless, untreated hypothyroidism is associated with adverse outcomes for mother and child, as evident from many studies 31.32.33.34 . A critical review of all pertinent studies on the diagnosis and management of thyroid diseases during pregnancy and postpartum have been published in recently updated guidelines 35,37,38 . In short, when hypothyroid women become pregnant and maintain the pregnancy, they carry an increased risk for early and late obstetrical complications, such as increased prevalence of abortion, anemia, gestational hypertension, placental abruption, and postpartum hemorrhages. These complications are more frequent with overt than with subclinical hypothyroidism and, most importantly, adequate thyroxine treatment greatly decreases the risk of a poorer obstetrical outcome. Untreated maternal overt hypothyroidism is associated with adverse neonatal outcomes including premature birth, low birth weight, and neonatal respiratory distress. Though less frequent than with overt hypothyroidism, complications have also been described in newborns from mothers with subclinical hypothyroidism. Last but not least, there appears to be a significant increased risk of impairment in neuropsychological developmental indices, IQ scores, and school learning abilities in the offspring of hypothyroid mothers. The subject is more fully discussed in chapter 14 on Thyroid Dysfunction in the Pregnant Patient.
9.5.10 ENDOCRINE SYSTEM
Anterior pituitary. Thyrotroph hyperplasia caused by primary hypothyroidism may result in sellar enlargement, particularly when the condition has remained untreated for a long period of time 1,2 . Rarely, such hyperplasia may give rise to a pituitary macroadenoma that shrinks after thyroxine replacement 3,4 . The serum prolactin concentration is elevated in approximately one-third of patients with primary hypothyroidism 5 . The hyperprolactinemia is modest in degree and is rarely associated with galactorrhea 6 ,39 . When present, it subsides with thyroid hormone replacement in conjunction with the reduction in the serum prolactin level. Since thyroid hormone decreases the mRNA for pre-pro TRH in the paraventricular nuclei, it is conceivable that hypothyroidism leads to increased TRH secretion with consequent hyperprolactinemia. The growth hormone response to insulin-induced hypoglycemia is blunted in hypothyroidism 7 . Growth hormone secretion is decreased in hypothyroidism related to an increase in hypothalamic somatostatinergic tone 8 , resulting in low serum IGF-1 concentrations 9 . It may cause growth retardation in hypothyroid children. Serum IGF-2, IGFBP-1 and IGFBP-3 also fall, whereas IGFBP-2 rises; these changes are reversible upon treatment 10 . Another study reports slightly different results: IGF-1 and IGFBP-3 in hypothyroid patients indeed were lower than in healthy volunteers but did not change upon replacement therapy with levothyroxine, whereas the raised levels of IGFBP-1 in hypothyroidism decreased significantly after therapy 36 . The latest study on this issue reports substantial increases of serum IGF-1, IGFBP-3 and the acid-labile subunit (ALS) after L-T4 replacement in primary hypothyroidism (37). In patients with spontaneous autoimmune hypothyroidism due to Hashimoto’s thyroiditis who are adequately treated with levothyroxine, the distribution of IGF-I serum concentrations is similar to that of controls (38). Hashimoto’s thyroiditis can be associated with lymphocytic hypophysitis, which may cause growth hormone deficiency. The prevalence of GH deficiency in Hashimoto patients is low, in the order of 0.4% (38).
Adrenal cortex. Adrenal steroid hormone production and metabolism are considerably affected. Serum cortisol levels are normal, but the turnover time is slowed. This slowing is principally due to a decrease in the rate of cortisol oxidation as a result of reduced 11-β -hydroxysteroid dehydrogenase activity 11 . Conjugation with glucuronic acid in the liver is normal 12 . Reflecting these alterations, urinary 17-hydroxycorticoid (as well as 17-ketosteroid) excretion is reduced 11 , 13 . The turnover rate of aldosterone is also decreased in hypothyroidism 11 . This reduction is probably due to an alteration in steroid reductases that tends to diminish the proportion of androsterone formed and reciprocally increases the level of the etiocholanolone metabolite 14 . The serum concentration of aldosterone is low or normal 15 . Renin activity is also often reduced, as is the sensitivity to angiotensin II 16 . Adrenal responsiveness to adrenocorticotrophic hormone (ACTH) may be reduced, or the response may be delayed until the second and third days of the standard ACTH test, with an actual augmentation of the total response 17 . The adrenal glands often atrophy. Pituitary responsiveness to the metyrapone test has been variable. Normal but delayed peak response 18 , impaired response 19 , or even lack of response 19 has been reported. Grossly impaired responses to the stimulation with lysine-8-vasopressin and a delayed increase in serum cortisol levels after insulin-induced hypoglycemia have also been observed 20,21,22 . A general picture of adrenal function in the hypothyroid patient who is not under stress seems clear. Adrenal steroid metabolism and production decrease. The decreased production is accomplished automatically by the pituitary through decreased ACTH secretion. The result is a normal concentration of free cortisol in the serum. Presumably, sufficient hormone is produced for the reduced needs of the hypothyroid subject. Whether steroid production can be augmented sufficiently in times of stress is not clear, but the provocative test results suggest that these patients usually have a mildly impaired hypothalamic-pituitary adrenal axis 23,24 . The specific association of primary autoimmune hypothyroidism and primary autoimmune adrenal insufficiency is called Schmidt’s syndrome (25). It may be part of polyglandular autoimmune syndromes (26,27). Untreated primary adrenal insufficiency may slightly increase serum TSH, which returns to normal upon glucocorticoid replacement (28).
Sympathoadrenal system. The plasma concentration of norepinephrine in hypothyroid humans is elevated and returns to normal with L-T4 treatment (29,30). The epinephrine concentration is normal. Excretion of catecholamines in the urine is normal, but a decrease in urinary dopamine excretion has been described (31). The circulatory response to injected epinephrine decreases in hypothyroidism but returns rapidly to normal after small doses of levothyroxine (32,33,34). The increased central sympathetic output seems to be compensatory to a reduced response to catecholamines in target tissues (35). Mechanisms involved include a decreased number of β1-adrenergic receptors in the heart. The physiological and clinical implications of the interactions between the sympathoadrenal system (including the sympathetic nervous system and the adrenal medulla) and thyroid hormones have been reviewed, with specific attention of its value in cold adaptation and in states needing high-energy output 40 .
Adipose tissue: see § 9.5.1. Gonads: see § 9.5.9. Parathyroid glands: see § 9.5.6. Posterior Pituitary: see § 9.5.8.
9.5.11 HEMATOPOIETIC SYSTEM
Erythrocytes. In hypothyroidism, plasma volume and red blood cell (RBC) mass are both diminished, and blood volume is decreased. Anemia of mild degree is commonly present, and the hemoglobin level may be as low as 8 to 9 g/dl. In two reports on a large series of patients with hypothyroidism from various causes, the incidence of anemia ranged from 32 percent (1) to as high as 84 percent (2). The anemia may be a result of a specific depression of marrow that lacks thyroid hormone (3), or may be due to blood loss from menorrhagia, to decreased absorption because of gastric achlorhydria, to coincident true Addisonian pernicious anemia, to a decreased absorption of vitamin B12 (which has been found to occur in certain patients with myxedema as a result of diminished intrinsic factor), or to diminished production of erythropoietin by the kidney. The erythropoietic effect of thyroid hormone is mediated through erythropoietin (4). This substance increases RBC production by stimulating the erythroid differentiation of the bone marrow, and its secretion by the kidney appears to be related to the oxygen tension of the tissue. Anemia caused by hypothyroidism per se may be normocytic or macrocytic and respond to thyroid therapy. If iron deficiency develops from menorrhagia, a hypochromic and microcytic anemia may occur. This condition usually responds to iron alone, but may respond optimally only to combined iron and thyroid hormone (5). Hypothyroidism per se causes diminished blood cell formation probably as a response to decreased oxygen demand (6). Plasma and RBC iron turnover are decreased, and the bone marrow is frequently hypoplastic. The relationship between hypothyroidism and pernicious anemia has been well established. Patients have been reported who developed pernicious anemia while hypothyroid, and who lost their need for parenteral vitamin B12 when hypothyroidism was treated. It is also known that some hypothyroid patients absorb oral vitamin B12 poorly, and the defect is sometimes corrected by intrinsic factor (7,8). After thyroid therapy, the absorption defect may disappear or may persist (8). The incidence of pernicious anemia is higher than normal in myxedematous persons (5,8). In Tudhope and Wilson's series of 73 patients with spontaneous primary hypothyroidism, 12.3 percent had true Addison's anemia that responded to vitamin B12 (8). They believe that macrocytic anemia in hypothyroidism should not be accepted as a manifestation of thyroid hormone lack per se, but that it is due instead to the increased coincidence of Addison's anemia. Half of the patients with Addisonian anemia have serum antibodies against the thyroid gland and half of the patients with Hashimoto's thyroiditis have antibodies against gastric cell cytoplasm, parietal cells or intrinsic factor. Megaloblastic anemia due to folic acid deficiency has also been demonstrated in hypothyroidism. Reduced intestinal absorption secondary to hypothyroidism may be responsible for this deficiency, as suggested by the changes observed in a patient given tritiated pteroylglutamate before and after treatment with thyroid hormone (9). Also, a peculiar RBC abnormality has been described in patients with untreated hypothyroidism (10): a small number of irregularly contracted RBCs resembling burr cells are present. The significance of this condition, which may be reversed by the administration of thyroid hormone, is unknown.The erythrocyte sedimentation rate may be elevated in uncomplicated hypothyroidism 11 .
Leucocytes and thrombocytes. Granulocyte, lymphocyte and platelet counts are usually normal in hypothyroidism. Leukopenia might indicate associated vitamin B12 or folic acid deficiency. Hypothyroidism is associated with enhancement of phagocytosis, increased levels of reactive oxygen species (ROS) and increased expression of pro-inflammatory molecules like interleukin-1β in monocytes and macrophages 20 . Mean platelet volume is positively correlated with serum TSH in healthy subjects and in subclinical hypothyroidism, whereas the increase in overt hypothyroidism was insignificant 21,22,23 .
Hemostasis. Hypothyroid patients may have bleeding symptoms such as easy bruising, menorrhagia, or prolonged bleeding after tooth extraction. A systematic review concludes that coagulation tests indicate a hypocoagulable state in overt hypothyroidism (in contrast, hyperthyroidism is associated with a hypercoagulable state, predisposing to thrombosis) (12). Observed defects in general hemostatic tests are prolonged bleeding time, prolonged activated partial thromboplastin time, prolonged prothrombin time, and prolonged clotting time (12,13). Coagulation tests in overt hypothyroidism reveal low or normal factor VIII activity, low von Willebrand factor antigen and activity, low or normal fibrinogen, and low ristocetin induced platelet agglutination (12,14). Fibrinolytic tests in overt hypothyroidism indicate a hyperfibrinolytic condition with reduced TAFIa (activated thrombin-activatable fibrinolysis inhibitor) dependent prolongation of clot-lysis time, despite unaltered TAFI levels (15). Acquired von Willebrand’s syndrome may be the main factor responsible for a bleeding diathesis in overtly hypothyroid patients with a prevalence of 33% (being moderately severe in 9% and mild in 23%) (16,24,25 Even if bleeding episodes are mainly mild and mucocutaneous, blood transfusion, drug administration, or surgical procedure may sometimes be required (16). Desmopressin rapidly reduces these abnormalities (17), and may be of value for the acute treatment of bleeding or as cover for surgery. Usually the clinical relevance of these abnormalities is limited, as illustrated by no excess blood loss or bleeding complications during and after surgery in a large series of hypothyroid patients (18). In patients with moderate hypothyroidism a hypofibrinolytic state has been found, which carries a risk of developing thrombosis (19). However, the concept of a hypercoagulable state in subclinical hypothyroidism is not supported by a systematic review of the existing literature (12,26,27).
9.6 COURSE OF THE DISEASE
Although technologically dated, one of the most charming and clear descriptions of a typical case of myxedema is that given by William M. Ord 1 in Allbutt's System of Medicine, published first in 1897. It is as follows:
THE PICTURE OF THE DISEASE
"Thirty years ago the writer of this article had occasion to investigate the case of a lady suffering from myxedema in a most definite form, and therefore offering complete opportunity of studying the symptoms and the relations of the disease. The patient, a lady of thirty-five, who had several children, presented an appearance suggestive of Bright's disease, yet, although she was greatly swollen on the whole of her body, on careful examination the swelling did not appear to be due to an ordinary dropsy. There was nowhere any pitting on pressure, and there was no albuminuria in the slightest amount. The diagnosis of chronic Bright's disease without albuminuria at first suggested itself, but on further examination many symptoms not known to be related with Bright's disease came under the eye. The face, very much swollen in all parts, was particularly swollen in the eyelids, upper and lower, in the lips, and in the alae nasi. There was a flush, very limited, over the malar bones, contrasting with a complete pallor over the orbital regions. The eyebrows were greatly raised by the effort to keep the lids apart. The skin of the face, and indeed of the whole body, was completely dry, rough and harsh to the touch; not exactly doughy, but giving a sensation of the loss of all elasticity or resilience. The hair was scanty, had no proper gloss, and was much broken. In the absence of all signs of visceral disease the condition of the nervous system was such as to attract much attention. The physiognomy was singularly placid at most times, less frequently heavy, with signs of somnolence, very rarely alert. In interviews the patient was imperturbably garrulous to a degree that could not fail to attract attention. For many minutes she would talk without cessation until obliged to stop and take a good breath. What she said was not altogether relevant, but it had to be said. All interrupting questions were disregarded. If, at the end of a small pause, she was asked to put her tongue, she ignored the request, but at the end of a varying time, when her breath became short, she would put out her tongue for a long time. She dealt in the same way with questions put to her in respect of the points raised by her statements. Her letters were frequent, voluminous, and, as regarded handwriting, very good. Her speech was slow and laboured. There was some difficulty in it, evidently due to the swelling of the lips, but was more than this: the words hung in a way that indicated nervous as well as physical difficulty, and inflexions of the voice were wanting. The tones of the voice were leathery, and suggested rather those of an automaton. The proper timbre was quite lost. Doubtless this was in part, again, due to obvious thickenings in the fauces and the larynx; but it did not in any way resemble the character of voice observed in ordinary swellings of those parts. Her temper was singularly equable, she was the most tender and solicitous of mothers, and in a long course of years during which she was under the writer's observation no word of unkindness or suspicion fell from her lips. Lethargy was an impressive part of her mental condition. Memory was slow, but correct. She thought slowly, performed all movements slowly, and was slow in receiving impressions. Her toilet, and she was no fashionable person, occupied hours. Her household duties could never be overtaken, and she had to seek assistance. Her gait presented a distinct ataxic quality. As her bulky body moved across a room, there occurred at each step forward a quiver running from the legs upwards, such as may be seen in people under the influence of great emotion, as in Lady Macbeth. This appeared to be due to a want of complete concert in the action of the flexors and extensors of the body, the flexors acting for the most part in advance. The interval between the action of the two sets of muscles was at some times extreme enough to determine falls, not in any way produced by obstacles. She fell forward on her knees, and, as a result, she sustained fracture of the patella on one side, and the patellar tendon on the other. Similar conditions existing in the head and neck produced excessive distress. From time to time the head would fall forward in spite of all voluntary effort to prevent it. The chin would then rest on the upper part of the sternum, as is seen in cretins. Sometimes by prolonged exertion of the will, sometimes with the assistance of the hands, the head would be raised, not always to good effect; for unless great care were exercised the head would fall backwards with a suddenness that was alarming. There was no obvious defect of the sense of touch, but it must be admitted that the speed of the reception of tactile sensations was not noted. After the establishment of the disease she bore two children; on both occasions severe postpartum haemorrhage occurred. She had no other haemorrhages. The first impression was, as I said above, that the case was one of Bright's disease without albuminuria. The urine was examined regularly for years without detection of albumin, and there were no such changes in the heart and arteries as belong to Bright's disease. After ten years, however, albumin appeared in the urine, and the patient died ultimately with symptoms of contracting granular kidney. A postmortem examination could not be obtained, and therefore the condition of the thyroid gland and of the kidneys cannot be recorded."
ONSET OF THE DISEASE
The onset of naturally occurring hypothyroidism is insidious. The patient is often unaware of it, as may be friends and relations. As the gland is gradually replaced by fibrous tissue, lymphocytic infiltration, or both, the serum hormone levels and metabolic rate begin slowly to fall. The first symptoms may be a decrease in sweating and dislike of cold. They may be present alone for a period of years before dramatic events occur. One of our patients gave a story of marked hypersensitivity to cold for 12 years, at the end of which time the picture of full-blown myxedema developed. Sometimes the presenting symptom may be a demand for a warmer room or more clothing. Sometimes a mere decrease in activity due to listlessness, lack of energy, or fatigue, is the first change noted. In other patients, mental dullness or drowsiness may be observed. We have also seen the opposite change, namely, nervousness and irritability, or even peevishness in the exceptional case.
Progressive constipation or increase in menstrual flow may occasionally be the first event. So, too, may any of the following: deafness, falling hair, thick speech, dizziness, puffiness of the face, headache, pallor, weight gain, or fatigue. When hypothyroidism occurs more suddenly, as after surgical thyroidectomy or RAI therapy, the symptoms may not be so insidious, and indeed may be quite upsetting to the patient. Musculoskeletal symptoms such as frequent cramps may be distressing, and acute depression or acute anxiety may appear. Thus, the clinical course may be much influenced by the cause of the hypothyroid state. Obvious symptoms and signs usually appear as the thyroxine (T4) level falls below normal. Of these symptoms, nonpitting edema, from which myxedema derives its name, is pathognomonic. It is a specific thyroprival sign, and when it develops, the disease is in the full-blown state. There may be little apparent change in the patient's appearance or condition for several years. During such a period the patient may be well off subjectively. The increased sensitivity to cold can be met by maintaining the living area at an unduly warm temperature. The decreased energy makes the person content to do little or nothing. The myxedematous state is characterized by an amazing placidity. The terminal stage may be called myxedematous cachexia.
MYXEDEMATOUS CACHEXIA
Myxedematous cachexia is characterized by an intensification of all symptoms and signs. There is great thickening of the tongue, thickness, dryness and coarseness of the skin, thickening and brittleness of the nails, falling and brittleness of the hair, progressive decrease in activity and responsiveness, and a closer and closer approach to a purely vegetative existence. Although the mucous edema persists - and indeed tends to increase - body fat may disappear, so that actual wasting takes place. After this stage has persisted for an indefinite period of months or even years, death takes place because of intercurrent infection, congestive heart failure, or both. The final symptom is coma, which may last for days. In the untreated patient, the length of time between the first symptoms and death may be as long as 15 years. It is, fortunately, seldom nowadays that one witnesses the natural termination of the disease. It is seen only when the patient is already moribund when he or she comes to the physician, the diagnosis previously having been overlooked or where severe myxedema is present in association with another serious illness. In the Report on Myxoedema, which was published before the discovery of the cure of the disease, the duration is given as 10 years or more. The evolution of the symptoms of myxedema is slowly progressive. If one compares patients with myxedema of 3, 6 or 12 years' duration, although all may have classic symptoms and identical thyroid function test results, the clinical picture will be more intense at 6 years than at 3 and still more at 12. The mental manifestations, and the integumentary changes in particular, intensify as the years pass. Such severe manifestations of hypothyroidism are rarely seen in the current era. Patients and their friends and relatives are often strangely unaware of evidence of myxedema. Often patients are identified during treatment for some entirely unrelated disorder. Myxedema has been called a "consultant's diagnosis", because the changes that appear as the disease develops are so subtle and gradual that they are often overlooked by the patient's family physician. This fact is becoming less true with the ready availability of objective diagnostic tests.
9.7 DIAGNOSIS OF HYPOTHYROIDISM
Evaluation of a patient suspected of hypothyroidism starts with obtaining conclusive evidence that thyroid hormone deficiency is absent or present. Clinical examination suffices to provide a definitive answer in very severe cases of thyroid hormone deficiency, but is less accurate in mild cases. Biochemical proof of thyroid hormone deficiency is thus required in the vast majority of patients. If hypothyroidism is demonstrated, the next question to be answered is which disease entity has caused the hypothyroid state (nosological diagnosis). Delineation of the cause of hypothyroidism is relevant for identification of patients with potentially reversible hypothyroidism; it migh also give a clue for the existence of other conditions associated with a specific cause. The diagnostic process is schematically represented in Table 9-9 .
Table 9-9
Schematic approach of the patient suspected of hypothyroidism.
9.7.1 CLINICAL EVALUATION (STAGE 1A)
Table 9-10 lists the relative frequency of symptoms and signs accumulated by Lerman in a study of 77 myxedematous patients in one thyroid clinic and by Murray in a study of 100 patients with primary hypothyroidism, 15 pituitary patients, and 100 normal control subjects. This analysis identifies the cardinal manifestations of the disease. It also discloses that a certain number of manifestations are occasionally found in overt myxedema that are somewhat more suggestive of hyperthyroidism than of hypothyroidism. Under this heading may be listed dyspnea, nervousness, palpitations, precordial pain, loss of weight, and emotional instability. These symptoms are also found in normal control subjects in nearly the same frequency.
Many symptoms typical of primary hypothyroidism are not commonly found in pituitary hypothyroidism - for example, coarse skin, thick tongue, coarseness of hair, peripheral edema, hoarseness, and paresthesias.
Table 9-10. I
ncidence of symptoms and signs in hypothyroidism.
The diagnosis of severe hypothyroidism is relatively straightforward on clinical grounds. All of the manifestations mentioned in the above discussion are present, and laboratory testing merely confirms the high index of clinical suspicion. However, severe hypothyroidism has become increasingly rare due to physicians' raised level of consciousness about the relatively high prevalence of this disease in women and the ease of making a laboratory diagnosis. Rather it is the more sublte or unusual presentations of hypothyroidism that may present difficulties 1 . Since laboratory confirmation of hypothyroidism is straightforward, the critical factor in successful diagnosis is maintaining a high degree of suspicion. If the diagnosis is not suspected in a patient with some of the typical manifestations of hypothyroidism at the first encounter, it may be several months before the physician reconsiders this explanation for the patient's complaints. Thus, hypothyroidism may be more readily diagnosed by a consultant who has not seen the patient before, since both the patient and the regular physician may have assumed that the many nonspecific symptoms are insignificant or at least unrelated to a specific organic disease.
There are certain symptoms or signs that should, irrespective of other factors, lead to a biochemical evaluation for possible hypothyroidism. In the child or adolescent, growth retardation is one of these. The presence of an enlarged thyroid should trigger a similar response. However, more subtle, less specific complaints, including depression or other organic mental syndromes, muscle cramps, paresthesias, carpal tunnel syndrome, hoarse voice, elevated cholesterol, pericardial effusion, arthritis, yellow skin (carotenemia), hyperkeratosis of the palms or soles, or menorraghia, can be manifestations of hypothyroidism. In addition, certain constellations of autoimmune disease occur in concert with hypothyroidism, including primary adrenal insufficiency, type I diabetes, and pernicious anemia. The presence of any of these should lead to search for primary thyroid dysfunction.
Statistical methods have been applied to the clinical diagnosis of hypothyroidism, based on the frequency of symptoms and signs in patients and controls. Well-known is the Billewicz score, composed of points given in a weighted manner for the presence or absence of 17 symptoms and signs 2 . Application of this score increases the pretest likelihood of hypothyroidism by 15-19% 3 . A newly developed clinical score is, however, easier to perform and more sensitive 4 (Table 9-11).
Table 9-11
Accuracy of 12 symptoms and signs in the diagnosis of primary hypothyroidism 122 .
† Add 1 point in women younger than 55 yr
§ Hypothyroid, 6 points; intermediate, 3-5 points; euthyroid, 2 points.
The positive predictive value of this new score for hypothyroidism is 96.9% at a score of 6 points or more; the negative predictive value for the exclusion of hypothyroidism is 94.2% at a score of 2 points or less. 62% of all overt hypothyroid and 24% of subclinical hypothyroid patients are classified as clinically hypothyroid by the new score, as opposed to 42% and 6% respectively by the Billewicz score Figure 9-4 . The diagnostic accuracy of these clinical scores is thus very low. In view of their poor performance, they should not be used for the diagnosis of hypothyroidism 12 .
Figure 9-4Assessment of hypothyroidism by a clinical score, composed of 12 symptoms and signs as listed in Table 3 (Reproduced with persmission(4)).
Age and smoking have been recognized as modifiers of the clinical expression of thyroid hormone deficiency. Elderly patients have a smaller number of clinical signs than younger patients 5 . Smokers have more severe manifestations of hypothyroidism than nonsmokers 6 .
9.7.2 LABORATORY EVALUATION (STAGE 1B)
The assay of TSH in serum has proven to be the best single test for the exclusion or detection of hypothyroidism 12 . Using the flow-chart of Figure 9-5 the following results can be obtained:
Figure 9-5Flow-diagram for the biochemical diagnosis of hypothyroidism.
(Figure 9-5) . Flow-diagram for the biochemical diagnosis of hypothyroidism.
1. TSH normal. Euthyroidism is almost certain, as primary hypothyroidism is excluded. However, two conditions will not be recognized. The first is the existence of central hypothyroidism. As isolated TSH deficiency is very rare, clinical examination of the patient will usually provide sufficient clues (symptoms and signs of a pituitary mass, of hypopituitarism, or of overproduction of pituitary hormones) to warrant further evaluation by a FT4 assay.The second is thyroid hormone resistance due to TRα mutations; this recently discovered and probably rare disease is characterized by low normal to slightyly low FT4 and high normal to slightly high T3 13,14 .
2. TSH elevated, FT4 decreases. This classical combination of test results indicates primary hypothyroidism. Test results are sometimes due to central hypothyroidism or nonthyroidal illness when TSH is slightly elevated (5-15 mU/l).
3. TSH elevated, FT4 normal. Test results indicate most often subclinical hypothyroidism, sometimes nonthyroidal illness.
4. TSH elevated, FT4 increased. A rarely encountered combination of test results, indicating either thyroid hormone resistance due to mutations in TRβ or TSH producing pituitary adenoma.
5. TSH decreased, FT4 decreased. Central hypothyroidism accounts for these test results, which, however, also can be observed in severe nonthyroidal illness and after recently instituted treatment for thyrotoxicosis (131I, surgery, antithyroid drugs) or recent discontinuation of excessive thyroid hormone medication.
6. TSH decreased, FT4 increased or normal. Hypothyroidism is excluded. Results indicate overt thyrotoxicosis or subclinical hyperthyroidism respectively.
The reference interval of serum TSH is about 0.4-4.0 mU/L. However, the lower normal limit is lower in pregnancy, and the upper normal limit increases with advancing age, reaching levels of 6.3 mU/L at the age of ≥80 yr 16. . Serum T3 should not be done for the diagnosis of hypothyroidism 12 .
9.7.3 NOSOLOGICAL DIAGNOSIS (STAGE 2)
The cause of the hypothyroid condition is in general easily established. Most informative are a careful clinical examination and determination of TPO antibodies in serum. Particularly relevant questions in the history taking are: family history of thyroid disease? recent delivery? previous thyroid surgery or 131I therapy? use of antithyroid drugs? exposure to iodine excess? Symptoms and signs of a pituitary mass or of hypopituitarism suggest the presence of central hypothyroidism. Physical examination may reveal a goiter (like the characteristic firm rubbery' goiter in goitrous Hashimoto's hypothyroidism), but many if not most hypothyroid patients have no palpable thyroid gland. High titers of TPO antibodies indicate chronic autoimmune thyroiditis, the most prevalent cause of hypothyroidism. Thyroid ultrasound can be helpful: the finding of a non-homogeneous hypoechogenic pattern indicates chronic autoimmune thyroiditis, which may be observed also in the absence of thyroid antibodies in serum 15 . Although most cases of hypothyroidism are permanent and require life-long treatment with thyroxine, a substantial minority is transient in nature due to the natural course of the underlying disease entity. Elimination of the causal factor is possible only in a few patients in whom the hypothyroid state is induced by antithyroid drugs or iodine excess. Table 9-12 provides the physician with possible clues for assessing the likelihood of reversible hypothyroidism in a particular patient. In selected cases further evaluation by thyroidal radioiodine uptake studies might be useful.
Table 9-12
Reversible causes of hypothyroidism.
9.8 TREATMENT OF HYPOTHYROIDISM
9.8.1 PHARMACOLOGY OF THYROID HORMONE REPLACEMENT PREPARATIONS
Levothyroxine.
L-thyroxine is prescribed as the sodium salt in order to enhance its absorption, which occurs along the entire small intestine 1,2 . Intestinal absorption of oral T4 is on average 80% 3 , and is greater in the fasting than in the fed state. Absorption is apparrently more complete and less eratic if the daily dose is taken in the fasting state 4 . Serum T4 concentrations peak 2 to 4 hours after an oral dose and remain above normal for approximately 6 hours in patients receiving daily replacement therapy 5,6 . The gradual conversion of T4 into T3 in various tissues increases serum T3 concentrations so slowly after thyroxine absorption that with daily levothyroxine administration, no significant changes in circulating free T3 are detectable. In North America, levothyroxine tablets are available in tablet strengths of 25, 50, 75, 88, 100, 112, 125, 137, 150, 175, 200 and 300 µg. The long half-life of thyroxine of about 7 days allows treatment with a singly daily tablet. Omission of an occasional tablet is of little relevance. If a patient misses a pill one day, they may take two the next. If they miss for two days, they may take three the next (7). Generic and brand-name levothyroxine preparations are mostly bioequivalent (8), but altered bioavailability has been reported due to changes in the formulation of preparations (9). Levothyroxine has a narrow therapeutic index with the potential for putting patients at risk for iatrogenic hyperthyroidism or hypothyroidism at doses only 25% less or greater than optimal, based on patient’s serum TSH (10). Available data suggest 15-29% of patients receiv.e inadequate doses of L-T4 and 18-24% receive excessive doses, based on serum TSH levels outside the reference range (11,12,46). It re-emphasizes the question whether the different marketed L-T4 formulations are mutually exchangeable: this would require bioequivalence between products. The FDA set criteria for testing bioequivalence of levothyroxine sodium tablets (13), and moved to approve generic L-T4 preparations as equivalent to branded preparations. This led to a joint statement by the American Thyroid Association, the Endocrine Society and the American Association of Clinical Endocrinologists in which they oppose the stand taken by the FDA (14). For pharmacokinetic studies designed to measure the bioavailability of L-T4 formulations, the FDA recommends that a single dose be administered to healthy subjects at a strength several times the normal therapeutic dose. With correction for the endogenous T4 pool, the method could distinguish the doses that differed by 25% and 33%, but not dosage strenghts that differed by 12.5% (15). The way the FDA proposes to measure the results of small changes in the T4 content of commercial preparations thus seems not very sensitive. Therefore it has been concluded that the type of T4 absorption studies the FDA uses as evidence, as opposed to serum TSH which thyroidologists rely on, is inappropriate (16). According to the FDA definition of bioequivalence, particular test formulations have found to be bioequivalent to the reference formulation of levothyroxine in healthy volunteers (17). However, a pharmacodynamic TSH-measurement bioequivalence protocol, using normal L-T4 replacement dosing in athyreotic volunteers, is likely to be more sensitive and safer than current FDA Guidance based on T4 pharmacokinetics (18). In the meantime the allowable potency range for L-T4 tablets has been tightened to 95-105%, a significant improvement of the original FDA-approved 90-110% range. A study comparing pH-dissolution profiles of selected commercial levothyroxine preparations, shows a considerable decrease in dissolution of L-T4 tablets with increase in pH, with differences in T4 dissolvement between the various preparations (19). Differential dissolution of T4 products can impact oral absorption and bioavailability of L-T4, and may result in bioequivalence problems. A survey among physicians in the USA managing patients with thyroid diseases that required the use of L-T4 preparations, indicated that 177 of 198 reports of adverse events associated with changes in TSH values, were associated with a change in the source of L-T4 (20). The exchanges were done by the patient’s pharmacy without the clinician’s knowledge in 92%. Fifty-four of the 198 reported cases resulted in serious adverse events, and 52 of these 54 cases were associated with a substitution of one L-T4 preparation with another. Against this background it is recommendable to continue to use the same L-T4 preparation once the appropriate L-T4 dose has been established.(27). The pharmacist should inform the clinician if a switch is being made to another L-T4 preparation.
Liothyronine.
After oral administration of L-triiodothyronine sodium (which is more readily absorbed than T4) peak levels of serum T3 are observed within 2 to 4 hours 21 .The serum T3 concentration may reach elevated values after a single dose of 50 µg or even 25 µg, sometimes associated with cardiac symptoms like palpitations 22 . The half-life of T3 is short (approximately one day) which requires several gifts per day.. Preparations of L-T3 can be useful in the short-term management of patients with thyroid cancer to shorten the period of hypothyroidism required for diagnosis and treatment of remaining tumor tissue with 131I, and in myxedema coma; they are not recommended for long-term replacement therapy in hypothyroidism. Pharmacodynamic equivalence of levothyroxine and liothyronine is achieved at a dose ratio of about 3:1 27
Desiccated thyroid.
Desiccated thyroid is prepared from porcine or bovine thyroid glands. In former days desiccated thyroid was standardized by the organic iodine content, which did not distinguish between iodotyrosines and iodothyronines 23 . Current guidelines stipulate that one grain (65 mg) of desiccated thyroid contains about 44 µg T4 and 9 µg of T3; the hormones are in the form of thyroglobulin 24,25 . In our experience, the biologic potency of a 1-grain desiccated thyroid tablet is about 75 to 88 µg T4. Because of the relatively high ratio of T3 to T4 in desiccated thyroid, patients receiving an amount of this medication adequate to normalize serum TSH generally have serum T4 concentrations in the lower half of the normal range. Serum T3 concentrations will vary in such patients, depending on the interval between ingestion of the medication and the time of blood sampling. The time course of the absorption of T3 is similar whether it is contained in thyroglobulin or free in the tablet, with peak levels approximately 2 to 4 hours after oral administration 21 .A recent randomized clinical trial compared levothyroxine replacement with desiccated thyroid extract (DTE, Armour Thyroid, of which each grain of 65 mg contained 38 μg L-T4 and 9 μg L-T3) 29 .Use of DTE relative to levothyroxine was associated with modest weight loss and greater patient preference; Serum T3 was higher and serum FT4 was lower during treatment with DTE than during levothyroxine treatment. Although DTE can provide satisfactory replacement therapy if TSH levels are maintained in the normal range, it is not recommended in current guidelines for treatment of hypothyroidism 27 .
Combinations of T3 and T4.
Liotrix, the only combination preparation currently available in the United States, contains 50 µg T4 and 12.5 µg T3/1 grain equivalent, but is biologically equivalent to a 65 mg (1 grain) tablet of desiccated thyroid. Recent studies in thyroidectomized rats have demonstrated that restoration of the euthyroid state in all tissues can only be restored by the combination of T4 and T3, and not by T4 alone 26 . This finding has aroused new interest in combinations of T3 and T4 in hypothyroid patients who are dissatisfied with the outcome of levothyroxine replacement therapy 33-37 (see 9.8.2).However, a meta-analysis of 11 randomized clinical trials found no evidence for superiority of L-T4 + L-T3 combination therapy over levothyroxine monotherapy: bodily pain, depression, anxiety,fatique and quality of life were not different between the two treatment modalities 30 . Some patients nevertheless do prefer combination therapy 31 , and it is hypothesized that particular genetic polymorphisms in thyroid hormone transporters and deiodinases are linked to this preference. Guidelines do not recommend combination therapy, and levothyroxine remains the standard treatment modality for hypothyroidism 27 . In patients with persistent complaints despite adequate levothyroxine replacement as evident from normal serum TSH levels, one may consider a trial of L-T4 + L-T3. This should be offered after exclusion of other conditions which might be responsible for the persistent complaints. Detailed instructions on selection of patients for combination therapy, calculation of levothyroxine and liothyronine doses, and monitoring are given in a recent guideline on this issue by the European Thyroid Association 32 ,
9.8.2 REPLACEMENT WITH THYROXINE
Of the available thyroid hormone replacement preparations, thyroxine is presently recommended as the drug of choice in view of its long half-life, ready quantitation in the blood, ease of absorption, and the availability of multiple tablet strenghts 1-4 .All guidelines state levothyroxine sodium is the standard treatment of hypothyroidism 51 .
Institution of therapy.
The rapidity with which normal thyroid hormone levels should be restored depends on a number of factors, including the age of patient, the duration and severity of the hypothyroidism, and the presence or absence of other disorders, particularly those of the cardiovascular system. Most patients under the age of 60 can immediately begin a complete replacement dose of 1.6 to 1.8 µg levothyroxine/kg ideal body weight (about 0.7 to 0.8 µg/1b). Requirements for children and infants are discussed separately and are higher than those for adults between the ages of 20 and 70. A randomized double-blind trial comparing a full starting dose (1.6 μg/kg L-T4) with a low starting dose (25 μg L-T4, increased every 4 weeks) in patients with newly diagnosed cardiac asymptomatic hypothyroidism observed that the full starting dose was safe and more convenient and cost-effective than a low starting dose regimen (47). Preference for gradual levothyroxine replacement and conservative dosage titration is, however, expressed widely (48). The standard advice is to take L-T4 tablets in the morning half an hour before breakfast. Interestingly, L-T4 taken at bedtime is associated with higher FT4 and T3 and lower TSH concentrations in serum compared to the same L-T4 dose taken in the morning, most likely due to better absorption of L-T4 during the night (49). Patients might be adviced to take their L-T4 tablets always either in the morning or in the late evening. Drinking espresso together while ingesting the L-T4 tablets, reduces intestinal absorpton of levothyroxine (50). The cause of hypothyroidism also influences replacement in that patients with total thyroidectomy or severe primary hypothyroidism have slightly higher requirements than do patients who become hypothyroid after radioiodine or surgical treatment for Graves' disease 5 . The latter group may have some residual thyroid function that is autonomous, and thus a complete replacement dose is excessive. For most women, a complete replacement dose will be between 100 and 150 µg per day and, for most men, between 125 and 200 µg per day. Pretreatment serum TSH predicts to a certain extent the daily maintenance dose of levothyroxine in patients with primary hypothyroidism (Figure 9-6) 6 . Individual L-T4 requirements are dependent on lean body mass. Age- and gender-related differences in L-T4 needs reflect different proportions of lean mass over the total body weight. An estimate of lean mass may be helpful to shorten the time required to attain a stable dose of L-T4, particularly in subjects with high body mass index values that may be due either to increased muscular mass or obesity (7).

Figure 9-6
Relationship between the optimal daily dose of levothyroxine sodium and the mean pretreatment serum TSH concentration in patients with primary hypothyroidism. Simple linear regressions are shown for two subgroups calculated according to the daily dose of L-T4 divided at the median dose of 125 µg; the intercept of these two correlation lines occurs at the TSH concentration of 36 mU/l. (Reproduced with permission) 6 .
Full replacement doses should not be administered initially to patients over the age of 60, to patients who have a history of coronary artery disease, or to patients with long-standing severe hypothyroidism. While levothyroxine improves cardiac function in patients with hypothyroidism and increases cardiac output and decreases systemic vascular resistance and end-diastolic volume, it also increases myocardial oxygen consumption. Thus, while patients with coronary artery disease and angina may benefit from reversal of their hypothyroid state, to avoid precipitating acute myocardial ischemia, the dose should be titrated, starting with 25µg a day and increased by increments of 25 µg at 8-week-intervals until serum TSH falls to normal or symptoms of angina worsen or appear. A similar slow approach is prudent in patients with long-standing, severe hypothyroidism, also because occasionally psychosis or agitation occurs during the initial phase of replacement in such cases 8,9,10 .
