Clinical Description
Fibrous dysplasia / McCune-Albright syndrome (FD/MAS) results from mosaic somatic activating pathogenic variants in GNAS, which encodes the cAMP pathway-associated G protein Gαs (Gs alpha subunit). Affected tissues can include those derived from ectoderm, mesoderm, and endoderm, and commonly include skin, skeleton, and certain endocrine organs. However, because Gαs signaling is present in virtually every tissue, additional sites may be affected.
The phenotypic spectrum of FD/MAS ranges from asymptomatic incidental findings to neonatal lethality. There is a high degree of variability between individuals, both in the number of affected tissues and the degree to which they are affected. Disease manifestations depend on the time during embryogenesis that the somatic pathogenic variant occurred, the tissue involved, and the role of Gαs in the affected tissue. Pathogenic variants occurring early in development lead to widespread disease, while those occurring later in development lead to limited disease.
Hyperpigmented macules are common and are usually the first manifestation of FD/MAS, apparent at or shortly after birth. There is no correlation between the size of the skin lesions and the extent of disease, nor between the distribution of skin lesions and the location of fibrous dysplasia.
Fibrous dysplasia (FD) demonstrates a mosaic pattern: it can involve any part or combination of the craniofacial, axial, and/or appendicular skeleton. The bones most commonly involved are the skull base and proximal femurs [Kelly et al 2008]. While there is generally a central-to-peripheral gradient, any combination of involved bones is possible. FD is widely variable, from an asymptomatic monostotic lesion discovered incidentally to severe, disabling polyostotic disease involving practically the entire skeleton and leading to loss of vision, hearing, and/or mobility.
Individual bone lesions typically manifest during the first few years of life and expand during childhood. The vast majority of clinically significant bone lesions are detectable by age ten years, with few new and almost no clinically significant bone lesions appearing after age 15 years [Hart et al 2007]. In adulthood, FD lesions typically become less active, likely related to apoptosis of pathogenic variant-bearing cells [Kuznetsov et al 2008].
The clinical presentation and course of FD depends on the location and extent of the affected skeleton:
Appendicular skeleton. Children with FD in the appendicular skeleton typically present with a limp, pain, and/or pathologic fractures. Recurrent fractures and progressive deformity may lead to difficulties with ambulation and loss of mobility.
Craniofacial region. FD may present as a painless "lump" or facial asymmetry. Expansion of craniofacial lesions may lead to progressive facial deformity (see ), and in rare instances (usually in association with growth hormone excess) loss of vision and/or hearing due to compromise of the optic nerves and/or external auditory canals [
Cutler et al 2006,
Boyce et al 2018].
Vertebrae. FD involving the vertebrae is common, and may lead to scoliosis, which in rare instances may be severe, progressive, and even lethal [
Berglund et al 2018]. Untreated, progressive scoliosis is one of the few features of FD that can lead to early morbidity.
Bone pain is a common complication of FD. Although bone pain may present at any age, it is common for bone pain to be absent in childhood, occur in adolescence, and progress into adulthood [Kelly et al 2008].
Aneurysmal bone cysts are rapidly expanding fluid-filled lesions that form within preexisting areas of FD. Such lesions are best detected by MRI. Affected individuals experience acute onset of severe pain, rapidly expanding localized deformity, and rarely – when cysts compress the optic nerve – rapid loss of vision. Aneurysmal bone cysts thus carry a high risk of morbidity (see Management).
Malignant transformation of FD lesions is a rare complication. Many instances of malignant transformation were reported in association with previous radiation treatment [Ruggieri et al 1994]. Growth hormone excess may be a predisposing factor [Salenave et al 2014].
Radiographic appearance of FD varies according to location.
Radiographs of the appendicular skeleton show expansive lesions with endosteal scalloping, thinning of the cortex, and a "ground-glass" appearance (see ).
FD in the craniofacial skeleton is typically expansile and appears sclerotic on radiographs, but demonstrates a typical "ground-glass" appearance on CT (see ).
With aging, FD lesions in the appendicular skeleton tend to become sclerotic on radiographs, and craniofacial FD lesions develop a "cystic" appearance (see ).
