Forged by natural selective forces, bacterial pathogens have developed a variety of sophisticated strategies to colonize a host and to ensure survival. Colonization by bacterial pathogens requires specific mechanisms, since commensal microorganisms dominate almost every part of the inner and outer surfaces of the host. Pathogenic Escherichia coli, Legionella spp., or Brucella spp. developed mechanisms to invade the host epithelial cells, gaining access to the deeper tissue, including the lymphatic system or the blood. Many enteric pathogens such as Salmonella and Yersinia species specifically target M cells to access inner compartments. M cells are differentiated cells that are scattered over the dome region of the Peyer's patches, a specialized lymphoid follicle of the intestinal tract. Once internalized by the M cells, the fate of the Salmonella and Yersinia species is different and depends on dedicated groups of virulence factors specific for each organism. Yersinia blocks phagocytosis by macrophages during the early phase of infection. Salmonella is phagocytosed but then prevents phagosome-lysosome fusions, allowing the bacterium to reside and replicate within the vacuole. Other pathogens, like Helicobacter pylori, remain extracellular with less frequent contact with the host epithelial cells. These more short-lived encounters are used to trigger signals to the host cell that interfere with basic cellular processes and may ultimately contribute to disease.
A commensal bacterium may evolve into a pathogen over considerable time. Bacterial evolution is not only a continuous process but may occur through the acquisition of segments of DNA by a process of horizontal transfer (8, 24, 25, 34). This term describes the acquisition of DNA from an unknown source such as a phage and integration into the chromosome or by insertion of large plasmids by homologous recombination. Blocks of newly integrated DNA, designated "islands," can encode a variety of functions such as iron-uptake systems, metabolic enzymes, or cell-specific adhesins. Pathogenicity islands (PAIs) are recognizable by unusual GC content and codon usage that suggest their origin from foreign sources. Other characteristics of PAIs are their instability, their integration at specific loci (for example, within tRNA genes), and the presence of direct repeats at the flanking regions. After initial acquisition, PAIs may be continuously optimized according to the requirements of the recipient in an amelioration process. Mobilization factors and cryptic genes are usually lost after inactivation by point mutations, and the GC content and the codon usage are progressively adapted to that of the host chromosome; thus, the relative age can be inferred by computational methods.
The cag Pathogenicity Island of H. pylori
H. pylori is a gram-negative spiral-shaped human pathogen that colonizes the antrum and the corpus of the stomach. Many virulence factors have been described in the past decade. These factors enable the bacterium to survive in the extreme acidic environment of the gastric tract, to reach the more neutral environment of the mucous layer, and to resist the human immune response, resulting in persistence. Most of the infections occur during childhood, with only a minority of all infections progressing to pathological states.
In this chapter we will focus on the functions that are encoded by a 40-kb chromosomal region with the features of a PAI (1, 9, 54). The H. pylori PAI was originally named cag (cytotoxin-associated gene) since it was thought to be associated with expression of the vacuolating toxin (VacA). However, it was later shown that both factors, VacA and the PAI, are independent of each other, even though cag-negative strains often do not express VacA (53). Interestingly, biopsies from patients with severe gastric diseases including chronic active gastritis, peptic ulcer disease, mucosa-associated lymphoid tissue lymphoma, and gastric cancer contain the cagA gene in more than 90% of all the cases, establishing a direct correlation of the presence of the cagA gene with disease (7, 13, 15). On the genetic level, the cag PAI is flanked by a 31-bp direct repeat, which contains the recombination site and corresponds to the last nucleotides of the glr gene (9) (Fig. 1). The module also forms the core of the left and right ends of an insertion sequence common in H. pylori, the IS605 element. Depending on the strain, different numbers of insertion sequences are associated with the PAI. Often, however, the PAI is split into subregions, cagI and cagII, which are interrupted by two IS605 elements. Between the two insertion sequences, intervening DNA is present. As a consequence, strains with many insertions often resemble type II strains (less virulent) more than type I (more virulent, PAI-containing) strains with respect to virulence attributes. Additional genetic instability of cag results from deletions and inversions (27). In some cases, the cag PAI can even be lost completely due to DNA transfer of an empty site from a cag-deficient strain into a type I strain and subsequent homologous recombination (30).