Monitoring.
In the patient given what is thought to be a complete replacement dose of levothyroxine, a TSH and free T4 should be measured about 2 months after therapy begins to establish that the estimated dose is appropriate for the patient. At that time, serum TSH may be still elevated, indicating the need for a modest increase in dose, or TSH may be suppressed, indicating that a reduction is in order. This is usually done in 12.5- to 25-µg increments, depending on the patient 11 . These studies should be repeated again in 2 months to titrate proper dosage. After proper dosage has been achieved, the test should be repeated yet again after the patient has been euthyroid for approximately 6 months. This is because in certain patients, normalization of thyroxine clearance may require more than 8 weeks, and a dose of levothyroxine that is adequate when the patient is metabolizing thyroxine more slowly may be inadequate when the patient is euthyroid. This dose should be continued and monitored on an annual basis. In patients with severe primary hypothyroidism, few adjustments will be required after the initial titration until the eight decade. However, patients with Graves' disease who have had radioactive iodine may require dosage adjustments up to as long as 5 to 10 years after treatment is begun. A similar course may be followed by patients who have had subtotal thyroidectomy for Graves' disease due to the slow deterioration of residual thyroid function. Therapy should be monitored with TSH measurements and estimates of free T4. As the goal of levothyroxine therapy is to normalize the thyroid status of the patient, and as serum TSH provides the most sensitive and readily quantification of thyroid status in the patient with primary hypothyroidism, one aims at TSH values in the low normal range. Serum FT4 concentrations will generally be above the middle of the normal range or slightly elevated if serum TSH concentrations are normalized, but serum T3 concentrations (predominantly derived from T4-5'-monodeiodination) will be in the low or midnormal range 12 . In 1,811 athyreotic patients under levothyroxine replacement, FT4 was higher than the upper normal limit in 7.2%, and FT3 was lower than the lower normal limit in 15.2%; the FT3/FT4 ratio was abnormally low in 29.6% (see Figure 9-7) 52 .
Figure 9-7
Frequency distribution curves of serum FT4, FT3 and FT3/FT4 ratio in 1,811 athyreotic patients under levothyroxine replacement. Shaded areas indicate normal range (2.5 and 97.5 percentiles) as calculated from 3,875 controls, with vertical lines indicating the median (reproduced with permission) 52 .
In patients with central hypothyroidism one should rely primarily on serum FT4, aiming at values in the mid-normal range 13,53 . The required replacement dose will frequently suppress serum TSH values to below 0.1 mU/l 14 .
Clinical response.
In general, serum thyroxine normalizes before serum TSH, and both may normalize before the disappearance of all of the symptoms of hypothyroidism. In the severely hypothyroid patient with long-standing disease, a number of profound alterations may occur as the hypothyroid state is corrected. Thus, loss of weight, primarily due to mobilization of interstitial fluid as the glycosaminoglycans are degraded, is prominent. The moon facies, coarse nasal voice, puffy fingers, deafness, and sleep apnea will all diminish. Many of nonspecific symptoms, such as fatigue or cold intolerance, will eventually reverse as well. Hair and skin abnormalities take longer to improve. Despite weight loss due to fluid loss, the obese patient should not expect more than a 10-pound weight change, particularly if serum TSH values are only modestly elevated. Virtually all of the weight loss in hypothyroidism is associated with mobilization of fluid, and significant decreases in body fat rarely occur. 54 While metabolic rate increases, in general, appetite increases as well, and a new equilibrium is established.
Treatment failures.
There are few compliant patients whose symptoms and signs do not resolve after thyroid hormone administration. Patients with thyroid hormone resistance sometimes present in this fashion. In patients whose symptoms do not improve with levothyroxine therapy, one should establish that they are taking and absorbing the medication and that it is effective in reducing TSH. The most common cause of treatment failure is poor compliance with ingestion of thyroxine tablets.Non-adherence to treatment can be assessed by a thyroxine absorption test 55 . Compliance might be enhanced by the (supervised) administration of thyroxine once weekly 14 . A slightly larger dose than 7 times the normal daily dose may be required; a singly weekly gift of 1000 µg T4 orally seems to be effective and well tolerated.
Dissatisfaction with treatment outcome
Whereas the vast majority of hypothyroid patients are satisfied with T4 replacement therapy, some are not. A Dutch study reports impaired cognitive functioning in T4 replaced hypothyroid patients relative to a reference population, as evident from worse scores on tests of cognitive motor speed, attention span, and learning and memory tasks (15). An English study reports a higher proportion of distressed subjects in T4-replaced hypothyroid patients than in controls, as evident from general health questionnaires (32.3 vs 25.6%) and thyroid symptom questionnaires (46.8 vs 35.0%)(p<0.01 after correction for age, sex, chronic diseases and chronic medication)(16). The significant difference with controls remained when only hypothyroid patients with TSH values between 0.4 and 4.0 mU/L were analyzed. Subtracting the proportion of distressed subjects in controls from that in the T4-replaced hypothyroid patients leaves us with an excess of 10% of distressed subjects among T4-replaced patients. How can we explain the dissatisfaction, assuming associated autoimmune diseases have been ruled out? It could be that simply being aware of having a chronic disease requiring lifelong treatment and regular control visits makes the patients feel unhappy and less healthy. A more specific explanation would be that L-T4 replacement therapy fails to mimic precisely the thyroidal secretion rates of T4 and T3 and the serum FT4 and FT3 concentrations of healthy subjects (17). A combination of L-T4 and L-T3 replacement therapy might reflect better physiological conditions. A number of randomized clinical trials ( RCTs) comparing T4 monotherapy with T4/T3 combination therapy have been performed to clarify this issue, also provoked by animal data indicating that restoration of the euthyroid state in all tissues of thyroidectomized rats is accomplished only by the combination of T4 and T3, and not by T4 alone. However, a meta-analysis published in 2006 of 11 randomized controlled trials with in total 1216 patients concluded that T4/T3 combination therapy used as replacement therapy for hypothyroid patients provided no advantage when compared with standard T4 monotherapy (18). Despite the outcome of the meta-analysis, the issue of potential benefit of T4/T3 combination therapy cannot be considered as closed in view of further developments: 1. A re-appraisal of the RCTs. The RCTs in the meta-analysis are heterogeneous with respect to the cause of hypothyroidism (thyroidectomy for thyroid cancer, 131I therapy for Graves’ hyperthyroidism, or spontaneous autoimmune hypothyroidism) and the study design (parallel or crossover), but this did not affect outcome (17). In 7 of the 11 RCTs (as well as in a more recent RCT)(19) 50 μg of the daily T4 dose was replaced by a fixed T3 dose (ranging from 10 to 25 μg T3) in patients randomized to combined T4/T3 treatment, giving rise to a wide variation between patients in each RCT in the ratio of the administered T4 to T3 dose (ranging from 20:1 to 1:1 by weight). This is a far cry from mimicking the ratio of T4 to T3 secretion by the human thyroid gland under physiological conditions, which is close to 13:1 by weight (20). Only four trials in the meta-analysis used a variable T3 dose in order to reach the same ratio of administered T4 to T3 dose in all study subjects; the fixed T4 to T3 ratio’s by weight in these studies were 5:1, 10:1, 15:1 and 19:1. Nevertheless, in these four trials, combination therapy was also judged not to be better than monotherapy with T4. To obtain TSH values similar to controls during T4 monotherapy, serum FT4 concentrations higher than controls are needed, whereas serum FT3 values are similar to those in controls (12) (see also figure 9.7) Indeed, the serum FT4 to FT3 ratio in patients randomized to receive T4 monotherapy in the meta-analysis ranged from 4.0 to 6.7, higher than the value of 3.3 observed in controls (21). The serum FT4 to FT3 ratio during combination therapy ranged from 2.2 to 4.8; in only two of the RCTs the ratio’s (3.3. and 3.4 respectively) were close to control values, but both studies still failed to demonstrate superiority of combination therapy over monotherapy (17). Applied outcome measurements in the various RCTs are a number of questionnaires on health-related quality-of-life, cognition, mood and thyroid symptoms, about the same in most but not all studies. The most recent RCT reports significantly better outcome of combination therapy in quality-of-life and depression scales (19), in contrast to all previous RCTs except two early biased ones (17) and one in which the beneft at 3 months was lost at 12 months (22). In this recent RCT when asking the patients themselves, 49% preferred combination therapy, 15% preferred monotherapy, and 36% had no preference (19). That 15% felt better in the period in which the same T4 dose was used as before entering the study, indicates a strong Hawthorne effect also observed in a previous study (23): patients feel better just by participating in a trial. If true, what might explain the preference for combination treatment? It could be loss of body weight: patients at the end of the combination therapy were on average 1.7 kg lighter than after monotherapy (19), and a similar loss of 1.7 kg was observed during combination therapy in another RCT in which patients also preferred the combination (23). In summary, each of the RCTs can be criticized and none is perfect. But these trials are very demanding to perform. If still further RCTs are required (17), special attention should be given to sample size calculation, T4 to T3 ratio’s in combination therapy, and dynamic monitoring of TSH in order to maintain a normal level by adjusting study medications if needed (24). 2. Over- or under-treatment in T4 monotherapy . Serum T3 levels during T4 treatment are mostly in the normal range, but the median is lower than in controls and 15.2% are below the lower normal limit (see figure 9.7) 52 .It is thus obvious that the capacity to generate T3 from exogenous levothyroxine is insufficient in many patients.Serum T3 concentrations similar to those prior to the development of hypothyroidism can be obtained according to a study in euthyroid patients before total thyroidectomy and after surgery when the patients were fully replaced with T4 (25). Nevertheless, the concentration ratio’s of FT4 to FT3 will still be nonphysiological, which may affect deiodinase activity and nuclear T3 receptor occupancy in target tissues. Another study with a double-blind randomized cross-over design investigated T4-replaced patients who were asked to continue with their usual T4 dose, or take 25 μg less or more (26). Mean T4 doses in each of the 6-week study periods were 100, 125 and 150 μg daily. It resulted in expected changes in serum FT4, TSH and cholesterol, but no changes were observed in scores of well-being, cognitive function, and quality-of-life and thyroid symptom questionnaires. It is thus unlikely that slight over- or under-treatment with levothyroxine provides a reasonable explabation for continuous dissatisfaction with T4 monotherapy.It appears more likely that the modality of levothyroxine replacement itself is involved (see 9.8.1). 3. Mode of T3 administration . During T4/T3 combination therapy, the T3 dose is given once or twice daily. It results in wide peak-to-trough variation in serum FT3, e.g FT3 increased by 42% in the first 4 h after T3 but did not change after T4 (27). A slow-release formula of T3 might circumvent the marked changes in serum FT3, and proof of principle of such a preparation has been obtained in a recent study (in which the serum FT4 to FT3 ratio was lower during T4 + slow-release T3 than during T4 monotherapy, but still higher than in controls)(28). Serum FT3 has –in contrast to FT4- a circadian rhythm: the FT3 acrophase occurs in early morning hours around 0300 h, about 90 minutes after the TSH acrophase (29). If one’s goal is to replicate the circadian T3 rhythm and to maintain a physiological ratio of serum FT4 to FT3 throughout 24 h in hypothyroid patients, replacement should provide constant FT4 levels and an early morning rise in serum FT3. This goal possibly can be reached by the administration of levothyroxine once daily in combination with a single nighttime dosing of a sustained-release T3 preparation. 4. Gene polymorphisms. Genetic polymorphisms in deiodinases and thyroid hormone transporters may not only affect serum thyroid hormone concentrations but also the biological availability of thyroid hormone in particular tissues (30). Single nucleotide polymorphisms (SNPs) in the gene encoding for deiodinase type 1 influence the serum FT4 to FT3 ratio, but do not have any association with psychological well-being in patients on thyroid hormone replacement (31,32). An early report did not find an association between the Thr92Ala polymorphism in the deiodinase type 2 gene and well-being, neurocognition or preference for T4/T3 combination therapy (33), but a study with a much larger sample size observed associations between the CC genotype of the D2 Thr92Ala polymorphism and worse baseline scores for general health and greater improvement on T4/T3 combination therapy (32). Lastly, several polymorphisms in the brain-specific thyroid hormone transporter OATP1C1 are associated with fatigue and depression in hypothyroid patients on levothyroxine, but not with neurocognitive functioning or preference for combination therapy (34). Taken together, it might well be that hypothyroid patients dissatisfied with levothyroxine monotherapy are frequent carriers of these polymorphisms, and might have a better response to T4/T3 combination therapy (17,35).One could envisage RCTs restricted to patients who are carriers of such polymorphisms.
Potential adverse effects of treatment.
Life-long treatment with thyroxine when properly monitored with annual assessments, seems to be free of complications. Long-term morbidity and mortality have reported to be normal 1-4 . Thyroxine treatment in TSH-suppressive doses, however, might give reason for some concern as it has been associated with detrimental effects on the heart and the bones. A TSH value of <0.1 mU/l has been identified as a risk factor for the development of atrial fibrillation 36 . Long-term levothyroxine therapy in TSH-suppressive doses may cause left ventricular hypertrophy 37 and increases the risk of ischemic heart disease in patients under the age of 65 years 38 . TSH-suppressive doses of levothyroxine have been associated with bone loss in some but not all studies. A recent extensive meta-analysis concluded that indeed bone mineral density was reduced in hypothyroid patients with a suppressed TSH due to excessive levothyroxine therapy, but only in postmenopausal women 39 . No or a minimal excess of bone fractures, however, has been observed in patients on levothyroxine even if TSH is suppressed 40,41,42,43,,44 A recent population-based study of all patients in Tayside, Scotland taking L-T4 replacement therapy (n = 17,684) considered fatal and nonfatal endpoints for cardiovascular disease, dysrhythmias, and fractures (45). Patients were categorized as having a suppressed TSH (≤ 0.03 mU/L), low TSH (0.04-0.4 mU/L), normal TSH (0.4-4.0 mU/L), or raised TSH (>4.0 mU/L). Cardiovascular disease, dysrhythmias, and fractures were increased in patients with a high TSH (adjusted hazards ratio 1.95, 1.80, and 1.83 respectively) and in patients with a suppressed TSH (1.37, 1.6, and 2.02 respectively) when compared to patients with a TSH in the laboratory reference range. Patients with a low TSH did not have an increased risk of any of these outcomes. Thus it may be safe for patients treated with T4 to have a low but not suppressed serum TSH.
9.8.3 SITUATIONS REQUIRING DOSE ADJUSTMENT
Table 9-13 lists a number of circumstances in which dosage requirements of levothyroxine may change in compliant patients.
Nonspecific absorption of L-T4 by dietary fibers decreases the bioavailability of T4 and necessitates a higher dose of L-T4 in patients with high intake of dietary fiber (whole-wheat bread, granola, bran) (1). A similar phenomenon may operate with the use of soy protein supplement (2).The timing of the L-T4 dose should be adjusted to take this into account. The association between dietary fiber and levothyroxine malabsorption could not be reproduced in healthy volunteers taking calcium polycarbophil or psyllium (3). Patients with impaired gastric acid secretion require a 22% to 34% higher than usual dose of L-T4 to suppress serum TSH (4), suggesting that normal gastric acid secretion is necessary for effective absorption of L-T4. Most likely this is due to suboptimal dissolution of L-T4 tablets in an environment with higher pH than usual (see section 9.8.1). The L-T4 requirement in autoimmune hypothyroidism is about 18% higher in parietal cell antibodies (PCA)-postive patients than in PCA-negative patients, and a significant positive correlation was found between L-T4 requirement and serum PCA levels (5). In patients with multinodular goiter on suppressive therapy, initiation of the proton-pump inhibitor omeprazole required a 37% increase in L-T4 dose to suppress TSH (4). Likewise, in patients with primary hypothyroidism commencing lansoprazole treatment, 19% of patients required dosage increases in levothyroxine (6). However, pharmacokinetic studies in healthy volunteers who were given L-T4 alone or L-T4 together with proton-pump inhibitors, did not observe significant differences in T4 absorption (7,8). The discrepancy between studies may be caused by differences in the duration of proton-pump treatment: one week in the healthy volunteers (7,8) but up to 6 months in the patients (6). Patients who develop clinical malabsorptive disorders like coeliac disease may require an increase in L-T4 dosage 9,10 . In view of the frequent occurrence of coeliac disease in patients with autoimmune thyroid disease, many authors suggest screening for coeliac disease with anti-gliadin antibodies in patients with hypothyroidism who require higher than expected doses of L-T4. Malabsorption may also occur in patients with short bowel syndrome 11-14 , and in particular cases of lactose intolerance (15) or chronic intestinal Giardiasis (16). Reduced drug absorption requiring higher doses pf levothyroxine may occur after bariatric surgery: it is observed rather frequently after jejunoileal bypass, less often after gastric bypass/gastroplasty, and rarely after biliopancreatic diversion (78.79). Oral liquid levothyroxine formula may be better absorbed compared to levothyroxine tablets following bariatric surgery (80). Interestingly, levothyroxine pharmacokinetics may improve after bariatric surgey: it occurred after sleeve gastrectomy and biliopancreatic diversion with long limps but not after Roux-en-Y gastric bypass (81). Bile acid sequestering agents bind –at least in vitro- large amounts of levothyroxine, and also interfere with the entero-hepatic circulation of thyroid hormone in which T4 and T3 conjugates are excreted in bile and partially deconjugated in the intestine with the release of small amounts of T4 and T3 for reabsorption. Treatment with colestipol or colestyramine may cause a slight increase in TSH in levothyroxine-treated patients, but not in normal subjects (17,18,19). The newer bile acid sequestrant colesevelam reduced absorption of levothyroxine by 96% in healthy subjects (20). An interval of at least 4-5 hours separating thyroxine and bile acid sequestrants is recommended to attain near-normal absorption of L-T4 (21). There are a number of other agents that bind L-T4 and thereby decrease the absorption of L-T4. As a result, serum TSH increases in some but not all levothyroxine-treated patients. The effect of these drug interactions is in general modest, and can be avoided largely by taking L-T4 and the other drug several hours apart (21); sometimes the L-T4 dose has to be increased. Sucralfate binds L-T4 in vitro (22), decreases absorption of L-T4 in healthy volunteers, and may cause resistant hypothyroidism in particular cases (23) but overall its interaction with levothyroxine in L-T4 treated patients seems to be limited (24). Aluminium may complex with levothyroxine, and a dose-related adsorption of levothyroxine with aluminium hydroxide has been demonstrated in vitro (25,26). Aluminium hydroxide treatment increases serum TSH to elevated levels in L-T4 replaced patients (25,26,27).Ferrous sulphate increases serum TSH to slightly elevated levels in hypothyroid patients on stable L-T4 replacement 28,29,30 . The basis of this interaction may be the formation of an insoluble complex by binding of Fe3+ to three T4 molecules. Calcium carbonate adsorbs L-T4 significantly in vitro (31), and it decreases the bioavailability of L-T4 in healthy volunteers (32). Calcium carbonate therapy in L-T4 replaced patients increases serum TSH, sometimes to above the normal range (31,33). Calcium carbonate is also used as a phosphate binder in chronic renal failure. Other phosphate binders may also adsorb L-T4, and both sevelamer and lanthanum carbonate reduce bioavailability of L-T4 in healthy subjects (34,35). The use of sevelamer in hemodialysis patients on levothyroxine is associated with significant increases of L-T4 dose after 6 months of therapy (36). Raloxifene and orlistat have also been reported to interfere with absorption of levothyroxine, but additional studies are required to confirm these observations 21,37,38,39 .
Estrogens increase the serum concentration of thyroxine-binding-globulin (TBG) through increased sialylation of TBG thereby slowing its clearance from the circulation by the liver (40). The effect of estrogens on TBG is dependent on the route of administration, the dose and the chemical structure of the estrogen (41). Transdermal administration of estradiol causes minimal changes in TBG, in contrast to oral estradiol which raises serum TBG by 50-70% (41). The contrast is due to high estrogen levels in the portal vessels and first-pass metabolism in the liver after oral administration. With regard to chemical structure of estrogens, ethinylestradiol in view of its limited liver metabolism causes a rise of serum TBG when administered either orally or transvaginally (42,43). Subjects with normal thyroid glands adapt quickly to the estrogen-induced changes in TBG, and reach a new steady state with no changes in serum FT4 and TSH (41,44). In contrast, estrogen therapy in hypothyroid subjects on stable levothyroxine replacement causes a decrease in serum FT4 and an increase in serum TSH; the effect is dose-dependent and is usually seen within 6 weeks after initiation of estrogens and reaches its peak at 12 weeks (45). Thyroid function tests therefore should be obtained 8-12 weeks after starting estrogen use, and the L-T4 dose adjusted accordingly. Selective estrogen receptor modifiers (SERM) may have similar effects: tamoxifen increases serum TBG by 24% and droloxifene by 41% (46,47). One would expect an increase in required L-T4 dose in hypothyroid patients initiating SERM therapy, but so far this has not been documented. An interesting case report describes the effect of loss of estrogens in a hypothyroid premenopausal women on levothyroxine: chemical castration by the GnRH analogue Goserelin caused an increase of serum FT4 and a decrease of serum TSH (48).
There is an increase in thyroxine requirement in pregnant patients with primary hypothyroidism, probably related to increased lean body mass and increased serum TBG 49,50 . In a review of four series comprising a total of 108 women, serum TSH increased in 58% and the mean L-T4 dose increased from 117 µg to 150 µg 51 . In a prospective study among women with hypothyroidism who were planning pregnancy, an increase in the L-T4 dose was necessary during 17 out of 20 pregnancies; the mean L-T4 requirement increased 47% during the first half of pregnancy (median onset of increase at gestational week 8) and plateaued by week 16, and the increased dose was required until delivery (52). The authors recommended that women should increase their L-T4 dose by the equivalent of two daily doses each week as soon as pregnancy is confirmed. Alternatively, the daily L-T4 dose might be increased by 25 to 50 μg,with thyroid function tests six weeks later (53). A recent study randomized 48 women with treated hypothyroidism seeking pregnancy in two groups: to increase L-T4 by either two tablets per week or three tablets per week (54). TSH suppression below 0.5 mU/L occurred less often at two tablets per week increase than at three tablets per week increase. A two-tablet per week increase in L-T4 initiated at confirmation of pregnancy significantly reduced the risk of maternal hypothyroidism and mimicked normal physiology. Monitoring TSH every four weeks through midgestation and less often thereafter was recommended. The L-T4 dose may be reinstituted at its pregestational level immediately after delivery. Timely adjustment of the thyroxine dose in early gestation might be relevant for infant development. Children at the age of 7-9 years have a lower intelligence quotient if their mother was hypothyroid (elevated TSH) during pregnancy (55).But also children of healthy women with normal TSH but FT4 levels below the 10th percentile (<10.4 pmol/l) at 12 weeks gestation have lower scores on a psychomotor developmental scale at 10 months of age, compared to children of mothers with higher FT4 values 56 ; psychomotor development was not related to maternal FT4 at 32 weeks gestation. A recent population-based study seems to confirm these findings: in pregnant women with normal TSH levels at 13 week gestation (> 2.5 mU/L), maternal hypothyroxinemia (FT4 below the 10 th percentile of 11.7 pmol/l) was associated with cognitive delay of children at 18 and 30 months of age 57 . The findings support the argument of Morreale de Escobar et al that not elevated maternal prenatal TSH levels but maternal hypothyroxinemia (low FT4 levels) is the principal factor leading to poor development of children 58 . For, the fetal brain is dependent on local deiodination of maternal T4 into T3 until the end of the first trimester when the hypothalamus-pituitary-thyroid axis of the fetus becomes functional. The data raise the issue of screening pregnant women for thyroid function disorders in the first trimester.
Several antiepileptic and tuberculostatic drugs induce mixed function oxygenases responsible for hepatic drug oxidation, which accelerates thyroxine clearance via pathways that do not lead to T3 production 59,60,61,62 . Under these circumstances, L-T4 dosage must be increased to compensate for this. Lastly, scattered reports indicate increased L-T4 requirements during treatment with amiodarone, sertraline or chloroquine 63,64,65 . The responsible mechanism is not well understood, although in the case of amiodarone it might have to do with inhibition of T4 transport and T4 deiodination into T3. A more recent study among hypothyroid patients on levothyroxine replacement, however, did not reveal changes in thyroid function tests upon randomization for treatment with sertraline or fluoxetine (both selective serotonin reuptake inhibitors used fro major depression) (83). Tyrosine kinase inhibitors can alter thyroid hormone regulation by mechanisms that apparently are specific to each molecule. Regular assessment of thyroid function is recommended before and during treatment with tyrosine kinase inhibitors (66).Sunitinib induces frequently hypothyroidism in euthyroid patients, but no data are available in levothyroxine-replaced hypothyroid subjects. Imatinib and motesanib do not affect serum TSH in euthyroid subjects with an intact thyroid gland. Imatinib in L-T4 treated hypothyroid patients increases serum TSH in all patients (towards five times the upper normal limit), and FT4 values are reduced by about 60% but remain within the normal range (67). The required L-T4 dose increased by 210% (68). The effect appears rapidly after initiation of therapy and is reversible, since TSH normalized after discontinuation of imatinib. Whereas changes in TBG or in deiodination are not observed, it has been hypothesized that these effects of imatinib are due to stimulation of T4 and T3 clearance by the induction of uridine diphosphate-glucuronosyltransferases. Motesanib in L-T4 treated hypothyroid patients is associated with TSH concentrations ten times higher than baseline on at least one occasion in 50% of patients. Hypothyroidism or TSH above the upper normal limit occurred in 22% of differentiated thyroid cancer and in 61% of medullary thyroid cancer, frequently necessitating a higher replacement dose of L-T4 (69,70). Sorafenib induces hypothyroidism in euthroid patients with intact thyroid glands in 18%. In L-T4 replaced hypothyroid patients sorafenib is associated with an increased TSH in 33% of patients (71). In another study sorafenib decreased serum FT4 and T3 by 11% and 18% respectively, whereas TSH levels increased (requiring a slight increase of L-T4 dose of 10%). The ratio’s of serum T3 to T4 and of T3 to reverse T3 decreased by 18% and 22% respectively, compatible with increased type 3 deiodination (72).In vitro studies demonstrate that several tyrosine kinase inhibitors inhibit cellular uptake of T3 and T4 mediated by the MCT8 thyroid hormone transporter; such a mechanism could also be operative in vivo (83)..
There are fewer conditions in which L-T4 dose requirements decrease. When discussing the effects of androgens on thyroid function, one should consider that synthetic androgens have variable degrees of aromatisation to estrogens in adipose tissue and liver (41). The 17β testosterone esters (given intramuscularly) still undergo significant hepatic metabolism. The 17α alkylated derivatives (given orally) are generally resistant to hepatic metabolism. In euthyroid subjects with an intact thyroid gland, administration of testosterone esters is associated with a decrease in TBG with 14% (44), whereas a nonaromatisable androgen caused a decrease in TBG with 50% (73); serum FT4 and TSH do not change. In hypothyroid patients on a stable dose of L-T4, fluoxymesterone (a non-aromatisable androgen used in breast cancer) caused a dramatic decrease in serum TBG associated with a rise in serum FT4 and a fall in TSH within 4 weeks after initiating therapy; the L-T4 dose had to be decreased by 25%-50% in order to maintain euthyroidism 73 . In patients over the age of 70, levothyroxine requirements are reduced about 25 percent, related to the decrease of lean body mass with age 74,75.76 . Hypothyroid patients with end-stage renal insufficiency need lower doses of T4 after renal transplantation 77 .
Table 9-13
Conditions requiring adjustment of the replacement dose of thyroxine for hypothyroidism.
9.8.4 INTERFERENCE WITH CO-EXISTENT CONDITIONS
Hypocortisolemia. The co-existence of thyroid hormone deficiency and glucocorticoid deficiency is not rare. Primary hypothyroidism due to chronic autoimmune thyroiditis is associated with primary adrenocortical insufficiency due to autoimmune adrenalitis. The very cause of central hypothyroidism in many instances will also result in ACTH deficiency and secondary adrenocortical insufficiency. If the two entities co-exist, it is important to replace glucocorticoid before starting thyroxine. For, treatment of hypothyroidism in patients with glucocorticoid deficiency may precipitate an adrenal crises because the adrenal is incapable to meet the increasing demand for cortisol induced by the rise of the metabolic rate 1 . Some patients with adrenal insufficiency have slightly elevated TSH levels without serological evidence of chronic autoimmune thyroiditis; TSH normalizes with glucocorticoid replacement therapy (2,3,4). It illustrates the small inhibitory effect of cortisol on TSH secretion (5).
Ischemic heart disease. Although treatment of hypothyroidism with levothyroxine will improve myocardial function and reduce peripheral vascular resistance, it will increase the need for oxygen in the myocardium 6,7,8 . In patients with an already compromised myocardial blood supply due to coronary atherosclerosis, thyroxine treatment may provoke anginal symptoms. In a large series of hypothyroid patients, new-onset angina occurred in 2% upon thyroxine treatment; pre-existent angina worsened in 16%, did not change in 46%, and improved in 38% 9 . Patients with preexisting angina should be evaluated for obstructive coronary lesions before thyroxine therapy begins. Retrospective studies suggest that the possibility of myocardial infarction is greater than is the possibility of an adverse event during angiography or angioplasty (10,11,12,13,14).However, it is quite surprising that major surgery, such as coronary artery bypass grafting, can be very easily withstood by the patient with even moderate hypothyroidism as long as attention is paid to reducing the level of analgesics, maintaining adequate ventilation, and controlling the administration of free water 12 . In a few patients, remediable lesions will not be present or, even with bypass grafting, complete correction of the hypothyroid state will not be possible. In such patients, submaximal amounts of levothyroxine supplemented by other agents to enhance myocardial function may be helpful in allowing the reestablishment of normal thyroid function 14 .
Drugs. The metabolism of many drugs is slowed in hypothyroidism, resulting in higher sensitivity to a loading dose and a lower maintenance dose. Marked respiratory depression can occur after a single small dose of morphine. An increase in the dose of digoxin or insulin is sometimes noticed once euthyroidism has been restored.
Growth hormone deficiency. A decrease in serum FT4 and an increase in T3 has been reported following growth hormone (GH) administration with or without a reduction in serum TSH, but literature data are not consistent about these changes (15). The significance of these changes is uncertain, although one study reports a good correlation between changes in serum T3 and resting energy expenditure and cardiac isovolumetric contraction time upon GH treatment (16). The changes in thyroid function can be transient and may revert to normal after a few months. However, in adult hypopituitary patients, GH replacement has been reported to unmask central hypothyroidism in 36%-47% of apparently euthyroid patients, necessitating thyroxine replacement (15,17). At highest risk are patients with organic pituitary disease or multiple pituitary hormone deficiencies. It is therefore prudent to monitor thyroid function in hypopituitary patients starting GH therapy.
Chronic renal failure. Subclinical hypothyroidism is a relatively common condition (approximately 18%) among patients with chronic kidney disease not requiring chronic dialysis, and it is independently associated with progressively lower glomerular filtration rates in unselected oupatient adults (18).
9.9 MYXEDEMA COMA
Definition and pathogenesis.
Myxedema coma is a rare, life-threatening clinical condition in patients with long-standing severe untreated hypothyroidism in whom adaptive mechanisms fail to maintain homeostasis. Most patients, however, are not comatose, and the entity rather represents a form of decompensated hypothyroidism 1 ,-5 . Usually a precipitating event disrupts homeostasis which is maintained in hypothyroid patients by a number of neurovascular adaptations. These adaptations include chronic peripheral vasoconstriction, diastolic hypertension and diminished blood volume; in this way a normal body core temperature is preserved. The hypothyroid heart also compensates by performing more work at a given amount of oxygen by better coupling of ATP to contractile events. In severely hypothyroid patients homeostasis might no longer be maintained if blood volume is reduced any further (e.g. by gastrointestinal bleeding or the use of diuretics), if respiration already compromised by a reduced ventilatory drive is further hampered by intercurrent pulmonary infection, or if CNS regulatory mechanisms are impaired by stroke, the use of sedatives or hyponatremia 2 .
Diagnosis .
The three key features of myxedema coma are 1 : 1. Altered mental status. The patient may be entirely obtruded or may be roused by stimuli. Usually lethargy and sleepiness have been present for many months. Sleep may have occupied 20 hours or more of the day and may have interfered even with eating. There may actually have been transient episodes of coma at home before a more complete variety developed.
2. Defective thermoregulation: hypothermia, or the absence of fever despite infectious disease. Usually coma comes on during the winter months. The severely myxedematous patient becomes essentially poikilothermic. With cold weather the body temperature may drop sharply. The temperature is subnormal, often much depressed: a temperature of 74 F (23º3 C) has been recorded. A thermometer reading lower than the usual 97 F must be used, or hypothermia may be missed.
3. Precipitating event: cold exposure, infection, drugs (diuretics, tranquillizers, sedatives, analgetics), trauma, stroke, heart failure, gastrointestinal bleeding.
Diagnosis on clinical grounds is relatively easy once the possibility is considered. Previous hypothyroidism had been diagnosed in 39% to 61% of all cases (6,7).The pulse is slow, and the absence of mild diastolic hypertension is a warning sign of impending myxedema coma 1 . Any patient with hypothermia and obtundation should be considered as having potential myxedema coma, especially if chronic renal insufficiency and hypoglycemia can be ruled out. The diagnosis can be confirmed by finding a reduced FT4 and marked elevation of serum TSH. However, TSH will not be elevated in myxedema coma due to central hypothyroidism (4% to 18% of cases (6,7,8). Sometimes serum TSH is just slightly elevated, possibly related to co-existent nonthyroidal illness 19 . Creatine phosphokinase is often elevated. Both hypoxia (80%)and hypercapnia (54%) may be present (7) . Hypothermia with a temperature less than 94ºF (34º4C) is seen in 88%.
Treatment. Myxedema coma is a medical emergency. Early diagnosis, rapid administration of thyroid hormones and adequate supportive measures (Table 9-14) are essential for the prognosis.
Table 9-14
Recommendations for the treatment of myxedema coma.
In view of the rarity of myxedema coma, it has been difficult to perform randomized studies to resolve the issue of whether T4 or T3 is the most appropriate treatment. There are advocates of T4 therapy alone 9 . T3 therapy alone 10,11 , and combinations thereof 12 . Differences in opinion about the optimal treatment are caused by a/ the lack of RCTs, b/ the precarious balance between the need to attain effective thyroid hormone levels in target tissues as fast as possible and the risk of precipitating fatal tachycardia or myocardial infarction, and c/ the impairment of T4 into T3 conversion associated with severe illness and inadequate caloric intake, which favours T3 therapy over T4. If T4 alone is used, it should be given parenterally in doses of 300 to 500 µg to replace the calculated T4 deficit 13 . Since the average volume of distribution of T4 in a 70-kg human is approximately 7 L, 420 µg should cause an increase of 77 nM/L in the serum T4 concentration.After this initial ‘loading’ dose, a maintenance L-T4 dose of 75-100 µg/day is given intravenously or orally if the patient is alert. Serum T4 with this schedule usually increase into the normal range within 24 hours, and an increase of serum T3 can also be observed (14). Initial L-T4 doses larger than 500 μg have no advantage and are associated with higher mortality (15). Mortality was 17% in patients selected at random to receive 500 μg T4 intavenously as bolus followed by 100 μg T4 daily, but 60% in patients treated with 100 μg T4 daily (8). However, the difference in mortality between both groups was not significant likely due to small sample size (n=11). If T3 alone is used, it may be given as a 10-20 μg intavenous bolus followed by 10 μg every 4 h for the first 24 h, and 10 μg every 6 h for days 2 and 3 (4).L-T3 doses larger than 75 μg per day are associated with higher mortality (15).The patient should be switched to oral therapy when possible. There has been one case report of a patient with myxedema-associated cardiogenic shock who did respond to T3 but not to T4 treatment (16) but exposing tissues to very high doses of T3 is not without risk. If T4 in combination with T3 is used, 200 to 300 µg of T4 and 10 to 25 µg T3 are given intravenously as an initial dose. After 24 h 100 μg T4 is given intravenously, followed by 50 μg T4 daily from the third day until the patient regains consciousness. Intravenous T3 is continued at a dose of 10 μg every 8-12 h until the patients is conscious and can take T4 maintenance dose 4,12 .
Intravenous glucocorticoid should also be administered during the first days of therapy, since in severe hypothyroidism pituitary-adrenal function is impaired, and the cortisol production rate is lower. While this low production is adequate when cortisol metabolism is reduced, as it is in hypothyroidism, the rapid restoration of a normal metabolic rate from the above treatment may precipitate transient adrenal insufficiency. In addition, the patient should be intubated and measures taken to retain body heat. Central warming may be attempted but peripheral warming should not, since it may lead to vasodilatation and shock. The cutaneous blood flow is markedly reduced in the hypothyroid patient in order to conserve body heat. Warming blankets will defeat this mechanism. Mechanical ventilation may be needed, particularly when obesity and myxedema are combined.Hyponatremia is characteristic and free water restriction and the use of isotonic sodium chloride will usually restore normal serum sodium, as will improved cardiovascular function, which is one cause of the impaired free water clearance.The new vasopressin antagonist conivaptan might be useful in treating hyponatremia as high vasopressin levels have been observed in myxedema coma, but so far no case report has been published in which this drug had been administered Serum glucose should be monitored. Supplemental glucose may be necessary, especially if adrenal insufficiency is present. Hypotension may develop, particularly if myxedema is severe. Volume expansion is usually required to remedy this, since patients are usually maximally vasoconstricted. Dopamine should be added if fluid therapy does not restore efficient circulation. Concomitantly, a vigorous search for precipitating factors should be instituted. Determining whether an infection is present should be a priority, since as many as 35 percent of patients with myxedema coma have infection. Since hypothyroid patients cannot mount an adequate temperature response, the usual signs of infection, including tachycardia, fever, and elevated white blood count, may be absent. Prophylactic antibiotics are indicated until infection can be ruled out; upper respiratory infection should be eliminated. While the hypothyroid patient withstands the stress of surgery in general very well 17 , inadvertently excessive narcotics, sedatives, and hypnotics can tip a severely hypothyroid patient into coma. History taking from family members can be very rewarding in detecting predisposing events. This is illustrated by a case report describing myxedema coma in an elderly woman who had been eating excessive amounts of raw bok choy (Chinese white cabbage) daily for several months in the belief that it would help control her diabetes; the goitrogenic action of compounds like thiocyanates and oxazolidines (generated by eating raw cabbage) was identified as the cause of her coma 20 .
Prognosis .
Most patients begin to show increases in body temperature within the first 24 hours of treatment. The absence of an increase in body temperature within 48 hours should lead to consideration of more aggressive therapy, specifically T3 therapy if it has not already been initiated. Most patients regain consciousness within a few days. Mortality is between 25% and 52%, and sepsis is the predominant cause of death (6,7). Predictors of mortality include hypotension and bradycardia at presentation, need for mechanical ventilation, hypothermia unresponsive to treatment, sepsis, intake of sedative drugs, lower Glasgow Coma Scale, high APACHE II score and high SOFA score (6). Clinical suspicion, early recognition, prompt thyroid hormone replacement, and appropriate support cares remain the key to successful tretament of this rare but often fatal emergency (18).