Endocrinopathies can include any of the following:
Precocious puberty is common in girls with FD/MAS (~85%) and is often the presenting feature. Recurrent ovarian cysts (see ) lead to intermittent estrogen production resulting in breast development, growth acceleration, and vaginal bleeding; during the intervals between cyst formation, breast tissue typically regresses and estrogen levels fall to prepubertal levels. Ovarian cysts typically continue into adulthood, leading to irregular menses. This has the potential to interrupt ovulatory cycles, which may increase the time to conception in adult women. Ovarian torsion has been seen rarely in girls and women with large and persistent cysts [
Clark et al 2000].
Precocious puberty is less common in boys with FD/MAS (~10%-15%), and is due to autonomous testosterone production [
Boyce et al 2012a], which leads to progressive pubertal development including growth acceleration, pubic and axillary hair, acne, and aggressive and/or inappropriately sexual behavior.
In both girls and boys, prolonged autonomous sex steroid production typically leads to activation of the hypothalamic-pituitary axis and the development of central precocious puberty.
Fertility. The effects of autonomous sex steroid production on pituitary-gonadal function and fertility in adults are not well characterized. Women with FD/MAS may have recurrent ovarian cysts leading to irregular menses in adulthood [
Lala et al 2007]. While many women in an NIH natural history study cohort have achieved successful pregnancies, it is possible that interruption of ovulatory cycles could decrease fertility and increase the time to conception [
Boyce et al 2019].
Testicular abnormalities. Testicular abnormalities are seen in the majority of boys and men with FD/MAS (~85%), and typically manifest as unilateral or bilateral macro-orchidism [
Boyce et al 2012a]. Ultrasound examination demonstrates discrete hyper- and hypoechoic lesions and microlithiasis, corresponding to areas of Leydig and/or Sertoli cell hyperplasia (see ).
The potential for malignant transformation of testicular lesions is unknown, but appears to be low [
Boyce et al 2012a].
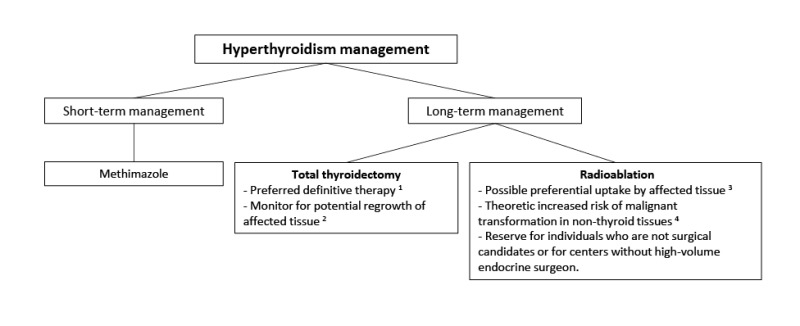
Thyroid disease. Thyroid involvement in FD/MAS is common. Approximately half of individuals with FD/MAS have ultrasound findings consistent with thyroid involvement, including mixed cystic and solid lesions interspersed with areas of normal-appearing tissue (see and ) [
Celi et al 2008,
Tessaris et al 2012].
Hyperthyroidism is present in 10%-30% of individuals with FD/MAS, and results from both increased hormone production and increased conversion of thyroxine (T
4) to triiodothyronine (T
3) [
Celi et al 2008]. Hyperthyroidism is typically mild to moderate, but may be severe, and if undetected can lead to thyroid storm during anesthetic induction for surgery [
Lawless et al 1992]. Uncontrolled hyperthyroidism may lead to bone age advancement, elevated bone turnover, and fractures.