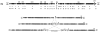
There are 31 open reading frames predicted within the cag region (Fig. 1). One of these open reading frames encodes the immunodominant antigen CagA, which is localized to the 3′ end of the island (12, 52). CagA was identified as the first protein of the PAI and appeared to be a major virulence factor (43). The molecular size of cagA is variable and depends on the number of repeats of a 102- to 108-bp motif that is repeated with specific strains. At the protein level, the absence of repeats corresponds to a molecular weight of 128 kDa, while every repeat up to a maximum of four repeats shifts the molecular weight of CagA by approximately 4 kDa (59). Comparison of strains with differences in the number of repeats isolated from patients with gastric cancer suggested that strains with more repeats were associated with higher levels of CagA antibody, more severe degrees of atrophy, and reduced survival in a low pH (pH 3). The repeats have a modular structure, which is a result of duplications and rearrangements of DNA from the central region of the cagA gene. However, the number, sequence, and position of the modules can vary, depending on the geographic region from which the strain was isolated. These local differences appear to be a result of frequent genetic recombination events and accumulation of mutations within the cagA gene that were maintained by geographic segregation. Besides CagA, the cag PAI encodes a helicobacter-specific type IV secretion system.
Type IV Secretion Systems and Their Functions
One of the most exciting research areas in microbial pathogenesis over the past decade has been the exploration of type III secretion systems. Many gram-negative pathogens including Salmonella, Yersinia, and Shigella species, pathogenic E. coli, and many plant pathogens use a type III system to export bacterial virulence proteins from the bacterial cytoplasm directly into the host cell (26, 31, 35). The exported molecules, called effectors, interfere with a variety of basic signaling pathways of the host cell (20). The effectors are species specific. We also know that at least one other secretion system, the type IV secretion system, is able to translocate virulence proteins into the host cells. It is important to note that the evolutionary origin of the two systems is different. Type III probably evolved from the flagellar system (37), while type IV systems originated from conjugative apparatuses (10, 11, 58). Whereas flagella are organelles of motility, the conjugation systems allow the exchange of DNA between bacteria. However, both systems also have important features in common. It is thought that both can transport proteins through a complex channel structure directly through the inner and outer bacterial membranes into the cytoplasm of the host cell. Furthermore, they use chaperones or chaperone-like molecules to stabilize the effectors and energize the protein export by ATP-hydrolysis; their polymerization is transient since it is induced upon cell contact. The variety of substrates and the secretion mechanism itself, however, seem to be less uniform for the type IV system. While type III systems exclusively export monomeric proteins, type IV machinery may export multisubunit and nucleoprotein complexes. In the case of the pertussis toxin, for example, sec-dependent transport through the inner membrane is coupled with type IV-driven transport through the outer membrane and substrate secretion occurs only into the supernatant.
Up to now, the group of pathogens encoding a type IV secretion system with demonstrated virulent functions is composed of five species: Agrobacterium tumefaciens (virB and virD4), Bordetella pertussis (ptl), Legionella pneumophila (icm/dot), Brucella suis (virB), and H. pylori (cag). Other type IV secretion systems with probable importance for pathogenesis were identified in the genome of Rickettsia prowazekii (3), Bartonella henselae, and Actinobacillus actinomycetemcomitans. The homologies among the Vir homologs in the different pathogens are mainly found in the structural core of the secretion apparatus. However, in contrast to type III secretion systems, the components of type IV systems are not interchangeable between the different bacterial species as shown by complementation experiments. Apart from the homologous components of the core, every system contains additional factors that are specific for each individual pathogen.