9.10 SUBCLINICAL HYPOTHYROIDISM
9.10.1 DIAGNOSIS AND ETIOLOGY
Subclinical hypothyroidism is defined as an increased serum TSH in the presence of a normal serum FT4 concentration. Increased refers to values above and normal to values within population-based reference ranges of these hormones. It is however not so simple to diagnose accurately subclinical hypothyroidism in day-to-day practice applying this biochemical definition. Diagnosis of subclinical hypothyroidism is hampered by uncertainty about what constitutes appropriate reference intervals, and by biologic variation in especially TSH. The upper limit of the TSH reference interval was 4.12 mU/L in the National Health and Nutrition Eamination Survey III (NHANES III) for a large reference population that was free of thyroid disease and representative of the U.S. population, in which subjects were excluded who had thyroid antibodies or were taking thyroid medications or other medications affecting thyroid measurements (1). Distribution curves of TSH are skewed to higher TSH concentrations, and under the assumption that this represents undetected thyroid disease the National Academy of Clinical Biochemistry suggested the upper normal limit of TSH should be 2.5 mU/L (2). This proposal has been very controversial, also because it would label as abnormal 10% to 20% of individuals of all ages and 35% of people older than 70 years (3,4,5). More recent population-based studies in which subjects were also excluded in case of abnormal thyroid ultrasonography, observed 97.5 th percentiles of TSH of 3.77 in Germany (6) and 4.1 mU/L in the USA (7), close to the original NHANES III value of 4.12 mU/L. The explanation for the skew in TSH distribution curves toward higher serum TSH is most likely that the upper normal limit of TSH increases with advanced age (8). When NHANES III data were reanalyzed by TSH distribution curves for specific age deciles, a progressive shift in the curves to higher TSH with age was observed, rather than a skew to higher values. E.g. the upper normal limit of TSH is 7.5 mU/L in subjects older than 80 years; 70% of subjects older than 80 years would have been labeled as having raised TSH when an upper normal limit of 4.5 mU/L is used. Age-specific reference ranges are thus recommended. Besides conceptual problems with reference ranges, the diagnosis of subclinical hypothyroidism is jeopardized by considerable biologic variation in TSH values. A particular study enrolled 21 patients with subclinical hypothyroidism (identified with serum TSH between 5 and 12 mU/L and normal T4, confirmed on two occasions 3 months apart) without former thyroid disease, who underwent monthly repeated measurements without intervention (9). In the one-year follow-upp period, one patient appeared to be euthyroid at all visits, and one patient developed profound overt hypothyroidism and was treated.The remaining patients had subclinical hypothyroidism at 74% of the visits, overt hypothyroidism at 22% and normal thyroid function tests at 4% of the visits. Diagnosis of overt hypothyroidism was highly dependent on T4 reference limits. In individual patients serum TSH was correlated to both TPO antibodies and to urinary iodine excretion, but not to hypothyroid symptoms and signs (10). The study shows how TSH and FT4 vary around the outer limits of their reference ranges, how limited the information is obtained from a single set of thyroid function tests, and how biologic variation may change the diagnosis from visit to visit. It is therefore recommended, especially If a slightly increased serum TSH is found, to take a second blood sample after 3-6 months in order to ascertain the diagnosis of subclinical hypothyroidism. If a normal TSH is found in the second blood sample, this can rarely be attributed to ultradian and circadian TSH rhythms (the relative risk of misjudging mean TSH serum levels by a single TSH determination between 07.00 and 17.00 hours is only 0.09% for values above 4.0 mU/l) 11 .
It is highly relevant after the biochemical diagnosis to establish a nosologic diagnosis to evaluate which condition is responsible for the elevated TSH. The llist of possible causes is very long (21). The most common causes of subclinical hypothyroidism are chronic autoimmune thyroiditis (Hashimoto’s disease), previous 131I therapy or thyroidectomy, and inappropriate dosage of thyroxine or antithyroid drugs. Loss-of-function mutations in the gene encoding for the TSH receptor are relatively common in isolated hyperthyrotropinemia, especially in children and adolescents (12,13). Other causes of an elevated TSH are interference of heterophilic TSH antibodies in TSH immunoassys, nonthyroidal illness syndrome (recovery phase), impaired renal function, untreated adrenal insufficiency (Addison’s disease) and obesity. Strictly speaking, the latter group should not be labeled as subclinical hypothyroidism because thyroid disease is absent and management is directed to the nonthytoidal cause. The case of obesity is illustrative in this respect (14). In patients with morbid obesity (BMI range of 30-67 kg/m²) TSH levels correlate positively with BMI (r=0.91), and the mean BMI change from 49 to 32 kg/m² after bariatric surgery is associated with a reduction in mean TSH levels from 4.5 to 1.9 mU/L; FT4 levels are not associated with BMI, and subclinical hypothyroidism observed in 10.5% disappears after weight reduction (15). Thyroid autoimmunity is not a major cause sustaining the high rate of an elevated TSH in morbid obesity (16). Thus it is important to establish if subclinical hypothyroidism is caused by an underlying thyroid disease. Apart from history and physical examination, this can be done easily by measuring TPO antibodies in serum.
It is highly relevant after the biochemical diagnosis to establish a nosologic diagnosis to evaluate which condition is responsible for the elevated TSH. The llist of possible causes is very long (21). The most common causes of subclinical hypothyroidism are chronic autoimmune thyroiditis (Hashimoto’s disease), previous 131I therapy or thyroidectomy, and inappropriate dosage of thyroxine or antithyroid drugs. Loss-of-function mutations in the gene encoding for the TSH receptor are relatively common in isolated hyperthyrotropinemia, especially in children and adolescents (12,13). Other causes of an elevated TSH are interference of heterophilic TSH antibodies in TSH immunoassys, nonthyroidal illness syndrome (recovery phase), impaired renal function, untreated adrenal insufficiency (Addison’s disease) and obesity. Strictly speaking, the latter group should not be labeled as subclinical hypothyroidism because thyroid disease is absent and management is directed to the nonthytoidal cause. The case of obesity is illustrative in this respect (14). In patients with morbid obesity (BMI range of 30-67 kg/m²) TSH levels correlate positively with BMI (r=0.91), and the mean BMI change from 49 to 32 kg/m² after bariatric surgery is associated with a reduction in mean TSH levels from 4.5 to 1.9 mU/L; FT4 levels are not associated with BMI, and subclinical hypothyroidism observed in 10.5% disappears after weight reduction (15). Thyroid autoimmunity is not a major cause sustaining the high rate of an elevated TSH in morbid obesity (16). Thus it is important to establish if subclinical hypothyroidism is caused by an underlying thyroid disease. Apart from history and physical examination, this can be done easily by measuring TPO antibodies in serum.
The prevalence of subclinical hypothyroidism in the general population is rather high in the order of 4% to 8%; it is higher in iodine-replete areas than in iodine-deficient areas (17,18) (see also section 9.2). In the classical population-based study among adults in the English county of Whickham the prevalence was 75 per 1000 women and 28 per 1000 men 19 . About 75% have TSH values between 5 and 10 mU/L, and 25% have TSH values greater than 10 mU/L (20). The higher prevalence of subclinical hypothyroidism in females than in males and in older than in younger subjects is in agreement with the higher prevalence of thyroglobulin and thyroid peroxidase (microsomal) antibodies in women and in elderly people.
9.10.2 NATURAL HISTORY
The natural history of subclinical hypothyroidism is reported in many studies, although it remains difficult to predict whether the increased TSH levels will return spontaneously to within the normal range, will remain stable, or will increase to higher values with development of overt hypothyroidism. In general it can be said that the higher the initial TSH, the higher the risk of progression; the presence of TPO antibodies potentiates the risk. Spontaneous normalization of increased TSH values in subclinical hypothyroidism is a well-known phenomenon, but the reported frequency of normalization differs markedly between studies from 4% up to 52% 1-10 . Reasons for the wide variation in normalization of TSH are differences in duration of follow-up, heterogeneity of study populations, and possibly age. In a prospective observational study normalization of TSH occurred at a median time of 18 months (range 6-60 months) (11). Some studies involve homogeneous populations (e.g.only subjects with proven autoimmune thyroiditis), whereas others do not specify the cause of subclinical hypothyroidism and consequently may harbour many subjects without underlying thyroid disease. E.g. studies in children and adolescents with subclinical hypothyroidism report stable, normalized or increasing TSH values in 31%, 31% and 38% respectively over a mean follow-up period of 41 months when all participants had Hashimoto’s thyroiditis (12), and 47%, 41% and 12% over a mean follow-up period of 24 months when all participants had idiopathic subclinical hypothyroidism (13).Normalization of TSH is more frequent in subjects with moderate TSH elevation up to 10 mU/L, with or without thyroid autoimmunity. Normalization of TSH might also be more common in old age. In a study of 107 subjects (mostly with autoimmune thyroiditis) with a mean age of 62 yr, 37% normalized TSH at a mean follow-up of 2.7 yr (9,11). In contrast, in a study of 21 subjects (with unspecified cause of elevated TSH) with a mean age of 85 yr, 52% normalized TSH at a mean follow-up of 3 yr (10).
Progression to overt hypothyroidism ranges from 7.8% to 17.8% in various studies 2,4,5 . According to the initial serum TSH concentrations (TSH 4-6, >6-12, >12 mU/L, Kaplan-Meier estimates of the incidence of overt hypothyroidism in subclinically hypothyroid women were 0%, 42.8%, and 76.9% respectively after 10 years (or 0%, 3%, and 11% respectively per year) 8 . The incidence of overt hypothyroidism was higher in patients with TPO antibodies ((58.5% vs 23.2%). The importance of thyroid antibodies is also evident from a Dutch study: 9.6% of 55-year old women with TPO antibodies had raised TSH levels 10 years later, in contrast to 3.2% of women without antibodies 14 . The most extensive data are from a 20-year follow-up in the participants of the Whickham survey 15 . The incidence of overt hypothyroidism was 4.1 per 1000 women per year and 0.6 per 1000 men per year. Odds ratio’s (with 95% CI) for development of spontaneous hypothyroidism in surviving women are 14 (9-24) for raised TSH regardless of thyroid antibody status, 13 (8-19) for positive thyroid antibodies regardless of TSH, and 38 (22-65) for raised TSH and positive thyroid antibodies combined. Odds ratio’s for men are higher: 44 (19-104) for raised TSH regardless of thyroid antibody status, 25 (10-63) for positive thyroid antibodies regardless of TSH, and 173 (81-370) for raised TSH and positive thyroid antibody status combined. Most interestingly, the risk for developing hypothyroidism in the Whickham survey already starts at TSH levels of 2.0 mU/L (see also section 9.2). Similar cutoff values of 2.5 mU/L for predicting hypothyroidism have been reported in a 5-yr follow-up study among Dutch healthy female relatives of patients with autoimmune thyroid disease (16), and in an Australian population-based study with a 13-yr follow-up (17). A hypoechographic pattern on thyroid ultrasound also increases the risk of progression, even in the absence of TPO antibodies in serum (18). In subjects ≥65 yr, persistence of subclinical hypothyroidism, after 2 and 4 yr was 56%; resolution of elevated TSH was more common with a TSH 4.5-6.9 mU/L (46% vs 10% with TSH 7-9.9 mU/L and 7% with TSH ≥10 mU/L) and with TPO-Ab negativity (48% vs 15% for positive TPO-Ab) (19). TSH ≥10 mU/L was independently associated with progression to overt hypothyroidism. Transitions between euthyroidism and subclinical hypothyroidism were more common between 2 and 4 yr; age and sex did not affect transitions. Taking a high-dose phytoestrogen dietary supplementation (30 g soy protein with 16 mg phytoestrogens, representative of a vegetarian diet) for 8 weeks increases 3-fold the risk in subjects with subclinical hypothyroidism to develop overt hypothyroidism (20).
9.10.3 SYSTEMIC MANIFESTATIONS
A multitude of papers have been published on alterations in subclinical hypothyroidism as compared to euthyroid subjects. The vast literature on this topic from 1990 through April 2007 has been nicely reviewed by Biondi and Cooper (1) and updated by Cooper and Biondi in 2012 62 . The abnormalities listed in Table 9-15 , have been reported in some but not all studies on subclinical hypothyroidism. The described abnormalities are in general minor, and more frequent in subjects with the highest TSH values 2 .
Effect on symptoms, quality-of-life (QoL), and cognitive function. Hypothyroid symptoms occured more often than in controls in some small studies and in the Colorado study 3,4,,5 , but not in a large population-based study 6 . A study among healthy females with a family history of thyroid disease, recruited by advertisement, indicated a higher lifetime frequency of depression in subjects with subclinical hypothyroidism (56%) than in euthyroid subjects (20%) 7 . But again large population-based studies did not reveal lower well-being, impaired QoL, or more depression and anxiety 6,8-12 . Impaired memory function has been reported in subclinical hypothyroidsm in some early small series of patients (13,14), but more recent large population-based studies have not corroborated this observation also not in the elderly: cognitive functions were not different from controls (6,8,9,10,12). In contrast, a study using functional MRI suggested that working memory (but not other memory functions) is impaired by subclinical hypothyroidism (15). Its findings are apparently confirmed by another small study showing that cognitive impairment in subclinical hypothyroidism appears predominantly mnemonic in nature, suggesting that the etiology is not indicative of general cognitive slowing (16).Subclinical hypothyroidism seems to be less symptomatic in the elderly (8,63,64). Subjects with subclinical hypothyroidism in their 8 th decade of life even had increased walking speed and retention of physical function as compared with their peers who were euthyroid (55).
Table 9-15
Abnormalities reported in some but not all studies on subclinical hypothyroidism.
Cardiac effects . The many cardiac effects are reviewed by Biondi and Cooper (1,62). Impaired left ventricular diastolic function at rest has been clearly demonstrated in subclinically hypothyroid subjects by Doppler echocardiography and radionuclide ventriculography: isovolumetric relaxation time is prolonged and time-to-peak filling rate is impaired as compared to controls. Left ventricular systolic function at rest was reported as normal, but using the more sensitive Doppler echocardiography seems to be impaired as documented by an increased preejection period (PEP) to left ventricular ejection time (LVET) ratio. Cardiac MRI, a most accurate procedure to evaluate cardiac volumes and function, demonstrates a significant decrease in preload (end-diastolic volume) and a significant increase in the afterload (systemic vascular resistance), thereby leading to impaired cardiac performance (17). Pulsed wave tissue Doppler imaging allows to measure velocities at any point of the ventricular wall during the cardiac cycle; myocardial time intervals were prolonged at both the posterior septum and the mitral annulus compared to controls (18). Ultrasonic myocardial texture analysis reveals altered myocardial composition, suggesting early myocardial structural changes(19). Recent studies suggest furthermore impaired coronary flow reserve (20,21). In summary, the most consistent cardiac abnormality in suclinical hypothyroidism is impaired left ventricular diastolic function, characterized by slowed myocardial relaxation and impaired ventricular filling. Impaired left ventricular systolic function is not consistently reported but has been identified with more sensitive techniques (1). Cardiac performance is also impaired during exercise (22,23).The cardiac changes in subclinical hypothyroidism are less severe but otherwise similar to those observed in overt hypothyroidism, suggesting a continuum in the cardiac effect of thyroid hormone.
Vascular effects. A higher prevalence of diastolic hypertension in subclinical hypothyroidism as compared to controls has been reported in some but not all studies (1,24,25). Three factors contribute to systemic hypertension in overt hypothyroidism: increased peripheral vascular resistance, increased arterial stiffness, and endothelial dysfunction. The same factors seem to operate in subclinical hypothyroidism. Systemic vascular resistance is increased in some studies but not in others (1,19). As T3 directly affects vascular smooth muscle cells promoting relaxation, subclinical hypothyroidism might affect vascular tone. Increased arterial stiffness has been identified as an independent risk factor for cardiovascular morbidity and mortality. Increased arterial stiffness has been demonstrated in several studies using various techniques like pulse wave velocity (24,26). The vascular endothelium regulates vascular smooth muscle function by diffusion of nitric oxide from the endothelium to the smooth muscle cells, inducing relaxation. Flow-mediated endothelium-dependent vasodilatation is significantly impaired in subclinical hypothyroidism compared to controls (27,28,29). The endothelium dysfunction is attributed to reduced nitric oxide availability (28). Low-grade chronic inflammation could be responsible for impaired nitric oxide availability by a cyclo-oxygenase 2-dependent pathway, increasing oxidative stress in subclinical hypothyroidism due to Hashimoto’s thyroiditis (30). Carotid intima-media thickness is useful in the early diagnosis of atherosclerosis and coronary heart disease. Patients with subclinical hypothyroidism have higher carotid intima-media thickness than age-and sex-matched controls in some but not all studies (29,31,32). The observed vascular changes potentially increase the risk of atherosclerosis and coronary artery disease.
Effects on biochemical tests. Serum lipids (total cholesterol, LDL cholesterol) may be increased in subclinical hypothyroidism, but the existing data in the literature are inconsistent (1). The lipid pattern is more abnormal with serum TSH > 10 mU/L. The Whickham Survey did not observe increased total cholesterol levels in subclinical hypothyroidism versus controls (33), in contrast to other population-based studies like the NHANES III (34) and the Busselton study (34) which did observe higher cholesterol levels in subclinical hypothyroidism; however, in the latter two studies the difference with controls disappeared almost completely after adjustment for age and sex. Other population-based studies report that serum TSH levels > 5.5 mU/L are associated with a rise in serum total cholesterol of 0.23 mmol/l (36), and that an increase of 1 mU/L of serum TSH is associated with an increase of serum cholesterol of 0.09 mmol/l in women and of 0.16 mmol/l in men (37). In older women LDL cholesterol was 13% higher and HDL cholesterol was 12% higher with TSH values > 5.5 mU/L compared to normal TSH values (38). The lipid peroxidation marker malondialdehyde is elevated in subclinical hypothyroidism compared to controls (39). Remnant lipoproteins are more often present in the fasting serum in subclinical hypothyroidism than in controls (40). Serum triglycerides are usually normal. No associations with Lp(a) have been observed. The relationship between TSH and LDL cholesterol may depend on other factors like the presence of insulin resistance (41) and smoking (42). The risk of hypercholesterolemia in subclinical hypothyroidism was restricted to smokers in one study (42). Fasting insulin levels in subclinical hypothyroidism are higher than in controls, and the homeostasis model of assessment (HOMA-IR) index suggests insulin resistance in one study but not in another (43,44). Homocysteine levels are apparently not related to subclinical hypothyroidism (31,45). Fasting insulin correlated positively with hsCRP (highly sensitive C-reactive protein), a strong predictor of cardiovascular risk (43,46).C-reactive protein levels are higher in subclinical hypothyroidism than in controls in some but not all studies (43,46,47).Another study observed higher hsCRP, total and LDL cholesterol, asymmetric dimethylarginine and arginine levels but lower nitric oxide levels in subclinical hypothyroidism than in controls (48). Alterations in coagulation parameters have also been reported (1). Global fibrinolytic capacity was lower in subclinical hypothyroidism than in controls. Subjects with subclinical hypothyroidism identified in a population based study, as compared to age- and sex-matched controls, had no changes in hemostatic factors but their factor VIIa levels were 10% lower (49). In summary, data on a potential association of subclinical hypothyroidism and traditional cardiovascular risk factors (like cholesterol) and nontraditional cardiovascular risk factors (C-reactive protein, coagulation parameters) are not consistent.
Other effects . In the central nervous system an abnormal stapedial reflex but no abnormalities in brainstem auditory evoked potentials has been observed (1,50). Discrete changes in peripheral nerve function are reported: conduction velocities are normal, but motor distal latencies are prolonged and amplitudes are decreased relative to controls 1,51 . Muscle metabolism is impaired: during exercise (but not at rest) blood lactate is higher in subclinical hypothyroidism than in controls, consistent with impaired mitochondrial oxidative function 52,53 . There exists lower exercise tolerance and less muscle strength (54). In contrast, a community-based study in 70-79 year old subjects does not demonstrate increased risk of mobility problems (tested by mean usual and rapid gait speed, cardiorespiratory fitness and walking ease) in subclinical hypothyroidism; in fact, those with TSH levels between 4.5 and 7.0 mU/L showed a slight functional advantage over euthyroid subjects (55). In another age- and sex-adjusted analysis, subclinical hypothyroidism was associated with lower vital capacity at rest and a lower work rate at the ventilator anaerobic threshold (23). Subclinical hypothyroidism is more common in patients with common bile duct stones as compared to nongallstone controls (56), but it was not analyzed if this association is independent of obesity. It has also been suggested that the prevalence of subclinical hypothyroidism is higher in patients with deep venous thrombosis (57). A study in postmenopausal women reports that subclinical hypothyroidsm affects not bone turnover but bone structure in the calcaneus (lower heel QUS) (58). Serum leptin concentrations are higher in subclinically hypothyroid than euthyroid postmenopausal women, even when controlling for body mass index (59). Plasma total and acylated ghrelin concentrations are not significantly changed by subclinical hypothyroidism (60). Fasting serum IGF-1 levels are lower in subclinical hypothyroidism than in controls (61). A prospective cohort study among community-dwelling US subjects ≥65 yr with a follow-up of 13 yr reported an association between endogenous subclinical hypothyroidism and incident hip fracture in men (hazard ratio 2.45, 95% CI 1.27-4.73) but not in women (65).
9.10.4. ASSOCIATIONS WITH CARDIOVASCULAR MORBIDITY AND MORTALITY.
Studies in the early 1970s suggested preclinical hypothyroidism as a risk factor for coronary heart disease, presumably via increased cholesterol levels 1,2 . Since then, many but not all studies have demonstrated a higher prevalence of classical (like hypercholesterolemia) and nonclassical risk factors for cardiovascular disease in subclinical hypothyroidism, as outlined in section 9.10.3. So the question has arisen whether or not subclinical hypothyroidism is associated with a higher prevalence or incidence of cardiovascular disease. To answer this question, a number of epidemiological studies have been performed. Because of inconsistencies in the obtained results of population-based studies, the data has been subjected to meta-analyses.
Population-based studies . We will review both cross-sectional (transversal) and follow-up (longitudinal) population-based studies in chronological order. In 1996, a 20-yr follow-up of the Whickham Survey in the UK among men and women of 18 yr and older, did not find an association between autoimmune thyroid disease at study entry (defined as treated hypothyroidism, positive thyroid antibodies and/or or elevated serum TSH) and subsequent development of ischemic heart disease or increased circulatory or all-cause mortality 3 . In 2000, a 4.5-yr follow-up of the Rotterdam Study among women of 55 yr and older (mean age 69±7.5 years), subclinical hypothyroidism was not associated with an increased incidence of myocardial infarction. However, at study entrance subclinical hypothyroidism was associated with a higher age-adjusted prevalence of aortic atherosclerosis (odds ratio 1.7, 95% CI 1.1 to 2.6) and myocardial infarction (odds ratio 2.3, 95% CI 1.3 to 4.0) 4 . Additional adjustment for body mass index, serum cholesterol, blood pressure, smoking and the use of β-blockers did not affect these estimates. The population attributable risk for subclinical hypothyroidism associated with myocardial infarction was 14%, within the range of that for known major risk factors for cardiovascular disease (hypercholesterolemia 18%, smoking 15%, hypertension 14%, diabetes 14%). In 2001, a 10-yr follow-up study in the UK among men and women of 60 yr and older, found no association of subclinical hypothyroidism and death from circulatory disease, but 40% of subclinically hypothyroid subjects developed overt hypothyroidism and started L-T4 therapy (5). In 2004, a 10-yr follow-up study among atomic bomb survivors from Nagasaki among men and women of 40 yr and older, found that subclinical hypothyroidism was associated with an increased mortality from all causes only in men after 3-6 years, but not after 10 years (6); the cross-sectional analysis at baseline showed an increased risk of ischemic heart disease. In 2004, a 4-yr follow-up study in Leiden among men and women of 85 yr, subclinical hypothyroidism was associated with greater longevity and a decreased risk of death from cardiovascular disease, attributed to a lower metabolic rate (7). In 2005, the 20-yr follow-up in the Busselton study in Western Australia among men and women of 17-89 yr, identified subclinical hypothyroidism as an independent predictor of coronary heart disease but not of death from cardiovascular disease (8). In the longitudinal analysis the increased risk was present at TSH levels of both 4-10 mU/L and >10 mU/L, but the risk at baseline in the cross-sectional analysis was only present at TSH levels >10 mU/L. in 2005, a 4-yr follow-up study of the Health, Aging and Body Composition Study in the USA among men and women of 70-79 yr, concluded that subclinical hypothyroidism is associated with an increased risk of congestive heart failure at TSH levels of 7.0 mU/L or greater, but not with other cardiovascular events and mortality (9). TSH levels between 4.5 and 6.9 mU/L carried no risk. By using TSH as a continuous variable, each standard deviation increase of 4.0 mU/L was associated with a 30% increase in congestive heart failure. In 2006, a 13-yr follow-up of the Cardiovascular Health Study in the USA among men and women of 65 yr and older, reported no association between subclinical hypothyroidism and cardiovascular disorders or mortality (10). In 2007, a 2.7-yr follow-up study in hospitalized patients admitted to the department of cardiology in Pisa among men and women of mean age 61 yr, observed lower survival rates for cardiac death and overall death in subclinical hypothyroidism than in euthyroidism with hazard ratio’s (after adjustment for several risk factors) of 2.40 (95% CI 1.36-4.21) and 2.01 (95% CI 1.33-3.04) respectively (11). In this study subclinical hypothyroidism was defined as TSH levels between 4.5 and 10 mU/l with FT4 and FT3 within the reference range; its cause was atrophic thyroiditis in 39%, Hashimoto’s thyroiditis in 36% and thyroidectomy or 131I therapy in 25%. In 2007, a cross-sectional population-based study in Tromso, Norway among men and women of 55-74 yr, found that subjects with subclinical hypothyroidism (TSH 3.5-10 mU/L) had no signs of cardiac dysfunction (12). In 2008, a 12-yr follow-up in the Cardiovascular Health Study in the USA among men and women 65 yr and older, indicated a greater incidence of heart failure in subclinically hypothyroid participants with TSH of 10 mU/L or greater compared with euthyroid participants with TSH values of 0.45 to 4.50 mU/l (adjusted hazard ratio 1.88, 95 CI 1.05-3.34); no increased risk of heart failure was observed in subclinical hypothyroidism with TSH values 4.5 to 9.9 mU/L (13). In 2010, a 10.6-yr follow-up in the EPIC-Norfolk study in the UK among men and women 45-79 yr, no association was found between subclinical hypothyroidism and the risk of coronary heart disease, despite the association between thyroid hormone levels and cardiovascular risk factors (14). In 2010, a 7.5-yr follow-up in the Japanese-Brazilian Thyroid Study among men and women 30 yr and older, observed subclinical hypothyroidism was associated with all-cause mortality (adjusted hazard ratio 2.3, 95% CI 1.2-4.4) but not with cardiovascular mortality (hazard ratio 1.6, 95% CI 0.6-4.2) (15). In 2010, the 20-yr follow-up of the Whickham Survey in the UK among men and women of 18 yr and older, was reanalyzed (16). Incident ischemic heart disease was significantly higher in the group with subclinical hypothyroidism vs. the euthyroid group (hazard ratio 1.76 , 95% CI 1.15-2.71), and incident ischemic heart disease mortality was also increased in subclinical hypothyroidism (hazardratio 1.79, 95% CI 1.02-3.56). Subsequent treatment of subclinical hypothyroidism with L-T4 appears to attenuate ischemic heart disease-related morbidity and mortality. In 2011 analysis of the PreCis database (from the Cleveland Clinic Preventive Cardiology Clinic) revealed an association between moderate (TSH 6.1-10 mU/L) but not with mild (TSH 3.1-6.0 mU/L) subclinical hypothyroidism and coronary heart disease prevalence and all-cause mortality in both genders in subjects under the age of 65 (but not in the age gourp older than 65 yr) (24). The PROSPER study among men and women aged 70-82 yr at high cardiovascular risk showed in 2012 an association between subclinical hypothyroidism and heart failure only at TSH >10 mU/L (25).Data from the MrOs cohort of men ≥65 yr followed for 8.3 yr did not show any relation between subclinical hypothyroidism and mortality, but only 8 men had TSH ≥10 mU/L (26). A study from Taiwan among the adult populationa with a 10-yr follow-up found an association between subclinical hypothyroidism and all-cause mortality and cardiovascular disease (27).The ‘oldest old’ from the Cardiovascular Health Study were retested after 13 yr (mean age 85 yr) ((28): there was a 13% increase in TSH, 1,7% increase in FT4 and 13% decrease in T3 over this period.There was no association between subclinical hypothyroidism or persistent TPO-Ab and death, but higher FT4 levels were associated with death. The findings raise concern for treatment of mildly elevated TSH levels in old age.In another report from the Cardiovascular Healthstudy among individuals ≥65 yr published in 2013, the 10-yr risk of incident coronary heart disease, heart failure and cardiovascular death was not related to persistent subclinical hypothyroidism (29).A nested case-control study among postmenopausal women within the Women’s Health Initiative cohort did not find an association between subclinical hypothyroidism and incident myocardial infarction (30). Among participants in the 3 rd NHANES, subclinical hypothyroidism was associated with greater mortality in those with congestive heart failure but not in those without (31). In summary , there are major discrepancies in epidemiological data about cardiovascular risk in subclinical hypothyroidism (1). This may be due to differences in the populations studied in terms of age, sex, race, life style and duration of follow-up, to differences in the extent of the TSH elevation, and to differences in the assessment of cardiovascular endpoints. Nevertheless,a general trend can be detected: the risk of adverse health outcomes is higher with higher TSH values (especially at TSH ≥ 10 mU/L), and is lower with advancing age (especially at age >65-70 yr).
Meta-analysis studies. In 2006, a meta-analysis of 14 observational studies published from 1996 to April 2005, indicated that subclinical hypothyroidism increased the risk of coronary heart disease (odds ratio 1.65, 95% CI 1.28-2.12) (17). The odds ratio’s varied little in analyses adjusted for various factors and analyses limited to various subgroups. In 2007, a meta-analysis of 4 studies published from 1966 to April 2007, revealed fthat subclinical hypothyroidism had a hazard ratio of 1.21 (95% CI 0.86-1.69) for circulatory mortality and of 1.25 (95% CI 1.03-1.53) for all-cause mortality (18). In 2008, a meta-analysis of 10 studies published up to January 2008, reports a relative risk for subclinical hypothyroidism for coronary heart disease of 1.20 (95% CI 0.97-1.49) (19). Risk estimates were higher among participants younger than 65 years (RR 1.51, 95% CI 1.09-2.09) for studies with mean participant age <65 yr, and the RR was1.05 (95% CI 0.90-1.22) for studies with mean participant age 65 yr and older. The RR was 1.18 (95% 0.98-1.42) for cardiovascular mortality and 1.12 (95% CI 0.99-1.26) for all-cause mortality. In 2008, a meta-analysis of 9 studies published up to July 2007, calculated a hazard ratio for subclinical hypothyroidism for all-cause mortality of 1.02 (95% CI 0.78-1.35) in cohorts from the community and of 1.76 (95% CI 1.36-2.30) in cohorts of participants with comorbidities (20). In 2008, a meta-analysis of 4 studies published in the period 2001-2005, showed a significant risk of subclinical hypothyroidism for coronary heart disease: the relative risk was 1.53 (95% CI 1.31-1.79) at baseline, and 1.19 (95% CI 1.02-1.34) at follow-up (21). The relative risk for all-cause mortality at follow-up was not significant, but the relative risk of cardiovascular mortality at follow-up was 1.28 (95% CI 1.02-1.60). In 2008, a meta-analysis of 15 studies published up to May 2007, concluded that incidence and prevalence of ischemic heart disease were higher in subclinically hypothyroid subjects compared with euthyroid subjects from studies including those younger than 65 yr, but not in studies of subjects aged older than 65 yr: odds ratio 1.57 (95% CI 1.19-2.06) vs 1.01 (95% CI 0.87-1.18), and 1.68 (95% CI 1.27-2.23) vs 1.02 (95% CI 0.85-1.22) (22). Cardiovascular/all-cause mortality was also elevated in participants from the younger than 65-yr studies, but not from the studies of older people: odds ratio 1.37 (95% CI 1.04-1.79) vs 0.85 (95% CI 0.56-1.29). Prevalent ischemic heart disease was higher in subclinically hypothyroid subjects of both genders, although this was significant only in women. These data suggest that increased vascular risk may only be present in younger individuals with subclinical hypothyroidism. In 2010, a meta-analysis based on individual participant data of 9 prospective cohort studies was presented, encompassing 41,685 participants with 381,647 person-years of follow-up (23). Subclinical hypothyroidism defined as TSH values of 4.5 to 19.9 mU/L with normal T4 concentrations was present in 2,621 subjects. Compared with euthyroidism, the hazard ratio for coronary heart disease events increased with higher TSH concentrations, from 1.07 (95% CI 0.84-1.35) for TSH 4.5-6.9 mU/L, 1,12 (95% CI 0.88-1.44) for TSH 7.0-9.9 mU/L, to 2.00 (95% CI 1.25-3,20) for TSH 10 mU/L and higher. Total mortality was not increased, but coronary heart disease mortality was increased only at TSH levels of 10 mU/L and higher (hazard ratio 1.64, 95% CI 1.11-2.42). Results were similar after further adjustment for traditional risk factors. Risks did not significantly differ by age, gender or preexisting cardiovascular disease.In 2012 the results were published of a pooled analysis of individual participant data from 6 prospective cohorts in which subclinical hypothyroidism was defined as TSh of 4.5-19.9 mU/L with normal FT4 (32). Risks of heart failure evnts were increased with higher TSH levels: hazard ratio was 1.01 (95% CI 0.81-1.26) for TSH 4.5-6.9 mU/L, 1.65 (0.84-3.23) for TSH 7.0-9.9 mU/L, and 1.86 (1.27-2.72) for TSH 10.0-19.9 mU/L (P for trend <0.01); risks remained similar after adjustment for cardiovascular risk factors. In summary , meta-analysis studies provide fair evidence that subclinical hypothyroidism is associated with cardiovascular morbidity and mortality. The risk on cardiovascular events is apparently dose-dependent: the higher the TSH, the greater the risk, with highest risk in subjects with TSH levels of ≥ 10 mU/L The risk is likely lower in subjects older than 65-70 yr.
9.10.5. TREATMENT
The many studies on the efffect of levothyroxine treatment in subclinical hypothyroidism have yielded inconsistent results. In order to get a more clear picture, a meta-analysis has been performed by the Cochrane Colloboration on studies published until May 2006 1 . The meta-analysis included 12 randomized controlled trials (RCTs) of 6-14 months duration involving 350 people. The daily average L-T4 dose required to normalize TSH in the active group varied between 67.5 to 85.5 μg (range 50 to 125 μg); subclinical hyperthyroidism at the end of the intervention was reported in 2 studies. In the placebo groups spontaneous normalization of TSH occurred in 42%, 25% and 24% of patients in 3 studies; these studies included patients without thyroid disease. The reader may consult the Cochrane review for references to studies evaluated in the meta-analysis 1 . Studies published after the meta-analysis will be referred to separately.
Treatment effects on symptoms and signs . The Cochrane meta-analysis did not observe statistically significant improvement in symptoms, mood or quality of life; one study showed a statistically significant improvement in cognitive function. More recent evaluations include a RCT among subjects of 65 yr and older in the UK (providing no evidence for improvement of cognitive function with L-T4) (2), and two non-RCTs in which attention and memory improved in L-T4 treated subjects relative to controls (3,4). A placebo-controlled randomized clinical trial in the UK demonstrated significant improvement in tiredness upon levothyroxine treatment (35) The Cochrane meta-analysis evaluated many parameters of systolic and diastolic heart function. Significant improvement after L-T4 treatment was observed for some parameters, like isovolumic relaxation time, index of myocardial performance, cycle variation index and left ventricular ejection time; systemic vascular resistance was not improved. Some studies report a lower systolic and diastolic blood pressure upon L-T4 treatment of subclinical hypothyroidism (5,6,7); one study reports a 6% reduction in supine mean arterial pressure (6). More recently, L-T4 treatment apparently improves arterial stiffness as evident from one RCT (8) and three non-RCTs (7,9,10). With regard to endothelium dysfunction, endothelial progenitor cells, expressing both endothelial and stem cell markers, are offered as a novel risk marker of cardiovascular disease. Subclinical hypothyroidism is associated with a lower number of endothelial progenitor cells in peripheral blood; the count increased after L-T4 treatment to values of healthy controls (11). Recent studies also indicate regression of the increased carotid intima-media thickness upon L-T4 treatment as evident from one RCT (12) and three non-RCTs (7,13,14). With respect to serum lipids, an early non-systematic review concluded that normalization of serum TSH in subclinical hypothyroidism decreases serum cholesterol on average by 0.4 mmol/l (95% CI 0.2-0.6 mmol/l) 16 . A subsequent meta-analysis also concluded that normalization of serum TSH decreases serum LDL-cholesterol by 0.26 mmol/l (95% CI 0.12-0.41 mmol/l) 17 , but the analysis has been criticized because inclusion of both observational and randomized studies with some of poor quality. The Cochrane meta-analysis found no significant improvement of total cholesterol upon L-T4 treatment. HDL- as well as LDL-cholesterol also did not reveal significant effects of L-T4 treatment, although a subgroup with LDL values >155 mg/dl showed significant effects. No differences between intervention groups were seen in the outcomes of triglycerides, apolipoprotein A and B and lipoprotein (a). A more recent RCT observed significant decreases in total and LDL cholesterol in the T4-treated subjects as compared to the placebo group (15) The reduction in serum total and LDL cholesterol was larger in individuals with TSH levels > 8.0 mU/L. The influence of subclinical hypothyroidism seems directly proportional to the degree of TSH elevation (18). Higher pretreatment cholesterol levels may also be associated with a greater reduction in total and LDL cholesterol 17,19 . It has been established from large trials outside the thyroid field that cardiovascular disease is reduced by 15% for each 10% reduction in plasma LDL cholesterol or 25% by a 38 mg/dl reduction in plasma total cholesterol. A 10% reduction in cholesterol may reduce the risk of cardiovascular mortality by 20% (1). Recent studies evaluated still other effects of L-T4 treatment in subclinical hypothyroidism. A RCT demonstrated increased cardiopulmonary exercise performance after L-T4 therapy in comparison to no treatment 20 . L-T4 treatment of subjects older than 70 yr with subclinical hypothyroidism did not document any benefit in terms of functional mobility 21 . A non-RCT shows normalization of reduced glomerular filtration rate and increased serum Cystatin-C levels upon L-T4 treatment of subclinical hypothyroidism (7). A RCT in iron-deficient subjects with subclinical hypothyroidism demonstrated greater increase in hemoglobin levels upon treatment with L-T4 plus iron than in treatment with iron alone (22). L-T4 treatment of subclinical hypothyroidism in pregnant women improves maternal and fetal outcomes of pregnancy, and is recommended 23 . A RCT in infertile women reports improved outcomes of in vitro fertilization upon L-T4 treatment as compared to placebo treatment 24 .
Treatment effects on cardiovascular morbidity and mortality, There are no placebo-controlled randomized clinical trials that assess the effect of long-term L-T4 treatment in subclinical hypothyroidism on cardiovascular morbidity or mortality. However, interesting data have emerged from the UK General Practitioner Research Database. Individuals with new subclinical hypothyroidism (TSH 5-10 mU/L) were identified in 2001 with outcomes analysed until March 2009 (34). After a median follow-up of 7.6 yr, 52.8% of the younger subjects (40-70 yr) and 49.9% of the older subjects (>70 yr) with subclinical hypothyroidism were treated with levothyroxine. Incident ischemic heart disease in the younger age group was observed in 4.2% in treated subjects and in 6.6% of the untreated subjects (multivariate-adjusted hazard ratio 0.61, 95% CI 0.39-0.95). In the older age group there were ischemic heart disease events in 12.7% in treated subjects and 10.7% in untreated subjects (hazard ratio 0.99, 95% CI 0.59-1.33). The data suggest that treatment of subclinical hypothyroidism with levothyroxine was associated with fewer ischemic heart disease events in younger individuals, but not in older people.