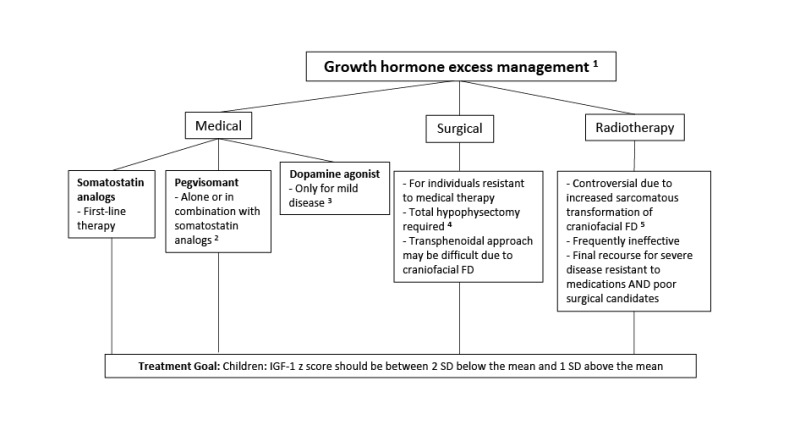
Growth hormone excess. Approximately 15%-20% of individuals with FD/MAS have
GNAS pathogenic variants in the anterior pituitary that can lead to autonomous growth hormone production; approximately 80% of affected individuals with autonomous growth hormone production also have hyperprolactinemia [
Salenave et al 2014].
Affected individuals typically present with linear growth acceleration, and may develop features of acromegaly. Clinically, growth hormone excess must be distinguished from precocious puberty and hyperthyroidism, which also present with growth acceleration.
Untreated growth hormone excess is associated with expansion of craniofacial FD, leading to macrocephaly and increased risk of vision and hearing loss [
Boyce et al 2013,
Boyce et al 2018] (see ).
Fibroblast growth factor 23 (FGF23)-mediated phosphate wasting. In the majority of individuals with FD, increased production of the phosphaturic hormone FGF23 in FD tissue results in a renal tubulopathy with some degree of phosphate wasting [
Collins et al 2001]. However, frank hypophosphatemia in persons with FD is infrequent, in part due to alterations in FGF23 processing that takes place in FD tissue and results in increased cleavage of FGF23 to its inactive fragments [
Bhattacharyya et al 2012]. The degree of FGF23 overproduction in FD correlates with disease severity and skeletal burden; thus, frank hypophosphatemia is only seen in individuals with a substantial FD burden [
Riminucci et al 2003].
In contrast to most other features of FD/MAS, hypophosphatemia may wax and wane over the course of a person's lifetime and become more severe during periods of rapid skeletal growth. Hypophosphatemia may resolve as persons with FD become older, likely reflecting the intrinsic changes in FD that occur with age [
Kuznetsov et al 2008].
Hypercortisolism. Infants with FD/MAS may rarely present with Cushing syndrome due to excess cortisol production from the fetal adrenal gland [
Brown et al 2010,
Carney et al 2011]. Clinical symptoms typically develop in the neonatal period and may be severe, leading to critical illness and death. Spontaneous regression has been reported in approximately half of survivors, presumably related to fetal adrenal involution.
Liver manifestations
Gastrointestinal manifestations
Pancreatic manifestations. Approximately 15% of individuals with FD/MAS have pancreatic complications:
Pancreatitis
Intraductal papillary mucinous neoplasms (IPMN) have been reported, which may present with variable grades of dysplasia [
Gaujoux et al 2014,
Wood et al 2017]. An individual with pancreatic carcinoma derived from an intestinal subtype of IPMN has been described [
Parvanescu et al 2014].
Myxomas. Intramuscular myxomas are benign, usually asymptomatic, and often found incidentally. There are case reports positing a potential association between myxomas and increased risk of malignant transformation of FD [Biazzo et al 2017]; however, this has not been reported in larger series [Majoor et al 2019b, Hagelstein-Rotman et al 2021] and may be an artifact of publication bias.
Hematologic manifestations. Bone and bone marrow are, to varying degrees, replaced by fibro-osseous tissue typically devoid of hematopoietic marrow. There have been reports of bone marrow failure with pancytopenia and extramedullary hematopoiesis requiring splenectomy in individuals with FD/MAS [Mahdi et al 2017, Robinson et al 2018a].
Breast cancer. The risk for breast cancer in women with FD/MAS may be increased and can occur at a younger age compared to the general population. However, pathogenic activating GNAS variants were identified in only half of the breast tumors from women with FD/MAS studied [Majoor et al 2018a].
Health-related quality of life. Several series have shown impaired physical functioning in individuals with FD/MAS, strongly correlated with disease severity. There is conflicting evidence pertaining to the impact of the condition on psychosocial functioning, and this requires further invesigation [Kelly et al 2005, Majoor et al 2018b, Konradi 2021, Meier et al 2022].