NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Panayiotopoulos CP. The Epilepsies: Seizures, Syndromes and Management. Oxfordshire (UK): Bladon Medical Publishing; 2005.
Idiopathic generalised epilepsies (IGEs) constitute one-third of all epilepsies.1–6 They are genetically determined and affect otherwise normal people of both sexes and all races. IGEs manifest with typical absences, myoclonic jerks and generalised tonic clonic seizures (GTCS), alone or in varying combinations and severity. Absence status epilepticus (ASE) is common. Most syndromes of IGE start in childhood or adolescence, but some have an adult onset. They are usually life long, though a few are age related. The EEG is the most sensitive test in the diagnosis and confirmation of IGE. EEG shows generalised discharges of spikes, polyspikes or spike/polyspike-wave either ictally or interictally. These discharges are often precipitated by hyperventilation, sleep deprivation and intermittent photic stimulation. Inconspicuous clinical manifestations become apparent on video EEG and with breath counting during hyperventilation. The EEG is unlikely to be normal in untreated patients. In suspected cases with normal routine awake EEG, an EEG during sleep and awakening should be obtained. Molecular genetic analyses have led to important breakthroughs in the identification of candidate genes and loci;7,8 genetic heterogeneity is common.8–11 Genetic mutations found in γ-aminobutyric acid (GABAA) receptor subunits strongly implicate the GABAA receptor in IGEs.12 Treatment of IGEs is demanding for two main reasons. Firstly, anti-epileptic drugs (AEDs) beneficial in focal epilepsies may be deleterious in IGEs.4,13 Secondly, efficacy of AEDs differs even within IGE seizures. This is because the generation of absences, for example, is due to a predominance of inhibitory activity, in contrast to generalised convulsive seizures in which an excess of excitatory activity is present.13 Most IGEs respond well to appropriate AEDs, but treatment is often life long. The fact that nearly 50% of patients with IGE are currently taking “ill-advised AED” medication14 is a grave problem that needs to be addressed.
Seizures of Idiopathic Generalised Epilepsies
The syndromes of IGEs manifest with three main types of seizures alone or in combination. These are:
- Typical absence seizures
- Myoclonic seizures
- Generalised tonic clonic seizures
ILAE Definition of Idiopathic Generalised Epilepsies
The ILAE Commission1 defined IGE as follows: “Idiopathic generalised epilepsies are forms of generalised epilepsies in which all seizures are initially generalised (absences, myoclonic jerks and generalised tonic clonic seizures), with an EEG expression that is a generalised bilateral, synchronous, symmetrical discharge (such as is described in the seizure classification of the corresponding type). The patient usually has a normal interictal state, without neurological or neuroradiologic signs. In general, interictal EEGs show normal background activity and generalised discharges, such as spikes, polyspike spike-waves, and polyspike-waves □ 3 Hz. The discharges are increased by slow sleep. The various syndromes of idiopathic generalised epilepsies differ mainly in age of onset. No aetiology can be found other than a genetic predisposition towards these disorders.”1
Typical Absence Seizures*
Typical absences (previously known as petit mal) are brief (lasting seconds) generalised epileptic seizures of abrupt onset and abrupt termination (Table 10.1). They have two essential components:
- a clinical component manifesting with impairment of consciousness (absence)
The absence seizures are fundamentally different and pharmacologically unique compared with any other type of seizure, which also makes their treatment different.4,16,17
The clinical and EEG manifestations of typical absences are extensive and syndrome-related.1–4,15,16,18
Clinical manifestations: Impairment of consciousness may be severe, moderate, mild or inconspicuous (and special cognitive testing may be required to detect it). It is often associated with other concomitant symptoms, such as myoclonia, automatisms and autonomic disturbances. Myoclonia may be rhythmic or random, mild or severe, regional (mouth or eyes) or widespread (head, limbs and trunk).
Typical absences are predominantly spontaneous, though they are precipitated by hyperventilation in around 90% of untreated patients. Other specific modes of precipitation include photic, pattern, video games and thinking (reflex absences).
Ictal EEG: The ictal EEG consists of generalised discharges with repetitive and rhythmic 3–4 Hz single or multiple spike-slow wave complexes (Figure 10.1).
These generalised spike-wave discharges (GSWD) may be brief (sometimes less than 3 s) or long (□ 30 s), and continuous or fragmented. The intradischarge frequency of the spike-wave may be relatively constant or may vary.
Typical absence seizures in IGE syndromes. Typical absences are severe in childhood absence epilepsy (CAE) and juvenile absence epilepsy (JAE), but mild or inconspicuous in other syndromes, such as juvenile myoclonic epilepsy (JME).
They may occur alone or in combination with other types of generalised seizures. IGE with absences may remit with age or be lifelong.
Typical ASE occurs in approximately one-third of patients who suffer from typical absence seizures.19
Clinical Manifestations of Typical Absence Seizures
The clinical manifestations of typical absence seizures vary significantly between patients.3,4,15,18,20–26 Impairment of consciousness may be the only clinical symptom, but it is often combined with other manifestations (Table 10.1). Typical absences are categorised as:
- simple absences with impairment of consciousness only
- complex absences when impairment of consciousness combines with other ictal motor manifestations.
Complex absences are far commoner than simple absences in children. Simple absences are commoner in adults. The same patient may have both simple and complex absences.
Absence with Impairment of Consciousness Only1
The classical27 and ILAE1 descriptions refer to absence seizures with severe impairment of consciousness as in CAE and JAE.
Patient note“Transient loss of consciousness without conspicuous convulsions. A patient stops for a moment whatever he or she is doing, very often turns pale, may drop whatever is in the hand.....There may be a slight stoop forward, or a slight quivering of the eyelids...The attack usually lasts only a few seconds. The return of the consciousness may be sudden and the patient after the momentary lapse, may be in just the same state as before the attack, may even continue a sentence or action which was commenced before it came on, and suspended during the occurrence.” W.R.Gowers (1885) 27
The hallmark of severe absence seizures is a sudden onset and interruption of ongoing activities, often with a blank stare. If the patient is speaking, speech is slowed or interrupted; if walking, he/she stands transfixed. Usually the patient will be unresponsive when spoken to. Attacks are often aborted by auditory or sensory stimulation.
In less severe absences, the patient may not stop his/her activities, though reaction time and speech may slow down. In their mildest form, absences may be inconspicuous to the patient and imperceptible to the observer (phantom absences), as disclosed by video EEG recordings showing errors and delays during breath counting or other cognitive tests during hyperventilation.
Absence with Clonic Components1
During the absence, as described above, clonic motor manifestations, rhythmic or arrhythmic and singular or repetitive, are particularly frequent at the onset. They may be continuous. They may also occur at any other stage of the seizure. The most common manifestations are clonic jerking of the eyelids, eyebrows and eyeballs, together or independently, as well as random or repetitive eye closures. Fast flickering of the eyelids is probably the most common ictal clinical manifestation, and may occur during brief GSWD without discernible impairment of consciousness. Myoclonias at the corner of the mouth and jerking of the jaw are less common. Myoclonic jerks of the head, body and limbs may be singular or rhythmical and repetitive, and they may be mild or violent. In some patients with absence seizures, single myoclonic jerks of the head and less often of the limbs may occur during the progression of ictus; in my opinion, these are indicative of a bad prognosis and may constitute an epileptic syndrome, but this needs further documentation.15,16
Absence with Atonic Components
Diminution of muscle tone is usual when absences are severe. This manifests with drooping of the head and, occasionally, slumping of the trunk, dropping of the arms and relaxation of the grip. Rarely, tone is sufficiently diminished to cause falls.
Absence with Tonic Components
Tonic seizures alone do not occur in IGEs. However, tonic muscular contractions are common concomitant manifestations during typical absence seizures. They mainly affect facial and neck muscles symmetrically or asymmetrically. The eyes and head may be drawn backwards (retropulsion) or to one side, and the trunk may arch. Tonic manifestations are prominent in myoclonic absence seizures.
Absence with Automatisms
Automatisms are common in typical absences when consciousness is sufficiently impaired, and they are more likely to occur 4–6 s after the onset of GSWD. They do not occur in mild absence seizures irrespective of duration as for example in ASE. Automatisms of typical absence seizures are simple and void of behavioural changes (see definitions in Chapter 12). They vary in location and character from seizure to seizure. Perioral automatisms, such as lip licking, smacking, swallowing or ‘mute’ speech movements, are the most common. Scratching, fumbling with clothes and other limb automatisms are also common. Automatisms can be evoked; passive movements, postural repositioning or other stimuli can change their pattern and distribution.24
Absence with Autonomic Components
Autonomic components consist of pallor and, less frequently, flushing, sweating, dilatation of the pupils and incontinence of urine. During absence seizures, pronounced changes in cerebral oxygenation occur with a decrease in oxygenated and an increase in deoxygenated haemoglobin. Oxygenation changes start several seconds after the EEG-defined onset of the absence and outlast the clinically defined event by 20–30 s.28
Absences with Focal Motor Components, Hallucinations and Other Manifestations of Neocortical or Limbic Symptomatology
During a typical absence seizure, patients frequently manifest with concomitant focal motor components (tonic or clonic) imitating focal motor seizures. Hallucinations and other manifestations such as concurrent epigastric sensations29 may occur; these are particularly more apparent during ASE.16
Patient noteI was in that state of confused mind. The surroundings were vertical and flat and I lost depth perception. People around me appeared as be wearing wigs in pastel shades.30
Electroencephalography
The ictal EEG is characteristic with regular and symmetrical 3–4 Hz GSWD (Figure 10.1). The intradischarge spike-wave frequency varies from onset to termination (Figure 10.2). It is usually faster and unstable in the opening phase (first second), becomes more regular and stable in the initial phase (first 3 s), and slows down towards the terminal phase (last 3 s).24 The intradischarge relationship between spike or multiple spike and slow wave frequently varies. The GSWD is often of higher amplitude in the anterior regions. A generalised discharge with an onset or a higher amplitude in the posterior regions may indicate a bad prognosis.31
Duration of the discharges commonly varies from 3 to 30 s (Figure 10.1).
The background interictal EEG is usually normal, though some paroxysmal activity (such as spikes or spike–wave complexes) may occur. Focal abnormalities or other asymmetries are common.32,33
Sleep EEG patterns are normal. GSWD are more likely to increase, but decrease during sleep. The discharges are often shorter and usually devoid of discernible clinical manifestations in sleep, even in those patients who have numerous clinical seizures with motor manifestations during the alert state.
Important Note
Though the EEG GSWD of typical absences is defined as symmetrical and synchronous, this is rarely the case at its onset. Commonly, the discharge starts with single or multiple spikes-slow waves that are asymmetrical and usually have a regional onset, mainly frontal (Figures 10.1 and 10.2). Often (but not always), there is an alternating side emphasis. Unilateral onset of the GSWD may be confused with secondary bilateral synchrony. The end of the discharge may be abrupt or consists of brief rhythmic or irregular slow waves (Figure 10.2). Sometimes more focal or fragmentary spikes occur, representing a ‘forme fruste’ of GSWD. These are more often recorded from the anterior regions, but other locations are also common.
Genetics
IGEs with typical absences are genetically determined, as indicated by the high incidence of similar disorders among families. However, the precise mode of inheritance and the genes involved remain largely unknown.7 Currently, various chromosomal loci have been identified for IGEs, as detailed in the description of individual syndromes. Furthermore, there is now evidence available to suggest that mutations in genes encoding GABA receptors34–36 or brain expressed voltage-dependent calcium channels37 may underlie CAE.
Genetic heterogeneity of the GSWD phenotype in animal models of absences favours a similar, and probably much wider, genetic heterogeneity in humans.7,13,17
Pathophysiology of Absence Seizures
The pathophysiological mechanisms of absence seizures have been studied in various animal models with GSWD associated with behavioural arrest.7,13,17,38,39 It appears that the GSWD are generated and sustained by highly synchronised abnormal oscillatory rhythms in thalamocortical networks that mainly involve neocortical pyramidal cells, the reticular thalamic nucleus and the relay nuclei of the thalamus. Neither the cortex nor the thalamus alone can sustain these discharges, indicating that both structures are involved in their generation.
The involvement of thalamus as the generator of GSWD is documented by the fact that:
- stimulation of the medial thalamus induces a cortical GSWD without leading to self-sustained activity
The relative importance of the cortex in the initiation and synchronisation of GSWD is mainly documented by the finding that, following thalamectomy, instigation of GSWD persists though the thalamus is required to maintain rhythmicity once the GSWD is established. More recently, in a rat model of absence, Meeren et al.43 showed that, during GSWD, cortical and thalamic interactions lag behind an initial burst of activity in the perioral region of the primary somatosensory cortex (S1po) during the first 500 ms of GSWD activity. These findings suggest that, in this animal model, a cortical focus within S1po is the dominant factor in initiating the paroxysmal oscillation within the corticothalamic loops, and that the large scale synchronisation is mediated by an extremely fast intracortical spread of seizure activity.43 This is also supported by experiments whereby microinfusion of ethosuximide into S1po produces an immediate cessation of GSWD.44
The basic intrinsic neuronal mechanisms involve low threshold T-type calcium currents elicited by activating the low threshold calcium channels. These channels are present in high densities in thalamic neurons and trigger regenerative burst firing that drive normal and pathological thalamocortical rhythms, including the GSWD of absence seizures. Ethosuximide exerts its anti-absence effect by either reducing thalamic low threshold calcium currents, probably by a direct channel blocking action that is voltage dependent,45 or through a potent inhibitory effect in the perioral region of the primary somatosensory cortex.13,44
Clinical noteIt is likely that the generation of absence seizures is due to a predominance of inhibitory activity, in contrast to generalised or focal convulsive seizures in which an excess of excitatory activity is present.13
Both inhibitory and excitatory neurotransmissions are involved in the genesis and control of absence seizures. This may be the result of excessive cortical excitability due to an imbalance between inhibition and excitation, or excessive thalamic oscillations due to abnormal intrinsic neuronal properties under the control of inhibitory GABAergic mechanisms. GABAB receptors play the most prominent role by eliciting long-standing hyperpolarisation required to drive low threshold calcium channels for the initiation of sustained burst firing. Typical absences are aggravated by GABAB agonists, such as baclofen, and suppressed by GABAB antagonists. GABAergic drugs (e.g. vigabatrin, tiagabine) are pro-absence substances; they interfere with the degradation of, and the re-uptake of, GABA.4,13 The only exception to GABAergic activation inhibiting absences is the reticular thalamic nucleus, which has exclusively GABAA receptors; it functions as a pacemaker to synchronise thalamocortical oscillations.46,47 Enhanced activation of GABAA receptors in this nucleus decreases the pacemaking capacity of these cells, thereby decreasing the likelihood of generating absence seizures.
Functional imaging using positron emission tomography (PET) demonstrates normal cerebral glucose metabolism and benzodiazepine receptor density in absence epilepsies, with diffuse hypermetabolism during 3 Hz GSWD.48,49 There is no evidence of any overall interictal abnormality of opioid receptors in IGE, but typical absences have been found to displace 11C-diprenorphine from the association areas of the neocortex. In contrast, binding of 11C-flumazenil to central benzodiazepine receptors has been shown to be unaffected by serial absences.49
Ictal single photon emission computed tomography (SPECT) shows an overall increase in cerebral blood flow50,51 and may be useful in detecting cases of frontal or other secondarily generalised absences.52 Interictally, relative hypoperfusion occurs in the frontal lobes, and may involve neighbouring parietal and temporal regions.51 Ictally, there is relative hyperperfusion in the same brain regions that are hypoperfused in the baseline study.51
Microdysgenesis and other cerebral structural changes were reported in some patients with CAE and JAE at autopsy53 and MRI54 studies. These results were not replicated in a more recent, blinded study.55 Microdysgenesis may be inconceivable for a benign, age-dependent and age-limited epileptic syndrome, such as CAE, though the current ion channel hypothesis for the pathogenesis of IGE does not preclude microscopic or ultramicroscopic abnormalities. Furthermore, recent quantitative MRI, PET and MRS studies have challenged the belief that IGEs are not associated with tissue pathology.42
Diagnosing Absences and Differential Diagnosis
Clinical noteThe brief duration of absence seizures with abrupt onset and abrupt termination of ictal symptoms, daily frequency and nearly invariable provocation by hyperventilation makes the diagnosis easy.4,16,18
The differential diagnosis of typical absence seizures with severe impairment of consciousness in children is relatively straightforward. The absences may be missed if mild or void of myoclonic components. Automatisms, such as lip smacking or licking, swallowing, fumbling or aimless walking, are common and should not be taken as evidence of complex partial (focal) seizures, which require entirely different management.
The EEG or, ideally, video EEG can confirm the diagnosis of typical absence seizures in more than 90% of untreated patients, mainly during hyperventilation.16 If not, the diagnosis of absences should be questioned.
In practical terms, a child with suspected typical absences should be asked to overbreathe for 3 minutes, counting his or her breaths while standing with hands extended in front. Hyperventilation will provoke an absence in more than 90% of those with typical absences. This procedure should preferably be videotaped to document the clinical manifestations. It may reveal features favouring a specific epileptic syndrome and, therefore, may determine the long-term prognosis and management. Video EEG documentation may be particularly useful if absences prove resistant to treatment, if other seizures develop, or for future genetic counselling. Focal spike abnormalities and asymmetrical onset of the ictal GSWD are common and may be a cause of misdiagnosis, particularly in resistant cases.15 If video EEG is not available, documentation of absences using a camcorder or modern digital means of recording is recommended.
The differentiation of typical from atypical absence seizures is shown in Table 7.4. Briefly, atypical absences differ from typical absences in the following ways:
- Atypical absences occur only in the context of mainly severe symptomatic or cryptogenic epilepsies of children with learning difficulties, who also suffer from frequent seizures of other types, such as atonic, tonic and myoclonic seizures.
- In atypical absences, onset and termination is not as abrupt as in typical absences, and changes in tone are more pronounced.
- The ictal EEG of atypical absence has slow (< 2.5 Hz) GSWD. These are heterogeneous, often asymmetrical, and may include irregular spike–wave complexes and other paroxysmal activity. Background interictal EEG is usually abnormal.
The differentiation of typical absences from complex focal seizures, detailed in Table 10.2, may be more difficult when the motor components of the absence are unilateral and in adults in whom absences are often misdiagnosed as temporal lobe seizures.56–58 Absences occur in 10% of adult patients with epileptic seizures.56–58
Misconceptions
Petit mal is seen almost exclusively in children, more rarely in adolescents and is a real curiosity in adults and the elderly.59
Contrary to the dominant view above, typical absence seizures occur in approximately 10% of adults with epilepsy.56,57 The alarming problem is that these are underdiagnosed or misinterpreted as focal seizures.
Myoclonic Jerks
Myoclonic jerks are shock-like, irregular and often arrhythmic, clonic-twitching movements that are singular or repetitive.21,60,61 They are of variable amplitude and force, ranging from mild and inconspicuous to sufficiently violent to make the patient fall on the ground, drop or throw things, or kick. Commonly, the same patients experience mild and violent jerks. Myoclonic jerks predominantly affect the eyelids, facial and neck muscles, the upper limbs more than the lower limbs and the body. Myoclonic jerks of IGE mainly occur on awakening. Precipitating factors are sleep deprivation, fatigue, excitement or distress and often photic stimulation. The patient is fully aware of myoclonic jerks unless they occur during impairment of consciousness in absence seizures. The location and extent of myoclonic jerks varies between IGE syndromes.
Polyspikes are the EEG accompaniment of myoclonic jerks (Figures 10.2, 10.3 and 10.5).
Diagnosing Myoclonic Jerks
Elicitation of the characteristic history of myoclonic jerks is something of an art. It is often necessary to physically demonstrate mild myoclonic jerks of the fingers and hands, and to inquire about morning clumsiness and tremors.62 Questions like ‘do you spill your morning tea?’ and ‘do you drop things in the morning?’, together with a simultaneous demonstration of how myoclonic jerks produce this effect, may be answered positively by patients who denied experiencing myoclonic jerks on direct questioning. Further elaboration is required to confirm that clumsiness was due to genuine myoclonic jerks. If the patient reports normal hypnagogic jactitations, it is reassuring that the concept of myoclonic jerks has been understood. Diagnostic yield may be improved by emphasising the close relationship between jerks and fatigue, alcohol and sleep deprivation. Some patients do not report their jerks, erroneously assuming that this is a self-inflicted normal phenomenon related to excess of alcohol and lack of sleep.
Generalised Tonic Clonic Seizures in IGEs
In IGEs, GTCS are primary (primarily) in the sense that they are generalised from onset without preceding auras or objective ictal focal symptoms, though they are often heralded by a series of myoclonic jerks or absences. This contrasts with secondarily GTCS of focal epilepsies that are often preceded by an aura or motor-sensory focal symptoms. Overall, primarily GTCS occur on awakening (17–53% of patients), diffusely while awake (23–36%) or during sleep (27–44%), or randomly (13–26%).63 The proportion of these patients who also have other generalised seizures, such as jerks or absences, is undetermined.
Differential Diagnosis of Primarily from Secondarily Generalised Tonic Clonic Seizures*
GTCS are dramatic in their presentation, which is the main reason for referral for medical consultation. This firstly demands careful exclusion of syncopal and other non-epileptic events. Once an unequivocal diagnosis of genuine epileptic GTCS has been established, the main differential diagnosis is between primarily and secondarily GTCS.
GTCS whether primarily (IGEs) or secondarily (focal epilepsies) are identical in their clinical presentation. Their differentiation, which is of immense clinical importance regarding overall management and AED treatment, is often easy (Table 10.3) based on:
- clinical history regarding other types of coexisting seizures, precipitating factors, circadian distribution and family history
- EEG manifestations
- brain imaging.
SPECT studies have shown activation of selective frontal, parietal and temporal networks after both spontaneous and induced primarily or secondarily GTCS, but thalamic cerebral blood flow is only increased after primarily GTCS.42
Status Epilepticus in Idiopathic Generalised Epilepsies
IGEs manifest with all types of generalised status epilepticus. ASE is probably the most common of all and the most likely to escape diagnosis or be misdiagnosed as focal status epilepticus or non-epileptic confusion, psychogenic or behavioural disorder.1,15,19,64–66
The new ILAE diagnostic scheme5 considers status epilepticus as a “continuous seizure” with two main categories (Table 1.2):
- Generalised status epilepticus
- Focal status epilepticus
The subcategories of generalised status epilepticus are relevant to this chapter:*
- Generalised tonic-clonic status epilepticus
- Clonic status epilepticus
- Absence status epilepticus
- Tonic status epilepticus
- Myoclonic status epilepticus
Definition of Status Epilepticus
There is no satisfactory definition of status epilepticus.64 Its definition is mainly influenced by the convulsive status epilepticus, because of its high morbidity, mortality and the need for early detection and early treatment. The World Health Organization defines status epilepticus as “a condition characterised by epileptic seizures that are sufficiently prolonged or repeated at sufficiently brief intervals so as to produce an unvarying and enduring epileptic condition”.67 By consensus, “sufficient length of time” was defined as being more than 30 minutes’ duration68 although more recent opinions argue for shorter periods of 5–10 minutes in defining convulsive status epilepticus.69 These short periods may not be applicable in other than convulsive types of status epilepticus.
The recent ILAE glossary makes no significant contribution in defining status epilepticus as “A seizure that shows no clinical signs of arresting after a duration encompassing the great majority of seizures of that type in most patients or recurrent seizures without interictal resumption of baseline central nervous system function”.70 Also, etymologically, status epilepticus is not a ‘continuous seizure’ as the ILAE Task Force proposed;5 it is a prolonged, enduring or rapidly repeated seizure which may also be ‘discontinuous’ and often stops without medical intervention.
Shorvons’ operational definition is more appealing “status epilepticus is condition in which epileptic activity persists for 30 minutes or more, causing a wide spectrum of clinical symptoms, and with a highly variable pathophysiological, anatomical and aetiological basis”.64 This definition implies that status is not simply a prolonged seizure or rapid repetition of seizures (in fact the word ‘seizure’ is no longer retained), but a condition (or group of conditions) in its own right with distinctive pathophysiological features.
Classification of Absence Status Epilepticus
Absence status epilepticus is divided into:
- typical ASE occurring in patients with IGEs who also have absence seizures
- atypical ASE occurring in patients with symptomatic epilepsies and epileptic encephalopathies (Figure 7.7)
- de novo or situation related absence status epilepticus occurring mainly in adults without a history of previous epileptic seizures commonly as the result of benzodiazepine or other drug discontinuation. 73,74 The most well-documented example is diazepine withdrawal. De novo ASE is often misdiagnosed as a psychotic state or dementia.66,73,74
Typical Absence Status Epilepticus in IGE
Typical (idiopathic) absence status epilepticus in IGEs is defined as a prolonged (> 30 minutes), generalised non-convulsive seizure of impairment of the content of consciousness (absence) and EEG generalised spike/polyspike-wave discharges.19,65,66,75,76
It should be emphasised that typical ASE, like absence seizures, is of many types with impairment of cognition as a shared common symptom. Impairment of consciousness may be mild or severe. It may occur alone (Figure 10.18) or more frequently be associated with other symptoms such as those listed in Table 10.1 for complex absence seizures. Motor manifestations such as myoclonic jerks, eyelid or perioral myoclonia, may predominate and be syndrome related (Figures 10.6 and 13.7).
Accordingly, typical (idiopathic) absence status epilepticus may be subdivided to:
- typical absence status epilepticus with impairment of consciousness only (Figure 10.18)
- myoclonic-atonic status epilepticus (Figure 10.6)
- myoclonic-absence status epilepticus (Figure 10.14)
- perioral myoclonic status epilepticus (Figure 10.16)
- eyelid myoclonic status epilepticus (Figure 13.8).
The ictal EEG is characteristic with usually regular and symmetrical 3 Hz (range 1–4 Hz) GSWD, which is continuous or repetitive (Figures 10.6, 10.14, 10.16, 10.18 and 13.7).
With the possible exception of CAE, all IGEs with typical absences may manifest with typical ASE, either as a spontaneous expression of their natural course or provoked by external factors or inappropriate treatment manoeuvres.
Impairment of Consciousness, Memory and Higher Cognitive Functions
The cardinal symptom shared by all cases of typical ASE is altered content of consciousness in a usually fully alert patient. Memory and higher cognitive intellectual functions, such as abstract thinking, computation and personal awareness, are the main areas of disturbance, which varies from very mild to very severe with intermediate states of severity occurring more often.
Mild disturbance is experienced as a state of slow reaction, behaviour and mental functioning:
Patient noteMy mind slows down and I am able to understand, but it takes longer to formulate answers.
I become slow, but can communicate verbally with others.
My behaviour slows down and I muddle up words.
It’s like being in a trance and missing pieces of conversation.
Moderate and severe impairment of consciousness manifest with varying degrees of confusion, global disorientation and inappropriate behaviour:
Patient noteConfused, cannot recognise people other than close relatives, disorientated in time and place, very quiet.
Disturbed, vague, uncooperative, confused.
Markedly confused, goes into a dreamy state, able to formulate some single word answers to simple questions, puts trousers on over pyjamas.
Confused, makes coffee twice, fades away mentally and physically, disoriented in time and place.
Usually, the patient is alert, attentive and cooperative. Verbal functioning is relatively well preserved, but is often slow with stereotypic and usually monosyllabic or monolectic answers. Movement and coordination are intact. Complete unresponsive is rare.
Behavioural Abnormalities and Experiential Phenomena
Though the most common behavioural changes refer to daily activities disturbed by the impairment of consciousness, some patients become depressed, agitated and, occasionally, hostile and aggressive. Experiential and sensational phenomena are more common than is usually appreciated and may include:
Patient noteSensation of viewing the world through a different medium and a feeling of not being in the same world as everyone else. Uncontrollable rush of thoughts. A feeling of fear of losing control of my mind.
A feeling of closeness.
A funny feeling that I can not elaborate.
A strange feeling of not being myself.
Edgy, worry and uncomfortable.
My character changes completely, I become extremely snappy, have a severe headache.
Weird.
Simple gestural and ambulatory automatisms, and automatic behavioural and fugue-like states may occur in the 20% of patients who also have severe impairment of consciousness:
Patient noteReplies yes to any question and fumbles with his clothes.
Myoclonic Jerks in Absence Status Epilepticus
Segmental myoclonic jerks, usually involving the eyelids or perioral region and less often the limbs, frequently occur during typical ASE, and vary in degree and severity. They are most likely to occur in syndromes that manifest with similar myoclonic phenomena during brief absences (see descriptions in the relevant sections of individual IGE syndromes).
GTCS Associated with Typical Absence Status Epilepticus
ASE ending with a GTCS is probably the rule irrespective of the syndrome. However, in one-third of patients, ASE always ends with GTCS. In the remaining two-thirds, it may also terminate spontaneously without GTCS. It is exceptional for GTCS to precede or be interspersed with ASE. It is also exceptional for more than one GTCS to occur following ASE.
Duration and Frequency of Typical Absence Status Epilepticus
ASE usually lasts for an average of 3–4 hours, rarely as little as half an hour, often exceeds 6–10 hours and occasionally lasts for 2–10 days. Frequency also varies from once in a lifetime to an average of 10–20 episodes/year, or be consistently catamenial. The duration and frequency depend on treatment strategies and syndromic classification.
Postictal State
Amnesia of the event is exceptional. Commonly patients are aware of what happens during the ASE and some are able to write down their experiences even when in status. Other patients have a patchy recollection of events and usually miss the last part prior to the GTCS. After a GTCS, the patient feels tired, has a headache and is confused for a varying duration of time.
Age at Onset and Sex
Mean age at onset of ASE is 29 years, with a range of 9–56 years. It is rare for ASE in IGE to start before the first decade of life. Other types of seizures, such as absences, myoclonic jerks and GTCS, may predate the first ASE by many years. ASE rarely is the first overt type of seizure.77
Precipitating Factors
Inappropriate use of AEDs, such as tiagabine,78,79 vigabatrin,66 carbamazepine and phenytoin,80 as well as discontinuation of proper anti-absence medication are the commonest precipitants of ASE. Sleep deprivation, stress and excess alcohol consumption, alone or usually in combination are common precipitating factors. Some women may have consistent catamenial precipitation.81–83
Differential Diagnosis
A confusional state lasting for hours and ending with GTCS creates significant diagnostic difficulties regarding its nature and cause. Causes to consider are intoxication, and psychogenic, metabolic or systemic diseases. If these are excluded, the differential diagnosis is between focal (complex, partial) or ASE.
Idiopathic ASE is commonly unrecognised or misdiagnosed. It is surprising how often physicians are deceived by the general good appearance, alertness and cooperation of the patient.66 Basic testing of memory and higher cognitive functions, essential for diagnosis, are rarely carried out. It is important to remember that more than half of patients are aware of the situation when entering or during ASE, which is of great practical significance with regard to termination of the ASE and prevention of the impending GTCS by self-administered appropriate medication.
Idiopathic ASE is easy to diagnose on the basis of proper identification of the IGE syndrome.
Differentiation of Generalised ASE from Complex Focal Status Epilepticus
Complex focal status epilepticus (including limbic status epilepticus) is rarer than generalised ASE. Patients frequently have recurring complex focal seizures with incomplete recovery between attacks, or a continuous ‘epileptic twilight state’ with cycling between unresponsiveness and partial responsiveness. The ictal EEG reveals recurrent epileptiform patterns consistent with those encountered in isolated complex focal seizures. The interictal EEG usually shows a unilateral or bilateral cortical focus. In complex focal status epilepticus, conscious levels fluctuate during the attack and patients experience postictal confusion and amnesia of the episode. Although automatisms may occur in both forms of status epilepticus, they are more complex and prolonged in complex focal status epilepticus.
The commonest reason for misdiagnosis between the two conditions is because absences are not recognised or misdiagnosed as complex focal seizures (Table 10.2). A previous or new EEG invariably shows generalised discharges in IGE. It may be normal or show specific focal spikes in focal epilepsies, mainly temporal lobe epilepsy. Ictal EEG with GSWD is diagnostic of IGE. Coexisting focal abnormalities should not be interpreted as evidence of focal epilepsy.
The Differentiation of Typical (Idiopathic) ASE from Atypical (Usually Symptomatic or Probably Symptomatic) ASE Is Also Easy
The major distinguishing feature of atypical ASE is that it occurs mainly in children with symptomatic or cryptogenic generalised epilepsies, who also have a plethora of other types of frequent seizures, such as atypical absences, tonic and atonic seizures, myoclonic jerks and GTCS. Most of these patients also have moderate or severe learning and physical handicaps. In addition, the interictal EEG is often very abnormal with slow background activity and frequent, brief or long runs of slow generalised spike–wave complexes, paroxysmal fast activity and paroxysms of polyspikes. It is often difficult to define the boundaries, onset and termination of atypical ASE, because these children frequently have alterations of behaviour and alertness as well as long interictal slow GSWD.
Atypical ASE is clinically characterised by fluctuating impairment of consciousness often with other ictal symptoms, such as repeated series of tonic or atonic seizures and segmental or generalised jerks. The ictal EEG pattern is of slow (< 2.5 Hz) GSWD. Both the clinical patterns and the EEG abnormalities are more variable than of the typical ASE.
Additional discriminating features of atypical ASE are:
- gradual onset and offset
- level of consciousness and other coexisting types of seizures tend to fluctuate sometimes for weeks, with little distinction between ictal and interictal phases
- initiation or termination with a GTCS is exceptional
- incontinence is common.
Epileptic Syndromes of Idiopathic Generalised Epilepsies
The recognised syndromes of IGEs are shown in Table 1.5 (1989 ILAE classification)1 and Table 1.6 (new ILAE classification scheme).5 Listed according to the age at onset, these are:
- Benign myoclonic epilepsy in infancy (Chapter 6)
- Epilepsy with myoclonic absences
- Epilepsy with myoclonic-astatic seizures
- Childhood absence epilepsy
- Idiopathic generalised epilepsies with variable phenotypes
- – Juvenile absence epilepsy
- – Juvenile myoclonic epilepsy
- – Epilepsy with generalised tonic-clonic seizures only
Other possible syndromes of IGE for consideration, which are not yet recognised by the ILAE Committees, are:1,5
- IGE with absences of early childhood
- Perioral myoclonia with absences
- Idiopathic generalised epilepsy with phantom absences
- Jeavons syndrome (eyelid myoclonia with absences)
- Benign adult familial myoclonic epilepsy
- Autosomal dominant cortical myoclonus and epilepsy
Considerations on the Classification of Idiopathic Generalised Epilepsies:
The classification of IGE is probably one of the most significant and debated issues. There are two schools of thought, with diversely opposing views:2 (a). IGE is one disease, (b). IGE comprises a large group of many distinct syndromes. The evidence so far is not conclusive in favour of one or the other, and any new classification should not take sides unreasonably. In practical terms, the view that ‘IGE is one disease’ would, overall, be an easy clinical diagnostic approach, but it would discourage the diagnostic precision required for genetic studies, prognosis and management decisions. The view that ‘IGE comprises a large group of many distinct syndromes’ would be more demanding diagnostically and occasionally require exhaustive clinical and video EEG data. However, this is often the price that we, as physicians, have to pay in pursuing an accurate diagnosis, which is is the golden rule in medicine. This view also satisfies: (a). “maximum practical application to differential diagnosis,”84 which is the main reason for reorganising the classification of epileptic syndromes in the forthcoming revisions; and (b). takes advantage of “significant advances in our understanding”85 of IGEs, which constitute one-third of cases of ‘epilepsy’. Similarly, there is no justification for the unification of ‘IGEs with onset in adolescence’ in a single syndrome as has been recently proposed.86 The major conceptual problem with this proposition is that it takes ‘onset in adolescence’ as the most significant almost defining factor, which is at variance with the definition of a syndrome.1 Further, the same IGE syndromes may start in childhood, adolescence and occasionally adult life.1 On the surface, syndromes of IGE may look alike if their clinical EEG manifestations are not properly analysed and synthesised. For example, JME and JAE both manifest with absences, myoclonic jerks and GTCS. However, severe absences are the main and most disturbing type of seizure in JAE, and myoclonic jerks may not occur or be randomly distributed.16 Conversely, myoclonic jerks on awakening is the defining symptom of JME; absences are mild and occur in only one-third of patients. The clinical-EEG features of typical absence seizures that may be syndrome-related are well described in video EEG studies.16,18 Unifying all typical absence seizures as a single type is of no benefit to any cause. Animal genetic studies have documented numerous syndromes of IGE17 and this is likely to be the case in humans.2
Recognised Syndromes of IGEs in the ILAE Classification of 1989 versus the Newly Proposed Diagnostic Scheme
The new ILAE diagnostic scheme5 has some significant differences in relation to the ILAE classification of 19891 regarding IGE of childhood and adolescence. These are:
(1). The syndromes of JAE, JME and IGE with GTCS only are considered as phenotypical variants of IGE of adolescence
(2). A new syndrome of ‘IGE with GTCS only’ has been proposed to replace ‘epilepsy with GTCS on awakening’
(3). ‘Epilepsy with myoclonic-astatic seizures’ and ‘epilepsy with myoclonic absences’ are included among idiopathic generalised epilepsies; these were previously categorised as symptomatic or cryptogenic generalised epilepsies.
(4). ‘Generalised epilepsy with febrile seizures plus’ is proposed as a new syndrome in development
Epilepsy with Myoclonic Absences
Clinical noteEpilepsy with myoclonic absences (MAE) is a rare syndrome of childhood, which demands scrupulous exclusion of other forms of symptomatic or probably symptomatic cases manifesting with the same seizure (myoclonic absences).173–177
Demographic Data
Age at onset varies from the first months of life to early teens with a median of 7 years. Boys (69%) predominate. MAE is a very rare disorder with an approximate prevalence of 0.5–1% among selected patients with epileptic disorders.173,177,178 I have seen only three cases (two of which were idiopathic MAE) over a period of 15 years out of nearly 200 patients with video EEG-recorded typical absence seizures.4,24,177
Clinical Manifestations
Myoclonic Absences
The myoclonic absences are the hallmark of MAE.173,178 They consist of impairment of consciousness, which varies from mild to severe and rhythmic myoclonic jerks, mainly of the shoulders, arms and legs with a concomitant tonic contraction. Eyelid twitching is practically absent, but perioral myoclonias are frequent. The jerks and the tonic contraction may be unilateral or asymmetrical, and head/body deviation may be a constant feature in some patients. The tonic contraction mainly affects the shoulder and deltoid muscles, and may cause elevation of the arms. Some patients maintain awareness of the jerks.
The duration of the absences varies from 8 to 60 s. Myoclonic absences occur many times a day.
Other Types of Seizure
Other types of seizure also occur in two-thirds of patients.173,174,179 These are infrequent GTCS and atonic seizures, which may precede or occur concurrently with the myoclonic absences. ASE is rare.
Precipitating Factors
Hyperventilation is the main precipitating factor.173,178 Photosensitivity is uncommon (14%).178 Non-photosensitive myoclonic absences precipitated by eye-closure or eye-opening have been described.173,178 Seizures induced by emotionally gratifying stimuli, such as cheek-kissing or after viewing pleasant or funny events have been reported in a child with inverted chromosomal 15 duplications.24
Neurological and Mental State
Neurological examination is usually normal, but nearly half of patients (45%) have impaired cognitive functioning prior to the onset of absences.175,178
Considerations on the Classification of Epilepsy with Myoclonic Absences
Myoclonic absences (the seizures) may feature either in normal or children with neurocognitive impairment.16,176 The 1989 ILAE Commission, discounting the idiopathic form, classified ‘epilepsy with myoclonic absences’ among ‘cryptogenic/symptomatic’ generalised epilepsies, that is in the same group of disorders as the Lennox-Gastaut syndrome and EM-AS.1 “The syndrome of epilepsy with myoclonic absences is clinically characterised by absences accompanied by severe bilateral rhythmical clonic jerks, often associated with a tonic contraction. On the EEG, these clinical features are always accompanied by bilateral, synchronous, and symmetrical discharge of rhythmical spike–waves at 3 Hz, similar to childhood absence. Seizures occur many times a day. Awareness of the jerks may be maintained. Associated seizures are rare. Age of onset is ~7 years, and there is a male preponderance. Prognosis is less favourable than in pyknolepsy owing to resistance to therapy of the seizures, mental deterioration, and possible evolution to other types of epilepsy such as Lennox-Gastaut syndrome or JME.”1 Contrary to this, the new ILAE diagnostic scheme considers only the idiopathic form (Table 1.7),5 which probably represents around one-third of the whole spectrum of epileptic disorders manifesting with myoclonic absences. The others are symptomatic or probably symptomatic cases.1
Aetiology
Only one-third of patients with myoclonic absences are idiopathic cases and only these belong to this syndrome of MAE. The other two-thirds with myoclonic absences (the seizures, not the syndrome) are due to symptomatic causes including chromosomal abnormalities, such as trisomy 12p, Angelman syndrome, inverted chromosomal 15 duplications179,180 and malformations of brain development.176,177,181
Diagnostic Procedures
By definition, in idiopathic MAE, all tests but the EEG should be normal. Brain MRI and chromosomal testing179 are needed to detect symptomatic cases.
Electroencephalography
Background EEG is usually normal at onset, but may deteriorate later or be abnormal in symptomatic cases. Interictal EEG shows brief generalised, focal or multifocal spike and slow waves in 50% of cases.173,177
Ictal EEG shows 3 Hz GSWD, even in those with unilateral or asymmetrical clinical manifestations. Polygraphic studies have revealed that each myoclonic jerk coincides with the spike component of the discharge.173,177
Differential Diagnosis
The differential diagnosis of MAE from other types of syndromes with absences is easy because of the characteristic type of myoclonic absences. The difficulty is between idiopathic and symptomatic/cryptogenic cases that manifest with the same seizure type (myoclonic absences). Symptomatic patients often have an abnormal neurological state, abnormal background EEG and abnormal brain MRI. Chromosomal abnormalities are common.179 Additionally, absences with rhythmic myoclonic jerking, but less than 2.5 Hz spike/polyspike–wave complexes and other characteristics of atypical absences may occur in epileptic encephalopathies173,178,182 and include some of the cases with chromosomal abnormalities.179
Prognosis
Myoclonic absences remit after an average of 5 years in one-third of patients.176,177 In the remaining (perhaps symptomatic) patients, absences continue into adult life together with other types of seizures, such as GTCS and atonic seizures, or they develop features of other types of epilepsy, such as Lennox-Gastaut syndrome or JME.
Nearly half of the children with MAE have impaired cognitive functioning prior to the onset of absences, but these are probably symptomatic cases. However, half of those who were normal prior to the onset of absences develop cognitive and behavioural impairment. This may mean that the EEG discharges have a deteriorating effect on cognition unless eliminated early with treatment.
Management
The aim is to stop myoclonic absence seizures as early as possible. Early control of absences may prevent subsequent cognitive deterioration and secure normal development.178
Myoclonic absences are often resistant to treatment. Treatment frequently requires high doses of valproate often combined with ethosuximide or lamotrigine.177 Interestingly, Tassinari et al.173 had a few cases that responded only to phenobarbitone alone or in combination with anti-absence drugs. Newer drugs, such as levetiracetam,183 or old drugs, such as clonazepam and acetazolamide, may be tried in resistant cases.
Baclofen a GABAB agonist used for the treatment of spasticity in neurologically impaired patients is contraindicated because of significant provocation of absence seizures.
See also AEDs contraidicated in IGEs (page 337).
Epilepsy with Myoclonic-Astatic Seizures Doose Syndrome
Clinical noteEpilepsy with myoclonic-astatic seizures (EM-AS)* or Doose syndrome** 87–97 is considered as an IGE in the new ILAE diagnostic sceme.5 Diagnosis of this syndrome requires careful application of inclusion and exclusion criteria. Its characteristic symptom, myoclonic-astatic seizures, is shared by many other childhood syndromes and particularly epileptic encephalopathies.
Demographic Data
Onset occurs between 7 months and 6 years, and peaks at 2–4 years. Two-thirds are boys. EM-AS accounts for about 1–2% of all childhood epilepsies.
Clinical Manifestations
Doose syndrome is characterised by myoclonic-astatic seizures that often occur together with atonic, myoclonic and absence seizures; myoclonic-astatic status epilepticus is common.
Children are normal prior to the onset of seizures. In two-thirds of children, febrile and afebrile generalised tonic clonic seizures appear first, several months prior to the onset of myoclonic-astatic seizures.
Considerations on the Classification of EM-AS
The 1989 ILAE Commission classifies EM-AS among ‘cryptogenic/symptomatic’ generalised epilepsies, that is in the same group of disorders as Lennox-Gastaut syndrome.1 It is defined as follows:
“Manifestations of myoclonic-astatic seizures begin between the ages of 7 months and 6 years (mostly between the ages of 2 and 5 years), with (except if seizures begin in the first year) twice as many boys affected. There is frequently hereditary predisposition and usually a normal developmental background. The seizures are myoclonic, astatic, myoclonic-astatic, absence with clonic and tonic components, and tonic-clonic. Status frequently occurs. Tonic seizures develop late in the course of unfavorable cases. The EEG, initially often normal except for 4–7 Hz rhythms, may have irregular fast spike-wave or polyspike wave. Course and outcome are variable.”1 Contrary to this, the ILAE Task Force considers EM-AS as IGE,5 a view which is similar to that of Doose (Table 10.4):98 “Myoclonic-astatic epilepsy belongs to the epilepsies with primarily generalized seizures and thus stands in one line with absence epilepsies, JME, as well as the infantile and juvenile idiopathic epilepsy with generalized tonic clonic seizures. Like these types of epilepsy, myoclonic-astatic epilepsy is polygenically determined with little nongenetic variability. The disease is characterized by the following criteria: genetic predisposition (high incidence of seizures and/or genetic EEG patterns in relatives); mostly normal development and no neurological deficits before onset; primarily generalized myoclonic, astatic or myoclonic-astatic seizures, short absences and mostly generalized tonic clonic seizures; no tonic seizures or tonic drop attacks during daytime (except for some rare cases with a most unfavourable course); generalized EEG patterns (spikes and waves, photosensitivity, 4–7 Hz rhythms), no multifocal EEG-abnormalities (but often pseudofoci).”98 The problem may reflect lack of specific diagnostic criteria and undefined boundaries of certain epileptic syndromes and particularly epileptic encephalopathies, which may manifest with myoclonic-astatic seizures. This particularly refers to Dravet, Lennox-Gastaut syndrome and atypical benign epilepsy of childhood. Cases of benign and severe myoclonic epilepsy in infants may have been included in EM-AS.89 Other myoclonic epilepsies with brief seizures reported as intermediate cases between EM-AS and Lennox-Gastaut syndrome probably prove this point.99
However, it is generally accepted that some children with myoclonic-astatic seizures are otherwise normal with no discernible causes other than a strong genetic epileptic background and probably represent the genuine ‘Doose syndrome’ of ‘idiopathic epilepsy with myoclonic-astatic seizures’. This point is exemplified in the study of Kaminska et al.,100 who found evidence that EM-AS is distinct from Lennox-Gastaut syndrome, and the distinction appears from the first year of the disorder.
A further exciting development is that myoclonic-astatic seizures frequently occur in patients with ‘generalised epilepsy with febrile seizures plus’101, a syndrome that also has strong genetic links with Dravet syndrome.
Myoclonic-astatic (in fact myoclonic-atonic) seizures are the defining symptoms (100% of cases).89 These manifest with symmetrical myoclonic jerks immediately followed by loss of muscle tone (post-myoclonic atonia) (Figure 10.5).
Atonic seizures of sudden, brief and severe loss of postural tone may involve the whole body or only the head. Attacks are brief, lasting 1–4 s and frequent. Generalised loss of postural tone causes a lightning-like fall. The patient collapses on the floor irresistibly. In brief and milder attacks, there is only head nodding or bending of the knees.
Myoclonic jerks may precede or less often intersperse with the atonic manifestations (Figures 10.5 and 10.6).
Brief absence seizures happen in more than 50% of cases. These often occur together with myoclonic jerks, facial myoclonias and atonic manifestations. Atonic and absence seizures may occur frequently, sometimes many times a day in the active period of the disease. Absence seizures alone are exceptional.
Tonic seizures are an exclusion criterion.
Myoclonic-atonic status epilepticus lasting for hours or even days (Figure 10.6) is common affecting one-third of patients. It manifests with varying degrees of usually severe cognitive impairment or cloudiness of consciousness interspersed with repetitive myoclonic and atonic fits. Facial myoclonus of eyelids and mouth may be continuous together with irregular jerks of the limbs and atonic seizures of head nodding or falls. Myoclonic-atonic status epilepticus may occur several times during a period of 1–2 years.
Aetiology
Doose syndrome may be genetically determined in a multifactorial polygenic fashion with variable penetrance.87–89 One-third of patients have familial seizure disorders and mainly IGEs.87–89 Of significant interest are the clinical and molecular studies in ‘generalised epilepsy with febrile seizures plus’ in which myoclonic-atonic seizures are common in some families.101 ‘Generalised epilepsy with febrile seizures plus’ also has strong genetic links with Dravet syndrome.
Diagnostic Procedures
By definition, all tests other than the EEG are normal.
Electroencephalography
Interictal EEG may be normal at the stage of febrile or afebrile GTCS. Rhythmic theta activity in the parasagittal regions may be the only significant abnormality. Subsequently, when myoclonic-atonic seizures appear, there are frequent clusters of 2–3 Hz GSWD interrupted by high amplitude slow waves in cases with predominant atonic or myoclonic-atonic seizures. In children with predominantly myoclonic seizures, paroxysms of irregular spikes or polyspike–wave complexes prevail.
The ictal EEG of myoclonic and atonic seizures manifests with discharges of irregular spike–wave or polyspike–wave complexes at a frequency of 2.5–3 Hz or more (Figures 10.5 and 10.6). Atonia is usually concurrent with the slow wave of a single or multiple spike–wave complex and the intensity of the atonia is proportional to the amplitude of the slow wave. Drop attacks are associated with diffuse EMG paucity indicating their true atonic nature.91 The myoclonus of Doose syndrome appears to be a primary generalised epileptic phenomenon, which differs from that of Lennox-Gastaut syndrome.102
In myoclonic-atonic status epilepticus, the EEG shows continuous or discontinuous and repetitive 2–3 Hz spike-wave complexes (Figure 10.6).
Differential Diagnosis
Differentiation of EM-AS is mainly between:
- benign myoclonic epilepsy in infancy
- Dravet syndrome
- Lennox-Gastaut syndrome
- late onset West syndrome.
In general, children with the Doose syndrome are normal prior to the development of seizures, have a strong family history of IGE, and the background EEG and brain imaging are normal.
Progressive myoclonic epilepsies, such as myoclonic epilepsy with ragged-red fibres, Lafora and Unverricht-Lundborg disease, may initially imitate Doose syndrome. However, the associated relevant neurological abnormalities and, sometimes, the relentless progression and deterioration will establish the diagnosis.
Atypical benign partial epilepsy of childhood may also imitate Doose syndrome, because of repeated falls, absences and diffuse slow spike–wave activity mainly in the sleep EEG.31,103–105 The main differentiating point is that these children also have nocturnal focal seizures similar to the Rolandic seizures (RS) that are often the presenting seizure symptom. Also, the EEG shows centrotemporal and other functional spikes in various locations.
Atypical evolutions of RS106,107 and Panayiotopoulos syndrome108,109 may have clinico-EEG features similar to those of Doose syndrome, but they are preceded by typical presentations of these syndromes (see Chapter 9). A similar, but reversible, clinico-EEG condition may be induced by carbamazepine,31 oxcarbazepine110 and lamotrigine111 in a few children with RS. This possibility should be considered in children with RS and dramatic deterioration after treatment with these AEDs.
Children with ‘epilepsy with continuous spike–wave complexes during slow-wave sleep’ may also have drop attacks due to atypical absences or negative epileptic myoclonus.
Non-epileptic myoclonus of many neurological disorders rarely raises a diagnostic problem with Doose syndrome, unless it is a symptom of a degenerative disease with associated epileptic features.90
Clinical noteDiagnostic tips
The diagnosis of Doose syndrome is probably safe if myoclonic-atonic seizures start in a previously normal child with pre-existing febrile or afebrile GTCS and familial seizure disorders.
Differential diagnostic problems from Lennox-Gastaut syndrome probably reflect ill-defined inclusion and exclusion criteria.
Prognosis
The prognosis is unclear probably because of different selection criteria. Half of the patients may achieve a seizure-free state and normal or near normal development.95 Myoclonic-atonic seizures remit within 1–3 years from onset despite initial resistance to treatment, but GTCS or clonic seizures tend to continue.95 These patients who have a good prognosis may correspond to the genuine Doose syndrome of the idiopathic form of EM-AS. Spontaneous remission with normal development has been observed in a few untreated cases, but these may belong to benign myoclonic epilepsy in infancy.
The others, probably belonging to symptomatic or probably symptomatic cases or other syndromes, may continue with seizures, severe impairment of cognitive functions and behavioural abnormalities. Ataxia, poor motor function, dysarthria and poor language development may emerge.
Management
Drug therapy is dictated by seizure type. Valproate, which is effective in myoclonic jerks, atonic seizures and absences, is the most efficacious. Add-on small doses of lamotrigine have a beneficial pharmacodynamic interaction with valproate. Topiramate reduces the frequency of atonic seizures.112 Levetiracetam may be an effective substitute AED for valproate.
In resistant cases, ketogenic diet, followed by adrenocorticotropin hormone (ACTH) and ethosuximide, have been found to be highly beneficial.95 Benzodiazepines, acetazolamide, sulthiame and even bromides are also used.
Carbamazepine, phenytoin, and vigabatrin are contraindicated.
In myoclonic-atonic status epilepticus, intravenous benzodiazepines are often efficacious, but rarely may precipitate tonic status.
Childhood Absence Epilepsy
Patient note“…a disease with an explosive onset between the ages of 4 and 12 years, of frequent short, very slight, monotonous minor epileptiform seizures of uniform severity, which recur almost daily for weeks, months, or years, are uninfluenced by anti-epileptic remedies, do not impede normal and psychical development, and ultimately cease spontaneously never to return. At most, the eyeballs may roll upwards, the lids may flicker, and the arms may be raised by a feeble tonic spasm. Clonic movements, however slight, obvious vasomotor disturbances, palpitations, and lassitude or confusion after the attacks are equivocal symptoms strongly suggestive of oncoming grave epilepsy, and for the present they should be considered as foreign to the more favourable disease.” 113
Patient noteW. J. Adie (1924) 113 defining CAE as an epileptic syndrome
Clinical noteCAE is the prototype IGE of typical absence seizures.7,15,16,18,114 It is genetically determined, age related and affects otherwise normal children.
Demographic Data
Onset is between 4 and 10 years of age, with a peak at 5–6 years.15,114–117 The onset of typical absence seizures in CAE before 4 years of age 118–121 and after 10 years of age118–121 is uncertain or at least exceptional.
Comments and Debate on the ILAE Definition of CAE
