A project was initiated to evaluate the structure-activity relationship of the (R)-phenethyl morpholine scaffold of the previously discovered VU0469118. This medicinal chemistry effort led to the identification of a potent and selective compound for D4.4 (130 nM) versus the other dopamine receptors (D1, D2S, D2L, D3, and D5, all >20 μM). Several analogs were evaluated and it was determined that amides, ureas, and sulfonamide substitutions were not tolerated on the morpholine nitrogen. But rather, benzylic substitution was well tolerated leading to three compounds with excellent antagonist activity against D4.4. From these studies, a novel 4-chlorobenzyl compound was identified and designated a Molecular Libraries Probe Production Centers Network (MLPCN) probe molecule, ML398, which shows >100-fold selectivity for the other dopamine receptors and is a potent antagonist with <100 nM (Ki = 36 nM).
Assigned Assay Grant #: 5U54 MH084659-05
Screening Center Name & PI: N/A
Chemistry Center Name & PI: Vanderbilt Specialized Chemistry Center for Accelerated Probe Development, Craig W. Lindsley
Assay Submitter & Institution: Craig W. Lindsley, Vanderbilt University
PubChem Summary Bioassay Identifier (AID): 743362

1. Recommendations for scientific use of the probe
ML398 (CID 72737723) is an antagonist against dopamine D4 receptor with excellent potency (IC50 = 130 nM) and binding (Ki = 36 nM). In addition, ML398 shows excellent selectivity (>100-fold) against the other dopamine receptor subtypes (D1,2S,2L,3,5; >20 μM). This represents the first potent and selective D4 antagonist that is not covered by patents from pharmaceutical companies, which, in some cases, can limit their utility. Also, most of these compounds contain chemotypes that have shown high promiscuity towards other dopamine subtypes. As the D4 receptor has been implicated in modifying cognitive impairment (such as those associated with Alzheimer's disease), cocaine addiction, and relieving L-DOPA induced dyskinesias (LIDs); the develop of this new probe molecule will allow researchers in these areas to validate these mechanisms with a small molecule antagonist. In addition, ML398 will be the starting point for the development of a PET tracer.
2. Materials and Methods
Dopamine Receptor Assays. All dopamine receptor assays (single point, IC50's and Ki's) were conducted by EuroFins (Luxembourg), as previously reported. In brief, compounds were tested either as a single point determination for radioligand displacement at 10 μM, or for IC50 determinations were tested at 5 concentrations by 3-fold serial dilutions starting at 20 μM. Human recombinant CHO cells were stably transfected with human D1, D2L, D2S, D3, D4.4 (CHO-K1 cells) or D5. Assay buffer was added 50 mM Tris-HCl, pH 7.4, 1.4 mM ascorbic acid, 0.001% BSA and 150 mM NaCl, and the compounds were diluted in 1% DMSO. The incubation time and temperature were 2 hours and 25 °C. The compounds were assayed using a radioligand binding method using the appropriate ligands (D1, 1.40 nM, [3H] SCH-23390; D2L and D2S, 0.16 nM [3H] Spiperone; D3, 0.70 nM [3H] Spiperone; D4.4, 1.20 nM [3H] Spiperone; D5, 2.0 nM [3H] SCH-23390). IC50 values were determined by a non-linear, least squares regression analysis using MathIQ™ (ID Business Solutions Ltd., UK).