Recommendations when to treat . Whether or not subclinical hypothyroidism should be treated was and still is hotly debated; there are strong defenders as well as strong opponents to levothyroxine treatment 25-28 . A 2004 scientific review by a panel of experts concluded that data supporting associations of subclinical thyroid disease with symptoms or adverse clinical outcomes or benefits of treatment are few, and that the consequences of subclinical thyroid disease are minimal 29 ; consequently, the panel recommended against routine treatment of subclinical hypothyroidism, albeit recognizing the possible need for treatment in selected individual cases. The 2007 Cochrane meta-analysis also could find no evidence supporting treatment (1). Subsequent meta-analyses of long-term follow-up population-based studies seem to indicate that subclinical hypothyroidism is indeed associated with a modest risk on cardiovascular morbidity and mortality (although this may be age-dependent), but proof that levothyroxine treatment decreases the risk is lacking. This would require appropriately powered, randomized,placebo- controlled, double-blinded interventional trials with long follow-up. The debate whether or not to treat, thus continues (30,31).Given the current state of affairs with a lack of controlled trials reporting on long-term outcome, the decision whether or not to treat has to be taken in the face of uncertainty. This is not rare in medical practice, and the physician copes with such problems by an individualized approach taking into account best available circumvential evidence and clinical judgment.
An algoritm for the individual management of subclinical hypothyroidism is given in figure 9-7.It is recommended to confirm the existence of subclinical hypothyroidism in a second blood sample taken about 3-6 months later. This recommendation in current guidelines is given in view of the high chance of spontaneous normalization of the elevated TSH value 36,37 .It might be useful to already order assay of TPO-Ab and serum lipids in the second sample, because it may be relevant for further management in case subclinical hypothyroidism turns out to be persisitent. If subclinical hypothyroidism is confirmed, a strong case can be made for levothyroxine treatment when TSH values are > 10 mU/L.. This is indeed recommended by one guideline 36 . The other guideline would recommend treatment at TSH >10 mU/L in subjects below the age of 70 yr, and in subjects older than 70 yr only in the presence of symptoms or high cardiovascular risk 37 . The discrepancy between the two guidelines is caused by uncertainty about the role of age: one meta-analysis concludes the association of subclinical hypothyroidism with cardiovascular morbidity and mortality is independent of age, whereas the other concludes the risk is only present in subjects younger than 65-70 yr. The observation that subclinical hypothyroidism might have some survival value in the elderly age group has led both guidelines to the recommendation not to treat elderly individuals with TSH values of 4-10 mU/L. In younger subjects with mild to moderate subclinical hypothyroidism (TSH values between 4 and 10 mU/L) one may opt to institute levothyroxine treatment in the presence of symptoms (in view of the chance that symptoms will improve), TPO-Ab (especially in case TPO-Ab concentration is high with the risk of imminent progression to overt hypothyroidism, or cardiovascular risk factors (in the hope based on circumstantial evidence obtained from population-based association studies and some observational intervention studies to diminish the risk of developing cardiovascular events). If these three conditions are absent, most will agree it is better not to treat. In case no treatment is given, follow-up with regular repeat TSH measurements is indicated. However there is certainly a role for clinical judgement in thes patients. Many practitioners will elect to try replacement therapy in patients with SCH who are symptomatic, especizally in patients under age 70, with careful attention to maintaining TSH in the normal range. For guidelines how to manage subclinical hypothyroidism in infertile women, in pregnant women or in women planning pregnancy, please consult the chapter on Thyroid and Pregnancy.

Figure 9-7
Algoritm for the individual management of subclinical hypothyroidism.(TPO-Ab = thyroid peroxidase antibodies; risk factors = cardiovascular risk factors).
Table 9-16
Summary of data on subclinical hypothyroidism.
9.11 SCREENING FOR HYPOTHYROIDISM
In view of the rather high prevalence of thyroid function disorders and the availability of a suitable screening test in the form of the sensitive TSH assay, the question arises if screening programs are warranted in the general adult population 1 . Case-finding strategies have been employed successfully: previously unknown hypothyroidism was found in 0.64% of middle-age women in connection with screening for cervical carcinoma 2 , and in 0.3% of women attending a primary care unit 3 ; the prevalence of subclinical hypothyroidism in the latter study was 1.2%.Case-finding in women over 40 years of age can be useful. Patients admitted to geriatric units also benefit from routine testing as 2% to 5% have treatable thyroid disease, but patients hospitalized with acute illness do not benefit from routine thyroid function tests due to frequent interference of test results by the sick euthyroid syndrome 4 . The cost-effectiveness of periodic screening for mild thyroid failure has been investigated using a state-transition computer decision model that account for case-finding, medical consequences of mild thyroid failure, and costs of care during 40 years of simulated follow-up 5 . The cost-effectiveness of screening 35-year old patients with a serum TSH assay every 5 years was $ 9223 per QALY (quality-adjusted life year) for women and $22595 for men. The cost-effectiveness compares favorably with other generally accepted prevention programs. The authors recommend screening in the general community for mild hypothyroidism with serum TSH (combined with serum cholesterol) every five years at the age of 35 years 5,6 . An update on screening for thyroid disease in the general adult population, however, argues that the evidence of the efficacy of treatment for subclinical thyroid dysfunction is inconclusive and that large randomized trials are needed to determine the likelihood that treatment will improve the quality of life in otherwise healthy subjects who have mildly elevated TSH levels 7 . On the other hand, the update favors office-based screening to detect overt thyroid dysfunction in women older than 50 years of age: in this group, 1 in 71 women screened would benefit from relief of symptoms. Taken together, the presently available data do not justify yet screening of the healthy adult population for hypothyroidism. It is not recommended by th US Preventive Services Task Forcealtough the American Thyroid Association recommend screening every 5 years in women and men older than 35 years.Case-finding, i.e. testing on patients visiting their physician for unrelated reasons, seems currently the best approach to detect previously unsuspected hypothyroidism; it is especially worthwhile in women over 40 years of age. Screening of all pregnant women on thyroid disorders has not yet been accepted, but is most likely beneficial in view of a recent report indicating better outcomes with universal screening as compared to case finding (12). Table 9-17 provides a useful list of indications for screening for hypothyroidism 8 .
Table 9-17. I
ndications for screening for hypothyroidism. (Reproduced with permission) 8
References
References for Section 9.1
9.1.1. Gull WW: On a cretinoid state supervening in adult life in women. Trans Clin Soc London 1874; 7: 180.
9.1.2. Ord WM: On myxedema, a term proposed to be applied to an essential condition in the "cretinoid" affection occasionally observed in middle-aged women. Medico-Chir Trans 1878; 61: 57.
9.1.3. Reverdin JL: In discussion. Société médicale de Genève. Rev Med Suisse Romande 1882; 2: 539.
9.1.4. Kocher T: Ueber Kropfexstirpation und ihre Folgen. Arch Klin Chir 1883; 29:254.
9.1.5. Report of a Committee of the Clinical Society of London to Investigate the Subject of Myxedema. London, Longmans. Green & Co. Ltd. 1888.
References for Section 9.2
9.2.1. Spencer CA, LoPresti JS, Guttler RB, et al.: Application of a new chemiluminescent thyrotropin assay to subnormal measurements. J Clin Endocrinol Metab 1990; 70: 453-460.
9.2.2. Wiersinga WM. Adult hypothyroidism and myxedema coma. In: DeGroot LJ, Jameson JL (eds): Endocrinology (5th ed.). Philadelphia, WB Saunders Company, 2004, ch. 107
9.2.3. Evered DC, Ormston BJ, Smith PA, et al.: Grades of hypothyroidism. Brit Med J 1973; 1: 657-662.
9.2.4. Ishii H, Inada M, Tanaka K, et al.: Induction of outer and inner ring monodeiodinases in human thyroid gland by thyrotropin. J Clin Endocrinol Metab 1983; 57: 500-505.
9.2.5. Laurberg P: Mechanisms governing the relative proportions of thyroxine and 3,5,3'-triiodothyronine in thyroid secretion.Metabolism 1984; 33: 379-392.
9.2.6. Lum SM, Nicoloff JT, Spencer CA, Kaptein EM: Peripheral tissue mechanism for maintenance of serum triiodothyronine values in a thyroxine-deficient state in man. J Clin Invest 1984; 73: 570-575.
9.2.7. Martino E, Bartalena L, Pinchera A. Central hypothyroidism. In: Braverman LE, Utiger RD (eds): The Thyroid: A Fundamental and Clinical Text (8th ed.). Philadelphia, JB Lippincott Williams & Wilkins, 2000, pp 762-773.
9.2.8. Tunbridge WMG, Evered DC, Hall R, et al.: The spectrum of thyroid disease in the community: the Whickham Survey. Clin Endocrinol 1977; 7: 481-493.
9.2.9. Vanderpump MPJ, Tunbridge WMG, French JM, et al.: The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham Survey. Clin Endocrinol 1995; 43: 55-68.
9.2.10. Dos Remedios LV, Weber PM, Feldman R, et al.: Detecting unsuspected thyroid dysfunction by the free thyroxine index. Arch Intern Med 1980; 140: 1045-1049.
9.2.11. Sawin CT, Castelli WP, Hershman JM, et al.: The aging thyroid: Thyroid deficiency in the Framingham study. Arch Intern Med 1985; 145: 1386-1388.
9.2.12. Okamura K. Ueda K, Sone H, et al.: A sensitive TSH assay for screening of thyroid functional disorder in elderly Japanese. J Am Geriatr Soc 1989; 37: 317-322.
9.2.13. Sundbeck G, Lundberg PA, Lindstedt G, et al.: Incidence and prevalence of thyroid disease in elderly women: Results from the longitudinal population study of elderly people in Gothenburg, Sweden. Age and Ageing 1991; 20: 291-298.
9.2.14. Wang C, Crapo LM: The epidemiology of thyroid disease and implication for screening. Endocrinol Metab Clin North Am 1997; 26: 189-218.
9.2.15. Geul KW, van Sluisveld ILL, Grobbee DE, et al.: The importance of thyroid microsomal antibodies in the development of elevated serum TSH in middle-aged women: Associations with serum lipids. Clin Endocrinol 1993; 39: 275-280.
9.2.16. Walsh JP, Bremner AP, Feddema P, et al. Thyrotropin and thyroid antibodies as predictors of hypothyroidism: a 13-year, longitudinal study of a community-based cohort using current immunoassay techniques. J Clin Endocrinol Metab 2010; 95: 1095-1104.
9.2.17. Asvold BO, Vatten LJ, Midthjell, Bjoro T. Serum TSH wthin the reference range as a predictor of future hypothyroidism: 11-year follow-up of the HUNT study in Norway. J Clin Endocrinol Metab 2012; 97: 93-99.
References for Section 9.3.1
9.3.1.1. Faglia G, Bitensky L, Pinchera A, et al.: Thyrotropin secretion in patients with central hypothyroidism: evidence for reduced biological activity of immunoreactive thyrotropin. J Clin Endocrinol Metab 1979; 48: 989-998.
9.3.1.2. Miura Y, Perkel VS, Papenberg KA, et al.: Concanavalin-A, lentil, and ricin lectin affinity binding characteristic of human thyrotropin: differences in the sialylation of thyrotropin in sera of euthyroid, primary, and central hypothyroid patients. J Clin Endocrinol Metab 1989; 69: 985-995.
9.3.1.3. Horimoto M, Nishikawa M, Ishihara T, et al.: Bioactivity of thyrotropin (TSH) in patients with central hypothyroidism: comparison between in vivo 3,5,3'-triiodothyronine response to TSH and in vitro bioactivity of TSH. J Clin Endocrinol Metab 1995; 80: 1124-1128.
9.3.1.4. Samuels MH, Lillehei K, Kleinschmidt-Demasters BK, et al.: Patterns of pulsatile glycoprotein secretion in central hypothyroidism and hypogonadism. J Clin Endocrinol Metab 1990; 70: 391-395.
9.3.1.5. Adriaanse R, Brabant G, Endert E, Wiersinga WM: Pulsatile TSH release in patients with untreated pituitary disease. J Clin Endocrinol Metab 1993; 77: 205-209.
9.3.1.6. Collu R, Tang J, Castagné J, et al.: A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab 1997; 82: 1361-1365.
9.3.1.7. Dacou-Voutetakis C, Feltquate DM, Drakopoulo M, et al.: Familial hypothyroidism caused by a nonsense mutation in the thyroid-stimulating hormone ß-subunit gene. Am J Hum Genet 1990; 46: 988-993.
9.3.1.8. Hayashizaki Y, Hiraoka Y, Tatsumi K: Deoxyribonucleic acid analyses of five families with familial inherited thyroid stimulating hormone deficiency. J Clin Endocrinol Metab 1990; 71: 792-796.
9.3.1.9. Persani L. Clinical review: Central hypothyroidism: pathogenic, diagnostic, and therapeutic challenges. J Clin Endocrinol Metab 2012; 97: 3068-3078.
9.3.1.10. Arafah BM. Reversible hypopituitarism in patients with large non-functioning pituitary adenomas. J Clin Endocrinol Metab 1986; 62: 1173-1179.
9.3.1.11. Constine LS, Woolf PD, Cann D, et al.: Hypothalamic-pituitary dysfunction after radiation for brain tumors. N Engl J Med 1993; 328: 87-94.
9.3.1.12. Snijder PJ, Fowble BF, Schatz NJ, et al.: Hypopituitarism following radiation therapy of pituitary adenomas. Am J Med 1986; 81: 457-462.
9.3.1.13. Edwards BM, Clark JDA: Post-traumatic hypopituitarism: six cases and a review of the literature. Medicine 1986; 65: 281-290.
9.3.1.14. Cosman F, Post KD, Holub D, Wardlaw SL, et al.: Lymphocytic hypophysitis. Report of 3 new cases and review of the literature. Medicine 1989; 68: 240-256.
9.3.1.15. Kaptein EM, Spencer CA, Kamile MB, Nicoloff JT: Prolonged dopamine administration and thyroid hormone economy in normal and critically ill subjects. J Clin Endocrinol Metab 1980; 51: 387-393.
9.3.1.16. Vagenakis AG, Braverman LE, Azizi F, et al.: Recovery of pituitary thyrotropic function after withdrawal of prolonged thyroid suppression therapy. N Engl J Med 1975; 293: 681-684.
9.3.1.17 . Sherman SI, Gopal J, Haugen BR, Chiu AC, Whaley K, Nowlakha P, Duvic M. Central hypothyroidism associated with retinoid X receptor-selective ligands. New Engl J Med 340:1075-1079, 1999.
9.3.1.18. Oliveira JHA, Persani L, Beck-Peccoz P, et al. Investigating the paradox of hypothyroidism and increased serum thyrotropin (TSH) levels in Sheehan’s syndrome: characterization of TSH carbohydrate content and bioactivity. J Clin Endocrinol Metab 2001; 86: 1694-1699.
9.3.1.19. Bonomi M, Proverbio MC, Weber G, et al. Hyperplastic thyroid gland, high serum glycoprotein α-subunit, and variable circulating thyrotropin (TSH) levels as hallmark of central hypothyroidism due to mutations of the TSHβ gene. J Clin Endocrinol Metab 2001; 86: 1600-1604.
9.3.1.20. Vuissoz JM, Deladoëy J, Buyukgebiz A et al. New autosomal recessive mutation of the TSHβ subunit gene causing central isolated hypothyroidism. J Clin Endocrinol Metab 2001; 86: 4468-4471.
9.3.1.21. Lando A, Holm K, Nysom K, et al. Thyroid function in survivors of childhood acute lymphoblastic leukemia: the significance of prophylactic cranial irradiation. Clin Endocrinol 2001; 55: 21-25.
9.3.1.22. Rose SR. Cranial irradiation and central hypothyroidism. Trends Endocrinol Metab 2001; 12:97-104.
9.3.1.23. Persani L, Ferretti E, Borgato S, et al. Circulating thyrotropin bioactivity in sporadic central hypothyroidism. J Clin Endocrinol Metab 2000; 85:3631.
9.3.1.24. Schmiegelow M, Feldt-Rasmussen U, Rasmussen AK, et al. A Population-based study of thyroid function after radiotherapy and chemotherapy for a childhood brain tumor. J Clin Endocrinol Metab 2003; 88:136-140.
9.3.1.25. Benvenga S, Campenmi A, Ruggeri RM, Trimarchi F. Hypopituitarism secondary to head trauma. J Clin Endocrinol Metab 2000; 85:1353-136.
9.3.1.26. Bellastella A, Bizzarro A, Coronella C, et al. Lymphocytic hypophysitis: a rare or underestimated disease? Eur J Endocrinol 2003; 149:363-376.
9.3.1.27. Schneider HJ, Kreitschmann-Andermahr I, Ghigo E, Stalla GK, Agha A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage. A systematic review. JAMA 2007; 298: 1429-1438.
9.3.1.28. Liu S, Ogilvie KM, Klausing K, et al. Mechanism of selective retinoid X receptor agonist-induced hypothyroidism in the rat. Endocrinology 2002; 143: 2880-2885.
9.3.1.29.Golden WM, Weber KB, Hernandez TL et al. Single-dose rexinoid rapidly and specifically suppresses serum thyrotropin in healthy subjects. J Clin Endocrinol Metab 2007; 92: 124-130.
9.3.1.30. Smit JW, Stokkel MP, Pereira AM et al. Bexarotene induced hypothyroidism: bexarotene stimulates the peripheral metabolism of thyroid hormones. J Clin Endocrinol Metab 2007; 92: 2496-2499.
9.3.1.31. Haugen BR. Drugs that suppress TSH or cause central hypothyroidism. Best Pract Res Clin Endocrinol Metab 2009; 23: 793-800.
9.3.1.32. Sun Y, Bak B, Schoenmakers N et al. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nat Genet 2012; 44: 1375-1381.
9.3.1.33. Joustra SD, Schoenmakers N, Persani L et al. The IGSF1 deficiency syndrome: characteristics of male and female patients. J Clin Endocrinol Metab 3013; Oct.9 [Epub ahead of print].
References for Section 9.3.2
9.3.2.1. Hayashi Y, Tamai H, Fukata S, et al.: A long-term clinical, immunological, and histological follow-up study of patients with goitrous chronic lymphocytic thyroiditis. J Clin Endocrinol Metab 1985; 61: 1172-1178.
9.3.2.2.. Michaelson ED, Young RL: Hypothyroidism with Graves' disease. JAMA 1970; 23: 1351.
9.3.2.3. Arikawa K, Ichikawa Y, Yoshida T, et al.: Blocking type antithyrotropin receptor antibody in patients with nongoitrous hypothyroidism: its incidence and characteristics of action. J Clin Endocrinol Metab 1985; 60: 953-959
9.3.2.4. Kraiem Z, Lahat N, Glaser B, et al.: Thyrotropin receptor blocking antibodies: incidence, characterization and in vivo synthesis. Clin Endocrinol 1987; 27: 409-421
9.3.2.5. Rieu M, Portos C, Lissak B, et al.: Relationship of antibodies to thyrotropin receptors and to thyroid ultrasonographic volume in euthyroid and hypothyroid patients with autoimmune thyroiditis. J Clin Endocrinol Metab 1996; 80: 641-645.
9.3.2.6. Tomer Y, Huber A. The etiology of autoimmune thyroid disease: a story of genes and environment. J Autoimmun 2009; 32: 231-239.
9.3.2.7. Jacobson EM, Tomer Y. The genetic basis of thyroid autoimmunity. Thyroid 2007; 17: 949-961.
9.3.2.8. Laurberg P, Pedersen KM, Hreidarsson A, et al.: Iodine intake and the pattern of thyroid disorders: a comparative epidemiological study of thyroid abnormalities in the elderly in Iceland and in Jutland, Denmark. J Clin Endocrinol Metab 1998; 83: 765-769.
9.3.2.9. Laurberg P, Cerqueira C, Ovesen L et al. Iodine intake as a determinant of thyroid disorders in populations. Best Pract Res Clin Endocrinol Metab 2010; 24: 13-27.
9.3.2.10. Sundrick RS, Bagchi N, Brown TR: The role of iodine in thyroid autoimmunity: from chickens to humans: a review. Autoimmunity 1992; 13: 61-68.
9.3.2.11. Effraimidis G, Tijssen JG, Wiersinga WM. Discontinuation of smoking increases the risk for developing thyroid peroxidase antibodies and/or thyroglobulin antibodies: a prospective study. J Clin Endocrinol Metab 2009; 94: 1324-1328.
9.3.2.12. Asvold BO, Bjoro T, Nilsen TI, Vatten LJ. Tobacco smoking and thyroid function: a population-based study. Arch Intern Med 2007; 167: 1428-1432.
9.3.2.13. Carle A, Bulow Pedersen I, Knudsen N et al. Smoking cessation is followed by a sharp but transient rise in the incidence of overt autoimmune hypothyroidism – a population-based, case-control study. Clin Endocrinol 2012; 77: 764-772.
References for Section 9.3.3.
9.3.3.1. Nikolai TF: Recovery of thyroid function in primary hypothyroidism. Am J Med Sci 1989; 297: 18-21.
9.3.3.2. Kasagi K, Iwata M, Misaki T, Konishi J. Effect of iodine restriction on thyroid function in patients with primary hypothyroidism. Thyroid 2003; 13: 561-567.
9.3.3.3. Okamura K, Sato K, Ikenoue H, et al.: Reevaluation of thyroidal radioactive iodine uptake test, with special reference to reversible primary hypothyroidism with elevated thyroid radioiodine uptake. J Clin Endocrinol Metab 1988; 67: 720-726.
9.3.3.4. Comtois R, Faucher L, Laflèche L: Outcome of hypothyroidism caused by Hashimoto's thyroiditis. Arch Int Med 1995; 155: 1404-1408.
9.3.3.5. Takasu N, Komiya I, Asawa T, et al.: Test for recovery from hypothyroidism during thyroxine therapy in Hashimoto's thyroiditis. Lancet 1990; 336: 1084-1086.
9.3.3.6. Takasu N, Yamada T, Takasu M, et al.: Disappearance of thyrotropin-blocking antibodies and spontaneous recovery from hypothyroidism in autoimmune thyroiditis. N Eng J Med 1992; 326: 513-518.
9.3.3.7. Kraiem Z, Baron E, Kahana L, et al.: Changes in stimulating and blocking TSH receptor antibodies in a patient undergoing three cycles of transition from hypo to hyperthyroi-dism and back to hypothyroidism. Clin Endocrinol 1992; 36: 211-216.
9.3.3.8. Gerstein HC: How common is postpartum thyroiditis? A methodologic overview of the literature. Arch Int Med 1990; 150: 1397-1400.
9.3.3.9. Kuypens JL, Pop VJ, Vader HL, et al.: Prediction of postpartum thyroid dysfunction: can it be improved? Eur J Endocrinol 1998; 139: 36-43.
9.3.310. Alvarez-Marfany M, Roman SH, Drexler AJ, et al.: Long-term prospective study of postpartum thyroid function in women with insulin dependent diabetes mellitus. J Clin Endocrinol Metab 1994; 79: 10-16.
9.3.3.11. Othman S, Phillips DI, Parkes AB, et al.: A long-term follow-up of postpartum thyroiditis. Clin Endocrinol 1990; 32: 559-564.
9.3.3.12. Kuypens JL, de Haan-Meulman M, Vader HL, et al.: Cell-mediated immunity and postpartum thyroid dysfunction: a possibility for the prediction of disease? J Clin Endocrinol Metab 1998; 83: 1959-1966.
9.3.3.13. Harris B, Othman S, Davies JA, et al.: Association between postpartum thyroid dysfunction and thyroid antibodies and depression. Brit Med J 1992; 305: 152-156.
9.3.3.14. Pop VJ, de Vries E, van Baar A, et al.: Maternal thyroid peroxidase antibodies during pregnancy: a marker of impaired child development? J Clin Endocrinol Metab 1995; 80: 3561-3566.
9.3.3.15. Pop VJ, Kuypens JL, Van Baar AL, et al.: Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol 1999; 50: 149-156.
9.3.3.16. Henrichs J, Bongers-Schokking JJ, Schenk JJ. Matrenal thyroid function during early pregnancy and cognitive functioning in early childhood: the Generation R study. J Clin Endocrinol Metab 2010; 95: (doi:10.2010/jc.2010-0415)
9.3.3.17. Vialettes B, Guillerand MA, Viens P, et al.: Incidence rate and risk factors for thyroid dysfunction during recombinant interleukin-2 therapy in advanced malignancies. Acta Endocrinol 1993; 129: 31-38.
9.3.3.18. Preziati D, La Rosa L, Covini G, et al.: Autoimmunity and thyroid function in patients with chronic active hepatitis treated with recombinant interferon alpha-2a. Europ J Endocrinol 1995; 132: 587-593.
9.3.3.19. Marazuela M, Garcia-Buey L, Gonzalez-Fernandez B, et al.: Thyroid autoimmune disorders in patients with chronic hepatitis C before and during interferon-a therapy. Clin Endocrinol 1996; 44: 635-642.
9.3.3.20. Prummel MF, Laurberg P. Interferon- α and autoimmune thyroid disease. Thyroid 2003; 13:547-551.
9.3.3.21. Mandac JC, Chaudrhy S, Sherman KE, Tomer Y. The clinical and physiological spectrum of interferon-alpha inducedthyroiditis: toward a new classification. Hepatology 2006; 43: 661-672.
9.3.3.22. Roti E, Minelli R, Giuberti T et al. Multiple changes in thyroid function in patients with chronic active HCV hepatitis treated with recombinant interferon-alpha. Am J Med 1996; 101: 482-487.
9.3.3.23. Tomer Y, Menconi F. Interferon induced thyroiditis. Best Pract Res Clin Endocrinol Metab 2009; 23: 703-712.
9.3.3.24. Akeno N, Blackard JT, Tomer Y. HCV E2 protein binds directly to thyroid cells and induces IL-8 production: a new mechanism for HCV induced thyroid autoimmunity. J Autoimmun 2008; 31: 339-344.
9.3.3.25. Baufin E, Marcellin P, Pouteau M et al. Reversibility of thyroid dysfunction induced by recombinant alpha interferon in chronic hepatitis C. Clin Endocrinol 1993; 39: 657-661.
9.3.3.26. Akeno N, Smith EP, Stefan M et al. IFN-α mediates the development of autoimmunity both by direct tissue toxicity and through immune cell recruitment mechanisms.J Immunol 2011; 186: 4693-4706.
References for Section 9.3.4.
9.3.4.1. Nofal MN, Beierwaltes WH, Patno ME: Treatment of hyperthyroidism with sodium iodide I-131, a 16-year experience. JAMA 1966; 197: 605-610.
9.3.4.2. Hagen GA, Ouellette RP, Chapman EM: Comparison of high and low dosage of 131I in the treatment of thyrotoxicosis. N Engl J Med 1967; 277: 559.
9.3.4.3. Roudebush CP, Hoye KE, DeGroot LJ: Compensated low-dose 131I therapy of Graves' disease. Ann Intern Med 1977; 87: 441.
9.3.4.4. Nygaard B, Hegedüs L, Gervil M, et al.: Influence of compensated 131I therapy on thyroid volume and incidence of hypothyroidism in Graves' disease. J Int Med 1995; 238: 491-497.
9.3.4.5. Stoffer SS, Hamburger JI: Inadvertent 131I therapy for hyperthyroidism in the first trimester of pregnancy. J Nucl Med 1976; 17: 146.
9.3.4.6. Huysmans DA, Corstens FH, Kloppenborg PW: Long-term follow-up in toxic solitary autonomous thyroid nodules treated with radioactive iodine. J Nucl Med 1991; 32: 27-30.
9.3.4.7. Nygaard B, Hegedüs L, Nielsen KG, et al.: Long-term effect of radioactive iodine on thyroid function and size in patients with solitary autonomously functioning toxic thyroid nodules. Clin Endocrinol 1999; 50: 197-202.
9.3.4.8. Smith RE, Adler RA, Clark P, et al.: Thyroid function after mantle irradiation in Hodgkin's disease. JAMA 1981; 245: 46-49.
9.3.4.9. Tell R, Sjödin H, Lundell G, et al.: Hypothyroidism after external radiotherapy for head and neck cancer. Int J Radiation Oncology Biol Phys 1997; 39: 303-308.
9.3.4.10. Smolarz K, Malke G, Voth E, et al. Hypothyroidism after therapy for larynx and pharynx carcinoma. Thyroid 2000; 10: 425-429.
9.3.4.11. Mercado G, Adelstein DJ, Saxton JP, et al. Hypothyroidism. A frequent event after radiotherapy and after radiotherapy with chemotherapy for patients with head and neck carcinoma. Cancer 2001; 92:2892-2897.
9.3.4.12. Ishiguro H, Yasuda Y, Tomita Y et al. Long-term follow-up of thyroid function in patients who received bone marrow transplantation during childhood and adolescence. J Clin Endocrinol Metab 2004; 89: 5981-5986.
References for Section 9.3.5.
9.3.5.1. Chau AM, Lynch MJG, Bailey JD et al.: Hypothyroidism in cystinosis: a clinical, endocrinologic and histologic study involving sixteen patients with cytinosis. Am J Med 1970; 48: 678.
9.3.5.2. Barsano CP: Other forms of hypothyroidism. In: Braverman LE, Utiger RD (eds): The Thyroid: A Fundamental and Clinical Text (7th ed.). Philadelphia, JB Lippincott, 1996, pp 768-778.
9.3.5.3. Paes JE, Burman KD, Cohen J et al. Acute bacterial suppurative thyroiditis: a clinical review and expert opinion. Thyroid 2010; 20: 247-256.
References for Section 9.3.7.
9.3.7.1. Wiersinga WM, Touber JL, Trip MD, van Royen EA: Uninhibited thyroidal uptake of radioiodine despite iodine excess in amiodarone-induced hypothyroidism. J Clin Endocrinol Metab 1986; 63: 485-491.
9.3.7.2. Braverman LE: Iodine and the thyroid: 33 years of study. Thyroid 1994; 4: 351-356.
9.3.7.3. Markou K, Georgopoulos N, Kyriazopoulou V, Vagenakis AG. Iodine-induced hypothyroidism. Thyroid 2001; 11: 501-510.
9.3.7.4. Trip MD, Wiersinga WM, Plomp TA: Incidence, predictability, and pathogenesis of amiodarone-induced thyrotoxicosis and hypothyroidism. Am J Med 1991; 91: 507-511.
9.3.7.5. Martino E, Safran M, Aghini-Lombardi F, et al.: Environmental iodine intake and thyroid dysfunction during chronic amiodarone therapy. Ann Int Med 1984; 101: 28-34.
9.3.7.6. Pedersen IB, Laurberg P, Knudsen N et al. An increased incidence of overt hypothyroidism after iodine fortification of salt in Denmark: a propsective population study. J Clin Endocrinol Metab 2007; 92: 3122-3127.
References for Section 9.3.8.
9.3.8.1. Braverman LE, Woeber KA, Ingbar SH: Induction of myxedema by iodide in patients euthyroid after radioiodine or surgical treatment of diffuse toxic goiter. N Engl J Med 1969; 281: 816.
9.3.8.2. Braverman LE, Ingbar SH, Vagenakis AG, et al.: Enhanced susceptibility to iodide myxedema in patients with Hashimoto's thyroiditis. J Clin Endocrinol 1971; 32: 515
9.3.8.3. Berens SC, Bernstein RS, Robbins J, Wolff J: Antithyroid effects of lithium. J Clin Invest 1970; 49: 1357.
9.3.8.4. Perrild H, Hegedüs L, Baastrup PC, et al.: Thyroid function and ultrasonically determined thyroid size in patients receiving long-term lithium treatment. Am J Psychiatry 1990; 147: 1508-1521.
9.3.8.5. Gaitan E, Cooksey RC, Legan J, et al.: Antithyroid and goitrogenic effects of coal-water extracts from iodine-sufficient goiter areas. Thyroid 1993; 3 49-53.
9.3.8.6. Langer P, Tajtakova M, Fodor G, et al.: Increased thyroid volume and prevalence of thyroid disorders in an area heavily polluted by polychlorinated biphenyls. Eur J Endocrinol 1998; 139: 402-409.
9.3.8.7. Braverman LE, He X, Pino S et al. The effect of perchlorate, thiocyanate, and nitrate on thyroid function in workers exposed to perchlorate long-term. J Clin Endocrinol Metab 2005; 90: 700-706.
9.3.8.8. Braverman LE, Pearce EN, He X et al. Effects of six months of daily low-dose perchlorate exposure on thyroid function in healthy volunteers. J Clin Endocrinol Metab 2006; 91: 2721-2724.
9.3.8.9. Pearce EN, Lazarus JH, Smyth PP et al. Perchlorate and thiocyanate exposure and thyroid function in first-trimester pregnant women. J Clin Endocrinol Metab 2010; 95: 3207-3215.
9.3.8.10. Illouz F, Laboureau-Soares S, Dubois S, Rohmer V, Rodien P. Tyrosine kinase inhibitors and modifications of thyroid function tests: a review. Eur J Endocrinol 2009; 160: 331-336.
9.3.8.11. Desai J, Yassa L, Marqusee E et al. Hypothyroidism after sunitinib treatment for patients with gastrointestinal stromal tumors. Ann Intern Med 2006; 145: 660-664.
9.3.8.12. Wolter P, Stefan C, Decallonne B et al. The clinical implications of sunitinib-induced hypothyroidism: a propsective evaluation. Brit J Cancer 2008; 99: 448-454.
9.3.8.13. Mannavola D, Coco P, Vannucchi G et al. A novel tyrosine kinase selective inhibitor, sunitinib, induces transient hypothyroidism by blocking iodine uptake. J Clin Endocrinol Mrtab 2007; 92: 3531-3534.
9.3.8.14. Salem AK, Fenton MS, Marion KM, Hershman JM. Effect of sunitinib on growth and function of FRTL-5 thyroid cells. Thyroid 2008; 18: 631-635.
9.3.8.15. Wong E, Rosen LS, Mulay M et al. Sunitinib induces hypothyroidism in advanced cancer patients and may inhibit thyroid peroxidase activity. Thyroid 2007; 17: 351-355.
9.3.8.16. Grossmann M, Premaratne E, Desai J, Davis ID. Thyrotoxicosis during sunitinib treatment for renal cell carcinoma. Clin Endocrinol 2008; 69: 669-672.
9.3.8.17. Rogiers A, Wolter P, Op de Beeck K, Thijs M, Decallone B, Schoffski P. Shrinkage of thyroid volume in sunitinib treated patients with renal cell carcinoma – a potential marker of irreversible thyroid dysfunction? Thyroid 2010; 20: 317-322.
9.3.8.18. Makita N, Miyakawa M, Fujita T, Iiri T. Sunitinib induces hypothyroidism with a markedly reduced vascularity. Thyroid 2010; 20: 323-326.
9.3.8.19. Hershman JM. How does sunitinib cause hypothyroidism? Thyroid 2010; 20: 243-244.
9.3.8.20. Tamaskar I, Bukowski R, Elson P et al. Thyroid function test abnormalities in patients with metastatic renal cell carcinoma treated with sorafenib. Ann Oncol 2008; 19: 265-268.
9.3.8.21. Miyake H, Kurahashi T, Yamanaka K et al. Abnormailties of thyroid function in Japanese patients with metastatic renal cell carcinoma treated with sorafenib: a prospective evaluation. Urol Oncol 2009; Nov.12 (Epub ahead of print)
9.3.8.22. Makita N, Iiri T. Tyrosine kinase inhibitor-induced thyroid disorders: a review and hypothesis. Thyroid 2013; 23: 151-159.
9.3.8.23. Torino F, Barnabei A, Paragliola R, Baldelli R, Appetecchia M, Corsello SM. Thyroid dysfunction as an unintended side effect of anticancer drugs. Thyroid 2013; 23: 1345-1366.
References for Section 9.3.9
9.3.9.1. Huang SA, Tu HM, Harney JW, et al. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. New Engl J Med 2000; 343: 185-189.
9.3.9.2. Huang SA, Fish SA, Dorfman DM et al. A 21-year-old woman with consumptive hypothyroidism due to a vascular tumor expressing type 3 iodothyronine deiodinase. J Clin Endocrinol Metab 2002; 87: 4457-4461.
9.3.9.3. Ruppe MD, Huang SA, Jan de Beur SM. Consumptive hypothyroidism caused by paraneoplastic production of type 3 iodothyroinine deiodinase. Thyroid 2005; 15: 1369-1372.
9.3.9.4. Howard D, La Rosa FG, Huang S et al. Consumptive hypothyroidism resulting from hepatic vascular tumors in an athyreotic adult. J Clin Endocrinol Metab 2011; 96: 1966-1970.
References for Section 9.4.
9.4.1. Smith TJ, Bahn RS, Gorman CA: Connective tissue, glycosaminoglycans, and diseases of the thyroid. Endocr Rev 1989; 10: 366-391.
9.4.2. Smith TJ, Horwitz AL, Refetoff S. The effect of thyroid hormone on glycosaminoglycan accumulation in human skin fibroblasts. Endocrinology 1981; 108: 2397.
9.4.3. Smith TJ, Murata Y, Korwitz AL, et al. Regulation of glycosaminoglycan synthesis by thyroid hormone in vitro. J Clin Invest 1982; 70: 1066.
9.4.4. Klion FM, Segal R, Schaffner F: The effect of altered thyroid function on the ultrastructure of the human liver. Am J Med 1971; 150: 137.
9.4.5. Wilkens L: Epiphysial dysgenesis associated with hypothyroidism. Am J Dis Child 1941; 61: 13.
9.4.6. Price TR, Netsky MG: Myxedema and ataxia: Cerebellar alterations and "neural myxedema bodies". Neurology 1966; 16: 957.
9.4.7. Rossman NP: Neurological and muscular aspects of thyroid dysfunction in childhood. Pediatr Clin North Am 1976; 23: 575.
9.4.8. Naeye RL: Capillary and venous lesions in myxedema. Lab Invest 1963; 12: 465.
9.4.9. Salomon MI, DiScala V, Grisham E et al: Renal lesions in hypothyroidism: a study based on kidney biopsies. Metabolism 1967; 16: 846.
9.4.10. Ezrin C, Swanson HE, Humphrey JG et al: The cells of the human adenohypophysis in thyroid disorders. J Clin Endocrinol Metab 1959; 19: 958.
9.4.11. Yamada T, Tsukui T, Ikejiri K et al: Volume of sella turcica in normal subjects and in patients with primary hypothyroidism and hyperthyroidism. J Clin Endocrinol Metab 1976; 42: 817.
9.4.12. Bloodworth JMB, Kirkendall WM, Carr TL: Addison's disease associated with thyroid insufficiency and atrophy (Schmidt syndrome). J Clin Endocrinol Metab 1954; 14: 540.
References for Section 9.5.1.
9.5.1.1. Diekman MJM, Romijn JA, Endert E, Sauerwein H, Wiersinga WM. Thyroid hormones modulate serum leptin levels: observations in thyrotoxic and hypothyroid women. Thyroid 1998; 8: 1081-1086.
9.5.1.2. Hsieh CJ, Wang PW, Wong ST et al. Serum leptin concentrations of patients with sequential thyroid function changes. Clin Endocrinol 2002; 57: 29-34.