Our knowledge of the localization of the Vir homologs within the secretion system is mainly obtained from the extensive work done on A. tumefaciens. On the basis of present knowledge, we divide individual components in several classes. VirD4, VirB4, and VirB11 homologs are localized in the inner bacterial membrane and encode proteins with ATPase activity. VirD4 is also thought to couple protein–protein interactions. Further classes involve (i) transglycosidases (VirB1) that may create holes in the murein sacculus to allow putative pilus components to assemble on the surface of the bacteria, (ii) periplasmic scaffolds consisting of the lipoprotein VirB7 associated with VirB9, (iii) cytoplasmic chaperones that bind and stabilize the substrate (VirD1 as chaperone for VirD2), (iv) the VirB2 pilus subunit (32), and (v) the substrates for translocation. In Agrobacterium sp. the type IV secretion system is formed on the bacterial surface after induction with phenolic compounds and at low temperature (6, 22). Three substrates are known to be translocated into the plant cell. VirE2 binds the 5′-end of the T-DNA and is injected into the plant cell as a nucleoprotein particle. Inside the host cell, an additional translocated protein, the single-strand DNA-binding protein VirE2, binds to the T-DNA and protects it from degradation. VirE2 and the F-box protein VirF are translocated independently of DNA and have a function within the plant cell that contributes to the tumor formation. This allows speculation about the origin of DNA transfer systems that might eventually have evolved from protein transfer systems. It was shown that the secretion signal for VirF and VirD2 is located in the C-terminal half of the proteins, where a common Arg-Pro-Arg motif was recognized (55). VirD2 and VirE2 carry nuclear targeting signals and mediate the transport of the T-DNA complex into the nucleus where the T-DNA integrates into the plant cell genome. The genetic information of the T-DNA is then translated into proteins, which interfere with the plant cell physiology and ultimately lead to plant cell tumors (crown gall).
As mentioned above, B. pertussis secretes the five-subunit pertussis toxin across the bacterial outer membrane into the supernatant using a type IV secretion apparatus (12, 14, 56). The toxin then binds to the receptor on the epithelial surface and is subsequently internalized. Inside the host cell, the toxin ADP-ribosylates heterotrimeric G proteins (Gi), thus interfering with receptor kinase signaling pathways.
The icm/dot genes of L. pneumophila, the causative agent of Legionnaire's disease and Pontiac fever, were identified during mutational studies to identify genes essential for intracellular growth and macrophage killing (49, 50). The substrates that are mediating these events are not yet described. The type IV secretion system of Legionella sp. contains only two genes, icmE and dotB, which share homology with the VirB homologs of Agrobacterium sp. Fourteen additional genes have homologs in bacterial conjugation systems, and not surprisingly, the Icm/Dot system has been shown to also mediate movement of the plasmid RSF1010. Thus, the type IV system of Legionella sp. has maintained both functions, one for DNA and one for protein export.
Homologs of most of the VirB proteins, including a VirB2 homolog, were recently discovered in B. suis (41). Mutants in virB5, virB9, or virB10 were highly attenuated in an in vitro infection model with human macrophages. This led to the conclusion that the virB region is essential for the intracellular survival and multiplication of B. suis (41). In H. pylori several homologs of VirB and VirD4 are present. These include the ATPases VirB4 (CagE) and VirB11 (HP525), which energize the transport of CagA and possible further substrates through the putative transenvelope channel. VirD4 (HP524) is localized in the inner membrane as well and might have a function in the substrate recognition. VirB7 (CagT) and VirB9 (HP528) are possibly located in the outer membrane. CagM might have a chaperone-like function, stabilizing VirB7 (33). VirB8 and VirB10 are probably part of the channel structure.
Pathogenic Functions of the cag Pathogenicity Island
When the genetic analysis of the cag PAI suggested the presence of a type IV secretion system, it was anticipated that this structure should mediate contact to the host cell and possibly induce signal transduction pathways that contribute to the virulence of H. pylori. At the present stage, we differentiate between two basic cag-mediated host cell signaling pathways that are induced upon attachment of H. pylori. One is induced by the immunodominant antigen of H. pylori, the CagA protein, while the second pathway is independent of CagA. Deconvolution microscopy, mutational analysis, and biochemical fractionation experiments have provided evidence that adherent H. pylori translocates CagA into host cells and that this process depends on a functional type IV secretion system (4, 5, 16, 42, 46, 51) (Fig. 2). Inactivation of all cag genes tested, except cagN, abolished CagA translocation.
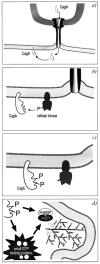
Inside the host cell, CagA becomes phosphorylated on a tyrosine residue by a yet unidentified host cell kinase (Fig. 2). Although c-src and crude host cell lysates were able to tyrosine-phosphorylate CagA in vitro, the significance of this finding under in vivo conditions remains unclear (4). Cell fractionations showed that the majority of intracellular CagA is found in the insoluble fraction, while a significant amount was associated with host cell membranes. This finding suggests that, following phosphorylation of tyrosine residues, CagA becomes part of an insoluble and probably membrane-associated complex inside the host cell. The CagA tyrosine-phosphorylation site was mapped to an EPIYA motif in the C-terminal half of the protein (Stein et al., unpublished observations). EPIYA is located immediately upstream and within the 102-bp repeat region in the C-terminal part of the protein.