DMPK Methods. In vitro: The metabolism of ML398 was investigated in human, rat and mouse hepatic microsomes (BD Biosciences, Billerica, MA) using substrate depletion methodology (% test article remaining). A potassium phosphate-buffered reaction mixture (0.1 M, pH 7.4) of test article (1 μM) and microsomes (0.5 mg/mL) was pre-incubated (5 min) at 37 °C prior to the addition of NADPH (1 mM). The incubations, performed in 96-well plates, were continued at 37 °C under ambient oxygenation and aliquots (80 μL) were removed at selected time intervals (0, 3, 7, 15, 25 and 45 min). Protein was precipitated by the addition of chilled acetonitrile (160 μL), containing glyburide as an internal standard (50 ng/mL), and centrifuged at 3000 rpm (4°C) for 10 min. Resulting supernatants were transferred to new 96-well plates in preparation for LC/MS/MS analysis. The in vitro half-life (t1/2, min, Eq. 1), intrinsic clearance (CLint, mL/min/kg, Eq. 2) and subsequent predicted hepatic clearance (CLhep, mL/min/kg, Eq. 3) were determined employing the following equations:
Plasma Protein Binding. Protein binding of ML398 was determined in human, rat and mouse plasma via equilibrium dialysis employing Single-Use RED Plates with inserts (ThermoFisher Scientific, Rochester, NY). Briefly plasma (220 μL) was added to the 96-well plate containing test article (5 μL) and mixed thoroughly. Subsequently, 200 μL of the plasma-test article mixture was transferred to the cis chamber (red) of the RED plate, with an accompanying 350 μL of phosphate buffer (25 mM, pH 7.4) in the trans chamber. The RED plate was sealed and incubated 4 h at 37 °C with shaking. At completion, 50 μL aliquots from each chamber were diluted 1:1 (50 μL) with either plasma (cis) or buffer (trans) and transferred to a new 96-well plate, at which time ice-cold acetonitrile (2 volumes) was added to extract the matrices. The plate was centrifuged (3000 rpm, 10 min) and supernatants transferred to a new 96-well plate. The sealed plate was stored at -20 °C until LC/MS/MS analysis.
Liquid Chromatography/Mass Spectrometry Analysis. In vitro experiments. ML398 was analyzed via electrospray ionization (ESI) on an AB Sciex API-4000 (Foster City, CA) triple-quadrupole instrument that was coupled with Shimadzu LC-10AD pumps (Columbia, MD) and a Leap Technologies CTC PAL auto-sampler (Carrboro, NC). Analytes were separated by gradient elution using a Fortis C18 2.1 × 50 mm, 3.5 μm column (Fortis Technologies Ltd, Cheshire, UK) thermostated at 40 °C. HPLC mobile phase A was 0.1% NH4OH (pH unadjusted), mobile phase B was acetonitrile. The gradient started at 30% B after a 0.2 min hold and was linearly increased to 90% B over 0.8 min; held at 90% B for 0.5 min and returned to 30% B in 0.1 min followed by a re-equilibration (0.9 min). The total run time was 2.5 min and the HPLC flow rate was 0.5 mL/min. The source temperature was set at 500°C and mass spectral analyses were performed using multiple reaction monitoring (MRM) utilizing a Turbo-Ionspray® source in positive ionization mode (5.0 kV spray voltage). LC/MS/MS analysis was performed employing a TSQ QuantumULTRA that was coupled to a ThermoSurveyor LC system (Thermoelectron Corp., San Jose, CA) and a Leap Technologies CTC PAL auto-sampler (Carrboro, NC). Chromatographic separation of analytes was achieved with an Acquity BEH C18 2.1 × 50 mm, 1.7 μm column (Waters, Taunton, MA).
2.1. Assays
- 2.1.1.
AID 743372 Discovery of Novel Antagonists for Human D4 Antagonists, Confirmatory Assay
- 2.1.2.
AID 743374 D4 Antagonists, CounterScreen Against Dopamine 5 DRD5 (Human)
- 2.1.3.
AID 743375 D4 Antagonists, CounterScreen Against Dopamine 3 DRD3 (Human)
- 2.1.4.
AID 743376 D4 Antagonists, CounterScreen Against Dopamine 2S, D2L (Human)
- 2.1.5.
AID 743377 D4 Antagonists, CounterScreen Against Dopamine 2L, D2L (Human)
- 2.1.6.
AID 743378 D4 Antagonists, CounterScreen Against Dopamine 1 (Human)
- 2.1.7.
AID 743362 Design and Development of D4 Antagonists, Summary Assay
- 2.1.8.
AID 743371 Primary Screen for Human D4 Antagonists Discovery of novel antagonists of the dopamine 4 (D4) human receptor
- 2.1.9.