The ILAE Commission of 1989 1 largely defined CAE by age at onset and frequency of absences: “Childhood absence epilepsy (pyknolepsy) occurs in children of school age (peak manifestation age 6–7 years), with a strong genetic predisposition in otherwise normal children. It appears more frequently in girls than in boys. It is characterised by very frequent (several to many per day) absences. The EEG reveals bilateral, synchronous symmetrical spike–waves, usually 3 Hz, on a normal background activity. During adolescence, GTCS often develop. Otherwise, absences may remit or more rarely, persist as the only seizure type.”1
The new ILAE diagnostic scheme also classifies CAE as IGE.5
The ILAE definition is a very broad and requires revision. Otherwise, any type of frequent absence seizures occurring in childhood would be erroneously equated with CAE. Because of this ambiguity, the epidemiology, genetics, age at onset, clinical manifestations, other types of seizures, long-term prognosis and treatment of CAE reviewed in this chapter may not accurately reflect the syndrome of CAE. It is also because of this ambiguity that some authors: (a.) have divided patients with childhood onset absence seizures into ‘subsyndromes’ including those who remit, those who persist into adolescence and develop GTCS, and those who develop both GTCS and myoclonic seizures during adolescence;138 and (b.) consider that patients with CAE ‘evolve’ into JAE or JME.139,140
The inclusion and exclusion criteria of Table 10.5 proposed by Loiseau and Panayiotopoulos114 for CAE should not be taken as an extreme position. They do not differ significantly from the ILAE (1989)1 criteria of CAE with:
- age at onset in childhood
- very frequent (several to many per day) absences presumably with severe impairment of consciousness
- ictal EEG with bilateral, synchronous and symmetrical 3 Hz GSWD, on a normal background activity (that presumably excludes fragmented, asymmetrical and asynchronous 3–5 Hz GSWD with intradischarge variations)
- GTCS accepted only if they develop later in adolescence.
Also, the Commission (1989)1 by accepting ‘epilepsy with myoclonic absences’ as a separate syndrome differentiates myoclonic absences from typical absences of CAE. It is along this line that Loiseau and Panayiotopoulos114 also considered eyelid myoclonia (which is a predominantly myoclonic and less of an absence syndrome) as an exclusion criterion. Whether, perioral myoclonia or single violent jerks during the ictus of an absence seizure is an exclusion criterion may be debatable. However, their presence indicates a worse prognosis.15,16,141 The same applies to multiple spikes (more than three spikes/wave), which also indicate a bad prognosis,138,142 and coexistent myoclonic jerks or GTCS.24
Further, the Commission (1989),1 by accepting ‘typical absence seizures consistently provoked by specific stimuli’ as a specific type of reflex seizures, indicates that these may be a separate group from CAE.16,114,136,143
Age at Onset of Absences Does Not Determine Syndromic Classification of CAE
We have studied 39 adults with IGE with typical absences starting before 10 years of age.144 All were older than 18 years (31.5±10.5; range 18–56) at the last follow-up and all had EEG (15 with video-EEG) recorded typical absence seizures. Typical absences had onset at 6.2±1.9 years (range 2–9) that persisted into adulthhood in 28 (71.8%) cases. GTCS occurred in 87.2% (onset 13±7.2 years; range 2–36). Myoclonic jerks occurred in 38.5% (onset 2.6±4.1 years; range 7–18). Women (82%) and photosensitivity (56.4%) markedly predominated. Eight patients have EMA, 5 JAE, 4 perioral myoclonia with absences, 3 JME, 3 absences with single myoclonic jerk, 1 CAE, 3 predominantly reflex absences. Twelve patients (8 with photosensitivity) could not be classified. Only 6 patients were free from all type of seizures (1 CAE, 2 JAE, 2 unclassified IGE with reflex absences). 144
Synonyms
Pyknolepsy. Many European clinicians use the word ‘pyknolepsy’ (from the Greek words pyknos = dense and epilepsy) either to define a high daily frequency of absences or as a synonym to CAE.
Two-thirds of patients are girls115,116,122,123 despite some studies indicating that boys and girls are equally affected.124,125
The prevalence is about 10%126–128 and the annual incidence is about 7/100,000129–131 in children with epileptic seizures who are less than 15–16 years of age. Recruitment bias explains the wide range in the reported incidence (1.9–8/100,000 of children < 16 years) and prevalence (2–37% of children with epileptic disorders) of CAE.124,129,130,132–137
Clinical Manifestations
Typical Absence Seizures
CAE manifests with the most characteristic and classical example of typical absence seizures, which are characterised by:
- short duration
- abrupt onset and abrupt termination
- severe impairment of consciousness
- high daily frequency.
Probably any other types of seizure are incompatible with this diagnosis. Mild impairment of consciousness in untreated patients is an exclusion criterion.114
Absences are severe and frequent, with from ten to hundreds per day, for which reason CAE is also known as pyknolepsy.113 They are of abrupt onset and abrupt termination (Figures 10.2, 10.7 and 10.8). Their duration varies from 4–20 s, though most last around 10 s. Clinically, the hallmark of the absence is abrupt, brief and severe impairment of consciousness with unresponsiveness and interruption of the ongoing voluntary activity, which is not restored during the ictus. The eyes open spontaneously, overbreathing, and speech and other voluntary activity stop within the first 3 s from the onset of the discharge. Simple automatisms occur in two-thirds of seizures, but are not stereotyped. The eyes stare or move slowly. Mild myoclonic elements of the eyes, eyebrows and eyelids may feature in CAE, but are usually mild and occur at the onset of the GSWD. However, more severe and sustained myoclonic jerks of the facial muscles may indicate other IGEs with absences.
The attack ends as abruptly as it commenced with sudden resumption of the pre-absence activity as if it was not interrupted.
Typical absence seizures are nearly invariably provoked by hyperventilation.
Other Types of Seizures
Clinical noteSeizures other than typical absences are not compatible with CAE. The only exceptions are (a). febrile seizures that may precede the onset of CAE and (b). solitary or infrequent GTCS that occur long after the onset of absence seizures and usually in adolescence after absences have remitted.
GTCS or myoclonic jerks preceding the onset of typical absences or concomitant with the stage of active absence seizures do not occur in CAE.15,114,143,145,146 However, about 10% of patients may later develop a few, solitary or infrequent GTCS in adolescence or adult life.145,146 This contrasts with the fact that focal seizures, myoclonic jerks, GTCS and other more bizarre fits have been described with CAE but these are probably other epileptic syndromes starting with absences in childhood.134
Status epilepticus, whether convulsive or non-convulsive, is incompatible with CAE. If this occurs, the diagnosis of CAE should be seriously challenged. Also, though absence status epilepticus may occur in 5–16% of patients with typical absence seizures starting before the age of 10 years,117,140,147 this is probably incompatible with CAE.15,19,77
Atonic falls do not occur in CAE.136
Seizure-Precipitating Factors
Typical absence seizures occur spontaneously, but they are also influenced by various other factors, mainly hyperventilation (Figures 10.2, and 10.6). Hyperventilation is the most potent precipitating factor that induces absences in more than 90% of the trials,148,149 ranging from 75%150 or 80%151 to 100%.143 A diagnosis of CAE should be seriously questioned in an untreated child who does not have an attack on hyperventilation.
Other precipitating or facilitating factors are emotional (anger, sorrow, fear, surprise, embarrassment), intellectual (lack of interest, release of attention, mealtimes for some children and school-time for others) and metabolic (hypoglycaemia).114 Typical absence seizures generally do not occur when the child is busy and stimulated by physical or mental activity, or has sustained attention.136 Emotional or conflicting situations, such as reading difficulties, may provoke absences.152 In a given patient, typical absence seizures are often triggered by the same factor.
There are no other precipitating factors in CAE, which is at variance with nearly all other forms of IGE. In particular, though photosensitivity is accepted according to the ILAE Commission’s definition,1 most other authors consider clinical photosensitivity as a significant exclusion criterion.15,136,143 Mild EEG photosensitivity or facilitation (not consistent provocation) of photoparoxysmal responses and absences may occur.
Aetiology
Although CAE is genetically determined, the precise mode of inheritance and the genes involved remain largely unidentified.7
In monozygotic twins, 84% had 3 Hz GSWD and only 75 % of pairs had clinical absence seizures. These events occurred 16 times less often in dizygotic twins.115 Currently, various chromosomal loci have been identified in families with absences of childhood onset (not necessarily equated with CAE). Linkage to chromosome 1 was found in families with absences starting in childhood and the later development of myoclonic jerks and GTCS, as in JME.9 Linkage analysis in five generations of a family in which affected patients had childhood absences and GTCS provided evidence of a locus on chromosome 8q24.9,138 The candidate region for this locus, designated ECA 1, has been refined, but a gene remains to be identified. According to the criteria proposed in this chapter, neither of these groups is CAE. There are also reports implicating chromosome 5q31.1 and 19p13.2 (see ref 7 for an excellent recent review).
Furthermore, there is now evidence available to suggest that mutations in genes encoding GABA receptors34–36 or brain-expressed voltage-dependent calcium channels37 may underlie CAE. Feucht et al.34 found a significant association between a polymorphism in GABAA receptor gamma 3 subunit in chromosome 15q11 in 50 families with CAE. Marini et al.36 found GABAA receptor gamma 2 subunit gene mutations on chromosome 5 in a large family with CAE and febrile seizures (including febrile seizures plus and other seizure phenotypes). This gene mutation segregated with febrile seizures and CAE, and also occurred in individuals with the other phenotypes. The clinical and molecular data suggested that the GABAA receptor subunit mutation alone could account for the febrile seizure phenotype, but an interaction of this gene with another gene or genes was required for the childhood absence phenotype in this family. Linkage analysis for a putative second gene contributing to the childhood absence phenotype suggested possible loci on chromosomes 10, 13, 14 and 15.36 Chen et al.37 found 68 variations, including 12 missense mutations in the calcium channel CACNA1H gene in CAE patients. The identified missense mutations occurred in the highly conserved residues of the T-type calcium channel gene.37 However, another study of 33 nuclear families, each with two or more individuals with CAE each provided conclusive evidence that the genes encoding GABAA and GABAB receptors, voltage-dependent calcium channels and the ECA1 region on chromosome 8q do not account independently for the childhood absence trait in a majority of the families.153
Acquired factors may play a facilitating role.
Diagnostic Procedures
In typical cases nothing but an EEG is needed.
Interictal EEG
The interictal EEG in CAE has normal background activity, with frequent rhythmic posterior delta activity (Figure 10.8). Benign functional spikes (mainly centrotemporal and less frequently occipital or frontal) may be seen and do not alter the prognosis.
Posterior rhythmic slow activity is considered a characteristic finding in CAE. This consists of long runs of bilateral rhythmic and fusiform, high amplitude slow waves at 3 Hz in the distribution regions of alpha rhythm. It is bilateral, often of higher amplitude on the right, but occasionally may be lateralised to one side or fluctuate from side to side in emphasis. It occurs in less than half of patients at a peak age of 6–10 years. Patients with this EEG finding tend to have longer absence seizures, a better prognosis and less often develop GTCS.154,155
Sleep EEG has not been studied systematically in pure forms of CAE. Generally, in IGE, GSWD are more likely to increase, but a reduction is also observed during sleep. The discharges are shorter and usually devoid of discernible clinical manifestations, even in those patients who have numerous clinical seizures during the alert state. However, clinical absence seizures that may awake the patient have been recorded during sleep. Sleep EEG patterns are normal.
Photic stimulation: Clinical photosensitivity or consistent provocation of typical absences by IPS is an exclusion criterion for CAE. However, IPS may act as a facilitatory factor in that these children may have more typical absences during IPS than in the resting EEG, but these are far less common than during hyperventilation.
Ictal EEG
Ictal EEG consists of high amplitude 3 Hz GSWD which are of higher voltage in the anterior regions. They are rhythmic at around 3 Hz (2.5–4 Hz) with a gradual and regular slowing down of the frequency by 0.5–1 Hz from the initial to the terminal phase of the discharge. The opening phase of the discharge, 1–2 s from the onset, is usually fast and unreliable for these measurements. There are no marked variations in the relationship of the spike to the slow wave, no fluctuations in the intradischarge frequency and certainly no fragmentations of the ictal discharges (Figure 10.7).
Clinical noteDiagnostic tips
Typical absence seizures of CAE are easy to diagnose and reproduce by hyperventilation. Any child with sudden, brief and frequent cessation of physical and mental activity should be tested clinically for absences. This is easily performed with the hyperventilation test. Ask the child to overbreathe for 3 minutes while counting his/her breaths and holding his/her hands in front. This will evoke an absence in more than 90% of children with childhood absence epilepsy.
It is essential that a video EEG is performed prior to initiating treatment for appropriate confirmation and documentation of the clinical EEG characteristics of the absences. If this is not possible, the clinical manifestations should be documented with home camcorders or modern digital recorders.
Differential Diagnosis
CAE should be the easiest type of epileptic syndrome to diagnose, because seizures have an abrupt onset and termination, high daily frequency and are nearly invariably provoked by hyperventilation. In practical terms, a child with suspected typical absences should be asked to overbreathe for 3 minutes while standing, counting his/her breaths and with the hands extended in front. This will provoke an absence in as many as 90% of those affected.
Important noteClarification
CAE is not synonymous with any type of absence seizures starting in childhood. Therefore, other epilepsy syndromes with absence seizures that may be life-long and have a worse prognosis should be meticulously differentiated from CAE.
Diagnosis should improve with heightened awareness and video EEG studies. Exclusion criteria for CAE are as important as inclusion criteria (Table 10.5). Automatisms have no significance in the diagnosis. They should not be taken as evidence of complex focal seizures (see Table 10.2), which require entirely different management.
Prognosis
Patient note“For myself I shall be well satisfied if I have made it appear probable to you that there does exist a form of epilepsy in children which is distinguishable by its clinical features and in which the prognosis is always good.” Adie (1924)113
Most of the studies on the prognosis of CAE are based on the seizure/symptom itself and not on the relevant syndrome. Absences and therefore their prognosis are syndrome related.15,24,141
Studies on the prognosis of typical absences with onset in childhood (which are not necessarily CAE) 115–117,145,147,151,154,156–169 give an overall rate of remission from 30–80%. Thus, 20–70 % of patients continue having typical absences in adult life and of these 90% will develop other generalised seizures such as GTCS and myoclonic jerks. Absences become less frequent and less severe with age. The diagnostic confusion is further indicated by the finding that some cases develop motor or versive seizures of frontal lobe epilepsies. If CAE is defined by age at onset alone, half of the patients develop GTCS later.168
The factors indicating a worse prognosis in these studies were:
- mental retardation, neurological and EEG background abnormalities (which by definition are against IGE)
- GTCS or other type of seizures preceding or coinciding with the onset of typical absences (an exclusion criterion for CAE)
- photosensitivity (exclusion criterion)
- onset in late childhood or adolescence (are these JAE? or JME?) or very early childhood (are these MAE? or EMA?).
- prominent ictal myoclonic components (are these absences with perioral or single myoclonic jerks?
- poor response to treatment.
Note on the Prognosis of Childhood Absence Epilepsy
In my opinion, based on prospective studies of children fulfilling the strict criteria of CAE (see Table 10.5) and retrospective evaluation of adults with absences15,56,57 the long-term prognosis of CAE is excellent. This implies complete remission of absences 2–6 years after onset and no more than 3% of patients developing infrequent GTCS in adult life. There is indirect, but not definite, evidence for this. In our series of 85 adult patients (> 16 years of age with mean age 32 years) with absences and syndromes of IGEs, none had CAE, though in 37 patients absences had started in childhood, before the age of 10 years.57 Most patients had JME (30 patients), but others had JAE (10 patients), IGE with phantom absences (12 patients), eyelid myoclonia with absences (11 patients), perioral myoclonia with absences (PMA; 7 patients), photosensitive absence syndromes (4 patients), absences with single myoclonic jerks (1 patient), and 10 patients were unclassified. Half of these patients would have been classified as CAE continuing in adult life, if the only criteria for diagnosis were age at onset and frequency of absences.
This optimistic view of mine is also supported by the results of long-term studies in which the remission rate for absences starting in childhood increased with the application of exclusion criteria.114,145,154
Loiseau and associates145,146 studied 53 patients over the age of 20 years at last follow-up. Inclusion criteria were age at onset (3–10 years) of daily and EEG-recorded typical absences as a presenting symptom of normal children, with no history of preceding seizures other than febrile convulsions. Patients with EEG multiple or irregular spike–wave complexes and/or photosensitivity were excluded. Absences persisted in five children (< 10%) and were the only type of seizure in two of them. GTCS occurred in 14 patients (26%), but were isolated or rare in 11 cases. GTCS were more common among patients with onset of absences at 9–10 years and without posterior delta rhythms. Response to treatment varied. Control was achieved in 12 patients within weeks, but in most cases absences persisted for years. In this scholarly study, the prognosis could probably have been improved if video EEG had been used to exclude patients who perhaps had other types of IGE with frequent absences from childhood. In other words, patients who did not remit may have had other epileptic syndromes, particularly
All the above studies involve long-term follow-up of patients with onset of absences in childhood or adolescence. Most were based on clinico-EEG documented ‘absences’, probably with severe impairment of consciousness, without reference to syndromic classification and without the help of video-EEG recordings. Myoclonic jerks or GTCS occurring independently of absences, severity of impairment of consciousness during the absence ictus, syndromic criteria of epilepsies and video-EEG documentation have not been utilised in these studies.
By applying strict diagnostic criteria, an excellent prognosis may be anticipated for CAE. At a time when no anti-absence drugs existed, Adie113 concluded that even if absence seizures in CAE (he called it pyknolepsy) persisted for a long time, they ultimately ceased, never to return. Adie considered only the pure form of CAE with severe pyknoleptic absences while with the advent of EEG this group was broaden to include any type absences starting in childhood.
A good prognosis is consistent with recent findings that absences of CAE, even if they persist for several years, they finally disappear with age in more than 90% of cases.18,154 In a Swedish population-based study, a 91% remission rate was found when patients with absence epilepsy had only absence seizures.162
Remission occurs before the age of 12 years. Less than 10% of patients develop infrequent or solitary GTCS in adolescence or adult life. It is exceptional for patients to continue having absence seizures when adults. Poor social adjustment has been reported in one-third of patients.122,147,154,160 This may be due to frequent absence seizures, particularly if they were not treated early at their onset, the attitudes of school mates and parents, or medication.
Management
Monotherapy either with valproate or ethosuximide controls absences in 80% of patients.4 Another option is lamotrigine monotherapy, though this is less effective and only nearly half of patients become seizure free.4,170,171
If monotherapy fails or unacceptable adverse reactions appear, replacement of one drug with the other is the alternative. Adding small doses of lamotrigine to valproate may be the best combination in resistant cases.
There are anecdotal reports that children may not respond to syrup of valproate despite adequate levels, but seizures stop if this is replaced with tablets of valproate. It is also anecdotal experience that, once seizure cessation has been achieved, valproate may be safely reduced to more moderate doses without relapses.4
Contraindicated drugs, which make seizures worse and may induce status epilepticus are: carbamazepine, gabapentin, oxcarbazepine, phenytoin, phenobarbitone, vigabatrin and tiagabine.
Withdrawing anti-epileptic medication: In the pure form of CAE, drug therapy can be gradually withdrawn (within 3–6 months) after 2–3 years free of seizures.
Clinical noteNote of practical significance
Clinical noteIn evaluating the efficacy of therapy or anticipating AED withdrawal, it should be remembered that:
Juvenile Absence Epilepsy
Clinical noteJAE is an IGE syndrome1,5,15 mainly manifesting with severe typical absence seizures; nearly all patients (80%) also suffer from GTCS and one-fifth from sporadic myoclonic jerks.15,16,24,184,185
Demographic Data
Usual age at onset is 9–13 years (70% of patients), but can range from 5 to 20 years.15,185 Myoclonic jerks and GTCS usually begin 1–10 years after the onset of absences. Rarely, GTCS may precede the onset of absences.15,184 Both sexes are equally affected.
The exact prevalence of JAE is unknown because of variable criteria. In adults over 20 years of age, the prevalence of JAE may be around 2–3% of all epilepsies and around 8–10% of IGEs.57,186
Clinical Manifestations
JAE manifests with severe typical absences.15,184,185 Nearly all patients also develop GTCS and one-fifth also suffer from mild myoclonic jerks.
Typical Absence Seizures
Frequent and severe typical absences are the defining seizure type of JAE.15,184,185 The seizures are similar to those of CAE, though they may be milder. The usual frequency of absences is approximately 1–10/day, but this may be much higher for some patients. The hallmark of the absence is abrupt, brief and severe impairment of consciousness with total or partial unresponsiveness. Mild or inconspicuous impairment of consciousness is not compatible with JAE. The ongoing voluntary activity usually stops at onset, but may be partly restored during the ictus. Automatisms are frequent, usually occurring 6–10 s after the onset of the EEG discharge (Figure 10.9). In JAE, mild myoclonic elements of the eyelids are common during the absence. However, more severe and sustained myoclonic jerks of the facial muscles may indicate other IGE with absences. Severe eyelid or perioral myoclonus, rhythmic limb jerking and single or arrhythmic myoclonic jerks of the head, trunk or limbs during the absence ictus are probably incompatible with JAE.
Considerations on Classification
The 1989 ILAE classification broadly defined JAE as follows:1 “The absences of JAE are the same as in pyknolepsy, but absences with retropulsive movements are less common. Manifestation occurs around puberty. Seizure frequency is lower than in pyknolepsy, with absences occurring less frequently than every day, mostly sporadically. Association with GTCS is frequent, and GTCS precede the absence manifestations more often than in childhood absence epilepsy, often occurring on awakening. Not infrequently, the patients also have myoclonic seizures. Sex distribution is equal. The spike-waves are often >3 Hz. Response to therapy is excellent.”
Age at onset (around puberty) and frequency of seizures (less frequent than CAE) are insufficient criteria for the categorisation of any syndrome.15 Thus, epidemiology, genetics, age at onset, clinical manifestations, other types of seizure, long-term prognosis and treatment may not accurately reflect the syndrome of JAE. Recently, JAE has been redefined based on a cluster of clinical and EEG manifestations studied in video EEG recordings (Table 10.6).15,24
The ILAE Task Force has not yet reached definite conclusions regarding the definition of JAE, though there is a tendency to consider JAE as part of a broader syndrome of IGE in adolescence.5,86
Age at Onset of Absences Does Not Determine Syndromic Classification of JAE
We have studied 71 adults with onset of typical absences after the age of 10 years (median 13 years). All were over 18 years of age and all had experienced typical absences, verified by EEG or video EEG. Two-thirds were women (43 patients). Mean age at last follow-up was 36 years.
In 65 patients (92%), absences continued during adulthood. All but two patients had GTCS with a mean age at onset of 19 years. A total of 33 patients (47%) also had myoclonic jerks with a mean age at onset of 16 years. One-third of patients (26 patients were clinically or EEG photosensitive. In terms of epileptic syndromes, 21 had JME, 13 phantom absences with GTCS, 11 JAE, 5 eyelid myoclonia with absences, 3 perioral myoclonia with absences, 2 purely photosensitive IGE, 2 GTCS on awakening and 1 absences with single myoclonic jerks; 13 patients could not be classified.
Patients with briefer, milder and later onset absence seizures had a worse prognosis.
Duration of the absences varies from 4–30 s, but it is usually long (about 16 s).
Generalised Tonic Clonic Seizures
GTCS are probably unavoidable in untreated patients. They occur in 80% of patients, mainly after awakening, though nocturnal or diurnal GTCS may also be experienced.15,57,168,184,185,187 GTCS are usually infrequent, but may also become severe and intractable.
Myoclonic Jerks
Myoclonic jerks occur in 15–25% of patients and are infrequent, mild and of random distribution. They usually occur in the afternoon when the patient is more tired than in the morning after awakening.15,66
Absence Status Epilepticus
ASE is truly generalised non-convulsive (without any type of jerks) and occurs in one-fifth of patients.19,66
Seizure-Precipitating Factors
Mental and psychological arousal is the main precipitating factor for absences. Conversely, sleep deprivation, fatigue, alcohol, excitement and lights, either alone or more usually in combination, are the main precipitating factors for GTCS.
Some authors have reported that 8% of JAE patients suffer from photosensitivity clinically or on the EEG.184 However, clinical photosensitivity that is consistent with provocation of seizures (absences, GTCS or jerks) may be incompatible with JAE. These patients may have another IGE syndrome.15 EEG photosensitivity (i.e. facilitation of absences by IPS) may not be uncommon.
Aetiology
JAE is determined by genetic factors, but mode of transmission and its relation to other forms of IGE, particularly CAE and JME, has not yet been established. A single Mendelian mode appears to be unlikely.
There is an increased incidence of epileptic disorders in families of patients with JAE and there are reports of monozygotic twins with JAE.24,185,188 A proband with JAE was found in 3 of 37 families selected, because at least three members were affected by IGE in one or more generations.189 However, only one sibling also had JAE, while other members mainly had GTCS.189
In various reports JAE has been linked to chromosome 8,190 21,191 1810 and probably 5.10 Heterogeneity may be common. Autopsy192 and MRI studies54 found microdysgenesis and other cerebral changes in patients with JAE.
Diagnostic Procedures
All tests apart from the EEG are normal.
Electroencephalography
In untreated patients, absences are easily elicited by hyperventilation (Figure 10.9), if not the diagnosis of JAE should be questioned.
The interictal EEG is normal or with mild abnormalities only. Focal epileptiform abnormalities and abortive asymmetrical bursts of spike/multiple spikes are common.
The ictal EEG shows 3–4 Hz GSWD. The frequency at the initial phase of the discharge is usually fast (3–5 Hz). There is a gradual and smooth decline in frequency from the initial to the terminal phase. The discharge is regular, with well-formed spikes and polyspikes, which retain a constant relation with the slow waves (Figure 10.9).
Differential Diagnosis
In general, and particularly in adults, absences are often misdiagnosed as complex focal seizures, though they are easy to differentiate (Table 10.2).56,57
The differentiation of JAE from other IGE with absences may be more difficult without appropriate video EEG evaluation.4,15,24 In children, it is often difficult to distinguish between CAE and JAE, because their features often overlap and manifestations are similar. In JAE, absences start later, usually they are less frequent and impairment of cognition is less severe.24 Automatisms are equally prominent in both. Limb myoclonic jerks (not during the absences) and/or GTCS in the presence of severe absences indicate JAE.
JAE is distinctly different from the Jeavons syndrome of very brief seizures marked with rapid eyelid myoclonia, PMA with rhythmic perioral myoclonia during the absence, and MAE with rhythmic myoclonic jerks during the absence. In adolescents, the differential diagnosis between JAE and JME should not be difficult. Severe absences are the major problem in JAE while myoclonic jerks are the main seizure type in JME (Table 10.7). Absences in JME are mild and often inconspicuous.
Prognosis
JAE is a lifelong disorder, though seizures can be controlled in 70–80% of patients. However, there is a tendency for the absences to become less severe, in terms of impairment of cognition, duration and frequency, with age and particularly after the fourth decade of life.168,184 GTCS are usually infrequent and are often precipitated by sleep deprivation, fatigue and alcohol consumption. Myoclonic jerks, if present, are not troublesome to the patient. However, one-fifth of patients may have frequent and sometimes intractable absences and GTCS, and this figure may be higher if appropriate treatment is not initiated in the early stages of JAE.
Management
In JAE, the consensus is that because of the frequent combination of absences and GTCS, the drug of choice is valproate, which controls all seizures in 70–80% of patients.4 Lamotrigine, though less effective than valproate, probably controls absences and GTCS in half of patients and may be a monotherapy option, particularly in women of childbearing age.193,194
If seizure control with valproate monotherapy is inadequate, add-on treatment with lamotrigine or ethosuximide (if absences persist) may control the situation. If patients are unwilling to receive treatment with valproate, combining lamotrigine with levetiracetam may prove a potent efficacious option based on their different seizure efficacy and mode of action. In one study of 5 patients with JAE all became seizure free with levetiracetam alone or as add-on therapy.183
Control of absences is usually associated with good control of GTCS (90% of cases).
Patients should be warned about the factors precipitating GTCS.
Treatment may be lifelong, because attempts to withdraw medication nearly invariably leads to relapses even after many years free of seizures.
Juvenile Myoclonic Epilepsy Janz Syndrome
Clinical noteJuvenile myoclonic epilepsy (JME) is one of the most important syndromes of IGE and is genetically determined.1,60,195–201
Demographic Data
The triad of absences, jerks and GTCS shows a characteristic age-related onset (Figure 10.10). Absences, when a feature, begin between the ages of 5 and 16 years. Myoclonic jerks follow between 1 and 9 years later, usually around the age of 14–15 years. GTCS usually appear a few months after, occasionally earlier, the myoclonic jerks. Exceptionally, JME may start or become clinically identifiable in adult life as ‘adult myoclonic epilepsy’.202 Both sexes are equally affected. The reported prevalence of JME in hospital-based clinics has increased since the syndrome was first described, from 2.7%,201 5.7%,203,204 8.7%,205 to 10.2%.60 In community-based studies,206,207 the reported prevalence is lower and probably reflects underdiagnosis, which may improve with heightened medical awareness.
Clinical Manifestations
Juvenile myoclonic epilepsy is characterised by the triad of:
- Myoclonic jerks on awakening (all patients)
- Generalised tonic clonic seizures (> 90% of patients)
- Typical absences (about one-third of patients).
Seizures have an age-related onset.
Myoclonic status epilepticus is common.
Patient noteLots of blanks and jerks; then I had a grand mal...I usually have fits when rushing after getting up; usually does not happen later in the day198
Classification and Definition of Juvenile Myoclonic Epilepsy
The 1989 ILAE Commission classified JME as a distinct syndrome of IGE and defined it as follows:1 “Juvenile myoclonic epilepsy (impulsive petit mal) appears around puberty and is characterised by seizures with bilateral, single or repetitive, arrhythmic, irregular myoclonic jerks, predominantly in the arms. Jerks may cause some patients to fall suddenly. No disturbance of consciousness is noticeable. The disorder may be inherited, and sex distribution is equal. Often, there are GTCS and, less often, infrequent absences. The seizures usually occur shortly after awakening and are often precipitated by sleep deprivation. Interictal and ictal EEG have rapid, generalized, often irregular spike–waves and polyspike–waves; there is no close phase correlation between EEG spikes and jerks. Frequently, the patients are photosensitive. Response to appropriate drugs is good.”
The ILAE Task Force has not yet reached definite conclusions regarding the definition of JME though there is a tendency to consider JME as part of a broader syndrome of IGE in adolescence.5,86
Myoclonic Jerks
Myoclonic jerks occurring after awakening are the most prominent and characteristic seizure type (Figures 10.3 and 10.11). They are shock-like, irregular and arrhythmic, clonic movements of proximal and distal muscles mainly of the upper extremities. They are often inconspicuous, restricted to the fingers, making the patient prone to drop things or look clumsy. They may be violent enough to cause falls. One-fifth of patients describe their jerks as unilateral, but video EEG shows that the jerks affect both sides (Figure 10.3).199,208
Some patients (< 10%) with mild forms of JME have myoclonic jerks only without developing GTCS.60,209
Typical Absence Seizures
One-third of patients have typical absences, which are brief with subtle impairment of consciousness. They are different from the absence seizures of childhood or JAE.24,25,60
Absences appearing before the age of 10 years may be more severe. They become less frequent and less severe with age.24,25,60
One-tenth of patients, do not perceive absences despite GSWD lasting more than 3 s.25,60 However, the GSWD seen on video EEG with breath counting during hyperventilation often manifest with mild impairment of cognition, eyelid flickering or both (Figures 10.12, and 10.13).4
Generalised Tonic Clonic Seizures
GTCS usually follow the onset of myoclonic jerks. Myoclonic jerks, which usually occur in clusters and often with an accelerating frequency and severity, may precede GTCS, a so-called clonic-tonic-clonic generalised seizure.199
Status Epilepticus
Myoclonic status epilepticus is probably more common than appreciated.60,210 It almost invariably starts on awakening and is associated with precipitating factors, such as sleep deprivation or missing medication. Consciousness may not be impaired, though in some patients absences are often interspersed with myoclonic jerks (myoclonic-absence status epilepticus) ( Figure 6.10 ).
Pure ASE with impairment of consciousness only is exceptional.19 Convulsive generalised tonic clonic status epilepticus is relatively rare.
Circadian Distribution
Seizures, principally myoclonic jerks, occurring within 30 min to 1 hour of awakening are characteristic of JME. Myoclonic jerks rarely occur at other times unless the patient is tired.
GTCS occur mainly on awakening, but may also be purely nocturnal or random. Absence seizures occur during any time of the day while the patient is awake.
Seizure-Precipitating Factors
Clinical noteSleep deprivation and fatigue, particularly after excessive alcohol intake, are the most powerful precipitants of jerks and GTCS in JME.
Sleep deprivation means a late night followed by a brief sleep suddenly interrupted by either compulsory early awakening in order to go to work or on a trip. An unscheduled telephone call early next morning may frequently have disastrous effects.
Photosensitivity is confirmed by the EEG in one-third of patients, but probably less than one-tenth of patients with JME have clinical seizures induced by photic stimulation in daily life (Figure 10.13).
Other common and prominent precipitants of seizures are mental stress, emotion and, in particular, excitement, concentration, mental and psychological arousal, failed expectations and frustration.60,211,212 Video games may precipitate seizures because of the photic/pattern effect, mental and psychological excitement, or both.60
Reading,213 writing214 and proprioceptive stimuli56 are exceptional precipitants of seizures in patients with typical features of JME.
Women often have their seizures premenstrually, particularly if other precipitating factors are also present.60
Hyperventilation is an effective precipitant of EEG generalised discharges, which may often be the only abnormality in a routine EEG.60
Frequently, patients with JME have seizures when these precipitating factors cluster together, such as during exams, trips or vacations.
Aetiology
JME is genetically determined.61,199,215,216 Between 50% and 60% of families of probands with JME report seizures in first- or second-degree relatives.216,217 Inheritance is probably complex.9,203,218,219 Families with autosomal recessive215 or dominant215,218 Mendelian inheritance have been described.
Susceptibility loci for JME have been found in chromosome 6p11–12 (EJM1)9 and 15q14 (EJM2).220,221 Genes C6orf33 222 or BRD2 (RING3)223 in the EJM1 region have been recently identified.
An association reported between JME and an HLA-DR allele224,225 has not been replicated.226
Genetic heterogeneity of JME is a possible explanation for such discordant observations.
Families with phenotypic overlap between JME and idiopathic photosensitive occipital epilepsy have been described.227
Magnetic resonance spectroscopy studies found reductions of N-acetyl aspartate (NAA) in the prefrontal and frontal brain regions of JME patients, but not in other forms of IGE.42 JME patients with reduced frontal NAA concentrations showed poor performance on neuropsychological tests of executive functions, but not JME patients with normal frontal NAA levels. Frontal glutamate plus glutamine concentrations were elevated, which suggests possible increased neuronal excitability.42
Diagnostic Procedures
All tests other than the EEG are normal. Using new MRI technologies, abnormalities of cerebral structure in some patients with JME involving mesiofrontal cortical structures have been reported.42,228
Electroencephalography25,32,60,198,229
Interictal EEG
Clinical noteA normal EEG in a patient suspected of having JME should prompt an EEG on sleep and awakening.
The EEG in untreated patients is usually abnormal, with generalised discharges of an irregular mixture of 3–6 Hz spike/polyspikes-slow waves, with intradischarge fragmentations and unstable intradischarge frequency (Figures 2.1, 10.12 and 10.13). One-third of patients have photoparoxysmal responses. Focal abnormalities are recorded in approximately one-third of patients.32 These consist of focal single spikes, spike–wave complexes or focal slow waves.
Ictal EEG
The typical EEG discharge of a myoclonic jerk is a generalised burst of multiple spikes of 0.5–2 s duration (Figures 10.3, and 10.11).
The ictal discharges of absences in JME are distinctly different from those in CAE and JAE.24,25 They consist of single, double, treble or multiple spikes usually preceding or superimposed on a slow wave. Multiple spikes consist of up to 8–10 spikes with a characteristic ‘worm-like’ or “compressed capital W” appearance (‘Ws’). The number and amplitude of the spikes shows considerable inter- and intradischarge variation. The intradischarge frequency of the GSWD varies from 2–10 Hz and is mainly 3–5 Hz. The frequency is often higher in the first second of onset. Fragmentations of the discharge are common and characteristic. Ws and fragmentation of discharges are observed in all patients, but vary quantitatively between patients and between discharges (Figures 2.1, 10.12, and 10.13).
Brief GSWD are far more common than long ones and most of them last for 1–4 s.
Photoparoxysmal discharges are evoked in 27% of patients (Figure 10.12).
Differential Diagnosis
JME is a typical example of a frequently misdiagnosed common epileptic syndrome resulting in avoidable morbidity.205,230 Failure to diagnose JME is a serious medical error, because JME defies all aspects of general advice regarding ‘epilepsy’. Diagnosis should improve with heightened medical awareness. Physicians should be ever alert to the possibility of JME, which is common.
Important noteHigh rate of misdiagnosis
The rate of misdiagnosis of JME is as high as 90%.205,230 Factors responsible include lack of familiarity with JME, failure to elicit a history of myoclonic jerks, misinterpretation of absences as complex focal seizures, misinterpretation of jerks as focal motor seizures, and a high prevalence of focal EEG abnormalities.
JME is easy to diagnose because of the characteristic clustering of myoclonic and other generalised seizures of IGEs, the circadian distribution, precipitating factors and EEG manifestations. Patients are otherwise normal and there is no mental or physical deterioration if properly diagnosed and treated early.
Of the other IGEs, JAE is more difficult to differentiate, because this syndrome may also manifest with similar clinical and EEG manifestations (Table 10.7). The main differentiating factor is that absences with severe impairment of consciousness, not the myoclonic jerks, are the main seizure type in JAE. Myoclonic jerks, if they occur, are mild and random often lacking the circadian distribution of JME.
Another formidable situation is when JME starts with absences in childhood prior to the development of myoclonic jerks. There are no prospective video EEG studies of these patients. Examining the EEG and clinical manifestations of these patients retrospectively, I am of the opinion that their absences are distinct from CAE or JAE in that they are usually shorter, milder and the ictal EEG often contains multiple spike–slow waves. Certainly, this situation is not CAE progressing to JME as some authors have reported:140 it is JME starting with absences prior to the development of myoclonic jerks.
Clinical noteDiagnostic tips for juvenile myoclonic epilepsy
GTCS, usually preceded by myoclonic jerks, are nearly pathognomonic of JME if they occur in the morning after:
Prognosis
All seizures are probably lifelong, though improve after the fourth decade of life. JME may vary in severity from mild myoclonic jerks to frequent and severe falls and GTCS if not appropriately diagnosed and treated.
Seizures are generally well controlled with appropriate medication in up to 90% of patients.60,198,199,231 Patients with all three types of seizures are more likely to be resistant to treatment.232
Accepted Practice for the Management of ‘Epilepsy' Is Often Inappropriate in JME
- “Treatment of epilepsy should not start after a first GTCS.” Withholding treatment in JME is an erroneous medical decision. A JME patient may have his/her first GTCS long after many myoclonic jerks on awakening, absences or both.
- “Treatment for epilepsy may be withdrawn after 2–3 years freedom of seizures.” Withdrawing treatment in JME is often an erroneous medical decision. More than 80% of patients will relapse.
Management
Lifestyle and Avoidance of Seizure Precipitants
Advice regarding circadian distribution, lifestyle and seizure precipitants may be as important as drug treatment. Avoidance of precipitating factors and adherence to long-term medication is essential to avoid seizures.
Some patients experience GTCS or myoclonic jerks only after encountering precipitating factors. Patients may have myoclonic jerks despite treatment only after excessively violating these factors and others may have seizures exclusively after awakening that may not affect their daily duties.
Advice on the risk of sleep deprivation and alcohol is mandatory; avoidance of alcohol indulgence and compensating for sleep deprivation is essential.
A young person can not probably do without late night entertainment. It is natural and understandable. However, sleeping longer the next morning may well compensate for staying up late at night. A glass of wine is acceptable providing that this is not followed by another five or more. Patients with JME are often intelligent. They understand these simple rules. There are some restrictions, but they do not deprive patients from their rights in life. Sometimes, they experiment against these rules. They find what is right or wrong for them by trial and error.
Anti-Epileptic Medication
The current state of knowledge of the drug treatment of JME is mainly based on clinical experience of prospective and retrospective studies with little evidence from randomised control trials (RCTs). There are no head-to-head comparisons between old and new AEDs.233 Thus, the assessment of new AED monotherapy or polytherapy in JME and, indeed all IGEs, is notoriously problematic. My evaluation of the AED treatment of JME is based on a thorough review of the literature including abstracts, exchanging views with eminent colleagues in the field and my own clinical research over the last 25 years. The following are the most pragmatic recommendations that I could reach. Briefly these are as follows:
Monotherapy: Valproate is unquestionably the most effective AED in JME but is humbled by serious adverse reactions in women. Other options include levetiracetam and lamotrigine. When cost is of concern, phenobarbitone is effective in around 60% of patients. In mild JME with myoclonic jerks only, clonazepam alone may be recommended.
Polytherapy: Valproate with small doses of clonazepam or small doses of lamotrigine is the most effective combination. When valproate is undesirable, combining levetiracetam with lamotrigine or lamotrigine with clonazepam (to counteract the pro-myoclonic effect of lamotrigine) may be the best option. Persisting absences are practically never a problem so ethosuximide may not have a place in the polytherapy of JME. Combination regimens with levetiracetam have the advantage that other comedications may be gradually withdrawn without seizure deterioration.234–236
Contraindicated or ineffective AEDs should be excluded and these are: carbamazepine, gabapentin, oxcarbazepine, phenytoin, tiagabine and vigabatrin.
Old Anti-Epileptic Drugs in JME
The indications and contraindications of old AEDs in JME have been established through numerous prospective and retrospective studies, and clinical experience over many years of use.
Valproate has been recognised as the most effective drug with full control of seizures in about 80% of patients with JME.4,237 Valproate monotherapy controls absence seizures in around 75%, GTCS in 70% and myoclonic jerks in 75%.4 The valproate dosage depends on the severity of JME. In a RCT, no difference in control of various types of JME could be demonstrated between 1000 mg and 2000 mg of valproate. 238
The usual dose is 500 mg twice daily. Resistant cases require higher doses of up to 1500 mg twice daily. Mild cases may be well controlled with smaller doses of 300 mg twice daily. The best way to find the most appropriate dose in individual patients is to start with a low dose and increase in small increments until all seizures stop. Similarly, any attempt at withdrawal should be tested with small decrements of approximately 200 mg daily every 3 months. Relapses may be heralded by myoclonic jerks indicating the need to reinstate drug treatment.
The major problem is that valproate is undesirable in women because of its teratogenic effects239–244 and its tendency to cause weight gain and polycystic ovary syndrome.245 Women exposed to valproate during pregnancy have a relatively small, but statistically significant increased risk of having a child with a major malformation (spina bifida, cardiac and kidney abnormalities, extra fingers and clubfoot) compared with mothers not exposed to AEDs. This risk with valproate was found to be 5% against a background risk of 1.62%243 or 5.9% versus 2.4%.244
Currently, media, internet and various other campaigns make it nearly impossible to prescribe valproate to women. There are increasing numbers of litigations against physicians and health authorities by parents of children with foetal valproate syndrome (even if the risks were appropriately explained to them) on a scale characterised as ‘bigger than thalidomide’.
Phenobarbitone is extensively used as a very effective form of monotherapy in JME by European neurologists.61,201,216 It is effective in controlling GTCS and myoclonic jerks, but may exacerbate absences. It is the AED of choice when cost is of concern. One dose of 100–200 mg of phenobarbitone prior to going to sleep is often sufficient. Some patients may be controlled with 60–90 mg nocte.
Teratogenicity and AEDs
The FDA categorization of drug risks to the foetus runs from “Category A” (safest) to “Category X” (known danger—do not use).
Valproate is a category D drug that is: there is positive evidence of human foetal risk, but the benefits from use in pregnant women may be acceptable despite the risk (e.g., if the drug is needed in a life-threatening situation or for a serious disease for which safer drugs cannot be used or are ineffective).
All newer AEDs are classified as Category C that is: either studies in animals have revealed adverse effects on the foetus (teratogenic or embryocidal or other) and there are no controlled studies in women, or studies in women and animals are not available. Drugs should be given only if the potential benefit justifies the potential risk to the foetus.
On preliminary basis from humans, gabapentin,248 lamotrigine249 and levetiracetam250 may be relatively safe but certainly their use in women of child-bearing age should be extremely cautious and their use should be based on the risk-benefit ratio.
See also Chapter 14, on pregnancy registries as a new method for assessing the foetal risks from exposures in pregnancy.251,252 Overall, the results are generally encouraging, in that of the total groups of all pregnancies, over 95% of offspring do not have a major congenital malformation.
Clonazepam is predominantly used as adjunctive treatment. It is one of the most effective anti-myoclonic drugs, but clonazepam alone may not suppress and may even precipitate GTCS. 253 Furthermore, clonazepam may deprive patients of the warning of an impending GTCS provided by the myoclonic jerks.60,253–255
Clonazepam should be given in small add-on doses (0.5–2 mg at night) when myoclonic jerks persist and are troublesome despite adequate monotherapy with another broad-spectrum AED. Monotherapy with clonazepam may be considered for mild cases of JME with myoclonic jerks only.
Clobazam has not been evaluated in JME, but it may be effective in some cases.233,256 Clobazam is a very useful AED in focal epilepsies as I described on pages 432. However, in IGEs, I found clobazam to be far inferior to clonazepam in controlling myoclonic jerks, and to valproate and ethosuximide in controlling absences.
Acetazolamide has been used for treating GTCS in cases resistant to conventional treatment, though its use may induce nephrolithiasis.257
New Anti-Epileptic Drugs in JME
The adverse effects of valproate and its lack of efficacy in 20% of patients with JME have prompted the search for alternatives. Of the new AEDs, only lamotrigine,4,193,258,259 levetiracetam,234,260–265 topiramate266,267 and possibly zonisamide268 appear to be therapeutic agents in JME. Levetiracetam, because of its efficacy in all seizure types234,260–265,269 and safer adverse reactions profile,270,271 appears to be the most promising substitute for valproate (Table 10.8).
Levetiracetam
Levetiracetam fulfils all expectations as probably the best new AED in the treatment of JME and it is the likely candidate to replace valproate in the treatment of this disorder, because of its high and sustained efficacy, fast action, good safety profile and lack of clinically meaningful interactions with other drugs.
The results of treatment with levetiracetam in JME are very impressive.234–236,272,273 In three independent studies, 62%,234 67%235 and 63%236 of patients with intractable JME became seizure free with levetiracetam monotherapy or polytherapy.
In a USA study, levetiracetam monotherapy was assessed in 24 patients with JME and GTCS.235
Clinical note“Sixteen (66.7%) had been free of GTCS; 3 of them had a single convulsive seizure after either stopping levetiracetam for 24 hours or reducing the dose to 500 mg per day but subsequently have remained free of convulsive seizures. Myoclonic seizures were effectively controlled in 22 of 24 patients.”235
In a UK case-note review of more than 30 patients with resistant JME, 62% became seizure free with levetiracetam.234
In a Dutch study of 16 intractable JME patients, levetiracetam had excellent effects achieving freedom from seizures in 10 (63%) and a greater than 50% seizure reduction in 2 patients (12%). No clinically meaningful change was seen in two patients (12%); one patient experienced an increase in myoclonic jerks (6%).236
A controlled study compared 12-week baseline and levetiracetam treatment periods in 55 patients with idiopathic generalised seizures (myoclonic, GTCS and absence seizures), in whom other anticonvulsants had failed. Three-quarters (76%) of patients had a greater than 50% seizure reduction with levetiracetam therapy and 40% became seizure free; 15% discontinued levetiracetam because of adverse events, mostly sedation.264
The high efficacy of levetiracetam in JME is consistent with its effectiveness against all types of epileptic seizure, epileptic and non-epileptic myoclonus and photosensitivity.
Levetiracetam has a potent antimyoclonic effect,254,255,274,275 even in severe myoclonic epilepsies, such as post-anoxic myoclonus and Unverricht-Lundborg disease,276 as well as non-epileptic myoclonus.262,277–279 This may be explained by its structural similarity to piracetam.
The prevalence of photosensitivity in IGE is high,280,281 with 30% of patients having EEG photoparoxysmal responses.60 Levetiracetam is the only new AED with well-established efficacy in EEG and clinical photosensitivity.263,282,283 Levetiracetam reduces or eliminates both the photoparoxysmal responses and the myoclonic jerks elicited by IPS.282
Clinical note“Long-term use of levetiracetam in some visually sensitive patients has shown a remarkably good suppressive effect for > 4 years; discontinuation of the drug before becoming pregnant resulted in a return of myoclonic jerks and an increase of IPS sensitivity.”283
Levetiracetam monotherapy was also studied in 18 patients aged 6–22 years with well-documented IGEs.261 The majority of patients (14) had the typical JME seizure triad (absences, myoclonic jerks and GTCS) and most were photosensitive. Eight patients (44.5%), five of whom were photosensitive, became clinically seizure free and the EEG normalised in seven patients. All but one of the other six patients improved with levetiracetam alone or in combination with valproate. One patient with absences and myoclonic jerks worsened with levetiracetam, but responded well to valproate.261
In a case series of highly refractory IGE (absence seizures, myoclonic jerks and GTCS), three patients in whom treatment with at least three AEDs had failed became seizure free when treated with levetiracetam monotherapy.260 Further evidence for the efficacy of levetiracetam in IGEs is that it reduces the density and duration of GSWD, which is documented with continuous EEG monitoring.265
In view of some conflicting evidence in regard to behavioural adverse reactions particularly in children treated with levetiracetam 284–287 it is advisable to apply the rule ‘start slow and go slow’. Thus, the starting dose of levetiracetam should be lower (250 mg) and titration should be slower (250 mg daily per week) than recommended by the manufacturers. Control of seizures is usually achieved at a maintenance dose of 1000–1500 mg in two divided doses daily. Higher doses may be unnecessary.
Lamotrigine
Lamotrigine is a very useful AED in IGEs, because of its efficacy in controlling GTCS and absence seizures. However, lamotrigine is often a pro-myoclonic AED that exaggerates myoclonic jerks in around 50% of patients. Cases with severe deterioration of JME are well reported.288,289 In view of the fact that JME is predominantly a myoclonic epileptic syndrome, it is unlikely that lamotrigine will be the successor to valproate, because of its weaker efficacy and the deterioration of myoclonic syndromes (Table 10.8). Lamotrigine monotherapy in JME is debatable and probably not recommended.4,290–292 Lamotrigine was the first of the new AEDs that appeared to be effective in JME.4,258,259 However, in most reports, the efficacy of lamotrigine in JME was evaluated not as monotherapy, but in polytherapy with valproate.4,258,259 Withdrawal of valproate often resulted in relapses.
Lamotrigine added to valproate is very effective in resistant cases,4,258 because of beneficial pharmacokinetic interactions.4,293 I should emphasise that small doses of lamotrigine (25–50 mg) added to adequate dosage of valproate are sufficient. This is clinically important for two reasons. Firstly, the combination of these drugs has an additive effect on adverse reactions associated with one or both of them.294 Secondly, on anecdotal evidence the beneficial pharmacodynamic interaction with valproate may be lost with increasing dosage of lamotrigine.295
With regard to photosensitivity, lamotrigine also appears to be effective;193 suppression of EEG photoparoxysmal discharges with lamotrigine has been reported in five patients, of whom four were also taking valproate.296
Small doses of lamotrigine added to valproate are very effective in resistant cases,4,258 because of pharmacokinetic interactions.4,293 Withdrawal of valproate often results in relapses even when lamotrigine dosage is increased.
In a recent report of 962 patients with various IGE syndromes (including JME), a 1-year period of remission was achieved mainly with valproate monotherapy (52.1%) with lower rates for lamotrigine (16.7%) and topiramate (34.6%).297 The combination of valproate and lamotrigine achieved a remission rate of 15.3%.297 In another report of JME patients who had also received phenytoin and carbamazepine often as initial treatment, poor outcome was more likely with lamotrigine than valproate; worsening of seizures occurred in 6% of patients with lamotrigine.291,292
Topiramate
Topiramate266,267,291,298 is another broad-spectrum AED that is effective in primarily GTCS,299–302 but with a weak anti-absence303 and anti-myoclonic action. Irrespective of efficacy, the major problem with topiramate is the high incidence of adverse reactions, some of which are very serious and include significant cognitive disturbances.304,305 Also, topiramate imposes severe drug–drug interactions, including with hormonal contraception.306
In IGE, including JME, topiramate appears to be much less effective than valproate, but more effective than lamotrigine;297 a 1-year period of remission was achieved in 34.6% of patients treated with topiramate compared with 52.1% with valproate.297 In one study, a poor outcome was less likely with valproate than topiramate, which worsened seizures in 5% of patients. Furthermore, tolerability of topiramate was low and worse than valproate.291
Topiramate polytherapy may be a remote option for the few patients in whom treatment with valproate, levetiracetam and lamotrigine treatment alone or in combination fails.291 Topiramate, which is less effective and has a worse safety profile than valproate, is unlikely to be useful as monotherapy in JME.
Zonisamide 307–309 is also a broad-spectrum AED, but its role in JME is largely unknown and probably weak.310
AEDs Contraindicated in JME
Carbamazepine is contraindicated in JME as 68% of patients suffer an increase in seizures,195,196,200,311,312 though it may improve control of GTCS in a few patients.313
Oxcarbazepine like carbamazepine is also contraindicated in JME.314
Phenytoin is often ineffective and may worsen seizures of JME. 312 In the original studies of Janz, it was significantly inferior to phenobarbitone.201
Gabapentin, tiagabine and vigabatrin are detailed on pages 337.
Prevention of GTCS and Termination of Myoclonic-Absence Status Epilepticus
It is important to remember that patients with JME often experience myoclonic jerks or myoclonic-absence status epilepticus long before terminating to a GTCS, which can be prevented by home administration of an appropriate benzodiazepine preparation. Rectal absorption of liquid diazepam is very rapid, reaches the brain within minutes and has a near-intravenous efficacy. Rectal tubes containing liquid diazepam are the most widely used formulation (Stesolid). Diazepam rectal gel is now available (Diastad). Buccal or nasal application of midazolam is another alternative.
Duration of AED Treatment and Withdrawal of Medication in JME
Lifelong AED treatment is usually considered necessary in patients with JME. Withdrawal of medication results in relapses, even in patients who have been seizure free for many years with an appropriate AED.60 In mild forms of JME, it may be safe to reduce the dose of medication slowly over months or years, especially after the fourth decade of life. Persistence or recrudescence of myoclonic jerks necessitates continuation of medication.
Addendum
