

NCBI Multiple Sequence Alignment Viewer Embedding API
- Introduction
- Simple Steps to Getting the Embed Code
- Including MSA Viewer on a web page
- Data Formats Supported
- MSA Viewer Parameters
- Events Sent by MSA Viewer
- Size and position parameters
- Embedding Demos
Introduction
The NCBI Multiple Sequence (MSA) Alignment Viewer is a general purpose tool for viewing multiple alignments of biological sequences. It can be embedded in a wide variety of web pages and with a large number of options. This document serves as a launching point to understand how to embed the Multiple Sequence Alignment Viewer in any context. For purposes of standardization in this document, all links presented include the common refrain https://www.ncbi.nlm.nih.gov.
For questions please contact the NCBI Help Desk.
Additional documentation can be found at: http://www.ncbi.nlm.nih.gov/tools/msaviewer
Simple Steps to Getting the Embed Code
This embedding workflow requires almost no JavaScript programming experience.
- Pick an alignment of interest and view it on the web version of MSA Viewer (https://www.ncbi.nlm.nih.gov/projects/msaviewer/).
- Set an anchor sequence/consensus if desired and adjust the coloring scheme.
- Use the "Link to View" option to show the URL, short-link URL, and embedding code.
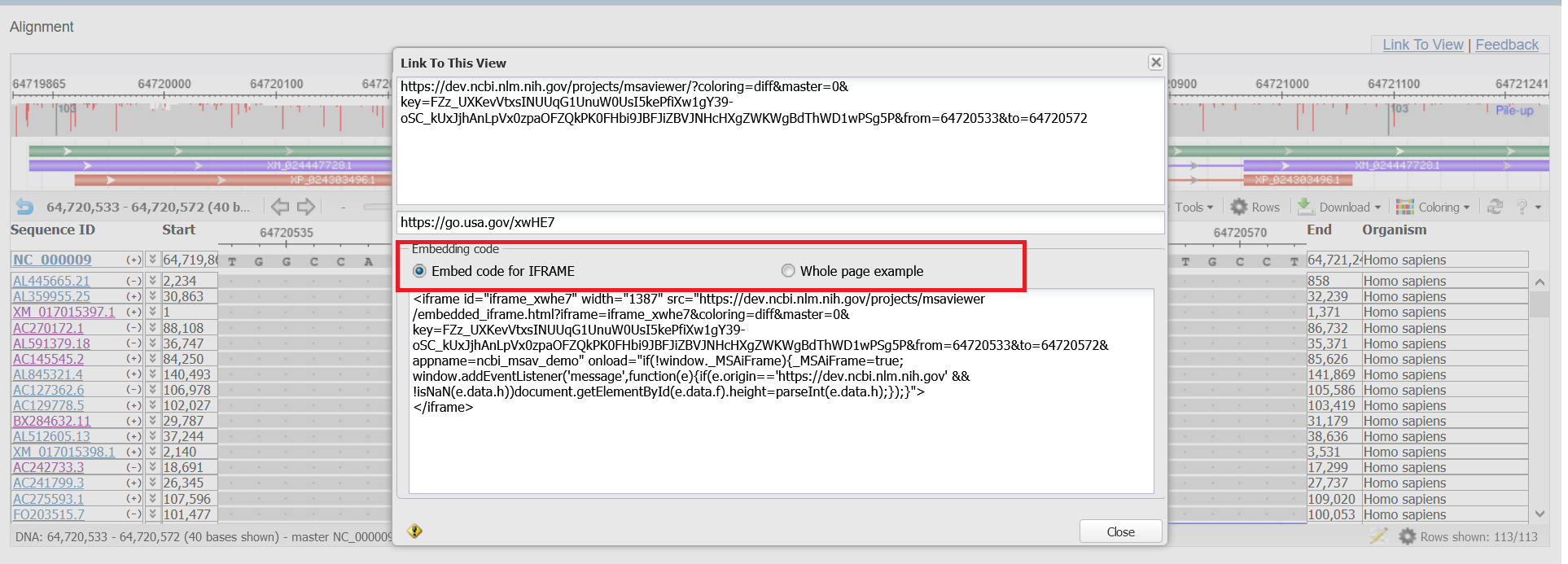
There are two types of embedding code. The first option, ‘Embed code for IFRAME’, can be used as is to embed the MSA Viewer by itself as an IFRAME. The second option,‘Whole page example,’ can be used to embed the MSA Viewer as a part of your page. This option has both the script in the HEAD element and DIV element with parameters.