AID 743435 Development of inhibitors for Dopamine D4 Receptors: Eurofin Panel Assay Results
2.2. Probe Chemical Characterization

Figure 1Chemical Characterization of ML398
Probe compound ML398 (CID 73737723, SID 172236217) was prepared according to the above scheme and had the following characterization. (R)-4-(4-chlorobenzyl)-2-phenethylmorpholine. 1H NMR (CDCl3, 400 MHz) δ (ppm): 1.73 (dm, J = 64 Hz, 2H), 1.88 (t, J = 10.4 Hz, 1H), 2.00 (dt, J = 3.1, 11.5 Hz, 1H), 2.59-2.71 (m, 3H), 2.73-2.84 (m, 1H), 3.45 (s, 2H), 3.45-3.53 (m, 1H), 3.65 (t, J = 11.3 Hz, 1H), 3.89 (d, J = 11.5 Hz, 1H), 7.15-7.22 (m, 3H), 7.23-7.32 (m, 6H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 31.5, 35.2, 53.1, 58.5, 62.4, 66.6, 74.8, 125.7, 128.3 (2C), 128.4, 130.3, 132.8, 136.3, 141.9. HRMS: C19H23NOCl, [M+H]+ calc = 316.1468, found = 316.1470. [α]D20 = +29.3 (c = 1.0, CH3OH, 92% ee).
Solubility. Solubility for ML398 in PBS was determined to be <5 μM, which is >35-fold higher than the cellular IC50 for D4 inhibition.
Stability. Stability was determined for ML398 at 23 °C in PBS (no antioxidants or other protectorants and DMSO concentration below 0.1%). After 48 hours, ∼6% of the initial concentration of ML398 remained in solution. The loss is most likely due to limited solubility in buffer. However, as evidenced by the in vivo PK study (vide infra), the compound is present in the plasma and brain, again suggesting solubility issues with the solution for this study.
Compounds added to the SMR collection (MLS#s): MLS005886361 (ML398, CID 73737723, 25.0 mg); MLS005886362 (CID 72737772, 5.4 mg); MLS005886363 (CID 72737750, 5.8 mg); MLS005886364 (CID 72737745, 5.8 mg); MLS005886365 (CID 72737730, 8.2 mg); MLS005886366 (CID 72737723, 8.6 mg)
2.3. Probe Preparation1

Figure 2Preparation of ML398
Probe compound ML398 (CID 73737723, SID 172236217) was prepared according to the above scheme and had the following characterization. (R)-4-(4-chlorobenzyl)-2-phenethylmorpholine.
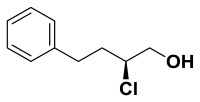
(S)-2-chloro-4-phenylbutan-1-ol. Suspended 4-phenylbutanal (147 mg, 0.992 mmol) in anhydrous dichloromethane (4.0 mL) and cooled to 0 °C. (2R,5R)-Diphenylpyrrolidine (22.0 mg, 0.099 mmol) added followed by N-chlorosuccinimide (172 mg, 1.29 mmol). The progress of this reaction was monitored by proton NMR (the starting material aldehyde peak being converted to the mono-chlorinated aldehyde). Once complete, methanol (4.0 mL) was added followed by slow addition of sodium borohydride (187 mg, 4.96 mmol). Continued stirring for 30 minutes before water was added and the solution was extracted with dichloromethane. After drying over sodium sulfate, filtering, and concentrating, the crude material was purified by column chromatography (0-15% ethyl acetate in hexanes). Pure chloro-alcohol was collected (158 mg, 86%) in 93% ee. 1H NMR (CDCl3, 400 MHz) δ (ppm): 1.99-2.09 (m, 3H), 2.73-2.80 (m, 1H), 2.87-2.94 (m, 1H), 3.67-3.82 (m, 2H), 3.96-4.02 (m, 1H), 7.20-7.33 (m, 5H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 32.3, 35.8, 64.0, 66.9, 126.2, 128.4, 128.5, 140.6. [α]D20 = -48.4 (c = 1.0, CH3OH). Spectral data matches that recorded by M. O'Reilly and C. Lindsley, Organic Letters, 14, 2910.