9.5.1.3. Iglesias P, Alvarez Fidalgo P, Codoceo R, Diez JJ. Serum concentrations of adipocytokines in patients with hyperthyroidism and hypothyroidism before and after control of thyroid function. Clin Endocrinol 2003; 59: 621-529.
9.5.1.4. Brackhik M, Marcisz C, Giebel S, Orzel A. Serum leptin and ghrelin levels in premenopausal women with stable body mass index during treatment of thyroid dysfunction. Thyroid 2008; 18: 545-550.
9.5.1.5. Gimenez-Palop O, Gimenez-Perez G, Mauricio D et al. Circulating ghrelin in thyroid dysfunction is related to insulin resistance and not to hunger, food intake or anthropometric changes. Eur J Endocrinol 2005; 153: 73-79.
9.5.1.6. Gjedde S, Vestergaard ET, Gormsen LC et al. Serum ghrelin levels are increased in hypothyroid patients and become normalized by L-thyroxine treatment. J Clin Endocrinol Metab 2008; 93: 2277-2280.
9.5.1.7. Altinova AE, Toruner FB, Akturk M et al. Adiponectin levels and cardiovascular risk factors in hypothyroidism and hyperthyroidism. Clin Endocrinol 2006; 65: 530-535.
9.5.1.8. Krassas GE, Pontikides N, Loustis K, Koliakos G, Constantinidis T, Kaltsas T. Resistin levels are normal in hypothyroidism and remain unchanged after attainment of euthyroidism: relationship with insulin levels and anthropometric parameters. J Endocrinol Invest 2006; 29: 606-612.
9.5.1.9. Kokkinos A, Mourouzis I, Kyriaki D, Pantos C, Katsilambros N, Cokkinos DV. Possible implications of leptin, adiponectin and ghrelin in the regulation of energy homeostasis by thyroid hormone. Endocrine 2007; 32: 30-32.
9.5.1.10. Lewallen CG, Rall JE, Berman M: Studies of iodoalbumin metabolism. II. The effects of thyroid hormone. J Clin Invest 1959; 38: 88.
9.5.1.11. Smith TJ, Murata Y, Horwitz AL, et al.: Regulation of glycosaminoglycan accumulation by thyroid hormone in vitro. J Clin Invest 1982; 70: 1066.
9.5.1.12. Smith TJ, Horwitz AL, Refetoff S: The effect of thyroid hormone on glycosaminoglycan accumulation in human skin fibroblasts. Endocrinology 1981; 108: 2397.
9.5.1.13. Crispell KR, Parson W, Hollifield G: A study of the rate of protein synthesis before and during the administration of L-triiodothyronine to patients with myxedema and healthy volunteers using N-15 glycine. J Clin Invest 1956; 35: 164.
9.5.1.14. Lamberg BA, Gräsbeck R: The serum protein pattern in disorders of thyroid function. Acta Endocrinol 1955; 19: 91.
9.5.1.15. Dimitriadis G, Mitrou P, Lambadiari V et al. Insulin action in adipose tissue and muscle in hypothyroidism. J Clin Endocrinol Metab 2006; 91: 4930-4937.
9.5.1.16. Brauman H, Corvilain J: Growth hormone response to hypoglycemia in myxedema. J Clin Endocrinol Metab 1968; 28: 301.
9.5.1.17. Shah JH, Motto GS, Papagiannes E, William GA: Insulin metabolism in hypothyroidism. Diabetes 1975; 24: 922.
9.5.1.18. Maratou E, Hadjidakis DJ, Kollias A et al. Studies of insulin resistance in patients with clinical and subclinical hypothyroidism. Eur J Endocrinol 2009; 160: 785-790.
9.5.1.19. Shah JH, Cerchio GM: Hypoinsulinemia of hypothyroidism. Arch Intern Med 1973; 132: 657.
9.5.1.20. Erdogan M, Canataroglu A, Ganidagli S, Kulaksizoglu M. Metabolic syndrome prevalence in subclinic and overt hypothyroid patients and the relation among metabolic syndrome parameters. J Endocrinol Invest 2010, Jul 22 (Epub ahead of priint).
9.5.1.21. Handisurya A, Pacini G, Tura A, Gessi A, Kautzky-Willer A. Effects of T4-replacement therapy on glucose metabolism in subjects with subclinical and overt hypothyroidism. Clin Endocrinol 2008; 69: 963-969.
9.5.1.22. Hecht A, Gershberg H: Diabetes mellitus and primary hypothyroidism. Metabolism 1968; 17: 108.
9.5.1.23. Abrams JJ, Grundy SM: Cholesterol metabolism in hypothyroidism and hyperthyroidism in man. J Lipid Res 1981; 22: 323.
9.5.1.24. Kritchevsky D: Influence of thyroid hormones and related compounds on cholesterol biosynthesis and degradation: a review. Metabolism 1960; 9: 984.
9.5.1.25. Kurland GS, Lucas JL, Freedberg AS: The metabolism of intravenously infused 14C-labeled cholesterol in euthyroidism and myxedema. J Lab Clin Med 1961; 57: 574.
9.5.1.26. Walton KW, Scott PJ, Dykes PW, Davies JWL: The significance of alterations in serum lipids in thyroid dysfunction. II. Alterations of the metabolism and turnover of 131I low-density lipoproteins in hypothyroidism and thyrotoxicosis. Clin Sci 1965; 29: 217.
9.5.1.27 Chait A, Bierman EL, Albers JJ: Regulatory role of triiodothyronine in the degradation of low density lipoprotein by cultered human skin fibroblasts. J Clin Endocrinol Metab 1979; 48: 887.
9.5.1.28. Scarabottolo L, Trezzi E, Roma P, Gatapano AL: Experimental hypothyroidism modulates the expression of the low density lipoprotein receptor by the liver. Atherosclerosis 1986; 59: 329.
9.5.1.29. Thompson GR, Soutar AK, Spengel FA, et al: Defects of receptor mediated low density lipoprotein catabolism in homozygous familial hypercholesterolemia and hypothyroidism in vivo. Proc Natl Acad Sci USA 1981; 78: 2591.
9.5.1.30. Diekman T, Demacker PNM, Kastelein JJP, et al.: Increased oxidizability of low-density lipoproteins in hypothyroidism. J Clin Endocrinol Metab 1998; 83: 1752-1755.
9.5.1.31. Pazos F, Alvarez JJ, Rubies-Prat J, et al.: Long term thyroid replacement therapy and levels of lipoprotein (a) and other lipoproteins. J Clin Endocrinol Metab 1995; 80: 562-566.
9.5.1.32. O'Brien T, Katz K, Hodge D, et al.: The effect of treatment of hypothyroidism and hyperthyroidism on plasma lipids and apolipoproteins AI, AII and E. Clin Endocrinol 1997; 46: 17-20.
9.5.1.33. Martinez-Triguero ML, Hernandez-Myares A, Nguyen TT, et al.: Effect of thyroid hormone replacement on lipoprotein (a), lipids, and apolipoproteins in subjects with hypothyroidism. Mayo Clin Proc 1998; 73: 837-841.
9.5.1.34. Dullaart RPF, Hoogenberg K, Groener JEM, et al.: The activity of cholesterol ester transfer protein is decreased in hypothyroidism: a possible contribution to alterations in high-density lipoproteins. Europ J Clin Invest 1990; 20: 581-587.
9.5.1.35. Tan KCB, Shiu SWM, Kung AWC: Plasma cholesteryl ester transfer protein activity in hyper- and hypothyroidism. J Clin Endocrinol Metab 1998; 83: 140-143.
9.5.1.36. Diekman MJM, Anghelescu N, Endert E, Bakker O, Wiersinga WM. Changes in plasma low-density lipoprotein (LDL)- and high-density lipoprotein cholesterol in hypo- and hyperthyroid patients are related to changes in free thyroxine, not to polymorphisms in LDL receptor or cholesterol ester transfer protein genes. J Clin Endocrinol Metab 2000; 85: 1857-1862.
9.5.1.37. Gjedde S, Gormsen LC, Rungby J et al. Decreased lipid intermediate levels and lipid oxidation rates despite normal lipolysis in patients with hypothyroidism. Thyroid 2010; 20: 843-849.
9.5.1.38. Tanaci N, Ertugrul DT, Sahin M et al. Postprandial lipemia as a risk factor for cardiovascular disease in patients with hypothyroidism. Endocrine 2006; 29: 451-456.
9.5.1.39. Makino M, Oda N, Miura N, et al. Effect of eicosapentaenoic acid ethyl ester on hypothyroid function. J Endocrinol 2001; 171: 259-265.
9.5.1.40. Ito M, Arishima T, Kudo T et al. Effect of levothyroxine replacement on non-high-density lipoprotein cholesterol in hypothyroid patients. J Clin Endocrinol Metab 2007; 92: 608-611.
9.5.1.41. Ito M, Takamutsu J, Matsuo T, et al. Serum concentrations of remnant-like particles in hypothyroid patients before and after thyroxine replacement. Clin Endocrinol 2003; 58:621-626.
9.5.1.42. Weintraub M, Grosskopf I, Trostanesky Y, et al. Thyroxine replacement enhances clearance of chylomicron remnants in patients with hypothyroidism. J Clin Endocrinol Metab 1999; 84: 2532-2536.
9.5.1.43. Pearce EN, Wilson PW, Yang Q, Vasan RS, Braverman LE. Thyroid function and lipid subparticle sizes in patients with short-term hypothyroidism and a population-based cohort. J Clin Endocrinol Metab 2008; 93: 888-894.
9.5.1.44. Baskol G, Atmaca H, Tanriverdi F, Baskol M, Kocer D, Bayram F. Oxidative stress and enzymatic antioxidant status in patients with hypothyroidism before and after treatment. Exp Clin Endocrinol Diab 2007; 115: 522-526.
9.5.1.45.Torun AN, Kulaksizoglu S, Kulaksizoglu M, Pamuk BO, Isbilen E, Tutuncu NB. Serum total antioxidant status and lipid peroxidation marker malondialdehyde levels in overt and subclinical hypothyroidism. Clin Endocrinol 2009; 70: 469-474.
9.5.1.46. Nanda N, Bobby Z, Hamide A, Koner BC, Sridhar MG. Association between oxidative stress and coronary lipid risk factors in hypothyroid women is independent of body mass index. Metabolism 2007; 56: 1350-1355.
9.5.1.47. Gjedde S, Gormsen LC, Riis AL et al. Reduced expression of uncoupling protein 2 in adipose tissue in patients with hypothyroidism. J Clin Endocrinol Metab 2010; 95: 3537-3541.
9.5.1.48. Teixeira PF, Cabral MD, Silva NA et al. Serum leptin in overt and subclinical hypothyroidism: effect of levothyroxine treatment and relationship to menopausal status and body composition. Thyroid 2009; 19: 443-450.
9.5.1.49. Kaplan O, Uzum AK, Aral H et al. Unchanged serum adipokine concentrations in the setting of short –term thyroidectomy-induced hypothyroidism. Endocr Pract 2012; 18: 887-893.
9.5.1.50. Kosowicz J, Baumann-Antczak A, Ruchala M, Grycznska M, Gurgul E, Sowinski J. Thyroid hormones affect plasma ghrelin and obestatin levels. Horm Metab Res 2011; 43: 121-125.
9.5.1.51. Guzel S, Seven A, Guzel EC, Buyuk B, Celebi A, Aydemir B. Visfatin, leptin, and TNF-α: interrelated adipokines in insulin-resistant clinical and subclinical hypothyroidism. Endocr Res 2013: [Epub ahead of print].
9.5.1.52. Goldberg IJ, Huang LS, Huggins LA et al. Thyroid hormone reduces cholesterol via a non-LDL receptor-mediated pathway. Endocrinology 2012; 153: 5143-5149.
9.5.1.53. Lin JZ, Martagon AJ, Hsueh WA et al. Thyroid hormone receptor agonists reduce serum cholesterol independent of the LDL receptor. Endocrinology 2012; 153: 6136-6144.
References for Section 9.5.2.
9.5.2.1. Smith TJ, Murata Y, Horwitz AL et al: Regulation of glycosaminoglycan accumulation by thyroid hormone in vitro. J Clin Invest 1982; 70 1066.
9.5.2.2. Aikawa JK: The nature of myxedema: Alterations in the serum electrolyte concentrations and radiosodium space and in the exchangeable sodium and potassium contents. Ann Intern Med 1956; 44: 30.
9.5.2.3. Crispell KR, Williams GA, Parson W, Hollifeld G: Metabolic studies in myxedema following administration of l-triiodothyronine: (1) Duration of negative nitrogen balance: (2) effect of testosterone proprionate: (3) comparison with nitrogen balance in a healthy volunteer. J Clin Endocrinol Metab 1957; 17: 221.
9.5.2.4. Smith TJ, Horwitz AL, Refetoff S: The effect of thyroid hormone on glycosaminoglycan accumulation in human skin fibroblasts. Endocrinology 1981; 108: 2397.
9.5.2.5. Smith TJ, Bahn RS, Gorman CA. Connective tissue, glycosaminoglycans, and diseases of the thyroid. Endocr Rev 1989; 10: 366-391.
9.5.2.6. Boelaert K, Newby PR, Simmonds MJ et al. Prevalence and relative risk of other autoimmune diseases in subjects with autoimmune thyroid disease. Am J Med 2010; 123: 183e1-183e9.
9.5.2.7. Artantas S, Gul U, Kilic A, Guler S. Skin findings in thyroid diseases. Eur J Intern Med 2009; 20: 158-161.
9.5.2.8. Levy Y, Segal N, Weintrob N, Danon YL. Chronic urticaria: association with thyroid autoimmunity. Arch Dis Child 2003; 88: 517-519.
References for Section 9.5.3.
9.5.3.1. Smith CD, Ain KB: Brain metabolism in hypothyroidism studied with 31P magnetic-resonance spectroscopy. Lancet 1995; 345: 619-620.
9.5.3.2. Dugbartey AT: Neurocognitive aspects of hypothyroidism. Arch Int Med 1998; 158: 1413-1418.
9.5.3.3. Jellinek EH, Kelly RE: Cerebellar syndrome in myxoedema. Lancet 1960; 2: 225.
9.5.3.4. Sanders V: Neurological manifestations of myxedema. N Engl J Med 1962; 266: 577, 599
9.5.3.5. Cremer GM, Goldstein NP, Paris J: Myxedema and ataxia. Neurology 1969; 19: 37.
9.5.3.6. Crevasse LE, Logue RB: Peripheral neuropathy in myxedema. Ann Intern Med 1959;50: 1433.
9.5.3.7. Nickel SN, Frame B: Nervous and muscular systems in myxedema. J Chronic Dis 1961; 14: 570.
9.5.3.8. Murray IPC, Simpson JA: Acroparaesthesia in myxoedema. Lancet 1958; 1: 1360.
9.5.3.9. Bland JH, Frymoyer JW: Rheumatoid syndrome of myxedema. N Engl J Med 1970; 282: 1171.
9.5.3.10. Frymoyer JW, Bland JH: Carpal-tunnel syndrome in patients with myxedematous arthropathy. J Bone Joint Surg 1973; 55A: 78.
9.5.3.11. Mra Z, Wax MK. Effects of acute thyroxin depletion on hearing in humans. The Laryngoscope 1999; 109: 343-350.
9.5.3.12. Cleare AJ, McGregor A, O'Keane V: Neuroendocrine evidence for an association between hypothyroidism, reduced central 5-HT activity and depression. Clin Endocrinol 1995; 43: 713-719.
9.5.3.13. Aronson R, Offman HJ, Joffe RT, Naylor CD: Triiodothyronine augmentation in the treatment of refractory depression. Arch Gen Psychiatry 1996; 53: 842-848.
9.5.3.14. Murray IPC: The reaction time in myxoedema.Lancet 1956; 2: 384.
9.5.3.15. Kapur VK, Koepsell TD, de Maine J, et al.: Association of hypothyroidism and obstructive sleep apnea. Am J Resp Crit Care Med 1998; 158: 1379-1383.
9.5.3.16. Tachman ML, Guthrie GP Jr: Hypothyroidism: diversity of presentation. Endocr. Rev 1984; 5: 456.
9.5.3.17. Loosen PT: Thyroid function in affective disorders and alcoholism. Endocrinol Metab Clin North Am 1988; 17: 55.
9.5.3.18. Clarnette RM, Peterson CJ: Hypothyroidism: does treatment cure dementia? J Geriatr Psychiat Neurol 1994; 7: 23-27.
9.5.3.19. Scheinberg P, Stead EA, Braman ES, Warren JV: Correlative observations on cerebral metabolism. J Clin Invest 1950; 29: 1139.
9.5.3.20. Sensenbach W, Madison L, Eisenberg S, Ochs L: The cerebral circulation and metabolism in hyperthyroidism and myxedema. J Clin Invest 1954; 33: 1434
9.5.3.21. O'Brien MD, Harris PWR: Cerebral-cortex perfusion rates in myxedema. Lancet 1965; 1: 1170.
9.5.3.22. Duyff RF, Bosch J vd, Laman DM, et al. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68: 750-755.
9.5.3.23. Gunarsson T, Sjöberg S, Eriksson M, Nordin C. Depressive symptoms in hypothyroid disorder with some observations on biochemical correlates. Neuropsychobiology 2001; 43: 70-74.
9.5.3.24. Kinuya S, Michigiski T, Tonami N, et al. Reversible cerebral hypoperfusion observed with Tc-99m HMPAO SPECT in reversible dementia caused by hypothyroidism. Clin Nucl Med 1999; 24: 666-668.
9.5.3.25. Constant EL, Volder AG de, Ivanoiu A, et al. Cerebral blood flow and glucose metabolism in hypothyroidism: a positron emission tomography study. J Clin Endocrinol Metab 2001; 86: 3864-3870.
9.5.3.26. Chong JY, Rowland LP, Utiger RD. Hashimoto encephalopathy: syndrome or myth? Arch Neurol 2003; 60: 164-171.
9.5.3.27. Fatourechi V. Hashimoto’s encephalopathy: myth or reality? An endocrinologist’s perspective. Best Pract Res Clin Endocrinol Metab 2005; 19: 53-66.
9.5.3.28. Burmeister LA, Ganguli M, Dodge HH, et al. Hypothyroidism and cognition: preliminary evidence for a specific defect in memory. Thyroid 2001; 11:1177-1185.
9.5.3.29. Miller KJ, Parsons TD, Whybrow PC et al. Verbal memory retrieval deficits associated with untreated hypothyroidism. J Neuropsychiatry Clin Neurosci 2007; 19: 132-136.
9.5.3.30. Anjana Y, Tandon OP, Vaney N, Madhu SV. Cognitive status in hypothyroid female patients: event-related evoked potential study. Neuroendocrinology 2008; 88: 59-66.
9.5.3.31. Correia N, Mullally S, Cooke G et al. Evidence for a specific defect in hippocampal memory in overt and subclinical hypothyroidism. J Clin Endocrinol Metab 2009; 94: 3789-3797.
9.5.3.32. Heinrich TW, Grahm G. Hypothyroidism presenting as psychosis: myxedema madness revisited. Prim Care Companion J Clin Psychiatry 2003; 5: 260-266.
9.5.3.33. Appelhof BC, Brouwer JP, van Dijck R et al. Triiodothyronine addition to paroxetine in the treatmentof major depressive disorder. J Clin Endocrinol Metab 2004; 89: 6271-6276.
9.5.3.34. Cooper-Kazaz R, Apter JT, Cohen R et al. Combined treatment with sertraline and liothyronine in major depression. A randomized, double-blind, placebo-controlled trial. Arch Gen Psychiatry 2007; 64: 679-688.
9.5.3.35. Thomsen AF, Kvist TK, Andersen PK, Kessing LV. Increased risk of developing affective disorder in patients with hypothyroidism: a register-based study. Thyroid 2005; 15: 700-707.
9.5.3.36. Bauer M, Silverman DH, Schlagenhauf F et al. Brain glucose metabolism in hypothyroidism: a positron emission tomography study before and after thyroid hormone replacement therapy. J Clin Endocrinol Metab 2009; 94: 2922-2929.
9.5.3.37. Comer DM, McConnell EM. Hypothyroid-associated sensorineuronal deafness. Ir J Med Sci 2010; 178: 621-622.
9.5.3.38. He XS, Ma N, Pan ZL et al. Functional magnetic resource imaging assessment of altered brain function in hypothyroidism during working memory processing. Eur J Endocrinol 2011; 164: 951-959.
9.5.3.39. Cooke G, Mullally S, Correia N, O’Mara S, Gibney J. Hippocampal volume is decreased in adult-onset hypothyroidism. Thyroid 2013; [Epub ahead of print].
9.5.3.40. Montero-Pedrazuela A, Fernandez-Lamo J, Alieva M, Pereda-Perez I, Venero C, Guadano-Ferraz A. Adult-onset hypothyroidism enhances fear memory and upregulates mineralocorticoid and glucocorticoid receptors in the amygdala. PLoS One 2011; 6: e26582.
9.5.3.41. Koromilas C, Liapi C, Schulpis KH, Kalafatakis K, Zarros A, Tsakiris S. Structural and functional alterations in the hippocampus due to hypothyroidism. Metab Brain Dis 2010; 3: 339-354.
9.5.3.42. Pilhatsch M, Marxen M, Winter C, Smolka MN, Bauer M. Hypothyroidism and mood disorders: integrating novel insights from brain imaging techniques. Thyroid Res 2011; 4 Suppl 1: S3.
References for Section 9.5.4.
9.5.4.1. Graettinger JS, Muenster JJ, Checchia CS et al.: A correlation of clinical and hemodynamic studies in patients with hypothyroidism. J Clin Invest 1958; 37: 502.
9.5.4.2. DeGroot WJ, Leonard JJ: The thyroid state and the cardiovascular system. Mod Concepts Cardiovasc Dis 1969; 38: 23.
9.5.4.3. Buccino RA, Spann JF Jr, Sonnenblock EH, Braunwald E: Effect of thyroid state on myocardial contractility. Endocrinology 1968; 82: 191.
9.5.4.4. Levey GS, Skelton L, Epstein SE: Decreased myocardial adenyl cyclase activity in hypothyroidism. J Clin Invest 1969; 48: 2244.
9.5.4.5. Santos AD, Miller RP, Puthenpurakal KM, et al: Echocardiographic characterization of the reversible cardiomyopathy of hypothyroidism. Am J Med 1980; 68: 675.
9.5.4.6. Fuller H Jr, Spittell JA Jr, McConahey WM, Schirger A: Myxedema and hypertension. Postgrad Med 1966; 40: 425.
9.5.4.7. Polikar R, Burger AG, Scherrer U, Nicod P: The thyroid and heart. Circulation 1993; 87: 1435.
9.5.4.8. Zondek H: Das Myxödemherz. Muenchen Med Wochenschr 1918; 65: 1180.
9.5.4.9. Ladenson PW, Sherman SI, Baughman KL et al.: Reversible alterations in myocardial gene expression in a young man with dilated cardiomyopathy and hypothyroidism. Proc Natl Acad Sci USA 1992; 89: 5251.
9.5.4.10. Ladenson PW: Recognition and management of cardiovascular disease related to thyroid dysfunction. Am J Med 1990; 88: 638.
9.5.4.11. Gordon AH: Pericardial effusion in myxedema. Trans Assoc Am Physians 1935; 50: 272.
9.5.4.12. Smolar EN, Rubin JE, Avramides A, Carter AC: Cardiac tamponade in primary myxedema and review of the literature. Am J Med Sci 1976; 272: 345.
9.5.4.13. Davis PJ, Jacobsen S: Myxedema with cardiac tamponade and pericardial effusion of "gold paint" appearance. Arch Intern Med 1967; 120: 615.
9.5.4.14. Hardisty CA, Naik DR, Munro DS: Pericardial effusion in hypothyroidism. Clin Endocrinol 1980; 13: 349.
9.5.4.15. Zondek H: The electrocardiogram in myxoedema. Br Heart J 1964; 26: 227.
9.5.4.16. Lee JK, Lewis JA: Myxoedema with complete A-V block and Adams-Stokes disease abolished with thyroid medication. Br Heart J 1962; 24: 253.
9.5.4.17. Cohen RD, Lloyd-Thomas HG: Exercise electrocardiogram in myxoedema. Br Med J 1966; 2: 327.
9.5.4.18. Fredlund B-O, Olsson SB: Long QT interval and ventricular tachycardia of "torsade de pointe" type in hypothyroidism. Acta Med Scand 1983; 213: 231.
9.5.4.19. Nesher G, Zion MM: Recurrent ventricular tachycardia in hypothyroidism - report of a case and review of the literature. Cardiology 1988; 75: 301.
9.5.4.20. Crowley WF, Ridgway EC, Bough EW et al.: Noninvasive evaluation of cardiac function in hypothyroidism: Response to gradual thyroxine replacement. N Engl J Med 1977; 296: 1.
9.5.4.21. Rodbard D, Fujita T, Rodbard S: Estimation of thyroid function by timing the arterial sounds. JAMA 1967; 201: 884.
9.5.4.22. Blumgart HL, Freedberg AS, Kurland GS: Hypercholesterolemia, myxedema, and atherosclerosis. Am J Med 1953; 14: 665.
9.5.4.23. Tielens E, Pillay M, Storm C, Berghout A. Cardiac function at rest in hypothyroidism evaluated by equilibrium radionuclide angiography. Clin Endocrinol 1999; 50: 497-502.
9.5.4.24. Tielens, Pillay M, Storm C, Berghout A. Changes in cardiac function at rest before and after treatment in primary hypothyroidism. Am J Cardiol 2000; 85: 376-380.
9.5.4.25. Steinberg AD: Myxedema and coronary artery disease - a comparative autopsy study. Ann Intern Med 1968; 68: 338.
9.5.4.26. Becker C: Hypothyroidism and atherosclerotic heart disease: pathogenesis, medical management, and the role of coronary artery bypass surgery. Endocr Rev 1985; 6: 432
9.5.4.27. Keating FR, Parkin TW, Selby JB, Dickinson LS: Treatment of heart disease associated with myxedema. Prog Cardiovasc Dis 1961; 3: 364.
9.5.4.28. Levine HD: Compromise therapy in the patient with angina pectoris and hypothyroidism: a clinical assessment. Am J Med 1980; 69: 411.
9.5.4.29. Diekman MJM, Harms MPM, Endert E, Wieling W, Wiersinga WM. Endocrine factors related to changes in total peripheral vascular resistance after treatment of thyrotoxic and hypothyroid patients. Eur J Endocrinol 2001; 144: 339-346.
9.5.4.30. Obuobie K, Smith J, Evans LM, et al. Increased central arterial stiffness in hypothyroidism. J Clin Endocrinol Metab 2002; 87: 4662-4666.
9.5.4.31. Dernellis J, Panaretou M. Effects of thyroid replacement therapy on arterial blood pressure in patients with hypertension and hypothyroidism. Am Heart J 2002; 143: 718-724.
9.5.4.32. Cappola AR, Ladenson PW. Hypothyroidism and atherosclerosis. J Clin Endocrinol Metab 2003; 88: 2438-2444.
9.5.4.33. Diekman MJM, van der Put NM, Blom HJ, et al. Determinants of changes in plasma homocysteine in hyperthyroidism and hypothyroidism. Clin Endocrinol 2001; 54: 197-204.
9.5.4.34. Christ-Crain M, Meier C, Guglielmetti M, et al. Elevated C-reactive protein and homocysteine values: cardiovascular risk factors in hypothyroidism? A cross-sectional and a double-blind placebo-controlled trial. Atherosclerosis 2003; 166: 379-386.
9.5.4.35. Razvi S, Weaver JU, Vanderpump MP, Pearce SH. The incidence of ischemic heart disease and mortality in people with subclinical hypothyroidism: reanalysis of the Whickham Survey cohort. J Clin Endocrinol Metab 2010; 95: 1734-1740.
9.5.4.36. Hak AE, Pols HA, Visser TJ, et al. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: the Rotterdam study. Ann Int Med 2000; 132: 270-278.
9.5.4.37. Kilein I, Danzi S. Thyroid disease and the heart. Circulation 2007; 116: 1725-1735.9.5.4.38. Danzi S, Klein I. Thyroid hormone and the cardiovascular system. Med Clin North Am 2012; 96: 257-268.
9.5.4.39. Pakula D, Marek B, Kajdaniuk D et al. Plasma levels of NT-pro-brain natriuretic peptide in patients with overt and subclinical hyperthyroidism and hypothyroidism. Endokrynol Pol 2011; 62: 523-528.
9.5.4.40. Biondi B. Mechanisms in endocrinology: heart failure and thyroid dysfunction. Eur J Endocrinol 2012; 167: 609-618.
9.5.4.41. Butala A, Chaudhari S, Sacerdote A. Cardiac tamponade as a presenting manifestation of severe hypothyroidism. BMJ Case Rep 2013;. Doi: 10.1136/bcr-12-2011-5281.
9.5.4.42. Thvilum M, Brandt F, Almind D, Christensen K, Brix TH, Hegedus L. Type and extent of somatic morbidity before and after the diagnosis of hypothyroidism. A nationwide register study. PLoS One 2013; 8: e75789. Doi: 10.371/journal.pone.0075789.
References for Section 9.5.5.
9.5.5.1. Wilson WR, Bedell GN: The pulmonary abnormalities in myxedema. J Clin Invest 1960; 39: 42.
9.5.5.2. Zwillich CW, Pierson DJ, Hofeldt FD et al.: Ventilatory control in myxedema and hypothyroidism. N Engl J Med 1975; 292: 662.
9.5.5.3. Nordqvist P, Dhunér KG, Stenberg K, Örndahl G: Myxedema coma and CO2 retention. Acta Med Scand 1960; 166; 189.
9.5.5.4. Weg JG, Calverly JR, Johnson C: Hypothyroidism and alveolar hypoventilation. Arch Intern Med 1965; 115: 302.
9.5.5.5. Orr WC, Males JL, Imes NK: Myxedema and obstructive sleep apnea. Am J Med 1981; 70: 1061
9.5.5.6. Ladenson PW, Goldenheim PD, Ridgway EC: Prediction and reversal of blunted ventilatory responsiveness in patients with hypothyroidism. Am J Med 1988: 84: 877-883.
9.5.5.7. Pelttari L, Rauhala E, Polo O, et al.: Upper airway obstruction in hypothyroidism. J Intern Med 1994; 236: 177-181.
9.5.5.8. George JT, Thow JC, Rodger KA, Mannion R, Jayagopal V. Reversibility of fibrotic appearance of lungs with thyroxine replacement therapy in patients with severe hypothyroidism. Endocr Pract 2009; 15: 720-724.
9.5.5.9. Mickelson SA, Lian T, Rosenthal L. Thyroid testing and thyroid hormone replacement in patients with sleep disordered breathing. Ear, Nose & Throut Journal 1999; 78: 768-775.
9.5.5.10. Attal P, Chanson P. Endocrine aspects of obstructive sleep apnea. J Clin Endocrinol Metab 2010; 95: 483-495.
References for Section 9.5.6.
9.5.6.1. Kaminsky P, Robin-Lherbier B, Brunotte F, et al.: Energetic metabolism in hypothyroid skeletal muscle, as studied by phosphorus magnetic resonance spectroscopy. J Clin Endocrinol Metab 1992; 74: 124-129.
9.5.6.2. Monzani F, Caraccio N, Siciliano G, et al.: Clinical and biochemical features of muscle dysfunction in subclinical hypothyroidism. J Clin Endocrinol Metab 1997; 82: 3315-3318.
9.5.6.3. Khaleeli AA, Griffith DG, Edwards RHT: The clinical presentation of hypothyroid myopathy. Clinical Endocrinology 1983; 19: 365.
9.5.6.4. Hurwitz LJ, McCormick D, Allen IV: Reduced muscle a-glucosidase (acid-maltase) activity in hypothyroid myopathy. Lancet 1970; 1: 67.
9.5.6.5. Waldstein SS, Bronsky D, Shrifter HB, Oester YT: The electromyogram in myxedema. Arch Intern Med 1958; 101: 97.
9.5.6.6. Debré R, Sémélaigne G: Syndrome of diffuse muscular hypertrophy in infants causing athletic appearance: Its connection with congenital myxedema. Am J Dis Child 1935; 50: 1351.
9.5.6.7. Thomasen E: Myotonia, Thomsen's Disease, Paramyotonia, Dystrophia Myotonica, Aarhus, Denmark, Universitetsforlaget, 1948.
9.5.6.8. Hsu I-H, Thadhani RI, Daniels GH: Acute compartment syndrome in a hypothyroid patient. Thyroid 1995; 5: 305-308.
9.5.6.9. Lambert EH, Underdahl LO, Beckett S, Mederos LO: A study of the ankle jerk in myxedema. J Clin Endocrinol Metab 1951; 11: 1186.
9.5.6.10. Famulski KS, Pilarska M, Wrzosek A, Sarzala MG: ATPase activity and protein phosphorylation in rabbit fast skeletal muscle sarcolemma. Eur J Biochem 1988; 171: 363.
9.5.6.11. Simonides WS, van Hardeveld C, Larsen PR: Identification of sequences in the promoter of the fast isoform of sarcoplasmic reticulum Ca-ATPase (SERCA1) required for transcriptional activation by thyroid hormone. Thyroid 1992; 2: S-102.
9.5.6.12. Bland JH, Frymoyer JW: Rheumatoid syndrome of myxedema. N Engl J Med 1970; 282: 1171.
9.5.6.13. Frymoyer JW, Bland JH: Carpal-tunnel syndrome in patients with myxedematous arthropathy. J Bone Joint Surg 1973; 55A: 78.
9.5.6.14. Bonakdarpour A, Kirkpatrick JA, Renzi A, Kendall N: Skeletal changes in neonatal thyrotoxicosis. Radiology 1972; 102: 149.
9.5.6.15. Krane S, Bronwell GL, Stanbury JB, Corrigan H: The effect of thyroid disease on calcium metabolism in man. J Clin Invest 1956; 35: 874.
9.5.6.16. Bouillon R, De Moor P: Parathyroid function in patients with hyper- or hypothyroidism. J Clin Endocrinol Metab 1974; 38: 999.
9.5.6.17. Lever EG: Primary hyperparathyroidism masked by hypothyroidism. Am J Med 1983; 74: 144.
9.5.6.18. Lave CE, Bird ED, Thomas WC: Hypercalcemia in myxedema. J Clin Endocrinol Metab 1962; 22: 261.
9.5.6.19. Bouillon R, Muls E, De Moor P: Influence of thyroid function on the serum concentration of 1,25-dihydroxy vitamin D3. J Clin Endocrinol Metab 1980; 51: 793.
9.5.6.20. Mundy GR, Shapiro JL, Bandelin JG, et al.: Direct stimulation of bone resorption by thyroid hormones. J Clin Invest 1976; 58: 529.
9.5.6.21. Eriksen EF: Normal and pathological remodeling of human trabecular bone: Three dimensional reconstruction of the remodeling sequence in normals and in metabolic bone disease. Endocr Rev 1986; 7: 379.
9.5.6.22. Duyff RF, Bosch J vd, Laman DM, et al. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68: 750-755.
9.5.6.23. Cakir M, Samanci N, Balci N, Balci MK. Musculoskeletal manifestations in patients with thyroid disease. Clin Endocrinol 2003; 59: 162-167.
9.5.6.24. Madariaga M. Polymyositis-like syndrome in hypothyroidism: review of cases reported over the past twenty-five years. Thyroid 2002; 12: 331-336.
9.5.6.25. Heemstra KA, Soeters MR, Fliers E et al. Type 2 iodothyronine deiodinase in skeletal muscle: effects of hypothyroidism and fasting. J Clin Endocrinol Metab 2009; 94: 2144-2150.
9.5.6.26. Vestergaard P, Mosekilde L. Fractures in patients with hyperthyroidism and hypothyroidism: a nationwide follow-up study in 16,249 patients. Thyroid 2002; 12: 411-419.
9.5.6.27. Vestergaard P, Rejnmark L, Mosekilde L. Influence of hyper- and hypothyroidism, and the effect of treatment with antithyroid drugs and levothyroxine on fracture risk. Calcif Tiss Int 2005; 77: 139-144.
References for Section 9.5.7.
9.5.7.1. Lerman J, Means JH: The gastric secretion in exophthalmic goitre and myxoedema. J Clin Invest 1932; 11: 167.
9.5.7.2. Donaich D, Roitt IM: An evaluation of gastric and thyroid auto-immunity in relation to hematologic disorders. Semin Hematol 1964; 1: 313.
9.5.7.3. Ebert EC. The thyroid and the gut. J Clin Gastroenterol 2010; 44: 402-406.
9.5.7.4. Lauritano EC, Bilotta AL, Gabrielli M et al. Association between hypothyroidism and small intestinal bacterial overgrowth. J Clin Endocrinol Metab 2007; 92: 4180-4184.
9.5.7.5. Rahman Q, Haboubi NY, Hudson PR, et al.: The effect of thyroxine on small intestinal motility in the elderly. Clin Endocrinol 1991; 35: 443-446
9.5.7.6. Hohl RD, Nixon RK: Myxedema ileus. Arch Intern Med 1965; 115: 145.
9.5.7.7. Nickerson JF, Hill SR, McNeil JH, Barker SB: Fatal myxedema with and without coma. Ann Intern Med 1960; 53: 475.
9.5.7.8. Tachman ML, Guthrie GP Jr: Hypothyroidism: diversity of presentation. Endocr Rev 1984; 5: 456.
9.5.7.9. Broitman SA, Bondy DC, Yachnin I et al.: Absorption and disabsorption of D-xylose in thyrotoxicosis and myxedema. N Engl J Med 1964; 270: 333.
9.5.7.10. Depczynski B, Ward R, Eisman J: The association between myxedematous ascites and extreme elevation of serum tumor markers. J Clin Endocrinol Metab 1996; 81: 4175.
9.5.7.11. Lorenzo y Losade H Jr, Staricoo EC, Cervino JM et al. Hypotonia of the galbladder of myxedematous origin. J Clin Endocrinol Metab 1957; 17: 133.
9.5.7.12. Cakir M, Kayacetin E, Toy H, Bozkurt S. Gallbladder motor function in patients with different thyroid hormone status. Exp Clin Endocrinol Diab 2009; 117: 395-399.
9.5.7.13. Fleisher G, McConahey W, Pankow M: Serum creatine kinase, lactic dehydrogenase, and glutamic-oxalacetic transaminase in thyroid diseases and pregnancy. Mayo Clinic Proc 1965; 40: 300.
9.5.7.14. Amino N, Kuro R, Yabu Y, et al.: Elevated levels of circulating carcinoembryonic antigens in hypothyroidism. J Clin Endocrinol Metab 1981; 52: 457.
9.5.7.15. Saha B, Maity C. Alterations of serum enzymes in primary hypothyroidism. Clin Chem Lab Med 2002; 40: 609-611.
9.5.7.16. Targher G, Montagnana M, Salvagno G et al. Association between serum TSH, free T4 and serum liver enzyme activities in a large cohort of unselected outpatients. Clin Endocrinol 2008; 68: 481-484.
References for Section 9.5.8.
9.5.8.1. Nedreb BG, Ericsson U-B, Nygård O, et al.: Plasma total homocysteine levels in hyperthyroid and hypothyroid patients. Metabolism 1998; 47: 89-93.