Including MSA Viewer on a web page
The Multiple Sequence Viewer is a component which can be embedded on internal NCBI as well as external Web page.
The Viewer is implemented using ExtJS JavaScript library version 5.0.1. There are some rules to follow when including the Viewer as a component on a page.
-
There is no need to declare or load ExtJS css files or js scripts. MSAV dynamically loads everything it needs from www.ncbi.nlm.nin.com website. On the other hand if ExtJS scripts are declared/loaded before multialign.js declaration MSAV omits their loading so this allows an embedding application to use a specific edition of the library (compatible to version 5.0.1). The following example demonstrate how to declare MSAV script:
<script type="text/javascript" src="https://www.ncbi.nlm.nih.gov/projects/msaviewer/js/multialign.js"></script> -
If an embedding application does not create MSAV instances dynamically MSAV application finds appropriate MultiAlignViewerApp declarations and create instances automatically (with data loading). For this the ‘div’ element should have class='MultiAlignViewerApp' and extra attribute ‘data-autoload’, and ‘a’ element inside ‘div’ with ‘href’ attribute containing MSA Viewer parameters. An example is:
<div id='some-id' class='MultiAlignViewerApp' data-autoload> <a href='?embedded=true&id=…'><a> </div>Please pay attention to the question mark at the beginning of ‘href’ argument. It is a syntax dictated by Web browsers and is obligatory.
Do not use this method if the Multiple Sequence Alignment Viewer div is initially hidden - it breaks all the logic as the Viewer needs to have real coordinate for proper rendering. Use method from point 3 to instantiate Multiple Sequence Alignment Viewer dynamically.
-
If an embedding application needs to create MSAV instances dynamically the following example demonstrates how to declare them and initiate MSAV instances creation and loading:
<div id=’msav1’></div> ... <script type='text/javascript'> MultiAlignViewOnReady(function() { var app = new MultiAlignView.App(‘msav1’); app.load(‘embedded=true&id=…’); } ); </script> -
Function MultiAlignViewOnReady(callback, [scope]) is designed to prevent MSAV usage before MultiAlignView class is initialized.
Parameters: callback - user callback function scope - callback function scope (if necessary)
Data Formats Supported
NCBI ASN.1 data containing alignment objects.
BLAST RID – request id of submission of a sequence to NCBI BLAST service.
MUSCLE – external format (to be implemented), result of run of MUSCLE tool ( https://www.drive5.com/muscle/ ).
MSA Viewer Parameters
Parameters may be passed to Multiple Sequence Alignment Viewer by putting them in the included <a href=''> link tag within the MSA Viewer <div> tag.
appname – it’s a way to identify who is using the component. It’s a mandatory parameter which should be unique for your application. If you plan to embed MSA Viewer on your page please email us at sviewer-service AT ncbi.nlm.nih.gov with suggested load so that we can plan our resource allocation.
embedded - defines the mode of MSA Viewer. Value 'true' does not include panorama view, value 'full' includes it. It is highly recommended to use this parameter as it changes processing of environment (e.g. it does not interpret URL string as a set of parameters) and provides better isolation of MSA Viewer component.