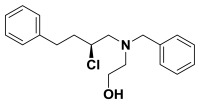
(S)-2-(benzyl(2-chloro-4-phenylbutyl)amino)ethanol. (S)-2-chloro-4-phenylbutan-1-ol (0.530 g, 2.87 mmol) was suspended in anhydrous dichloromethane (28.0 mL) and to this 2,6-lutidine (3.3 mL, 28.7 mmol) was added. This solution was cooled to -78 °C before triflic anhydride (0.63 mL, 3.73 mmol) was added dropwise. This was stirred 30 minutes and the triflate was confirmed to be formed by proton NMR. N-benzylethanolamine (2.1 mL, 14.3 mmol) in anhydrous dichloromethane (2.0 mL) was added to the solution dropwise. The reaction was allowed to warm to room temperature slowly overnight and the consumption of the triflate was established by NMR. Diethyl ether (100 mL) and water (100 mL) was added and the organic layer was separated from the aqueous, dried over sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (0-20% ethyl acetate in hexanes) to yield the chloro-aminoalcohol (0.715 g, 78%). 1H NMR (CDCl3, 400 MHz) δ (ppm): 1.88-1.99 (m, 1H), 2.07-2.18 (m, 1H), 2.63-2.96 (m, 7H), 3.53-3.76 (m, 4H), 3.90-3.99 (m, 1H), 7.20-7.40 (m, 10H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 32.3, 37.6, 56.3, 58.8, 59.3, 60.3, 61.1, 126.1, 127.4, 128.4, 128.5, 128.5, 129.0, 138.2, 140.8. [α]D20 = -15.1 (c = 1.0, CH3OH). Spectral data matches that recorded by M. O'Reilly and C. Lindsley, Organic Letters, 14, 2910.
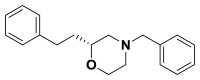
(R)-4-benzyl-2-phenethylmorpholine. Suspended (S)-2-(benzyl(2-chloro-4-phenylbutyl) amino)ethanol (0.660 g, 2.08 mmol) in anhydrous acetonitrile (100 mL) and cooled to -20 °C. Potassium tert-butoxide (1.17 g, 10.4 mmol) was added and the progress of the reaction was followed by TLC. Upon consumption of starting material, water and diethyl ether were added to the reaction and it was extracted with ether, dried over sodium sulfate, filtered, and concentrated. The crude material was purified by column chromatography (0-40% ethyl acetate in hexanes) to give the morpholine (0.316 g, 54%). 1H NMR (CDCl3, 400 MHz) δ (ppm): 1.63-1.74 (m, 1H), 1.78-1.89 (m, 1H), 1.92 (t, J = 10.7 Hz, 1H), 2.21 (td, J = 3.2, 11.1 Hz, 1H), 2.63-2.86 (m, 4H), 3.50-3.58 (m, 3H), 3.70 (td, J = 2.4, 11.1 Hz, 1H), 3.92 (d, J = 11.1 Hz, 1H), 7.18-7.24 (m, 3H), 7.25-7.39 (m, 7H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 31.5, 35.3, 53.2, 58.6, 63.3, 66.7, 74.8, 125.7, 127.1, 128.2, 128.3, 128.4, 129.1, 137.7, 142.0. [α]D20 = +30.3 (c = 1.0, CH3OH). Spectral data matches that recorded by M. O'Reilly and C. Lindsley, Organic Letters, 14, 2910.
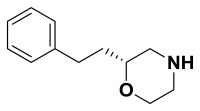
(R)-2-phenethylmorpholine. Suspended (R)-4-benzyl-2-phenethylmorpholine (88.0 mg, 0.313 mmol) in anhydrous methanol (6.3 mL). The flask was purged three times alternating vacuum and argon. Added 10% Pd/C (6.6 mg, 0.063 mmol) then purged the flask three times alternating vacuum and hydrogen gas. The reaction was followed by TLC and LCMS until it was complete. The solution was then filtered through celite with methanol, concentrated, and carried on crude.