A just recently completed control study confirmed the efficacy and tolerability of levetiracetam (3000 mg/day) when given as adjunctive treatment in adolescents (□ 12 years) and adults (□ 65 years) suffering from idiopathic generalized epilepsy with myoclonic seizures.395 This was a double-blind, multicenter, randomized, placebo-controlled trial in which 122 patients were randomized. To be eligible, patients had to experience at least 8 days with myoclonic seizures during the 8-week baseline period and had to be treated with one concomitant AED. Patients were uptitrated over 4 weeks and treated at a stable dose over 12 weeks (evaluation period). Seizure activity was recorded by the patients on a seizure diary and evaluated by the investigators using the ILAE classification. Tolerability was assessed using adverse event reporting, ECG, standard clinical examinations and laboratory analyses including plasma concentrations of AEDs and levetiracetam. Entry into a long-term follow-up trial was offered to patients who had benefited from the treatment. The primary efficacy endpoint was the responder rate (□ 50% reduction in days with myoclonic seizures during the treatment period versus baseline).
One hundred and twenty one patients were included in the intention to treat analysis (placebo: n=60; levetiracetam: n=61). Responder rate was 58.3% under levetiracetam and 23.3% under placebo (p=0.0002). The corresponding odds ratio [95% CI] was 4.77 [2.12; 10.77]. Thirteen patients on levetiracetam versus 2 patients on placebo were seizure-free during the evaluation period. The most common adverse effect was headache (23.3% in placebo; 21.6% in levetiracetam). One placebo and 2 levetiracetam patients prematurely discontinued due to adverse reactions prior to the end of the evaluation period.395
Based on these results the authors concluded that “Levetiracetam proved to be highly efficacious in the treatment of refractory patients with idiopathic generalized epilepsy experiencing myoclonic seizures. Levetiracetam’s outstanding tolerability profile was also confirmed”.395
Idiopathic Generalised Epilepsy with Generalised Tonic Clonic Seizures Only
Clinical note‘Idiopathic generalised epilepsy with GTCS only’ is a newly proposed IGE syndrome of undetermined definition and boundaries.5 All patients suffer from primarily GTCS occurring at any time in wakefulness, sleep or awakening. Thus, this syndrome is to include ‘epilepsy with GTCS on awakening’ (EGTCSA) which has been extensively studied by Janz. 315–317
Demographic Data
Age at onset varies from 6 to 47 years with a peak at 16–17 years; 80% of patients have their first GTCS in the second decade of life. Men (55%) predominate slightly, probably because of differences in alcohol exposure and sleep habits to women.
The prevalence of ‘IGE with GTCS only’ is unknown. In my experience, it is very rare if strict criteria are applied (GTCS only). Of 1000 patients with one or more afebrile seizures, 356 patients (35.6%) had various syndromes of IGEs, but only 9 patients (0.9%) had GTCS only, though this was often the reason for referral. The low yield of ‘IGE with GTCS only’ in this sample reflects both the fact that we methodically question patients and witnesses regarding the occurrence of minor seizures and that we make long video EEG recordings, including video EEG during sleep and on awakening. As a result of this approach, for example, we found that 14 patients mainly referred for late onset GTCS had ‘IGE with phantom absences’.58,319
In the experience of other authors, the prevalence of ‘IGE with GTCS only’ varies from 10–15%314,320–322 to as high as 62%14 among IGEs. Of 253 patients with IGEs, 30 (12%) had EGTCSA and 39 (15%) had “a mild form of IGE characterised by infrequent GTCS and generalised interictal EEG discharges of spike wave”.320 Of 1033 patients with IGEs, 138 (13%) had GTCS only with onset between 3 and 18 years of age.321 Of 101 patients with IGE beginning in adolescence, 10 had GTCS only, but neither on awakening nor in the evening period of relaxation.322
The reported prevalence of epilepsy with GTCS on awakening also varies from 0%207 to as high as 17%315 in patients with epileptic seizures.
ILAE Definition and Considerations on Classification
The 1989 ILAE Commission1 recognised a syndrome of ‘epilepsy with GTCS on awakening’ among IGEs and defined it as follows:
“Epilepsy with GTCS on awakening is a syndrome with onset occurring mostly in the second decade of life. The GTCS occur exclusively or predominantly (> 90% of the time) shortly after awakening regardless of the time of day or in a second seizure peak in the evening period of relaxation. If other seizures occur, they are mostly absence or myoclonic, as in JME. Seizures may be precipitated by sleep deprivation and other external factors. Genetic predisposition is relatively frequent. The EEG shows one of the patterns of idiopathic generalised epilepsy. There is a significant correlation with photosensitivity.”1
The new ILAE diagnostic scheme 5,86 broadens this to a syndrome of ‘IGE-GTCS only’ in which ‘epilepsy with GTCS on awakening (EGTCSA)’ is incorporated though there is some evidence that this is genetically different.318 ‘IGE with GTCS only’ has not been defined by the ILAE Task Force.5 Its name implies that it includes only those patients with GTCS alone (i.e. without absences and/or jerks) and that these may occur at any time. However, it is more likely that it is a broader category (rather than a syndrome) of ‘IGE with predominantly GTCS’ (which also includes patients with mild absences, myoclonic jerks or both). If this is so, it is undetermined what proportion of patients also has other generalised seizures (jerks or absences) and there may be significant overlap with other syndromes of IGEs.
GTCS commonly feature in IGEs and occur predominantly on awakening.319 GTCS are the most severe form of epileptic seizures, while absences and myoclonic jerks may be mild and sometimes inconspicuous to the patient and imperceptible to the observer.58 They are often detected only by taking a meticulous history or video EEG. A patient with a first GTCS has often suffered from minor seizures (absences, myoclonic jerks or both), sometimes for many years prior to a GTCS.
Clinical Manifestations
GTCS are the defining clinical manifestations. 63,316,317,319
In EGTCSA, GTCS occur within 1–2 hours of awakening from either nocturnal or diurnal sleep. The seizure may occur while the patient is still in bed, having his breakfast or on arriving at work. Seizures may also occur during relaxation or leisure.
However, GTCS in the syndrome of ‘IGE with GTCS only’ may also occur at any other time during sleep, wakefulness or awakening. Overall, GTCS are reported to occur on awakening in 17–53% of patients, diffusely while awake in 23–36%, during sleep in 27–44% or randomly in 13–26%.63
With age, GTCS tend to increase in frequency and become more unpredictable. Janz described patients with EGTCSA as unreliable, unstable and prone to neglect.315,316 Their sleep patterns are particularly unstable and modifiable by external factors (i.e. AEDs), and patients may suffer from chronic sleep deficit.63,316,317
Precipitating Factors
Sleep deprivation, fatigue and excessive alcohol consumption are the main precipitants of seizures. Shift work, changes in sleep habits, particularly during holidays and celebrations, predispose to GTCS on awakening. A few patients may be clinically photosensitive.
Aetiology
There is a high incidence of epileptic disorders in other family members.63,316 Recently, a link to the EJM-1 locus has been reported in EGTCSA while no such link was found in adolescent-onset idiopathic epilepsy with GTCS at any time while awake.318 Microdysgenesis has been reported in pathological specimens of EGTCSA.323
Diagnostic Procedures
By definition, all tests apart from the EEG are normal.
Electroencephalography
The EEG shows generalised discharges of 3–4 Hz spike/multiple spike–wave complexes (GSWD) in 50% of patients with pure EGTCSA (Figure 10.15) and 70% of those with additional absences or myoclonic jerks.
A normal routine EEG should prompt a video EEG during sleep and on awakening. Myoclonic jerks or, more frequently, brief absences will often be revealed.
Focal EEG abnormalities occur in one-third of patients but these are exceptional in the absence of GSWD. Photoparoxysmal responses are reported in 17% of females and 9% of males with EGTCSA.63
Differential Diagnosis
The differential diagnosis is mainly from other IGEs that share with EGTCSA the same propensity to seizures after awakening and the same precipitating factors. JME, JAE and eyelid myoclonia with absences are examples of IGE syndromes that may cause diagnostic difficulties (see Chapter 14). Symptomatic and focal epileptic seizures with secondary generalisation may also occur, predominantly on awakening.
Prognosis
As in all other types of IGEs with onset in the mid-teens, EGTCSA is probably a lifelong disease with a high incidence of relapse (83%) on withdrawal of treatment.316 Characteristically, the intervals between seizures become shorter with time, the precipitating factors less obvious and GTCS may become more random (diurnal and nocturnal), either as a result of the evolution of the disease or drug-induced modifications.63,316,317
Management
Patients should be warned of the common seizure precipitants, namely sleep deprivation with early awakening and alcohol consumption, and when possible should avoid occupational night shifts. Patients, after adjusting their lifestyles, may become seizure free.
Treatment is with AEDs that control primarily GTCS, such as valproate, phenobarbitone, lamotrigine, levetiracetam and topiramate. In the pure forms of ‘GTCS only’, carbamazepine, oxcarbazepine and phenytoin may be used, but require exclusion of other types of seizures that may be exacerbated by these AEDS.
Primarily GTCS in AED RCT
Results of RCTs of AEDs in primarily GTCS present significant difficulties in interpretation:
- secondarily GTCS may contaminate the selected populations for the active AED and the comparator agent (established AED or placebo)
- patients with primarily GTCS and absences or myoclonic jerks are often included though these seizures respond differently to AEDs
- inappropriate AEDs for IGEs, such as carbamazepine, have been used in one-quarter of IGE patients as a comparator agent.
Important noteThe results of such studies are of uncertain value and should be interpreted with caution.
IGE with Absences of Early Childhood
Clinical noteTypical absence seizures starting in early childhood (between a few months and 4 years of age)187,324–327 are not a specific expression of a distinct syndrome. This may be the first manifestation of various syndromes of IGEs with absences or more severe forms of generalised epilepsies. By excluding all these conditions, it is realistic to accept that there is a syndrome of IGE that starts in early childhood primarily manifesting with absences, often combined with GTCS and possibly with myoclonic jerks.
Doose,325,326 having studied 140 cases with onset of absences in early childhood, rightly concluded that:
“this is an heterogeneous subgroup within IGE. There is a distinct overlap with early childhood epilepsy with GTCS and myoclonic-astatic epilepsy on the one side and with childhood absence epilepsy on the other. Thus it should not be regarded as a special syndrome”. 325,326
I am in complete agreement with this statement. Age at onset of absence seizures alone can not define an epileptic syndrome. However, with improved diagnostic skills, applying inclusion (e.g. including absences and GTCS) and exclusion criteria (e.g. excluding CAE and symptomatic cases), it appears that there is a rare IGE, which needs a precise definition.
This is an IGE syndrome (occurring in otherwise normal children). 325,326
- Onset of absences occurs between 1–5 years of age. Absences are markedly different from CAE. Clinically, they are less severe and less frequent. Ictal EEG 2.5–4 Hz GSWD is very irregular and termination is not abrupt, but often fades with spike–wave complexes.
- GTCS are common (affecting two-thirds of patients) and are often the first seizure type. Boys are more likely to suffer GTCS than girls.
- Myoclonic jerks and myoclonic-astatic seizures occur in 40% of patients.
- Absence status epilepticus may lead to cognitive impairment.
- Background EEG shows a moderate excess of slow waves.
- Long-term prognosis is worse than in CAE.
- There is a strong family history of IGE and GSWD in the EEG of unaffected members, particularly mothers.
Perioral Myoclonia with Absences
Clinical noteTypical absences with ictal motor symptoms of perioral myoclonia is a type of seizure.16,26,328–331 However, this is often combined with a clustering of other clinical and EEG features probably constituting an interesting IGE syndrome of perioral myoclonia with absences (PMA).16,18,328,331
Demographic Data
Age at onset covers a wide range from 2–13 years (median 10 years). Girls are far more frequently affected than boys.
The syndrome is uncommon in children (< 1% with typical absences) but, because it fails to remit, is relatively common in adults (9.3%) with typical absence seizures.16,328,331
Clinical Manifestations
Typical absence seizures with perioral myoclonia are the defining symptom. The characteristic feature is perioral myoclonia, which consists of rhythmic contractions of the orbicularis oris muscle that cause protrusion of the lips, contractions of the depressor anguli oris resulting in twitching of the corners of the mouth or, rarely, more widespread involvement, including the muscles of mastication producing jaw jerking (Figure 10.16). Impairment of consciousness varies from severe to mild. Most patients are usually aware of the perioral myoclonia. Duration is usually brief, lasting a mean of 4 s (range 2–9 s). Absences with perioral myoclonia may be very frequent and occur many times a day or 1–2 times a week, or they are rare.
Generalised tonic clonic seizures occur in all patients. GTCS often start before or soon after the onset of clinically apparent absences. Exceptionally, GTCS may start many years after the onset of absences. GTCS are usually infrequent (ranging from once in a lifetime to 12/year) and are often heralded by clusters of absences or ASE.
Absence status epilepticus is very common in PMA (57%) and frequently ends with GTCS (Figure 10.16). It is more common than in any other syndrome of IGE with typical absences.19 Perioral myoclonia may be more apparent than impairment of consciousness or vice versa.
Aetiology
Half of patients with PMA have first degree relatives and mainly siblings with IGE and absences.141
Diagnostic Procedures
All tests apart from the EEG are normal.
Electroencephalography
Interictal EEG frequently shows: (a). abortive bursts or brief less than 1 s generalised discharges of 4–7 Hz spikes/multiple spikes-waves, which are usually asymmetrical and may give the impression of a localised focus; and (b). focal abnormalities, including single spikes, spike–wave complexes and theta waves with variable side emphasis.
Ictal EEG consists of 3–4 Hz GSWD with frequent irregularities in terms of the number of spikes in the spike–wave complex, the fluctuations in spike amplitude and the occurrence of fragmentations (Figure 10.16).
There is no photosensitivity.
Differential Diagnosis
Patients with PMA are frequently erroneously diagnosed as having focal motor seizures because: (a). the prominent motor features of the absences, which are often reported or sometimes recorded as unilateral; and (b). the presence of interictal focal EEG abnormalities.
However, this error is unlikely to happen if EEG is properly recorded and interpreted. Also, patients with focal motor seizures are unlikely to suffer ASE, which is common in PMA.
Considerations on Classification
Absences with perioral myoclonia despite unequivocal documentation with video EEG16,328,331 have not been recognised by the ILAE as a seizure type.1,5 Perioral myoclonia may occur in typical and atypical absence seizures of idiopathic and symptomatic epilepsies. Therefore, absences with perioral myoclonia alone can not be taken as evidence of any syndrome. However, they often occur together with other symptoms that cluster in a non-fortuitous manner thus constituting the main manifestation of a syndrome within the broad spectrum of IGE, which we proposed to call perioral myoclonia with absences. 16,328 Other manifestations of this syndrome include GTCS, which often start early prior or together with the absences, frequent occurrence of ASE, resistance to treatment and persistence in adult life.16,328,331
The main differential diagnosis is from other syndromes of IGEs such as CAE, JAE and MAE depends on the age at onset. Video EEG invariably reveals perioral myoclonia that sometimes, and particularly in treated patients, may be subtle. Onset of GTCS before or at the same age as typical absences, the relatively brief duration of the absences with the concurrent perioral myoclonia and the frequent occurrence of ASE are useful clinical indicators in favour of PMA and against childhood, juvenile or other forms of IGE. PMA may be difficult to differentiate from MAE, particularly if the latter presents with mild myoclonic jerks localised in the face.26 However, MAE is often symptomatic and is rarely associated with generalised non-convulsive status epilepticus.177
Prognosis
Absences and GTCS may be resistant to medication, unremitting and possibly lifelong.16,328,331
Management
Treatment is with valproate alone or combined with ethosuximide, small doses of lamotrigine or clonazepam. Levetiracetam may be effective, because of the myoclonic elements of the absences.
ASE with perioral myoclonia, of which most patients are aware, should be terminated with immediate self-administered medication of oral midazolam or rectal diazepam.
Idiopathic Generalised Epilepsy with Phantom Absences
Clinical noteThe syndrome of IGE with phantom absences58 is characterised by the triad of:
Demographic Data
The first overt clinical manifestations of GTCS appear in adult life, though absences may have started much earlier. ASE as the first overt symptom in childhood is rare.77 Men and women are equally affected. The prevalence was estimated to be 15% among IGE with typical absences, 10% of IGE and 3% of 410 consecutive patients over 16 years of age with epileptic seizures.58 Genton et al.338 reported that, among 253 consecutive cases of IGE, 32 (15.4%) patients had rare GTCS with GSWD in the interictal EEG. It is possible that these patients suffered from IGE with GTCS only and/or IGE with phantom absences.
Definition of Phantom Absences
‘Phantom absences’ denote typical absence seizures, which are so mild that they are inconspicuous to the patient and imperceptible to the observer.16,58
Why the name phantom absences? Is this synonymous with ‘subclinical or larval absences’?
We coined the term ‘phantom absences’ because of their clinically elusive and inconspicuous character.58 Cognitive impairment during ‘subclinical, larval’ GSWD are well documented332,333 and phantom absences may be a good example of them. However, it should be emphasised that patients with phantom absences have, by definition,1,15 active, clinical absence seizures that manifest with mild impairment of cognition, as demonstrated by errors and discontinuation during breath counting on video EEG (Figure 10.17).334 It should also be emphasised that phantom absences in these adults do not represent aborted past childhood or juvenile absences modified by age or medical treatment.58 Furthemore, phantom absences is not synonymous with the EEG pattern of abortive “phantom 4–6 Hz spike and slow wave”, which is a non-specific EEG abnormality of no clinical significance.335–337
Considerations on Classification
Phantom absences, or mild absence seizures, have not been categorised as such by the ILAE.1,5 The absences are simple, brief (usually 2–4 s) causing only inconspicuous impairment of cognition, which is not clinically disturbing to the patient. Though not classical, they fulfil the criteria of typical absences with more than 2.5 Hz GSWD.58
There is reasonable evidence to suggest that phantom absences are not only discrete seizures, but may also constitute the main symptom of a syndrome within the broad spectrum of IGE. There is non-fortuitous clustering of other symptoms, such as GTCS of usually late onset, frequent occurrence of ASE and persistence in adult life.58 That these patients have IGE is beyond any doubt, as they all are of normal intelligence and physical state, high resolution MRI is normal, the EEG shows GSWD, and the seizures are generalised.
The syndrome of IGE with phantom absences has not been recognized by the ILAE.1,5 Accordingly, these cases are probably orphaned or probably categorised among undefined IGE or other syndromes of IGE.
Clinical Manifestations
Phantom absences are the defining and consistent symptom in these patients.58 Phantom absences manifest with mild, but definite, impairment of cognition documented by video EEG (Figure 10.17). There are no other clinical symptoms except eyelid flickering that consistently occurs in some patients. However, patients are not usually aware, even retrospectively, that absences interfere with their daily life, even when driving or in demanding professions, such as computer programming, civil engineering, major business and administration.58
Patients may retrospectively admit to momentary lack of concentration and forgetfulness, which in their opinion was of no practical significance.58 Rarely patients or witnesses also become aware of some minor motor manifestations during the absence:
Patient noteoccasional, very brief episodes of quick flickering of the eyeballs upwards accompanied by a brief lack of concentration
Phantom absences are common in patients with IGEs, but are often unrecognised. These absences are impossible to detect without breath counting and video EEG.
Generalised tonic clonic seizures occur in all adult patients that seek medical attention. They are usually the first overt clinical manifestation.58 They are of late onset, infrequent and without consistent circadian distribution or specific precipitating factors.
Absence status epilepticus occurs in 50% of patients. This often lasts for many hours alone or prior to GTCS (Figure 10.18).58,76 It manifests with cognitive impairment, which is usually of mild or moderate severity.
Patient noteAt the age of 28, this patient was able to drive for 2–3 hours while in ASE. This was on Christmas Eve with no precipitating factors. For the whole day he was feeling unwell, “tired, unable to compute conversations, clouded mind, missing words, as if sleep waking, slow mind”. Despite this, he had his haircut and then drove alone on a 1-hour trip through busy roads to relatives, offering the wrong Christmas gifts to the wrong persons. He then drove back home for another hour, picked up his wife and drove again towards a friend’s house. His wife noticed that he appeared “slow and very quiet during the drive, but I though he was just tired. I was not alerted because he was driving very well, not making any errors”. She did not notice any abnormal movements, including gestural automatisms or jerking. He became completely silent for 1–2 minutes prior to a GTCS, which occurred while parking; his wife managed to stop the car and avoid an accident. No further seizures occurred in the next 5 years while on valproate 1000 mg daily.
Impaired cognition during ASE varies. Most patients often communicate poorly and slowly, feel strange and confused, make errors at work and look depressed, but do not become unresponsive (Figures 10.17, and 10.18). Experiential, mental and sensational symptoms are more common than is usually appreciated.30 Frequently, they have a good recollection of the ictal events and may be able to write down their experiences while in ASE.30,58
Neuropsychological examination under video EEG monitoring during ASE in 2 patients revealed only mild attentional and executive disturbances.339 GSWD were associated with selective impairment in the initiation of response and self-generated action, whereas short-term storage of external information was fully preserved.339
Patients with recurrent ASE are often aware of the impeding GTCS and try to find a safe place to have it.
Aetiology
IGE with phantom absences is probably genetically determined.58 It is difficult to explain the high frequency of ASE in these patients, with such brief and mild absences and infrequent GTCS. It is possible that, under the influence of precipitating factors that are not fully understood, phantom absences might cluster and evolve into ASE facilitated either by lack of or inappropriate treatment.
Vuilleumier et al.339 proposed that a predominant involvement of frontomesial thalamocortical circuitry may underlie an ‘inconspicuous’ disorder of consciousness, as seen in phantom absences with selective loss of initiation and goal-oriented behaviour, whereas involvement of more lateral frontal areas in typical absences may also disrupt working memory processes.339
Diagnostic Procedures
All tests apart from the EEG are normal.
Interictal EEG: The background activity is normal, 50% of patients have EEG focal paroxysmal abnormalities consisting of short transients of localised slow, sharp waves or spikes, or both, occurring either independently or in association with brief GSWD. 58,340 EEG photosensitivity is exceptional.
Ictal EEG consists of 3–4 Hz GSWD with occasional fragmentations (Figure 10.17). They are typically brief lasting of no more than 5 s. Mild cognitive impairment manifested with hesitation, discontinuation and errors in breath counting is the only clinical ictal symptom during GSWD. A few may also have mild ictal eyelid fluttering.58,340 Hyperventilation is a major provocative factor.
During ASE, the EEG shows continuous, generalised, mainly 3 Hz spike/multiple spike slow wave activity (Figure 10.18).
Differential Diagnosis
The diagnostic and management errors involved in adult patients with IGE and typical absence seizures have been well reported.56,58 The magnitude of the problem is worse in IGE with phantom absences, in which the absences are very mild, ASE is confused with non-epileptic events or temporal lobe epilepsy, and GTCS are of late onset. This is compounded by frequent EEG focal abnormalities and the current practice of most EEG departments to not test cognition appropriately during brief GSWD.
The main problems to consider in IGE with phantom absences are:
- the first overt unprovoked GTCS appears in adult life
- absence status epilepticus
- differentiation from other syndromes of IGE.
It is essential to take a careful clinical history and to interpret symptoms correctly, which may be suggestive of typical absences and ASE. A history of altered consciousness preceding GTCS should not be taken as evidence of complex focal seizures, depression or an unspecified seizure prodrome.
Other forms of the so-called ‘adult onset IGE’ may be otherwise typical examples of JME, JAE or other IGE syndromes that start or become clinically identifiable after the age of 20 years.202,341–344 Some of the patients described may suffer from IGE with phantom absences.
Prognosis
IGE with phantom absences may be a lifelong propensity to seizures, which is of undetermined onset and remission. Patients are of normal intelligence, which does not show any signs of deterioration. Further, phantom absences, though frequent, do not appear to affect daily activity.
Management
There are many unanswered questions as to whether patients with phantom absences need treatment. All patients with IGE with phantom absences had a normal life without medication until their first GTCS, probably many years after the onset of frequent daily mild absence seizures. We do not know how many people there are in the general population with the same problem but without GTCS or conspicuous ASE. If treatment is considered necessary (driving a car is a significant factor), valproate, levetiracetam and lamotrigine are AEDs to consider.
A Reminder of “Adult Onset Idiopathic Generalised Epilepsies”
Recently, there has been increasing interest in the so-called adult-onset IGE.202,341–344 Seizures consist of GTCS, myoclonic jerks and absences, alone or in combination, which first become clinically detectable after the age of 20 years. I emphasise clinically detectable, because these patients may have had minor seizures (as is the case with phantom absences) long before seeking medical advice, which is usually prompted by a GTCS. Thus, adult onset IGE appears to consist of classical syndromes of IGE, but with delayed onset or delayed identification. Other than age at the time of the first overt seizure, all other clinical and EEG manifestations are typical of JME, JAE or IGE with GTCS only. For example, ‘adult myoclonic epilepsy’ is identical to JME.202 When grouped together on the basis of age at onset, clinical manifestations, EEG and family history of epilepsies, there is a similarity between ‘adult onset IGE’ and ‘IGE of classical late childhood or adolescence onset’.342–344 However, an exemption exists with ‘benign familial adult myoclonic epilepsy’,345 which is a distinct autosomal epileptic syndrome with seizures first appearing in adult life (page 332).
Syndromes of autosomal recessive inheritance such as ‘familial infantile myoclonic epilepsy’356,357 have also been described and may be considered in ILAE revisions.
Familial (Autosomal Dominant) Generalised Epileptic Syndromes
Clinical noteThere is an increasing number of reports of familial idiopathic generalised epilepsies which have not been recognised by the ILAE yet. These include:
Benign Familial Adult Myoclonic Epilepsy
Benign familial adult myoclonic epilepsy (FAME)345 is probably the most common of the autosomal epileptic syndromes not yet recognised by the ILAE. FAME has been initially identified in Japanese346,347,352,355 and more recently in European families.351,353 FAME is a relatively benign non-progressive autosomal dominant idiopathic generalised epileptic syndrome with high penetrance.
Clinically, FAME is characterised by adult-onset cortical myoclonus and tremor of the fingers; 80% of patients also have infrequent GTCS in periods of worsening myoclonus. Movement and emotional stress intensify the myoclonus. Age at onset varies from 30 to 60 years (mean 40 years). Families with members having concurrent migraine or blindness have been reported.
The EEG shows generalised polyspikes and waves, photosensitivity and giant somatosensory evoked potentials. Consistent with cortical myoclonus, long-loop C reflexes are enhanced and there is a preceding wave on jerk-locked back EEG averaging.
The genes for FAME have been mapped to 8q24 in Japanese families346,347,352,355 and 2p11.1-q12.2 in European351,353 families. There may be allelism with ‘autosomal dominant cortical myoclonus and epilepsy syndrome’.
Autosomal Dominant Cortical Myoclonus and Epilepsy
Autosomal dominant cortical myoclonus and epilepsy (ADCME) with complex focal and generalised seizures is based on a study by Guerrini et al354 of a pedigree, in which eight individuals presented with a non-progressive disorder with onset between the ages of 12 and 50 years.354 It was characterised by predominantly distal, semicontinuous rhythmic myoclonus (all patients), GTCS (all patients) and complex focal seizures (three patients). Most patients had suffered infrequent seizures and had a normal cognitive level, but three patients with intractable seizures had mild mental retardation. The pattern of inheritance was autosomal dominant with high penetrance. All patients had frontotemporal as well as generalised interictal EEG abnormalities. Back-averaging analysis and other neurophysiological studies of the myoclonus suggested a cortical origin. The C-reflex at rest was enhanced and somatosensory, and visual evoked potentials were of high amplitude. The resting motor threshold intensity in response to transcranial magnetic stimulation was significantly reduced and the post-motor evoked potential silent period was significantly shortened compared with the controls. These clinical and neurophysiological characteristics suggest diffuse cortical hyperexcitability and a high propensity for intra-hemispheric and inter-hemispheric cortical spread, as well as rhythmic myoclonic activity. The disease has been linked to chromosome 2p11.1-q12.2.354
Treatment of Idiopathic Generalised Epilepsies
Consider the following facts:
Patient note“A majority (48%) of patients with idiopathic generalised epilepsy initially receive ill-advised AED, which cause IGE to appear intractable.” Benbadis et al.14
Patient note“Carbamazepine is still being inappropriately prescribed to children (47%) with typical absence syndromes to their detriment.” Parker et al.358
Patient note“In a class 1 control AED study, one quarter (27%) of patients with IGE, including JME, were treated with carbamazepine (a contra-indicated drug in IGE) which was the comparator drug on the basis of the physicians’ ‘intention to treat’.” Privitera et al.301
These are striking, recent examples of commonly occurring and disturbing inappropriate medications in patients with IGEs. The most important of the multiple reasons for this continuing error are:
- diagnostic misclassification of IGEs as focal epilepsies233
- sparse or methodologically ambiguous AED RCTs 233
- official guidelines and publications paying scant attention to important management aspects of IGEs 359
- Formal national formularies are conspicuously void of warning against established pro-epileptic action of certain AEDs in certain types of seizures and syndromes of IGEs.
I take some examples from the British National Formulary (BNF), which are more or less the same everywhere:
Patient note“Indications for carbamazepine: partial and secondary generalised tonic-clonic seizures and some primary generalised seizures.”
Thus, physicians may reasonably prescribe carbamazepine for absences and myoclonic jerks (which are primary generalised seizures as also defined in the BNF) with disastrous effects.
Patient note“Indications for tiagabine or vigabatrin: adjunctive treatment for partial seizures with or without secondary generalisation.”
There is no warning that these AEDs are deleterious for IGEs, which are often misdiagnosed as partial epilepsies.
IGEs demand different treatment strategies to focal epilepsies.4,14,310,360–363 Ignoring this fact results in avoidable morbidity and sometimes mortality. There is an urgent need for clear and unequivocal guidelines in the use of old and new AEDs in IGEs and its various types of seizures. Practising physicians have a colossal task in not only properly diagnosing IGE, but also in deciding which of the many old and new AEDs is the most suitable and which is contraindicated for the seizures and preferably the syndromes of IGEs.
The methodology followed in this book regarding treatment recommendations for AEDs is detailed in Chapter 4. The treatment of each individual IGE syndrome has been detailed in the appropriate chapter.
Recent reviews and publications on the AED treatment of IGEs are recommended reading.14,233,310,314,360,362,363
Important established documentation to remember is that:
- Certain AEDs that are beneficial in focal epilepsies are ineffective or even contraindicated in IGE. 4,276,292,364,365
- Tiagabine and vigabatrin are major pro-absence agents.
- Carbamazepine, oxcarbazepine and phenytoin exacerbate absences and myoclonic jerks.
- Gabapentin is ineffective in all types of idiopathic epileptic seizures and may exacerbate some of them.
- A drug efficacious in one type of generalised seizure may be ineffective or exaggerate another type of generalised seizure.
- Clonazepam is the best choice of drug for myoclonic jerks, but is ineffective in GTCS. Ethosuximide is highly efficacious only for absence seizures and negative epileptic myoclonus, but is ineffective or may exaggerate GTCS.
- Carbamazepine and oxcarbazepine are effective in primarily GTCS, but often aggravate myoclonic jerks and absences.
- Lamotrigine is effective in primarily GTCS and absences, but may exacerbate myoclonic jerks.
- If a drug is found to be efficacious in ‘generalised’ childhood epileptic encephalopathies, it does not mean that this is also the case in IGEs.
- Vigabatrin is the drug of first choice in West syndrome, but it is contraindicated in IGE.
- A drug found to be efficacious in secondarily GTCS, may be ineffective in primarily GTCS or deleterious in IGEs.
- Gabapentin, an AED licensed for the treatment of focal and secondarily GTCS is ineffective in primarily GTCS and may aggravate other types of IGE seizures.
- Tiagabine, an AED licensed for the treatment of focal and secondarily GTCS, is a potent pro-absence agent that induces absence seizures and provokes ASE often ending with GTCS in IGE.
- IGEs are often easily treatable which means that a small dose of an appropriate AED is as good as a large dose . 233
- No difference in control of various types of JME could be demonstrated between 1000 mg and 2000 mg of valproate. 238
Diagnosis and Treatment of Newly Identified IGEs
Diagnosis should first establish that the patient suffers from genuine epileptic seizures and then define:
- this is IGE and not focal epilepsy
- the types of seizures that the patient suffers
- if possible, the IGE syndrome.
In choosing the first AED to be recommended from Table 10.8 efficacy and adverse reactions have to be carefully balanced, because treatment is often lifelong.
Old Anti-Epileptic Drugs in IGEs
Briefly, prior to the introduction of new AEDs, the position was as follows.
Valproate has superior efficacy in all seizures and syndromes of IGEs but its use in certain populations and particularly in women of reproductive age is highly problematic and sometimes impossible.
Clonazepam, even in small doses of 0.5 –1 mg, is probably the most potent antimyoclonic drug with some anti-absence effect;4 it may deteriorate GTCS or deprive patients from the warning symptoms of an impeding GTCS in JME (see page 317).253–255
Ethosuximide is a potent drug against absences. It may improve myoclonic seizures (particularly negative epileptic myoclonus), but is ineffective and may worsen GTCS.4
Phenytoin is effective in primarily GTCS, but deteriorates absences and possibly myoclonic jerks; it is often ineffective or worsens JME.312
Carbamazepine is effective in primarily GTCS, but aggravates absences and myoclonic jerks.
Phenobarbitone was historically the preferred drug in JME and is still used in Europe and developing countries; it worsens absences.61,201
Evolving Treatment of IGE in the Era of New Anti-Epileptic Drugs
The adverse effects of valproate and its lack of efficacy in 20% of patients has prompted the search for alternatives. Half of the new AEDs appear to be either ineffective or contraindicated (vigabatrin, tiagabine, oxcarbazepine, gabapentin)366 in IGE (Table 4.1). Of the other four new AEDs, lamotrigine,4,258,259 levetiracetam,234,260–265 topiramate266,267,298 and zonisamide268 appear to be therapeutic agents. Levetiracetam, because of its efficacy in all types of seizure234,260–265 and safer adverse reactions profile,270,271 appears to be the most promising substitute for valproate. Lamotrigine is effective in GTCS and absence seizures, but often aggravates myoclonic jerks. It has important beneficial synergistic interactions with valproate. Topiramate is mainly effective in GTCS, but its adverse effect profile and drug–drug interactions are possibly worse than those of valproate. Zonisamide may have a weak therapeutic effect.
Prescribing Errors in IGEs
The error of prescribing tiagabine or vigabatrin in patients with absence seizures may be of the same magnitude as prescribing a gluten-rich diet in the treatment of coeliac disease. 4 Prescribing carbamazepine in patients with JME is a similar situation.
New Anti-Epileptic Drugs Useful in IGEs
These have been detailed on pages 315–18 in the treatment of JME. Briefly, in order of preference:
Levetiracetam,4,367 so far fulfils all expectations as probably the best new AED in the treatment of IGEs and is the likely candidate to replace valproate in at least JME, with its high and sustained efficacy, fast action, excellent safety profile, lack of clinically meaningful interactions with other drugs and no need for laboratory tests. Its role in syndromes with predominant absence seizures (CAE and JAE) is yet unclear though it has a unique mode of action in animal models of absence seizures.368 In a recent report 3 of 4 patients with CAE and 5 of 5 patients with JAE became seizure free with levetiracetam alone or in combination with other AEDs. 183
Lamotrigine has proven efficacy in controlling GTCS and absence seizures in at least 50% of patients, but may exaggerate myoclonic jerks. It is inferior to valproate in terms of efficacy, but superior with respect to adverse reactions despite a high incidence of idiosyncratic reactions that can occasionally be fatal. Efficacy and adverse reactions have to be carefully balanced in these cases, because treatment is often lifelong.
Lamotrigine is recommended in the treatment of CAE and JAE either alone170,171 or in combination with other anti-absence AEDs.4,170,171,194,246,247,271 However, lamotrigine monotherapy in JME is probably not recommended but small doses of lamotrigine added to valproate are probably the most effective combination treatment for resistant cases.
Topiramate 266,298,369 is another broad-spectrum AED that is effective in primary GTCS299–302 with a weak anti-absence303 and anti-myoclonic action.291,297 In IGEs, including JME, it appears to be much less effective than valproate, but more effective than lamotrigine.297
In a well-cited class 1 study, topiramate has been found to be effective in primarily GTCS.299,301,302 Methodological drawbacks of these studies included: (a). patients with symptomatic generalised seizures were enrolled; (b). one fourth (27%) of patients with IGEs were assigned to carbamazepine as a comparator AED; and (c). carbamazepine was statistically decreased in the topiramate group thus favouring the results in this group for at least the ‘subtypes of generalised seizures’, such as absences and myoclonic jerks that deteriorate with carbamazepine.
Topiramate is unlikely to achieve monotherapy status in the long-term treatment of IGEs mainly because of its many short and long term adverse effects and drug-drug interactions.
Zonisamide 307–310 is also a broad-spectrum AED, but its role in IGEs is largely unknown and probably weak. In the long experience of eminent Japanese colleagues that I consulted, zonisamide is effective in the treatment of focal seizures, secondarily GTCS, epileptic encephalopathies (West syndrome responds much better than Lennox-Gastaut syndrome) and progressive myoclonic epilepsies. Zonisamide appears to be much less effective in IGE, primarily GTCS, absences and jerks, though a few patients may have an excellent response. In children, cognitive adverse reactions may be troublesome. Currently in Japan, zonisamide is a second-line AED for focal epilepsies after carbamazepine and for symptomatic generalised epilepsy after valproate, clonazepam and clobazam.
Polytherapy
Clinical noteFor patients in whom monotherapy with valproate fails, the combination of valproate with small doses of lamotrigine (25–50 mg) appears to be the most effective.4 In those with persistent myoclonic jerks, clonazepam is the best add-on drug to valproate.4 Ethosuximide should be added only for uncontrolled absence seizures. However, in all these scenarios, valproate is the principal AED. The situation is likely to change dramatically with use of levetiracetam instead of valproate but this needs RCTs.
The consensus is that IGEs have a better prognosis with, and a more favourable response to, appropriate AEDs than symptomatic and focal epilepsies. “Most patients with IGE are easily controlled with appropriate medication, refractory patients are rare.”246,247 However, there is no reliable figure regarding the prevalence of intractable IGE, which may be in the order of 10–30%.370 A factor contributing to this uncertainty and probably high incidence of intractable IGEs is that IGEs are often inappropriately treated with AEDs that are either ineffective or contraindicated. Table 4.1 lists AEDs that are indicated and contraindicated in IGE seizures.
Management of patients with intractable IGEs, providing that they truly suffer from epileptic seizures, should take the following steps:
- Based on clinical and EEG evidence, establish the type or types of seizures (absences, myoclonic jerks and GTCS alone or in combination), and make sure that these are primarily and not secondarily generalised. Previous EEGs particularly, in untreated stages, are invaluable.
- Establish precipitating factors and circadian distribution as well as their effect regarding intractability.
- List in chronological order, all AEDs used, and in what doses and combinations. Establish which drugs were beneficial and which made the situation worse.
- Consider thoroughly the current situation regarding: (a). seizures – which are the more predominant and more disturbing; and (b). AEDs – which are definitely or possibly effective, ineffective or contraindicated with respect to seizures and adverse reactions.
- Consider thoroughly all the above, including compliance, in making a definite plan of which AEDs with adverse effects (seizure efficacy and patient tolerability) should be withdrawn and which of the indicated AEDs should be increased in dosage or added to the scheme.
- Of the new AEDs, those which are likely to be effective as monotherapy (Table 4.1 and Table 10.8) are also the most likely to be suitable in polytherapy. The order of priority as determined by efficacy, safety, drug–drug interactions and other parameters are levetiracetam, lamotrigine, topiramate and zonisamide. All other new AEDs –tiagabine, oxcarbazepine and gabapentin –are unsuitable.
New AEDs Contraindicated in IGEs
Gabapentin: all relevant studies have shown that gabapentin is, at least, ineffective in IGEs including primary GTCS, and it may exacerbate absences and myoclonic jerks.194,246,247,271,371–373
Oxcarbazepine appears to have a similar seizure profile to carbamazepine. In IGEs, like carbamazepine, oxcarbazepine mainly aggravates absences and myoclonic jerks; GTCS may also worsen.365 In a case series, all six IGE patients showed significant deterioration in terms of absences and myoclonic jerks related to oxcarbazepine treatment; GTCS also worsened in three patients.365
Vigabatrin and tiagabine are contraindicated. These are pro-absence AEDs, because of their GABAergic action, which explains the high incidence of drug-induced ASE and the appearance of new types of seizures.4
Drug Withdrawal
In CAE, treatment may be slowly withdrawn 1–3 years after controlling all absences.4 All other IGE syndromes are probably lifelong and confront the usual textbook advice of withdrawal of medication after 2–3 years from the last seizure. Relapses are probably unavoidable. However, if seizures are mild and infrequent, drug withdrawal may be attempted. This should be done in small decrements, probably over years, warning the patient that re-emergence of even minor seizures, such as absences or myoclonic jerks, mandates continuation of treatment. EEG confirmation of the seizure-free state is needed during the withdrawal period.4
First-Line Drugs
Valproate (the most effective of all but often unsuitable for women)
Levetiracetam (but needs RCTs)
Lamotrigine (may exaggerate myoclonic jerks)
Ethosuximide (only for absences and negative myoclonus; may exaggerate GTCS)
Clonazepam (only for pure myoclonic syndromes; may exaggerate GTCS)
Second-Line or Adjunctive Drugs
Topiramate (but with serious adverse reactions and drug–drug interactions)
Zonisamide (needs further documentation regarding efficacy and adverse reactions)
Phenobarbitone (may be first line AED for those without absence seizures if cost is a major issue)
Acetazolamide (only for absences)
Contraindicated Drugs
Carbamazepine, oxcarbazepine and phenytoin (though they may control primarily GTCS if added to first-line drugs)
Gabapentin (ineffective in primarily GTCS and may exacerbate absences and myoclonic jerks)
Tiagabine and vigabatrin (pro-absence drugs with a high incidence of induced ASE)
Treatment of Status Epilepticus in Idiopathic Generalised Epilepsy
Idiopathic generalised ASE with all its variations (generalised non-convulsive status epilepticus) has a high prevalence in IGE, though it is often unrecognised, because symptoms may be mild. Generalised tonic clonic status epilepticus is less common in IGE than symptomatic and probably symptomatic focal or generalised epilepsies.
The emergency treatment of convulsive or non-convulsive status epilepticus is the same irrespective of causes, idiopathic or symptomatic, in focal or generalised epilepsies. There are recent major publications and reviews for more extensive reading.363,371,374–379,379–388
Convulsive Status Epilepticus
Convulsive status epilepticus (CSE) is a medical emergency and should be managed urgently and properly according to established and well-publicised protocols.371,374–379 Following any of these protocols results in appropriate and rapid management of CSE, which reduces morbidity and mortality.377 The mortality of CSE is around 10%. The commonest cause of CSE is withdrawal of anti-epileptic medication, which should be reintroduced as soon as possible after the onset of CSE. The longer CSE lasts, the harder it is to treat and the greater the morbidity and mortality.
The aim of treatment is early termination of CSE in order to prevent neuronal damage caused by systemic and metabolic disturbances and by the direct excitotoxic effect of electrical seizure discharges. Control of overt and electrical seizures is imperative. The risk of brain damage increases progressively if continuous CSE persists for more than 30 minutes and particularly after 1–2 hours. This is because compensatory mechanisms to prevent brain damage are relatively satisfactory during the first 30 minutes. Subsequently, compensatory mechanisms break down with increasing speed if the seizures are not stopped during the first 30 minutes. Neuronal damage leads to transient or permanent neurological, epileptic and cognitive sequelae or even death.
Drug Treatment of CSE Can Be Divided into Three Stages
- In the early stage (first 30 minutes), treatment comprises intravenous administration of a fast-acting benzodiazepine of which diazepam, lorazepam or midazolam are the most effective agents. Diazepam is the traditional drug, then lorazepam came to prominence as the drug of first choice377 and, recently, midazolam infusion has become increasingly popular as an effective and well-tolerated therapeutic agent.389 Most cases are controlled with this approach. If CSE is not controlled at this stage, the patient enters into the second stage of established status epilepticus, which carries an appreciable morbidity.
- In the stage of established CSE, first-line drug options are intravenous phenytoin or fosphenytoin. If these are ineffective, subanaesthetic doses of phenobarbitone are used. If seizures are not controlled at this stage the patient enters refractory status epilepticus.
- The last stage of refractory status epilepticus requires general anaesthesia and a continuous infusion of AEDs, such as pentobarbital, midazolam or propofol, and concomitant EEG monitoring of seizure or EEG background suppression.381 Despite these measures, the mortality of refractory CSE is more than 20%.381
Absence (Generalised Non-Convulsive) Status Epilepticus
Absence status epilepticus19,66,390 occurs in 10–20% of cases of IGE and in as many as 50% of cases of some syndromes of IGE such as those manifesting with phantom absences or perioral myoclonia. Additionally, nearly all patients are fully aware of this epileptic status and know that it may inevitably lead to a GTCS, though it is avoidable:
Patient noteIt is the same feeling of: “slowing down”, “uncontrollable rush of thoughts”, “losing control of my mind”, “taking me much longer to formulate my response which occasionally is inappropriate and bumbled. Then I know that I will have the fit. If I can, I just go to a private place and wait for it.”
This stage is unlikely to be considered as a genuine status epilepticus by the physicians in accident and emergency departments. Therefore, advice to the patient regarding therapeutic options for self-administration of drugs is imperative. Benzodiazepines and, mainly, diazepam, lorazepam or midazolam are the most effective agents.
Self-administration: Rectal diazepam (10–20 mg for adults and 0.5 mg/kg for children) as soon as the first symptoms appear may stop ASE and prevent an impending GTCS. Rectal absorption of liquid diazepam is very rapid, reaches the brain within minutes and has a near-intravenous efficacy. Proprietary rectal tubes (Stesolid) containing ready-made liquid diazepam are the most widely used formulation. Suppositories of diazepam are not useful, because of their slow absorption. Diazepam rectal gel is now available in the USA and some other countries.391 However, adult patients, either because of embarrassment or inconvenience, rarely use rectal preparations. An oral bolus dose of valproate (usually twice the daily prophylactic dose) from onset of symptoms is often effective in terminating the ASE and preventing GTCS (most patients prefer this to rectal preparations of drugs).
Buccal392,393 or intranasal394 application of midazolam may be the best practical and effective therapeutic option. Midazolam buccal administration has equal efficacy and rapidity of action to rectal diazepam. It is more convenient and less traumatising than rectal preparations of diazepam. Midazolam (0.3 mg/kg for children and 10–20 mg for adults) drawn up from an ampoule is dissolved with peppermint (otherwise it smells and tastes terrible) and should be swirled around the mouth for 4–5 min and then spat out.
Clonazepam (1–4 mg) may be given orally at the onset of generalised non-convulsive status epilepticus and is the preferred option in patients with mainly myoclonic jerks.
Patient noteThis helps me to go to sleep and when I wake up I am fine.
Hospital management: With intravenous administration of any type of the benzodiazepines discussed above, generalised non-convulsive status epilepticus usually stops abruptly. The problem is that this condition is not recognised and the patients are not believed when they seek such treatment even when they produce a relevant letter from their treating physician, which clearly explains their situation and the need for urgent attention.
Patient noteShe started doing the same silly things. I recognised it. The doctor told me that she is OK. No, no I said, she is going to have a fit.
References
- 1.
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389–99. [PubMed: 2502382]
- 2.
- Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic generalised epilepsies. London: John Libbey & Company Ltd; 1994.
- 3.
- Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995.
- 4.
- Panayiotopoulos CP. Treatment of typical absence seizures and related epileptic syndromes. Paediatr Drugs. 2001;3:379–403. [PubMed: 11393330]
- 5.
- Engel J Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42:796–803. [PubMed: 11422340]
- 6.
- Panayiotopoulos CP, editor. Idiopathic generalised epilepsies:A review and modern approach. Epilepsia. 2005;(Suppl) (in press) [PubMed: 16302869]
- 7.
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002;3:371–82. [PubMed: 11988776]
- 8.
- Gourfinkel-An I, Baulac S, Nabbout R, Ruberg M, Baulac M, Brice A, et al. Monogenic idiopathic epilepsies. Lancet Neurol. 2004;3:209–18. [PubMed: 15039033]
- 9.
- Delgado-Escueta AV, Medina MT, Serratosa JM, Castroviejo IP, Gee MN, Weissbecker K, et al. Mapping and positional cloning of common idiopathic generalized epilepsies: juvenile myoclonus epilepsy and childhood absence epilepsy. Adv Neurol. 1999;79:351–74. [PubMed: 10514826]
- 10.
- Durner M, Keddache MA, Tomasini L, Shinnar S, Resor SR, Cohen J, et al. Genome scan of idiopathic generalized epilepsy: evidence for major susceptibility gene and modifying genes influencing the seizure type. Ann Neurol. 2001;49:328–35. [PubMed: 11261507]
- 11.
- Noebels JL. Exploring new gene discoveries in idiopathic generalized epilepsy. Epilepsia. 2003;44(Suppl 2):16–21. [PubMed: 12752457]
- 12.
- Jones-Davis DM, Macdonald RL. GABA(A) receptor function and pharmacology in epilepsy and status epilepticus. Curr Opin Pharmacol. 2003;3:12–8. [PubMed: 12550736]
- 13.
- Manning JP, Richards DA, Bowery NG. Pharmacology of absence epilepsy. Trends Pharmacol Sci. 2003;24:542–9. [PubMed: 14559407]
- 14.
- Benbadis SR, Tatum WO, Gieron M. Idiopathic generalized epilepsy and choice of antiepileptic drugs. Neurology. 2003;61:1793–5. [PubMed: 14694051]
- 15.
- Panayiotopoulos CP. Absence epilepsies. In: Engel JJ, Pedley TA, editors. Epilepsy: A comprehensive Textbook. Philadelphia: Lippincott-Raven Publishers; 1997. pp. 2327–46.
- 16.
- Panayiotopoulos CP. Typical absence seizures. In: Gilman S, editor. Medlink Neurology. San Diego SA: Arbor Publishing Corp; 2004.
- 17.
- Snead OC III, Depaulis A, Vergnes M, Marescaux C. Absence epilepsy: advances in experimental animal models. Adv Neurol. 1999;79:253–78. [PubMed: 10514819]
- 18.
- Loiseau P, Panayiotopoulos CP, Hirsch E. Childhood absence epilepsy and related syndromes. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3. London: John Libbey & Co Ltd; 2002. pp. 285–304.
- 19.
- Agathonikou A, Panayiotopoulos CP, Giannakodimos S, Koutroumanidis M. Typical absence status in adults: diagnostic and syndromic considerations. Epilepsia. 1998;39:1265–76. [PubMed: 9860061]
- 20.
- Penry JK, Porter RJ, Dreifuss RE. Simultaneous recording of absence seizures with video tape and electroencephalography. A study of 374 seizures in 48 patients. Brain. 1975;98:427–40. [PubMed: 1182486]
- 21.
- Commission of Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia. 1981;22:489–501. [PubMed: 6790275]
- 22.
- Stefan H. Epileptic absences. Studies on the structure, pathophysiology and clinical course of the seizure. Fortschr Med. 1983;101:996–8. [PubMed: 6409768]
- 23.
- Stefan H, Snead OC III. Absence seizures. In: Engel JJ, Pedley TA, editors. Epilepsy:A comprehensive textbook. Philadelphia: Lippincott-Raven Publishers; 1997. pp. 579–90.
- 24.
- Panayiotopoulos CP, Obeid T, Waheed G. Differentiation of typical absence seizures in epileptic syndromes. A video EEG study of 224 seizures in 20 patients. Brain. 1989;112:1039–56. [PubMed: 2505885]
- 25.
- Panayiotopoulos CP, Obeid T, Waheed G. Absences in juvenile myoclonic epilepsy: a clinical and video-electroencephalographic study. Ann Neurol. 1989;25:391–7. [PubMed: 2496640]
- 26.
- Capovilla G, Rubboli G, Beccaria F, Lorenzetti ME, Montagnini A, Resi C, et al. A clinical spectrum of the myoclonic manifestations associated with typical absences in childhood absence epilepsy. A video-polygraphic study. Epileptic Disord. 2001;3:57–62. [PubMed: 11431166]
- 27.
- Gowers WR. Their causes, symptoms and treatment. London: Churchill,J.A; 1881. Epilepsies and other chronic convulsive diseases.
- 28.
- Buchheim K, Obrig H, Pannwitz W, Muller A, Heekeren H, Villringer A, et al. Decrease in haemoglobin oxygenation during absence seizures in adult humans. Neurosci.Lett. 2004;354:119–22. [PubMed: 14698453]
- 29.
- Wiest R, Schindler K, Kollar M, Donati F. Epigastric sensations as an unusual manifestation of adult absence epilepsy. Epileptic Disord. 2001;3:13–6. [PubMed: 11313217]
- 30.
- Ferner RE, Panayiotopoulos CP. ‘Phantom’ typical absences, absence status and experiential phenomena. Seizure. 1993;2:253–6. [PubMed: 8162390]
- 31.
- Panayiotopoulos CP. Benign childhood partial seizures and related epileptic syndromes. London: John Libbey & Company Ltd; 1999.
- 32.
- Aliberti V, Grunewald RA, Panayiotopoulos CP, Chroni E. Focal electroencephalographic abnormalities in juvenile myoclonic epilepsy. Epilepsia. 1994;35:297–301. [PubMed: 8156947]
- 33.
- Lombroso CT. Consistent EEG focalities detected in subjects with primary generalized epilepsies monitored for two decades. Epilepsia. 1997;38:797–812. [PubMed: 9579907]
- 34.
- Feucht M, Fuchs K, Pichlbauer E, Hornik K, Scharfetter J, Goessler R, et al. Possible association between childhood absence epilepsy and the gene encoding GABRB3. Biol Psychiatry. 1999;46:997–1002. [PubMed: 10509183]
- 35.
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. [PubMed: 11326275]
- 36.
- Marini C, Harkin LA, Wallace RH, Mulley JC, Scheffer IE, Berkovic SF. Childhood absence epilepsy and febrile seizures: a family with a GABA(A) receptor mutation. Brain. 2003;126:230–40. [PubMed: 12477709]
- 37.
- Chen Y, Lu J, Pan H, Zhang Y, Wu H, Xu K, et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol. 2003;54:239–43. [PubMed: 12891677]
- 38.
- Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog Neurobiol. 1998;55:27–57. [PubMed: 9602499]
- 39.
- Futatsugi Y, Riviello JJ Jr. Mechanisms of generalized absence epilepsy. Brain Devel. 1998;20:75–9. [PubMed: 9545175]
- 40.
- Blumenfeld H. From molecules to networks: cortical/subcortical interactions in the pathophysiology of idiopathic generalized epilepsy. Epilepsia. 2003;44(Suppl 2):7–15. [PubMed: 12752456]
- 41.
- Bernasconi A, Bernasconi N, Natsume J, Antel SB, Andermann F, Arnold DL. Magnetic resonance spectroscopy and imaging of the thalamus in idiopathic generalized epilepsy. Brain. 2003;126:2447–54. [PubMed: 12902313]
- 42.
- Koepp MJ, Duncan JS. Epilepsy. Curr Opin Neurol. 2004;17:467–74. [PubMed: 15247544]
- 43.
- Meeren HK, Pijn JP, van Luijtelaar EL, Coenen AM, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–95. [PMC free article: PMC6757554] [PubMed: 11850474]
- 44.
- Manning JP, Richards DA, Leresche N, Crunelli V, Bowery NG. Cortical-area specific block of genetically determined absence seizures by ethosuximide. Neuroscience. 2004;123:5–9. [PubMed: 14667436]
- 45.
- Coulter DA. Antiepileptic drug cellular mechanisms of action: where does lamotrigine fit in? J Child Neurol. 1997;12(Suppl 1):S2–9. [PubMed: 9429123]
- 46.
- Gibbs JW3, Schroder GB, Coulter DA. GABAA receptor function in developing rat thalamic reticular neurons: whole cell recordings of GABA-mediated currents and modulation by clonazepam. J Neurophysiol. 1996;76:2568–79. [PubMed: 8899628]
- 47.
- Hosford DA, Lin FH, Wang Y, Caddick SJ, Rees M, Parkinson NJ, et al. Studies of the lethargic (lh/lh) mouse model of absence seizures: regulatory mechanisms and identification of the lh gene. Adv Neurol. 1999;79:239–52. [PubMed: 10514818]
- 48.
- Ryvlin P, Mauguiere F. Functional imaging in idiopathic generalized epilepsy. Rev Neurol (Paris). 1998;154:691–3. [PubMed: 9846339]
- 49.
- Duncan JS. Positron emission tomography receptor studies. Adv Neurol. 1999;79:893–9. [PubMed: 10514872]
- 50.
- Yeni SN, Kabasakal L, Yalcinkaya C, Nisli C, Dervent A. Ictal and interictal SPECT findings in childhood absence epilepsy. Seizure. 2000;9:265–9. [PubMed: 10880286]
- 51.
- Kapucu LO, Serdaroglu A, Okuyaz C, Kose G, Gucuyener K. Brain single photon emission computed tomographic evaluation of patients with childhood absence epilepsy. J Child Neurol. 2003;18:542–8. [PubMed: 13677580]
- 52.
- Iannetti P, Spalice A, De Luca PF, Boemi S, Festa A, Maini CL. Ictal single photon emission computed tomography in absence seizures: apparent implication of different neuronal mechanisms. J Child Neurol. 2001;16:339–44. [PubMed: 11392518]
- 53.
- Meencke HJ. Pathological findings in childhood absence epilepsy. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 122–32.
- 54.
- Woermann FG, Sisodiya SM, Free SL, Duncan JS. Quantitative MRI in patients with idiopathic generalized epilepsy. Evidence of widespread cerebral structural changes. Brain. 1998;121:1661–7. [PubMed: 9762955]