Although the function of CagA on the molecular level is still unknown, it was suggested that CagA is involved in the rearrangements of the actin cytoskeleton, which results in the formation of cup-like structures underneath the adherent bacteria (47, 51). On the molecular level, those changes are mediated by activation of Arp2/3 complexes via small GTPases and N-WASP or N-WASP-like proteins (57). Listeria sp., Shigella sp. (19), and vaccinia virus use variations of this mechanism to move on the tip of actin tails within the host cells (36). Attaching and effacing pathogens, including enteropathogenic and enterohemorrhagic E. coli, remain extracellular and cause actin rearrangements in a similar manner through the host cell membrane (28, 29). The resulting protrusions on the cell surface (pedestals) somehow resemble the cup-like structures induced by H. pylori, although they are clearly different structures.
A second kind of change in the host cell morphology also depends on CagA translocation. A type I wild-type strain, but not the cagA isogenic mutant, induced a scatter factor-like phenotype, which was called the "hummingbird'' phenotype and probably results from activation of small GTPases of the Rho family. It was also shown that cag-positive strains affect gastric epithelial cell proliferation, diminish cell viability, and attenuate apoptosis in vivo (Fig. 3). The fact that cag interferes with the cell cycle might offer a possible explanation for the increased risk of gastric cancer (44). Whether these events also depend on CagA or on additional injected components is still not yet understood.

Among the CagA-independent pathways, the induction of interleukin-8 (IL-8) secretion by gastric epithelial cells was the first signaling event that was connected to infection with type I strains (18). Mutational analysis demonstrated that most of the cag genes were essential for increased IL-8 secretion. Only four cag mutants, cagA, cagF, cagN, and the virD4 homolog did not show any difference in their IL-8 levels when compared to the wild-type strain (9, 17, 18, 39, 48).
IL-8 is the major chemokine that causes the infiltration of neutrophils to the infection site. The result of these processes is a strong inflammatory response in the gastric mucosa. Since H. pylori is one of the few pathogens that chronically infect the host, a continuous inflammation may thus be an important factor for the development of gastric disease. Two cag-dependent pathways were described that lead to the induction of IL-8 secretion in H. pylori. The first one depends on the activation of transcription factor nuclear factor-kappa B (NF-κB) via p21-activated kinase 1 (PAK1). PAK1 associates with and phosphorylates NF-κB interacting kinase (NIK), which then leads to the degradation of the inhibitory subunit IκBa from the NF-κB complex. The released NF-κB dimerizes and enters the nucleus, where it binds and positively regulates the IL-8 promoter. cag mutants that do not activate NF-κB also fail to induce IL-8 secretion (21, 23). A second transcription factor that positively regulates the IL-8 promoter is activator protein 1 (AP-1) (40). The molecular mechanism of AP-1 activation is initiated through small GTPases of the Rho family that activate ERK/MAP kinase cascades. Pathways further downstream involve MKK4 and JNK, and their activation results in phosphorylation of c-Jun and Elk-1 (38, 40). Elk-1 then upregulates c-fos transcription. Homo- and heterodimers of the proto-oncogenes c-Fos and c-Jun finally form a complex with AP-1 that can bind the IL-8 promoter and induce transcription of IL-8. The bacterial molecules that are mediating these inflammatory events are not yet identified. We can imagine that either the contact of the type IV system with the host cell is sufficient to trigger the response or the type IV secretion system exports a factor that mediates the IL-8 secretion.
Recent observations characterized the involvement of the cag PAI in phagocytosis of H. pylori. Mononuclear phagocytes were shown to engulf H. pylori within approximately 4 minutes, but the internalized bacteria were not killed if type I strains were used for the infection. Instead, H. pylori induced homotypic phagosome fusions leading to the formation of large vacuoles. These so-called megasomes contained multiple organisms that were viable for at least 24 h. The process was not induced by cag-negative mutants and was dependent on intact microtubules and bacterial protein synthesis (2). In a different study, a functional type IV secretion system and de novo protein biosynthesis, but not CagA, were required to block phagocytosis through polymorphonuclear leukocytes and monocytes. This effect resembled antiphagocytosis by Yersinia enterocolitica and prevented the engulfment not only of H. pylori but also of latex beads and cells of Neisseria gonorrhoeae (45). This finding suggests that CagA is not the only molecule that is injected into the host cell, but that further cag-encoded factors are also translocated and may be responsible for induction of the antiphagocytotic response.