One of the following parameters MUST be present as a source of alignment data:
id – id of the sequence which has alignment data, can be gi or accession
rid – BLAST RID for already finished job
url – URL pointing to the data file
key – key for the data stored at NCBI after user data upload. Usually visible in URL produced by applications
There are two alternative ways to set a visible range for the view:
f – from, 1-based position of the beginning of the view
t – to, 1-based position of the end of the view
or
v – view range, 1-based range in the format from:to
master – the id of a row to be selected as anchor
collapse_pan – adding this parameter (without values) hides annotated features under MSA Panorama View
coloring – coloring method, abbreviated value, see setColoring below
coloring_opt – options for coloring methods (currently implemented only for "cons" method). Conservation method uses two colors – red and blue – to designate highly conserved or less conserved regions. The threshold between these colors is defined by this options and is expressed as bit score. To set this option strings ‘one_bit’, ‘two_bits’, ‘three_bits’, ‘four_bits’, ‘five_bits’, ‘identity’ are used. The value defines the “information content” of the column threshold. It is calculated by the formula \sum_{i} (f(i) * log_2 (f(i)/q(i)), where i is over all residue types, f(i) is observed residue frequency, and q(i) is background/relative residue frequency which is defined by BLOSUM62 matrix. The Identity setting uses red font only in columns that contain the same residue in all of the sequence rows displayed. All other aligned columns are colored in blue and unaligned columns are shown in grey .
datasource - multiple values, 'id', 'local' separated by comma. E.g. datasource=local,id - first look for sequences/annotations in the provided ASN.1 file, if not available check ID. datasource=id,local - try to get data from ID, if not available use the local file. datasource=local - use the alignment ASN.1 file as the only datasource.
identical - true/false value defining should we show bases/AAs identical to consensus or anchor as letters. Default - false, meaning we show bases/AAs identical to consensus as dots.
consensus - true/false value defining whether we show consensus when anchor is not set. Default - true.
cds_alignment - true/false value, provides viewer with additional info about alignment and that we should color non-synonymous changes. Default - false.
columns - allows to re-arrange columns including adjusting column width, disabling some default columns, enabling optional ones and changing columns order.
Format examples:
- columns=d:80,b:30,e:30,o:100 - adjusting widths of default columns
- columns=e:0,b:0,is,cntr:80 - disables default columns “Start” and “End”, enables optional columns “Isolation Source” with default width and “Country”
- columns=d,o:100,cd,aln,h - adding “aln” column to the list allows to change columns order
The letters corresponding to the default columns as follows: d - Sequence ID, b - Start, e - End, o – Organism. Optional columns can be designated by the following acronyms/letters: cd – Collection Date, h – Host, is – Isolation Source, cntr – Country, pi - Identity, c - Coverage, mm - Mismatches, gs - Gene symbol. Special column – aln stands for the graphical section of alignments.
sort - allows to sort data by column in ascending or descending order. Format: sort=column_name:sort_order, where column_name is one of the following: d - Sequence ID, b - Start, e - End, o - Organism; sort_order: a - ascending, d - descending, sort_order may be omitted with default = a. Examples: sort=o:d – descending sort by organism sort=b – ascending sort by Start
Events Sent by MSAV
An application that uses MSAV can subscribe to events that MSAV will send in case of a user interaction with MSAV. Here is the list of supported events with parameters passed in each event.
'graphical_image_loaded' - fired when graphical image is loaded to view view – view object of the just loaded image
'visible_range_changed' - fired when visible range is changed (for example, zoom or shift) range – array with [from, to, len] of the sequence or alignment visible
'selection_changed' - fired when the selection is changed rows – array of row ids selected
'master_changed' - fired when the anchor is set/unset row_id – row id of new anchor or null if anchor is unset
'coloring_changed' - fired when coloring of the alignment is changed coloring – what specific coloring is applied
Functions
MultiAlignView.App.getApps() – get array with all app objects
MultiAlignView.App.findAppByIndex(num) – find an app by its ordinal number
MultiAlignView.App.findAppByDivId(id) – find an app instantiated in div with given id
App Methods
reload(params) - reloads the application with MSA Viewer Parameters
getRows(fSelected) – get array of all or only selected rows
selectRow(row_id, fSelect) – select or unselect row with row_id
mapCoords(coords, source, target, callback) - translates an array of coordinates from a source alignment to a target alignment, where: coords - array of translated coordinates, source - current anchor ID or string "alignment" if no anchor is set, target - target alignment row ID, callback(response) - a callback function processing results of mapping, where response is an array of translated coordinates.
