Clinical Description
Mandibulofacial dysostosis with microcephaly (MFDM) is a multiple malformation syndrome comprising craniofacial skeletal anomalies, microcephaly, developmental delay / intellectual disability, abnormalities of the ears and hearing, and, in some instances, extracranial malformations (esophageal atresia, congenital heart defects, thumb anomalies), and/or short stature.
To date, 126 individuals have been identified with a pathogenic variant in EFTUD2 [Gordon et al 2012, Lines et al 2012, Need et al 2012, Luquetti et al 2013, Voigt et al 2013, Lehalle et al 2014, Deml et al 2015, Gandomi et al 2015, Sarkar et al 2015, Smigiel et al 2015, Huang et al 2016, Vincent et al 2016, Bick et al 2017, Matsuo et al 2017, McDermott et al 2017, Rengasamy Venugopalan et al 2017, Williams et al 2017, Paderova et al 2018, Yu et al 2018, Lacour et al 2019, Silva et al 2019, Wu et al 2019]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Mandibulofacial Dysostosis with Microcephaly: Frequency of Select Features
View in own window
| Feature | % of Persons
w/Feature | Comment |
|---|
Facial
structural
differences
| Malar hypoplasia | 92% | |
Micrognathia / Mandibular
hypoplasia | 93% | |
| Cleft palate | 43% | |
| Choanal atresia | 30% | |
| Facial asymmetry | 58% | |
|
Microcephaly
| 87% | Occipitofrontal circumference ≥2 SD below mean |
|
Developmental delay / Intellectual disability
| 97% | Severity varies (may be mild, moderate, or severe; critical sequelae (e.g. neonatal airway compromise, cardiac anomalies) may affect developmental outcome. |
Ear
malformations
& hearing loss
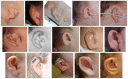
| Microtia / Dysplastic pinna(e) | 97% | |
| Auditory canal atresia or stenosis | 68% | |
| Preauricular tag | 50% | |
| Hearing loss | 83% | |
|
Other findings
| Cardiac anomalies | 35% | Typically atrial &/or ventricular septal defect |
| Thumb anomalies | 34% | Typically proximally placed; uncommonly, preaxial polydactyly or hypoplasia |
Esophageal atresia /
Tracheoesophageal fistula | 33% | |
| Short stature | 30% | |
| Spine anomalies | 28% | Incl scoliosis, kyphosis, hemivertebrae, & cervical segmentation anomalies |
| Epilepsy | 26% | |
Mandibulofacial dysostosis is characterized by malar and maxillary hypoplasia.
Accompanying findings in MFDM include micrognathia/mandibular hypoplasia, cleft palate, and/or choanal abnormality.
Cleft palate in MFDM occurs as a Pierre Robin sequence, characterized by a midline bony defect without accompanying cleft lip. Submucous cleft has also been described. Choanal atresia is generally osseous, being either unilateral or bilateral; choanal stenosis is also frequent.
Zygomatic arch cleft has been identified in ten of 19 individuals assessed (best done with cranial CT with 3-D reconstruction).
Characteristic dysmorphic features (), distinct from those seen in the other mandibulofacial and acrofacial dysostoses (see Differential Diagnosis), are recognizable by early childhood. In addition to malar and maxillary hypoplasia, microcephaly, and the typical ear anomalies described in this section, features include metopic ridge, prominent glabella, broad nasal bridge with prominent ridge and bulbous tip, large oral aperture, everted lower lip, and/or (frequently) facial asymmetry.
Typical craniofacial features of MFDM. These include micrognathia, malar hypoplasia, a relatively high nasal root with prominent ridge, everted lower lip, and (frequently) facial asymmetry. Characteristic ear malformations, present in essentially all (more...)
Microcephaly is present in about 87% of reported individuals (n=33; median -3.5 SD; range -0.2 SD to -6.5 SD) [Huang et al 2016]. Cephalic growth curves for MFDM are published [Huang et al 2016]. In some instances, individuals have exhibited apparent cephalic "catch-up" growth, resulting in a normal adult occipitofrontal circumference despite microcephaly in childhood. Individuals whose head circumference falls within the normal range have also been reported to have intellectual disability [Luquetti et al 2013, Lehalle et al 2014].
Developmental delay and/or intellectual disability are present in almost all individuals. Among 30 persons on whom data are available, the degree of intellectual disability was reported as "mild" (~40%), "moderate" (~50%), or "severe" (~10%) [Gordon et al 2012, Lines et al 2012, Luquetti et al 2013, Voigt et al 2013].
Affected children are ambulatory but show delayed motor development, taking first steps at a median age of 26 months (n=38; range 13-60 months) [Huang et al 2016].
Among those who are verbal, the median reported age at first words is 27 months (n=32; range 12 months to 5.6 years); some affected persons remain nonverbal into adult life [Huang et al 2016]. Assessment of language skills may be confounded by the presence of hearing loss and/or cleft palate.
To date there have been no detailed or cross-sectional studies of long-term neuropsychological outcomes in MFDM. Developmental data in the few affected adults identified to date suggest a broad range of outcomes, with some affected persons achieving semi-independent living with paid employment [Huang et al 2016], whereas others are nonverbal and require extensive assistance with daily activities [Authors, unpublished data].
Ear malformations and hearing loss
External ear malformations. External ears are anomalous in virtually all affected individuals. Typical findings (see ) include microtia (grades I-III), deficiency of the superior helix and antihelix, preauricular tags, and auditory canal atresia/stenosis. The posterior-inferior margin of the lobule may have a right-angle ("squared-off") configuration.
Hearing loss. Hearing loss is typically conductive (~60%) as opposed to sensorineural or mixed, and is likely to result from malformation or absence of the middle ear ossicles, auditory canal atresia, or both.
Other relatively common findings
Cardiac anomalies are present in 35% of individuals. Hemodynamically insignificant atrial and ventricular septal defects are the most common; tetralogy of Fallot, patent ductus arteriosus, and aortic arch abnormalities (e.g., coarctation) have also been reported [
Need et al 2012,
Lehalle et al 2014].
Thumb anomalies (proximally placed, duplicated, or hypoplastic thumbs) are seen in about 35% of individuals.
Esophageal atresia / tracheoesophageal fistula (EA/TEF) is present in about 35% of affected individuals. EA/TEF is typically type C (the most common type), in which the upper esophageal pouch ends blindly and the lower esophageal pouch connects abnormally to the trachea (distal tracheoesophageal fistula). Laryngotracheal anomalies (tracheomalacia, posterior laryngotracheoesophageal clefts) may be seen in association with EA/TEF. It may be suspected antenatally because of polyhydramnios or absent stomach echolucency, or neonatally in the context of unexplained respiratory distress and/or failed nasogastric tube placement.
Short stature is present in 30% of individuals. Height growth curves for MFDM are published [
Huang et al 2016].
Spine anomalies include scoliosis, kyphosis, hemivertebrae, and cervical segmentation anomalies.
Epilepsy is present in 26% of individuals. Detailed clinical data regarding the type of epilepsy have not been specifically reported.
Matsuo et al [2017] report one individual with recurrent seizures for which EEG demonstrated occasional spike discharges originating from the right frontal area.
Additional malformations (low frequency)