Conclusions
The function of the cag PAI in H. pylori-associated disease has been the subject of controversy ever since its discovery in 1993. Clinical studies with different outcomes have been performed to test whether the presence of the cag PAI is connected with peptic ulcer disease or gastric cancer. Often these studies were based on PCR amplifications of the cagA gene, which was used as a marker for the cag PAI. In some of these studies, clear proof of the involvement of cag in severe forms of gastric diseases often was not provided. The problem in some of these studies was that cag was examined as a static DNA region (cagA present or not) rather than as a DNA sequence encoding a sophisticated secretion apparatus with the function of injecting virulence factors into the host cell (Fig. 4). The work of various laboratories during the past year therefore has provided better insight into the biological complexity of H. pylori-host interactions and the molecular function of the cag PAI in pathogenesis. Future approaches have to consider that the presence of the cagA gene alone is not equal to virulence. A single point mutation anywhere in the 40-kbp region may abolish the function of the type IV secretion system and CagA transloction into the host cell (Fig. 4).
We may assume that the majority of type I strains are de facto less virulent due to an abrogated function of the secretion system or of the cagA gene. Additionally, we have to consider that bacteria are just one term of the equation. No matter how many virulence factors they possess, the host has receptors and targets for these factors and allelic polymorphism may dictate resistance. If we restrict our attention to the group of patients with peptic ulcer disease and who are infected with H. pylori, we will find that more than 90% of the individuals carry type I strains. On the basis of the nature of the cag-encoded factors, it is most likely that the translocation of virulence proteins including CagA through the type IV secretion system is one important molecular mechanism by which H. pylori can, in the long term, influence the clinical outcome of the infection.
Acknowledgments
We thank C. Montecucco and W. Pansegrau for helpful suggestions. We also acknowledge S. Guidotti for the unpublished observations on cag deletion. We gratefully acknowledge G. Corsi for the illustrations and C. Mallia for editorial assistance.
References
- 1.
- Akopyanz N., Clifton S. W., Kersulyte D., Crabtree J. E., Youree B. E., Reece C. A., Bukanov N. O., Drazek E. S., Roe B. A., Berg D. E. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol. Microbiol. 1998;28:37–53. [PubMed: 9593295]
- 2.
- Allen L. A., Schlesinger L. S., Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J. Exp. Med. 2000;191:115–128. [PMC free article: PMC2195807] [PubMed: 10620610]
- 3.
- Andersson S. G., Zomorodipour A., Andersson J. O., Sicheritz-Ponten T., Alsmark U. C., Podowski R. M., Naslund A. K., Eriksson A. S., Winkler H. H., Kurland C. G. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. [PubMed: 9823893]
- 4.
- Asahi M., Awuma T., Ito S., Ito Y., Suto H., Nagai Y., Tsubokawa M., Tohyama Y., Maeda S., Omata M., Suzuki T., Sasakawa C. Helicobacter pylori CagA protein can be tyrosine phosphoylated in the gastric epithelial cells. J. Exp. Med. 2000;191:593–602. [PMC free article: PMC2195829] [PubMed: 10684851]
- 5.
- Backert S., Ziska E., Brinkmann V., Zimmy-Arndt U., Fauconnier A., Jungblut P. R., Naumann M., Meyer T. F. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell. Microbiol. 2000;2:155–164. [PubMed: 11207572]
- 6.
- Baker B., Zambryski P., Staskawicz B., Dinesh-Kumar S. P. Signaling in plant-microbe interactions. Science. 1997;276:726–733. [PubMed: 9115193]
- 7.
- Blaser M. J. Helicobacter pylori and gastric diseases. Br. Med. J. 1998;316:1507–1510. [PMC free article: PMC1113159] [PubMed: 9582144]
- 8.
- Blum G., Ott M., Lischewski A., Ritter A., Imrich H., Tschape H., Hacker J. Excision of large DNA regions termed pathogenicity islands from tRNA-specific loci in the chromosome of an Escherichia coli wild-type pathogen. Infect. Immun. 1994;62:606–614. [PMC free article: PMC186147] [PubMed: 7507897]
- 9.