- 55.
- Opeskin K, Kalnins RM, Halliday G, Cartwright H, Berkovic SF. Idiopathic generalized epilepsy: lack of significant microdysgenesis. Neurology. 2000;55:1101–6. [PubMed: 11071485]
- 56.
- Panayiotopoulos CP, Chroni E, Daskalopoulos C, Baker A, Rowlinson S, Walsh P. Typical absence seizures in adults: clinical, EEG, video-EEG findings and diagnostic/syndromic considerations. J Neurol Neurosurg Psychiatr. 1992;55:1002–8. [PMC free article: PMC1015282] [PubMed: 1469393]
- 57.
- Panayiotopoulos CP, Giannakodimos S, Chroni E. Typical absences in adults. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 289–99.
- 58.
- Panayiotopoulos CP, Koutroumanidis M, Giannakodimos S, Agathonikou A. Idiopathic generalised epilepsy in adults manifested by phantom absences, generalised tonic-clonic seizures, and frequent absence status. J Neurol Neurosurg Psychiatr. 1997;63:622–7. [PMC free article: PMC2169820] [PubMed: 9408104]
- 59.
- Gastaut H. The epilepsies:Electro-clinical correlations. Springfield,Illinois; CC Thomas: 1954.
- 60.
- Panayiotopoulos CP, Obeid T, Tahan AR. Juvenile myoclonic epilepsy: a 5-year prospective study. Epilepsia. 1994;35:285–96. [PubMed: 8156946]
- 61.
- Janz D, Durner M. Juvenile myoclonic epilepsy. In: Engel JJ, Pedley TA, editors. Epilepsy: A comprehensive Textbook. Philadelphia: Lippincott-Raven Publishers; 1997. pp. 2389–400.
- 62.
- Diagnosing juvenile myoclonic epilepsy [editorial] Lancet. 1992;340:759–60. [PubMed: 1356182]
- 63.
- Wolf P. Epilepsy with grand mal on awakening. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey & Company; 1992. pp. 329–41.
- 64.
- Shorvon SD. Status epilepticus:its clinical features and treatment in children and adults. Cambridge: Cambridge University Press; 1994.
- 65.
- Shorvon S. Absence status epilepticus. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 263–74.
- 66.
- Panayiotopoulos CP. Absence status epilepticus. In: Gilman S, editor. Medlink Neurology. San Diego SA: Arbor Publishing Corp; 2004.
- 67.
- Gastaut H. Part I:Definitions. Geneva: World Health Organisation; 1973. Dictionary of epilepsies.
- 68.
- Treatment of convulsive status epilepticus. Recommendations of the Epilepsy Foundation of America’s Working Group on Status Epilepticus. JAMA. 1993;270:854–9. [PubMed: 8340986]
- 69.
- Bleck TP. Management approaches to prolonged seizures and status epilepticus. Epilepsia. 1999;40(Suppl 1):S59–S63. [PubMed: 10421562]
- 70.
- Blume WT, Luders HO, Mizrahi E, Tassinari C, van Emde BW, Engel J Jr. Glossary of descriptive terminology for ictal semiology: report of the ILAE task force on classification and terminology. Epilepsia. 2001;42:1212–8. [PubMed: 11580774]
- 71.
- Kaplan PW. Assessing the outcomes in patients with nonconvulsive status epilepticus: nonconvulsive status epilepticus is underdiagnosed, potentially overtreated, and confounded by comorbidity. J Clin Neurophysiol. 1999;16:341–52. [PubMed: 10478707]
- 72.
- Kaplan PW. Behavioral Manifestations of Nonconvulsive Status Epilepticus. Epilepsy Behav. 2002;3:122–39. [PubMed: 12609414]
- 73.
- Thomas P, Andermann F. Late-onset absence status epilepticus is most often situation-related. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic generalized epilepsies. London: John Libbey & Company Ltd; 1994. pp. 95–109.
- 74.
- Thomas P. Absence status epilepsy. Rev Neurol (Paris). 1999;155:1023–38. [PubMed: 10637922]
- 75.
- Snead OC III, Dean JC, Penry JK. Absence status epilepticus. In: Engel JJ, Pedley TA, editors. Epilepsy:a comprehensive textbook. Philadelphia: Lippincott-Raven Publishers; 1997. pp. 701–7.
- 76.
- Baykan B, Gokyigit A, Gurses C, Eraksoy M. Recurrent absence status epilepticus: clinical and EEG characteristics. Seizure. 2002;11:310–9. [PubMed: 12076103]
- 77.
- Panayiotopoulos CP, Ferrie CD, Koutroumanidis M, Rowlinson S, Sanders S. Idiopathic generalised epilepsy with phantom absences and absence status in a child. Epileptic Disord. 2001;3:63–6. [PubMed: 11431167]
- 78.
- Skardoutsou A, Voudris KA, Vagiakou EA. Non-convulsive status epilepticus associated with tiagabine therapy in children. Seizure. 2003;12:599–601. [PubMed: 14630501]
- 79.
- Schapel G, Chadwick D. Tiagabine and non-convulsive status epilepticus. Seizure. 1996;5:153–6. [PubMed: 8795133]
- 80.
- Osorio I, Reed RC, Peltzer JN. Refractory idiopathic absence status epilepticus: A probable paradoxical effect of phenytoin and carbamazepine. Epilepsia. 2000;41:887–94. [PubMed: 10897162]
- 81.
- Agathonikou A, Koutroumanidis M, Panayiotopoulos CP. Fixation-off-sensitive epilepsy with absences and absence status: video-EEG documentation. Neurology. 1997;48:231–4. [PubMed: 9008523]
- 82.
- Ming X, Kaplan PW. Fixation-off and eyes closed catamenial generalized nonconvulsive status epilepticus with eyelid myoclonic jerks. Epilepsia. 1998;39:664–8. [PubMed: 9637610]
- 83.
- Foldvary-Schaefer N, Falcone T. Catamenial epilepsy: pathophysiology, diagnosis, and management. Neurology. 2003;61:S2–15. [PubMed: 14504304]
- 84.
- Engel J Jr. Classifications of the International League Against Epilepsy: time for reappraisal. Epilepsia. 1998;39:1014–7. [PubMed: 9738683]
- 85.
- Engel J Jr. Classification of Epileptic Disorders. Epilepsia. 2001;42:316. [PubMed: 11442146]
- 86.
- Andermann F, Berkovic SF. Idiopathic generalized epilepsy with generalised and other seizures in adolescence. Epilepsia. 2001;42:317–20. [PubMed: 11442147]
- 87.
- Doose H. Das akinetische petit mal. Arch Psychiatr Nervenkr. 1965;205:638–54. [PubMed: 14334557]
- 88.
- Doose H, Gerken H, Leonhardt T, Volz E, Volz C. Centrencephalic myoclonic-astatic petit mal. Clinical and genetic investigation. Neuropadiatrie. 1970;2:59–78. [PubMed: 5001125]
- 89.
- Doose H. Myoclonic-astatic epilepsy. Epilepsy Research-Supplement. 1992;6:163–8. [PubMed: 1418479]
- 90.
- Aicardi J. Myoclonic-astatic epilepsy. In: Wallace S, editor. Epilepsy in Children. London: Chapman & Hall; 1996. pp. 263–70.
- 91.
- Oguni H, Fukuyama Y, Tanaka T, Hayashi K, Funatsuka M, Sakauchi M, et al. Myoclonic-astatic epilepsy of early childhood—clinical and EEG analysis of myoclonic-astatic seizures, and discussions on the nosology of the syndrome. Brain Dev. 2001;23:757–64. [PubMed: 11701290]
- 92.
- Dulac O, Dreifuss F. Myoclonic-astatic epilepsy of childhood. In: Gilman S, editor. Medlink Neurology. San Diego SA: Arbor Publishing Corp; 2004.
- 93.
- Weber P, Tillmann B, Minet JC, Blauenstein U. Myoclonic-astatic epilepsy in early childhood: review of clinical signs, EEG features, etiology, and therapy. Klin Padiatr. 2002;214:279–84. [PubMed: 12235543]
- 94.
- Guerrini R, Parmeggiani A, Kaminska A, Dulac O. Myoclonic astatic epilepsy. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3. London: John Libbey & Co Ltd; 2002. pp. 105–12.
- 95.
- Oguni H, Tanaka T, Hayashi K, Funatsuka M, Sakauchi M, Shirakawa S, et al. Treatment and long-term prognosis of myoclonic-astatic epilepsy of early childhood. Neuropediatrics. 2002;33:122–32. [PubMed: 12200741]
- 96.
- Guerrini R, Aicardi J. Epileptic encephalopathies with myoclonic seizures in infants and children (severe myoclonic epilepsy and myoclonic-astatic epilepsy). J Clin Neurophysiol. 2003;20:449–61. [PubMed: 14734934]
- 97.
- Aicardi J. Myoclonic-astatic epilepsy. In: Wallace SJ, Farrell K, editors. Epilepsy in childen. London: Arnold; 2004. pp. 163–8.
- 98.
- Doose H. Myoclonic-astatic epilepsy. Epilepsy Res Suppl. 1992;6:163–8. [PubMed: 1418479]
- 99.
- Aicardi J, Levy Gomes A. Clinical and electroencephalographic symptomatology of the ‘genuine’ Lennox-Gastaut syndrome and its differentiation from other forms of epilepsy of early childhood. Epilepsy Res-Suppl. 1992;6:185–93. [PubMed: 1418482]
- 100.
- Kaminska A, Ickowicz A, Plouin P, Bru MF, Dellatolas G, Dulac O. Delineation of cryptogenic Lennox-Gastaut syndrome and myoclonic astatic epilepsy using multiple correspondence analysis. Epilepsy Res. 1999;36:15–29. [PubMed: 10463847]
- 101.
- Scheffer IE, Wallace R, Mulley JC, Berkovic SF. Clinical and molecular genetics of myoclonic-astatic epilepsy and severe myoclonic epilepsy in infancy (Dravet syndrome). Brain Dev. 2001;23:732–5. [PubMed: 11701287]
- 102.
- Bonanni P, Parmeggiani L, Guerrini R. Different neurophysiologic patterns of myoclonus characterize Lennox-Gastaut syndrome and myoclonic astatic epilepsy. Epilepsia. 2002;43:609–15. [PubMed: 12060020]
- 103.
- Aicardi J. Epilepsy in Children. New York: Raven Press; 1994.
- 104.
- Aicardi J, Chevrie JJ. Atypical benign partial epilepsy of childhood. Devel Med Child Neurol. 1982;24:281–92. [PubMed: 6807733]
- 105.
- Arzimanoglou A, Guerrini R, Aicardi J. Aicardi’s epilepsy in children. Philadelphia: Lippincott Williams & Wilkins; 2004.
- 106.
- Fejerman N, Caraballo R, Tenembaum SN. Atypical evolutions of benign localization-related epilepsies in children: are they predictable? Epilepsia. 2000;41:380–90. [PubMed: 10756401]
- 107.
- Fejerman N. Atypical evolution of benign partial epilepsy in children. Rev Neurol. 1996;24:1415–20. [PubMed: 8974748]
- 108.
- Ferrie CD, Panayiotopoulos CP. Idiopathic generalised epilepsy with generalised tonic clonic seizures on awakening. In: Wallace SJ, Farrell K, editors. Epilepsy in Children. 2. London: Edward Arnold (Publishers) Limited; 2002.
- 109.
- Caraballo RH, Astorino F, Cersosimo R, Soprano AM, Fejerman N. Atypical evolution in childhood epilepsy with occipital paroxysms (Panayiotopoulos type). Epileptic Disord. 2001;3:157–62. [PubMed: 11679309]
- 110.
- Chapman K, Holland K, Erenberg G. Seizure exacerbation associated with oxcarbazepine in idiopathic focal epilepsy of childhood. Neurology. 2003;61:1012. [PubMed: 14557585]
- 111.
- Catania S, Cross H, De Sousa C, Boyd S. Paradoxic reaction to lamotrigine in a child with benign focal epilepsy of childhood with centrotemporal spikes. Epilepsia. 1999;40:1657–60. [PubMed: 10565596]
- 112.
- Jayawant S, Libretto SE. Topiramate in the treatment of myoclonic-astatic epilepsy in children: A retrospective hospital audit. J.Postgrad Med. 2003;49:202–6. [PubMed: 14597780]
- 113.
- Adie WJ. Pyknolepsy:a form of epilepsy occurring in children with a good prognosis. Brain. 1924;47:96–102. [PMC free article: PMC2201422] [PubMed: 19983793]
- 114.
- Loiseau P, Panayiotopoulos CP. Childhood absence epilepsy. In: Gilman S, editor. Medlink Neurology. San Diego SA: Arbor Publishing Corp; 2004.
- 115.
- Lennox WG, Lennox MA. Epilepsy and related disorders. Boston: Little,Brown & Co; 1960.
- 116.
- Currier RD, Kooi KA, Saidman J. Prognosis of pure petit mal:a follow-up study. Neurology. 1963;13:959–67. [PubMed: 14079956]
- 117.
- Livingston S, Torres J, Pauli LL, Rider RV. Petit mal epilepsy: results of a prolonged follow-up study of 117 patients. JAMA. 1965;194:227–32. [PubMed: 4953647]
- 118.
- Beaumanoir A. Problèmes de diagnostic et de traitement. Bâle: Editions Roche; 1976. Les épilepsies infantiles.
- 119.
- Cavazzuti GB, Ferrari F, Galli V, Benatti A. Epilepsy with typical absence seizures with onset during the first year of life. Epilepsia. 1989;30:802–6. [PubMed: 2512115]
- 120.
- Darra F, Fontana E, Scaramuzzi V, Santorum E, Zoccante L, Zulumi E, et al. Typical absence seizures in the first three years of life: electroclinical study of 31 cases. Epilepsia. 1996;37(Suppl 4):95.
- 121.
- Aicardi J. Typical absences in the first two years of life. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 284–8.
- 122.
- Hertoft P. The clinical, electroencephalographic and social prognosis in petit mal epilepsy. Epilepsia. 1963;4:298–314. [PubMed: 14122548]
- 123.
- Bergamini L, Bram S, Broglia S, Riccio A. L’insorgenza tardiva di crisi Grande Male nel Piccolo Male puro. Studio catamnestico di 78 casi. Arch Suisses Neurol Neurochir Psychiatr. 1965;96:306–17. [PubMed: 4956413]
- 124.
- Rocca WA, Sharbrough FW, Hauser WA, Annegers JF, Schoenberg BS. Risk factors for absence seizures: a population-based case-control study in Rochester, Minnesota. Neurology. 1987;37:1309–14. [PubMed: 3112608]
- 125.
- Hollowack J, Thurston DL, O’Leary J L. Petit mal epilepsy. Pediatrics. 1962;60:893–901. [PubMed: 13961432]
- 126.
- Callenbach PM, Geerts AT, Arts WF, van Donselaar CA, Peters AC, Stroink H, et al. Familial occurrence of epilepsy in children with newly diagnosed multiple seizures: Dutch Study of Epilepsy in Childhood. Epilepsia. 1998;39:331–6. [PubMed: 9578054]
- 127.
- Berg AT, Levy SR, Testa FM, Shinnar S. Classification of childhood epilepsy syndromes in newly diagnosed epilepsy: interrater agreement and reasons for disagreement. Epilepsia. 1999;40:439–44. [PubMed: 10219269]
- 128.
- Berg AT, Shinnar S, Levy SR, Testa FM, Smith-Rapaport S, Beckerman B. How well can epilepsy syndromes be identified at diagnosis? A reassessment 2 years after initial diagnosis. Epilepsia. 2000;41:1269–75. [PubMed: 11051121]
- 129.
- Olsson I. Epidemiology of absence epilepsy. I. Concept and incidence. Acta Paediatr Scand. 1988;77:860–6. [PubMed: 3144826]
- 130.
- Loiseau J, Loiseau P, Guyot M, Duche B, Dartigues JF, Aublet B. Survey of seizure disorders in the French southwest. I. Incidence of epileptic syndromes. Epilepsia. 1990;31:391–6. [PubMed: 2369875]
- 131.
- Blom S, Heijbel J, Bergfors PG. Incidence of epilepsy in children: a follow-up study three years after the first seizure. Epilepsia. 1978;19:343–50. [PubMed: 100316]
- 132.
- Olsson I, Hagberg G. Epidemiology of absence epilepsy. III. Clinical aspects. Acta Paediatr Scand. 1991;80:1066–72. [PubMed: 1750340]
- 133.
- Olsson I, Hedstrom A. Epidemiology of absence epilepsy. II. Typical absences in children with encephalopathies. Acta Paediatr Scand. 1991;80:235–42. [PubMed: 1903572]
- 134.
- Berkovic SF. Childhood absence epilepsy and juvenile absence epilepsy. In: Wyllie E, editor. The treatment of epilepsy:Principles and practice. Philadelphia: Lea & Febiger; 1993. pp. 547–51.
- 135.
- Berkovic SF. Generalised absence seizures. In: Wyllie E, editor. The treatment of epilepsy:Principles and practice. Philadelphia: Lea & Febiger; 1993. pp. 401–10.
- 136.
- Loiseau P. Childhood absence epilepsy. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey & Company; 1992. pp. 135–50.
- 137.
- Sander JWAS. The epidemiology and prognosis of typical absence seizures. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 135–44.
- 138.
- Fong GC, Shah PU, Gee MN, Serratosa JM, Castroviejo IP, Khan S, et al. Childhood absence epilepsy with tonic-clonic seizures and electroencephalogram 3–4-Hz spike and multispike-slow wave complexes: linkage to chromosome 8q24. Am J Hum Genet. 1998;63:1117–29. [PMC free article: PMC1377498] [PubMed: 9758624]
- 139.
- Wirrell EC. Natural history of absence epilepsy in children. Can J Neurol Sci. 2003;30:184–8. [PubMed: 12945939]
- 140.
- Wirrell EC, Camfield CS, Camfield PR, Gordon KE, Dooley JM. Long-term prognosis of typical childhood absence epilepsy: remission or progression to juvenile myoclonic epilepsy. Neurology. 1996;47:912–8. [PubMed: 8857718]
- 141.
- Panayiotopoulos CP. Typical absences are syndrome related. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 304–10.
- 142.
- Fakhoury T, Abou-Khalil B. Generalized absence seizures with 10–15 Hz fast discharges. Clin Neurophysiol. 1999;110:1029–35. [PubMed: 10402089]
- 143.
- Hirsch E, Marescaux C. What are the relevant criteria for a better classification of epileptic syndromes with typical absences? In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic generalised epilepsies. London: John Libbey & Company Ltd; 1994. pp. 87–93.
- 144.
- Agathonikou A, Giannakodimos S, Koutroumanidis M, Parker APJ, Ahmed Sharoqi I, Rowlinson S, et al. Idiopathic GeneralisedEpilepsies in Adults with Onset of Typical Absences before the Age of 10 Years. Epilepsia. 1997;38(Suppl 3):213.
- 145.
- Loiseau P, Duche B, Pedespan JM. Absence epilepsies. Epilepsia. 1995;36:1182–6. [PubMed: 7489694]
- 146.
- Loiseau P, Duche B. Childhood absence epilepsy. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 152–60.
- 147.
- Dieterich E, Baier WK, Doose H, Tuxhorn I, Fichsel H. Longterm follow-up of childhood epilepsy with absences. I. Epilepsy with absences at onset. Neuropediatrics. 1985;16:149–54. [PubMed: 3930988]
- 148.
- Panayiotopoulos CP. Lamictal (lamotrigine) monotherapy for typical absence seizures in children. Epilepsia. 2000;41:357–9. [PubMed: 10403222]
- 149.
- Panayiotopoulos CP. Typical absence seizures and their treatment. Arch Dis Child. 1999;81:351–5. [PMC free article: PMC1718096] [PubMed: 10490445]
- 150.
- Wirrell EC, Camfield PR, Gordon KE, Camfield CS, Dooley JM, Hanna BD. Will a critical level of hyperventilation-induced hypocapnia always induce an absence seizure? Epilepsia. 1996;37:459–62. [PubMed: 8617175]
- 151.
- Dalby MA. Epilepsy and 3 per second spike and wave rhythms. A clinical,electroencephalographic and prognostic analysis of 346 patients. Acta Neurol Scand. 1969;40(Suppl ):1–183. [PubMed: 4979890]
- 152.
- Bureau M, Guey J, Dravet C, Roger J. Etude de la répartition des absences chez l’enfant en fonction de ses activités. Rev Neurol. 1968;118:493–4. [PubMed: 4972884]
- 153.
- Robinson R, Taske N, Sander T, Heils A, Whitehouse W, Goutieres F, et al. Linkage analysis between childhood absence epilepsy and genes encoding GABAA and GABAB receptors, voltage-dependent calcium channels, and the ECA1 region on chromosome 8q. Epilepsy Res. 2002;48:169–79. [PubMed: 11904235]
- 154.
- Loiseau P, Pestre M, Dartigues JF, Commenges D, Barberger-Gateau C, Cohadon S. Long-term prognosis in two forms of childhood epilepsy: typical absence seizures and epilepsy with rolandic (centrotemporal) EEG foci. Ann Neurol. 1983;13:642–8. [PubMed: 6410975]
- 155.
- Niedermeyer E, Lopes da Silva F. Electroencephalography. Basic principles,clinical applications, and related fields. 4. Baltimore: Williams & Wilkins; 1999.
- 156.
- Lees F, Liversedge LA. The prognosis of petit mal and minor epilepsy. Lancet. 1962;i:797–82.
- 157.
- Charlton MH, Yahr M D. Long term follow up of patients with petit mal. Arch Neurol. 1967;16:595–8. [PubMed: 4961019]
- 158.
- Gibberd FB. The prognosis of petit mal. Brain. 1966;89:531–8. [PubMed: 4958588]
- 159.
- Gibberd F B. The prognosis of petit mal in adults. Epilepsia. 1972;3:171–5. [PubMed: 4622569]
- 160.
- Lugaresi E, Pazzaglia PP, Franck L, Roger J, Bureau-Paillas M, Ambrosetto G, et al. Evolution and prognosis of primary generalised epilepsy of petit mal absence type. In: Lugaressi E, Pazzaglia PP, Tassinari CA, editors. Evolution and prognosis of epilepsy. Bologna: Auto Gaggi; 1973. pp. 3–22.
- 161.
- Gastaut H, Zifkin BG, Mariani E, Puig JS. The long-term course of primary generalized epilepsy with persisting absences. Neurology. 1986;36:1021–8. [PubMed: 3090476]
- 162.
- Hedstrom A, Olsson I. Epidemiology of absence epilepsy: EEG findings and their predictive value. Pediat Neurol. 1991;7:100–4. [PubMed: 1905542]
- 163.
- Sato S, Dreifuss FE, Penry JK. Prognostic factors in absence seizures. Neurology. 1976;26:788–96. [PubMed: 821006]
- 164.
- Sato S, Dreifuss FE, Penry JK, Kirby DD, Palesch Y. Long-term follow-up of absence seizures. Neurology. 1983;33:1590–5. [PubMed: 6417557]
- 165.
- Covanis A, Skiadas K, Loli N, Lada C, Theodorou V. Absence epilepsy: early prognostic signs. Seizure. 1992;1:281–9. [PubMed: 1344778]
- 166.
- Dieterich E, Doose H, Baier WK, Fichsel H. Longterm follow-up of childhood epilepsy with absences. II. Absence-epilepsy with initial grand mal. Neuropediatrics. 1985;16:155–8. [PubMed: 3930989]
- 167.
- Bartolomei F, Roger J, Bureau M, Genton P, Dravet C, Viallat D, et al. Prognostic factors for childhood and juvenile absence epilepsies. Eur Neurol. 1997;37:169–75. [PubMed: 9137927]
- 168.
- Oller L. Prospective study of the differences between the syndromes of infantile absence epilepsy and syndromes of juvenile absence epilepsy. Rev Neurol. 1996;24:930–6. [PubMed: 8755355]
- 169.
- Trinka E, Baumgartner S, Unterberger I, Unterrainer J, Luef G, Haberlandt E, et al. Long-term prognosis for childhood and juvenile absence epilepsy. J Neurol. 2004;251:1235–41. [PubMed: 15503104]
- 170.
- Frank LM, Enlow T, Holmes GL, Manasco P, Concannon S, Chen C, et al. Lamictal (lamotrigine) monotherapy for typical absence seizures in children. Epilepsia. 1999;40:973–9. [PubMed: 10403222]
- 171.
- Coppola G, Licciardi F, Sciscio N, Russo F, Carotenuto M, Pascotto A. Lamotrigine as first-line drug in childhood absence epilepsy: a clinical and neurophysiological study. Brain Dev. 2004;26:26–9. [PubMed: 14729411]
- 172.
- Browne TR, Dreifuss FE, Penry JK, Porter RJ, White BG. Clinical and EEG estimates of absence seizure frequency. Arch Neurol. 1983;40:469–72. [PubMed: 6409062]
- 173.
- Tassinari CA, Bureau M, Thomas P. Epilepsy with myoclonic absences. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey & Company; 1992. pp. 151–60.
- 174.
- Manonmani V, Wallace SJ. Epilepsy with myoclonic absences. Arch Dis Child. 1994;70:288–90. [PMC free article: PMC1029780] [PubMed: 8185360]
- 175.
- Verrotti A, Greco R, Chiarelli F, Domizio S, Sabatino G, Morgese G. Epilepsy with myoclonic absences with early onset: a follow-up study. J Child Neurol. 1999;14:746–9. [PubMed: 10593554]
- 176.
- Tassinari CA, Rubboli G, Michellluchi R. Epilepsy with myoclonic absences. In: Gilman S, editor. Medlink Neurology. San Diego SA: Arbor Publishing Corp; 2004.
- 177.
- Tassinari CA, Rubboli G, Gardella E, Michellluchi R. Epilepsy with myoclonic absences. In: Wallace SJ, Farrell K, editors. Epilepsy in childen. London: Arnold; 2004. pp. 189–94.
- 178.
- Tassinari CA, Michelucci R, Rubboli G, Passarelli D, Riguzzi P, Parmeggiani L, et al. Myoclonic absence epilepsy. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 187–95.
- 179.
- Elia M, Guerrini R, Musumeci SA, Bonanni P, Gambardella A, Aguglia U. Myoclonic absence-like seizures and chromosome abnormality syndromes. Epilepsia. 1998;39:660–3. [PubMed: 9637609]
- 180.
- Aguglia U, Le Piane E, Gambardella A, Messina D, Russo C, Sirchia SM, et al. Emotion-induced myoclonic absence-like seizures in a patient with inv-dup(15) syndrome: a clinical, EEG, and molecular genetic study. Epilepsia. 1999;40:1316–9. [PubMed: 10487199]
- 181.
- Kimura S, Adachi K. Localization-related epilepsy mimicking epilepsy with myoclonic absence in a patient with pachygyria. Pediatr.Int. 2002;44:171–3. [PubMed: 11896877]
- 182.
- Ferrie CD, Giannakodimos S, Robinson RO, Panayiotopoulos CP. Symptomatic typical absence seizures. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 241–52.
- 183.
- Fattouch J, Di Bonaventura C, Mari F, Egeo G, Vaudano AE, Mascia A, et al. Role of levetiracetam in the treatment of primary generalised epilepsy. Epilepsia. 2004;45(Suppl 3):142.
- 184.
- Wolf P. Juvenile absence epilepsy. In: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey & Company; 1992. pp. 307–12.
- 185.
- Obeid T. Clinical and genetic aspects of juvenile absence epilepsy. J Neurol. 1994;241:487–91. [PubMed: 7964917]
- 186.
- Osservatorio Regionale per L’Epilessia (OREp), Lombardy. Osservatorio Regionale per L’Epilessia (OREp) L. ILAE classification of epilepsies: its applicability and practical value of different diagnostic categories. Epilepsia. 1996;37:1051–9. [PubMed: 8917054]
- 187.
- Doose H, Volzke E, Scheffner D. Verlaufsformen kindlicher epilepsien mit spike wave-absencen. Arch Psychiatr Nervenkr. 1965;207:394–415. [PubMed: 4960128]
- 188.
- Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins. In: Wolf P, editor. Epileptic seizures and syndromes. London: John Libbey & Company Ltd; 1994. pp. 157–64.
- 189.
- Bianchi A. the Italian LAE Collaborative Group. Study of concordance of symptoms in families with absence epilepsies. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 328–37.
- 190.
- Durner M, Zhou G, Fu D, Abreu P, Shinnar S, Resor SR, et al. Evidence for linkage of adolescent-onset idiopathic generalized epilepsies to chromosome 8-and genetic heterogeneity. Am J Hum Genet. 1999;64:1411–9. [PMC free article: PMC1377879] [PubMed: 10205274]
- 191.
- Sander T, Hildmann T, Kretz R, Furst R, Sailer U, Bauer G, et al. Allelic association of juvenile absence epilepsy with a GluR5 kainate receptor gene (GRIK1) polymorphism. Am J Med Genet. 1997;74:416–21. [PubMed: 9259378]
- 192.
- Meencke HJ, Janz D. The significance of microdysgenesia in primary generalized epilepsy: an answer to the considerations of Lyon and Gastaut. Epilepsia. 1985;26:368–71. [PubMed: 4006898]
- 193.
- Gericke CA, Picard F, Saint-Martin A, Strumia S, Marescaux C, Hirsch E. Efficacy of lamotrigine in idiopathic generalized epilepsy syndromes: a video-EEG-controlled, open study. Epileptic Disord. 1999;1:159–65. [PubMed: 10937148]
- 194.
- French JA, Kanner AM, Bautista J, Abou-Khalil B, Browne T, Harden CL, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new onset epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1252–60. [PubMed: 15111659]
- 195.
- Thomas P, Genton P, Wolf P. Juvenile myoclonic epilepsy. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editors. Epileptic syndromes in infancy, childhood and adolescence. 3. London: John Libbey & Co Ltd; 2002. pp. 335–56.
- 196.
- Genton P, Gelisse P. Juvenile myoclonic epilepsy. Arch Neurol. 2001;58:1487–90. [PubMed: 11559326]
- 197.
- Panayiotopoulos CP. Juvenile myoclonic epilepsy: an uderdiagnosed syndrome. In: Wolf P, editor. Epileptic seizures and syndromes. London: John Libbey & Company Ltd; 1994. pp. 221–30.
- 198.
- Grunewald RA, Panayiotopoulos CP. Juvenile myoclonic epilepsy. A review. Arch Neurol. 1993;50:594–8. [PubMed: 8503795]
- 199.
- Delgado-Escueta AV, Enrile-Bacsal F. Juvenile myoclonic epilepsy of Janz. Neurology. 1984;34:285–94. [PubMed: 6422321]
- 200.
- Schmitz B, Sander T. Juvenile myoclonic epilepsy: the Janz syndrome. Petersfield, UK: Wrightson Biomedical Publishing Ltd; 2000.
- 201.
- Janz D, Christian W. Impulsiv-Petit mal. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Zeitschrift f Nervenheilkunde. Idiopathic generalised epilepsies. Vol. 176. London: John Libbey & Company Ltd; 1957. 1957. pp. 346–386.pp. 229–51. (Translated in English by Genton,P.)
- 202.
- Gilliam F, Steinhoff BJ, Bittermann HJ, Kuzniecky R, Faught E, Abou-Khalil B. Adult myoclonic epilepsy: a distinct syndrome of idiopathic generalized epilepsy. Neurology. 2000;55:1030–3. [PubMed: 11061264]
- 203.
- Tsuboi T, Christian W. On the genetics of the primary generalised epilepsy with sporadic myoclonus of impulsive petit mal type. Humangenetik. 1973;19:155–82. [PubMed: 4200688]
- 204.
- Tsuboi T, Christian W. Epilepsy:A clinical, electroencephalographic and statistical study of 466 patients. Berlin: Springer; 1976. [PubMed: 973125]
- 205.
- Grunewald RA, Chroni E, Panayiotopoulos CP. Delayed diagnosis of juvenile myoclonic epilepsy. J Neurol Neurosurg Psychiatry. 1992;55:497–9. [PMC free article: PMC1014908] [PubMed: 1619419]
- 206.
- Sander JW, Hart YM, Johnson AL, Shorvon SD. National General Practice Study of Epilepsy: newly diagnosed epileptic seizures in a general population. Lancet. 1990;336:1267–71. [PubMed: 1978113]
- 207.
- Manford M, Hart YM, Sander JW, Shorvon SD. The National General Practice Study of Epilepsy. The syndromic classification of the International League Against Epilepsy applied to epilepsy in a general population. Arch Neurol. 1992;49:801–8. [PubMed: 1524512]
- 208.
- Oguni H, Mukahira K, Oguni M, Uehara T, Su YH, Izumi T, et al. Video-polygraphic analysis of myoclonic seizures in juvenile myoclonic epilepsy. Epilepsia. 1994;35:307–16. [PubMed: 8156949]
- 209.
- Jain S, Padma MV, Maheshwari MC. Occurrence of only myoclonic jerks in juvenile myoclonic epilepsy. Acta Neurol Scand. 1997;95:263–7. [PubMed: 9188899]
- 210.
- Salas Puig J, Tunon A, Vidal JA, Mateos V, Guisasola LM, Lahoz CH. Janz’s juvenile myoclonic epilepsy: a little-known frequent syndrome. A study of 85 patients. Med.Clin.(Barc). 1994;103:684–9. [PubMed: 7808074]
- 211.
- Matsuoka H. A clinical and electroencephalographic study of juvenile myoclonic epilepsy: its pathophysiological considerations based on the findings obtained from neuropsychological EEG activation. Seishin Shinkeigaku Zasshi. 1989;91:318–46. [PubMed: 2510209]
- 212.
- Matsuoka H, Takahashi T, Sasaki M, Yoshida S, Numachi Y, Sato M. The long-term course of seizure susceptibility in two patients with juvenile myoclonic epilepsy. Seizure. 2002;11:126–30. [PubMed: 11945100]
- 213.
- Mayer T, Wolf P. Reading epilepsy: clinical and genetic background. In: Berkovic SF, Genton P, Hirsch E, Picard F, editors. Genetics of focal epilepsies. London: John Libbey and Company Ltd; 1999. pp. 159–68.
- 214.
- Chifari R, Piazzini A, Turner K, Canger R, Canevini MP, Wolf P. Reflex writing seizures in two siblings with juvenile myoclonic epilepsy. Acta Neurol Scand. 2004;109:232–5. [PubMed: 14763964]
- 215.
- Panayiotopoulos CP, Obeid T. Juvenile myoclonic epilepsy: an autosomal recessive disease. Ann Neurol. 1989;25:440–3. [PubMed: 2505665]
- 216.
- Canevini MP, Mai R, Di Marco C, Bertin C, Minotti L, Pontrelli V, et al. Juvenile myoclonic epilepsy of Janz: clinical observations in 60 patients. Seizure. 1992;1:291–8. [PubMed: 1344779]
- 217.
- Janz D. Juvenile myoclonic epilepsy. Epilepsy with impulsive petit mal. Cleve Clin J Med. 1989;56(Suppl Pt 1):S23–33. S40. [PubMed: 2498010]
- 218.
- Serratosa JM, Delgado-Escueta AV, Medina MT, Zhang Q, Iranmanesh R, Sparkes RS. Clinical and genetic analysis of a large pedigree with juvenile myoclonic epilepsy. Ann Neurol. 1996;39:187–95. [PubMed: 8967750]
- 219.
- Delgado-Escueta AV, Greenberg D, Weissbecker K, Liu A, Treiman L, Sparkes R, et al. Gene mapping in the idiopathic generalized epilepsies: juvenile myoclonic epilepsy, childhood absence epilepsy, epilepsy with grand mal seizures, and early childhood myoclonic epilepsy. Epilepsia. 1990;31(Suppl 3):S19–29. [PubMed: 2121470]
- 220.
- Elmslie FV, Rees M, Williamson MP, Kerr M, Kjeldsen MJ, Pang KA, et al. Genetic mapping of a major susceptibility locus for juvenile myoclonic epilepsy on chromosome 15q. Hum Mol Genet. 1997;6:1329–34. [PubMed: 9259280]
- 221.
- Taske NL, Williamson MP, Makoff A, Bate L, Curtis D, Kerr M, et al. Evaluation of the positional candidate gene CHRNA7 at the juvenile myoclonic epilepsy locus (EJM2) on chromosome 15q13–14. Epilepsy Res. 2002;49:157–72. [PubMed: 12049804]
- 222.
- Suzuki T, Ganesh S, Agarwala KL, Morita R, Sugimoto Y, Inazawa J, et al. A novel gene in the chromosomal region for juvenile myoclonic epilepsy on 6p12 encodes a brain-specific lysosomal membrane protein. Biochem Biophys Res Commun. 2001;288:626–36. [PubMed: 11676489]
- 223.
- Pal DK, Evgrafov OV, Tabares P, Zhang F, Durner M, Greenberg DA. BRD2 (RING3) is a probable major susceptibility gene for common juvenile myoclonic epilepsy. Am J Hum Genet. 2003;73:261–70. [PMC free article: PMC1180366] [PubMed: 12830434]
- 224.
- Greenberg DA, Durner M, Shinnar S, Resor S, Rosenbaum D, Klotz I, et al. Association of HLA class II alleles in patients with juvenile myoclonic epilepsy compared with patients with other forms of adolescent-onset generalized epilepsy. Neurology. 1996;47:750–5. [PubMed: 8797474]
- 225.
- Obeid T, el Rab MO, Daif AK, Panayiotopoulos CP, Halim K, Bahakim H, et al. Is HLA-DRW13 (W6) associated with juvenile myoclonic epilepsy in Arab patients? Epilepsia. 1994;35:319–21. [PubMed: 8156951]
- 226.
- Le Hellard S, Neidhart E, Thomas P, Feingold J, Malafosse A, Tafti M. Lack of association between juvenile myoclonic epilepsy and HLA- DR13. Epilepsia. 1999;40:117–9. [PubMed: 9924913]
- 227.
- Taylor I, Marini C, Johnson MR, Turner S, Berkovic SF, Scheffer IE. Juvenile myoclonic epilepsy and idiopathic photosensitive occipital lobe epilepsy: is there overlap? Brain. 2004;127:1878–86. [PubMed: 15201194]
- 228.
- Woermann FG, Free SL, Koepp MJ, Sisodiya SM, Duncan JS. Abnormal cerebral structure in juvenile myoclonic epilepsy demonstrated with voxel-based analysis of MRI. Brain. 1999;122:2101–8. [PubMed: 10545395]
- 229.
- Montalenti E, Imperiale D, Rovera A, Bergamasco B, Benna P. Clinical features, EEG findings and diagnostic pitfalls in juvenile myoclonic epilepsy: a series of 63 patients. J Neurol Sci. 2001;184:65–70. [PubMed: 11231034]
- 230.
- Panayiotopoulos CP, Tahan R, Obeid T. Juvenile myoclonic epilepsy: factors of error involved in the diagnosis and treatment. Epilepsia. 1991;32:672–6. [PubMed: 1915175]
- 231.
- Penry JK, Dean JC, Riela AR. Juvenile myoclonic epilepsy: long-term response to therapy. Epilepsia. 1989;30(Suppl 4):S19–23. S24. [PubMed: 2506007]
- 232.
- Gelisse P, Genton P, Thomas P, Rey M, Samuelian JC, Dravet C. Clinical factors of drug resistance in juvenile myoclonic epilepsy. J Neurol Neurosurg Psychiatry. 2001;70:240–3. [PMC free article: PMC1737198] [PubMed: 11160477]
- 233.
- Faught E. Clinical trials for treatment of primary generalized epilepsies. Epilepsia. 2003;44(Suppl 7):44–50. [PubMed: 12919339]
- 234.
- Greenhill L, Betts T, Smith K. Effect of levetiracetam on resistant juvenile myoclonic epilepsy. Epilepsia. 2002;43(Suppl 7):179.
- 235.
- Resor SR, Resor LD.Levetiracetam monotherapy in the treatment of convulsive seizures in juvenile myoclonic epilepsy. American Academy Neurology, 54th annual meeting; April 2002;
- 236.
- Jongsma M, Janssen G, Engelsman M, Haan D. Promising results of levetiracetam in juvenile myoclonic epilepsy. Epilepsia. 2002;43(Suppl 8S):153.
- 237.
- Calleja S, Salas-Puig J, Ribacoba R, Lahoz CH. Evolution of juvenile myoclonic epilepsy treated from the outset with sodium valproate. Seizure. 2001;10:424–7. [PubMed: 11700996]
- 238.
- Sundqvist A, Nilsson BY, Tomson T. Valproate monotherapy in juvenile myoclonic epilepsy: dose-related effects on electroencephalographic and other neurophysiologic tests. Ther Drug Monit. 1999;21:91–6. [PubMed: 10051060]
- 239.
- Morrell MJ. Guidelines for the care of women with epilepsy. Neurology. 1998;51:S21–S27. [PubMed: 9818920]
- 240.
- Crawford P, Appleton R, Betts T, Duncan J, Guthrie E, Morrow J. Best practice guidelines for the management of women with epilepsy. The Women with Epilepsy Guidelines Development Group. Seizure. 1999;8:201–17. [PubMed: 10452918]
- 241.
- Yerby MS. Management issues for women with epilepsy: neural tube defects and folic acid supplementation. Neurology. 2003;61:S23–S26. [PubMed: 14504306]
- 242.
- Tatum WO, Liporace J, Benbadis SR, Kaplan PW. Updates on the treatment of epilepsy in women. Arch Intern Med. 2004;164:137–45. [PubMed: 14744836]
- 243.
- Wyszynski DF, Holmes LB. The AED (antiepileptic drug) pregnancy registry: a six year experience. Epilepsia. 2004;45(Suppl 3):56. [PubMed: 15148143]
- 244.
- Morrow JI, Russell AJC, Irwin B, Guthrie E, Morrison P, et al. The safety of antiepileptic drugs in pregancy: results of the UK epilepsy and pregnancy register. Epilepsia. 2004;45(Suppl 3):57.
- 245.
- Isojarvi JI, Tauboll E, Tapanainen JS, Pakarinen AJ, Laatikainen TJ, Knip M, et al. On the association between valproate and poycystic ovary syndrome:A response and an alternative view. Epilepsia. 2001;42:305–10. [PubMed: 11442144]
- 246.
- French JA, Kanner AM, Bautista J, Abou-Khalil B, Browne T, Harden CL, et al. Efficacy and tolerability of the new antiepileptic drugs II: treatment of refractory epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1261–73. [PubMed: 15111660]
- 247.
- French JA, Kanner AM, Bautista J, Abou-Khalil B, Browne T, Harden CL, et al. Efficacy and Tolerability of the New Antiepileptic Drugs, II: Treatment of Refractory Epilepsy: Report of the TTA and QSS Subcommittees of the American Academy of Neurology and the American Epilepsy Society. Epilepsia. 2004;45:410–23. [PubMed: 15101822]
- 248.
- Montouris G. Gabapentin exposure in human pregnancy: results from the Gabapentin Pregnancy Registry. Epilepsy Behav. 2003;4:310–7. [PubMed: 12791334]
- 249.
- Tennis P, Eldridge RR. Preliminary results on pregnancy outcomes in women using lamotrigine. Epilepsia. 2002;43:1161–7. [PubMed: 12366730]
- 250.
- Long L. Levetiracetam monotherapy during pregnancy: a case series. Epilepsy Behav. 2003;4:447–8. [PubMed: 12899868]
- 251.
- Tomson T, Perucca E, Battino D. Navigating toward fetal and maternal health: the challenge of treating epilepsy in pregnancy. Epilepsia. 2004;45:1171–5. [PubMed: 15461670]
- 252.
- Cunnington MC. The international lamotrigine pregnancy registry update for the epilepsy foundation. Epilepsia. 2004;45:1468. [PubMed: 15509254]
- 253.
- Obeid T, Panayiotopoulos CP. Clonazepam in juvenile myoclonic epilepsy. Epilepsia. 1989;30:603–6. [PubMed: 2507306]
- 254.
- Van Zandijcke M. Treatment of myoclonus. Acta Neurol Belg. 2003;103:66–70. [PubMed: 12891998]
- 255.
- Wheless JW, Sankar R. Treatment Strategies for Myoclonic Seizures and Epilepsy Syndromes with Myoclonic Seizures. Epilepsia. 2003;44(Suppl 11):27–37. [PubMed: 14641568]
- 256.
- Canadian Study Group for Childhood Epilepsy. Clobazam has equivalent efficacy to carbamazepine and phenytoin as monotherapy for childhood epilepsy. Epilepsia. 1998;39:952–9. [PubMed: 9738674]
- 257.
- Resor SR Jr, Resor LD. Chronic acetazolamide monotherapy in the treatment of juvenile myoclonic epilepsy. Neurology. 1990;40:1677–81. [PubMed: 2122276]
- 258.
- Buchanan N. The use of lamotrigine in juvenile myoclonic epilepsy. Seizure. 1996;5:149–51. [PubMed: 8795132]
- 259.
- Isojarvi JI, Rattya J, Myllyla VV, Knip M, Koivunen R, Pakarinen AJ, et al. Valproate, lamotrigine, and insulin-mediated risks in women with epilepsy. Ann Neurol. 1998;43:446–51. [PubMed: 9546324]
- 260.
- Cohen J. Levetiracetam monotherapy for primary generalised epilepsy. Seizure. 2003;12:150–3. [PubMed: 12651079]
- 261.
- Covanis A, Katsalouli M. Levetiracetam monotherapy in generalised epilepsy and photosensitivity. Epilepsia. 2003;44(Suppl 8):80.
- 262.
- Gelisse P, Crespel A, Genton P, Baldy-Moulinier M. Dramatic effect of levetiracetam on epileptic negative myoclonus. Acta Neurol Scand. 2003;107:302–3. [PubMed: 12675706]
- 263.
- Kasteleijn-Nolst Trenite DG, Hirsch E. Levetiracetam: preliminary efficacy in generalized seizures. Epileptic Disord. 2003;5(Suppl 1):S39–S44. [PubMed: 12915340]
- 264.
- Krauss GL, Betts T, Abou-Khalil B, Bergey G, Yarrow H, Miller A. Levetiracetam treatment of idiopathic generalised epilepsy. Seizure. 2003;12:617–20. [PubMed: 14630506]
- 265.
- Gallagher MJ, Eisenman LN, Brown KM, Erbayat-Altay E, Hecimovic H, Fessler AJ, et al. Levetiracetam Reduces Spike-Wave Density and Duration during Continuous EEG Monitoring in Patients with Idiopathic Generalized Epilepsy. Epilepsia. 2004;45:90–1. [PubMed: 14692914]
- 266.
- Nieto BM. Characteristics and indications of topiramate. Rev Neurol. 2002;35(Suppl 1):S88–S95. [PubMed: 12373660]
- 267.
- Prasad A, Knowlton RC, Mendez M, Martin R, Kuzniecky R, Faught E. A comparison of lamotrigine and topiramate in juvenile myoclonic epilepsy. Epilepsia. 2002;43(Suppl 7):198–9.
- 268.
- Wallace SJ. Myoclonus and epilepsy in childhood: a review of treatment with valproate, ethosuximide, lamotrigine and zonisamide. Epilepsy Res. 1998;29:147–54. [PubMed: 9477147]
- 269.
- Fattouch J, Di Bonaventura C, Mari F, Egeo G, Vaudano AE, Mascia A, Giallonardo AT, Manfredi M. Role of levetiracetam in the treatment of primary generalised epilepsy. Epilepsia. 2004;45(Suppl 3):142.
- 270.
- Arroyo S, Crawford P. Safety profile of levetiracetam. Epileptic Disord. 2003;5(Suppl 1):S57–S63. [PubMed: 12915343]
- 271.
- French JA, Kanner AM, Bautista J, Abou-Khalil B, Browne T, Harden CL, et al. Efficacy and Tolerability of the New Antiepileptic Drugs, I: Treatment of New-Onset Epilepsy: Report of the TTA and QSS Subcommittees of the American Academy of Neurology and the American Epilepsy Society. Epilepsia. 2004;45:401–9. [PubMed: 15101821]
- 272.
- Czapinski PP, Czapinska EM. The effectiveness of levetiracetam in drug-resistant juvenile myoclonic epilepsy. Epilepsia. 2004;45(Suppl 3):141.
- 273.
- La Neve A, Boero G, Specchio N, Santasabato M, De Paolo A, et al. Levetiracetam is effective in juvenile myoclonic epilepsy. Epilepsia. 2004;45(Suppl 3):141.
- 274.
- Agarwal P, Frucht SJ. Myoclonus. Curr Opin Neurol. 2003;16:515–21. [PubMed: 12869812]
- 275.
- Crest C, Dupont S, LeGuern E, Adam C, Baulac M. Levetiracetam in progressive myoclonic epilepsy: an exploratory study in 9 patients. Neurology. 2004;62:640–3. [PubMed: 14981187]
- 276.
- Genton P, Gelisse P. Antimyoclonic effect of levetiracetam. Epileptic Disord. 2000;2:209–12. [PubMed: 11174151]
- 277.
- Frucht SJ, Louis ED, Chuang C, Fahn S. A pilot tolerability and efficacy study of levetiracetam in patients with chronic myoclonus. Neurology. 2001;57:1112–4. [PubMed: 11571347]
- 278.
- Krauss GL, Bergin A, Kramer RE, Cho YW, Reich SG. Suppression of post-hypoxic and post-encephalitic myoclonus with levetiracetam. Neurology. 2001;56:411–2. [PubMed: 11171914]
- 279.
- Schauer R, Singer M, Saltuari L, Kofler M. Suppression of cortical myoclonus by levetiracetam. Mov Disord. 2002;17:411–5. [PubMed: 11921136]
- 280.
- Guerrini R, Genton P. Epileptic syndromes and visually induced seizures. Epilepsia. 2004;45(Suppl 1):14–8. [PubMed: 14706039]
- 281.
- De Bittencourt PR. Photosensitivity: the magnitude of the problem. Epilepsia. 2004;45(Suppl 1):30–4. [PubMed: 14706043]
- 282.
- Kasteleijn-Nolst Trenite DG, Marescaux C, Stodieck S, Edelbroek PM, Oosting J. Photosensitive epilepsy: a model to study the effects of antiepileptic drugs. Evaluation of the piracetam analogue, levetiracetam. Epilepsy Res. 1996;25:225–30. [PubMed: 8956920]
- 283.
- Covanis A, Stodieck SR, Wilkins AJ. Treatment of photosensitivity. Epilepsia. 2004;45(Suppl 1):40–5. [PubMed: 14706045]
- 284.
- Cramer JA, De Rue K, Devinsky O, Edrich P, Trimble MR. A systematic review of the behavioral effects of levetiracetam in adults with epilepsy, cognitive disorders, or an anxiety disorder during clinical trials. Epilepsy Behav. 2003;4:124–32. [PubMed: 12697136]
- 285.
- Huber B, Bommel W, Hauser I, Horstmann V, Liem S, May T, et al. Efficacy and tolerability of levetiracetam in patients with therapy-resistant epilepsy and learning disabilities. Seizure. 2004;13:168–75. [PubMed: 15010054]
- 286.
- Mula M, Trimble MR, Sander JW. Psychiatric adverse events in patients with epilepsy and learning disabilities taking levetiracetam. Seizure. 2004;13:55–7. [PubMed: 14741183]
- 287.
- Besag FM. Behavioral aspects of pediatric epilepsy syndromes. Epilepsy Behav. 2004;5(Suppl 1):S3–13. [PubMed: 14725841]
- 288.
- Biraben A, Allain H, Scarabin JM, Schuck S, Edan G. Exacerbation of juvenile myoclonic epilepsy with lamotrigine. Neurology. 2000;55:1758. [PubMed: 11113246]
- 289.
- Carrazana EJ, Wheeler SD. Exacerbation of juvenile myoclonic epilepsy with lamotrigine. Neurology. 2001;56:1424–5. [PubMed: 11376212]
- 290.
- Genton P, Bauer J, Duncan S, Taylor AE, Balen AH, Eberle A, et al. On the association between valproate and poycystic ovary syndrome. Epilepsia. 2001;42:295–304. [PubMed: 11442143]
- 291.
- Prasad A, Kuzniecky RI, Knowlton RC, Welty TE, Martin RC, Mendez M, et al. Evolving antiepileptic drug treatment in juvenile myoclonic epilepsy. Arch Neurol. 2003;60:1100–5. [PubMed: 12925366]
- 292.
- Grunewald RA, Genton P, Salas Puig J, Panayiotopoulos CP. Evolving antiepileptic drug treatment in juvenile myoclonic epilepsy. Arch Neurol. 2004;61:1328–9. [PubMed: 15313858]
- 293.
- Ferrie CD, Robinson RO, Knott C, Panayiotopoulos CP. Lamotrigine as an add-on drug in typical absence seizures. Acta Neurol Scand. 1995;91:200–2. [PubMed: 7793236]
- 294.
- Reutens DC, Duncan JS, Patsalos PN. Disabling tremor after lamotrigine with sodium valproate. Lancet. 1993;342:185–6. [PubMed: 8101290]
- 295.
- Panayiotopoulos CP. Beneficial effect of relatively small doses of lamotrigine. Epilepsia. 1999;40:1171–2. [PubMed: 10448836]
- 296.
- Binnie CD, van Emde BW, Kasteleijn-Nolste-Trenite DG, de Korte RA, Meijer JW, Meinardi H, et al. Acute effects of lamotrigine (BW430C) in persons with epilepsy. Epilepsia. 1986;27:248–54. [PubMed: 3698937]
- 297.
- Nicolson A, Appleton RE, Chadwick DW, Smith DF. The relationship between treatment with valproate, lamotrigine, and topiramate and the prognosis of the idiopathic generalised epilepsies. J Neurol Neurosurg Psychiatry. 2004;75:75–9. [PMC free article: PMC1757463] [PubMed: 14707312]
- 298.
- Salas PX, Calleja S, Jimenez L, Gonzalez DM. Juvenile myoclonic epilepsy. Rev Neurol. 2001;32:957–61. [PubMed: 11424054]
- 299.
- Biton V, Montouris GD, Ritter F, Riviello JJ, Reife R, Lim P, et al. A randomized, placebo-controlled study of topiramate in primary generalized tonic-clonic seizures. Topiramate YTC Study Group. Neurology. 1999;52:1330–7. [PubMed: 10227614]
- 300.
- Waugh J, Goa KL. Topiramate: as monotherapy in newly diagnosed epilepsy. CNS Drugs. 2003;17:985–92. [PubMed: 14533950]
- 301.
- Privitera MD, Brodie MJ, Mattson RH, Chadwick DW, Neto W, Wang S. Topiramate, carbamazepine and valproate monotherapy: double-blind comparison in newly diagnosed epilepsy. Acta Neurol Scand. 2003;107:165–75. [PubMed: 12614309]
- 302.
- Wheless JW, Neto W, Wang S. Topiramate, carbamazepine, and valproate monotherapy: double-blind comparison in children with newly diagnosed epilepsy. J Child Neurol. 2004;19:135–41. [PubMed: 15072107]
- 303.
- Cross JH. Topiramate monotherapy for childhood absence seizures: an open label pilot study. Seizure. 2002;11:406–10. [PubMed: 12160672]
- 304.
- Aldenkamp AP, Baker G, Mulder OG, Chadwick D, Cooper P, Doelman J, et al. A multicenter, randomized clinical study to evaluate the effect on cognitive function of topiramate compared with valproate as add-on therapy to carbamazepine in patients with partial-onset seizures. Epilepsia. 2000;41:1167–78. [PubMed: 10999556]
- 305.
- Kockelmann E, Elger CE, Helmstaedter C. Significant improvement in frontal lobe associated neuropsychological functions after withdrawal of topiramate in epilepsy patients. Epilepsy Res. 2003;54:171–8. [PubMed: 12837568]
- 306.
- Patsalos PN, Perucca E. Clinically important drug interactions in epilepsy: interactions between antiepileptic drugs and other drugs. Lancet Neurol. 2003;2:473–81. [PubMed: 12878435]
- 307.
- Jain KK. An assessment of zonisamide as an anti-epileptic drug. Expert Opin Pharmacother. 2000;1:1245–60. [PubMed: 11249491]
- 308.
- Glauser TA, Pellock JM. Zonisamide in pediatric epilepsy: review of the Japanese experience. J Child Neurol. 2002;17:87–96. [PubMed: 11952083]
- 309.
- Newmark ME, Dubinsky S. Zonisamide monotherapy in a multi-group clinic. Seizure. 2004;13:223–5. [PubMed: 15121129]
- 310.
- Sullivan JE, Dlugos DJ. Idiopathic Generalized Epilepsy. Curr Treat Options Neurol. 2004;6:231–42. [PubMed: 15043806]
- 311.
- Panayiotopoulos CP. Juvenile myoclonic epilepsy. In: Panayiotopoulos CP, editor. A clinical guide to epileptic syndromes and their treatment. Oxford: Bladon Medical Publishing; 2002. pp. 139–45.
- 312.
- Genton P, Gelisse P, Thomas P, Dravet C. Do carbamazepine and phenytoin aggravate juvenile myoclonic epilepsy? Neurology. 2000;55:1106–9. [PubMed: 11071486]
- 313.
- Knott C, Panayiotopoulos CP. Carbamazepine in the treatment of generalised tonic clonic seizures in juvenile myoclonic epilepsy. J Neurol Neurosurg Psychiatr. 1994;57:503. [PMC free article: PMC1072885] [PubMed: 8164005]
- 314.
- Gelisse P, Genton P, Kuate D, Pesenti A, Baldy-Moulinier M, Crespel A. Worsening of seizures by oxcarbazepine in juvenile idiopathic generalized epilepsies. Epilepsia. 2004;45:1282–8. [PubMed: 15461683]
- 315.
- Janz D. Die epilepsien:Spezielle pathologie and therapie. Stuttgart: Georg Thieme; 1969.
- 316.
- Janz D. Pitfalls in the diagnosis of grand mal on awakening. In: Wolf P, editor. Epileptic seizures and syndromes. London: John Libbey & Company Ltd; 1994. pp. 213–20.
- 317.
- Janz D. Epilepsy with grand mal on awakening and sleep-waking cycle. Clin Neurophysiol. 2000;111(Suppl 2):S103–S110. [PubMed: 10996562]
- 318.
- Greenberg DA, Durner M, Resor S, Rosenbaum D, Shinnar S. The genetics of idiopathic generalized epilepsies of adolescent onset: differences between juvenile myoclonic epilepsy and epilepsy with random grand mal and with awakening grand mal. Neurology. 1995;45:942–6. [PubMed: 7746411]
- 319.
- Ferrie CD, Panayiotopoulos CP. Idiopathic generalised epilepsy with generalised tonic clonic seizures on awakening. In: Wallace SJ, Farrell K, editors. Epilepsy in Children. 2. London: Edward Arnold (Publishers) Limited; 2004. pp. 219–30.
- 320.
- Roger J, Bureau M, Oller Ferrer-Vidal L, Oller-Daurella L, Saltarelli A, Genton P. Clinical and electroencephalographic characteristics of idiopathic generalised epilepsies. In: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, editors. Idiopathic generalised epilepsies. London: John Libbey & Company Ltd; 1994. pp. 7–18.
- 321.
- Oller-Daurella LF-V, Oller L. Clinica y evolucion. Barcelona: Ciba-Geigy; 1994. 5000 epilepticos.
- 322.
- Reutens DC, Berkovic SF. Idiopathic generalized epilepsy of adolescence: are the syndromes clinically distinct? Neurology. 1995;45:1469–76. [PubMed: 7644043]
- 323.
- Meencke HJ, Janz D, Cervos-Navarro J. Neuropathology of primary generalized epilepsies with awakening grand mal. Acta Neuropathol Suppl (Berl). 1981;7:378–80. [PubMed: 6784442]
- 324.
- Baier WK, Doose H. Petit mal-absences of childhood onset: familial prevalences of migraine and seizures. Neuropediatrics. 1985;16:80–3. [PubMed: 3925365]
- 325.
- Doose H. Absence epilepsy of early childhood—genetic aspects. Eur J Pediatr. 1994;153:372–7. [PubMed: 8033930]
- 326.
- Doose H. Absence epilepsy of early childhood. In: Wolf P, editor. Epileptic seizures and syndromes. London: John Libbey & Company Ltd; 1994. pp. 133–5.
- 327.
- Chaix Y, Daquin G, Monteiro F, Villeneuve N, Laguitton V, Genton P. Absence epilepsy with onset before age three years: a heterogeneous and often severe condition. Epilepsia. 2003;44:944–9. [PubMed: 12823578]
- 328.
- Panayiotopoulos CP, Ferrie CD, Giannakodimos S, Robinson RO. Perioral myoclonia with absences: a new syndrome. In: Wolf P, editor. Epileptic seizures and syndromes. London: John Libbey & Company Ltd; 1994. pp. 143–53.
- 329.
- Hirsch E. Perioral myoclonia with absences. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. p. 230.
- 330.
- Clemens B. Perioral myoclonia with absences? A case report with EEG and voltage mapping analysis. Brain Dev. 1997;19:353–8. [PubMed: 9253489]
- 331.
- Bilgic B, Baykan B, Gurses C, Gokyigit A. Perioral myoclonia with absence seizures: a rare epileptic syndrome. Epileptic Disord. 2001;3:23–7. [PubMed: 11313219]
- 332.
- Aarts JH, Binnie CD, Smit AM, Wilkins AJ. Selective cognitive impairment during focal and generalized epileptiform EEG activity. Brain. 1984;107:293–308. [PubMed: 6421454]
- 333.
- Kasteleijn-Nolst Trenite DG. Transient cognitive impairment during subclinical epileptiform electroencephalographic discharges. Semin Pediatr Neurol. 1995;2:246–53. [PubMed: 9422252]
- 334.
- Giannakodimos S, Ferrie CD, Panayiotopoulos CP. Qualitative and quantitative abnormalities of breath counting during brief generalized 3 Hz spike and slow wave ‘subclinical’ discharges. Clin Electroencephalogr. 1995;26:200–3. [PubMed: 8575099]
- 335.
- Marsall C. Some clinical correlates of the wave and spike phantom. Electroencephalogr Clin Neurophysiol. 1955;7:633–6. [PubMed: 13270697]
- 336.
- Tharp BR. The 6-per-second spike and wave complex. The wave and spike phantom. Arch Neurol. 1966;15:533–7. [PubMed: 5955947]
- 337.
- Silverman D. Phantom spike-waves and the fourteen and six per second positive spike pattern: a consideration of their relation. Electroencephalograph Clin Neurophysiol. 1967;23:207–13. [PubMed: 4167919]
- 338.
- Genton P. Epilepsy with 3Hz spike-and-waves without clinically evideent absences. In: Duncan JS, Panayiotopoulos CP, editors. Typical absences and related epileptic syndromes. London: Churchill Communications Europe; 1995. pp. 231–8.
- 339.
- Vuilleumier P, Assal F, Blanke O, Jallon P. Distinct behavioral and EEG topographic correlates of loss of consciousness in absences. Epilepsia. 2000;41:687–93. [PubMed: 10840400]
- 340.
- Panayiotopoulos CP. Epilepsy with generalised tonic-clonic seizures on awakening. In: Wallace S, editor. Epilepsy in Children. London: Chapman & Hall; 1996. pp. 349–53.
- 341.
- Cutting S, Lauchheimer A, Barr W, Devinsky O. Adult-onset idiopathic generalized epilepsy: clinical and behavioral features. Epilepsia. 2001;42:1395–8. [PubMed: 11879340]
- 342.
- Marini C, King MA, Archer JS, Newton MR, Berkovic SF. Idiopathic generalised epilepsy of adult onset: clinical syndromes and genetics. J Neurol Neurosurg Psychiatry. 2003;74:192–6. [PMC free article: PMC1738270] [PubMed: 12531947]
- 343.
- Yenjun S, Harvey AS, Marini C, Newton MR, King MA, Berkovic SF. EEG in adult-onset idiopathic generalized epilepsy. Epilepsia. 2003;44:252–6. [PubMed: 12558583]
- 344.
- Nicolson A, Chadwick DW, Smith DF. A comparison of adult onset and “classical” idiopathic generalised epilepsy. J Neurol Neurosurg Psychiatry. 2004;75:72–4. [PMC free article: PMC1757447] [PubMed: 14707311]
- 345.
- Okino S. Familial benign myoclonus epilepsy of adult onset: a previously unrecognized myoclonic disorder. J Neurol Sc. 1997;145:113–8. [PubMed: 9073039]
- 346.
- Mikami M, Yasuda T, Terao A, Nakamura M, Ueno S, Tanabe H, et al. Localization of a gene for benign adult familial myoclonic epilepsy to chromosome 8q23.3–q24.1. Am J Hum Genet. 1999;65:745–51. [PMC free article: PMC1377981] [PubMed: 10441581]
- 347.
- Plaster NM, Uyama E, Uchino M, Ikeda T, Flanigan KM, Kondo I, et al. Genetic localization of the familial adult myoclonic epilepsy (FAME) gene to chromosome 8q24. Neurology. 1999;53:1180–3. [PubMed: 10522869]
- 348.
- Saka E, Saygi S. Familial adult onset myoclonic epilepsy associated with migraine. Seizure. 2000;9:344–6. [PubMed: 10933990]
- 349.
- Labauge P, Amer LO, Simonetta-Moreau M, Attane F, Tannier C, Clanet M, et al. Absence of linkage to 8q24 in a European family with familial adult myoclonic epilepsy (FAME). Neurology. 2002;58:941–4. [PubMed: 11914412]
- 350.
- Manabe Y, Narai H, Warita H, Hayashi T, Shiro Y, Sakai K, et al. Benign adult familial myoclonic epilepsy (BAFME) with night blindness. Seizure. 2002;11:266–8. [PubMed: 12027575]
- 351.
- de Falco FA, Striano P, de Falco A, Striano S, Santangelo R, Perretti A, et al. Benign adult familial myoclonic epilepsy: genetic heterogeneity and allelism with ADCME. Neurology. 2003;60:1381–5. [PubMed: 12707452]
- 352.
- Shimizu A, Asakawa S, Sasaki T, Yamazaki S, Yamagata H, Kudoh J, et al. A novel giant gene CSMD3 encoding a protein with CUB and sushi multiple domains: a candidate gene for benign adult familial myoclonic epilepsy on human chromosome 8q23.3–q24.1. Biochem Biophys Res Commun. 2003;309:143–54. [PubMed: 12943675]
- 353.
- Striano P, Chifari R, Striano S, De Fusco M, Elia M, Guerrini R, et al. A New Benign Adult Familial Myoclonic Epilepsy (BAFME) Pedigree Suggesting Linkage to Chromosome 2p11.1–q12.2. Epilepsia. 2004;45:190–2. [PubMed: 14738428]
- 354.
- Guerrini R, Bonanni P, Patrignani A, Brown P, Parmeggiani L, Grosse P, et al. Autosomal dominant cortical myoclonus and epilepsy (ADCME) with complex partial and generalized seizures: A newly recognized epilepsy syndrome with linkage to chromosome 2p11.1–q12.2. Brain. 2001;124:2459–75. [PubMed: 11701600]
- 355.
- Sano A, Mikami M, Nakamura M, Ueno S, Tanabe H, Kaneko S. Positional candidate approach for the gene responsible for benign adult familial myoclonic epilepsy. Epilepsia. 2002;43(Suppl 9):26–31. [PubMed: 12383276]
- 356.
- Zara F, Gennaro E, Stabile M, Carbone I, Malacarne M, Majello L, et al. Mapping of a locus for a familial autosomal recessive idiopathic myoclonic epilepsy of infancy to chromosome 16p13. Am J Hum Genet. 2000;66:1552–7. [PMC free article: PMC1378007] [PubMed: 10741954]
- 357.
- de Falco FA, Majello L, Santangelo R, Stabile M, Bricarelli FD, Zara F. Familial infantile myoclonic epilepsy: clinical features in a large kindred with autosomal recessive inheritance. Epilepsia. 2001;42:1541–8. [PubMed: 11879364]
- 358.
- Parker AP, Agathonikou A, Robinson RO, Panayiotopoulos CP. Inappropriate use of carbamazepine and vigabatrin in typical absence seizures. Dev Med Child Neurol. 1998;40:517–9. [PubMed: 9746003]
- 359.
- Panayiotopoulos CP, Benbadis SR, Covanis A, Dulac O, Duncan JS, Eeg-Olofsson O, et al. Efficacy and tolerability of the new antiepileptic drugs; commentary on the recently published practice parameters. Epilepsia. 2004;45:1646–9. [PubMed: 15571526]
- 360.
- Bourgeois BF. Chronic management of seizures in the syndromes of idiopathic generalized epilepsy. Epilepsia. 2003;44(Suppl 2):27–32. [PubMed: 12752459]
- 361.
- Posner EB, Mohamed K, Marson AG. Ethosuximide, sodium valproate or lamotrigine for absence seizures in children and adolescents. Cochrane Database Syst Rev. 2003:CD003032. [PubMed: 12917940]
- 362.
- Mattson RH. Overview: idiopathic generalized epilepsies. Epilepsia. 2003;44(Suppl 2):2–6. [PubMed: 12752455]
- 363.
- Wheless JW. Acute management of seizures in the syndromes of idiopathic generalized epilepsies. Epilepsia. 2003;44(Suppl 2):22–6. [PubMed: 12752458]
- 364.
- Hirsch E, Genton P. Antiepileptic drug-induced pharmacodynamic aggravation of seizures: does valproate have a lower potential? CNS Drugs. 2003;17:633–40. [PubMed: 12828499]
- 365.
- Gelisse P, Genton P, Kuate D, Pesenti A, Baldy-Moulinier M, Crespel A. Worsening of seizures by oxcarbazepine in juvenile idiopathic generalised epilepsies. Epilepsia. 2004;45:1282–8. [PubMed: 15461683]
- 366.
- Genton P. When antiepileptic drugs aggravate epilepsy. Brain Dev. 2000;22:75–80. [PubMed: 10722956]
- 367.
- Kasteleijn-Nolst Trenite DG, Marescaux C, Stodieck S, Edelbroek PM, Oosting J. Photosensitive epilepsy: a model to study the effects of antiepileptic drugs. Evaluation of the piracetam analogue, levetiracetam. Epilepsy Res. 1996;25:225–30. [PubMed: 8956920]
- 368.
- Klitgaard H, Matagne A, Gobert J, Wulfert E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur.J.Pharmacol. 1998;353:191–206. [PubMed: 9726649]
- 369.
- Montouris G, Biton V, Rosenfeld W. the Topiramate YTC/YTCE study group. Nonfocal generalized tonic-clonic seizures:Response during long-trem topiramate treatment. Epilepsia. 2000;41(Suppl 1):S77–S81. [PubMed: 10768306]
- 370.
- Siren A, Eriksson K, Jalava H, Kilpinen-Loisa P, Koivikko M. Idiopathic generalised epilepsies with 3 Hz and faster spike wave discharges: a population-based study with evaluation and long-term follow-up in 71 patients. Epileptic Disord. 2002;4:209–16. [PubMed: 12446224]
- 371.
- Shorvon S. Handbook of epilepsy treatment. Oxford: Blackwell Science; 2000.
- 372.
- Chadwick D, Leiderman DB, Sauermann W, Alexander J, Garofalo E. Gabapentin in generalized seizures. Epilepsy Res. 1996;25:191–7. [PubMed: 8956916]
- 373.
- Trudeau V, Myers S, LaMoreaux L, Anhut H, Garofalo E, Ebersole J. Gabapentin in naive childhood absence epilepsy: results from two double-blind, placebo-controlled, multicenter studies. J Child Neurol. 1996;11:470–5. [PubMed: 9120226]
- 374.
- Scott RC, Neville BG. Pharmacological management of convulsive status epilepticus in children. Dev Med Child Neurol. 1999;41:207–10. [PubMed: 10210254]
- 375.
- Smith BJ. Treatment of status epilepticus. Neurol Clin. 2001;19:347–69. [PubMed: 11358748]
- 376.
- Lowenstein DH, Alldredge BK, Allen F, Neuhaus J, Corry M, Gottwald M, et al. The prehospital treatment of status epilepticus (PHTSE) study: design and methodology. Control Clin Trials. 2001;22:290–309. [PubMed: 11384791]
- 377.
- Shorvon S. The management of status epilepticus. J Neurol Neurosurg Psychiatry. 2001;70(Suppl 2):II22–II27. [PMC free article: PMC1765558] [PubMed: 11385046]
- 378.
- Alldredge BK, Gelb AM, Isaacs SM, Corry MD, Allen F, Ulrich S, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. NEJM. 2001;345:631–7. [PubMed: 11547716]
- 379.
- Hirsch LJ, Claassen J. The current state of treatment of status epilepticus. Curr Neurol Neurosci Rep. 2002;2:345–56. [PubMed: 12044254]
- 380.
- Appleton R, Martland T, Phillips B. Drug management for acute tonic-clonic convulsions including convulsive status epilepticus in children. Cochrane Database Syst Rev. 2002:CD001905. [PubMed: 12519562]
- 381.
- Claassen J, Hirsch LJ, Emerson RG, Mayer SA. Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: a systematic review. Epilepsia. 2002;43:146–53. [PubMed: 11903460]
- 382.
- Claassen J, Hirsch LJ, Mayer SA. Treatment of status epilepticus: a survey of neurologists. J.Neurol Sci. 2003;211:37–41. [PubMed: 12767495]
- 383.
- Gaitanis JN, Drislane FW. Status epilepticus: a review of different syndromes, their current evaluation, and treatment. Neurolog. 2003;9:61–76. [PubMed: 12808369]
- 384.
- Manno EM. New management strategies in the treatment of status epilepticus. Mayo Clin.Proc. 2003;78:508–18. [PubMed: 12683704]
- 385.
- Ruegg SJ, Dichter MA. Diagnosis and Treatment of Nonconvulsive Status Epilepticus in an Intensive Care Unit Setting. Curr Treat Options Neurol. 2003;5:93–110. [PubMed: 12628059]
- 386.
- Sirven JI, Waterhouse E. Management of status epilepticus. Am Fam Physician. 2003;68:469–76. [PubMed: 12924830]
- 387.
- Walker MC. Status epilepticus on the intensive care unit. J Neurol. 2003;250:401–6. [PubMed: 12700903]
- 388.
- Lowenstein DH. Treatment options for status epilepticus. Curr Opin Pharmacol. 2003;3:6–11. [PubMed: 12550735]
- 389.
- De Negri M, Baglietto MG. Treatment of status epilepticus in children. Paediatr Drugs. 2001;3:411–20. [PubMed: 11437186]
- 390.
- Walker MC. Diagnosis and treatment of nonconvulsive status epilepticus. CNS Drugs. 2001;15:931–9. [PubMed: 11735613]
- 391.
- Fitzgerald BJ, Okos AJ, Miller JW. Treatment of out-of-hospital status epilepticus with diazepam rectal gel. Seizure. 2003;12:52–5. [PubMed: 12495650]
- 392.
- Scott RC, Besag FM, Neville BG. Buccal midazolam and rectal diazepam for treatment of prolonged seizures in childhood and adolescence: a randomised trial. Lancet. 1999;353:623–6. [PubMed: 10030327]
- 393.
- Kutlu NO, Dogrul M, Yakinci C, Soylu H. Buccal midazolam for treatment of prolonged seizures in children. Brain Dev. 2003;25:275–8. [PubMed: 12767460]
- 394.
- Lahat E, Goldman M, Barr J, Bistritzer T, Berkovitch M. Comparison of intranasal midazolam with intravenous diazepam for treating febrile seizures in children: prospective randomised study. BMJ. 2000;321:83–6. [PMC free article: PMC27427] [PubMed: 10884257]
- 395.
- Verdru P, Wajgt A, Schiemann Delgado J, Noachtar S. Efficacy and safety of levetiracetam 3000 mg/d as adjunctive treatment in adolescents and adults suffering from idiopathic generalized epilepsy with myoclonic seizures. Epilepsia. 2005;46(Suppl 6):56–7.
Footnotes
- *
The term ‘typical’ is used not to characterise them as ‘classical’, but to differentiate them from ‘atypical’ absence seizures.
- *
The terminological differences between primarily as opposed to primary and secondarily as opposed to secondary have been detailed in Chapter 1.
- *
Non-convulsive status epilepticus is a term that has been rightly discarded in the new diagnostic scheme,5 because it encompasses heterogeneous conditions which may be focal, such as limbic status epilepticus, or generalised, such as ASE.71,72 Convulsive elements and particularly myoclonic jerks are common in generalised non-convulsive status epilepticus as, for example, in eyelid or perioral status epilepticus. Non-convulsive status epilepticus is not synonymous with ASE. If this term is used, the distinction between ‘focal non-convulsive’ and ‘generalised non-convulsive’ should be made for clinical and management purposes.
- *
Syndrome in development
- *
Astatic is not synonymous with atonic seizures. The term astatic seizures is abandoned in the new ILAE diagnostic scheme, which uses only the term ‘atonic’ or ‘myoclonic-atonic’ seizures.5
- **
I use the eponymic nomenclature ‘Doose syndrome’ only for the pure form of ‘idiopathic epilepsy with myoclonic-astatic seizures’ to exclude cases of symptomatic cause manifesting with myoclonic-astatic seizures.
Figures