9.5.8.2. Yount E, Little JM: Renal clearance in patients with myxedema. J Clin Endocrinol Metab 1955; 15: 343.
9.5.8.3. Discala VA, Kinney MJ: Effects of myxedema on the renal diluting and concentrating mechanism. Am J Med 1971; 50: 325
9.5.8.4. Ford RV, Owens JC, Curd GW Jr et al.: Kidney function in various thyroid states. J Clin Endocrinol Metab 1961; 21: 548.
9.5.8.5. Moses AM, Gabrilove JL, Soffer LJ: Simplified water loading test in hypoadrenocorticism and hypothyroidism. J Clin Endocrinol Metab 1958; 18: 1413.
9.5.8.6. Crispell KR, Parson W, Sprinkle P: A cortisone-resistant abnormality in the diuretic response to ingested water in primary myxedema. J Clin Endocrinol Metab 1954; 14: 640.
9.5.8.7. Bleifer KH, Belsky JL, Saxon L, Papper S: The diuretic response to administered water in patients with primary myxedema. J Clin Endocrinol Metab 1960; 20: 409.
9.5.8.8. Davies CE, MacKinnon J, Platts MM: Renal circulation and cardiac output in "low-output" heart failure and in myxoedema. Br Med J 1952; 2: 595.
9.5.8.9. Pettinger WA, Talner L, Ferris TF: Inappropriate secretion of antidiuretic hormone due to myxedema. N Engl J Med 1965; 272: 361.
9.5.8.10. Showsky WR, Kikuchi TA: The role of vasopressin in the impaired water excretion of myxedema. Am J Med 1978; 64: 613.
9.5.8.11. Iwasaki Y, Oiso Y, Yamauchi K, et al.: Osmoregulation of plasma vasopressin in myxedema. J Clin Endocrinol Metab 1990; 70: 434.
9.5.8.12. Hanna FWF, Scanlon MF: Hyponatraemia, hypothyroidism and role of arginine-vasopressin. Lancet 1997; 350: 755-756.
9.5.8.13. Montenegro J, Gonzalez O, Saracho R, et al.: Changes in renal function in primary hypothyroidism. Am J Kidney Dis 1996; 27: 195-198.
9.5.8.14. Leeper RD, Benua RS, Brener JL, Rawson RW: Hyperuricemia in myxedema. J Clin Endocrinol Metab 1960; 20: 1457.
9.5.8.15. Jones JE, Desper PC, Shane SR, Flink EB: Magnesium metabolism in hyperthyroidism and hypothyroidism. J Clin Invest 1966; 45: 891.
9.5.8.16. Bernstein R, Midtb K, Urdal P, et al.: Serum N-terminal pro-atrial natriuretic factor 1-98 before and during thyroxine replacement therapy in severe hypothyroidism. Thyroid 1997; 7: 415-419.
9.5.8.17. Diekman MJM, Put NM vd, Blom HJ, Tijssen JGP, Wiersinga WM. Determinants of changes in plasma homocysteine in hyperthyroidism and hypothyroidism. Clin Endocrinol 2001; 54: 197-204.
9.5.8.18. Sahun M, Villabona C, Rosel P, et al. Hypothyroidism is associated with plasma hypo-osmolality and impaired water excretion that is vasopressin-independent. J Endocrinol 2001; 168:435-445.
9.5.8.19. Goede DL, Wiesli P, Brandle M et al. Effects of thyroxine replacement on serum creatinine and cystatin C in patients with primary and central hypothyroidism. Swiss Med Wkly 2009; 139: 339-344.
9.5.8.20. Schrier RW. Vasopressin and aquaporin 2 (AQP2) in clinical disorders of water homeostasis. Semin Nephrol 2008; 28: 289-296.
9.5.8.21. Chen Y-C, Cadnapaphomchai MA, Yang J et al. Nonosmotic release of vasopressin and renal aquaporins in impaired urinary dilution in hypothyroidism. Am J Physiol Renal Physiol 2005; 289: F672-F678.
9.5.8.22. Iglesias P, Diez JJ. Thyroid dysfunction and kidney disease. Eur J Endocrinol 2009; 160: 503-515.
9.5.8.23. Kimmel M, Braun N, Alscher MD. Influence of thyroid function on different kidney function tests. Kidney Blood Press Res 2012; 35: 9-17.
9.5.8.24. Ye Y, Gai X, Xie H, Jiao L, Zhang S. Impact of thyroid function on serum cystatin C and estimated glomerular filtration rate: a cross-sectional study. Endocr Pract 2013; 19: 397-403.
9.5.8.25. Tsuda A, Inaba M, Ichii M et al. Relationship between serum TSH levels and intrarenal hemodynamic parameters in euthyroid subjects. Eur J Endocrinol 2013; 169: 45-50.
9.5.8.26. Schwarz C, Leichtle AB, Arampatzis S et al. Thyroid function and serum electrolytes: does an association really exists? Swiss Med Wkly 2012; 142: w13669. Doi: 10.4414/smw.2012.13669.
9.5.8.27. Hammami MM, Almogbel F, Hammami S, Faifi J, Alqahtani A, Hashem W. Acute severe hypothyroidism is not associated with hyponatremia even with increased water intake: a prospective study in thyroid cancer patients. BMC Endocr Disord 2013; 13:27 [Epub ahead of print].
9.5.8.28. Peri A. Clinical review: the use of vaptans in clinical endocrinology. J Clin Endocrinol Metab 2013; 98: 1321-1332.
9.5.8.29. Karmisholt J, Andersen S, Laurberg P. Weight loss after therapy of hypothyroidism is mainly caused by excretion of excess body water associated with myxoedem. J Clin Endocrinol Metab 2011; 96: E99-E103.
References for Section 9.5.9.
9.5.9.1. Krassas GE, Poppe K, Glinoer D. Thyroid function and human reproductive health. Endocrine Rev 2010; 31: doi:10.1210/er.2009-0041.
9.5.9.2. Tagawa N, Takano T, Fukata S et al. Serum concentrations of androstenediol and androstenediol sulfate in patients with hyperthyroidism and hypothyroidism. Endocr J 2001; 48: 345-354.
9.5.9.3. Gordon GG, Southren AL: Thyroid-hormone effects on steroid-hormone metabolism. Bull NY Acad Med 1977; 53: 241.
9.5.9.4. Copinschi G, Leclercq R, Bruno OD, Cornil A: Effects of altered thyroid function upon cortisol secretion in man. Horm Metab Res 1971; 3: 437.
9.5.9.5. Gordon GG, Southern AL, Tochimoto S, et al.: Effect of hyperthyroidism and hypothyroidism on testosterone and androstenedione in man. J Clin Endocrinol Metab 1969; 29: 164.
9.5.9.6. De La Balze FA, Arrillaga F, Mancini RE, et al.: Male hypogonadism in hypothyroidism: a study of six cases. J Clin Endocrinol Metab 1962; 22: 212.
9.5.9.7. Hopwood NJ, Lockhart LH, Bryan GT: Acquired hypothyroidism with muscular hypertrophy and precocious testicular enlargement. J Pediatr 1974; 85: 233
9.5.9.8. Bruder JM, Samuels MH, Bremner WJ, et al.: Hypothyroidism-induced macroorchidism: use of a gonadotropin-releasing hormone agonist to understand its mechanism and augment adult stature. J Clin Endocrinol Metab 1995; 80: 11-16.
9.5.9.9. Anasti JN, Flack MR, Froehlich J, et al.: A potential novel mechanism for precocious puberty in juvenile hypothyroidism. J Clin Endocrinol Metab 1995; 80: 276-279.
9.5.9.10. Carani C, Isidori AM, Granata A et al. Multicenter study on the prevalence of sexual symptoms in male hypo- and hyperthyroid patients. J Clin Endocrinol Metab 2005; 90: 6472-6479.
9.5.9.11. Krassas GE, Tziomalos K, Papadopoulou F, Pontikides N, Perros P. Erectile dysfunction in patients with hyper- and hypothyroidism: how common and should we treat? J Clin Endocrinol Metab 2008; 93: 1815-1819.
9.5.9.12. Corrales Hernandez JJ, Miralles Garcia JM, Garcia Diez LC. Primary hypothyroidism and human spermatogenesis. Arch Androl 1990; 25: 21-27.
9.5.9.13. Jaya Kumar B, Khurana ML, Ammini AC, Kamarkar MG, Ahuja MM. Reproductive endocrine functions in men with primary hypothyroidism: effect of thyroxine replacement. Horm Res 1990; 34: 215-218.
9.5.9.14. Krassas GE, Papadopoulou F, Tziomalos K, Zeginiadou T, Pontikides N. Hypothyroidism has an adverse effect on human spermatogenesis: a prospective, controlled study. Thyroid 2008; 18: 1255-1259.
9.5.9.15. Longcope C, Abend S, Braverman LE, Emerson CH. Androstenedione and estrone dynamics in hypothyroid women. J Clin Endocrinol Metab 1990; 70: 903-907.
9.5.9.16. Costin G, Kershnar AK, Kogut MD, Turkington RW. Prolactin activity in juvenile hypothyroidism and precocious puberty. Pediatrics 1972; 50: 881.
9.5.9.17. Goldsmith RE, Sturgis SH, Lerman J, Stanbury JB: The menstrual pattern in thyroid disease. J Clin Endocrinol Metab 1952; 12: 846.
9.5.9.18. Samuels MH, Veldhuis JD, Henry P, Ridgway EC: Pathophysiology of pulsatile and copulsatile release of thyroid-stimulating hormone, luteinizing hormone, follicle-stimulating hormone, and alpha-subunit. J Clin Endocrinol Metab 1990; 71: 425-432
9.5.9.19. Krassas GE, Pontikides N, Kaltsas Th et al. Disturbances of menstruation in hypothyroidism. Clin Endocrinol 1999; 50: 655-659.
9.5.9.20. Benson RC, Dailey ME. The menstrual pattern in hyperthyroidism and subsequent posttherapy hypothyroidism. Surg Gynecol Obstet 1955; 100: 19-26.
9.5.9.21. Goldsmith RE, Sturgis SH, Lerman J, Stanbury JB. The menstrual pattern in thyroid disease. J Clin Endocrinol Metab 1952; 12: 846-855.
9.5.9.22. Scott Jr JC, Mussey E. Menstrual patterns in myxedema. Am J Obstet Gynecol 1964; 90: 161-165.
9.5.9.23. Joshi JV, Bhandarkar SD, Chadha M, Balaiah D, Shah R. Menstrual irregularities and lactation failure may precede thyroid dysfunction or goitre. J Postgrad Med 1993; 39: 137-141.
9.5.9.24. Ross F, Nusynowitz ML. A syndrome of primary hypothyroidism, amenorrhea and galactorrhea. J Clin Endocrinol Metab 1968; 28: 591.
9.5.9.25. Davis LE, Leveno KJ, Cunningham FG: Hypothyroidism complicating pregnancy. Obstet Gynecol 1988; 72: 108.
9.5.9.26. Potter JD: Hypothyroidism and reproductive failure. Surg Gynecol Obstet 1980; 150: 251.
9.5.9.27. Montoro M, Collea JV, Frasier SD, Mestman JH: Successful outcome of pregnancy in women with hypothyroidism. Ann Intern Med 1981; 94: 31.
9.5.9.28. Lincoln SR, Ke RW, Kutteh WH. Screening for hypothyroidism in infertile women. J Reprod Med 1999; 44: 455-457.
9.5.2.29. Cramer DW, Sluss PM, Powers RD et al. Serum prolactin and TSH in an in vitro fertilization population: is there a link between fertilization and thyroid function? J Assist Reprod Genet 2003; 20: 210-215.
9.5.9.30. Abalovich M, Gutierrez S, Alcaraz G, Maccallini G, Garcia A, Levalle O. Overt and subclinical hypothyroidism complicating pregnancy. Thyroid 2002; 12: 63-68.
9.5.9.31. Liu H, Momotani N, Noh JY et al.: Maternal hypothyroidism during early pregnancy and intellectual development of progeny. Arch Intern Med 1994; 154: 785.
9.5.9.32. Man EB, Brown JF, Serunian SA: Maternal hypothyroxinemia: Psychoneurological deficits of progeny. Ann Clin Lab Ser 1991; 21: 227.
9.5.9.33. Smallridge RC, Ladenson PW. Hypothyroidism in pregnancy: consequences to neonatal health. J Clin Endocrinol Metab 2001; 86: 2349-2353.
9.5.9.34. Haddow JE, Palomaki GE, Allan WC, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl. J Med 1999; 341: 549-555.
9.5.9.35. Abalovich M, Amino N, Barbour LA et al. Management of thyroid dysfunction during pregnancy and postpartum. An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2007; 92 (Supplement): S1-S47.
9.5.9.36. Krassas GE, Poppe K, Glinoer D. Thyroid function and human reproductive health. Endocr Rev 2010; 31: 702-755.
9.5.9.37. Stagnaro-Green A, Abalovich M, Alexander E et al. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid diseases during pregnancy and postpartum. Thyroid 2011; 21: 1081-1125.
9.5.9.38. De Groot L, Abalovich M, Alexander EK et al. Management of thyroid dysfunction during pregnancy and postpartum: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2012; 97: 2543-2565.
References for Section-9.5.10.
9.5.10.1. Yamada T, Tsukui T, Ikejiri K, et al.: Volume of sella turcica in normal subjects and in patients with primary hypothyroidism and hyperthyroidism. J Clin Endocrinol Metab 1976; 42: 817.
9.5.10.2. Yamamoto K, Saito K, Takai T, et al.: Visual field defects and pituitary enlargement in primary hypothyroidism. J Clin Endocrinol Metab 1983; 57: 283.
9.5.10.3. Vagenakis AG, Dole K, Braverman LE: Pituitary enlargement, pituitary failure, and primary hypothyroidism. Ann Intern Med 1976; 85: 195.
9.5.10.4. Sarlis NJ, Brucker-Davis F, Doppman JL, Skarulis MC: MRI-demonstrable regression of a pituitary mass in a case of primary hypothyroidism after a week of acute thyroid hormone therapy. J Clin Endocrinol Metab 1997; 82: 808-811.
9.5.10.5. Honbo KS, Van Herle AJ, Kellett KA: Serum prolactin levels in untreated primary hypothyroidism. Am J Med 1978; 64: 782.
9.5.10.6. Onishi T, Miyai K, Aono T, et al.: Primary hypothyroidism and galactorrhea. Am J Med 1977; 63: 373.
9.5.10.7. Brauman H, Corvilain J: Growth hormone response to hypoglycemia in myxedema. J Clin Endocrinol Metab 1968; 28: 301.
9.5.10.8. Valcavi R, Valente F, Dieguez C, et al.: Evidence against depletion of the growth hormone (GH)-releasable pool in human primary hypothyroidism: studies with GH-releasing hormone, pyridostigmine, and arginine. J Clin Endocrinol Metab 1993; 77: 616-620.
9.5.10.9. Miell JP, Taylor AM, Zini M, et al.: Effects of hypothyroidism and hyperthyroidism on insulin-like growth factors (IGFs) and growth hormone- and IGF-binding proteins. J Clin Endocrinol Metab 1993; 76: 950.
9.5.10.10. Miell JP, Zini M, Quin JD, et al.: Reversible effects of cessation and recommencement of thyroxine treatment on insulin-like growth factors (IGFs) and IGF-binding proteins in patients with toal thyroidectomy. J Clin Endocrinol Metab 1994; 79: 1507-1512.
9.5.10.11. Gordon GG, Southren AL: Thyroid-hormone effects on steroid-hormone metabolism. Bull NY Acad Med 1977; 53: 241.
9.5.10.12. Brown H, Englert E, Wallach S: Metabolism of free and conjugated 17-hydroxycorticosteroids in subjects with thyroid disease. J Clin Endocrinol Metab 1958; 18: 167.
9.5.10.13. Copinschi G, Leclercq R, Bruno OD, Cornil A: Effects of altered thyroid function upon cortisol secretion in man. Horm Metab Res 1971; 3: 437.
9.5.10.14. Hellman L, Bradlow HL, Zumoff B, et al.: Thyroid-androgen interrelations and the hypocholesteremic effect of androsterone. J Clin Endocrinol Metab 1959; 19: 939.
9.5.10.15. Ogihara T, Yamamoto T, Miyai K, Kumahara Y: Plasma renin activity and aldosterone concentration of patients with hyperthyroidism and hypothyroidism. Endocrinol Jpn.
9.5.10.16. Saruta T, Kitajima W, Hayashi M, et al.: Renin and aldosterone in hypothyroidism: relation to excretion of sodium and potassium. Clin Endocrinol 1980; 12: 483.
9.5.10.17. Felber JP, Reddy WJ, Selenkow HA, Thorn GW: Adrenocortical response to the 48-hour ACTH test in myxedema and hyperthyroidism. J Clin Endocrinol Metab 1959; 19: 895.
9.5.10.18. Liddle GW, Estep HL, Kendall JW Jr, et al.: Clinical application of a new test of pituitary reserve. J Clin Endocrinol Metab 1959; 19: 875.
9.5.10.19. Gold EM, Kent JR, Forsham PH: Clinical use of a new diagnostic agent, methopyrapone (SU-4885), in pituitary and adrenocortical disorders. Ann Intern Med 1961; 54: 175.
9.5.10.20. Lessof MH, Maisey MN, Lyne C, Sturge RA: Effect of thyroid failure on the pituitary-adrenal axis. Lancet 1969; I: 642.
9.5.10.21. Ridgway EC, McCammon JA, Benotti J, Maloof F: Acute metabolic responses in myxedema to large doses of intravenous L-thyroxine. Ann Intern Med 1972; 77: 549.
9.5.10.22. Bigos ST, Ridgway EC, Kourides JA, Maloof F: Spectrum of pituitary alterations with mild and severe thyroid impairment. J Clin Endocrinol Metab 1978; 46: 317.
9.5.10.23. Kamilaris TC, DeBold CR, Pavlou SN, et al.: Effect of altered thyroid hormone levels on hypothalamic-pituitary-adrenal function. J Clin Endocrinol Metab 1987; 65: 994.
9.5.10.24. Clausen N, Lins PE, Adamson U, et al.: Couterregulation of insulin-induced hypoglycemia in primary hypothyroidism. Acta Endocrinol 1986; 111: 516.
9.5.10.25. Carpenter CCJ, Solomon N, Silverberg SG, et al.: Schmidt's syndrome (thyroid and adrenal insufficiency): a review of the literature and a report of fifteen new cases including ten instances of coexistent diabtes mellitus. Medicine 1964; 43: 153.
9.5.10.26. Salvi M, Fukazawa H, Bernard N, et al.: Role of autoantibodies in the pathogenesis and association of endocrine autoimmune disorders. Endocr Rev 1988; 9: 450.
9.5.10.27. Lack EE: Lymphoids "hypophysitis" with end organ insufficiency. Arch Pathol 1975; 99: 215.
9.5.10.28. Gharib H, Gastineau CF, Hodgson SF, et al.: Reversible hypothyroidism in Addison's disease. Lancet 1972; 2:734.
9.5.10.29. Christiansen CJ: Increased levels of plasma noradrenalin in hypothyroidism. J Clin Endocrinol Metab 1972; 35: 359.
9.5.10.30. Stoffer SS, Jiang N-S, Gorman CA, Pikler GM: Plasma catecholamines in hypothyroidism and hyperthyroidism. J Clin Endocrinol Metab 1973; 36: 587.
9.5.10.31. Heemstra KA, Burggraaf J, van der Klaauw AA, Romijn JA, Smit JW, Corssmit EP. Short-term overt hypothyroidism induces sympathovagal imbalance in thyroidectomized differentiated thyroid carcinoma patients. Clin Endocrinol 2010; 72: 417-421.
9.5.10.32. Raab W: Epinephrine tolerance of the heat altered by thyroxine and thiouracil. J Pharmacol Exp Ther 1944; 82: 330.
9.5.10.33. Raab W: Diminution of epinephrine sensitivity of the normal human heart through thiouracil. J Lab Clin Med 1945; 30: 774.
9.5.10.34. Schneckloth RE, Kurland GS, Freedberg AS: Effect of variation in thyroid function on the pressor response to norepinephrine in man. Metabolism 1953; 2: 546.
9.5.10.35. Polikar R, Kennedy B, Maisel A, et al.: Decreased adrenergic sensitivity in patients with hypothyroidism. J Am Coll Cardiol 1990; 15: 94-98.
9.5.10.36. Iglesias P, Bayon C, Mendez J, et al. Serum insulin-like growth factor type 1, insulin-like growth factor-binding protein-1, and insulin-like growth factor-binding protein-3 concentrations in patients with thyroid dysfunction. Thyroid 2001; 11: 1043-1048.
9.5.10.37. Schmid C, Zwimpfer C, Brandle M, Krayenbuhl PA, Zapf J, Wiesli P. Effect of thyroxine replacement on serum IGF-1, IGFBP-3, and the acid-labile subunit in patients with hypothyroidism and hypopituitarism. Clin Endocrinol 2006; 65: 706-711.
9.5.10.38. Eskes SA, Endert E, Fliers E, Wiersinga WM. Prevalence of growth hormone deficiency in Hashimoto’s thyroiditis. J Clin Endocrinol Metab 2010; 95: 2266-2270.
9.5.10.39. Raber W, Gesol A, Nowotny P, Vierhapper H. Hyperprolactinaemia in hypothyroidism: clinical significance and impact of TSH normalization. Clin Endocrinol 2003; 58: 185-191.
9.5.10.40. Silva JE, Bianco SD. Thyroid-adrenergic interactions: physiological and clinical implications. Thyroid 2008; 18: 157-165.
References for Section 9.5.11.
9.5.11.1. Watanakunakorn C, Hodges RE, Evans TC: Myxedema: a study of 400 cases. Arch Intern Med 1965; 116: 183.
9.5.11.2. Stern B, Altshule MD: Hematological studies in hypothyroidism following total thyroidectomy. J Clin Invest 1936; 15: 633.
9.5.11.3. Bomford R: Anaemia in myxoedema: and the role of the thyroid gland in erythropoiesis. Q J Med 1938; 7: 495.
9.5.11.4. Shalet M, Coe D, Reisman KR: Mechanism of erythropoietic action of thyroid hormone. Proc Soc Exp Biol Med 1966; 123: 443.
9.5.11.5. Tudhope GR, Wilson GM: Anemia in hypothyroidism. Q J Med 1960; 29: 513.
9.5.11.6. Muldowney FP, Crooke J, Wayne EJ: The total red cell mass in thyrotoxicosis and myxoedema. Clin Sci 1957; 16: 309.
9.5.11.7. Leithold SL, David D, Best WR: Hypothyroidism with anemia demonstrating abnormal vitamin B12 absorption. Am Jmed 1958; 24: 535.
9.5.11.8. Tudhope GR, Wilson GM: Deficiency of vitamin B12 in hypothyroidism. Lancet 1962; 1: 703.
9.5.11.9. Hines JD, Halstead CH, Criggs RC, Harris JW: Megaloblastic anemia secondary to folate deficiency associated with hypothyroidism. Ann Intern Med 1968; 68: 792.
9.5.11.10. Wardrop C, Hutchinson HE: Red cell shape in hypothyroidism. Lancet 1969; 1: 1243.
9.5.11.11. Lillington GA, Gastineau CF, Underdahl LO: The sedimentation rate in primary myxedema. Proc Staff Meetings Mayo Clinic 1959; 34: 605.
9.5.11.12. Squizzato A, Roumadi E, Buller HR, Gerdes VEA. Thyroid dysfunction and effects on coagulation and fibrinolysis: a systematic review. J Clin Endocrinol Metab 2007; 92: 2415-2420.
9.5.11.13. Ford HC, Carter JM: Haemostasis in hypothyroidism. Postgrad Med J 1990; 66: 280-284.
9.5.11.14. Hofbauer LC, Heufelder A: Coagulation disorders in thyroid disease. Europ J Endocrinol 1997; 136: 1-7.
9.5.11.15. Verkleij CJ, Stuijver DJ, van Zaane B et al. Thrombin-activatable fibrinolysis inhibitor in hypothyroidism and hyperthyroxinemia. Thromb Haemost 2013; 109: 214-220.
9.5.11.16. Manfredi E, van Zaane B, Gerdes VEA, Brandjes DPM, Squizzato A. Hypothyroidism and acquired von Willebrand’s syndrome: a systematic review. Haemophilia 2008; 14: 423-433.
9.5.11.17. Erfurth EM, Ericsson U-BC, Egervalh K, Lethagen SR: Effect of acute desmopressin and of long-term thyroxine replacement on haemostasis in hypothyroidism. Clin Endocrinol 1995; 42: 373-378.
9.5.11.18 Showsky WR, Kikuchi TA: The role of vasopressin in the impaired water excretion of myxedema. Am J Med 1978; 64: 613.
9.5.11.19. Chadarevian R, Bruckert E, Leenhardt L, Giral Ph, Ankri A, Turpin G. Components of the fibrinolytic system are differently altered in moderate and severe hypothyroidism. J Clin Endocrinol Metab 2001; 86: 732-737.
9.5.11.20. De Vito P, Incerpi S, Pedersen JZ, Luly P, Davis FB, Davis PJ. Thyroid hormones as modulators of immune activities at the cellular level. Thyroid 2011; 21: 879-890.
9.5.11.21. Kim JH, Park JH, Kim SY, Bae HY. The mean platelet volume is positively correlated with serum thyrotropin concentrations in a population of healthy subjects and subjects with unsuspected subclinical hypothyroidism. Thyroid 2013; 23: 31-37.
9.5.11.22. Erikci AA, Karagoz B, Ozturk A et al. The effect of subclinical hypothyroidism on platelet parameters. Hematology 2009; 14: 115-117.
9.5.11.23. Torun AN, Uzum AK, Aksoy N. Overt and mild subclinical hypothyroidism do not influence mean platelet volume in premenopausal women having low cardiac risk. Clin Appl Thromb Hemost 2012; 18: 312-315.
9.5.11.24. Stuijver DJ, Plantanida E, van Zaane B et al. Acquired von Willebrand syndrome in patients with overt hypothyroidism: a prospective cohort study. Haemophilia 2013 [Epub ahead of print]. Doi: 10.1111/hae.12275.
9.5.11.25. Yango J, Alexopoulou O, Eeckhoudt S, Hermans C, Daumerie C. Evaluation of the respective influence of thyroid hormones and TSH on blood coagulation parameters after total thyroidectomy. Eur J Endocrinol 2011; 164: 599-603.
9.5.11.26. Vescovi PP, Favorolo EJ, Lippi G et al. The spectrum of coagulation abnormalities in thyroid disorders. Semin Thromb Hemost 2011; 37: 7-10.
9.5.11.27. Marongiu F, Barcellona D, Mameli A, Mariotti S. Thyroid disorders and hypercoagulability. Semin Thromb Hemost 2011; 37: 11-16.
References for Section 9.6.
9.6.1. Ord WM: In Allbutt TC (ed): System of Medicine. New York. The Macmillan Co, 1897.
References for Sections 9.7.1-9.7.3.
9.7.1. Tachman ML, Guthrie GP: Hypothyroidism: diversity of presentation. Endocr Rev 1984; 5: 456-465.
9.7.2. Billewicz WL, Chapman RS, Crooks J, et al.: Statistical methods applied to the diagnosis of hypothyroidism. Q J Med (New Series) 1969; 38: 255-266.
9.7.3. Seshadri MS, Samuel BU, Kanagasabapathy AS, Cherian AM: Clinical scoring system for hypothyroidism: is it useful? J Gen Intern Med 1989; 4: 490-492.
9.7.4. Zulewski H, Müller B, Exer P, et al.: Estimation of tissue hypothyroidism by a new clinical score: evaluation of patients with various grades of hypothyroidism and controls. J Clin Endcrinol Metab 1997; 82: 771-776.
9.7.5. Doucet J, Trivalle Ch, Chassagne Ph, et al.: Does age play a role in clinical presentation of hypothyroidism? J Am Geriatr Soc 1994; 42: 984-986.
9.7.6. Müller B, Zulewski H, Huber P, et al.: Impaired action of thyroid hormone associated with smoking in women with hypothyroidism. New Engl J Med 1995; 333: 964-969.
9.7.7. Nikolai TF: Recovery of thyroid function in primary hypothyroidism. Am J Med Sci 1989; 297: 18-21.
9.7.8. Comtois R, Faucher L, Laflèche L: Outcome of hypothyroidism caused by Hashimoto's thyroiditis. Arch Int Med 1995; 155: 1404-1408.
9.7.9. Okamura K, Sato K, Ikenoue H, et al.: Reevaluation of thyroidal radioactive iodine uptake test, with special reference to reversible primary hypothyroidism with elevated thyroid radioiodine uptake. J Clin Endocrinol Metab 1988; 67: 720-726.
9.7.10. Takasu N, Komiya I, Asawa T, et al.: Test for recovery from hypothyroidism during thyroxine therapy in Hashimoto's thyroiditis. Lancet 1990; 336: 1084-1086.
9.7.11. Wiersinga WM, Touber JL, Trip MD, van Royen EA: Uninhibited thyroidal uptake of radioiodine despite iodine excess in amiodarone-induced hypothyroidism. J Clin Endocrinol Metab 1986; 63: 485-491.
9.7.12. Garber JR, Cobin RH, Gharib H, et al. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Thyroid 2012; 22: 1200-1235.
9.7.13. Bochukova E, Schoenmakers N, Agostini M, et al: A mutation in the thyroid hormone receptor alpha gene. . N Engl J Med 2012; 366: 243-249.
9.7.14. Van Mullem A, Van Heerebeek R, Chrysis D et al: Clinical phenotype and mutant TRα1. N Engl J Med 2012; 366: 1451-1453.
9.7.15. Pedersen OM, Aardal NP, Larssen TB, Varhaug JE, Myking O, Vik-Mo H: The value of ultrasonography in predicting autoimmune thyroid disease. Thyroid 2000; 10: 251-259.
9.7.16. Boucai L, Hollowell JG, Surks MI. An approach for development of age-, gender-, and ethnicity=specific thyrotropin reference limits. Thyroid 2011; 21: 5-11.
References for Section 9.8.1.
9.8.1.1. Hays MT: Localization of human thyroxine absorption. Thyroid 1991; 3: 241.
9.8.1.2. Hays MT: Thyroid hormone and the gut. Endocr Res 1988; 14: 203.
9.8.1.3. Fish LH, Schwartz HL, Cavanaugh J, et al.: Replacement dose, metabolism, and bioavailability of levothyroxine in the treatment of hypothyroidism. Role of triiodothyronine in pituitary feedback in humans. New Engl J Med 1987; 316: 764-770.
9.8.1.4. Bach-Huynh TG, Nayak B, Loh J et al.: Timing of levothyroxine administration affects serum thyrotropin concentration. J Clin Endocrinol Metab 2009; 94: 3905-3912.
9.8.1.5. Wennlund A: Variation in serum levels of T3, T4, FT4 and TSH during thyroxine replacement therapy. Acta Endocrinol 1986; 113: 47.
9.8.1.6. Browning MCK, Bennet WM, Kirkaldy AJ, Jung RT: Intra-individual variation of thyroxine, triiodothyronine, and thyrotropin in treated hypothyroid patients: Implications for monitoring replacement therapy. Clin Chem 1988; 34: 696.
9.8.1.7. Daniels GH. Response to “How do you approach the problem of TSH elevation in a patient on high-dose thyroid hormone replacement?” Clin Endocrinol 2009; 71: 603.
9.8.1.8. Dong BJ, Hauck WW, Gambertoglio JG et al. Bioequivalence of generic and brand-name levothyroxine products in the treatment of hypothyroidism. JAMA 1997; 227: 1205-1213.
9.8.1.9. Olveira G, Almaraz MC, Soriguer F et al. Altered bioavailability due to changes in the formulation of a commercial preparation of levothyroxine in patients with differentiated carcinoma. Clin Endocrinol 1997; 46: 707-711.
9.8.1.10. Carr D, McLeod DT, Parry G, Thornes HM. Fine adjustment of thyroxine replacement dosage: Comparison of the thyrotropin releasing hormone test using a sensitive thyrotropin assay with measurement of free thyroid hormones and cli nical assessment. Clin Endocrinol 1988; 28: 325-333.
9.8.1.11. Canaris GJ, Manowitz NR, Mayor GM, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Int Med 2000; 160: 526-534.
9.8.1.12. Hollowell JG, Staehling NW, Flanders WD et al. Serum TSH, T4, and thyroid antibodies in the United States Population (1988 to 1994): National Hea;th and Nutritional Examination Survey (NHANES III). J Clin Endocrinol Metab 2002; 87: 489-499.
9.8.1.13. Food and Drug Administration, Center for Drug Evaluation and Research 2000 Guidance for Industry: Levothyroxine sodium tablets – In vivo pharmacokinetic and bioavailability studies and in vitro dissolution testing. www.fda.gov/cder/guidance/364fnl.htm
9.8.1.14. American Thyroid Association, Endocrine Society, American Association of Clinical Endocrinologists. Joint statement on the U.S. Food and Drug Administration’s decision regarding bioequivalence of levothyroxine sodium. Thyroid 2004; 14: 486.
9.8.1.15. Blakesley V, Awni W, Locke C, Ludden T, Granneman GR, Braverman LE. Are bioequivalence studies of levothyroxine sodium formulations in euthyroid volunteers reliable? Thyroid 2004; 14: 191- 200.
9.8.1.16. Stockigt J. Testing the bioavailability of oral L-thyroxine by studying its absorption: smoke or mirrors? Thyroid 2004; 14: 167-168.
9.8.1.17. Di Girolamo G, Keller GA, de Los Santos AR, Schere D, Gonzalez CD. Bioequivalence of two levothyroxine tablet formulations without and with mathematical adjustment for basal thyroxine levels in healthy Argentinian volunteers: a single-dose, randomized, open-label, crossover study. Clin Ther 2008; 30: 2015-2023.
9.8.1.18. Eisenberg M, DiStefano JJ. TSH-based protocol, tablet instability, and absorption effects on L-T4 bioequivalence. Thyroid 2009; 19: 103-110.
9.8.1.19 .Pabla D, Akhlaghi F, Zia H. A comparative pH-dissolution profile study of selected commercial levothyroxine products using inductively coupled mass spectrometry. Eur J Pharm Biopharm 2009; 72: 105-110.
9.8.1.20. Hennessey JV, Malabanan AO, Haugen BR, Levy EG. Adverse event reporting in patients treated with levothyroxine: results of the pharmacovigilance task force survey of the American Thyroid Association, American Association of Clinical Endocrinologists and The Endocrine Society. Endocr Pract 2010; 11: 1-41.
9.8.1.21. LeBoff MS, Kaplan MM, Silva JE, Larsen PR: Bioavailability of thyroid hormones from oral replacement preparations. Metabolism 1982; 31: 900.
9.8.1.22. Jackson S, William E, Cobb E: Why does anyone still use desiccated thyroid USP? Am J Med 1978; 64: 284-288.
9.8.1.23. Blumberg KR, Mayer WJ, Parikh DK, Schnell LA: Liothyronine and levothyroxine in Armour thyroid. J Pharm Sci 1993; 76: 346.
9.8.1.24. Rees-Jones RW, Rolla AR, Larsen PR: Hormone content of thyroid replacement preparations. JAMA 1980; 243: 549.
9.8.1.25. Rees-Jones RW, Larsen PR: Triiodothyronine and thyroxine content of desiccated thyroid tablets. Metabolism 1977; 26: 1213.
9.8.1.26. Escobar-Morreale HF, Escobar del Ray F, Obregon MJ, Morreale de Escobar G: Only the combined treatment with thyroxine and triiodothyronine ensures euthyroidism in all tissues of the thyroidectomized rat. Endocrinology 1996; 137: 2490-2502.
9.8.1.27. Garber JR, Cobin RH, Gharib H et al. Clinical practice guidelines for hypothyroidism in adults: cosponsored by th American Association of Clinical endocrinologists and the American Thyroid association. Thyroid 2012; 22: 1200-1235.
9.8.1.28. Celi FS, Zemskova M, Linderman JD et al. The pharmacodynamic equivalence of levothyroxine and liothyronine: a randomized, double blind, cross-over study in thyroidectomized patients. Clin Endocrinol 2010; 72: 709-713.
9.8.1.29. Hoang TD, Olsen CH, Mai VQ, Clyde PW, Shakir MKM. Desiccated thyroid extract compared with levothyroxine in the treatment of hypothyroidism: a randomized, double-blind, crossover study. J Clin Endocrinol Metab 2013; 98: 2013.
9.8.1.30. Grozinsky-Glasberg S, Fraser A, Nahshoni E, Weizman A, Leibovici L. Thyroxine-triiodothyronine combination therapy versus thyroxine monotherapy for clinical hypothyroidism: meta-analysis of randomized controlled trials. J Clin Endocrinol Metab 2006; 91: 2592-2599.
9.8.1.31. Nygaard B, Jensen EW, Kvetny J, Jarlov A, Faber J. Effect of combination therapy with thyroxine (T4) and 3,5,3’-triiodothyronine versus T4 monotherapy in patients with hypothyroidism, a double-blind, randomized cross-over study. Eur J Endocrinol 2009; 161: 895-902.
9.8.1.32. Wiersinga WM, Duntas L, Fadeyev V, Nygaard B, Vanderpump MP. 2012 ETA guidelines: the use of L-T4 + L-T3 in the treatment of hypothyroidism. Eur Thyroid J 2013; 1: 55-71.
9.8.1.33. Bunevicius R, Kazanavicius G, Zalinkevicius R, Prange AJ: Effects of thyroxine as compared with thyroxine plus triiodothyronine in patients with hypothyroidism. New Engl J Med 1999; 340: 424-429.
9.8.1.34. Toft AD: Thyroid hormone replacement - one hormone or two? New Engl J Med 1999; 340: 469-470.
9.8.1.35. Bunevicius R, Prange AJ. Mental improvement after replacement therapy with thyroxine plus triiodothyronine: relationship to cause of hypothyroidism. Int J Neuropsychopharmacol 2000; 3: 167-174.
9.8.1.36. Walsh JP, Shiels L, Lim EM, et al. Combined thyroxine/liothyronine treatment does not improve well-being, quality of life, or cognitive function compared to thyroxine alone: a randomized controlled trial in patients with primary hypothyroidism. J Clin Endocrinol Metab 2003; 88: 4543-4550.
9.8.1.37. Sawka AM, Gerstein HC, Marriott MJ, et al. Does a combination regimen of thyroxine (T4) and 3, 5,3’-triiodothyronine improve depressive symptoms better than T4 alone in patients with hypothyroidism? Results of a double-blind, randomized, controlled trial. J Clin Endocrinol Metab 88:4551-4555, 2003.
References for Section 9.8.2.
9.8.2.1. Roti E, Minelli R, Gardini E, Braverman LE: The use and misuse of thyroid hormone. Endocr Rev 1993; 14: 401-423.
9.8.2.2. Oppenheimer JH, Braverman LE, Toft A, et al.: Thyroid hormone treatment: when and what? J Clin Endocrinol Metab 1995; 80: 2875-2883.
9.8.2.3. Singer PA, Cooper DS, Levy EG, et al.: Treatment guidelines for patients with hyperthyroidism and hypothyroidism. JAMA 1995; 273: 808-812.
9.8.2.4. Vanderpump MPJ, Ahlquist JAO, Franklyn JA, Clayton RN: Consensus statement for good practice and audit measures in the management of hypothyroidism and hyperthyroidism. Brit Med J 1996; 313: 539-544.
9.8.2.5. Bearcroft CP, Toms GC, Williams SJ et al: Thyroxine replacement in post-radioiodine hypothyroidism. Clin Endocrinol 1991; 34: 115.