- Censini S., Lange C., Xiang Z., Crabtree J. E., Ghiara P., Borodovsky M., Rappuoli R., Covacci A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA. 1996;93:14648–14653. [PMC free article: PMC26189] [PubMed: 8962108]
- 10.
- Christie P. Agrobacterium tumefaciens T-complex transport apparatus: a paradigm for a new family of multifunctional transporters in eubacteria. J. Bacteriol. 1997;179:3085–3094. [PMC free article: PMC179082] [PubMed: 9150199]
- 11.
- Christie P. J. The cag pathogenicity island: mechanistic insights. Trends Microbiol. 1997;5:264–265. [PubMed: 9234504]
- 12.
- Covacci A., Censini S., Bugnoli M., Petracca R., Burroni D., Macchia G., Massone A., Papini E., Xiang Z., Figura N., Rappuoli R. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA. 1993;90:5791–5795. [PMC free article: PMC46808] [PubMed: 8516329]
- 13.
- Covacci A., Falkow S., Berg D. E., Rappuoli R. Did the inheritance of a pathogenicity island modify the virulence of Helicobacter pylori? Trends Microbiol. 1997;5:205–208. [PubMed: 9160510]
- 14.
- Covacci A., Rappuoli R. Pertussis toxin export requires accessory genes located downstream from the pertussis toxin operon. Mol. Microbiol. 1993;8:429–434. [PubMed: 8326857]
- 15.
- Covacci A., Rappuoli R. Helicobacter pylori: molecular evolution of a bacterial quasi-species. Curr. Opin. Microbiol. 1998;1:96–102. [PubMed: 10066468]
- 16.
- Covacci A., Rappuoli R. Tyrosine-phosphorylated bacterial proteins: Trojan horses for the host cell. J. Exp. Med. 2000;191:587–592. [PMC free article: PMC2195833] [PubMed: 10684850]
- 17.
- Crabtree J. E., Kersulyte D., Li S. D., Lindley I. J., Berg D. E. Modulation of Helicobacter pylori induced interleukin-8 synthesis in gastric epithelial cells mediated by cag PAI encoded VirD4 homologue. J. Clin. Pathol. 1999;52:653–657. [PMC free article: PMC501539] [PubMed: 10655985]
- 18.
- Crabtree J. E., Xiang Z., Lindley I. J., Tompkins D. S., Rappuoli R., Covacci A. Induction of interleukin-8 secretion from gastric epithelial cells by a cagA negative isogenic mutant of Helicobacter pylori. J. Clin. Pathol. 1995;48:967–969. [PMC free article: PMC502959] [PubMed: 8537502]
- 19.
- Egile C., Loisel T. P., Laurent V., Li R., Pantaloni D., Sansonetti P. J., Carlier M. F. Activation of the CDC42 effector N-WASP by the Shigella flexneri lcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell. Biol. 1999;146:1319–1332. [PMC free article: PMC2156126] [PubMed: 10491394]
- 20.
- Falkow S. Who speaks for the microbes? Emerg. Infect. Dis. 1998;4:495–497. [PMC free article: PMC2640304] [PubMed: 9716983]
- 21.
- Foryst-Ludwig, A., and M. Naumann. 2000. PAK1 activates the NIK-IKK NF-kappaB pathway and proinflammatory cytokines in H. pylori infection. J. Biol. Chem. epub ahead of print. [PubMed: 11016939]
- 22.
- Fullner K. J., Lara J. C., Nester E. W. Pilus assembly by agrobacterium T-DNA transfer genes. Science. 1996;273:1107–1109. [PubMed: 8688097]
- 23.
- Glocker E., Lange C., Covacci A., Bereswill S., Kist M., Pahl H. L. Proteins encoded by the cag pathogenicity island of Helicobacter pylori are required for NF-kappaB activation. Infect. Immun. 1998;66:2346–2348. [PMC free article: PMC108202] [PubMed: 9573128]
- 24.
- Groisman E. A., Ochman H. Pathogenicity islands: bacterial evolution in quantum leaps. Cell. 1996;87:791–794. [PubMed: 8945505]
- 25.
- Hacker J., Blum-Oehler G., Muhldorfer I., Tschape H. Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol. Microbiol. 1997;23:1089–1097. [PubMed: 9106201]
- 26.
- Hueck C. J. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 1998;62:379–433. [PMC free article: PMC98920] [PubMed: 9618447]
- 27.