Figure 10.1
Examples from video EEG recorded generalised discharges of 3 Hz spikes or multiple spikes-waves of typical absence seizures. These seven patients had different syndromes of idiopathic and symptomatic absence epilepsies. Note that the GSWD may be brief or prolonged, with or without polyspikes and of regular or irregular sequence. Also, note that the intradischarge frequency of the spike–wave complexes may show significant diversity. Though there are significant variations between different syndromes, the GSWD is not itself pathognomonic of any syndrome. The syndromic diagnosis requires homogeneous clustering of symptoms

Figure 10.2Girl, Aged 7 Years, with Childhood Absence Epilepsy.
The seizure occurred during hyperventilation with breath counting. She stopped counting (black arrow), opened her eyes (open arrow) and became unresponsive. Marked perioral automatisms occurred later (*). She abruptly recovered at the end of the discharge (O.K.).
The GSWD is artificially divided into the opening phase (first 1 s of the GSWD), the initial phase of stabilisation (following 3 s) and terminal phase (last 3 s).24 Note that the frequency of the regular spike–wave complexes gradually slows down from 3 Hz in the initial phase to 2 Hz in the terminal phase. The opening phase is asymmetrical and asynchronous with faster or irregular spike wave frequencies.
See also Figures 2.3 and 10.1.