9.8.2.6. Kabadi UM, Jackson T: Serum thyrotropin in primary hypothyroidism. A possible predictor of optimal daily levothyroxine dose. Arch Intern Med 1995; 155: 1046-1048.
9.8.2.7. Santini F, Pinchera A, Marsili A et al. Lean body mass is a major determinant of levothyroxine dosage in the treatment of thyroid diseases. J Clin Endocrinol Metab 2005; 90: 124-127.
9.8.2.8. Hall RCW: Psychiatric effects of thyroid hormone disturbance. Psychosomatics 1983; 24: 7.
9.8.2.9. Josephson AM, Mackenzie TB: Thyroid-induced mania in hypothyroid patients. Br J Psychiatr 1980; 137: 222.
9.8.2.10. Josephson AM, Mackenzie TB: Appearance of manic psychosis following rapid normalization of thyroid status. Am J Psychiatr 1979; 136: 846.
9.8.2.11. Carr K, McLeod DT, Parry G, Thornes HM: Fine adjustment of thyroxine replacement dosage : comparison of the thyrotrophin releasing hormone tests using a sensitive thyrotrophin assay with measurement of free thyroid hormones and clinical assessment. Clin Endcrinol 1988; 28: 325.
9.8.2.12. Fish LH, Schwartz HL, Cavanaugh J, et al.: Replacement dose, metabolism, and bioavailability of levothyroxine in the treatment of hypothyroidism. Role of triiodothyronine in pituitary feedback in humans. New Engl J Med 1987; 316: 764-770.
9.8.2.13. Ferretti E, Persani L, Jaffrain-Rea M-L, Giambona S, Tamburrano G, Beck-Peccoz P. Evaluation of the adequacy of levothyroxine replacement therapy in patients with central hypothyroidism. J Clin Endocrinol Metab 1999; 84: 924-929.
9.8.2.14. Shimon I, Cohen O, Lubetsky A, Olchovsky D. Thyrotropin suppression by thyroid hormone replacement is correlated with thyroxine level normalization in central hypothyroidism. Thyroid 2002; 12: 823-827.
9.8.2.14. Grebe SKG, Cooke RR, Ford HC, et al.: Treatment of hypothyroidism with once weekly thyroxine. J Clin Endocrinol Metab 1997; 82: 870-875.
9.8.2.15. Wekking EM, Appelhof BC, Fliers E et al. Cognitive functioning and well-being in euthyroid patients on thyroxine therapy for hypothyroidism. Eur J Endocrinol 2005; 153: 747-753.
9.8.2.16. Saravanan P, Chau WF, Roberts N, Vedhara K, Greenwood R, Dayan CM. Psychological well-being in patients on ‘adequate ’ doses of L-thyroxine: results of a large, controlled community-based questionnaire study. Clin Endocrinol 2002; 57: 577-585.
9.8.2.17. Wiersinga WM. Do we need still more trials on T4 and T3 combination therapy in hypothyroidism? Eur J Endocrinol 2009; 161: 955-959.
9.8.2.18. Grozinsky-Glasberg S, Fraser A, Nahshoni E, Weizman A, Leibovici L. Thyroxine-triiodothyronine combination therapy versus thyroxine monotherapy for clinical hypothyroidism: meta-analysis of randomized controlled trials. J Clin Endocrinol Metab 2006; 91: 2592-2599.
9.8.2.19. Nygaard B, Jensen EW, Kvetny J, Jarlov A, Faber J. Effect of combination therapy with thyroxine (T4) and 3,5,3’-triiodothyronine (T3) versus T4 monotherapy in patients with hypothyroidism, a double-blind, randomized cross-over study. Eur J Endocrinol 2009;161: 895-902.
9.8.2.20. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev 2002; 23: 38-89.
9.8.2.21. Escobar-Morreale HF, Botella-Carretero JI, Escobar del Rey F, Morreale de Escobar G. Review. Treatment of hypothyroidism with combinations of levothyroxine plus liothyronine. J Clin Endocrinol Metab 20005; 90: 4946-4954.
9.8.2.22. Saravanan P, Simmons DJ, Greenwood R, Peters TJ, Dayan CM. Partial substitution of thyroxine (T4) with triiodothyronine in patients on T4-replacement therapy: results of a large community-based randomized controlled trial. J Clin Endocrinol Metab 2005; 90: 805-812.
9.8.2.23. Appelhof BC, Fliers E, Wekking EM et al. Combined therapy with levothyroxine and liothyronine in two ratios, compared with levothyroxine monotherapy in primary hypothyroidism: a double-blind, randomized, controlled clinical ytrial. J Clin Endocrinol Metab 2005; 90: 2666-2674.
9.8.2.24. Kaplan MM, Sarne DH, Schneider AB. In search of the impossible dream? Thyroid hormone replacement that treats all symptoms in all hypothyroid patients. J Clin Endocrinol Metab 2003; 88: 4540-4542.
9.8.2.25. Jonklaas J, Davidson B, Bhagat S, Soldin SJ. Triiodothyronine levels in athyreotic individuals during levothyroxine therapy. JAMA 2008; 299: 769-777.
9.8.2.26. Walsh JP, Ward LC, Burke V et al. Small changes in thyroxine dosage do not produce measurable changes in hypothyroid symptoms, well-being, or quality-of-life: results of a double-blind, randomized clinical trial. J Clin Endocrinol Metab 2006; 91: 2624-2630.
9.8.2.27. Saranavan P, Siddique H, Simmons DJ, Greenwood R, Dayan CM. Twenty-four hour hormone profiles of TSH, free T3 and free T4 in hypothyroid patients on combined T3/T4 therapy. Exp Clin Endocrinol Diab 2007; 115: 261-267.
9.8.2.28. Henneman G, Docter R, Visser TJ, Postema PT, Krenning EP. Thyroxine plus low-dose, slow-release triiodothyronine replacement in hypothyroidism. Thyroid 2004; 14: 271-275.
9.8.2.29. Russell W, Harrison RF, Smith N, Darzy K, Shalet S, Weetman AP, Ross RJ. Free triiodothyronine has a distinct circadian rhythm that is delayed but parallels thyrotropin levels. J Clin Endocrinol Metab 2008; 93: 2300-2306.
9.8.2.30. Dayan CM, Panicker V. Novel insights into thyroid hormones from the study of common genetic variation. Nature Reviews Endocrinology 2009; 5: 211-218.
9.8.2.31. Saravanan P, Visser TJ, Dayan CM. Psychological well-being correlates with free thyroxine but not free 3,5,3’-triiodothyronine levels in patients on thyroid hormone replacement. J Clin Endocrinol Metab 2006; 91: 3389-3393.
9.8.2.32. Panicker V, Saravanan P, Vaidya B et al. Common variation in the DIO 2 gene predicts baseline psychological well-being and response to combination thyroxine plus triiodothyroinine therapy in hypothyroid patients. J Clin Endocrinol Metab 2009; 94: 1623-1629.
9.8.2.33. Appelhof BC, Peeters RP, Wiersinga WM et al. Polymorhisms in type 2 deiodinase are not associated with well-being, neurocognitive functioning, and preference for combined thyroxine/3,5,3’-triiodothyronine therapy. J Clin Endocrinol Metab 2005; 90: 6296-6299.
9.8.2.34. van der Deure WM, Appelhof BC, Peeters RP et al. Polymorphisms in the brain-specific thyroid hormone transporter OATPC1 are associated with fatigue and depression in hypothyroid patients. J Clin Endocrinol Metab 2008; 69: 804-811.
9.8.2.35. Kim BW, Bianco AC. For some, L-thyroxine replacement might not be enough: a genetic rationale. J Clin Endocrinol Metab 2009; 94: 1521-1523.
9.8.2.36. Sawin CT, Geller A, Wolf PA, et al.: Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. New Engl J Med 1994; 331: 1249-1252.
9.8.2.37. Biondi B, Fazio S, Carella C, et al.: Cardiac effects of long term thyrotropin-suppressive therapy with levothyroxine. J Clin Endocrinol Metab 1993; 77: 334-338.
9.8.2.38. Leese GP, Jung RT, Guthrie C, et al.: Morbidity in patients on L-thyroxine: a comparison of those with a normal TSH to those with a suppressed TSH. Clin Endocrinol 1992; 37: 500-503.
9.8.2.39. Uzzan B, Campos J, Cucherat M, et al.: Effects on bone mass of long term treatment with thyroid hormones: a meta-analysis. J Clin Endocrinol Metab 1996; 81: 4278-4289
9.8.2.40. Leese GP, Jung RT, Guthrie C, et al.: Morbidity in patients on L-thyroxine: a comparison of those with a normal TSH to those with a suppressed TSH. Clin Endocrinol 1992; 37: 500-503.
9.8.2.41. Solomon BL, Wartofsky L, Burman KD: Prevalence of fractures in postmenopausal women with thyroid disease. Thyroid 1993; 3: 17-23.
9.8.2.42. Shimon I, Cohen O, Lubetsky A, Olchovsky D. Thyrotropin suppression by thyroid hormone replacement is correlated with thyroxine level normalization in central hypothyroidism. Thyroid 2002; 12:823-827.
9.8.2.43. Vestergaard P, Weeke J, Hoeck HC, et al. Fractures in patients with primary idiopathic hypothyroidism. Thyroid 2000; 10:335-340.
9.8.2.44. Sheppard MC, Holder R, Franklyn J. Levothyroxine treatment and occurrence of fracture of the hip. Arch Int Med 2002; 162: 338-343.
9.8.2.45. Flynn RW, Bonellie SR, Jung RT, MacDonald TM, Morris AD, Leese GP. Serum thyroid-stimulating hormone concentration and morbidity from cardiovascular disease and fractures in patients on long-term thyroxine therapy. J Clin Endocrinol Metab 2010; 95: 186-193.
9.8.2.46. Somwaru LL, Amold AM, Joshi N, Fried LP, Cappola AR. Thyroid function test abnormalities are common in elderly people taking thyroid hormone replacement medications. J Clin Endocrinol Metab 2009; 94: 1342-1345.
9.8.2.47. Roos A, Linn-Rasker SP, van Domburg RT, Tijssen JP, Berghout A. The starting dose of levothyroxine in primary hypothyroidism treatment. A prospective, randomized, double-blind trial. Arch Int Med 2005; 165: 1714-1720.
9.8.2.48. Wartofsky L. Levothyroxine therapy for hypothyroidism. Should we abandon conservative dosage titration? Arch Int Med 2005; 165: 1683-1684.
9.8.2.49, Bolk N, Visser TJ, Kalsbeek A, van Domburg RT, Berghout A. Effects of evening vs morning thyroxine ingestion on serum thyroid hormone profiles in hypothyroid patients. Clin Endocrinol 2007; 66: 43-48.
9.8.2.50. Benvenga S, Bartolone L, Pappalardo MA et al. Altered intestinal absorption of L-thyroxine caused by coffee. Thyroid 2008; 18: 293-301.
9.8.2.51. Garber JR, Cobin RH, Gharib H et al. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Thyroid 2012; 22: 1200-1235.
9.8.2.52. Gullo D, Latina A, Frasca F, Le Moli R, Pelegritti G, Vigneri R. Levothyroxine monotherapy cannot guarantee euthyroidism in all athyreotic patients. PLoS ONE 2011; 6: e22552.
9.8.2.53. Beck-Peccoz P. Treatment of central hypothyroidism. Clin Endocrinol 2011; 74: 671-672.
9.8.2.54. Karmisholt J, Andersen S, Laurberg P. Weight loss after therapy of hypothyroidism is mainly caused by excretion of excess body water associated with myxoedem. J Clin Endocrinol Metab 2011; 96: E99-E103.
9.8.2.55. Walker JN, Shillo P, Ibbotson V et al. A thyroxine absorption test followed by weekly thyroxine administration: a method to assess non-adherence to treatment. Eur J Endocrinol 2013; 169: 913-917.
References for Section 9.8.3.
9.8.3.1. Topliss DJ, Wright JA, Volpe R: Increased requirement for thyroid hormone after a jejunal bypass operation. Can Med Assoc J 1980; 123: 765-766.
9.8.3.1. Liel Y, Harman-Boehm I, Shany S: Evidence for a clinically important adverse effect of fiber-enriched diet on the availability of levothyroxine in adult hypothyroid patients. J Clin Endocrinol Metab 1996; 81: 857-859.
9.8.3.2. Bell DSH, Ovalle F. Use of soy protein supplement and resultant need for increased dose of levothyroxine. Endocr Pract 2001; 7: 193-194.
9.8.3.3. Chiu AC, Sherman SI. Effects of pharmacological fiber supplements on levothyroxine absorption. Thyroid 1998; 8: 667-671,
9.8.3.4. Centanni M, Gargano L, Canettieri G et al. Thyroxine in goiter, Helicobacter infection, and chronic gastritis. N Engl J Med 2006; 354: 1787-1795.
9.8.3.5. Checchi S, Montanaro A, Pasqui L et al. L-thyroxine requirement in patients with autoimmune hypothyroidism and parietal cell antibodies. J Ckin Endocrinol Metab 2008; 93: 465-469.
9.8.3.6. Sachmechi I, Reich DM, Aninyei M et al. Effect of proton pump inhibitors on serum thyroid-stimulating hormone level in euthyroid patients treated with levothyroxine for hypothyroidism. Endocr Pract 2007; 13: 345-349.
9.8.3.7.Dietrich JW, Gieselbrecht K, Holl RW et al. Absorption kinetics of levothyroxine is not altered by proton-pump inhibitor therapy. Horm Metab Res 2006; 38: 57-59.
9.8.3.8. Ananthakrishnan S, Braverman LE, Levin RM et al. The effect of famotidine, esomeprazole, and ezetimibe on levothyroxine absorption. Thyroid 2008; 18: 493-498.
9.8.3.9. D’Esteve-Bonetti L, Bennet AP, Malet D et al. Gluten-induced enteropathy (coeliac disease) revealed by resistance to treatment with levothyroxine and alfacalcidol in a sixty-eight-year old patient: a case report. Thyroid 2002; 12: 633-636.
9.8.3.10. McDermott JH, Coss A, Walsh CH. Celiac disease presenting as resistant hypothyroidism. Thyroid 2005; 15: 386-388.
9.8.3.11. Azizi F, Belur R, Albano J. Malabsorption of thyroid hormones after jejunoileal bypass for obesity. Ann Intern Med 1979; 90: 941-942.
9.8.3.12. Topliss DJ, Wright JA, Volpe R. Increased requirement for thyroid hormone after a jejunal bypass operation. Can Med Assoc J 1980; 123: 765-766.
9.8.3.13. Smyrniotis V, Vaos N, Arkadopoulos N, Kostopanagiotou G, Theodoraki K, Lambrou A. Severe hypothyroidism in patients dependent on prolonged thyroxine infusion through a jejunostomy. Clin Nutr 2000; 19: 65-67.
9.8.3.14. Stone E, Leiter LA, Lambert JR, Silverberg JDH, Jeejeebhoy KN, Burrow GN. L-thyroxine absorption in patients with short bowel. J Clin Endocrinol Metab 1984; 59: 139-141.
9.8.3.15. Munoz-Torres M, Varsavsky M, Alonso G. Lactose intolerance revealed by severe resistance to treatment with levothyroxine. Thyroid 2006; 16: 1171-1173.
9.8.3.16. Seppel T, Rose F, Schlaghecke R. Chronic intestinal Giardiasis with isolated thyroxine malabsorption as reason for severe hypothyroidism – implications for localization of thyroid hormoine absorption iin the gut. Exp Clin Endocrinol Diab 1996; 104: 180-182.
9.8.3.17. Witztum JL, Jacobs LS, Schonfeld G: Thyroid hormone and thyrotropin levels in patients placed on colestipol hydrochloride. J Clin Endocrinol Metab 1978; 46: 838-840.
9.8.3.18. Northcutt RC, Stiel JN, Hollifield JW et al. The influence of cholestyramine on thyroxine absorption. JAMA 1969; 208: 1857-1861.
9.8.3.19. Harmon SM, Seifert CF: Levothyroxine-cholestyramine interaction reemphasized. Ann Int Med 1991; 115: 658-659.
9.8.3.20. Weitzman SP, Ginsburg KC, Carlson HE. Colesevelam hydrochloride and lanthanum carbonate interfere with the absorption of levothyroxine. Thyroid 2009; 19: 77-79.
9.8.3.21. Liwanpo L, Hershman JM. Conditions and drugs interfering with thyroxine absorption. Best Pract Res Clin Endocrinol Metab 2009; 23: 781-792.
9.8.3.22. Havrankova J, Lahaie R: Levothyroxine binding by sucralfate. Ann Int Med 1992; 147: 445-446 [Erratum 1993; 118:398].
9.8.3.23. Sherman SI, Tielens ET, Ladenson PW. Sucralfate causes malabsorption of L-thyroxine. Am J Med 1994; 90: 531-535.
9.8.3.24. Campbell JA, Schmidt BA, Bantle JP. Sucralfate and the absorption of L-thyroxine. Ann Int Med 1994; 121: 152.
9.8.3.25. Liel Y, Sperber AD, Shany S. Nonspecific intestinal adsorption of levothyroxine by aluminium hydroxide. Am J Med 1994; 97: 363-365.
9.8.3.26. Mersebach H, Rasmussen AK, Kirkegaard L et al. Intestinal adsorption of levothyroxine by antacids and laxatives: case stories and in vitro experiments. Pharmacol Toxicol 1999; 84: 107-109.
9.8.3.27. Sperber AD, Liel Y: Evidence for interference with the intestinal absorption of levothyroxine sodium by aluminium hydroxide. Arch Int Med 1992; 152: 183-184.
9.8.3.28. Campbell NRC, Hasinoff BB, Stalts H, et al.: Ferrous sulfate reduces thyroxine efficacy in patients with hypothyroidism. Ann Int Med 1992; 117: 1010-1013.
9.8.3.29. Shakir KM, Chute JP, Aprill BS et al. Ferrous sulfate-induced increase in requirement for thyroxine in a patient with primary hypothyroidism. South Med J 1997; 90: 637-639.
9.8.3.30. Leger CS, Ooi TC. Ferrous fumarate-induced malabsorption of thyroxine. Endocrinologist 1999; 9: 493-495.
9.8.3.31. Singh N, Singh PN, Hershman JM. Effect of calcium carbonate on the absorption of levothyroxine. JAMA 2000; 283: 2822-2825.
9.8.3.32. Singh N, Weisler SL, Hershman JM. The acute effect of calcium carbonate on the intestinal absorption of levothyroxine. Thyroid 2001; 11: 967-971.
9.8.3.33. Csako G, McGriff NJ, Rotman-Pikielny P, Sarlis NJ, Pucino F. Exaggerated levothyroxine malabsorption due to calcium carbonate supplementation in gastrointestinal disorders. Ann Pharmacother 2001; 35: 1578-1583.
9.8.3.34. John-Kalarickal J, Pearlman G, Carlson HE. New medications which decrease levothyroxine adsorption. Thyroid 2007; 17: 763-765.
9.8.3.35. Weitzman SP, Ginsburg KC, Carlson HE. Colesevelam hydrochloride and lanthanum carbonate interfere with the absorption of levothyroxine. Thyroid 2009; 19: 77-79.
9.8.3.36. Diskin CJ, Stokes TJ, Dansby LM et al. Effect of phosphate binders upon TSH and L-thyroxine dose in patients on thyroid replacement. Int Urol Nephrol 2007; 39: 599-602.
9.8.3.37. Siraj ES, Gupta MK, Reddy SS. Raloxifene causing malabsorption of levothyroxine. Arch Int Med 2003; 163: 1367-1370.
9.8.3.38. Garwood CL, van Schepen KA, McDonough RP et al. Increased thyroid-stimulating hormone levels associated with concomitant administartion of levothyroxine and raloxifene. Pharmacotherapy 2006; 26: 881-885.
9.8.3.39. Madhava K, Hartley A. Hypothyroidism in thyroid carcinoma follow-up: orlistat may inhibit the absorption of thyroxine. Clin Oncol 2005; 17: 492.
9.8.3.40. Ain KB, Mori Y, Refetoff S. Reduced clearance rate of thyroxine-binding globulin (TBG) with increased sialylation: a mechanism for estrogen induced elevation of serum TBG concentration. J Clin Endocrinol Metab 1987; 65: 689-696.
9.8.3.41. Tahboub R, Arafah BM. Sex steroids and the thyroid. Best Pract Res Clin Endocrinol Metab 2009; 23: 769-780.
9.8.3.42. Sitruk-Ware R, Plu-Bureau G, Menard J et al. Effects of transvaginal ethinyl estradiol on hemostatic factors and hepatic proteins in a randomized, cross over study. J Clin Endocrinol Metab 2007; 92: 2074-2079.
9.8.3.43. Goebelsman IJ, Mashchak CA, Mishell Jr DR. Comparison of hepatic impact of oral and vaginal administration of ethinyl estradiol. Am J Obstetr Gynecol 1985; 151: 868-877.
9.8.3.44. Bisschop PH, Toorians AW, Endert E et al. The effects of sex-steroid administration on the pituitary-thyroid axis in transsexuals. Eur J Endocrinol 2006; 155: 11-16.
9.8.3.45. Arafah BM. Increased need for thyroxine in women with hypothyroidism during estrogen therapy. N Eng J Med 2001; 344:1743-1749.
9.8.3.46. Anker GB, Lonning PE, Aakvaag A et al. Thyroid function in postmenopausal breast cancer patients treated with tamoxifen. Scand J Clin Lab Invest 1998; 58: 103-107.
9.8.3.47. Marqusee E, Braverman LE, Lawrence J et al. The effect of droloxofene and estrogen on thyroid function in postmenopausal women. J Clin Endocrinol Metab 2000; 85: 4407-4410.
9.8.3.48. Van Bon AC, Wiersinga WM. Goserelin-induced transient thyrotoxicosis in a hypothyroid women on L-thyroxine replacement. Neth J Med 2008; 66: 256-258.
9.8.3.49. Mandell SJ, Larson PR, Seely EW, Brent GA: Increased need for thyroxine during pregnancy in women with rpimary hypothyroidism. New Engl J Med 1990; 323: 91-96.
9.8.3.50. Kaplan MM: Monitoring thyroxine treatment during pregnancy. Thyroid 1992; 2: 147-152.
9.8.3.51. Smallridge RC, Ladenson PW. Hypothyroidism in pregnancy: consequences to neonatal health. J Clin Endocrinol Metab 2001; 86: 2349-2353.
9.8.3.52.Alexander EK, Marqusee E, Lawrence J, Jarolim P, Fischer GA, Larsen PR. Timing and magnitude of increases in levothyroxine requirements during pregnancy in women with hypothyroidsm. N Engl J Med 2004; 351: 241-249.
9.8.3.53. Toft A. Increased levothyroxine requirements in pregnancy – Why, When, and How much? N Engl J Med 2004; 351: 292-294.
9.8.3.54. Yassa L, Marqusee E, Fawcett R, Alexander EK. Thyroid hormone early adjustment in pregnancy (the THERAPY) trial. J Clin Endocrinol Metab 2010; 95: 3234-3241.
9.8.3.55. Haddow JE, Palomaki GE, Allen WC et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med 1999; 341: 549-555.
9.8.3.56. Pop VJ, Kuypens JL, Baar AL van, et al. Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol 1999; 50: 149-155.
9.8.3.57. Henrichs J, Bongers-Schokking JJ, Schenk JJ. Maternal thyroid function during early pregnancy and cognitive functioning in early childhood: the Generation R study. J Clin Endocrinol Metab 2010; 95 (doi:10.2010/jc.2010-0415) .
9.8.3.58. Morreale de Escobar G, Obregon MJ, Escobar del Rey F. Is neuropsychological development related to maternal hypothyroidism or maternal hypothyroxinemia? J Clin Endocrinol Metab 2000; 85: 3975-3987.
9.8.3.66.Illouz F, Laboureau-Soares S, Dubois S, Rohmer V, Rodien P. Tyrosine kinase inhibitors and modifications of thyroid function. Eur J Endocrinol 2009; 160: 331-336.
9.8.3.67. de Groot JW, Zonnenberg BA, Plukker JT, van der Graaf WT, Links TP, Imatinib induces hypothyroidism in patients receiving levothyroxine. Clin Pharmacol Ther 2005; 78: 433-438.
9.8.3.68. de Groot JW, Zonnenberg BA, van Ufford-Mannesse PQ et al. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab 2007; 92: 3466-3469.
9.8.3.69. Pacini F, Sherman S, Schlumberger M et al. Exacerbation of postsurgical hypothyroidism during treatment of advanced differentiated (DTC) or medullary (MTC) thyroid carcinoma with AMG 706. Horm Res 2007; 68:29.
9.8.3.70. Sherman S, Wirth L, Droz JP et al. Motesanib diphosphate in progressive differentiated thyroid cancer. N Engl J Med 2008; 359: 31-42.
9.8.3.71. Gupta-Abramson V, Troxel AB, Nellore A et al. Phase II trial of sorafenib in advanced thyroid cancer. J Clin Oncol 2008; 26: 4714-4719.
9.8.3.72. Abdulrahman RM, Verloop H, Hoftijzer H et al. Sorafenib-induced hypothyroidism is associated with increased type 3 deiodination. J Clin Endocrinol Metab 2010; 95: 3758-3762..
9.8.3.59. Curran PG, DeGroot LJ: The effect of hepatic enzyme-inducing drugs on thyroid hormones and the thyroid gland. Endocr Rev 1991; 12:135.
9.8.3.60. Faber J, Lumholtz IB, Kirkegaard C et al.: The effects of phenytoin on the extrathyroidal turnover of thyroxine, 3,5,3'-triiodothyronine, 3,3',5'-triiodothyronine, and 3',5'-diiodothyronine in man. J Clin Endocrinol Metab 1985; 61: 1093.
9.8.3.61. DeLuca F, Arrigo T, Pandullo E et al.: Changes in thyroid function tests induced by 2 month carbamazepine treatment in L-thyroxine-substituted hypothyroid children. Eur J Pediatr 1986; 145: 77.
9.8.3.62. Isley WL: Effect of rifampin therapy on thyroid function tests in a hypothyroid patient on replacement L-thyroxine. Ann Int Med 1987; 107: 517-518
9.8.3.63. Figge J, Dluhy RG: Amiodarone-induced elevation of thyroid stimulating hormone in patients receiving levothyroxine for primary hypothyroidism. Ann Int Med 1990; 113: 563-565.
9.8.3.64. McCowen KC, Garber JR, Spark R: Elevated serum thyrotropin in thyroxine-treated patients with hypothyroidism given sertraline. New Engl J Med 1997; 337: 1010-1011.
9.8.3.65. Munera Y, Hugues FC, Le Jeunne C, Pays IF: Interaction of thyroxine sodium with antimalarial drugs. Brit Med J 1997; 314: 1593.
9.8.3.73. Arafah BM: Decreased levothyroxine requirement in women with hypothyroidism during androgen therapy for breast cancer. Ann Int Med 1994; 121: 247-251.
9.8.3.74. Cunningham JJ, Barzel NS: Lean body mass is a predictor of the daily requirement of thyroid hormone in older men and women. J Am Geriatr Soc 1984; 32: 204-207.
9.8.3.75. Rosenbaum RL, Barzel US. Levothyroxine replacement dose for primary hypothyroidism decreases with age. Ann Int Med 1982; 96: 53-55.
9.8.3.76. Griffin JE: Hypothyroidism in the elderly. Am J Med Sci 1990; 299: 334-345.
9.8.3.77. Thomas MC, Mathew TH, Russ GR. Changes in thyroxine requirements in patients with hypothyroidism undergoing renal transplantation. Am J Kidney Dis 2002; 39: 354-357.
9.8.3.78. Padwal R, Brocks D, Sharma AM. A systematic review of drug absorption following bariatric surgery and its theoretical implications. Obes Rev 2010; 11: 41-50.
9.8.3.79. Rubio IG, Gairao AL, Santo MA, Zanini AC, Medeiros-Neto G. Levothyroxine absorption in morbidly obese patients before and after Roux-en-Y gastric bypass (RYGB) surgery. Obes Surg 2012; 22: 253-258.
9.8.3.80. Pirola I, Formenti AM, Gandossi E et al. Oral liquid L-thyroxine (L-T4) may be better absorbed compared to L-T4 tablets. Obes Surg 2013; 23: 1493-1496.
9.3.8.81. Gkotsina M, Michelaki M, Mamali I et al. Improved levothyroxine pharmacokinetics after bariatric surgery. Thyroid 2013; 23: 414-419.
9.3.8.82. Braun D, Kim TD, le Coutre P, Kohrle J, Hershman JM, Schweizer U. Tyrosine kinase inhibitors noncompetitively inhibit MCT8-mediated iodothyronine transport. J Clin Endocrinol Metab 2012; 97: E100-E105.
9.8.3.83. de Carvalho GA, Bahls SC, Boeving A, Graf H. Effects of selective serotonin reuptake inhibitors on thyroid function in depressed patients with primary hypothyroidism or normal thyroid function. Thyroid 2009; 19: 691-697.
References for Section 9.8.4.
9.8.4.1, Murray JS, Jayarajasingh R, Perros P. Deterioration of symptoms after start of thyroid hormone replacement. Brit Med J 2001; 323: 332-333.
9.8.4.2. Topliss DJ, White EL, Stockigt JR. Significance of thyrotropin excess in untreated primary adrenal insufficiency. J Clin Endocrinol Metab 1980; 50: 52-56.
9.8.4.3. Shigemasa C, Kouchi T, Ueta Y et al. Evaluation of thyroid function in patients with isolated adrenocorticotropin deficiency. Am J Med Sci 1992; 304: 279-284.
9.8.4.4. Abdullatif HD, Ashraf AP. Reversible subclinical hypothyroidism in the presence of adrenal insufficiency. Endocr Pract 2006; 12: 572-575.
9.8.4.5. Samuels MH. Effects of variations in physiological cortisol levels on thyrotropin secretion in subjects with adrenal insufficiency: a Clinical Research Center Study. J Clin Endocrinol Metab 2000; 85: 1388-1393.
9.8.4.6. Santos AD, Miller RP, Puthenpurakal KM et al.: Echocardiographic characterization of the reversible cardiomyopathy of hypothyroidism. Am J Med 1980; 68: 675.
9.8.4.7. Polikar R, Burger AG, Scherrer U, Nicod P: The thyroid and heart. Circulation 1993; 87: 1435.
9.8.4.8. Ladenson PW: Recognition and management of cardiovascular disease related to thyroid dysfunction. Am J Med 1990; 88: 638.
9.8.4.9. Keating FR, Parkin TW, Selby JB, Dickinson LS: Treatment of heart disease associated with myxedema. Prog Cardiovasc Dis 1961; 3: 364.
9.8.4.10. Hay ID, Duick DX, Vlietstra RE et al.: Thyroxine therapy in hypothyroid patients undergoing coronary revascularization: a retrospective analysis. Ann Intern Med 1981: 95: 456.
9.8.4.11. Weinberg AD, Brennan MD, Gorman CA et al.: Outcome of anesthesia and surgery in hypothyroid patients. Arch Intern Med 1983; 143: 893.
9.8.4.12. Ladenson PW, Levin AA, Ridgway EC, Daniels GH: Complications of surgery in hypothyroid patients. Am J Med 1984; 77: 261.
9.8.4.13. Sherman SI, Ladenson PW: Percutaneous transluminal coronary angioplasty in hypothyroidism. Am J Med 1991; 90: 367-370.
9.8.4.14. Levine HD: Compromise therapy in the patient with angina pectoris and hypothyroidism: a clinical assessment. Am J Med 1980; 69: 411.
9.8.4.15. Behan LA, Monson JP, Agha A. The interaction between growth hormone and the thyroid axis in hypopituitary patients. Clin Endocrinol 2010; Apr 23 [Epub ahead of print].
9.8.4.16. Martins MR, Doin FC, Komatsu WR, Barros-Neto TL, Moises VA, Abucam J. Growth hormone replacement improves thyroxine biological effects: implications for management of central hypothyroidism. J Clin Endocrinol Metab 2007; 92: 4144-4153.
9.8.4.17. Porretti S, Giavoli C, Ronchi C, et al. Recombinant human GH replacement therapy and thyroid function in a large group of adult GH-deficient patients. When does L-T4 therapy becomes mandatory? J Clin Endocrinol Metab 2002; 87: 2042-2045.
9.8.4.18. Chonchol M, Lippi G, Salvagno G, Zoppini G, Muggeo M, Targher G. Prevalence of subclinical hypothyroidism in patients with chronic kidney disease. Clin J Am Soc Nephrol 2008; 3: 1296-1300.
References for Section 9.9.
9.9.1. Nicoloff JT, LoPresti JS: Myxedema coma. A form of decompensated hypothyroidism. Endocrinol Metab Clin North Am 1993; 22: 279-290.
9.9.2. Jordan RM: Myxedema coma. Pathophysiology, therapy, and factors affecting prognosis. Med Clin North Am 1995; 79: 185-194.
9.9.3. Fliers E, Wiersinga WM. Myxedema coma. Rev Endocr Metab Dis 2003; 4: 137-141.
9.9.4. Wartofsky L. Myxedema coma. Endocrinol Metab Clin N Am 2006; 35: 687-698.
9.9.5. Kwaku MO, Burman Kd. J Intensive Care Med 2007; 22: 224-231.
9.9.6. Dutta P, Bhansali A, Masoodi SR, Bhadada S, Sharma N, Rajput R. Predictors of outcome in myxedema coma: a study from a tertiary care centre. Crit Care 2008; 12: R1
9.9.7. Reinhardt W, Mann K. Incidence, clinical picture, and treatment of hypothyroid coma: results of a survey. Med Klin 1997; 92: 521-524.
9.9.8. Rodriguez I, Fluiters E, Perez-<emdez LF, Luna R, Paramo C, Garcia-Mayor RV. Factors associated with mortality of patients with myxedema coma: prospective study in 11 cases treated in a single institution. J Endocrinol 2004; 180: 347-350.
9.9.9. Holvey DN, Goodner CJ, Nicoloff JT, Dowling JT: Treatment of myxedema coma with intravenous thyroxine. Arch Intern Med 1964; 113: 139.
9.9.10. Blackburn CM, McConahey WM, Keating RF, Albert A: Calorigenic effect of single intravenous doses of L-triiodothyronine and L-thyroxine in myxedematous persons. J Clin Invest 1954; 33: 819.
9.9.11. MacKerrow SD, Osborn LA, Levy H et al.: Myxedema-associated cardiogenic shock treated with intravenous triiodothyronine. Ann Intern Med 1992; 117: 1014.
9.9.12. Hylander B, Rosenquist U: Treatment of myxedema coma - factors associated with fatal outcome. Acta Endocrinol 1985; 108: 65.
9.9.13. Arlot S, Debussche X, Lalau JD et al.: Myxedema coma: response of thyroid hormones with oral and intravenous high-dose L-thyroxine treatment. Intensive Care Med 1991; 17: 16.
9.9.14. Ridgway EC et al. Metabolic responses of patients with myxedema to large doses of intravenous L-thyroxine. Ann Int Med 1972; 77: 549-555.
9.9.15. Yamamoto T, Fukuyama J, Fujiyoshi A. Factors associated with mortality of myxedema coma: report of eight cases and literature survey. Thyroid 1999; 9: 1167-1174.
9.9.16. McKerrow SD, Osborn LA, Levy H, Eaton P, Economon O. Myxedema-associated cardiogenic shock treated with triiodothyronine. Ann Int Med 1992; 117: 1014-1015.
9.9.17. Ladenson PW, Levin AA, Ridgway EC, Daniels GH. Complications of surgery in hypothyroid patients. Am J Med 1984; 77: 261.
9.9.18. Sheu CC, Cheng MH, Tsai JR, Hwang JJ. Myxedema coma: a well-known but unfamiliar medical emergency. Thyroid 2007; 17: 371-372.9.9.19. Malliphedi A, Vali H, Okosieme O. Myxedema coma in a patient with subclinical hypothyroidism. Thyroid 2011; 21: 87-89.
9.9.20. Chu M, Seltzer TF. Myxedema coma induced by ingestion of raw bol choy. New Engl J Med 2010; 362: 1945-1946.
References for Section 9.10.1.
9.10.1.1. Hollowell JG, Staehling NW, Flanders WD et al. Serum TSH, T4 and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab 2000; 87: 489-499.
9.10.1.2. National Academy of Clinical Biochemistry. NACB laboratory medicine practice guidelines. Available at: http://www.nacb.org/lmpg/main.stm
9.10.1.3. Wartofsky L, Dickey RA. The evidence for a narrower thyrotropin reference range is compelling. J Clin Endocrinol Metab 2005; 90: 5483-5488.
9.10.1.4. Surks MI, Goswami G, Daniels GH. The thyrotropoin reference range should remain unchanged. J Clin Endocrinol Metab 2005; 90: 5489-5496.
9.10.1.5. Brabant G, Beck-Peccoz P, Jarzab B et al. Is there a need to redefine the upper normal limit of TSH? Eur J Endocrinol 2006; 154: 633-637.
9.10.1. 6. Kratzsch J, Fielder GM, Leichtle A et al. New reference intervals for thyrotropin and thyroid hormones based on National Academy of Clinical Biochemistry criteria and regular ultrasonography of the thyroid. Clin Chem 2005; 51: 1480-1486.
9,10.1.7. Hamilton TE, Davis S, Onstad L. Kopecky KJ. Thyrotropin levels in a population with no clinical, antibody, or ultrasonographic evidence of thyroid disease: implications for the diagnosis of subclionical hypothyroidism. J Clin Endocrinol Metab 2008; 93: 1224-1230.
9.10.1.8. Surks MI, Hollowell JG. Age-specific distribution of serum thyrotropin and thyroid antibodies in the US population: implications for the prevalence of subclinical hypothyroidism. J Clin Endocrinol Metab 2007; 92: 4575-4582.
9.10.1.9. Karmisholt J, Andersen S, Laurberg P. Interval between tests and thyroxine estimation method influence outcome of monitoring of subclinical hypothyroidism. J Clin Endocrinol Metab 2008; 93: 1634-1640.
9.10.1.10. Karmisholt J, Laurberg P. Serum TSH and serum thyroid peroxidase antibody fluctuate in parallel and high urinary iodine excretion predicts subsequent thyroid failure in a 1-year study of patients with untreated subclinical hypothyroidism. Eur J Endocrinol 2008; 158: 209-215.
9.10.1.11. Brabant G, Dogu E, Kausche H, Prank K, von zur Mühlen A, Adriaanse R, Romijn JA, Wiersinga WM. Temporal pattern of TSH secretion and its significance in evaluating thyroid status from single TSH determinations. Exp Clin Endocrinol 1994; 102: 49-56.
9.10.1.12. De Marco G, Agretti P, Camilot M et al. Functional studies of new TSH receptor (TSHr) mutations identified in patients affected by hypothyroidism or isolated hyperthyrotrophinaemia. Clin Endocrinol 2009; 70: 335-338.
9.10.1.13. Nicoletti A, Bal M, De Marco G et al. Thyrotropin-stimulating hormone receptor gene analysis in pediatric patients with non-autoimmune subclinical hypothyroidism. J Clin Endocrinol Metab 2009; 94: 4187-4194.
9.10.1.14. Rapa A, Monzani A, Moia S et al. Subclinical hypothyroidism in children and adolescents: a wide range of clinical, biochemical and genetic factors involved. J Clin Endocrinol Metab 2009; 94: 2414-2420.
9.10.1.15. Chikunguwo S, Brethauer S, Niruyogi V et al. Influence of obesity and surgical weight loss on thyroid hormone levels. Surg Obes Relat Dis 2007; 6: 631-635.