(R)-4-(4-chlorobenzyl)-2-phenethylmorpholine. In a microwave vial, suspended (R)-2-phenethylmorpholine (0.100 g, 0.523 mmol) in anhydrous acetonitrile (2.6 mL). Potassium carbonate (0.361 g, 2.61 mmol) and 4-chlorobenzyl bromide (0.107 g, 0.523 mmol) were added. This was heated under microwave conditions to 120 °C for 10 minutes. Water was added and the product was extracted with ethyl acetate. The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo. The crude material was purified by column chromatography (0-30% ethyl acetate in hexanes) to provide pure product (0.103 mg, 61%, >98% pure). 1H NMR (CDCl3, 400 MHz) δ (ppm): 1.73 (dm, J = 64 Hz, 2H), 1.88 (t, J = 10.4 Hz, 1H), 2.00 (dt, J = 3.1, 11.5 Hz, 1H), 2.59-2.71 (m, 3H), 2.73-2.84 (m, 1H), 3.45 (s, 2H), 3.45-3.53 (m, 1H), 3.65 (t, J = 11.3 Hz, 1H), 3.89 (d, J = 11.5 Hz, 1H), 7.15-7.22 (m, 3H), 7.23-7.32 (m, 6H). 13C NMR (CDCl3, 100 MHz) δ (ppm): 31.5, 35.2, 53.1, 58.5, 62.4, 66.6, 74.8, 125.7, 128.3 (2C), 128.4, 130.3, 132.8, 136.3, 141.9. HRMS: C19H23NOCl, [M+H]+ calc = 316.1468, found = 316.1470. [α]D20 = +29.3 (c = 1.0, CH3OH, 92% ee).
3. Results
3.1. Dose Response Curves
.&p=BOOKS&id=280047_ml398f9.jpg "Click on image to zoom")
Figure 3Concentration-response curves for ML398 in the dopamine D4.4 assay (EuroFins)
ML398 is a selective compound D4 antagonist exhibiting >100-fold selectivity over the other dopamine receptors (D1,2L,2S,3,5).
3.2. Cellular Activity
The primary screening assay (dopamine D4) is a cell-based assays, indicating that ML398 can gain access to its molecular target when applied to cells.
3.3. Profiling Assays
To more fully characterize this potent, selective D4 antagonist, ML398 was tested using EuroFins (formerly MDS Pharma's) Lead Profiling Screen (binding assay panel of 68 GPCRs, ion channels and transporters screened at 10 μM). Included in the EuroFins screening panel are a number of ion channels (Calcium Channel, L-Type and N-Type; Potassium channel [KATP]; Potassium channel [hERG]) and class A GPCRs (D1-5, H1-3, etc…). ML398 was found to not significantly interact with 63 out of the 68 assays conducted (inhibition of radio ligand binding > 50% at 10 μM). ML398 did have activity against several targets, however, it should be pointed out that these are only single-point values and that functional selectivity may be significantly better than suggested by these “% activities.”
4. Discussion
4.1. Comparison to existing art and how the new probe is an improvement
ML398 (CID 72737723) is a potent dopamine D4.4 receptor antagonist (IC50 = 130 nM), which excellent binding affinity for the receptor (Ki = 36 nM) and is selective against the other dopamine receptors (>20 mM, D1,2L,2S,3,5). The existing art all possess scaffolds that are known to be privileged structures for GPCRs and thus do not possess the appropriate selectivity for probing the dopamine D4 receptor function. With the selectivity of ML398 for the dopamine receptors and rather clean ancillary pharmacology (as assessed by the EuroFins Lead Profiling screen), ML398 represents an improvement on the compounds in the literature. ML398 is highly brain penetrant with a total B:P ∼2.0. In addition, ML398 will be freely available to anyone in the scientific community, another improvement over the previous compounds, as many (if not all) are covered in the patent literature.
4.2. Mechanism of Action Studies
ML398 is a competitive antagonist of the dopamine D4.4 receptor.
4.3. Efficacy in Cell-Based Assays
The primary dopamine D4 receptor assay is a cell based functional assay.
5. References
- 1.
- O'Reilly MC, Lindsley CW. A general, enantioselective synthesis of protected morpholines and piperazines. Org. Lett. 2012;14:2910–2913. [PMC free article: PMC3369273] [PubMed: 22616979]
Publication Details
Author Information and Affiliations
Authors
Cynthia B. Berry, Charles W. Locuson, J. Scott Daniels, Craig W. Lindsley, and Corey R. Hopkins .
.Affiliations
Corresponding author.Publication History
Received: April 15, 2014; Last Update: January 16, 2015.
Copyright
Publisher
National Center for Biotechnology Information (US), Bethesda (MD)
NLM Citation
Berry CB, Locuson CW, Daniels JS, et al. Discovery and characterization of ML398, a potent and selective chiral morpholine based antagonist of the dopamine 4 (D4) receptor. 2014 Apr 15 [Updated 2015 Jan 16]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.