Figure 10.3
Samples from the video EEG of a woman with JME on carbamazepine. Violent myoclonic jerks occur with generalised multiple spike discharges (see also Figure 10.14 of the same patient).

Figure 10.5Samples from a Video EEG of a 6-Year-Old Normal Boy with Doose Syndrome
The background activity was normal, but there were frequent (at least every 10 s) generalised discharges of high-voltage spike/polyspike–wave complexes at 3–6 Hz with an anterior maximum. They were brief lasting 1–4 s and were frequently associated with single jerks of mainly the shoulders, but occasionally of the hands, thumbs, eyelids or elsewhere. The jerks occurred simultaneously with the first or the second polyspike–wave complex of the GSWD. Some jerks were followed by atonic attacks. The EEG also showed brief (< 0.5 s) abortive generalised discharges of polyspikes at around 15 Hz with anterior maximum and alternating, but not consistent, side emphasis. There were no clinical manifestations.
The paroxysmal discharges occurred with the eyes open and closed, spontaneously and during overbreathing. Intermittent photic stimulation (IPS) did not evoke photoparoxysmal responses.

Figure 10.6Samples of a Video EEG of a Boy in Myoclonic-Atonic Status Epilepticus
Top: The EEG showed electrical status epilepticus during wakefulness and sleep. This consisted of nearly continuous generalised discharges of spikes (or double spikes) and slow wave complexes at 2.5–3Hz. This pattern occasionally alternated with relatively normal background activity lasting less than 30 s. The main clinical manifestations were frequent facial subtle myoclonias (eyelid fluttering, upwards deviation of the eyes with spontaneous eye opening associated with fast eyelid fluttering, subtle facial twitches) and a few massive myoclonic jerks occasionally with some atonic components. Clinically, there was no apparent impairment of consciousness.
Middle: Details of the EEG in the top of the Figure. Magnified frontal EEG channels and increased time scale.
Bottom: The video EEG 1 month later was normal during wakefulness with a few myoclonic jerks only during sleep.