- Jiang Q., Hiratsuka K., Taylor D. E. Variability of gene order in different Helicobacter pylori strains contributes to genome diversity. Mol. Microbiol. 1996;20:833–842. [PubMed: 8793879]
- 28.
- Kalman D., Weiner O. D., Goosney D. L., Sedat J. W., Finlay B. B., Abe A., Bishop J. M. Enteropathogenic E. coli acts through WASP and Arp2/3 complex to form actin pedestals. Nat. Cell. Biol. 1999;1:389–391. [PMC free article: PMC2828053] [PubMed: 10559969]
- 29.
- Kenny B., DeVinney R., Stein M., Reinscheid D. J., Frey E. A., Finlay B. B. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell. 1997;91:511–520. [PubMed: 9390560]
- 30.
- Kersulyte D., Chalkauskas H., Berg D. E. Emergence of recombinant strains of Helicobacter pylori during human infection. Mol. Microbiol. 1999;31:31–43. [PubMed: 9987107]
- 31.
- Kubori T., Matsushima Y., Nakamura D., Uralil J., Lara-Tejero M., Sukhan A., Galan J. E., Aizawa S. I. Supra-molecular structure of the Salmonella typhimurium type III protein secretion system. Science. 1998;280:602–605. [PubMed: 9554854]
- 32.
- Lai E. M., Kado C. I. Processed VirB2 is the major subunit of the promiscuous pilus of Agrobacterium tumefaciens. J. Bacteriol. 1998;180:2711–2717. [PMC free article: PMC107224] [PubMed: 9573157]
- 33.
- Lange, C., A. Covacci, and R. Rappuoli. 1999. The CagT of cag, the pathogenicity island of Helicobacter pylori, is a lipoprotein that assembles into a core system and requires CagM for stabilization. Submitted for publication.
- 34.
- Lee C. A. Pathogenicity islands and the evolution of bacterial pathogens. Infect. Agents Dis. 1996;5:1–7. [PubMed: 8789594]
- 35.
- Lee C. A. Type III secretion systems: machines to deliver bacterial proteins into eukaryotic cells? Trends Microbiol. 1997;5:148–156. [PubMed: 9141189]
- 36.
- Loisel T. P., Boujemaa R., Pantaloni D., Carlier M. F. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. [PubMed: 10524632]
- 37.
- Macnab R. M. Genetics and biogenesis of bacterial flagella. Annu. Rev. Genet. 1992;26:131–158. [PubMed: 1482109]
- 38.
- Meyer-Ter-Vehn T., Covacci A., Kist M., Pahl H. L. Helicobacter pylori activates mitogen-activated protein kinase cascades and induces expression of the proto-oncogenes c-fos and c-jun. J. Biol. Chem. 2000;275:16064–16072. [PubMed: 10747974]
- 39.
- Munzenmaier A., Lange C., Glocker E., Covacci A., Moran A., Bereswill S., Baeuerle P. A., Kist M., Pahl H. L. A secreted/shed product of Helicobacter pylori activates transcription factor nuclear factor-kappa B. J. Immunol. 1997;159:6140–6147. [PubMed: 9550415]
- 40.
- Naumann M., Wessler S., Bartsch C., Wieland W., Covacci A., Haas R., Meyer T. F. Activation of activator protein 1 and stress response kinases in epithelial cells colonized by Helicobacter pylori encoding the cag pathogenicity island. J. Biol. Chem. 1999;274:31655–31662. [PubMed: 10531374]
- 41.
- O'Callaghan D., Cazevieille C., Allardet-Servent A., Boschiroli M. L., Bourg G., Foulongne V., Frutos P., Kulakov Y., Ramuz M. A homologue of the Agrobacterium tumefaciens VirB and Bordetella pertussis Ptl type IV secretion systems is essential for intracellular survival of Brucella suis. Mol. Microbiol. 1999;33:1210–1220. [PubMed: 10510235]
- 42.
- Odenbreit S., Puls J., Sedlmaier B., Gerland E., Fischer W., Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. [PubMed: 10688800]
- 43.
- Parsonnet J., Friedman G. D., Orentreich N., Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut. 1997;40:297–301. [PMC free article: PMC1027076] [PubMed: 9135515]
- 44.