getMaster – get currently set anchor or null if unset
setMaster(row_id) – set anchor
getRowInfo(row_id) – returns object with following fields (can be extended in the future): id – sequence id for the row, start – start of the sequence in the alignment, end – end of the sequence in the alignment
setColoring(coloring) – set coloring, one of the following:
Value Description:
disable - Disable alignment score coloration
fbd – DNA/Protein: Score residues based on their frequency in the column
diff – DNA/Protein: Score residues based on match/mismatch to a master/anchor row
na – DNA: Assign color based on residue (A – Red, G – Blue, C – Yellow, T – Green)
cq_na – DNA: Score residues based on how well a particular residue agrees with the others in a column
cq_aa – Protein: Score residues based on how well a particular residue agrees with the others in a column.
rasmol – Protein: Used by the application RasMol to group amino acids with similar properties
shapely – Protein: Used by the application RasMol to group amino acids with similar shapes using a standard color table
blosum45 – Protein: Matrix made by matblas from blosum45.iij BLOSUM Clustered Scoring Matrix in 1/3 Bit Units
blosum62 – Protein: Matrix made by matblas from blosum62.iij BLOSUM Clustered Scoring Matrix in 1/2 Bit Units
blosum80 – Protein: Matrix made by matblas from blosum80.iij BLOSUM Clustered Scoring Matrix in 1/2 Bit Units
hydropathy – Protein: Side chain hydropathy, corrected for solvation
membrane – Protein: Membrane-buried preference parameters
signal – Protein: Signal sequence helical potential
size – Protein: Amino acid size
cons – Displays degrees of conservation among aligned residues
nss - nonsynonymous substitution. Colors triplets of nucleotides which translate into different amino-acid than consensus or anchor sequence. For this coloring method to work sequences participating in an alignment should be complete CDSs starting with start codon, and consensus should be coded unambiguously. When this parameter present, menu item 'Nonsynonymous substitutions' is added to 'Coloring' menu.
getColoring() – returns current coloring
getColoringMethods() - returns an available list the coloring methods
setColumns(params) - re-arranges columns with params described in MSA Viewer Parameters.
Size and position parameters
You can specify the height of the MSAV view.
Fixed height
Define CSS value height either directly in the MSAV div element:
<div id='some-id' class='MultiAlignViewerApp' data-autoload style='height:400px'>
<a href='?embedded=true&...'></a>
</div>
or in a CSS class
<style type='text/css'>
.my-msa-view {height: 400px;}
</style>
...
<div id='some-id' class='MultiAlignViewerApp my-msa-view' data-autoload>
<a href='?embedded=true&...'></a>
</div>
Adjustable height
Specify CSS values max-height and min-height directly
<div id='some-id' class='MultiAlignViewerApp' data-autoload style='min-height:200px;max-height:500px'>
<a href='?embedded=true&...'></a>
</div>
or via a class:
<style type='text/css'>
.my-msa-view {min-height: 200px;max-height:500px}
</style>
...
<div id='some-id' class='MultiAlignViewerApp my-msa-view' data-autoload>
<a href='?embedded=true&...'></a>
</div>
Max margin to the bottom of the page
Add data-bottom attribute as the margin in pixels (you can, optionally, specify min-height as well):
<div id='some-id' class='MultiAlignViewerApp' data-autoload data-bottom=80 style='min-height:200px'>
<a href='?embedded=true&...'></a>
</div>
Embedding Demos
Interaction with Sequence Viewer Demo
Table of Contents
- Multiple Sequence Alignment Viewer application
- Documentation home
- General
- Help
- Demo Pages
- Related Resources