9.10.1.16. Rotondi M, Leporati P, La Manna A et al. Raised serum TSH levels in patients with morbid obesity: is it enough to diagnose subclinical hypothyroidism? Eur J Endocrinol 2009; 160: 403-408.
9.10.1.17. Szabolcs I, Podoba J, Feldkamp J et al. Comparative screening for thyroid disorders in old age in areas of iodine deficiency, long-term iodine prophylaxis and abundant iodine intake. Clin Endocrinol 1997; 47: 87-92.
9.10.1.18. Laurberg P, Pedersen KM, Hreidarsson A, Sigfusson N, Iversen E, Knudsen PR. Iodine intake and the pattern of thyroid disorders: a comparative epidemiological study of thyroid abnormalities in the elderly in Iceland and in Jutland, Denmark. J Clin Endocrinol Metab 1998; 83: 765-769.
9.10.1.19. Tunbridge WMG, Evered DC, Hall R et al. The spectrum of thyroid disease in the community: the Whickham survey. Clin Endocrinol 1977; 7: 481-493.
9.10.1.20. Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado Thyroid Disease Prevalence Study. Arch Intern Med 2000; 160: 526-534.
9.10.1.21. Cooper DS, Biondi B. Subclinical thyroid disease. Lancet 2012; 379: 1142-1154.
References for Section 9.10.2.
9.10.2.1 Tunbridge WMG, Brewis M, French JM, et al.: Natural history of autoimmune thyroiditis. Brit Med J 1980; 282: 258-262.
9.10.2.2. Gordin A, Lamberg BA. Spontaneous hypothyroidism in symptomless thyroiditis. A long-term follow-up study. Clin Endocrinol 1981; 15: 537- 543.
9.10.2.3. Cooper DS, Halpern R, Wood LC, Levin AA, Ridgway EC. L-thyroxine therapy in subclinical hypothyroidism. A double-blind, placebo-controlled trial. Ann Int Med 1984; 101: 18-24.
9.10.2.4. Nyström E, Caidahl K, Fager G, Wikkelso C, Lundberg PA, Lindstedt G. A double-blind cross-over 12-month study of L-thyroxine treatment of women with “subclinical hypothyroidism”. Clin Endocrinol 1988; 29: 63-75.
9.10.2.5. Parle JV, Franklyn JA, Cross KW, et al.: Prevalence and follow-up of abnormal thyrotropin (TSH) concentrations in the elderly in the United Kingdom. Clin Endocrinol 1993; 34: 77-83.
9.10.2.6. Jaeschke R, Guyatt G, gerstein H et al. Does treatment with L-thyroxine influence health status in middle-aged and older adults with subclinical hypothyroidism? J Gen Intern Med 1996; 11: 744-749.
9.10.2.7. Kong WM, Sheikh MH, Lumb PJ et al. A 6-month randomized trial of thyroxine treatment in women with mild subclinical hypothyroidism. Am J Med 2002; 112: 348-354.
9.10.2.8. Huber G, Staub JJ, Meier C et al. Prospective study of the spontaneous course of subclinical hypothyroidism: prognostic factors of thyrotropin, thyroid reserve, and thyroid antibodies. J Clin Endocrinol Metab 2002; 87: 3221-3226.
9.10.2.9. Diez JJ, Iglesias P. Spontaneous subclinical hypothyroidism in patients older than 55 years: an analysis of natural course and risk factors for the development of overt thyroid failure. J Clin Endocrinol Metab 2004; 87: 4890-4897.
9.10.2.10. Gussekloo J, van Exel E, de Craen AJM, Meinders AE, FrohlichM, Westendorp RGJ. Thyroid status, disability and cognitive function, and survival in old age. JAMA 2004; 292: 2591-2599.
9.10.2.11. Diez JJ, Iglesias P, Burman KD. Spontaneous normalization of thyrotropin concentrations in patients with subclinical hypothyroidism. J Clin Endocrinol Metab 2005; 90: 4124-4127.
9.10.2.12. Demirbilek H, Kandemir N, Gonc EN, Ozon A, Alikasifoglu A. Assessment of thyroid function during the long course of Hashimoto’s thyroiditis in children and adolescents. Clin Endocrinol 2009; 71: 451-454.
9.10.2.13. Wasniewska M, Salermo M, Cassio A et al. Prospective evaluation of the natural course of idiopathic subclinical hypothyroidism in childhood and adolescence. Eur J Endocrinol 2009; 160: 417-421.
9.10.2.14. Geul KW, van Sluisveld ILL, Grobbee DE, et al.: The importance of thyroid microsomal antibodies in the development of elevated serum TSH in middle-aged women: Associations with serum lipids. Clin Endocrinol 1993; 39: 275-280.
9.10.2.15. Vanderpump MPJ, Tunbridge WMG, French JM, et al.: The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham Survey. Clin Endocrinol 1995; 43: 55-68.
9.10.2.16. Strieder TG, Tijssen JG, Wenzel BE, Endert E, Wiersinga WM. Prediction of progression to overt hypothyroidism or hyperthyroidism in female relatives of patients with autoimmune thyroid disease using the Thyroid Events Amsterdam (THEA) score. Arch Int Med 2008; 168: 1657-1663.
9.10.2.17. Walsh JP, Bremner AP, Feddema P, Leedman PJ, Brown SJ, O’Leary P. Thyrotropin and thyroid antibodies as predictors of hypothyroidism: a 13-year, longitudinal study of a community-based cohort using current immunoassay techniques. J Clin Endocrinol Metab 2010; 95: 1095-1104.
9.10.2.18. Rosario PW, Bessa B, Valadao MM, Purisch S. Natural history of mild subclinical hypothyroidism: prognostic value of ultrasound. Thyroid 2009; 19: 9-12
9.10.2.19. Somwaru LL, Rariy CM, Arnold AM, Cappola AR. The natural history of subclinical hypothyroidism in the elderly: the cardiovascular health study. J Clin Endocrinol Metab 2012; 97: 1962-1969..
9.10.2.20. Sathyapalan T, Manuchehri AM, Thatcher NJ et al. The effect of soy phytoestrogen supplementation on thyroid status and cardiovascular risk markers in patients with subclinical hypothyroidism: a randomized, double-blind, crossover study. J Clin Endocrinol Metab 2011; 96: 1442-1449.
References for Section 9.10.3.
9.10.3.1. Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev 2008; 29: 76-131.
9.10.3.2. Staub JJ, Althaus BU, Engler H, et al.: Spectrum of subclinical and overt hypothyroidism: effect on thyrotropin, prolactin, and thyroid reserve, and metabolic impact on peripheral target tissues. Am J Med 1992; 92: 631-641.
- Cooper DS, Halpern R, Wood Lc, Levin AA, Ridgway EC. Thyroxine therapy in subclinical hypothyroidism. A double-blind, placebo-controlled trial. Ann Int Med 1984; 101: 18-24.
- Zulewski H, Muller B, Exer P, Miserez AR, Staub JJ. Estimation of tissue hypothyroidism by a new clinical score: evaluation of patients with various grades of hypothyroidism and controls. J Clin Endocrinol Metab 1997; 82: 771-776.
- Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Int Med 2000; 160: 526-534.
9.10.3.6. Grabe HJ, Volzke H, Ludemann J et al. Mental and physical complaints in thyroid disorders in the general population. Acta Psychiatr Scand 2006; 145: 573-581.
9.10.3.7. Haggerty JJ, Stern RA, Mason GA, et al.: Subclinical hypothyroidism: a modifiable risk factor for depression? Am J Psychiatry: 150; 508-510.
9.10.3.8. Gussekloo J, van Exel E, de Craen AJM, Meinders AE, FrohlichM, Westendorp RGJ. Thyroid status, disability and cognitive function, and survival in old age. JAMA 2004; 292: 2591-2599.
9.10.3.9. Roberts LM, Pattison H, Roalfe A et al. Is subclinical thyroid dysfunction in the elderly associated with depression or cognitive dysfunction? Ann Int Med 2006; 145: 573-581.
9.10.3.10. Jordes R, Waterloo K, Storhaug H et al. Neuropsychologic ial function and symptoms in subjects with subclinical hypothyroidism and the effect of thyroxine treatment. J Clin EndocrinolMetab 2006; 91: 145-153.
9.10.3.11. Bell RJ, Rivera-Woll L, Davidson SL, Topliss DJ, Donath S, Davis SR. Well-being, health-related quality of life and cardiovascular risk profile in women with subclinical thyroid disease – a community –based study. Clin Endocrinol 2007; 66: 548-556,
9.10.3.12. Park YJ, Lee EJ, Lee YJ et al. Subclinical hypothyroidism (SCH) is not associated with metabolic derangement, cognitive impairment, depression or poor quality of life (QoL) in elderly subjects. Arch Gerontol Geriatr 2010; 50: e68-e73.
9.10.3.13. Monzani F, Del Guerra P, Caraccio N, et al.: Subclinical hypothyroidism: neurobehavioral features and beneficial effect of L-thyroxine treatment. Clin Investig 1993; 71: 367-371.
9.10.3.14. Baldini IM, Vita A, Mauri MC et al. Psychopathological and cognitive features in subclinical hypothyroidism. Prog Neuropsychopharmacol Biol Psychiatry 1997; 21: 925-935.
9.10.3.15. Zhu DF, Wang ZX, Zhang DR et al. fMRI revealed neural substrate for reversible working memory dysfunction in subclinical hypothyroidism. Brain 2006; 129: 2923-2930.
9.10.3.16. Correia N, Mullally S, Cooke G et al. Evidence for a specific defect in hippocampal memory in overt and subclinical hypothyroidism. J Clin Endocrinol Metab 2009; 94: 3789-3797.
9.10.3.17. Ripoli A, Pingitore A, Favilli B et al. Does subclinical hypothyroidism affect cardiac pump performance? Evidence from a magnetic resonance imaging study. J Am Coll Cardiol 2005; 45: 439-445.
9.10.3.18. Vitale G, Galderisi M, Lupoli GA et al. Left ventricular myocardial impairment in subclinical hypothyroidism assessed by a new ultrasound tool: pulsed tissue Doppler. J Clin Endocrinol Metab 2002; 87: 4350-4355.
9.10.3.19. Aghini-Lombardi F, Di Bello Vitantonio, Enrica T et al. Early textural and functional alterations of left ventricular myocardium in mild hypothyroidsm. Eur J Endocrinol 2006; 155: 3-9.
9.10.3.20. Baycan S, Erdogan D, Caliskan M et al. Coronary flow reserve is impaired in subclinical hypothyroidism. Clin Cardiol 2007; 30: 562-566.
9.10.3.21. Biondi B, Galderisi M, Pagano L et al. Endothelial-mediated coronary flow reserve in patients with mild thyroid hormone deficiency. Eur J Endocrinol 2009; 161: 323-329.
9.10.3.22. Brenta G, Mutti LA, Schnitman M, Fretes O, Pezzone A, Matute ML. Assessment of left ventricular diastolic function by radionuclide ventriculography at rest and exercise in subclinical hypothyroidism, and its response to L-thyroxine therapy. Am J Cardiol 2003; 91: 1327-1330.
9.10.3.23. Kahaly GJ. Cardiovascular and atherogenic aspects of subclinical hypothyroidism. Thyroid 2000; 10: 665-679.
9.10.3.24. Nagasaki T, Inaba M, Kumeda Y et al. Increased pulse wave velocity in subclinical hypothyroidism. J Clin Endocrinol Metab 2006; 91: 154-158.
9.10.3.25. Nagasaki T, Inaba M, Kumeda Y et al. Central pulse wave velocity is responsible for increased brachial-ankle pulse wave velocity in subclinical hypothyroidism. Clin Endocrinol 2007; 66: 304-308.
9.10.3.26. Owen PJD, Rajiv C, Vinereanu D, Mathew T, Fraser AG, Lazarus JH. Subclinical hypothyroidsm, arterial stiffness and myocardial reserve. J Clin Endocrinol Metab 2006; 91: 2126-2132.
9.10.3.27. Lekakis J, Papamichael C, Alevizaki M, Piperingos G, Marafelia P, Mantzos J. Flow-mediated, endothelium-dependent vasodilatation is impaired in subjects with hypothyroidism, borderline hypothyroidism, and high-normal serum thyrotropin (TSH) values. Thyroid 1997; 7: 411-414.
9.10.3.28. Taddei S, Caraccio N, Virdis A et al. Impaired endothelium-dependent vasodilatation in subclinical hypothyroidism: beneficial effect of levothyroxine therapy. J Clin Endocrinol Metab 2003; 88: 3731-3737.
9.10.3.29. Cikim AS, Oflaz H, Ozbey N et al. Evaluation of endothelial function in subclinical hypothyroidism and subclinical hyperthyroidism. Thyroid 2004; 14: 605-609.
9.10.3.30. Taddei S, Caraccio N, Virdis A et al. Low-grade systemic inflammation causes endothelial dysfunction in patients with Hashimoto’s thyroiditis. J Clin Endocrinol Metab 2006; 91: 5076-5082.
9.10.3.31. Monzani F, Caraccio N, Kozakowa M et al. Effect of levothyroxine replacement on lipid profile and intima-media thickness in subclinical hypothyroidism: a double-blind, placebo-controlled study. J Clin Endocrinol Metab 2004; 89: 2009-2106.
9.10.3.32. Nagasaki T, Inaba M, Henmi Y et al. Decrease in carotid intima-media thickness in hypothyroid patients after normalization of thyroid function. Clin Endocrinol 2003; 59: 607-612.
9.10.3.33. Tunbridge WM, Evered DC, Hall R et al. Lipid profiles and cardiovascular disease in the Whickham area with particular reference to thyroid failure. Clin Endocrinol 1977; 7: 495-508.
9.10.3.34. Hueston WJ, Pearson WS. Subclinical hypothyroidism and the risk of hypercholesterolemia. Ann Fam Med 2004; 2: 351-355.
9.10.3.35. Walsh JP, Bremmer AP, Bulsara MK et al. Thyroid dysfunction and serum lipids: a community-based study. Clin Endocrinol 2005; 63: 670-675.
9.10.3.36. Kanaya AM, Harris F, Volpato S, Perez-Stable EJ, Harris T, Bauer DC. Association between thyroid dysfunction and total cholesterol level in an older biracial population: the health, aging and body composition study. Arch Int Med 2002; 162: 773-779.
9.10.3.37. Bindels AJ, Westendorp RG, Frolich M, Seidell JC, Blokstra A, Smelt AH. The prevalence of subclinical hypothyroidism at different total plasma cholesterol levels in middle aged men and women: a need for case-finding? Clin Endocrinol 1999; 50: 217-220.
9.10.3.38. Bauer DC, Ettinger B, Browner WS. Thyroid function and serum lipids in older women: a population-based study. Am J Med 1998; 104: 546-551.
9.10.3.39. Torun AN, Kulaksizoglu S,Kulaksizoglu M, Pamuk BO, Isbilen E, Tutuncu NB. Serum total antioxidant status and lipid peroxidation marker malondialdehyde levels in overt and subclinical hypothyroidism. Clin Endocrinol 2009; 70: 469-474.
9.10.3.40. Brenta G, Berg G, Zago V et al. Proatherogenic mechanisms in subclinical hypothyroidism: hepatic lipase activity in relation to the VLDL remnant IDL. Thyroid 2008; 18: 1233-1236.
9.10.3.41. Bakker SJ, ter Maaten JC, Popp-Snijders C, Slaets JP, Heine RJ, Gans RO. The relationship between thyrotropin and low density lipoprotein cholesterol is modified by insulin sensitivity in healthy euthyroid subjects. J Clin Endocrinol Metab 2001; 86: 1206-1211.
9.10.3.42. Muller B, Zulewski H, Huber P, Ratcliffe JG, Staub JJ. Impaired action of thyroid hormone associated with smoking in women with hypothyroidism. N Engl J Med 1995; 333: 964-969.
9.10.3.43. Tuzcu A, Bahceci M, Gokalp D, Tuzun Y, Gunes K. Subclinical hypothyroidism may be associated with elevated highsensitive C-reactive protein (low grade inflammation) and fasting hyperinsulinemia. Endocr J 2005; 52: 89-94.
9.10.3.44. Maratou E, Hadjidakis DJ, Kollias A et al. Studies of insulin resistance in patients with clinical and subclinical hypothyroidism. Eur J Endocrinol 2009; 160: 785-790.
9.10.3.45. Christ-Crain M, Meier C, Guglielmetti M et al. Eleveated C-reactive protein and homocysteine values: cardiovascular risk factors in hypothyroidism? A cross-sectional and a double-blind, placebo-controlled trial. Atherosclerosis 2003; 166: 379-386.
9.10.3.46. Kvetny J, Heldgaard PE, Bladbjerg EM, Gram J. Subclinical hypothyroidism is associated with a low-grade inflammation, increased triglyceride levels and predicts cardiovascular disease in males below 50 years. Clin Endocrinol 2004; 61: 232-238.
9.10.3.47. Hueston WJ, King DE, Geesey ME. Serum biomarkers for cardiovascular inflammation in subclinical hypothyroidism. Clin Endocrinol 2005; 63: 582-587.
9.10.3.48. Ozcan O, Cakir E, Yaman H, et al. The effects of thyroxine replacement on the levels of serum asymmetric dimethylarginine (ADMA) and other biochemical cardiovascular risk markers in patients with subclinical hypothyroidism. Clin Endocrinol 2005; 63: 203-206.
9.10.3.49. Jorde R, Figenschau Y, Hansen JB. Haemostatic function in subjects with mild subclinical hypothyroidism. The Tromso study. Thromb Haemost 2006; 95: 750-751.
9.10.3.50. Goulis DG, Tsimpiris N, Delaroudis S et al.: Stapedial reflex: A biological index found to be abnormal in clinical and subclinical hypothyroidism. Thyroid 1998; 8: 583.
9.10.3.51. Misiunas A, Niepominiscze H, Ravera B et al. Peripheral neuropathy in subclinical hypothyroidsm. Thyroid 1995; 5: 283-286.
9.10.3.52. Monzani F, Caraccio N, Siciliano G et al.: Clinical and biochemical features of muscle dysfunction in subclinical hypothyroidism. J Clin Endocrinol Metab 1997; 82: 3315-3318.
9.10.3.53. CaraccioN, Natali A, Sironi A et al. Muscle metabolism and exercise tolerance in subclinical hypothyroidsm: a controlled trial of levothyroxine. J Clin Endocrinol Metab 2005; 90: 4057-4062.
9.10.3.54. Reuters VS, Teixeira PF, Vigario PS et al. Functional capacity and muscular abnormalities in subclinical hypothyroidism. Am J Med Sci 2009; 338: 259-263.
9.10.3.55. Simonsick EM, Newman AB, Ferrucci L et al. Subclinical hypothyroidism and functional mobility in older adults. Arch Int Med 2009; 169: 2011-2017.
9.10.3.56. Laukkarinen J, Kiudelis G, Lempinen M et al. Increased prevalence of subclinical hypothyroidism in common bile duct stones. J Clin Endocrinol Metab 2007; 92: 4260-4264.
9.10.3.57. Squizzato A, Romualdi E, Piantanida E et al. Subclinical hypothyroidism and deep venous thrombosis. Thromb Haemost 2007; 97: 803-806.
9.10.3.58. Nagata M, Suzuki A, Sekiguchi S et al. Subclinical hypothyroidism is related to lower heel QUS in postmenopausal women. Endocr J 2007; 54: 625-630.
9.10.3.59, Texeira PF, Cabral MD, Silva NA et al. Serum leptin in overt and subclinical hypothyroidism: effect of levothyroxine treatment and relationship to menopausal status and body composition. Thyroid 2009; 19: 443-450.
9.10.3.60. Tanda ML, Lombardi V, Genovesi M et al. Plasma total and acylated ghrelin concentrations in patients with clinical and subclinical thyroid dysfunction. J Endocrinol Invest 2009; 32: 74-78.
9.10.3.61. Akin F, Yaylali GF, Turgut S, Kaptanoglu B. Growth hormone/insulin-like growth factor axis in patients with subclinical thyroid dysfunction. Growth Horm IGF Res 2009; 19: 252-255.
9.10.3.62. Cooper DS, Biondi B. Subclinical thyroid disease. Lancet 2012; 379: 1142-1154.
9.10.3.63.de Jongh RT, Lips P, van Schoor NM, et al. Endogenous subclinical thyroid disorders, physical and cogniitive function, depression, and mortality in older individuals. Eur J Endocrinol 2011; 165: 545-554.
9.10.3.64. Lindeman RD, Schade DS, LaRue A, et al. Subclinical hypothyroidism in a biethnic, urban community. J Am Geriatr Soc 1999; 47: 703-709.
9.10.3.65. Lee JS, Buzkova P, Fink HA, et al. Subclinical thyroid dysfunction and incident hip fracture in older adults. Arch Intern Med 2012; 170: 1876-1883.
References for Section 9.10.4.
9.10.4.1. Bastenie PA, Vanhaelst L, Bonnyns M et al.: Preclinical hypothyroidism: a risk factor for coronary heart disease. Lancet 1971; i: 203-204
9.10.4.2. Bastenie PA, Vanhaelst L, Goldstein J et al.: Asymptomatic autoimmune thyroiditis and coronary heart disease. Cross-sectional and prospective studies. Lancet 1977; ii: 155-158.
9.10.4.3. Vanderpump MPJ, Tunbridge WMG, French JM, et al.: The development of ischemic heart disease in relation to autoimmune thyroid disease in a 20-year follow-up study of an English community. Thyroid 1996; 6: 155-160.
9.10.4.4. Hak AE, Pols HA, Visser TJ, Drexhage HA, Hofman A, Witteman JC. Subclinical hypothyroidism is an independent risk factor for atheroslerosis and myocardial infarction in elderly women: the Rotterdam study. Ann Intern Med 2000; 132: 270-278.
9.10.4.5. Parle JV, Maisonneuve P, Sheppard MC, Boyle P, Franklyn JA. Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: a 10-year cohort study. Lancet 2001; 358: 861-865.
9.10.4.6. Imaizumi M, Akahoshi M, Ichimaru S et al. Risk for ischemic heart disease and all-cause mortality in subclinical hypothyroidism. J Clin Endocrinol Metab 2004; 89: 3365-3370.
9.10.4.7. Gussekloo J, van Exel E, de Craen AJ, Meinders AE, Frolich M, Westendorp RG. Thyroid status, disability and cognitive function, and survival in old age. JAMA 2004; 292: 2591-2599.
9.10.4.8. Walsh JP, Bremner AP, Bulsara MK et al. Subclinical thyroid dysfunction as a risk factor for cardiovascular disease. Arch Intern Med 2005; 165: 2467-2472.
9.10.4.9. Rodondi N, Newman AB, Vittinghoff E et al. Subclinical hypothyroidism and the risk of heart failure, other cardiovascular events, and death. Arch Intern Med 2005; 165: 2460-2466.
9.10.4.10. Cappola AR, Fried LP, Arnold AM et al. Thyroid status, cardiovascular risk, and mortality in older adults: the cardiovascular health study. JAMA 2006; 295: 1033-1041.
9.10.4.11. Iervasi G, Molinaro S, Landi P et al. Association between increased mortality and mild thyroid dysfunction in cardiac patients. Arch Intern Med 2007; 167: 1526-1532.
9.10.4.12. Iqbal A, Schimer H, Lunde P, Figenschau Y, Rasmussen K, Jorde R. Thyroid stimulating hormone and left ventricular function. J Clin Endocrinol Metab 2007; 92: 3504-3510.
9.10.4.13. Rodondi N, Bauer DC, Cappola AR et al. Subclinical thyroid dysfunction, and the risk of heart failure. The Cardiovascular Health Study. J Am Coll Cardiol 2008; 52: 1152-1159.
9.10.4.14. Boekholdt SM, Titan SM, Wiersinga WM et al. Initial thyroid status and cardiovascular risk factors; the EPIC-Norfolk propsective population study. Clin Endocrinol 2010; 72: 404-410.
9.10.4.15. Sgarbi JA, Matsumura LK, Kasamatsu TS, Ferreira SR, Maciel RM. Subclinical thyroid dysfunctions are independent risk factors for mortality in a 7.5-year follow-up: the Japanese-Brazilian thyroid study. Eur J Endocrinol 2010; 162: 569-577.
9.10.4.16. Ravzi S, Weaver JU, Vanderpump MP, Pearce SH. The incidence of ischemic heart disease and mortality in people with subclinical hypothyroidism: reanalysis of the Whickham Survey cohort. J Clin Endocrinol Metab 2010; 95: 1734-1740.
9.10.4.17. Rodondi N, Aujesky D, Vittinghoff, Cornuz J, Bauer DC. Subclinical hypothyroidism and the risk of coronary heart disease: a meta-analysis. Am J Med 2006; 119: 541-551.
9.10.4.18. Volzke H, Schwahn, Wallaschoski H, Dorr M. Review: The association of thyroid dysfunction with all-cause and circulatory mortality: is there a causal relationship? J Clin Endocrinol Metab 2007; 92: 2421-2429.
9.10.4.19. Ochs N, Auer R, Bauer DC et al. Meta-analysis: subclinical thyroid dysfunction and the risk for coronary heart disease and mortality. Ann Intern Med 2008; 148: 832-845.
9.10.4.20. Haentjens P, Van Meerhaeghe A, Poppe K, Velkeniers B. Subclinical thyroid dysfunction and mortality: an estimate of relative and absolute excess all-cause mortality based on time-to-event data from cohort studies. Eur J Endocrinol 2008; 159: 329-341.
9.10.4.21. Singh S, Duggal J, Molnar J, Maldonado F, Barsano CP, Arora R. Impact of subclinical disorders on coronary heart disease, cardiovasdcular and all-cause mortality: a meta-analysis. Int J Cardiol 2008; 125: 41-48.
9.10.4.22. Ravzi S, Shakoor A, Vanderpump M, Weaver JU, Pearce SHS. The influence of age on the relationship between subclinical hypothyroidism and ischemic heart disease: a metaanlysis. J Clin Endocrinol Metab 2008; 93: 2998-3007.
9.10.4.23. Rodondi N, Den Elzen W, Bauer DC et al. Subclinical hypothyroidism and the risk of coronary disease and mortality. JAMA 2010; 304: 1365-1374.
9.10.4.24. McQuade C, Skugor M, Brennan DM, Hoar B, Stevenson C, Hoogwerf BJ. Hypothyroidism and moderate subclinical hypothyroidism are associated with increased all-cause mortality independent of coronary heart disease risk factors: a PreCIS database study. Thyroid 2011; 21: 837-843.
9.10.4.25. Nanchen D, Gussekloo J, Westerdorp RG, et al. Subclinical thyroid dysfunction and the risk of heart failure in older persons at high cardiovascular risk. J Clin Endocrinol Metab 2012; 97: 852-861.
9.10.4.26. Waring AC, Harrison S, Samuels MH, et al. Thyroid function and mortality in older men: a prospective study. J Clin Endocrinol Metab 2012; 97: 862-870.
9.10.4.27. Tseng FY, Lin WY, Lin CC, et al. Subclinical hypothyroidism is associated with increased risk for all-cause and cardiovascular mortality in adults. J Am Coll Cardiol 2012; 60: 730-737.
9.10.4.28. Waring AC, Arnold AM, Newman AB, Buzkova P, Hirsch C, Cappola AR. Longitudinal changes in the oldest old and survival: the cardiovascular health study all-stars study. J Clin Endocrinol Metab 2012; 97: 3944-3950.
9.10.4.29. Hyland KA, Arnold AM, Lee Js, Cappola AR. Persisitent subclinical hypothyroidism and cardiovascular risk in the elderly: the cardiovascular health study. J Clin Endocrinol Metab 2013; 98: 533-540.
9.10.4.30. LeGrys VA, Funk MJ, Lorenz CE, et al. Subclincial hypothyroidism and risk for incident myocardial infarction among postmenopausal women. J Clin Endocrinol Metan 3013; 98: 2308-2317. 9.10.4.31. Rhee CM, Curhan GC, Alexander EK, Bhan I, Brunelli SM. Subclinical hypothyroidism and survival: the effects of heart failure and race. J Clin Endocrinol Metab 2013; 98: 2326-2336.
9.10.4.32. Gencer B, Collet TH, Virgini V, et al. Subclinical thyroid dysfunction and the risk of heart failure events: an individual participant data analysis from 6 prospective cohorts. Circulation 2012; 126: 1040-1049.
References for Section 9.10.5
9.10.5.1. Villar HC, Saconato H, Valente O, Atallah AN. Thyroid hormone replacement for subclinical hypothyroidism. Cochrane Database Syst Rev 2007; issue 3: CD003419.
9.10.5.2. Parle J, Roberts L, Wilson S et al. A randomized controlled trial of the effect of thyroxine replacement on cognitive function in community-living elderly subjects with subclinical hypothyroidism: the Birmingham Elderly Thyroid Study. J Clin Endocrinol Metab 2010; 95: May 25 (Epub ahead of print).
9.10.5.3. Baldini M, Colasanti A, Orsatti A, Airaghi L, Mauri MC, Cappellini MD. Neuropsychological functions and metabolic aspects in subclinical hypothyroidism: the effects of L-thyroxine. Prog Neuropsychopharmacol Biol Psychiatry 2009; 33: 854-859.
9.10.5.4. Correia N, Mullally S, Cooke G et al. Evidence for a specific defect in hippocampal memory in overt and subclinical hypothyroidism. J Clin Endocrinol Metab 2009; 94: 3789-3797.
9.10.5.5. Biondi B, Palmieri EA, Lombardi G, Fazio S. Effects of subclinical thyroid dysfunction on the heart. Ann Int Med 137:904-914, 2002.
9.10.5.6. Faber J, Petersen L, Wiinberg N et al. Hemodynamic changes after levothyroxine treatment in subclinical hypothyroidism. Thyroid 2002; 12: 319-324.
9.10.5.7. Adrees M, Gibney J, El-Saeity N, Boran G. Effects of L-T4 replacement in women with subclinical hypothyroidism. Clin Endocrinol 2009; 71: 298-303.
9.10.5.8. Nagasaki T, Inaba M, Yamada S et al. Decrease of brachial-ankle pulse wave velocity in female subclinical hypothyroid patients during normalization of thyroid function: a double-blind, placebo-controlled study. Eur J Endocrinol 2009; 160: 409-415.
9.10.5.9. Owen PJ, Rajiv C, Vinereanu D, Mathew T, Fraser AG, Lazarus JH. Subclinical hypothyroidism, arterial stiffness, and myocardial reserve. J Clin Endocrinol Metab 2006; 91: 2126-2132.
9.10.5.10. Peleg RK, Efrati S, Benbassat C, Fygenzo M, Golik A. The effect of arterial stiffness and lipid profile in patients with subclinical hypothyroidism. Thyroid 2008; 18: 825-830.
9.10.5.11. Shakoor SK, Aldibbiat A, Ingoe LE et al. Endothelial progenitor cells in subclinical hypothyroidism: the effect of thyroid hormone replacement therapy. J Clin Endocrinol Metab 2010; 95: 319-322.
9.10.5.12. Duman D, Demirtunc R, Sahin S, Esertas K. The effects of simvastatin and levothyroxine on intima-media thickness of carotid artery in female normolipemic patients with subcliniccal hypothyroidism: a prospective, randomized-controlled study. J Cardiovasc Med 2007; 8: 1007-1011.
9.10.5.13. Kim SK, Kim SH, Park KS, Park SW, Cho YW. Regression of increased common carotid artery-intima media thickness in subclinical hypothyroidism after thyroid hormone replacement. Endocr J 2009; 56: 753-758.
9.10.5.14. Kebapcilair L, Comlekci A, Tuncel P et al. Effect of levothyroxine replacement therapy on paraoxonase-1 and carotid intima-media thickness in subclinical hypothyroidism. Med Sci Monit 2010; 16: CR41-47.
9.10.5.15. Teixeira PF, Reuters VS, Ferreira MM et al. Treatment of subclinical hypothyroidism reduces atherogenic lipid levels in a placebo-controlled double-blind trial. Horm Metab Res 2008; 40: 50-55.
9.10.5.16. Tanis BC, Westendorp RGJ, Smelt AHM: Effect of thyroid substitution on hypercholesterolaemia in patients with subclinical hypothyroidism: a reanalysis of intervention studies. Clin Endocrinol 1996; 44: 643-649.
9.10.5.17. Danese MD, Ladenson PW, Meinert CL, Powe NR. Effect of thyroxine therapy on serum lipoproteins in patients with mild thyroid failure: a quantitative review of the literature. J Clin Endocrinol Metab 2000; 85: 2993-3001.
9.10.5.18. Duntas LH, Wartofsky L. Cardiovascular risk factors and subclinical hypothyroidism: focus on lipids and new emerging risk factors. What is the evidence? Thyroid 2007; 17: 1075-1084.
9.10.5.19. Meier C, Staub J-J, Roth C-B, et al. TSH-controlled L-thyroxine therapy reduces cholesterol levels and clinical symptoms in subclinical hypothyroidism: a double-blind, placebo-controlled trial (Basel Thyroid Study). J Clin Endocrinol Metab 2001; 86: 4860-4866
9.10.5.20. Mainenti MR, Vigario PS, Teixeira PF, Maia MD, Oliveira FP, Vaisman M. Effect of levothyroxine replacement on exercise performance in subclinical hypothyroidism. J Endocrinol Invest 2009; 32: 470-473.
9.10.5.21. Simonsick EM, Newman AB, Ferrucci L et al. Health ABC study. Subclinical hypothyroidism and functional mobility in older adults. Arch Intern Med 2009; 169: 2011-2017.
9.10.5.22. Cinemre H, Bilir C, Gokosmanoglu F, Bahcebasi T. Hematologic effects of levothyroxine in iron-deficient subclinical hypothyroid patients: a randomized, double-blind, controlled study. J Clin Endocrinol Metab 2009; 94: 151-156.
9.10.5.23. Abalovich M, Amino N, Barbour LA et al. Thyroid dysfunction during pregnancy and and postpartum: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2007; 92: Supplement S1-S47.
9.10.5.24. Rahman AH, Abbassy HA, Elatif Abbassy AA. Improved IVF outcomes after treatment of subclinical hypothyroidism in infertle women. Endocr Pract 2010; 29: 1-17.
9.10.5.25. Dermott MT, Ridgway EC. Subclinical hypothyroidism is mild thyroid failure and should be treated. J Clin Endocrinol Metab 2001; 86: 4585-4590.
9.10.5.26. Owen PJD, Lazarus JH. Subclinical hypothyroidism: the case for treatment. Trends Endocrinol Metab 2003; 14: 257-261.
9.10.5.27. Chu JW, Crapo LM. The treatment of subclinical hypothyroidism is seldom necessary. J Clin Endocrinol Metab 2001; 86: 4591-4599.
9.10.5.28. Vanderpump M. Subclinical hypothyroidism: the case against treatment. Trends Endocrinol Metab 2003; 14: 262-266.
9.10.5.29. Surks MI, Ortiz E, Daniels GH et al. Subclinical thyroid disease. Scientific review and guidelines for diagnosis and management. JAMA 2004; 291: 228-238.
9.10.5.30. Ladenson PW. Cardiovascular consequences of subclinical thyroid dysfunction: more smoke but no fire. Ann Intern Med 2008; 148; 880-881.
9.10.5.31. Klubo-Gwiezdzinska J, Wartofsky L. Thyrotropin blood levels, subclinical hypothyroidism, and the elderly patient. Arch Intern Med 2009; 169: 1949-1951.
9.10.5.32.. Cooper DS. Subclinical hyperthyroidism. N Engl J Med 2001; 345: 260-265.
9.10.5.33. Gussekloo J, van Exel E, de Craen AJ, Meinders AE, Frolich M, Westendorp RG. Thyroid status, disability and cognitive function, and survival in old age. JAMA 2004; 292: 2591-2599.
9.10.5.34. Razvi S, Weaver JU, Butler TJ, Pearce SH. Levothyroxine treatment of subclinical hypothyroidism, fatal and nonfatal cardiovascular events, and mortality. Arch Intern Med 2012; 172: 811-817.
9.10.5.35. Ravzi S, Ingoe L, Keeka G, Outes C, McMillan C, Weaver JU. The beneficial effect of L-thyroxine on cardiovascular risk factors, endothelial function, and quality of life in subclinical hypothyroidism: randomized, crossover trial. J Clin Endocrinol Metab 2007; 92: 1715-1723.
9.10.5.36 Garber JR, Cobin RH, Gharib H, et al: Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid association. Thyroid 2012; 22:1200-1235.
9.10.5.37 Pearce SHS, Brabant G, Duntas LH, et al: 2013 ETA guideline: Management of subclinical hypothyroidism. Eur Thyroid , 2013; 2:
References for Section 9.11.
9.11.1. Wang C, Crapo LM: The epidemiology of thyroid disease and implication for screening. Endocrinol Metab Clin North Am 1997; 26: 189-218.
9.11.2. Kågedal B, Månson JC, Norr A, et al.: Screening for thyroid disorders in middle-age women by computer-assisted evaluation of a thyroid hormone panel. Scand J Clin Lab Invest 1981; 41: 403-408.
9.11.3. Meier Ch, Staub JJ, Roth CB, et al.: Screening for new thyroid dysfunction in women attending a primary care unit using a third generation TSH assay. Schweiz Med Wochenschr 1998; 128: 128: 250-253
9.11.4. Helfand M, Capro LM: Screening for thyroid disease. Ann Int Med 1990; 112: 840-849.
9.11.5. Danese MD, Powe NR, Sawin CT, Ladenson PW: Screening for mild thyroid failure at the periodic health examination: A decision and cost-effectiveness analysis. JAMA 1996; 276: 285-292.
9.11.6. Powe NR, Danese MD, Ladenson PW: Decision analysis in endocrinology and metabolism. Endocrinol Metab Clin North Am 1997; 26: 89-112.
9.11.7. Helfand M, Redfern CC: Screening for thyroid disease: an update. Ann Intern Med 1998; 129: 144-158
9.11.8. Weetman AP: Hypothyroidism: screening and subclinical disease. Brit Med J 1997; 314: 1175-1178.
9.11.9. Vanderpump MPJ, Ahlquist JAO, Franklyn JA, Clayton RN: Consensus statement for good practice and audit measures in the management of hypothyroidism and hyperthyroidism. BMJ 1996; 313: 539-544.
9.11.10. Oomen HAPC, Schipperijn AMJ, Drexhage HA: The prevalence of affective disorder and in particular of a rapid cycling of bipolar disorder in patients with abnormal thyroid function tests. Clin Endocrinol 1996; 45: 215-223.
9.11.11. Giani C, Fierabracci P, Bonacci R, et al.: Relationship between breast cancer and thyroid disease: relevance of autoimmune thyroid disorders in breast malignancy. J Clin Endocrinol Metab 1996; 81: 990-994.
9.11.12. Negro R, Schwartz A, Gismondi R, Tinelli A, Mangieri T, Stagnaro-Green A. Universal sreening versus case finding for detection and treatment of thyroid hormonal dysfunction during pregnancy. J Clin Endocrinol Metab 2010; 95: 1699-1707.
Publication Details
Author Information and Affiliations
Publication History
Last Update: March 28, 2014.
Copyright
This electronic version has been made freely available under a Creative Commons (CC-BY-NC-ND) license. A copy of the license can be viewed at http://creativecommons.org/licenses/by-nc-nd/2.0/.
Publisher
MDText.com, Inc., South Dartmouth (MA)
NLM Citation
Wiersinga WM. Adult Hypothyroidism. [Updated 2014 Mar 28]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.