Figure 10.18Video EEG of a Highly Intelligent Woman, Aged 79 Years, with Phantom Absences.
ASE and GTCS (case 1 in ref 58). She had her first overt seizure at the age of 30 years. She was moderately confused for 12 hours prior to a GTCS. Since then she had 1–3 similar episodes of ASE every year. She was misdiagnosed as temporal lobe epilepsy and was on primidone and sulthiame for nearly 40 years. Retrospectively, she admits brief episodes of mild impairment of cognition: “The absences lasted a couple of seconds; the other state (ASE) lasts much longer, for 24 hours or more. They may be linked I suppose.” No further seizures of any type occurred in the next 11 years of follow-up on monotherapy with valproate 1000 mg daily Upper: ASE with continuous spike and occasional polyspike-slow wave activity mainly at 3 Hz. Note also slower or faster components and some topographic variability of the discharge. The patient was fully alert, attentive and cooperative. Movements and speech were normal. There were no abnormal ictal symptoms other than severe global memory deficit and global diminution of content of consciousness. She was unable to remember her name, how many children she had, the date and her location. She could not perform simple calculations, but could repeat up to five numbers given to her. She could read text correctly and she correctly wrote her address, though she could not remember it on verbal questioning. She did not know where she was, but given the choice between various locations, she correctly recognised that she was in a hospital. This ASE was successfully interrupted with intravenous administration of diazepam.
Bottom: Interictal video-EEG showing brief 3 Hz GSWD lasting 2–3 s without apparent clinical manifestations.