- Peek R. M., Blaser M. J., Mays D. J., Forsyth M. H., Cover T. L., Song S. Y., Krishna U., Pietenol J. A. Helicobacter pylori strain-specific genotypes and modulation of the gastric epithelial cell cycle. Cancer Res. 1999;59:6124–6131. [PubMed: 10626802]
- 45.
- Ramarao N., Gray-Owen S. D., Backert S., Meyer T. F. Helicobacter pylori inhibits phagocytosis by professional phagocytes involving type IV secretion components. Mol. Microbiol. 2000;37:1389–1404. [PubMed: 10998171]
- 46.
- Segal E. D., Cha J., Lo J., Falkow S., Tompkins L. S. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. USA. 1999;96:14559–14564. [PMC free article: PMC24475] [PubMed: 10588744]
- 47.
- Segal E. D., Falkow S., Tompkins L. S. Helicobacter pylori attachment to gastric cells induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins. Proc. Natl. Acad. Sci. USA. 1996;93:1259–1264. [PMC free article: PMC40067] [PubMed: 8577751]
- 48.
- Segal E. D., Lange C., Covacci A., Tompkins L. S., Falkow S. Induction of host signal transduction pathways by Helicobacter pylori. Proc. Natl. Acad. Sci. USA. 1997;94:7595–7599. [PMC free article: PMC23867] [PubMed: 9207137]
- 49.
- Segal G., Russo J. J., Shuman H. A. Relationships between a new type IV secretion system and the icm/dot virulence system of Legionella pneumophila. Mol. Microbiol. 1999;34:799–809. [PubMed: 10564519]
- 50.
- Segal G., Shuman H. A. How is the intracellular fate of the Legionella pneumophila phagosome determined? Trends Microbiol. 1998;6:253–255. [PubMed: 9717210]
- 51.
- Stein M., Rappuoli R., Covacci A. Tyrosine-phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc. Natl. Acad. Sci. USA. 2000;97:1263–1268. [PMC free article: PMC15590] [PubMed: 10655519]
- 52.
- Tummuru M. K., Cover T. L., Blaser M. J. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect. Immun. 1993;61:1799–1809. [PMC free article: PMC280768] [PubMed: 8478069]
- 53.
- Tummuru M. K., Cover T. L., Blaser M. J. Mutation of the cytotoxin-associated cagA gene does not affect the vacuolating cytotoxin activity of Helicobacter pylori. Infect. Immun. 1994;62:2609–2613. [PMC free article: PMC186552] [PubMed: 8188385]
- 54.
- Tummuru M., Sharma S. A., Blaser M. J. Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Mol. Microbiol. 1995;18:867–876. [PubMed: 8825091]
- 55.
- Vergunst A. C., Schrammeijer B., Dulk-Ras A., de Vlaam C. M. T., Regensburg-Tuink T. J. G., Hooykaas P. J. J. VirB/D4-dependent protein translocation from agro-bacterium into plant cells. Science. 2000;290:979–982. [PubMed: 11062129]
- 56.
- Weiss A. A., Johnson F. D., Burns D. L. Molecular characterization of an operon required for pertussis toxin secretion. Proc. Natl. Acad. Sci. USA. 1993;90:2970–2974. [PMC free article: PMC46218] [PubMed: 8464913]
- 57.
- Welch M. D. The world according to arp: regulation of actin nucleation by the Arp2/3 complex. Trends Cell. Biol. 1999;9:423–427. [PubMed: 10511705]
- 58.
- Winans S. C., Burns D. L., Christie P. J. Adaptation of a conjugal transfer system for the export of pathogenic macromolecules. Trends Microbiol. 1996;4:64–68. [PMC free article: PMC4848025] [PubMed: 8820569]
- 59.
- Xiang Z., Censini S., Bayeli P. F., Telford J. L., Figura N., Rappuoli R., Covacci A. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect. Immun. 1995;63:94–98. [PMC free article: PMC172962] [PubMed: 7806390]
Publication Details
Author Information and Affiliations
Authors
Markus Stein, Rino Rappuoli, and Antonello Covacci.Affiliations
Copyright
Publisher
ASM Press, Washington (DC)
NLM Citation
Stein M, Rappuoli R, Covacci A. The cag Pathogenicity Island. In: Mobley HLT, Mendz GL, Hazell SL, editors. Helicobacter pylori: Physiology and Genetics. Washington (DC): ASM Press; 2001. Chapter 31.