Figure 10.14Myoclonic-absence status epilepticus in JME.
Video EEG of a woman with classical JME, but on carbamazepine at the time of this recording (see also Figure 10.3 from the same patient). The patient was mildly confused with continuous jerking of the hands (middle) and rarely the eyelids. The ictal EEG consisted of repetitive and discontinuous frequent GSWD interrupted by brief intervals of relative normality. Each GSWD consisted of varying numbers of polyspikes/spikes-wave in various combinations and morphologies.

Figure 10.16Diagnostic Errors in PMA
Top: Video EEG recording of an 18-year-old woman with perioral myoclonia with absences (case 6 in ref 328). She was referred because of ‘focal motor seizures and secondarily GTCS’. She had onset of GTCS and absences at 11 years of age, which continued despite treatment with various combinations of valproate, ethosuximide, clonazepam, lamotrigine and acetazolamide. Absences were frequent, often in daily clusters and consisted of brief (about 5 s) moderate impairment of consciousness with violent rhythmic jerking of the jaw. GTCS occurred 1–10 times a year, usually after awakening, preceded by clusters of absences with the jaw myoclonus spreading to limb jerks prior to generalised convulsions. She was more concerned about the absences because they interfered with her daily life “everyone notices the jerks of my jaw” and less about the GTCS, which usually occurred at home. The initial misdiagnosis of ‘focal motor seizures’ was because the jaw jerking was perceived and described by her mother as unilateral.
Bottom: EEG of 23-year-old woman while in perioral status epilepticus (case 2 in ref 328). She was mildly confused with continuous perioral twitching. This ended with a GTCS. Initial presentation at 11 years of age was with GTCS. PMA was noted at the same time and was diagnosed as focal motor seizures. Despite medication with valproate, she continues having infrequent episodes of ASE terminated with self-administered benzodiazepines.

Figure 10.4Samples from a video EEG showing idiopathic and symptomatic myoclonic absence seizures. The EEG GSWD are similar with no apparent differentiating features or asymmetry in the symptomatic patient.
Top: Video EEG of a boy aged 8 years with myoclonic absences from the age of 6 years. These were frequent, hundreds per day, and manifested with rhythmic myoclonic jerks and unresponsiveness. Valproate failed to achieve control, but absences ceased completely when ethosuximide was added at the age of 8 years. Medication was withdrawn at 11 years of age. No further seizures occurred in the next 8 years of follow-up and he is academically successful.
The absence is terminated by somatosensory stimulation (tapping his shoulder).
Bottom: Video EEG of a girl aged 15 months with myoclonic absences due to birth anoxia. Myoclonic absences started at 15 months of age, occurred many times a day, lasted for 8–10 s and were characterised by synchronous and bilateral myoclonic jerks of the limbs and head with apparent loss of consciousness. Subsequently, she developed frequent, mainly nocturnal GTCS while myoclonic absences continued. These were intractable to any appropriate medication. At 8 years of age, she had severe learning difficulties and spastic quadraparesis.
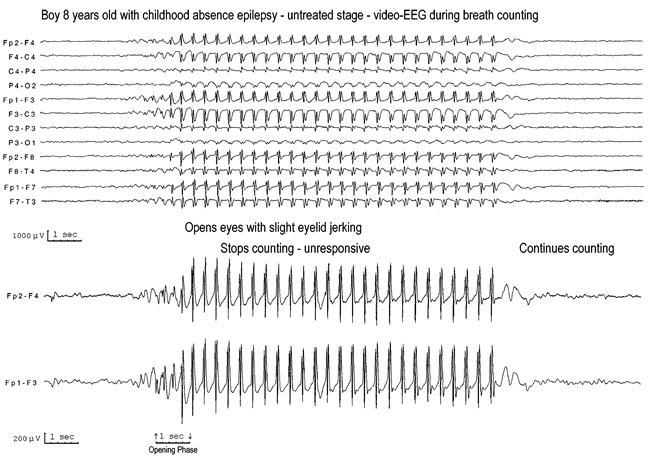
Figure 10.7Video EEG of an 8-year-old boy with classical CAE.
He was referred for EEG because of “blanks and day dreaming”; the diagnosis had been overlooked despite frequent absences for 2 years, which also interfered with his scholastic performance.
The seizure had all the characteristic features of typical absences in CAE. Initially there was some brief eyelid flickering in the opening phase of the GWSD followed by opening of the eyes, eyes and head deviating upwards and to the right, and simultaneous cessation of breath counting. He remained unresponsive during the rest of the GSWD. Counting was restored immediately after the end of the GSWD.
Note the fast and asymmetrical onset of the GSWD in the opening phase during the first second. Subsequently, the GSWD is entirely rhythmical and regular with abrupt termination to clinical and EEG normality.24

Figure 10.8Girl Aged 6 Years with Untreated Childhood Absence Epilepsy
Left: Long runs of bilateral, rhythmic and fusiform, high amplitude 3 Hz slow waves appear in the posterior regions (between arrows).
Right: Onset of a typical absence seizure.

Figure 10.9Video EEG of a 26-Year-Old Normal Woman.
She started having frequent typical absence seizures (tens of seizures each day) with severe impairment of consciousness at the age of 11 years, but medical attendance was sought only after her first GTCS at the age of 14 years. Severe and lengthy absences of 20–30 s occur 5–10 times/day. Also she has had 3–5 GTCS every year, mostly after awakening and mainly premenstrually. GTCS are preceded by clusters of absences. Occasionally, she also has random, infrequent and mild limb myoclonic jerks, which started at the age of 20 years. Treatment with various appropriate anti-absence drugs resulted in minor improvement, but compliance varies.
From Panayiotopoulos (2004)16 with the permission of the Editor of Medlink.

Figure 10.10Age at onset of absences, myoclonic jerks and GTCS in 66 consecutive patients with JME.
Modified from Panayiotopoulos et al60 with the permission of the Editor of Epilepsia

Figure 10.11Violent Myoclonic Jerks of the Hands Associated with Typical EEG Manifestations.
From Panayiotopoulos et al. (1994)60 with the permission of the Editor of Epilepsia.

Figure 10.12Video EEG of two patients with JME.
Top: GSWD are not associated with apparent clinical manifestations (but these may have been revealed if breath counting was performed during hyperventilation).
Bottom: GSWD are associated with mild impairment of cognition.
Modified from Panayiotopoulos et al60 with the permission of the Editor of Epilepsia

Figure 10.13Sample from a video EEG of a woman aged 28 years who had suffered from JME since the age of 9 years.
This woman was referred for a routine EEG because she had experienced “probable IGE-absences from age 9 until age 18 years. Myoclonus as a teenager when sleep deprived. Recently suffered her first ever GTCS following sleep deprivation. No absence or myoclonus for 10 years. Treatment with valproate was withdrawn at age 18 years despite continuing myoclonic jerks and absences.” The video EEG documented that she still had brief absences, which manifest with mild impairment of cognition and eyelid jerks. These were spontaneous, or induced by hyperventilation (top) and intermittent photic stimulation (bottom).
Note the multiple spike–wave of the discharges and the irregular intradischarge frequency (magnification on the bottom). Also note the bifrontal spike-slow wave discharges (upper right).
Figure 10.15Asymptomatic GSWD on video EEG of a 19-year-old university student who had two GTCS at 14 and 18 years of age. They both occurred half an hour after awakening from a brief sleep during exam periods. There was no clinical history of any other type of seizures and there were no other symptoms preceding either of the GTCS.
Note that the discharges may start from right or left. Also note focal spikes at various locations.

Figure 10.17Video EEG of two patients suffering from IGE with phantom absences.
The numbers denote the actual breath counting during hyperventilation. Note that errors, when they occur, are only related to these brief GSWD.
Errors consist of hesitation in pronouncing the next consecutive number, repetitions and erroneous counting sequence.
Tables
Table 10.1
Clinical and EEG manifestations of typical absence seizures21
| Typical absence seizures: clinical manifestations |
| The hallmark of the absence attack is a sudden onset, interruption of ongoing activities, a blank stare, possibly a brief upward rotation of the eyes. If the patient is speaking, speech is slowed or interrupted, if walking, he stands transfixed; if eating, the food will stop on his way to the mouth. Usually the patient will be unresponsive when spoken to. In some, attacks are aborted when the patient is spoken to. The attack lasts from a few seconds to half a minute and evaporates as rapidly as it commenced. |
| Clinical seizure type |
|
| Absence EEG manifestations |
Ictal EEG
|
From the Commission on Classification and Terminology of the ILAE21 with the permission of the Commission and the editor of Epilesia
Table 10.2
Differential diagnosis of typical absences from complex focal seizures
| Clinical criteria | Typical absences | Complex focal seizures |
|---|---|---|
| Duration for less than 30 seconds | As a rule | Exceptional |
| Duration for more than 1 minute | Exceptional | As a rule |
| Non-convulsive status epilepticus | Frequent | Rare |
| Daily frequency | As a rule | Rare |
| Simple automatisms | Frequent | Frequent |
| Complex behavioural automatisms | Exceptional | Frequent |
| Simple and complex hallucinations or illusions | Exceptional | Frequent |
| Bilateral facial myoclonic jerks or eyelid closures | Frequent | Exceptional |
| Evolving to other focal seizure manifestations | Never | Frequent |
| Sudden onset and termination | As a rule | Frequent |
| Postictal symptoms | Never | Frequent |
| Reproduced by hyperventilation | As a rule | Exceptional |
| Elicited by photic stimulation | Frequent | Exceptional |
| EEG criteria | ||
| Ictal generalised 3–4 Hz spike and wave | Exclusive | Never |
| Interictal generalised discharges | Frequent | Exceptional |
| Interictal focal abnormalities of slow waves | Exceptional | Frequent |
| Normal EEG in untreated state | Exceptional | Frequent |
The primary differences are shown in red.
From Panayiotopoulos (2002)16 with the permission of the Editor of Medlink.
Table 10.3
Differentiation of primarily versus secondarily GTCS
| Primarily GTCS | Secondarily GTCS | |
|---|---|---|
| GTCS with other clinically evident seizures | About 90% | About 90% |
| Typical absences | About 40% | None |
| Myoclonic jerks | About 60% | None |
| Focal seizures | None | About 90% |
| GTCS without other clinically evident seizures | About 10%** | About 10%** |
| Precipitating factors | > 60% | < 10% |
| Consistently on awakening | Common | Uncommon |
| Family history of similar epilepsies | Common | Uncommon |
| EEG in untreated patients | ||
| Generalised discharges | About 80% | Exceptional |
| Focal abnormalities alone | About 10% | About 60% |
| Generalised discharges and focal abnormalities | About 30% | Exceptional |
| High resolution brain imaging | ||
| Focal abnormalities | Exceptional | About 60% |
| Normal | By definition | About 40% |
- **
It is these patients, who make up about 10% of each category, without clinically apparent other types of seizures that constitute the main problem in the differential diagnosis between primarily and secondarily GTCS. However, other features, such as precipitating factors, circadian distribution, EEG and brain imaging, are often of diagnostic significance.
Table 10.4
Diagnostic criteria for Doose syndrome (idiopathic EM-AS)91
Inclusion criteria
|
Exclusion criteria
|
Table 10.5
Criteria for childhood absence epilepsy
Inclusion criteria
|
Exclusion criteriaThe following may be incompatible with
childhood absence epilepsy:
|
Table 10.6
Main inclusion and exclusion criteria for juvenile absence epilepsy (JAE)
Inclusion criteria
|
| Exclusion criteria |
| The following may be incompatible with juvenile absence epilepsy: |
Clinical exclusion criteria
|
EEG exclusion criteria
|
Table 10.7
Main differences between JME and JAE
| JME | JAE | |
|---|---|---|
| Main type of seizures | Myoclonic jerks | Typical absences |
| Circadian distribution | Mainly on awakening | Any time during the day |
| Typical absences | Mild and often imperceptible; they occur in one-third of patients | Defining seizure type; they are very severe and occur in all patients |
| Myoclonic jerks | Defining seizure type; they occur in all patients and mainly on awakening | Mild; they occur in one-third of patients and are random |
| GTCS | They mainly occur after a series of myoclonic jerks on awakening | They mainly occur independently or less commonly after a series of absence seizures |
| EEG | Brief (1–3 s) 3–6 Hz GSWD which are usually asymptomatic | Lengthy (8–30 s) 3–4 Hz GSWD, which are usually associated with severe impairment of consciousness. |
Table 10.8
Efficacy and safety of new AEDs in the JME triad of seizures and photosensitivity in comparison with valproate
| Myoclonic jerks | GTCS | Absences | Photosensitivity | Serious adverse reactions 194;271 | Titration | Drug-drug interactions | |
|---|---|---|---|---|---|---|---|
| Patients (%) | 100% | 90% | 30% | 30% | |||
| Valproate | Very effective | Very effective | Very effective | Very effective | Yes | Optional (2–4 weeks) | Mainly with lamotrigine |
| Levetiracetam | Very effective | Very effective | Effective | Very effective | No | Optional (1–2 weeks) | None |
| Lamotrigine | Exaggerates in 50% | Very effective | Very effective | Probably effective | Yes | Mandatory (6–8 weeks) | Many |
| Topiramate | Probably effective | Very effective | Weakly effective | Undetermined | Yes | Mandatory (6–8 weeks) | Many |